
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and multi-scale properties of PuO2 nanoparticles: recent advances and open questions
Matthieu
Virot
 *a,
Thomas
Dumas
b,
Manon
Cot-Auriol
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
*a,
Thomas
Dumas
b,
Manon
Cot-Auriol
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
aICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: matthieu.virot@cea.fr
bCEA, DEN, DMRC, Univ Montpellier, Marcoule, France
First published on 24th October 2022
Abstract
Due to the increased attention given to actinide nanomaterials, the question of their structure–property relationship is on the spotlight of recent publications. Plutonium oxide (PuO2) particularly plays a central role in nuclear energetics and a comprehensive knowledge about its properties when nanosizing is of paramount interest to understand its behaviour in environmental migration schemes but also for the development of advanced nuclear energy systems underway. The element plutonium further stimulates the curiosity of scientists due to the unique physical and chemical properties it exhibits around the periodic table. PuO2 crystallizes in the fluorite structure of the face-centered cubic system for which the properties can be significantly affected when shrinking. Identifying the formation mechanism of PuO2 nanoparticles, their related atomic, electronic and crystalline structures, and their reactivity in addition to their nanoscale properties, appears to be a fascinating and challenging ongoing topic, whose recent advances are discussed in this review.
1. Introduction
During the last few decades, nanocrystalline oxide materials attracted outstanding attention given their potential applications and related innovations in most of the modern technological domains.1–6 Their nanoscale dimensions, with a characteristic length between 1 and 100 nm, have often been demonstrated to give rise to specific physico-chemical properties in comparison to their bulk counterparts. Useful developments and applications have come out for instance in catalysis, solar cells, sensors, coatings and magnetic data storage. The control and optimization of the nanomaterial properties have been reported to strongly depend on the nanoparticle size, shape and local structure also explaining the strong interest for uncommon morphologies such as nano-rods, -ribbons, -tubes, etc.7,8 Size-dependent properties have been reported for nanomaterials exhibiting dimensions in the nanometer range, typically below 10 nm. Shrinking the materials within these dimensions provides a much higher contribution of surface atoms in comparison to bulk ones (surface to volume ratio, Fig. 1). As a consequence, an increase of the specific surface area is observed in addition to structural disorder reflected by a higher density of edge and corner sites thus explaining the often observed particular activity. Structural defects, strains and distortion can therefore contribute to the modification of the nanomaterial properties, so as the increasing surface curvature radius that may also play a significant role at the interface including for instance adsorption or group transfer reactions.9,10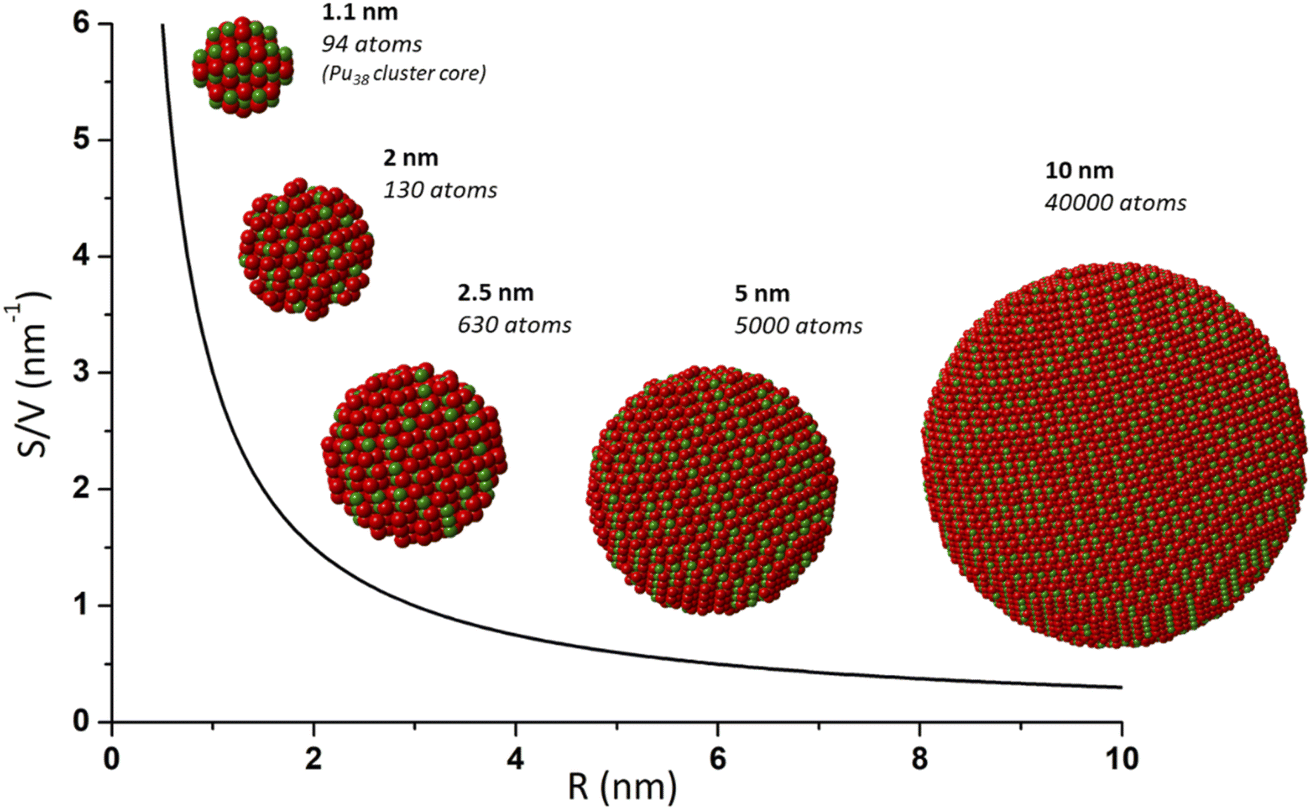 | ||
| Fig. 1 Scheme illustrating the increasing contribution of surface atoms (surface to volume ratio) toward a shrinking nanoparticle. This effect contributes to providing specific properties to a nanoparticle and is generally observed for a size lower than 10–20 nm. Nanoparticles and calculations of atoms (Pu in green and O in red) were generated with crystal maker software from bulk PuO2 and Pu38 CIF file from Sigmon et al.16 | ||
Almost all of the s-, p- and d- elements of the periodic table have been reported to have a nanotechnological impact with some of them (∼30) in high demand for the preparation of nanostructured materials.11 Similarly, the control of nanoparticle synthesis and the related structure–property effects are well described in the literature for transition metal oxides. Actinide-based nanoparticles and the relationship of their size vs. their physico-chemical properties and reactivity are far less explored in comparison.12–14 This assessment can be mostly attributed to the radioactive nature and high chemical toxicity of the elements of the actinide series, which involves difficulties in accessing and handling these elements but also results from safety rules appearing much more restrictive than with any other chemical elements. Nevertheless, significant advances have been revealed in the recent literature. The increased attention given to actinide nanoparticles and nanomaterials essentially results from environmental issues owing to their undisputable presence in the geosphere and their recently confirmed mobility, but also to their fundamental screening and technological interest related to the nuclear fuel cycle including fuel preparation, recycling and its related behaviour under operation.12 Furthermore, it has been demonstrated that the performance of heterogeneous catalysts based on actinide oxides is highly dependent on the nanoscale dimensions of the material.10 Important developments dealing with nanomaterials of the f-block elements have also focused on their detection, separation and recovery for therapeutics and bioimaging.15
Among the actinide series, the element plutonium arouses particular interest for military applications, space conquest and industrial nuclear power applications. Plutonium is an anthropogenic element almost exclusively formed in nuclear reactors with all of its isotopes being strongly radioactive. It is worth noting that very small amounts of stable 244Pu (t1/2 = 81 million years) naturally exist on earth as a result of the rapid neutron capture process that occurs during nucleosyntheses (r-process). Neutron capture reactions resulting from the spontaneous fission of natural 238U are also reported to generate naturogenic 239Pu.17,18 The fissile 239Pu (t1/2 = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110 years) is the most common isotope formed by neutron capture from 238U in typical nuclear power reactors, which further presents an energetic interest and enters into the composition of MOX (Mixed Oxide) fuel after separation and recycling steps. MOX is a ceramic made out of a mixture of depleted uranium and plutonium dioxides both crystallizing in the fluorite structure of the face-centered cubic system (Fm
110 years) is the most common isotope formed by neutron capture from 238U in typical nuclear power reactors, which further presents an energetic interest and enters into the composition of MOX (Mixed Oxide) fuel after separation and recycling steps. MOX is a ceramic made out of a mixture of depleted uranium and plutonium dioxides both crystallizing in the fluorite structure of the face-centered cubic system (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group).19,20 Current MOX fuel used in some PWR reactors has a plutonium mass level (Pu/(U+Pu)) lower than 10%, whereas future nuclear reactors, such as sodium-cooled fast reactors, would burn more Pu, necessitating a 30% mass level in nuclear fuel.21 Current fuel preparation, based upon the precipitation and calcination of oxalate precursors, can lead to localized fuel heterogeneities responsible for the decrease of the fuel performance during operation (release of fission gas, fuel swelling, dislocation, etc.) but also to difficulties in its reprocessing due to enriched local areas in Pu which already require drastic chemical conditions to be dissolved. Much effort is currently focused on the preparation of an advanced nuclear fuel in which the control of the arrangement of the materials at the atomic scale would tailor specific properties, particularly for compacting, sintering or dissolving steps.14,22–25 Recently, the report of the first synthesis of nanocrystalline MOX U1−xPuxO2(+y) (up to x = 46) has been described by Kauric et al. offering interesting perspectives for the preparation of a homogeneous nuclear fuel.26 A recent survey clearly demonstrates the challenges and viable nanotechnological options exhibiting a positive and direct impact on nuclear performance and safety.25,27
m space group).19,20 Current MOX fuel used in some PWR reactors has a plutonium mass level (Pu/(U+Pu)) lower than 10%, whereas future nuclear reactors, such as sodium-cooled fast reactors, would burn more Pu, necessitating a 30% mass level in nuclear fuel.21 Current fuel preparation, based upon the precipitation and calcination of oxalate precursors, can lead to localized fuel heterogeneities responsible for the decrease of the fuel performance during operation (release of fission gas, fuel swelling, dislocation, etc.) but also to difficulties in its reprocessing due to enriched local areas in Pu which already require drastic chemical conditions to be dissolved. Much effort is currently focused on the preparation of an advanced nuclear fuel in which the control of the arrangement of the materials at the atomic scale would tailor specific properties, particularly for compacting, sintering or dissolving steps.14,22–25 Recently, the report of the first synthesis of nanocrystalline MOX U1−xPuxO2(+y) (up to x = 46) has been described by Kauric et al. offering interesting perspectives for the preparation of a homogeneous nuclear fuel.26 A recent survey clearly demonstrates the challenges and viable nanotechnological options exhibiting a positive and direct impact on nuclear performance and safety.25,27
Microstructural defects and fissuring are known to occur in the nuclear oxide fuel during burn-up. In addition, neutron irradiation and temperature gradients may provoke significant fuel restructuring with the formation of nanometric grains and micrometric pores referred to as high-burn up structures (HBS).28,29 This nanoscale organization is mostly situated in the ceramic rim and has been demonstrated to increase in thickness with the burning rate. Nanostructuration also goes with lattice parameter modifications and the accumulation of point defects.28,30,31 HBS have been reported to be formed earlier in Pu enriched areas and their close surroundings in comparison to UO2.28,32 The fundamental question of the mechanism (polygonization or recrystallization) responsible for this nanoscale restructuring process remains open.28,29 Since experimental investigations on nuclear fuel are technically difficult to perform, the possible preparation of HBS simulants using reproducible and controlled actinide oxide nanoparticles as a starting material represents an undisputable interest.25,29,30,33–35 Furthermore, very high burn-ups will be considered for advanced nuclear fuels and recent publications discuss the advantages of nanocrystalline oxide fuels in comparison to conventional large-grain ceramics. A possible increase in fission gas retention in the HBS pores, increased thermal conductivity, and improved plasticity with greater tolerance to irradiation-induced mechanical damage are all mentioned.28,29,34,36,37 Nevertheless, the formation of stable colloidal PuO2 nanoparticles during hydrometallurgical processes may contribute to mass balance troubles and hinder some steps related to the preparation or recycling of nuclear fuel or the dismantling of nuclear facilities (safety, criticality, separation, mass balance, etc.). For instance, foaming has been reported as a result of colloid formation during the concentration step by evaporation as well as emulsification during solvent extraction.38
The mastering of nuclear energy by human beings through nuclear testing, space conquest, industrial development but also accidents (e.g. Fukushima), resulted in significant amounts of plutonium being released into the environment.17,39–41 The comprehensive survey provided by Geckeis et al. indicated that atmospheric and underground nuclear testing contributed to the release of more than 3.4 and 2.8 metric tons of Pu into the environment, respectively. From nuclear electricity production, the global inventory of plutonium reached 2630 metric tons in 2014, and 70–90 additional metric tons are expected each year. The management of such an amount has to be considered as well.17 “Hot particles” generally refer to radioactive contaminant particles ranging in size from sub-micrometers to fragments. Their speciation is highly dependent on the source and release scenario, but also on the weathering conditions. Hot particles resulting from explosions and fire are mostly composed of very poorly insoluble PuO2.17,41 The central role played by PuO2 in modern actinide chemistry is more precisely related to the recent evidence of the kilometre-scale transport of Pu in the environment through the formation of nanometric colloidal species.12,40–43 Pseudo-colloids consist of natural colloids of organic, inorganic or microbial origin, where Pu has been incorporated or adsorbed, whereas intrinsic colloids result from the exceptional property of Pu to form its own and extremely stable colloid through hydrolysis and subsequent condensation reactions, even under acidic conditions.40,42,44–46 The proximity of the redox potential of the three redox couples (Pu(V)/Pu(IV), Pu(VI)/Pu(V), Pu(IV)/Pu(III)) around 1.0 V/SHE provides the possibility of the element Pu to exist simultaneously under acidic to near neutral conditions in four different oxidation states (from Pu(III) to Pu(VI)) thus increasing its complex behaviour in aqueous solution. Until recent evidence, Pu was thought to be immobile in the environment due to its strong sorption properties and low solubility in aquatic systems. The presence of suspended colloidal particles in high concentrations may however exceed the thermodynamic solubility calculated for plutonium under such conditions and enhance its mobility in aquifers. The question of their transport processes under geochemical conditions and their interaction with the surroundings is also of particular interest.47,48 This assertion is strengthened by the reported high physical and chemical stability of Pu colloids even when increasing the ionic strength.49–51
Focusing on the structure, recent studies have demonstrated that intrinsic colloids exhibit a PuO2-like structure differing from their bulk counterparts in terms of local environment, structural disorder and a possible contribution of different oxidation states in the oxide.40,45 Pu speciation is strongly dependent upon the oxidation state, dispersion media and redox conditions. In addition, the contamination scenario and storage history have been suggested to significantly affect the formation and fate of PuO2 nanoparticles in the environment.39 There is clear evidence for the contribution of pseudo-colloids to the mobility of actinides in aquifers whereas migration through the formation of intrinsic colloids is not yet clear and this possibility has only been postulated.12,42,52,53 Nevertheless, the stabilization of Pu ions onto environmentally relevant mineral phases (e.g. muscovite, quartz, hematite) has been demonstrated to occur by the precipitation of PuO2-like nanoparticles thus strengthening the interest of the community.39,54–61 Because the migration ability of An(IV) colloids is highly related to the particle size, stability, and chemical reactivity, there is an obvious interest in their rigorous characterization.53 Evidence of Pu nanoparticles as colloidal or particle fractions has also been reported in oceans, seawaters and marine sediments.40,41,62–65 In natural seawaters, Pu generally shows a (+IV) oxidation state and exhibits a PuO2 colloid structure ranging from oxo–hydroxo species to more crystalline PuO2 colloidal particles which become denser and less soluble when aging.66,67 This overall environmental topic also includes the safety questions related to the potential intrusion and leaching behavior of water in the case of contact with the spent nuclear fuel under surface or deep underground storage.68–70
This analysis explains the current, ongoing and exciting research dealing with the synthesis and characterization of PuO2 nanoparticles. Understanding and predicting the behaviour of Pu nanoparticles in the environmental context or under storage conditions require identifying their formation mechanism, deciphering their structure and evaluating their reactivity. In accordance with the quickly evolving nano-world observed outside the nuclear sphere, reducing the size of plutonium oxide to the nanoparticle state must provide specific physico-chemical properties differing from the bulk counterpart ones. A better understanding of how plutonium oxide is made and how its structure and properties are linked at the nanoscale could have important scientific, technological, and environmental outcomes. Some of the global challenging topics requiring answers include the safe storage and corrosion of spent nuclear fuel, the decommissioning of polluted sites, the next generation of nuclear fuels and reactors, and the behaviour of radionuclides in the environment. The current review aims to focus on the recent relevant progresses observed in PuO2 nanoparticle syntheses and characterizations including the recently deciphered electronic, structural and physico-chemical properties. For additional information going from the fundamental aspects of plutonium chemistry to environmental issues and applications, the reader is encouraged to refer to the excellent and comprehensive overviews provided by other authors.12,17,40,42,45,71–73
2. Preparation of PuO2 nanoparticles and related formation mechanisms
2.1. General aspects and terminology
From an environmental perspective, it is worth noting that the International Atomic Energy Agency (IAEA) defines radioactive particles as “localized aggregates of radioactive atoms that give rise to an inhomogeneous distribution of radionuclides significantly different from that of the matrix background”.17,74 Three subcategories are discriminated according to their size: “fragments” are used for entities showing a diameter greater than 2 mm, “particle” when the size is between 0.45 μm and 2 mm, and “radioactive colloids or pseudo-colloids” when the size ranges between 1 nm and 0.45 μm. Radioactive particles observed under air conditions are classified according to their aerodynamic diameters from submicronic size to fragments. Particles smaller than 10 μm in size are considered respiratory.17,74The terminology used for nanomaterials is not straightforward even if several groups have worked towards a uniform approach for their description outside the nuclear sphere. Although nanomaterials are generally defined by nanoscale characteristics (generally in the 1–100 nm range), they can be categorized based on several parameters including their size, shape, origin, surface properties and composition, but also with respect to the concerned research field, authority or scientific community.75 In their technical IUPAC report, Gubala et al. provided a thorough overview of the proposed classifications for the nanomaterials.76 In general, nanomaterials refer to materials having a size between 1 and 100 nm for at least one dimension whose characteristics are often correlated with size-dependent properties.76,77 Nanoparticles may refer to nano-objects exhibiting nanoscale dimensions in all 3 directions (ISO and IUPAC definitions). When the lengths of the longest and shortest axes of the object differ significantly, other terms such as nanofiber, nanorod, nanotube, nanowire or nanoplate are recommended.75,76 The assembly of these nanoparticles may lead to the formation of nanostructured materials which have been defined by Gleiter as materials having a microstructure with a characteristic length scale in the order of a few nanometers, typically in the 1–10 nm range.78 In addition, nanoparticles can associate to form agglomerated or aggregated clusters of particles differing by the type of forces holding them together (weak or strong forces, respectively).76
The properties of nanomaterials are strongly dependent upon their size, shape, chemical composition and interfacial properties. The latter can particularly influence the interactions between the nanoparticles in the colloidal state and their surroundings dictating for instance their solubility, adsorption properties, or assembly.77 In consequence, it appears relevant to discriminate associated nanoparticles (agglomerated or aggregated nanoparticle precipitates) from colloidal dispersions which refer to a continuous phase in which mono- or polymolecular particles are dispersed and have at least in one direction a dimension ranging from the nm to the μm (IUPAC).79 In general, these particles do not settle as a result of mutual repulsion and Brownian movement.17 In the literature dedicated to An nanoparticles, both terms are often used interchangeably; their distinction appears important since it can create confusion and misunderstanding. The examination of reports dedicated to PuO2 nanomaterials, in conjunction with definitions available in the literature, allows for the classification of PuO2 nanoparticles into several categories, as shown in Fig. 2. The distinction between associated nanoparticles, nanostructured materials, and colloidal dispersions appears relevant since their nanoscale characteristics may give rise to different atomic arrangements and different properties as already evidenced for other elements.75,76,80 Focus is particularly given to Pu intrinsic colloids which have been demonstrated to exhibit a PuO2-like structure (also called polymers or eigen colloids in the literature) and result from the hydrolysis properties of Pu(IV) and its ability to form its own colloid. They differ from pseudo-colloids which are associated with the sorption of Pu onto or into natural colloids and are not the purpose of this review. Table 1 presents a survey related to the synthesis approaches, properties and characterizations reported in the literature for PuO2 nanoparticles.
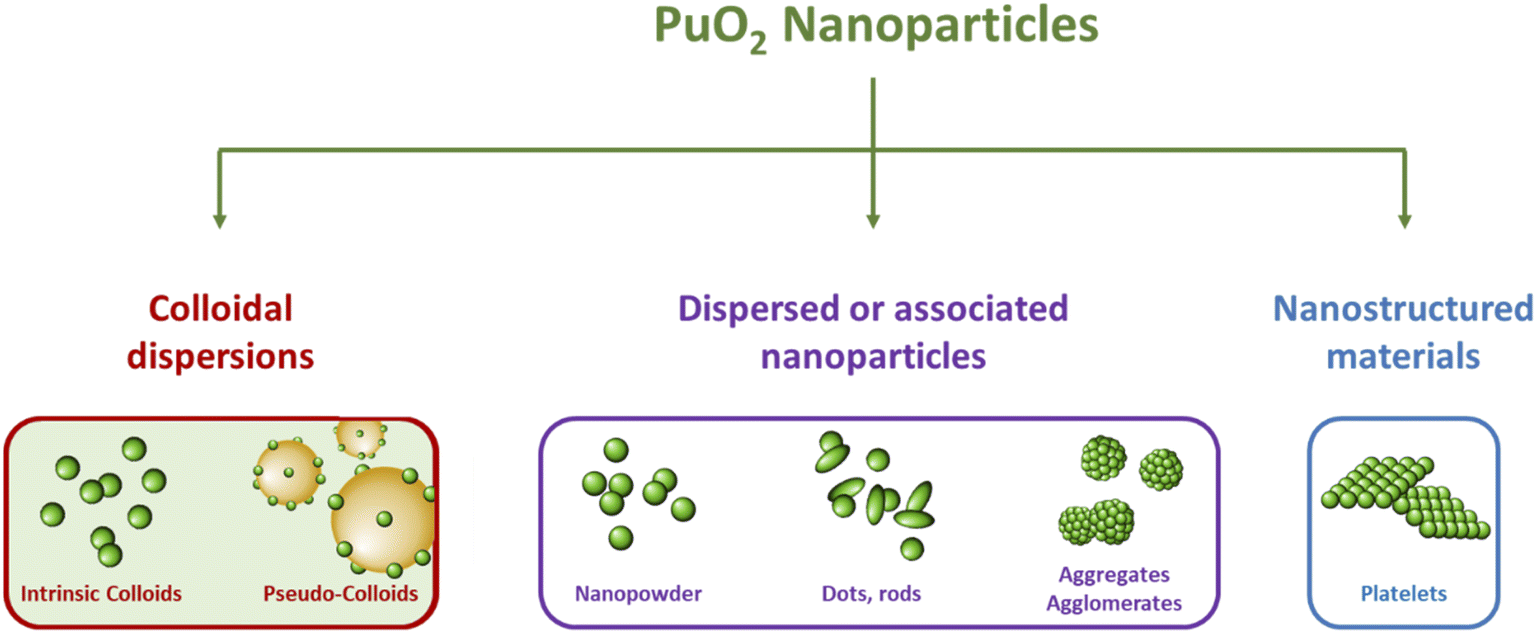 | ||
| Fig. 2 Scheme illustrating the diversity of PuO2 nanoparticles reported in the literature with their classification into three subcategories. | ||
| NPs | Synthesis approach (characteristics of NPs) | Used precursors (experimental conditions) | Particle diameter sizes (lattice parameter) | Characterization techniques | Isotopy (when given) | Ref. |
|---|---|---|---|---|---|---|
| a RT: room temperature, PEG: polyethylene glycol, BnOBn: benzyl ether, OA: oleic acid, OAm: oleylamine, XRD: X-ray diffraction, (HR-)TEM: (high-resolution) transmission electron microscopy, HERFD-XANES: X-ray absorption near edge structure in high energy resolved fluorescence detection mode, EXAFS: extended X-ray absorption fine structure, HEXS: high energy X-ray scattering, AFM: atomic force microscopy, IR: infrared spectroscopy, UV-Vis: UV-visible absorption spectroscopy, SQUID: superconducting quantum interference device, SAXS: small angle X-ray scattering, NEXAFS: near-edge X-ray absorption fine structure, LIBD: laser-induced breakdown detection, ESI-MS: electrospray ionization-mass spectrometry, STXM: scanning transmission X-ray microscopy. SSVNIR: solid-state visible-near infrared reflectance spectroscopy, APS: autocorrelation photon spectroscopy. | ||||||
| Stable intrinsic colloids | Controlled hydrolysis | Pu(IV) in HNO3 solution (dilution in pure water) | 2.6 nm (SAXS) 2.9 nm (HR-TEM) | HR-TEM, UV-vis, SAXS Pu-L3 XANES Pu-L3 EXAFS O–K NEXAFS-STXM | Ref. 96: 96.9% 239Pu 2.98% 240Pu 0.04% 241Pu 0.06% 242Pu | 94 and 96 |
| Neutralization of an acidic solution | Addition of NaOH to a Pu(IV) solution in HCl | 5 nm (LIBD) | UV-vis, LIBD, ESI-MS Pu-L3 XANES Pu-L3 EXAFS | Ref. 115: 94.4% 239Pu 5.5% 240Pu 0.1% 241Pu | 46 and 115 | |
| Extraction of nitric acid by n-hexanol | Addition of n-hexanol to a Pu(IV) solution (NO3/Pu mole ratio = 1) | <10 nm (TEM) | TEM, UV-vis | — | 99 | |
| Ultrafiltration of a Pu(IV) solution | Ultrafiltration (10 kD) from Pu(IV) sol. In 1 M HClO4 | <5 nm (TEM) | HR-TEM, UV-vis Pu-L3 XANES, Pu-L3 EXAFS Pu-L3 HERFD-XANES Pu-M5 HERFD-XANES | — | 124 | |
| Liquid-state auto-radiolysis | Pu(III) solution | 32 nm | APS | — | 125 | |
| Liquid-state auto-radiolysis | Pu(VI) solution | 2.9 nm (HR-TEM) | HR-TEM, UV-vis | Ref. 97: 96.9% 239Pu 2.98% 240Pu 0.04% 241Pu 0.06% 242Pu | 97, 108 and 109 | |
| Sonochemistry | Sonication of PuO2 nanopowder (pure water, Ar/(10%)CO, 30 h) | 7.1 nm (HR-TEM) 5.7× > 30 nm2 (SAXS) | HR-TEM, UV-vis, SAXS Pu-L3 XANES Pu-L3 EXAFS O–K NEXAFS-STXM | Ref. 96: 96.9% 239Pu 2.98% 240Pu 0.04% 241Pu 0.06% 242Pu | 94 and 96 | |
| Dispersed or associated nanoparticles | Thermal decomposition (PuO2 nanopowder) | Pu(IV) hydroxides precipitated by adding PEG and ammonia (485 °C, 2 h) | 5.1 nm (XRD) 4.6 nm (HR-TEM) (5.4014 Å) | XRD, HR-TEM Pu-L3 XANES Pu-L3 EXAFS | 96.9% 239Pu 2.99% 240Pu 0.06% 241Pu 0.02% 238Pu 0.06% 242Pu | 116 |
| Hydrothermal decomposition in hot compressed water (agglomerated nanoparticles) | Pu(IV) oxalates (95 °C, 120 h, autogenic pressure 1–2 bar) | 3.7 nm (XRD) (5.404 Å) 2.6 nm (TEM) (5.397 Å) | XRD, TEM | — | 14, 118 and 126 | |
| Neutralization of acidic Pu solution (monodisperse PuO2) | Addition of NH3·H2O to a Pu solution (degree III, IV, V or VI) | 2.5 nm | XRD, HR-TEM, SAXS | 99.99% 239Pu 0.01% 238Pu | 112 | |
| Neutralization of acidic Pu solution (monodisperse PuO2) | Addition of NH3·H2O to a Pu solution (degree III, IV, V or VI) | 2.3–3.2 nm (HR-TEM) 1.6–2.4 nm (XRD) (5.398–5.407 Å) 1.3–2.07 nm (HEXS) (5.38–5.402 Å) | XRD, HR-TEM, HEXS Pu-L3 EXAFS Pu-L3 XANES Pu-L3 HERFD-XANES Pu-M4 HERFD-XANES | Ref. 111: 99.74% 242Pu | 111, 113 and 114 | |
| Solvolysis in BnOBn/OA/OAm mixture (mixture of nano-dots and -rods) | Thermal decomposition of [PuO2(NO3)2]·3H2O (280 °C, 30 min, N2) | 2 nm (XRD) 3.2(9) nm (TEM) (5.3940 Å) | XRD, TEM, IR, Raman UV-vis, SQUID Pu-L3 XANES Pu-L3 EXAFS | 97.7% 239Pu 2.2% 240Pu 0.1% (238Pu, 241Pu, 242Pu) | 117 | |
| Arc vaporization | Pu metal wire | 1 nm (TEM) | TEM | — | 127 and 128 | |
| Stabilization onto environmentally relevant mineral surfaces | Low concentration Pu solutions | 1.5 nm (HR-TEM) 1–10 nm (HR-AFM) | AFM, HR-TEM Pu-L3 XANES Pu-L3 EXAFS | — | 54 and 56 | |
| Solid-state auto-radiolysis | Pu(IV) and Pu(III) oxalates (auto-degradation in air at RT over several months) | Few nm | SEM, XRD, IR, SSVNIR | 94% 239Pu, 6% 240Pu | 120 | |
| Nanostructured oxides | Thermal decomposition of oxalate precursors | Pu(IV) or Pu(III) oxalates (485 °C, 2–12 h) | 5.2–200.0 nm (HR-TEM) 7.4–193.0 (XRD) (5.3938–5.4021 Å) | XRD, HR-TEM Pu-L3 XANES Pu-L3 EXAFS | 96.9% 239Pu 2.99% 240Pu 0.06% 241Pu 0.02% 238Pu 0.06% 242Pu | 116 |
2.2. Preparation of PuO2 colloidal nanoparticles
Kraus already discussed the hydrolytic behaviour of tetravalent plutonium during the Manhattan project and described the observation of an opalescent green solution when sodium hydroxide or ammonia was added to nitric and sulfuric Pu(IV) solutions.81 After centrifugation, the author reported the observation of a strong Tyndall cone suggesting the formation of a colloidal dispersion. The solutions of typical deep green colour are found to be very stable with reference to sedimentation. This has been prevented in industrial processing by maintaining plutonium flux in acidic media. From the plutonium chemistry perspective, the formation of such species is related to the intricate hydrolysis properties exhibited by Pu(IV) in aqueous solutions. This phenomenon can even occur under acidic conditions and also competes with the disproportionation properties of Pu(IV).45,82–84 The first step related to the formation of Pu(IV) hydrolytic species can be described with eqn (1). The degree of cation hydrolysis depends on the ionic potential of the cation (charge to radius ratio) and several parameters resulting from the solution media (pH, temperature, composition, etc.).72 The ripening of these species can lead to precipitates or stabilized nanoparticles (referred to as Pu(IV) intrinsic colloids) which can even form under very acidic conditions and at low concentrations (Pu > 10−6 M).46,72 The related condensation processes may occur through olation or oxolation reactions described in eqn (2) and (3). For −log[H+] = 0.6 and a 0.5 M ionic strength, Rothe et al. indicated that a plutonium solution is about 80% in the form of hydrolyzed species Pu(OH)3+ or Pu(OH)22+.83 They suggested that [Pu(OH)2]n2n+, possibly with a coordination number of 8 for Pu, contributes as a reactant in the formation of PunOp(OH)4n−2p(H2O)z (0 < p < 2n) oligomers that can further condensate to yield colloids exhibiting a Pu–O–Pu basic fcc structure whose order increases with the colloid size (eqn (4)).| [Pu(OH2)m]n+ → [Pu(OH2)m−h(OH)h](n−h)+ + hH+ | (1) |
| Pu–OH + Pu–OH2 → Pu–OH–Pu + H2O | (2) |
| Pu–OH + Pu–OH → Pu–O–Pu + H2O | (3) |
| 2Pu(OH)2(H2O)62+ + xH2O ↔ Pu2Op(OH)8−2p(H2O)6+p + 4H+ + (2 + x)H2O | (4) |
Pu colloids or precipitates formed in aqueous solution as a result of Pu(IV) hydrolysis have been long-time thought to be composed of polymeric and amorphous hydrous oxo-hydroxides until isolated single crystals of a Pu38 oxo-cluster were reported to exhibit similar UV-vis absorption spectra to the one acquired for Pu(IV) intrinsic colloids.46,72,85–87 The resolved structure for these species, which were obtained by the neutralization of acidic Pu(IV) solutions with various alkali hydroxides, corresponded to the formula [Pu38O56Cl54(H2O)8]14− including a distorted PuO2 core structure that differs from the ill-defined and often suggested hydrous polymers. Importantly, the PuO2-like nanocluster does not have Pu–OH moieties in its structure thus favouring a mechanism based on oxolation reactions for the condensation of Pu(IV).72,85 Recently, Sigmon and Hixon synthesized Pu oxide nanoclusters formed with 16, 22 and 38 Pu atoms capped with inorganic water and chlorine molecules limiting the formation of bigger particles. The structures of all of these clusters only contain Pu in the tetravalent state. Careful examination of the resolved structures demonstrated their relationship and the authors postulated that the bigger Pu38 cluster is formed with smaller Pu16 and Pu22 building blocks. More interestingly, all of these nanoclusters share a distorted PuO2 core in which Pu(IV) cations coordinate with 8 O atoms via μ4-oxo bonds (the related structures are shown in Fig. 3). The evidence for hydroxo groups in these nanoclusters (also including the oxohydroxo hexanuclear Pu(IV) entities (Pu6) and hydroxo-bridged Pu(IV) dimers88–92), and the exclusive presence of oxo-bridges in the Pu38 building block which shares structural similarities with Pu(IV) intrinsic colloids suggest that the latter could be formed by a succession of olation and oxolation mechanisms, most likely through the stacking of these low nuclearity polynuclear structures.
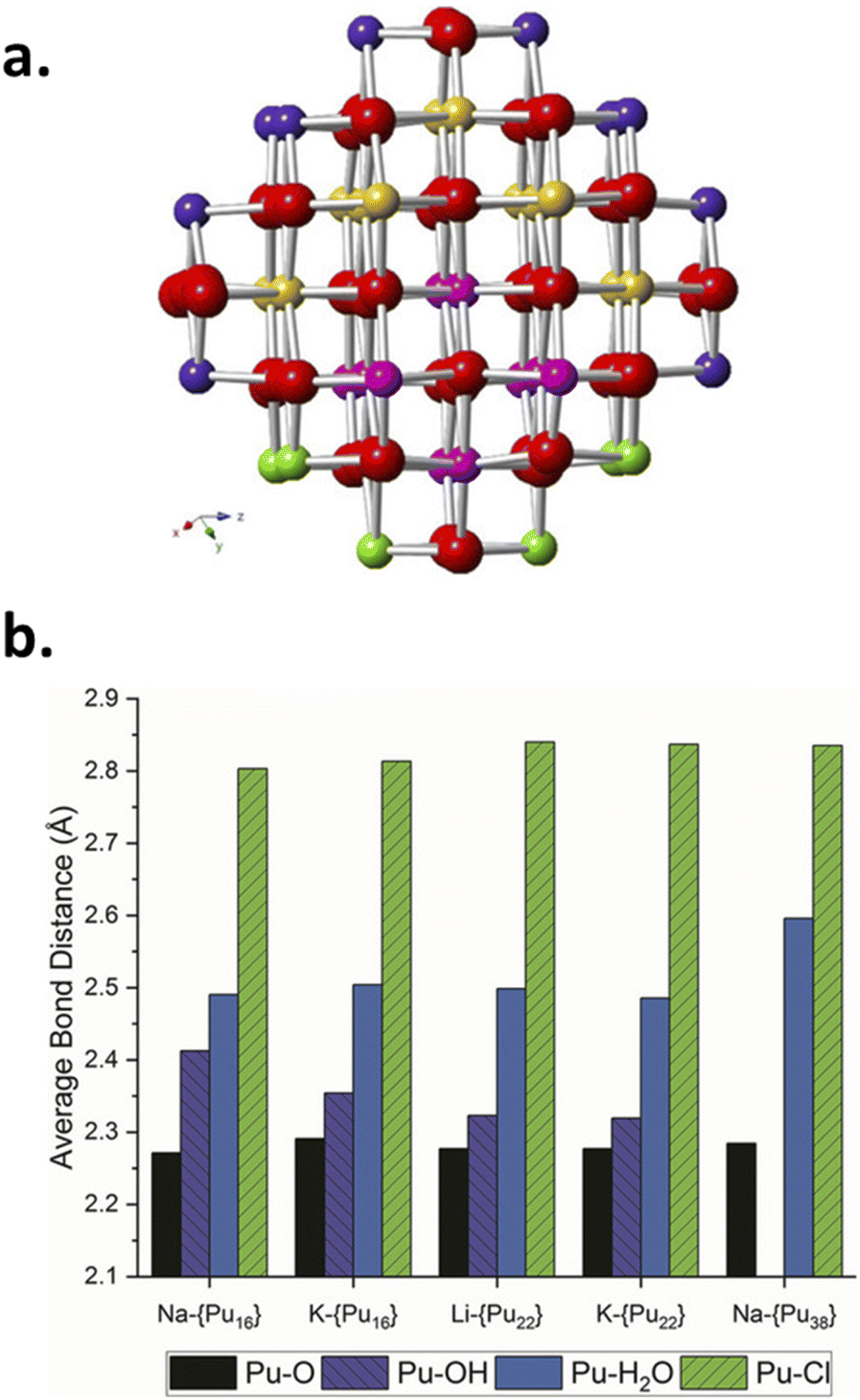 | ||
| Fig. 3 (a) Ball and stick model of the core of the Pu38 nanocluster postulated to be formed by smaller Pu16 and Pu22 nanoclusters. Red represents oxygen atoms whereas yellow, pink, purple and green are used for Pu. The Pu16 core is represented using pink and green Pu atoms whereas the Pu22 core is made with pink and yellow ones. (b) Diagram of the Pu–O, Pu–OH, Pu–H2O and Pu–Cl bond lengths observed in the Pu clusters evidencing the similar core for the various structures and the presence/absence of OH moieties in the structure of the smallest/largest cluster(s), respectively. Reproduced from Sigmon et al.16 with permission from John Wiley and Sons, copyright 2019. | ||
Several methods for producing stable dispersions of Pu(IV) intrinsic colloids have been proposed (Table 1).40,45 The simplest method involves diluting Pu(IV) solutions previously stabilized in acid media with water to cause an increase in the pH of the solution.38,93–96 Well-defined and spherical nanoparticles of 2.9 nm showing a PuO2-like crystal structure (using High-Resolution Transmission Electron Microscopy, HR-TEM) have been obtained using this approach (Fig. 4).97 Such synthesis conditions lead to a competition between hydrolysis (eqn (1) or eqn (5)) and disproportionation (eqn (6)–(8)) reactions for Pu(IV) that can influence the formation kinetics of the colloidal dispersions (along with solution pH or composition and Pu concentration). Typically, UV-Vis absorption spectroscopy can be used to evaluate the conversion of soluble plutonium ions into colloids. The spectral signature of the latter is characteristic and shows a strong absorption in the near UV range (<550 nm) attributed to Mie scattering and resulting from the presence of dispersed nanoparticles in the medium (Fig. 4). Other typical absorption bands can be found at λ = 579 nm, ca. 614 nm (large band), 684 nm and 736 nm. In comparison to aqueous ionic forms of Pu, intrinsic Pu(IV) colloids do not show sharp absorption bands and exhibit a very low molar extinction coefficient. As discussed previously by Lloyd and Haire, the spectrum of the colloid is retained when concentrating the solution by evaporation to dryness and is not significantly modified when suspending or peptizing the colloid after denitration.98,99 Disproportionation and redox reactions (eqn (6), slow step; eqn (7), rapid step; both reactions are summarized in eqn (8)) particularly yield Pu(VI) observed at ca. 830 nm in the UV-Vis absorption spectra, which is very often observed as a mixture with hydrolytic colloidal dispersions. Its concentration varies with time as colloids form and ripen. Although technically simple, the preparation of stable and concentrated dispersions of Pu(IV) intrinsic colloids under such conditions requires mother Pu solutions exhibiting high Pu concentrations with low acidity in order to significantly increase the pH during dilution.
| xPu4+ + yH2O ⇄ Pux(OH)y4x−y + yH+ | (5) |
| 2Pu4+ + 2H2O ⇄ Pu3+ + PuO2+ + 4H+ | (6) |
| Pu4+ + PuO2+ ⇄ Pu3+ + PuO22+ | (7) |
| 3Pu4+ + 2H2O ⇄ 2Pu3+ + PuO22+ + 4H+ | (8) |
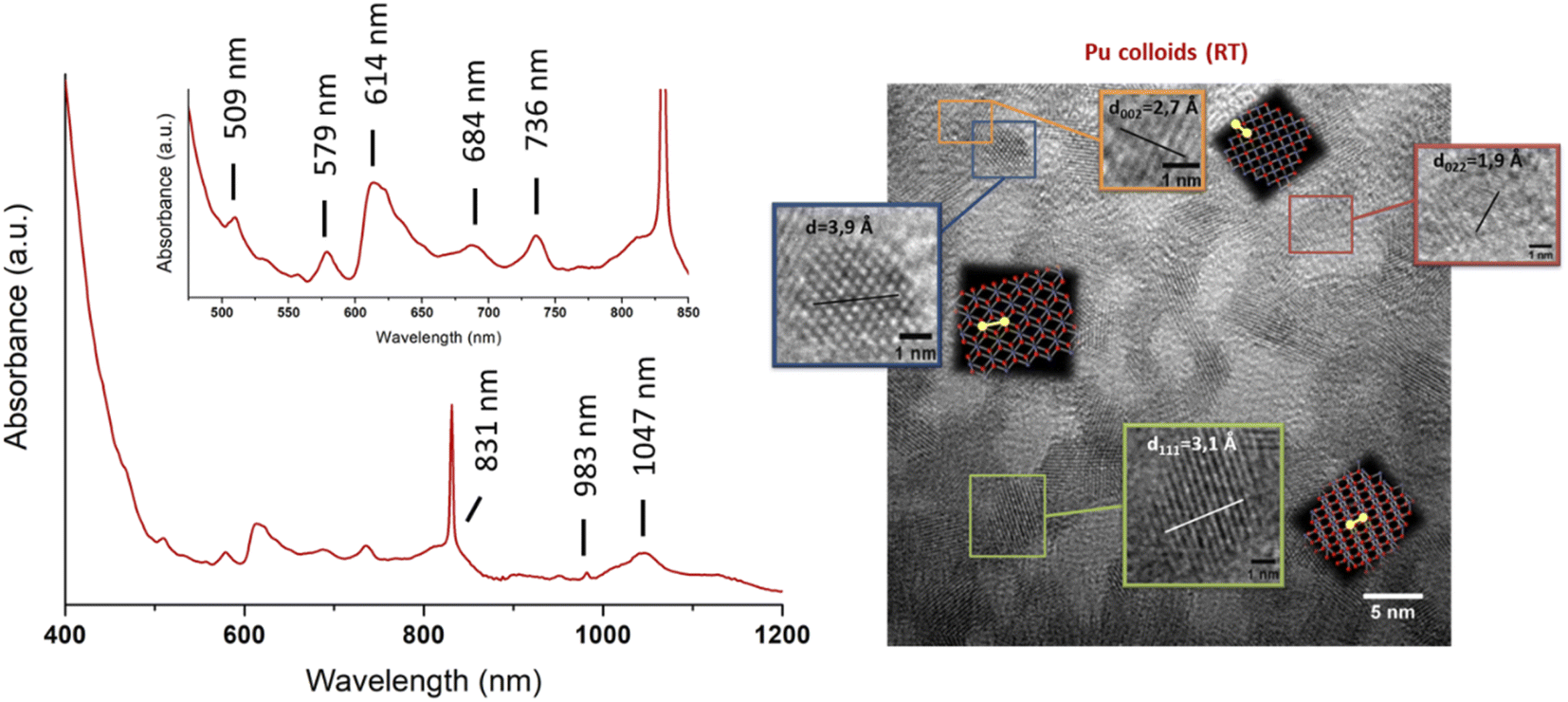 | ||
| Fig. 4 Left: UV-vis-NIR absorption spectrum acquired on intrinsic Pu(IV) colloids prepared by dilution in pure water of a Pu(IV) aliquot previously stabilized in nitric medium (RT, [Pu] = 9.2 mM). The spectrum shows the additional presence of Pu(VI) absorbing at 830 nm as a mixture with the colloids. Right: HR-TEM image acquired on intrinsic Pu(IV) colloids. Adapted from Dalodiere PhD thesis.97 | ||
Thiyagarajan reported the quantitative formation of polymers (i.e. colloids) by heating a Pu(IV) nitric acid solution (pH = 1.3–1.4) in a container with restricted air access at 85–95 °C. The potential presence of Pu(VI) could involve the addition of nitrite to reduce it into Pu(IV) and favour the hydrolysis of the latter.100 Zhao and colleagues recently prepared 2–3 nm Pu(IV) intrinsic colloids based on the work of Haire et al. by heating a 1.6 × 10−3 M Pu(IV) solution stabilized in 0.1 M HNO3 to 60–80 °C for 30 min. The authors reported an instantaneous colour variation from dark brown to emerald green upon heating.101,102 Several authors described the precipitation of particles after the addition of NH4OH to Pu(IV) solutions initially stabilized in nitric media, followed by a washing step (to remove ionic Pu species and counter-ions, generally nitrates) or an aging step under temperature. Then, the precipitate was treated in acid media to obtain plutonium colloidal dispersions (peptization).99,103–105 A possible additional concentration step followed by baking and resuspension in water has also been described. Lloyd and Haire used this approach to prepare 1–3 M concentrated Pu colloids exhibiting a poor concentration of nitrates (0.1–0.15 M).98 Another approach consisted in extracting HNO3 from Pu solutions stabilized in nitric media with a long-chain alcohol (typically, n-hexanol).106 Hence, the authors described the preparation of low nitrate colloidal dispersions (NO3−/Pu mole ratio = 0.1–0.2) with crystalline nanoparticles of 6–7 nm.106
Maillard and Adnet reported the observation of Pu(IV) polymers (i.e. intrinsic colloids) when adding H2O2 to Pu(VI) aqueous solutions in the 0.1–1 M range under weakly acidic conditions.107 Radiolytic transformation of plutonium ions in solution (auto-radiolysis) can also yield intrinsic colloids.108,109 The primary products observed during radiolysis of aqueous solutions are generally defined in agreement with eqn (9) and involve several species responsible for redox processes occurring with Pu ions.109 Newton et al. observed the formation of Pu(IV) intrinsic colloids by auto-radiolysis of 10−4 M Pu(VI) solutions with pH ranging from 1.3 to 2.2. The proposed mechanism involves the alpha-radiolytic reduction of Pu(VI) into Pu(V) followed by the disproportionation of the latter (eqn (10) and (11)). Fig. 5 illustrates the behaviour of a Pu(VI) solution prepared by the oxidation of Pu(IV) with HClO4 under thermal treatment followed by dilution in pure water. The storage of the as-prepared Pu(VI) solution in an aerobic atmosphere over several months in a glove box allowed its slow transformation from an orange into a characteristic green colloidal solution. HR-TEM analyses confirmed the crystalline nature of the particles crystallizing in a cubic face centered structure similar to PuO2 bulk with a nanoparticle size of ca. 2.9 ± 0.5 nm.96,97 Auto-radiolysis of Pu(III) solutions has also been reported to yield intrinsic Pu(IV) colloids.110 Interestingly, investigations carried out by autocorrelation photon spectroscopy concluded a particle diameter of 31.6 nm whereas particle diameter determined for other methods involving dilution with pure water, neutralization with NaOH and temperature aging are more generally in the ca. 1–10 nm range. Peptization led to the observation of a mixture of two different populations of about 13.6 and 370 nm.110 Other but less detailed preparation techniques have also been reported by Clark et al. including for instance the reduction of Pu(V), slow oxidation of Pu(III) with O2 and the addition of a moderately acidic solution of Pu(IV) in boiling water.45
| H2O → e−aq, H, OH˙, H2, H2O2, H+, OH− | (9) |
| PuO22+ + 1/2H2O → PuO2+ + H+ + 1/4O2 | (10) |
| 2PuO2+ ↔ PuO22+ + PuO2(coll) | (11) |
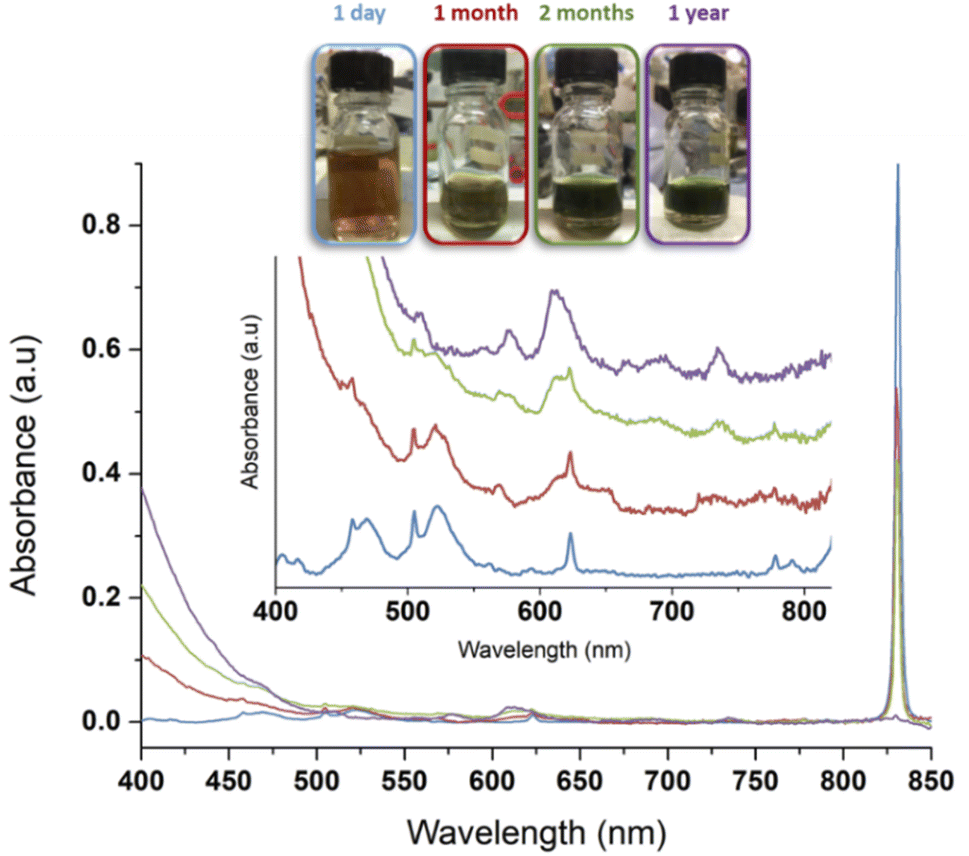 | ||
| Fig. 5 Evolution of a 1 mM Pu(VI) solution stored at room temperature in a glove box. Pu(VI) (main band at 831 nm) decreases with time while Pu(IV) colloids accumulate in solution (main band at ca. 614 nm). One can observe increase of the absorption in the near UV range attributed to Mie scattering and the presence of nanoparticles dispersed in the medium. After one year, the conversion is complete. Note: the decrease of the volume in the flask is attributed to solution sampling during aging. Adapted from ref. 97. | ||
Dalodière et al. reported the preparation of intrinsic Pu(IV) colloids by applying an ultrasound treatment to bulk PuO2 powder dispersed in pure water (Fig. 6).96 The originality of the technique relies on the unexpected reactivity of refractory PuO2 in pure water which allows the preparation of very stable colloids without addition of any electrolyte or capping agent. Absorption spectroscopy demonstrated strong similarities when comparing sonolytic and hydrolytic (i.e. by dilution of Pu(IV) in pure water) colloids. Nevertheless, a stronger absorption in the near UV range for the sonolytic colloids was attributed to the presence of bigger particles which was confirmed using HR-TEM (2.9 nm and 7.1 nm, respectively).96 Also, whereas hydrolytic colloids are always observed with Pu(VI) traces (strong molar coefficient allowing the detection of tiny traces), sonolytic colloids never show this oxidation state which was attributed to a different formation mechanism avoiding disproportionation reactions (note that the technique does not guarantee the potential contribution of other oxidation states in the solid nanoparticles). The authors demonstrated enhanced kinetics and yields in a reducing Ar/(10%)CO atmosphere while an oxidizing atmosphere prevented their formation. At first, the formation of the colloids was suggested to result from the fragmentation and surface hydrolysis of bulk PuO2 particles driven by the acoustic cavitation (i.e., the nucleation, growth and rapid implosive collapse of gas and vapour filled micro-bubbles). This step was proposed to be followed by the reductive dissolution of the dispersed and fragmented PuO2 particles into aqueous Pu(III) by H2 gas produced inside the cavitation bubbles. The re-oxidation of Pu(III) into Pu(IV) and its subsequent hydrolysis was suggested to slowly accumulate Pu(IV) colloids showing typical green colour and electronic signature.
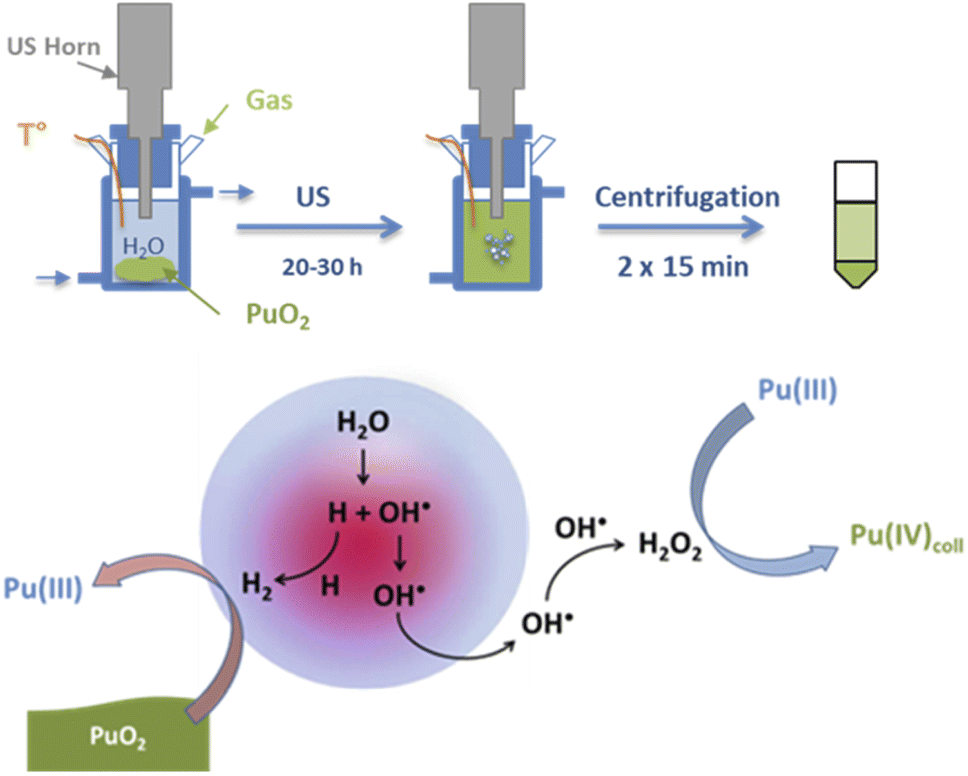 | ||
| Fig. 6 Schemes of the sonochemical protocol used to prepare stable dispersions of Pu(IV) intrinsic colloids (top) and their corresponding formation mechanism (bottom). | ||
2.3. Preparation of PuO2 nanoparticle precipitates
A conventional preparation route for PuO2 nanoparticles involves their rapid precipitation under alkaline conditions through the addition of ammonia or sodium hydroxide to Pu solutions.50,101,111 The mechanism driving the precipitation of the nanoparticles is generally discussed analogically with the one observed for the preparation of colloids. The green precipitates thus obtained from Pu(III), Pu(IV) or Pu(V) aqueous solutions by Romanchuk et al. were in accord with the formation of monodisperse and crystalline PuO2 nanoparticles of ca. 2.5 nm (XRD and HR-TEM) showing that the particle nature and size are more dependent on the chemical conditions rather than the initial plutonium redox state.112 By comparing the pH of the resulting solution after addition of ammonia to Pu(IV) solutions, Gerber et al. concluded that crystalline PuO2 nanoparticles of 2 nm can be observed in the 1–10 pH range. An additional significant amount of ionic Pu(III) and Pu(VI) in solution was observed at the lowest pH = 1.113 When adding aliquots of Pu(VI) solutions to 3 M NH3·H2O, the precipitation process required to yield the PuO2 nanoparticles was much longer and involved the formation of an intermediate product identified to be a metastable solid phase of pentavalent Pu.114 Ekberg et al. described the synthesis of a green colloidal dispersion by the addition of 0.19 M NaOH solutions of Pu(IV) stabilized in 1 M HCl solutions.115The alkaline precipitation procedure using Pu(IV) was recently modified with the addition of an organic polymer (polyethylene glycol, 3000 g mol−1, 2.5 wt%) during the synthesis step to avoid the growth and agglomeration of the nucleated nanoparticles. The thermal conversion of the precursors at 485 °C (2 h, air) leads to well-defined crystalline PuO2 nanoparticles of about 4.6 nm (HR-TEM, Fig. 7).116 As for Hudry and co-workers, the preparation of PuO2 nanoparticles was achieved through the decomposition of plutonyl nitrate precursors [PuO2(NO3)2]·3H2O in a coordinating organic media composed of benzyl ether, oleic acid and oleylamine.117 The thermal treatment of the mixture at 280 °C for 30 min led to the formation of highly crystalline PuO2 particles composed of a mixture of spherical nanoparticles (ca. 2 nm) and nanodots (ca. 0.9 × 4 nm2) as evidenced in Fig. 7. The latter is particularly interesting since it constitutes, to our knowledge, the only reported anisotropic shape observed for PuO2 nanoparticles. The role of the geometry of the trans-dioxo molecular Pu(VI)O22+ ions in the formation of these anisotropic particles appears to be important.
 | ||
| Fig. 7 (HR-)TEM images acquired on PuO2 nanoparticle precipitates obtained by (a) thermal conversion (485 °C, 2 h) of Pu powder obtained while pouring ammonia in a Pu(IV)/PEG solution, (b and c) hydrothermal treatment of Pu(IV) oxalates at 95 °C for 120 h under autogenic pressure, (d and e) solvothermal decomposition of plutonyl nitrate precursors in a mixture of benzyl ether, oleic acid and oleylamine. Adapted from ref. 116 and 118 with permission from the Royal Society of Chemistry (copyrights 2018 and 2020) and from ref. 117 with permission from John Wiley and Sons (copyright 2014). | ||
Hydrothermal conditions have been alternatively demonstrated to be an interesting route for the preparation of Pu oxide nanoparticles (Fig. 7). PuO2 nanocrystals of high crystallinity (4.7 nm according to XRD and 2.5 nm through HR-TEM, a0 = 5.397(1)) have been prepared by hydrothermal decomposition of Pu(IV) oxalates at 95 °C for 120 h under autogenic pressure.118 The preparation of these nanocrystals occurred at a much lower temperature than the one required for analogous Th or U indicating a potential application of hydrothermal conditions for separation purposes.14 The decomposition of the oxalate ligands in hot compressed water apparently does not involve redox transformation of Pu and the observed morphology of the precipitates differs from the squared platelets usually obtained by the oxalate route (Fig. 7vs.8).118 The as-obtained powder rather shows spherical agglomerates of 100–200 nm composed of nanometric crystallites (see Fig. 7b).14,118 In comparison to what is usually observed with the thermal decomposition of the oxalate precursor in an air atmosphere, the investigations suggested a different decomposition scheme under hydrothermal conditions, involving the contribution of water as evidenced by isotopic exchange reactions probed by nuclear magnetic resonance (magic-angle spinning MAS-NMR) and Raman spectroscopy techniques.
More generally, this synthesis approach has been performed with Th, U, Np and Pu elements and showed several advantages including the reproducibility and time required for the quantitative synthesis, the high crystallinity of the powder and its homogeneity but also its good behavior during sintering (not shown using Pu to our knowledge).14,26,118,119 In addition, the destructuring of the plate-like aggregates typical of oxalates is advantageous with a direct effect on the sintering properties of the powders without residual carbon impurities. Recently, Kauric et al. described promising results using this preparation route, with the low-temperature preparation of nanometric mixed oxide U1−xPuxO2(+y) particles (solid solution) and measuring between 7 and 19 nm according to X-ray diffraction (XRD) as a function of the composition of the oxide (7 to 33 nm with HR-TEM).26 While it is well known that plutonium oxalates radiolytically decompose with time during storage, poor information is available about the speciation of the intermediate and finally formed products. Recently, the formation of crystalline PuO2 nanoparticles of a few nm has been reported by auto-radiolytic decomposition of solid Pu(III) and Pu(IV) oxalate compounds. The hydrated oxalate precursors were stored in air for several years at room temperature and pressure.120
2.4. Preparation of nanostructured PuO2
Thermal decomposition of oxalate precursors is the most widespread method used for the preparation of PuO2.71,121 This synthesis route has been highly used at the industrial scale for the production of nuclear fuel or in-lab purification of solutions. Hydrated precursors can be easily precipitated when mixing Pu(III) or Pu(IV) solutions with oxalic acid at room temperature. After separation and washing, their thermal decomposition at elevated temperatures leads to PuO2 through a series of intermediate decomposition reactions coupled with the generation of gaseous H2O, CO and CO2 products.121 While the technique generally enables holding the microscopic morphology of the initial precursors, the preparation step and firing conditions can significantly modify the properties of the resulting oxide.122 The general thermal decomposition mechanisms are described in eqn (12) and (13) for both Pu(IV) and Pu(III) oxalates, respectively. According to HR-TEM investigations, Pu(III) oxalates fired in a 485 °C air atmosphere for 2 hours resulted in the formation of nanostructured elongated objects composed of 6.2 ± 1.3 nm nanoparticles (7.3 ± 1.9 nm when fired for 12 hours), whereas Pu(IV) ones fired under similar conditions allowed the observation of micrometric squared platelets composed of 5.1 ± 1.1 nm nanoparticles (Fig. 8). For both samples, nanoparticles were found to be monodispersed, quasi-spherical and crystalline, agreeing with the PuO2 bulk structure.116,123 The preparation of nanostructured actinide oxide architectures has also been demonstrated by the thermal conversion of Th and U oxalates.33 Such materials are often used as simulants for Pu compounds and address structural trends within the actinide series (also often including Ce as another simulant of Pu). Time-dependent observations acquired during Th oxalate annealing using HR-TEM and AFM revealed the nanoscale organization of the obtained oxide and the effect of the firing conditions (duration and time) on the formation properties of the nanostructured oxides. Of particular interest, AFM demonstrated the absence of nanostructuration for the oxalate precursor and its formation during thermal treatment before grains begin to grow and coalesce (Fig. 8).| Pu(IV)(C2O4)2·xH2O → Pu(IV)O2 + 2CO + 2CO2 + xH2O | (12) |
| Pu2(III)(C2O4)3·xH2O → 2Pu(IV)O2 + 4CO + 2CO2 + xH2O | (13) |
 | ||
| Fig. 8 (Top) Selection of images acquired with SEM or HR-TEM for PuO2 powder precipitates exhibiting a nanoscale architecture (nanopowder and nanostructured oxides). (Bottom) AFM images acquired on Th oxalate crystal and the corresponding nanostructured oxide after calcination at 485 °C (4 h). AFM evidences the absence of nanostructuration at the initial stage. Adapted from personal data and from Bonato et al.116 with permission from the Royal Society of Chemistry, copyright 2020. | ||
3. Probing the size, morphology and crystalline nature of PuO2 nanoparticles
The reliable preparation of PuO2 nanoparticles is closely connected to their characterization which represents a difficult task due to the nanoscale nature of the considered samples and the chemical toxicity and strong radioactivity of the element Pu. As a consequence, advanced structural characterizations generally require dedicated devices (i.e. nuclearized) and often need large or very large instruments (e.g., HR-TEM, synchrotron radiations…) usually conditioned by the writing and acceptance of proposals. Some analytical techniques gave strong insights into the formation mechanisms and structural properties of PuO2 nanoparticles. Comprehensive overviews dedicated to analytical techniques allowing the speciation and characterization of actinide complexes, clusters or nanostructures in solution or in the solid state can be found in the literature.12,45,129 For most of the plutonium oxide nanoparticles recently reported, HR-TEM investigations evidenced the presence of monodispersed and small crystalline nanoparticles (Table 1 and Fig. 4 as examples). Selected area electron diffraction (SAED), often used to complete HR-TEM characterizations, also supports the presence of polycrystalline nanoparticles exhibiting an FCC cubic structure in accordance with bulk PuO2 (Fig. 7, for example).55,96,111,116Strong similarities have been reported when comparing electron absorption spectra acquired in the solid or liquid states for intrinsic PuO2 colloids, the Pu38 cluster and bulk or nanoscale PuO2.116,130,131 Absorption and diffuse reflectance spectroscopies indeed demonstrated similar energy transitions between these samples evidencing structural similarities.131 A similar conclusion was given by Toth and Friedman when they compared FTIR spectra acquired from PuO2 and polymers (i.e. colloidal PuO2 nanoparticles) prepared by the addition of NaOH to acidic Pu(IV) solutions.105 Raman spectra recently acquired on PuO2 nanoparticles showed an enlargement and red shift of the T2g band which is usually located at 476 cm−1 for bulk PuO2 whose mode corresponds to the symmetric stretching of the oxygen network (Fig. 9).132 These Raman features result from phonon confinement and provide a fingerprint of the nanocrystalline nature of the studied PuO2 samples. A similar behaviour has been observed with nanoparticles of the structural analogue ThO2. The possibility of using these characteristics against a calibration curve for the latter has been proposed to determine the nanoparticle size.33,116,133 Alternatively, the technique allowed the demonstration of the greater structural stability of restructured zones in high burn up structures (irradiated nuclear fuel) composed of plutonium enriched agglomerates.134 Relevant studies have been reported about the use of Raman spectroscopy for the characterization of PuO2 ranging from the micrometric to the bulk scales concerning the aging and temperature-dependence of defects, its behaviour under the laser beam, or for the preparation of reference particles for safeguard purposes for instance.135–139 Nevertheless, this powerful technique remains poorly applied to nanosized PuO2 and further insights are expected in this domain.
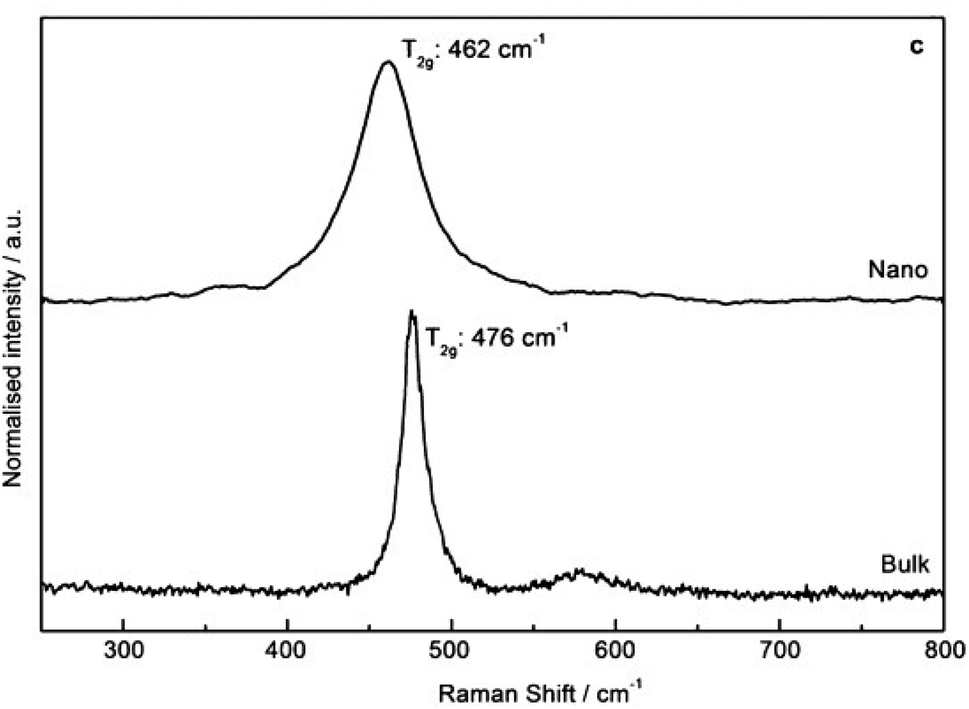 | ||
| Fig. 9 Raman spectra (488 nm, 1 mW) acquired on PuO2 nanoparticles (top) and bulk (bottom) in the solid state demonstrating the enlargement and red shift of the T2g band as evidence of nanoscale materials. Adapted from ref. 117 with permission from John Wiley and Sons, copyright 2014. | ||
X-ray diffraction (XRD), which is based on the measurement of constructive interferences occurring between a monochromatic X-ray beam and a crystalline sample, has provided interesting structural information about the crystalline structure, lattice parameter and size of PuO2 nanoparticles. The technique provides statistically relevant and average information acquired on a large volume of generally powdered and/or dried sample.140 The reflections observed on the reported XRD diagrams of PuO2 nanoparticles agree with the cubic fluorite structure of their bulk counterparts (JCPDS file 00-041-1170, Fmm space group).111,112,116,117 In comparison to bulk PuO2 patterns, the low intensity and increased width of the diffraction peaks reported by the various authors confirm the presence of nanoscale oxides.111,112,117,118 Gerber et al. and Romanchuk et al. combined HR-TEM and XRD data acquired with synchrotron radiation to evidence the monodisperse and crystalline nature of ca. 2.5 nm PuO2 nanoparticles prepared by ammonia neutralization of aqueous acidic solutions of Pu of different oxidation states.111,112 It is worth noting that the preparation step required for XRD measurements might induce chemical and/or structural modifications of the nanoparticles during precipitation and drying. To avoid such potential effects of drying on the structure of the Pu nanoparticles, Romanchuk and co-workers successfully modified their set-up to acquire XRD patterns in the aqueous media.112
In another study, by fitting the complete experimental diffraction pattern acquired on powdered PuO2 nanoparticles using Rietveld refinement, the size of the coherent domains composing the samples (crystallites) has been correlated with the nanoparticle sizes determined by the HR-TEM approach by Bonato et al.116 The size of the nanoparticles that composed the PuO2 samples was then correlated with the firing conditions and durations of the various precursors. Popa et al. used HR-TEM and XRD techniques to determine the size of PuO2 nanoparticles formed by the decomposition of the corresponding Pu oxalate precursors in hot compressed water (2.6 and 3.7 nm, with a0 = 5.397 Å, respectively).118 In comparison with other studied oxides, it is worth noting that PuO2 nanoparticles are obtained at much lower temperature (95 °C) resulting in smaller particle sizes. The investigated variations of the size and lattice parameter of the as-obtained nanoparticles at different temperatures demonstrated a rather limited increase for PuO2 until ca. 750 °C. A linear growth was then observed as a result of thermal expansion (agreeing with the a0 increase).118 The temperature-dependent growth of the nanoparticles was found to be intermediate between the ones observed for CeO2 < UO2 and NpO2 < ThO2 nanoparticles. The growth of the PuO2 nanocrystalline particles was then evaluated using isothermal XRD measurements which allowed the authors to evaluate the activation energy to be ca. 351 kJ mol−1. The results suggested that particles grow in the 820–1100 °C range via surface diffusion (pore controlled kinetics mechanism).126
The High Energy X-ray Scattering (HEXS) approach was initially applied for the characterization of the structural properties of the Pu38 oxo-cluster by Soderhom et al.,85 and more recently for PuO2 nanoparticles by Gerber et al.111 HEXS is a total scattering method useful for understanding the coordination environment of a metal centre without long-range order. The technique is based on the collection of hard X rays as a function of the scattering angle and wavelength of radiation providing a distribution probability of atoms as a function of distance (pair distribution function, PDF).129,141,142 Long- and short-range orders extracted by the technique indicated strong similarities between the nanoparticles and PuO2 bulk material. By contrast, the damping differences of the signals observed on the reduced pair distribution functions G(r) for each sample allowed the author to determine the size of the PuO2 nanoparticles (1.3–2.07 nm) that were found to be in reasonable agreement with HR-TEM (2.3–3.0 nm) and XRD (1.6–2.4 nm) investigations.111
Laser induced breakdown detection (LIBD) is another technique that has been used to determine the size and the concentration of Pu particles in solution. The technique is based on the detection of a plasma selectively created between a colloidal dispersion and a focused pulsed laser. Using this technique, very small concentrations of colloids (below 10−8 M) can be probed, providing important insights into the formation mechanism, solubility, and properties of PuO2 intrinsic colloids.46,83 A series of Pu(IV) colloid samples with a total Pu concentration of about 1.5 × 10−4 M were prepared by the addition of an aqueous solution or NaOH to Pu(IV) acidic solutions. LIBD analyses performed one day after the sample preparation did not allow the detection of colloids up to pH 0.7 whereas entities of about 5 nm in diameter were observed in the 0.9–1.4 pH range with a total concentration of approx. 10−7 M. The sample prepared at pH = 1.8 under similar conditions was characterized by the observation of 400 nm particles which aggregated to particle sizes of 1 μm after two years, also accounting for more than 90% of the Pu(IV) in solution. It is interesting to note that the provided and corresponding UV-vis absorption spectra converged to the typical Pu(IV) colloid spectra upon oversaturation (highest pH) and aging.46 It is worth noting that the detection limit of the technique, indicated to be about 5 nm, raises questions since colloidal PuO2 nanoparticles have been very often measured below this size limit when using other techniques.
Small-Angle X-ray or Neutron Scattering techniques (SAXS and SANS, respectively) are complementary techniques which are both able to probe structures at the nanoscale. When entering in contact, elastically scattered radiations of X-ray or neutrons can be quantified to provide information about the size, the shape and the nature of interactions (or also, orientations) occurring between the components of a sample.143,144 These techniques are non-intrusive and allow studying samples without specific preparation, providing in situ information averaged over a large sample volume and directly in the native environment, which appear clearly advantageous for colloidal systems. Scattering techniques are dependent upon the contrast difference existing in the studied sample. X-ray scattering cross-section increases with the electron density and is therefore sensitive to the atomic number Z, whereas neutron scattering depends on fluctuations of the density of nuclei which vary randomly across the periodic table but appears highly sensitive towards isotopes.143,144 Thiyagarajan et al. used the scattering properties of neutrons to probe Pu polymers (i.e. colloids) which revealed the presence of a polydispersed and elongated assembly of nanoparticles measuring more than 190 nm in length with a diameter of ca. 4.7 nm (Fig. 10a).100 This size is much larger than the one reported with other techniques for Pu nanoparticles prepared using similar approaches. Scattering patterns further allowed the observation of Bragg diffraction lines agreeing with a PuO2-like periodic structure. By varying the parameters, the authors highlighted the influence of the preparation method on the morphology of the particles including the effect of the temperature. In addition, the scattering patterns demonstrated that the presence of uranyl ions during Pu colloid formation retards the rate of their formation thus agreeing with previous studies.145 The latter was attributed to the interaction of U(VI) with Pu hydrolyzed species during condensation reactions.
 | ||
| Fig. 10 (a) SANS diagram acquired on Pu colloids prepared in aqueous solution (total Pu 0.12 M) which measured 4.7 nm × 190 nm according to data simulations. Adapted from ref. 100 with permission from the American Chemical Society, copyright 1990. (b) SAXS diagrams acquired for 10 mM hydrolytic and 0.8 mM sonolytic colloids. Black lines indicate the slope power laws. The slope observed for the hydrolytic colloid is typical for 3D objects and compact particles (2.6 nm according to simulations). The one observed for the sonolytic colloid is characteristic of elongated objects (e.g. thin disks or lamellas measuring 5.7 nm x > 30 nm according to simulations). Reproduced from ref. 94 with permission from the Royal Society of Chemistry, copyright 2020. | ||
SAXS has been used by other authors to probe plutonium oxide nanoparticles prepared by ammonia neutralization of aqueous Pu solutions of different oxidation states which indicated agglomerates of 2.5 nm PuO2 nanoparticles.112 More recently, a combination of synchrotron SAXS and XAS (X-ray Absorption Spectroscopy) techniques was developed to probe the multi-scale structural properties of actinide compounds (i.e., from the local structure to the size, morphology and interactions of the nanoparticles in solution).94 Normalized SAXS diagrams acquired using this approach for Pu(IV) intrinsic colloids prepared by hydrolysis or sonolysis evidenced strong differences in the particle shapes and sizes of both samples (Fig. 10b). The spherical shape of the hydrolytic colloid nanoparticles was confirmed by SAXS pattern simulations which indicated a particle diameter of 2.6 nm in agreement with HR-TEM investigations. By contrast, the sonolytic colloids were better considered as elongated lamellas or disks with 5.7 nm of thickness and >30 nm length, which is much larger than what observed by HR-TEM. Such striking difference is most likely because of an aggregation phenomenon occurring with aging and resulting from the absence of counter ions in the system (such morphology reminds us of the one observed by Thiyagarajan et al., Fig. 10a100).
In combination with HR-TEM and EXAFS experiments that evidenced the PuO2-like structure for the colloidal particles, the observed significant variations of the scattering length density profile for the intrinsic Pu(IV) hydrolytic colloid allowed describing them as spherical nanoparticles composed of a PuO2 core surrounded by a disordered Pu–O shell possibly solvated with nitrates originating from the mother solution.94 The significant variations observed for both intrinsic and sonolytic colloids highlighted the strong influence of the preparation conditions on the structural properties of the PuO2 nanoparticles. It is worth noting that combined SAXS/XAS synchrotron techniques recently allowed probing the size, shape and local structure of a water-soluble plutonium hexanuclear cluster.146 The stabilization of these very small and low concentration aggregates with the DOTA ligand (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) allowed their characterization with a good signal-to-noise ratio, thus demonstrating the technical capacity of the mutual approach. Scattering techniques are obviously strongly considered in modern actinide science147–149 but remain poorly applied for trans-uranium oxide nanoparticles and we expect a stronger interest for the technique in the near future. The results recently provided by Zhai et al. on the formation and structure of ThO2 colloidal nanoparticles could be helpful for understanding the properties of analogous nanosized An oxides.149 After the addition of NaOH to aqueous solutions of Th(NO3)4·4H2O, the dispersions formed at different pH values (5.5–8.0) above the precipitates were characterized by SAXS. The authors demonstrated that dispersions aged for 5 months were composed of a mixture of polymeric colloidal structures measuring 20 nm and more compact particles of 3–4 nm (diameters). The dispersions aged for 18 months were found to be composed of crystalline ThO2 particles of ca. 5 nm forming agglomerates of ca. 12 nm in diameter.149
It is important to emphasize that the various analytical techniques used to probe the PuO2 nanoparticles may offer several advantages and drawbacks thus highlighting the observed interest of most of the authors in the combination of approaches when characterizing PuO2 nanoparticles. For instance, slight variations in the nanoparticle sizes are generally noticed when comparing both XRD and TEM techniques. Such a difference can be attributed to nanocrystal boundary effects that cannot be well discriminated or probed by XRD, but also to intrinsic limitations of the HR-TEM technique which do not allow quantifying a large number of particles (statistics and human eye errors).140 Combining the analytical techniques to evaluate the size of the particle appears to be relevant. Note that lattice strains or instrumental effects are additional parameters that can participate in the broadening of the diffraction peaks and thus deviate the values extracted from the diffractograms. Variations in the determined crystallite sizes have also been demonstrated to depend upon the computational approach; for instance, Kumar et al. observed differences when they investigated the size of nanocrystalline cobalt ferrite particles by extracting structural data from XRD patterns with Scherrer's formulae, Williamson–Hall plot, or Rietveld method.150 Alternatively, TEM experiments have been reported to contribute to the relaxation of the lattice contraction of ceria nanoparticles during their analyses under vacuum, thus explaining the larger values obtained in comparison to room temperature XRD measurements.151 Analysis of liquid-borne particles may force their precipitation and drying, which can dramatically impact their surface chemistry and chemical structure. In particular, synchrotron radiation measurements should be considered with caution regarding the beam-induced reactions (e.g. redox processes) or damages (e.g. precipitation of (nano-)particles in the cell or on the kapton windows of the sample holders) thus requiring as much as possible the additional control of the samples with alternative techniques during and/or after the measurements.124,152,153
4. Impact of the nanoparticle size on the crystalline structure of PuO2
4.1. Nanoscale-induced structural variations for PuO2
Plutonium crystallizes as the dioxide in the fluorite CaF2 structure of the face-centered cubic system (Fmm space group). The fluorite lattice exhibits a symmetrical environment illustrated in Fig. 11 and consists of a cubic system regularly occupied by tetravalent Pu cations whereas eight O2− ligands are located at the tetrahedral interstitial positions. In this structure, each oxygen is surrounded by four Pu cations and each of them is coordinated by eight oxygen atoms at the corner of a cube forming infinite arrays of AnO8 polyhedra sharing their edges. At room temperature and pressure, the distances of Pu–O and Pu–Pu are 2.337 and 3.817 Å, respectively, which are similar within this structure giving a lattice parameter a0 = 5.396 Å and a density d = 11.46 g cm−3.20,115 In the oxide, the second O shell (CN = 24) surrounds the Pu atom at a distance of 4.476 Å. The fluorite configuration further allows the formation of vacant octahedral interstitial sites located at the center and middle of the edge of the cubic system thus accommodating a relatively open crystal structure.154 This property particularly provides a strong resistance of the ceramic to radiation damage and good stability in the nuclear reactor, without deformation of the structure and surrounding cladding that could lead to migration of matter in the exchangers.
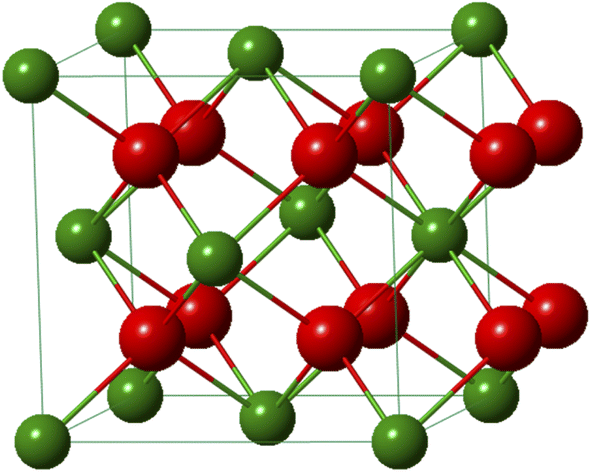 | ||
| Fig. 11 Cell of the PuO2 structure. Pu4+ and O2− are shown in green and red, respectively. In the structure, each O is surrounded by 4 Pu atoms forming tetrahedral sites and each Pu is coordinated by 8 O atoms forming infinite arrays of PuO8 cubes. | ||
A general consensus supported by the above described techniques (and in particular, the diffraction lines acquired with XRD measurements) allows the community to confirm the similarity between bulk PuO2 and the plutonium oxide nanoparticles described in this review (Table 1). When prepared under ambient conditions, the latter very often measure 2–5 nm in diameter and the preparation of bigger objects appears less common and often requires non-conventional approaches such as autoxidation of Pu(III), sonochemistry, high temperatures or more exotic procedures.45,96,116,125,126 Due to the shrinking dimensions and the related higher proportion of surface atoms exhibiting a different chemical environment, the average coordination number observed for a nanoparticle (particularly for the Pu–Pu coordination shell) is expected to be smaller compared to the bulk ones. This phenomenon can be even more pronounced for the smallest particles and also depends upon the particle shape. Significant structural modifications can accompany these observations in agreement with what was observed for d-block oxides. In particular, the contraction or expansion of the bond length of the surface and subsurface atoms has been reported for several materials, which is suggested to be enhanced for curved surfaces where facets, corners, steps and edge atoms can be observed.6,155,156 Strain variations have been reported to depend upon the synthesis conditions and nanoparticle treatments, in addition to their interaction with the environment. Such features can be related to differences in the surrounding media and local symmetry of the surface atoms, effects of which can also propagate within the core of the particle and are directly related to the cell dimensions within a nanoparticle. As a result, surface energy and physico-chemical properties of the oxide can be significantly varied (e.g. reactivity, mechanical properties, electronic structure, stability, etc.).4,6,155,157,158 Recent simulations carried out on metal nanoparticles also demonstrated the influence of the displacement of surface atoms to the interatomic spacing of subsurface ones.158
It is important to emphasize that a 3.0 nm PuO2 nanoparticle exhibiting an a0 parameter of 5.396 Å contains only ca. 90 unit cells in the volume and only 5–6 lattices are required to go through the particles. This calculation decreases to 50 units for a 2.5 nm particle and 26 units for a 2.0 nm particle, meaning that an extreme mechanical stress should be addressed to the whole particle when the particle size and shape are modified in addition to surface interactions with the surroundings. The order of magnitude of atoms involved in a nanoparticle is schematized in Fig. 1 demonstrating that the particle tends to be almost composed of surface atoms when shrinking to a nanometric size. Note also that an increase in the coordination number from 6 to 8 for Pu(IV) allows increasing its ionic radii from 0.86 to 0.96 Å, respectively.159
The size vs. crystallographic property relationship has been poorly addressed for actinide oxides in comparison to lighter elements including TiO2, ZrO2, CeO2, Co3O4, Fe3O4, etc.6,160–164 Nanoscale oxides generally show a negative surface stress (i.e. an increase in the lattice parameter) whereas metals or halides rather exhibit a positive surface stress (i.e. a decrease in the lattice parameter).160,164,165 Size-induced lattice variations have been noticed for nanosized CeO2 which is an isostructural oxide crystallizing in the cubic fluorite structure and is very often used as a PuO2 simulant for laboratory studies. A 0.45% increase of the cell parameter has been observed for CeO2 nanoparticles when decreasing in size with a reported value of 5.4087 Å for micrometric bulk particles against 5.4330 Å for 6.1 nm nanoparticles.166 Some authors reported both lattice expansion and contraction for shrinking ceria nanocrystals and attributed this observation to a balance between the concentration of surface strains or anion vacancies in the nanocrystal and the pressure derived from the nanoparticle surface tension.151 The size dependent crystallographic and electronic properties have been recently demonstrated for CeO2 nanoparticles.167 The authors observed an increase in the lattice parameter from 5.411 Å (i.e., the value reported for bulk CeO2) to 5.456 Å when the particle size decreased from 91 to 2.0 nm. The strongest variations were observed for particles measuring less than 5 nm. The contribution of Ce(III) in the nanoparticles and the associated formation of oxygen vacancies was ruled out by combining XPS spectroscopy with synchrotron hard and soft X-ray absorption experiments at the L3 and M5 edges, as well as computational methods. A tensile effect occurring at the nanoparticle surfaces was rather invoked to result from the contribution of carbonate and hydroxyl surface moieties that were probed with FTIR and TGA.167,168
Although much less considered, several authors probed the lattice parameter of PuO2 nanoparticles, more generally using XRD patterns which provide the advantage of being statistically relevant when compared to HR-TEM. The simulation of the diffraction patterns provides a good description of the long range ordering of the coherent domains in the studied sample without emphasizing on the material heterogeneities.169 The lattice parameters given in the literature for PuO2 nanoparticles are gathered in Table 1 and are correlated with their particle sizes acquired using various approaches (XRD, HEXS, HR-TEM, etc.) as shown in Fig. 12. Although the correlation does not provide a clear trend, significant variations in the cell parameters reported for PuO2 nanoparticles can be pointed out when compared to the bulk PuO2 reference reported at 5.396 Å.45 Furthermore, one can note that the reference value is almost exclusively lower than the ones reported for the nanoparticles, strengthening the observation of a potential negative surface stress for nanosized PuO2. A similar, but more pronounced phenomenon, has been recently reported for ThO2 nanoparticles (another isostructural simulant of PuO2). Combining XRD, Th L3-XAS and HR-TEM, the authors observed an increase by about 1.1% for 2.5 nm ThO2 nanoparticles (5.66 Å) in comparison to bulk materials (5.60 Å).165 The unit cell parameter dependence on the ThO2 particle size was found to be a power law relationship. Since Th is only expected to exist in the (+IV) oxidation state, the formation of vacancies was refuted in agreement with the observations carried out with XAS experiments. A combination of thermogravimetry coupled to mass spectrometry analyses allowed explaining the negative stress observed for the ThO2 nanoparticles by a tensile effect exerted by the presence of surface hydroxyl and carbonate groups. The adsorption of H2O or CO2 can be explained by the high charge of the nanoparticle surface resulting from the breaking of the surface ionic bonds.
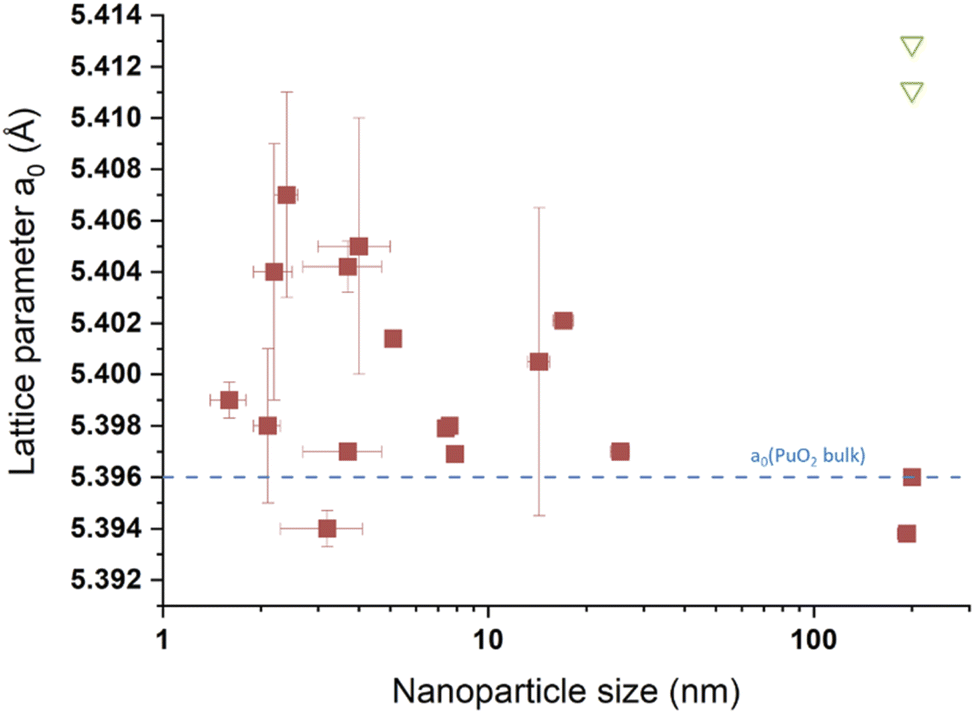 | ||
| Fig. 12 Survey of the size vs. lattice parameters extracted from the literature for PuO2 nanoparticles. The blue line corresponds to the theoretical a0 value expected for PuO2.00 (a0 = 5.396 Å) according to ref. 45. The green triangles transcribe values taken from the literature for aged PuO2 samples prepared by thermal conversion of oxalate precursors at 600 °C.138 200 nm has been used as an arbitrary value for the 4 points given for this size. | ||
4.2. Structural variations induced by PuO2 nanoparticle surface adsorbates
At the nanoscale, the higher proportion of surface atoms contributes to the increase of the surface energy which may result in particular reactivity and properties. Nanoparticle surfaces may therefore undergo the physico-chemical influence of their surrounding medium or atmosphere.170 As previously described, significant variations of the lattice have been reported in the presence of adsorbates or the formation of surface hydroxyl groups for various oxides, including ThO2 and CeO2.160,164,165,167,171 Adsorption properties of species on the PuO2 nanoparticle surface have been poorly addressed in comparison. Nevertheless, some interesting observations can be revealed when considering the Pu38 clusters which have been evidenced to share some structural properties with Pu(IV) intrinsic colloids according to spectroscopic investigations. In particular, the cores of the former have been demonstrated to be composed of distorted PuO2. Fig. 13 presents the second layer of Pu–Pu bond distances (or third coordination shell) extracted from Pu38 cluster structures refined by Soderholm and Sigmon and co-workers,16,85 and the theoretical lattice parameter value of 5.396 Å typical of bulk PuO2 (red line in the figure). Although both Pu38 clusters share a similar [Pu38O56]40+ core derivative of the Fmm fluorite structure, one can observe slight variations between both structures in addition to a strong splitting of the Pu–Pu distances. Such an observation can be explained by a difference in the ratio of chloride and water molecules serving as capping ligands at the cluster surface. Similar phenomena are expected for PuO2 particles shrunk to a nanometric size. Nevertheless, it becomes very difficult to assign a property such as a lattice parameter to an individual PuO2 nanoparticle.
 | ||
| Fig. 13 Pu–Pu bond distances occurring at a distance close to the a0 parameter extracted from the resolved Pu38 cluster published by Soderholm et al. and Sigmon et al.16,85 The red line corresponds to the theoretical a0 value given for PuO2 bulk. | ||
Recent studies demonstrate the strong affinity of the PuO2 bulk surface for H2O or CO2 molecules. By using ab initio modelling, Moxon et al. recently demonstrated the strong interaction of the surface of PuO2 with CO2 forming carbonate species.172 Toth and Friedman reported that a dried polymer (i.e. PuO2 nanoparticles) prepared by NaOH addition on a Pu(IV) acidic solution shows a great affinity for CO2 forming carbonates which was confirmed by FTIR bands at 1500, 1350 and 890 cm−1. More interestingly, the carbon content of the polymer equilibrated with air reached a value of 1.4 wt% which would approximately account for one carbonate for every three plutonium atoms.105 FTIR spectra recently acquired on PuO2 nanoparticles prepared by auto-radiolytic decomposition of Pu(IV) and Pu(III) oxalates in the air atmosphere were also suggested to be possibly associated with carbonate moieties.120 Computational calculations allowed the estimation of the surface energy and predicted that the (111) surface is the most stable surface for PuO2 in comparison to the (110) and (100) indexed ones. It is expected that the selective adsorption of ligands on the desired surface would also control the morphology of nanocrystals that are formed.19,173 Simulations performed by Zhang et al. indicated that interactions between the PuO2(111) surface and H2O are higher than those observed with CO2 under similar (P,T) conditions.174
Both molecular and dissociative adsorption of water molecules have been demonstrated on the PuO2 surface using simulations. Particularly, it has been demonstrated that water can be present as adsorbed hydroxyl groups on the (110) and (100) surfaces, even at elevated temperatures and pressures.19,173 In contrast, the interaction of H2O with PuO2 bulk surface has been experimentally reported to generate H2 gas as well as an overstoichiometric oxide PuO2+x containing Pu(VI) moieties.175 Using X-ray and ultra-violet photoelectron spectroscopies (XPS and UPS, respectively), Gouder et al. investigated the behaviour of a PuO2 thin film surface in contact with water. At room temperature, they observed the formation of a hydroxyl layer in addition to small amounts of adsorbed water molecules without significant redox variation of Pu. In the presence of UV-light, the warming of a PuO2 surface in contact with a water ice film induced the reduction of the Pu surface which was interpreted as a photochemically driven interface reaction.176,177 Recently, the comparison of the experimental EXAFS spectra acquired for Pu(IV) intrinsic colloids offering particle size variations in combination with their HR-TEM characterizations allowed the description of the species as core–shell nanoparticles made out of a PuO2 core recovered by a hydrolyzed layer shell.96 Understanding the nature of the surface of PuO2 nanoparticles and its interaction with the environment appears complex but remains challenging due to obvious environmental issues and should lead to exciting investigations in the near future.
4.3. Comments on structural variations induced by radiolysis for PuO2 nanoparticles
In the course of research activities dedicated to the observation of PuO2+X species, Sarsfield and coworkers have observed the growth of vibration bands with solid phase Raman spectroscopy during PuO2 aging.138 They attributed this phenomenon to the formation of structural defects as a result of Pu alpha-decay without changes in the O/Pu ratio. In comparison to the theoretical cell parameter of 5.396 Å reported for bulk PuO2, the lattice parameters of the aged samples reached 5.4111 Å and 5.4129 Å (green triangles in Fig. 11), which are higher than the values reported for the nanoparticle counterparts discussed in this review. The maximum value of 5.404 Å associated with PuO2.26 by Haschke et al. for a0 was found to be possibly reached within 1 year of storage for the reprocessed material and 450 days for pure 239PuO2.138 Several studies have described the alpha-decay dependency of the lattice parameter increase for PuO2 bulk powders as a function of isotopy.20,178–180 With regard to PuO2 nanoparticles, Ekberg et al. suggested that alpha decay could affect the crystallinity of Pu(IV) colloids leading to more amorphous and/or smaller particles thus explaining EXAFS fit differences.115 From Table 1 and literature data, and besides auto-radiolysis effects reported for Pu solutions, it appears that the isotopic composition of the Pu solutions taken to prepare PuO2 nanoparticles is not a highly discussed topic and surely deserve more attention. The isotopic composition is not often detailed in publications and appears to be overlooked, as is the question of solution purification.It is important to emphasize that the presence of Pu-238 in PuO2 particles aged in water solution has been suggested to significantly impact the structure of the oxide and modify its apparent solubility.101,181 An increase in the rate of dissolution of Pu-238-based oxides by about 200 times under neutral conditions has been indicated in comparison to Pu-239 ones exhibiting similar characteristics.182–184 The increase in apparent solubility in the presence of Pu-238 has not been correlated with chemical kinetics but rather suggested to result from nuclei recoiling as a result of alpha decay thus inducing particle fragmentation. By using the nuclear track method, it was indicated that 300 nm monodisperse PuO2 particles may undergo fragmentation in pure water into smaller particles containing 60 to 104 Pu atoms.183,184 The local formation of very small particles can therefore be expected as a result of radiolysis. Modifications of the physico-chemical properties of the nanoparticles as a result of the radiolytic modification of the surroundings should also be considered. Along with this observation, the amount of plutonium-241 used during the preparation of PuO2 nanoparticles is almost never addressed. Pu-241 exhibits a short half-life (14.3 years) leading to the formation of americium-241 by β− decay.185 After a few years, the latter can therefore be significantly concentrated in plutonium oxide solids. The contribution of Am in the oxide, in addition to the damages induced by the various radioactive decays, should impact the intrinsic structure of a nanosized oxide. Investigations dealing with the local environment and valences of the cations in (Pu,Am)O2−x indicated that Am occupies the Pu sites in the oxide and will reduce to Am(III) prior to any reduction of Pu(IV).154,186 In comparison to the theoretical a0 values of 5.396 Å and 5.373 Å for PuO2 and AmO2, respectively, it was demonstrated that the presence of Am in (Pu1−y, Amy)O2−x may induce an increase in the lattice parameter for the oxide studied (a0 = 5.400 and 5.411 Å for Am/(Pu+Am) = 0.008 and 0.2, respectively).187 At high temperature, low Am concentration and low oxygen-to-metal ratio, Am is mainly found in the +III state.186 What is the influence of Am and the potential formation of oxygen vacancies on the crystalline structure of PuO2 nanoparticles?
These examples and observations demonstrate that, in addition to synthesis conditions, both the size and the surface properties of PuO2 nanoparticles may contribute to the variations of their structural properties and also to the data dispersion observed in Fig. 12 and 13. Reports dealing with the radiolysis effect on Pu nanoparticle formation and solution behavior are very few; their effects on the synthesis kinetics, aging, stability and crystalline properties remain a fair question.180 It is worth noting that alpha particles roughly travel less than 10 cm in air when emitted with an energy of 4–10 MeV and range from 20 to 100 μm in pure water.188 As the size of a nanoparticle is significantly smaller than the distance travelled by alpha particles, radiolysis is obviously concerned with the nanoparticle core, the surface and the surrounding medium at the same time. In analogy with the recent report on oxalate ligands by Corbey et al., the observed radiolysis effect on ligands potentially complexing Pu aqueous species but also PuO2 nanoparticles under environmental conditions appears relevant.120 Size-dependent crystallographic properties of PuO2, or AnO2 nanoparticles to a large extent therefore represent a complex topic requiring a detailed understanding and more rationalized investigations.
5. Probing the atomic and electronic local structures
5.1. General considerations
Whatever the Pu oxide nanoparticle size, aging and synthesis approach are, the above discussion demonstrates the consensus reached with respect to their general structural description agreeing with the basic structure of PuO2. As described above, this has been evidenced by several techniques including, for instance, XRD, HR-TEM, SAED, HEXS, but also EXAFS which appears as a technique of choice when probing the local structure of PuO2 nanoparticles. In general, Pu oxide nanoparticles exhibit common features evidenced by EXAFS spectroscopy that will be detailed hereinafter and can be summarized by (i) the observation of two peaks on the Fourier Transform (FT) of the Pu L3 edge EXAFS spectra matching with the PuO2 spectra; (ii) a rather stable Pu sublattice agreeing with PuO2 (often correlated with other characterization techniques). In contrast with PuO2, FT spectra acquired for PuO2 nanoparticles generally show a strong dampening of both Pu–O and Pu–Pu shells as a result of an increased local disorder, and a possible distortion, broadening and splitting of the Pu–O shell as a result of a complex oxygen distribution in the structure. This latter point allowed some reports to demonstrate strong similarities between PuO2 nanoparticles and PuO2+x EXAFS spectra.40,45It has long been thought that PuO2 was the highest oxide phase that plutonium could reach. The reported formation of higher oxides when bulk PuO2 is exposed to water vapour at temperatures ranging from room temperature to 350 °C has shaken the understanding of the structure of plutonium oxide and raised concerns about industrial nuclear strategies in many countries.83,175,189 The report of the oxidation of PuO2 into PuO2+x (with x ≤ 0.25) has been argued to be accompanied by the generation of hydrogen gas and a significant contribution of higher Pu oxidation states to the crystal structure in agreement with eqn (14). The final product retains the fluorite structure with a slight expansion of the lattice parameter correlated with an increase in the O/Pu ratio in analogy with UO2+x.190,191 Conradson et al. demonstrated the possible diversity of Pu L3 edge EXAFS spectra that can be observed for PuO2 by comparing a dozen of XRD-determined phase pure samples.192 The differences were attributed to a large distribution of Pu–O distances (up to 3.4 Å), even in high-fired PuO2, and explained by the presence of aperiodically distributed plutonyl distances in the 1.83–1.91 Å range, as well as hydrolysis moieties, which could be stabilized as non-diffracting nanoscale domains in the lattice.20,192 The formation of hyperstoichiometric PuO2 in the bulk state remains controversial and the question of its occurrence at the nanoscale appears highly relevant. The topic is indeed of paramount interest for the long-term storage and environmental behavior of Pu, particularly because of the undesirable generation of hydrogen gas under storage conditions and the potential easier leaching of higher oxidation states of Pu in the poorly reactive oxide material.115,190
| PuO2 (s) + xH2O (g) → PuO2+x (s) + xH2 (g) | (14) |
The Pu L3 edge EXAFS spectra acquired on PuO2 nanoparticles generally show low period oscillations corresponding to the short Pu–O shell at low k values (2–10 Å−1) and higher period oscillations corresponding to Pu–Pu interactions at longer distances for high k values (8–16 Å−1). Both features agree with what was reported for EXAFS characterization of bulk PuO2.96,115,193,194 The FT observed for those signals show two peaks located at ca. R+φ = 1.8 Å and 3.7 Å (uncorrected for the phase-shift) and agreeing with the Pu–O and Pu–Pu coordination shells observed in bulk PuO2 with corrected distances located at 2.33 and 3.81 Å, respectively. Table 2 provides the structural parameters obtained from EXAFS fit for pure phase PuO2 bulk oxides used in select studies. The fit of the second-nearest-neighbor shell generally obtained for Pu oxide nanoparticles agrees with distances of ca. 3.8 Å typically observed in PuO2 thus demonstrating the preservation of the Pu–Pu pair distribution in the nanoparticles. Interestingly, this value appears to be poorly influenced by the nanoparticle properties, origin and history (size, synthesis approach, state, O-shell contribution, etc.) thus confirming that a rather rigid Pu–O–Pu skeleton dominates in the nanoparticle structure.83,193 However, the resolution of the EXAFS technique does not make it possible to account for the fine effects observed in the cell parameters with XRD (e.g.Fig. 12).
| PuO2 NP category | PuO2 NP characteristics (synthesis route) | Characteristics of the best fits | Pu–O | Pu–Pu | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| R Pu–O (Å) | CN Pu–O | DWFPu–O (×10−3 Å2) | R Pu–Pu (Å) | CN Pu–Pu | DWFPu–Pu (×10−3 Å2) | ||||
|
a Fixed values.
b Taken from a series of hydrolytic colloids showing different Pu:HNO3 ratios (5–10 mM:21–42 mM).
|
|||||||||
| Bulk reference | PuO2.00 (thermal conversion of Pu(IV) oxalates, 650 °C, 1 h, 10–20 m2.g−1) | Single O-shell | 2.33(1) | 8 | 6.5(4) | 3.81(1) | 12 | 4.9(1) | 196 |
| S 0 2 = 0.9, k = 2.4–14 Å−1 | |||||||||
| PuO2 ref (thermal conversion of oxalates, 650 °C) | S 0 2 = 0.9, k = 2.5–15 Å−1 | 2.33a | 8a | 5.9 | 3.81(1) | 10.5(19) | 3.5(6) | 94 | |
| Colloids and nanoclusters | Pu38 nanocluster | — | 2.33a | 8a | 14.9(12) | 3.80(1) | 4.4(14) | 10(1) | 94 |
| PuO2 in contact with H2O | Single O-shell | 2.321(4) | 8 | 9.0(5) | 3.816(2) | 12 | 3.5(2) | 115 | |
| Pu(IV) intrinsic colloids (addition of NaOH to Pu(IV) solution) | Single O-shell | 2.294(8) | 8 | 8.1(5) | 3.793(3) | 3.9(5) | 3.6(9) | ||
| Aged Pu(IV) intrinsic colloids (addition of NaOH to Pu(IV) solution) | Single O-shell | 2.267(3) | 8 | 15.2(6) | 3.779(4) | 1.7(3) | 1.9(13) | ||
| Pu(IV) intrinsic colloids (controlled hydrolysis)b | Single O-shell | 2.33a | 8a | 14.9(7)–16.7(10) | 3.80(1)–3.81(1) | 5.0(17)–5.7(13) | 5.9(7)–6.7(8) | 94 | |
| Fourier filtered S02 = 0.9, k = 2.5–15 Å−1 | |||||||||
| Pu(IV) intrinsic colloids (sonication of PuO2 nanopowder) | Single O-shell | 2.33a | 8a | 7.8(6) | 3.82(1) | 8.6(18) | 3.7(10) | 94 | |
| Fourier filtered S02 = 0.9, k = 2.5–15 Å−1 | |||||||||
| Pu(OH)4 (Am) (addition of NaOH to Pu(IV) solution) | Single O-shell | 2.32 | 4.0 | 10.4 | 3.87 | 2.4 | 6.6 | 83 | |
| S 0 2 = 1.0, k = 2.7–12.5 Å−1 | |||||||||
| R f = 0.016 | |||||||||
| Pu(IV) intrinsic colloids (addition of H2O, NaOH or NaCl solution to Pu(IV) solution) | Two O-shells | 2.20–2.24 | 0.7–3.3 | 0.4–8 | 3.86–3.9 | 2.1–4.9 | 7.5–9.0 | 83 | |
| S 0 2 = 1.0, k = 2.7–12.5 Å−1 | 2.38–2.42 | 0.8–5.6 | 5.9–13.6 | ||||||
| R f = 0.004–0.013 | |||||||||
| Dispersed or associated nanoparticles | PuO2 nanopowder (thermal conversion of hydroxides, 485 °C) | Single O-shell | 2.33a | 2.41a | 8.9(10) | 3.80(2) | 9.5(13) | 4.9(7) | 116 |
| S 0 2 = 0.9, k = 2.0–14 Å−1 | |||||||||
| R f = 0.045 | |||||||||
| Nano-dots and nano-rods (solvolysis in BnOBn/OA/OAm mixture) | Single O-shell | 2.31(1) | 6.6(0.8) | 9(2) | 3.81(1) | 8.1(15) | 2(1) | 117 | |
| S 0 2 = 0.9, k = 2.8–10.9 Å−1 | |||||||||
| R f = 0.025 | |||||||||
| Monodisperse PuO2 (addition of NH3·H2O to Pu(III–V) solutions) | Single O-shell | 2.30(1)–2.32(1) | 7.2–7.8 | 10a | 3.79(1)–3.81(1) | 3.2–4.5 | 4.3(5)–7.1(5) | 111 | |
| S 0 2 = 0.95, k = 2.0–14 Å−1 | |||||||||
| Two O-shells | 2.26(1)–2.30(1) | 5.5–7.4 | 7.6(5)–9.7(5) | 3.79(1)–3.81(1) | 3.2–4.5 | 4.3(5)–7.1(5) | 111 | ||
| S 0 2 = 0.95, k = 2.0–14 Å−1 | 2.39(1)–2.44(1) | 0.6–2.5 | 1.0(5)–4.3(5) | ||||||
| Three O-shells | 1.81(1)–1.84(1) | 0.2–0.5 | 1.0(5) | 3.79(1)–3.81(1) | 3.2–4.5 | 4.3(5)–7.1(5) | 111 | ||
| S 0 2 = 0.95, k = 2.0–14 Å−1 | 2.28(1)–2.29(1) | 3.7–6.1 | 3.2(5)–8.3(5) | ||||||
| 2.42(1)–2.44(1) | 1.5–2.3 | 1.0(5)–5.3(5) | |||||||
| Nanostructured oxides | Nanostructured PuO2 powder (thermal conversion of oxalates, 485 °C) | Single O-shell | 2.33a | 2.41a | 7.0(11) | 3.81(1) | 11.8(18) | 5.6(6) | 116 |
| S 0 2 = 0.9, k = 2.0–14 Å−1 | |||||||||
| R f = 0.045 | |||||||||
5.2. Probing the Pu–O coordination shell with EXAFS
The Pu–O shell observed for the nanoparticles generally matches the general description of its PuO2 counterpart but is often characterized by a distorted peak suggesting Pu–O bond length splitting in some reports. The presence of multiple sites for the Pu–O distances remains the subject of discussions due to a certain analogy reported for some nanoparticles and PuO2+x. Particularly, the observation of a shoulder at R+φ = 1.9 Å in the FT of the Pu L3 edge EXAFS spectra of PuO2 colloids suggested structural similarities and the potential contribution of other Pu oxidation states to the structure of the nanoparticles.40,45,192 Also, different oxidation states might be observed in XAS data without necessarily coming from the nanoparticle itself but due to a potential contribution of soluble ionic species from the solution.113 The interpretation of the Pu–O shell is difficult and challenging because several external parameters can affect the data treatment around that distance involving the generation of artefacts or misinterpretations which complicates the comparison between the authors. Particularly, the chosen wavevector range, the influence of the Pu–Pu coordination sphere on the spline removal or the double electron excitation signal observed in the L3 edge spectra of An have been suggested to spuriously modify the Pu–O shell contribution to the EXAFS signal.94,156,195 These external parameters are added to the intrinsic properties of the studied nanomaterials which are also known to significantly impact the EXAFS spectra.156 The observation and interpretation of the atomic scale variations of the nanoparticles are therefore tricky and must be considered with caution.Several approaches have been proposed in the literature to fit the L3 EXAFS spectra of nanosized Pu oxides. The simplest model consists of using a single Pu–O shell assuming a PuO2-like structure with Pu–O and Pu–Pu distances respectively located at 2.31 and 3.81 Å. This model is often favored because only two distances are used to fit the data and the potential contribution of other distances is not explained but rather counterbalanced with an increased Debye Waller factor (DWF) parameter resulting in an important local disorder around the absorbing atom when compared to the bulk PuO2 reference. Such an approach has been successfully used to fit high-fired oxides with rational and consistent structural parameters (see for instance Martin et al.,196Table 2). By using a single Pu–O shell, Ekberg et al. obtained good similarity between the fitted structural parameters they obtained for bulk PuO2 and their nanoparticles with quite a high Debye–Waller factor coefficient of 8.1 × 10−3 Å2 for the Pu–O shell which increased to 15.2 × 10−3 Å2 when the particles were aging (see Table 2 for the structural parameters). This way, the Debye Waller factor provides overall information about the local structural disorder resulting from both the thermal (i.e., dynamic or vibrational) and static (i.e., configurational) displacements of atoms from their ideal lattice positions.197,198 The presence of lattice strains and defects including, for instance, dislocations, steps, insertions, vacancies, etc. would contribute to the increase of this parameter.
Hudry et al. reported structural similarities between L3 EXAFS spectra acquired for bulk and nanoscale PuO2 with fitted distances located at 2.31(1) and 3.81(1) Å and coordination numbers at 6.6(8) and 8.1(15) for the Pu–O and Pu–Pu shells, respectively (Table 2).117 The decrease in the peak amplitudes and the high DWF parameters obtained for the Pu–O shell (9 × 10−3 Å2) in comparison to the bulk were attributed to a nanoparticle size effect resulting from a higher proportion of probed surface atoms in comparison to the bulk. This effect is well known for other materials and explains the lower coordination and higher degree of disorder. Gerber et al. confirmed this effect by analyzing the structural parameters obtained with a single Pu–O shell (Table 2) on PuO2 nanoparticles synthesized by ammonia addition into Pu(III) to Pu(V) aqueous solutions (Fig. 14). Note, the absence of Pu–O shell splitting was mentioned by the authors. Although the model well reproduced the data, very high DWF values were observed for the Pu–O shells of the various samples (10 × 10−3 Å2 as an upper constrained limit). The lower coordination numbers and higher DWF observed for both shells were also attributed to a nanoparticle effect when compared to bulk PuO2.111 More generally and in combination with other techniques, these examples confirm the observation of PuO2 nanoparticles with a high degree of local disorder as evidenced by the decreased coordination numbers and strong DWF parameters.
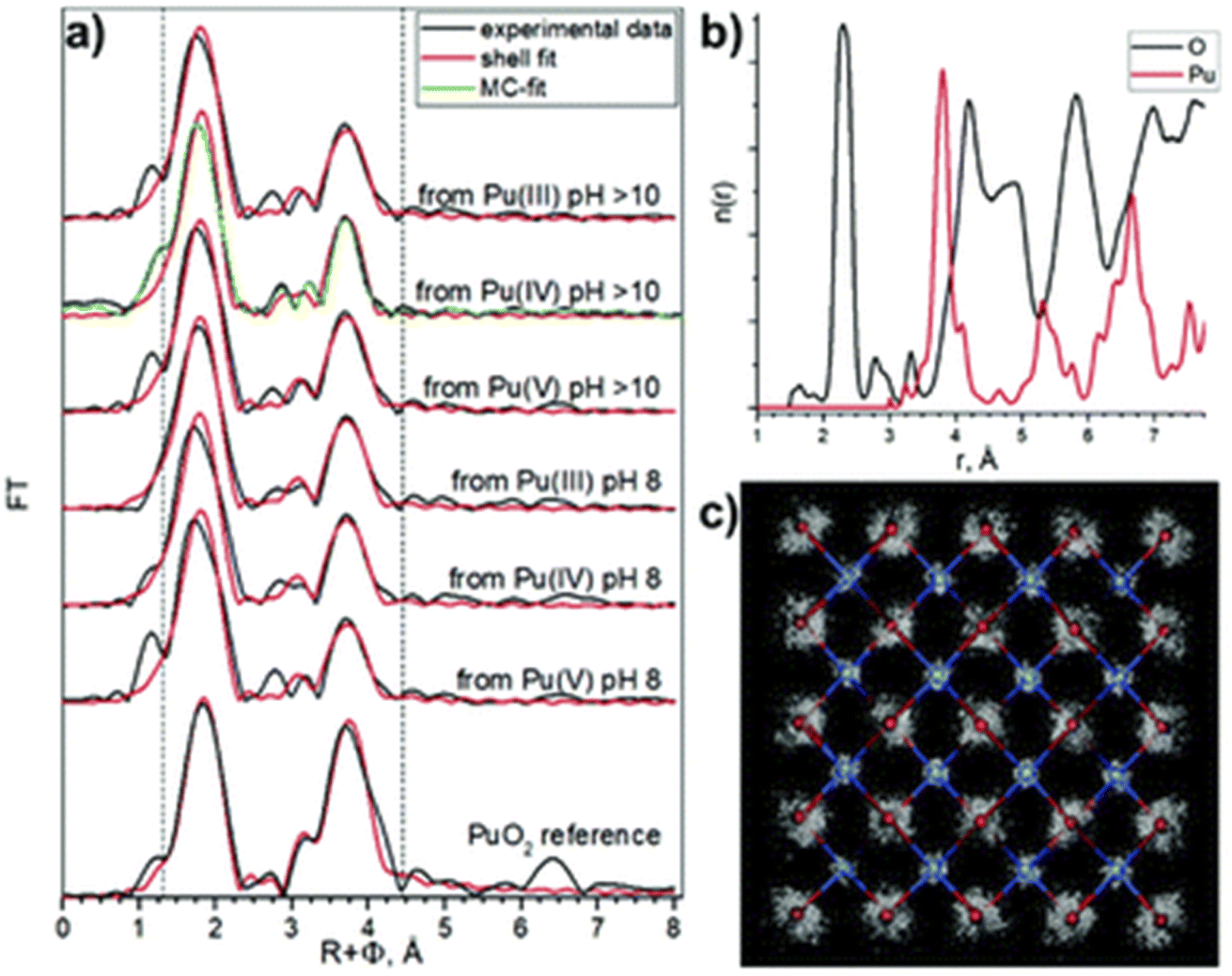 | ||
| Fig. 14 (a) FT magnitude of experimental Pu L3 EXAFS spectra (black) acquired on PuO2 nanoparticles prepared by ammonia addition into Pu(III)–Pu(V) aqueous solutions. χ(R) fit results obtained with one Pu–O and one Pu–Pu shell fit are represented in red and Monte-Carlo simulation fits are represented in green. The two vertical dashed lines represent the area of the shell fit. (b) Radial particle distribution function (n(r)) for Pu–O and Pu–Pu obtained for Monte-Carlo simulation and (c) 3D structural refinement based on bulk PuO2 structure. Reproduced from ref. 111 with permission from the Royal Society of Chemistry, copyright 2020. | ||
In comparison to the single Pu–O shell fit, Gerber et al. also experienced two and three Pu–O shell models to fit their data. The three shell model was based on the contribution of Pu(IV)–O (R = 2.43 Å) and Pu(IV)–OH (R = 2.28 Å) distances, in addition to a short Pu(V)–O one standing at R = 1.83 Å that would add approximately 15% of Pu(V) in the nanoparticles according to the structural data obtained from the fit. The latter model was rejected due to inconsistencies with the XANES results acquired in High Energy Resolved Fluorescence Detection (HERFD) mode with respect to the oxidation states in the nanoparticles. The two shell model excluded the Pu(V)–O path with good results but the high dispersion of the obtained structural parameters and the inconsistencies observed with the fit acquired with a similar approach for bulk PuO2 forced the authors to also exclude this approach. The report finally concluded that the best model to fit the data acquired from their nanoparticles is a PuO2 structure with only one Pu–O and one Pu–Pu scattering paths. Monte-Carlo EXAFS simulations agreed with this approach (Fig. 14). From the Monte-Carlo simulations, the PuO2 disorder measured by EXAFS could only be attributed to structural distortion in the PuO2 nanoparticle core since the surface effect is not taken into account.
Alternative studies were based on a multisite model to describe Pu(IV) colloids based upon the formula PuO2+x−y(OH)2y·zH2O derived from the perfectly ordered stoichiometric PuO2 which would only exist in the total absence of H2O and at low activity of oxygen.193 Here, Pu(IV) colloids are described as Pu(IV,V) oxyhydroxide compounds having identical diffraction patterns to PuO2 with evidence of disorder and exhibiting a possible slight increase of the lattice parameter (when x > 0).192,199 The multisite distribution is supported by several discrete Pu–O distances reported to range from 2.25 Å typical of terminal Pu–OH hydroxo moieties to more than 3 Å while keeping the Pu–Pu distances identical to bulk PuO2.200 When necessary, the excess of oxygen is explained by the addition of Pu(V)–O groups with observed distances in the 1.83–1.91 Å range and a multisite distribution in the first Pu–O shell which is retained even if x = 0. The formation mechanism of the colloids would result from irregular surface- nucleated precipitates. The formation of bridges through hydroxyl, water molecules and oxide bonds to accommodate the as-induced strains would result in a disordered structure with a Pu–Pu distribution agreeing with PuO2. The protons from hydroxo and aquo ligands would be introduced at the origin of intrinsic vacancies or cation substitutions in the structure.200
Comparing a 1 mM Pu(IV) solution hydrolyzed in 0.5 M NaCl (pH = 0.56) with Pu precipitates obtained by the addition of NaOH led to the observation of a strong decrease in the coordination numbers for the Pu–O shell correlated with quite high DWF measuring 9.4–10.4 × 10−3 Å2.83 For a solution prepared by the hydrolysis of almost similar concentrations of Pu(IV) with H2O, NaOH or NaCl solutions, Rothe et al. observed that the increase of the pH of the solution was associated with an increase followed by a decrease in the degree of splitting for the Pu–O shell. This behavior was correlated with an increase in the coordination numbers for the second neighboring Pu–Pu coordination shell.83 Nevertheless, the Pu–O shell was treated with two shell models that highlighted two contributions in the 2.20–2.24 Å and 2.38–2.42 Å range distances. The splitting of the Pu–O shell was reported to be reduced for the sample prepared at the highest pH (1.75) with moderate addition of NaOH. The Pu–O shell disorder was explained by different oxygen-based ligands including oxo O2−, hydroxo HO- and H2O moieties that could not be discriminated by the technique. The corresponding Pu–Pu distances only varied by 0.05–0.09 Å indicating a poor influence of the Pu–O shell disorder on them and a rigid Pu–O–Pu basic structure for the nanoparticles. However, the increase in the Pu coordination number for the second shell with increasing pH and particle size suggested an increasing order in the colloid thus supporting the proposed mechanism involving the condensation of Pu oligomers.
The characterization of PuO2 intrinsic colloids and nanoparticles exhibiting different sizes by L3 edge EXAFS spectroscopy revealed FT with different degrees of distortion and splitting for the Pu–O shell.96 Dalodiere et al. observed that these features were found to be strengthened for the smallest PuO2 nanoparticles (Fig. 15). The fit of the EXAFS data was inspired by and constrained to match with the Pu38 cluster model that involves 3 Pu–O distances: short (1.93–2.23 Å), medium (2.23–2.63 Å) and long (2.63–3.13 Å) Pu–O distances. The resulting coordination numbers observed for Pu–Pu and Pu–O at distances typical of bulk PuO2 (i.e., the medium ones located at 2.23–2.63 Å) were found to decrease with the decreasing size of the nanoparticles, whereas the short Pu–O distances were found to gradually increase using this model. These observations were combined with the absence of significant contributions from oxidation states differing with Pu(IV) using L3 XANES, which allowed attributing the short Pu–O distances to a surface effect most probably resulting from terminal Pu–OH moieties and/or μ3-O atoms that may reside on the edge of the particle. In accord with the HR-TEM experiments which evidenced the crystalline nature of the particles in agreement with the PuO2 fluorite structure, these results allowed describing these nanoparticles (intrinsic colloids and nanostructured oxide) as core–shell particles made out of a crystalline PuO2 core recovered by a thin layer of hydrolyzed and μ3-O moieties. In addition, a stronger influence of hydrated and/or hydrolyzed forms of Pu(IV) was suggested by oxygen K-edge NEXAFS spectroscopy for the smallest nanoparticles whereas the biggest colloids resembled bulk PuO2 to a greater extent.96 A change in the surface to core atomic ratio was proposed to explain these results. Concomitant to the O-shell, the S/V ratio was also found to be correlated with the Pu–Pu coordination number which decreased with the nanoparticle size as observed in Fig. 15.
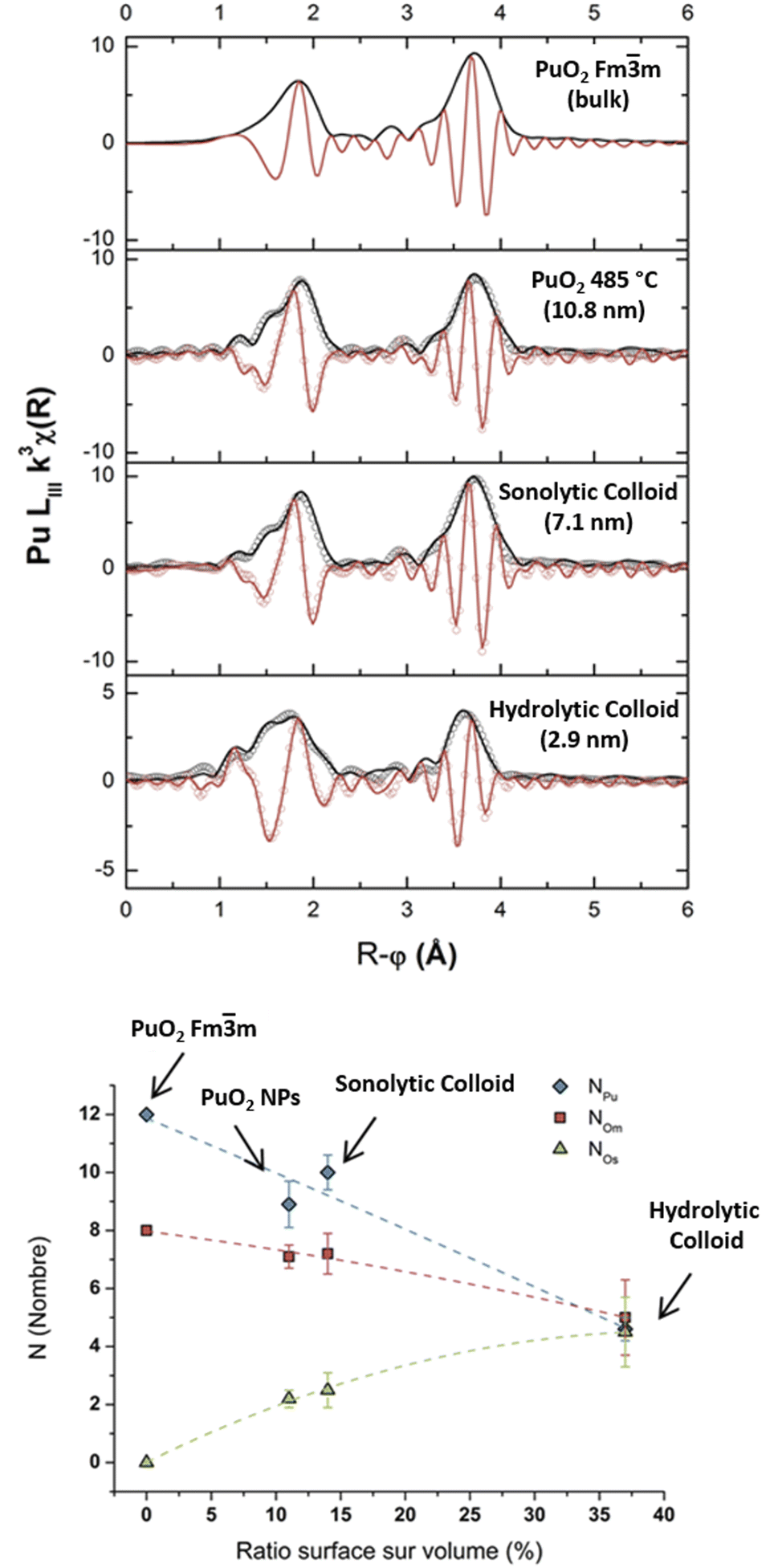 | ||
| Fig. 15 Top: FT magnitudes of experimental k3-weighted EXAFS spectra acquired at the Pu L3 edge on PuO2 nanoparticles in comparison to a reference (black curve, data obtained in the 2–14 Å−1 range). Bottom: correlation of the coordination numbers obtained for the three Pu–O distances used to fit the Pu–O shell as a function of the S/V ratio of the nanoparticle. Adapted from ref. 96 with permission from Nature Publishing Group, copyright 2017. | ||
The recent EXAFS comparison of some PuO2 and ThO2 nanoparticles gave strong evidence for the absence of the contribution of other oxidation states of Pu in the selected PuO2 nanoparticles.116 In analogy with PuO2, ThO2 crystallizes in the fluorite structure of the cubic face centered system (Fmm space group) and therefore shows comparable FT for the EXAFS signal (i.e. An–O and An–An shells). Despite their ionic radii difference (1.05 Å for Th(IV) against 0.96 Å for Pu(IV) in 8-fold coordination), using Th for comparison is interesting since this element exists under these conditions only in the (+IV) oxidation state and the potential contribution of redox reactions (in particular higher oxidation states) is therefore excluded. ThO2 and PuO2 nanoparticles as aggregated nanoparticles, nanostructured materials or colloids were studied with XAS. Both samples generally exhibited FT EXAFS spectra that agreed with their bulk counterparts. As previously described, both oxides additionally showed a gradual decrease in the EXAFS shell intensities coupled to a distortion of the An–O coordination sphere with the shrinking nanoparticles composing the samples (Fig. 16a).116
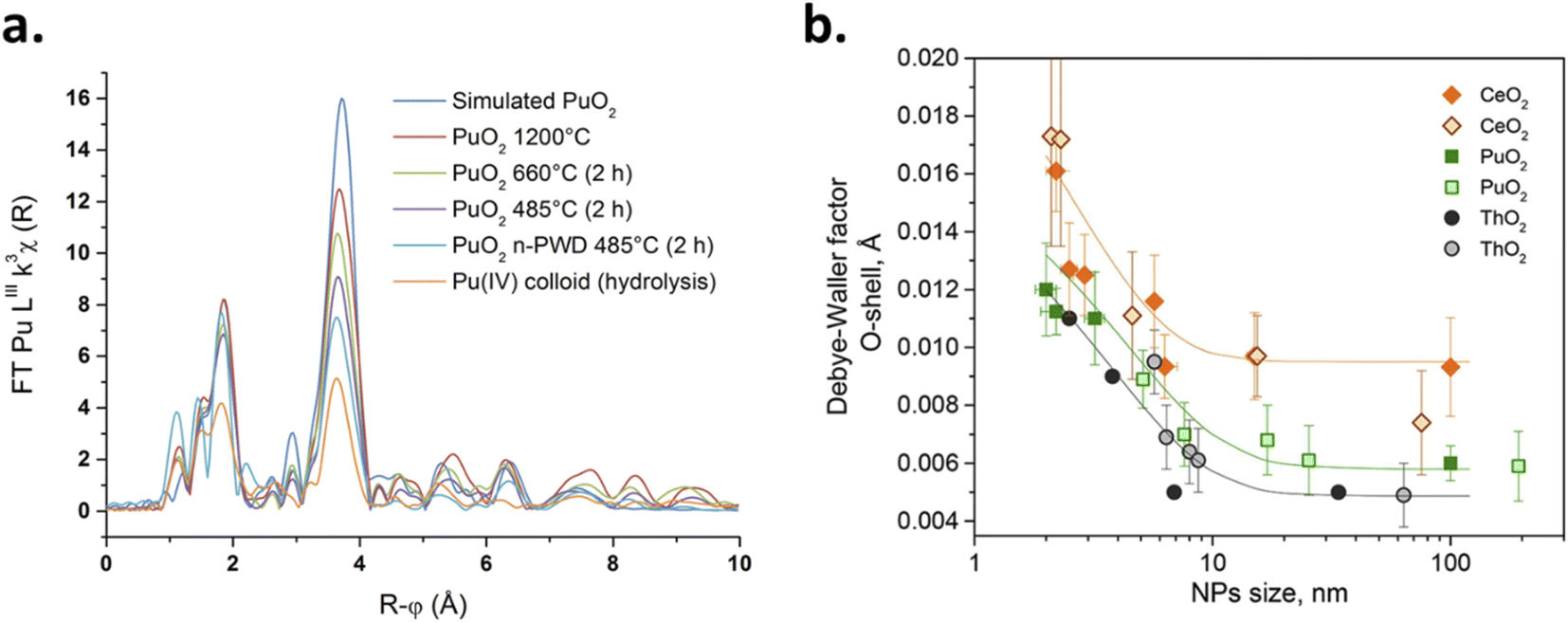 | ||
| Fig. 16 (a) FT of the k3-weighted EXAFS spectra observed for a selection of PuO2 samples exhibiting different sizes. Reproduced from ref. 116 with permission from the Royal Society of Chemistry, copyright 2020. (b) Observation of the dependency of the DWF of the Pu–O sphere as a function of the nanoparticle size for CeO2, PuO2 and ThO2 samples. A literature survey was used by the author to plot the figure. Adapted from ref. 201 with permission from the International Union of Crystallography, copyright 2022. | ||
The data treatment procedure was standardized in order to facilitate the comparison between the samples and avoid the generation of artefacts. Briefly, the An–O shell was Fourier filtered and a single An–O scattering path was used to fit the DWF parameter for an ideal 8 coordinated An–O shell. Such an approach provided An–O distances agreeing with the respective oxides in addition to the observation of a striking correlation between the size-decreasing nanoparticles and the extracted DWF parameters, which were the only floating parameters for this shell and allows probing the overall local disorder around the absorbing atom.116 The trend of the DWF was found to correlate with the nanoparticle size and was very similar for both PuO2 and ThO2 oxides. The modification of the intensity of the EXAFS peaks was therefore correlated with a local structural disorder driven by the nanoparticle shrinking. In agreement with the XRD, TEM and EXAFS results which confirmed the AnO2 structure of the nanoparticles, this phenomenon was explained by a nanoparticle surface effect. Since Th oxidation state is only (+IV) under these conditions, the gradual modification of the An–O sphere observed for the AnO2 samples (decrease in intensity, distortion and splitting for the smallest particles) was attributed to the contribution of surface atomic sites and not to high-valence atoms in the measured samples, thus excluding the contribution of other Pu oxidation states in the selected PuO2 nanoparticles.116 Very recently, the above described procedure was repeated and it confirmed the dependency of the DWF parameter on the size of PuO2 and ThO2 nanoparticle samples prepared by different groups and using different synthesis routes (Fig. 16b).201 This effect was confirmed to be dramatically increased for particles smaller than 10 nm; moreover, the authors applied the fitting approach to CeO2 samples (isostructural oxide) and reached a similar conclusion. It is worth noting that the DWF followed the trend ThO2 < PuO2 < CeO2 (with a reported strong splitting of the Ce–O shell for the smallest particles).
5.3. Probing the Pu–Pu coordination shell with EXAFS
By comparing PuO2 nanoparticles of different sizes, recent investigations further supported the correlated variation of the coordination numbers determined for the Pu–Pu shell. Coordination numbers observed for stable intrinsic colloids ranged between 5.0–5.7 and 8.6 for ca. 2 nm and 5.7× > 30 nm2 particles, respectively, whose values were found to be framed between the ones observed for bulk PuO2 (CN = 10.5) and the Pu38 cluster (CN = 4.4) with homogeneous Pu–Pu distances observed at 3.81 ± 1 Å. This observation was attributed to a size-dependent surface effect for the probed samples (S/V atomic ratio) which can also be correlated with the increasing DWF parameter (i.e. the local structural disorder) when particles shrink.94,116 However, a comparison with the theoretical data for the Pu38 cluster and the bulk PuO2 references revealed an underestimation of the CN for the Pu–Pu sphere (Fig. 17). In particular, the structural theoretical parameters observed for the Pu38 cluster indicate CN = 7.58 (i.e. much higher than CN = 4.4 obtained by fitting the corresponding simulated spectra with a single Pu–Pu shell) with Pu–Pu interatomic distance in the 3.53–3.92 Å range. A comparison of the variation of the CN (Pu–Pu) experimentally observed for various PuO2 nanoparticles with theoretical calculations built from geometric models coming from SAXS observations demonstrated similar trends.This offset observed between the experimental and theoretical data highlights the limits of the model used to fit the Pu–Pu coordination sphere for nanoscale materials (Fig. 17a). In particular, CN lower than 6 should not be observed for spherical particles since it is the lowest value supposed to be observed for surface atoms in agreement with the Pu38 cluster. In addition to moieties that may decrease the Pu coordination numbers at the nanoparticle surface (e.g. μ3-oxo bonds, OH groups or adsorbed water molecules), several hypotheses were proposed to explain such a difference including a possible size polydispersity, surface roughness or the possible contribution of smaller Pu clusters.83,96,156,168,202 In particular, the latter (e.g. Pu6, Pu16, Pu22) have been reported to exhibit much lower coordination numbers for the Pu–Pu sphere (CN = 4, 5.23, and 5.36, respectively) and could contribute to the formation mechanism of larger clusters or particles.16,88,89 Finally, it is also worth noting that the distance distribution of the Pu–Pu bonds can significantly split for nanomaterials in agreement with Fig. 13. The average EXAFS oscillation therefore coming from core and surface probed atoms may interfere and decrease the amplitude of the total oscillations when measured with a single shell.
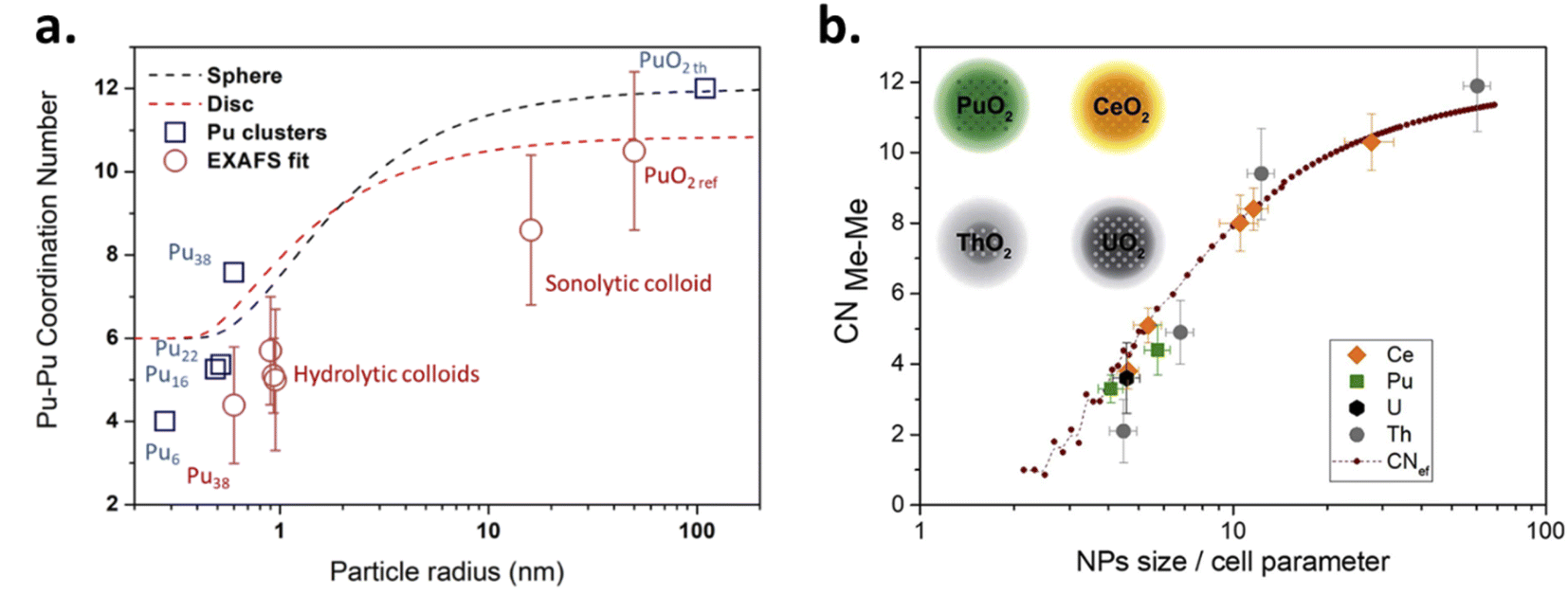 | ||
| Fig. 17 (a) Comparison of the size dependence of the Pu–Pu coordination sphere when comparing EXAFS experimental data with calculations from geometrical models for PuO2 nanoparticles. In the figure, red circles represent the experimental data: SAXS vs. EXAFS fit, whereas the blue squares result from theoretical calculations in agreement with the published crystallographic structures. Reproduced from Micheau et al.94 with permission from the Royal Society of Chemistry, copyright 2020. (b) Similar approach using a structural model described by the equation eqn (15) for PuO2 and CeO2 samples. Reproduced from ref. 201 with permission from the International Union of Crystallography, copyright 2022. | ||
The absence of correlation between experimental EXAFS and geometrical calculation approaches was later confirmed for the Pu–Pu coordination shell.201 A general structural model for MeO2 (Me = Ce, Th, U or Pu) was used to take into account this difference while considering core–shell structures for the nanoparticles and calculating an effective Me–Me coordination number (CNef) based on eqn (15) where Ncore and Ntotal are the number of Me atoms in the core and total structure, respectively (Fig. 17b). Using this approach, the authors were able to observe a very good agreement between the experimental and theoretical data, further suggesting a better EXAFS sensitivity of the ordered core in comparison to surface atoms. In addition, it allowed the authors to estimate the size of the core for each plotted nanoparticle, which was found to approximately correlate with the lattice parameter of the studied oxides (ca. 0.4 nm). Finally these correlations (Fig. 17b) confirmed similar structural properties for the various studied oxides CeO2, ThO2, UO2 and PuO2.165,201,203 It is worth noting that slight differences in comparison to the other oxides in terms of CN for the An–An sphere and DWF for the An–O sphere were explained by the more amorphous nature of ThO2 nanoparticles.
 | (15) |
5.4. General comments about EXAFS interpretations of PuO2 nanoparticles
In view of the above, multisite distribution models generally allow good reproducibility of the experimental data with however a very high number of structural parameters to interpret which goes with an increased uncertainty. The increasing number of variables requires a deep questioning of the subjacent crystallography without further evidence. Although the fitted distances generally agree with PuO2, the observed significant decrease in the intensity of both the Pu–O and Pu–Pu coordination spheres is most often interpreted by a decrease in the coordination numbers of the shells that can be explained by an increased local disorder. Most of the authors conclude that this increased disorder is related to the Pu–O coordination sphere and attribute it to the effect of the nanoparticle surface that increases when shrinking in agreement with the PuO2 crystalline structure of the nanoparticles revealed by other techniques (including HR-TEM).83,124 The transition between Pu clusters and bigger particles is also important to consider since the smallest objects appear to impact the Pu–Pu distances significantly, despite the rigid Pu–O–Pu structure supported by all of the recent results. Indeed, recent findings about PuO2 nanoparticles and small Pu(IV) clusters support a progressive distortion of Pu–Pu distances from the core to the surface.16,85 The crystallographic description of plutonium clusters well depicts what could be the atomic distribution in PuO2 nanoparticles. First of all, in the Pu38 structure the fluorite-like arrangements of Pu and O atoms in the core are preserved. Second, the surface plutonium atoms don't necessarily bond to hydroxo moieties but rather form μ3-oxo bridges that are shorter than the oxo bonds in the particle core by approximately 0.15 Å. Third, water molecules may bond to the surface plutonium atoms at longer distances. These surface oxobonds and water molecules therefore appear as good candidates to explain the distance distribution often observed for the Pu–O shell of PuO2 nanoparticles (either through a large DWF or a multi-shell model).Furthermore, it is interesting to point out the strong differences visually and structurally observed by the various authors in the EXAFS spectra acquired for the variety of available PuO2 nanoparticles (e.g. splitting of the Pu–O shell, differences in the CN or DWF). Ockenden demonstrated the influence of the solution aging and preparation route on the electronic absorption spectra observed for Pu(IV) colloids.65 In their relevant review, Walther and Denecke indicated that colloids prepared at low pH differ from those obtained under basic conditions with the observation of less ordered structures when the acidic Pu(IV) solution is quickly stabilized.40 Nevertheless, Gerber et al. observed that the addition of ammonia to Pu(IV) solutions yielded 2 nm spherical PuO2 nanoparticles whatever the pH of the resulting solution in the 1–10 pH range. HERFD at the Pu M4 edge demonstrated that Pu(IV) dominates among all of these nanoparticles whereas an additional contribution of Pu(III) and Pu(VI), originating from the solution, was noticed when the nanoparticles were formed under the lowest pH conditions (pH = 1).113
Zhao et al. recently investigated the chemical behaviour of PuO2 nanoparticles prepared by alkaline (neutralization of Pu(IV) solution by the addition of NaOH) and acidic (precipitated from heated Pu(IV) acidic solutions) routes.101 Although both types of particles offered comparable properties according to TEM (size from 2 to 4.5 nm with an estimated specific surface area in the 150–210 m2 g−1 range), the particles obtained under acidic conditions were found to form more ordered aggregates indicative of higher crystallinity which resulted in better stability of the particles regarding their dissolution in the presence of montmorillonite, a mineral of environmental significance. Such an observation strengthened the relevance of the nanoparticle formation conditions to their physico-chemical properties including the long term stability of intrinsic Pu colloids at environmentally relevant timescales.101 Interestingly, the authors also mentioned that the improved stability generally known to be provided by the thermal treatment for bulk PuO2 also appears to be true at the nanoscale for particles exhibiting similar size properties.
5.5. XANES spectroscopy
XANES spectroscopy has been demonstrated to be sensitive to the charge and the local geometry of absorbing atoms. For plutonium in nanosized objects, the absorption edge observed by Pu L3 XANES spectroscopy (which results from dipole allowed transitions occurring between the 2p3/2 core level and the empty 6d states) depends on several factors. The position and form of the edge as well as the form of the post-edge region may be modified by the redox state of the absorbing Pu ion, the presence of uncoordinated atoms and the strong contribution of the surface of the nanoparticles and their geometry (morphology, defects, edges, corners, etc.). Oxide nanomaterials can therefore be assumed to show significant variations from their bulk counterparts and display particular features in the XANES spectra as a result of size-induced structural and electronic properties.112,124,156,202,204,205 The energy position of the white line for Pu(III), Pu(IV), Pu(V) and Pu(VI) aqueous species when probed at the L3 edge has been determined at 18060.1, 18063.2, 18062.6 and 18064.8 eV, respectively. The difference between them is significant enough to discriminate the main Pu oxidation state in the PuO2 nanoparticles. However, since Pu L3 edge probes the 6d states which are identical for Pu(IV), Pu(V), and Pu(VI), it is not possible to affirm the exclusive contribution of only one of them, particularly for Pu(V) and Pu(VI).205 Also, the interaction of ligands with the nanoparticles, in addition to modifications of the local geometry, have been reported to possibly affect the edge energy.117,192 Nevertheless, the strong predominance of Pu(IV) oxidation state in PuO2 nanoparticles has been highlighted by most of the authors using L3 XANES spectroscopy in agreement with the work of Vitova et al. illustrated in Fig. 18 (left image).83,94,111,116,124 | ||
| Fig. 18 Pu L3 XANES (left) and HERFD-XANES (right) spectra comparing Pu(III)–Pu(VI) aqueous solutions with Pu(IV) colloids. Adapted from Vitova et al.124 with permission from the Royal Society of Chemistry, copyright 2018. | ||
The comparison of fresh and 5 years hydrolyzed clusters prepared by the NaOH neutralization of a Pu(IV) acidic solution by Ekberg et al. did not show any trace of Pu(VI).115 A strong decrease in the CN obtained for the Pu–Pu coordination sphere, along with a decrease in the mean Pu–O distance (CN = 8, with almost identical DWF), was linked to particle size and aging. This was explained by an evenly distributed oxidation of Pu(IV) without the formation of plutonyl species. These results were compared to PuO2 bulk particles (oxalic route) in contact with water, which showed a significant decrease of the fitted Pu–O distances (both first and second shells) in comparison to PuO2 references and was attributed to the potential oxidation of the surface. Rothe et al. studied the valence and electronic structure of Pu colloids obtained by dilution of acidic Pu solutions in aqueous solution and by precipitation with the addition of NaOH to acidic Pu(IV) solutions.83 Comparable L3 XANES profiles and WL positions were observed for all of the studied samples. The absence of significant energy shift or extra features that could be responsible for the presence of Pu(V) or Pu(VI) in the spectra supported the tetravalent state of Pu in these samples. Colloidal samples exhibited WL intensities that were intermediate between the one observed for Pu(IV) aqueous ion described by more localized 6d final states (molecular-like orbitals), and the one of the precipitate described with band-like structures. The decrease of the WL amplitude was therefore correlated with the pH increase of the solution expressed by a particle size effect in which the density of state tends towards the one observed for bulk PuO2 samples when particles grow.83 Several authors evidenced the damping of the WL and post-edge region intensities in EXAFS when analyzing PuO2 nanoparticles.117 Walther et al. have also observed a slight decrease in the Pu L3 XANES WL intensity during the aging of intrinsic colloids. EXAFS spectroscopy indicated the ripening of these colloids into crystalline PuO2 structures strengthened with a decrease in the Pu–Pu distances and an increase in the coordination numbers related to the Pu–O and Pu–Pu shells.46 Note that some authors reported an inverse effect when comparing Pu colloids with bulk PuO2 references with a higher intensity of the WL for the latter.94,117 Therefore, this L3-edge intensity difference observed between PuO2 nanoparticles and the bulk remains difficult to explain since other experimentally dependent parameters including self-absorption may contribute to the intensity variations on the absorption edge.
Recent investigations demonstrated the dramatic increase of the resolution of the obtained spectra by monitoring XANES in High-Energy Resolution Fluorescence Detection (HERFD) mode. The enhanced quality of the data results from the elimination of the core–hole lifetime broadening due to the reduction of the experimental spectral width. Detailed information about the oxidation state, electronic structure and local geometry can be obtained using this approach.206 A recent review describes the relevant state of the art of the technique.207Fig. 18 (right image) provides HERFD-XANES spectra for Pu(IV) colloids acquired at the Pu L3 edge against Pu(III)–Pu(VI) in aqueous solutions. In comparison to conventional XANES spectra, HERFD mode allowed the observation of an increase in the intensity of the WL and better resolution.124 The rigorous comparison of the Pu L3 XANES and HERFD-XANES experimental results confirmed the absence of energy shift between Pu(IV) colloids and aqueous Pu(IV) thus confirming the strong predominance of the tetravalent state for the former. Using additional computational data to calculate the XANES spectra, the authors further explained the WL intensity reduction and asymmetric shape observed for the Pu(IV) colloid by a condensation effect of the Pu 6d states. The results also suggest the splitting of the Pu d states as a result of the cubic crystal field thus explaining the band-like structure observed for the Pu(IV) colloids contrasting with the narrower and better defined d state observed for aqueous Pu(IV).124
While the Pu L3 edge is measured in the hard X-ray regime (∼17–18 keV), the emerging possibility of probing the actinides at the M4,5 edges (3.5–4 keV) recently allowed the authors to obtain very interesting information on PuO2 nanoparticles. In comparison to the L3 edge (which allows probing empty 6d states), measuring Pu XANES spectra in the tender X-ray range has been reported to be more instructive about the electronic structure and oxidation state of An. Indeed, measuring M edges allows probing the 5f density of states (3d–5f electronic transitions) which are closer to the Fermi levels.111,114,141,207 Significant differences have been observed when comparing the shape of the HERFD-XANES spectra acquired for PuO2 nanoparticles at the M4 or M5 edges.114 This has been attributed to dipole selection rules leading to the reaching of different 5f electronic levels by the excited electrons (M5: J = 5/2 or 7/2; M4: J = 5/2 only). Fig. 19a displays HERFD-XANES spectra acquired at the Pu M5 edge for Pu(IV) colloid, bulk PuO2 and an aqueous Pu(IV) solution contaminated with ab. 6% Pu(VI). The significant contribution of Pu(VI) traces can be noticed on the aqueous Pu(IV) spectra with a characteristic high energy shift (feature A) and the formation of a characteristic feature (B) specific to Pu(VI). The absence of the latter in the colloid spectra strongly suggested the absence of Pu oxidation states other than (IV). The analysis in the post-edge region (feature C) for bulk PuO2 and Pu colloid confirms structural similarities for both samples. Nevertheless, the observation of a 0.5 eV energy shift for the WL was attributed to a nanoparticle surface effect possibly related to the presence of a tiny amount of Pu(V) or Pu(VI), surface ionic bonds (e.g. Pu–OH, Pu–OH2), or O vacancies.124 It is important to note that the preparation of pure Pu solutions (aqueous or colloidal) generally used as references is tricky and the resulting solutions described in the literature are sometimes contaminated by a few percent of other Pu oxidation states thus potentially disturbing the results and interpretations.
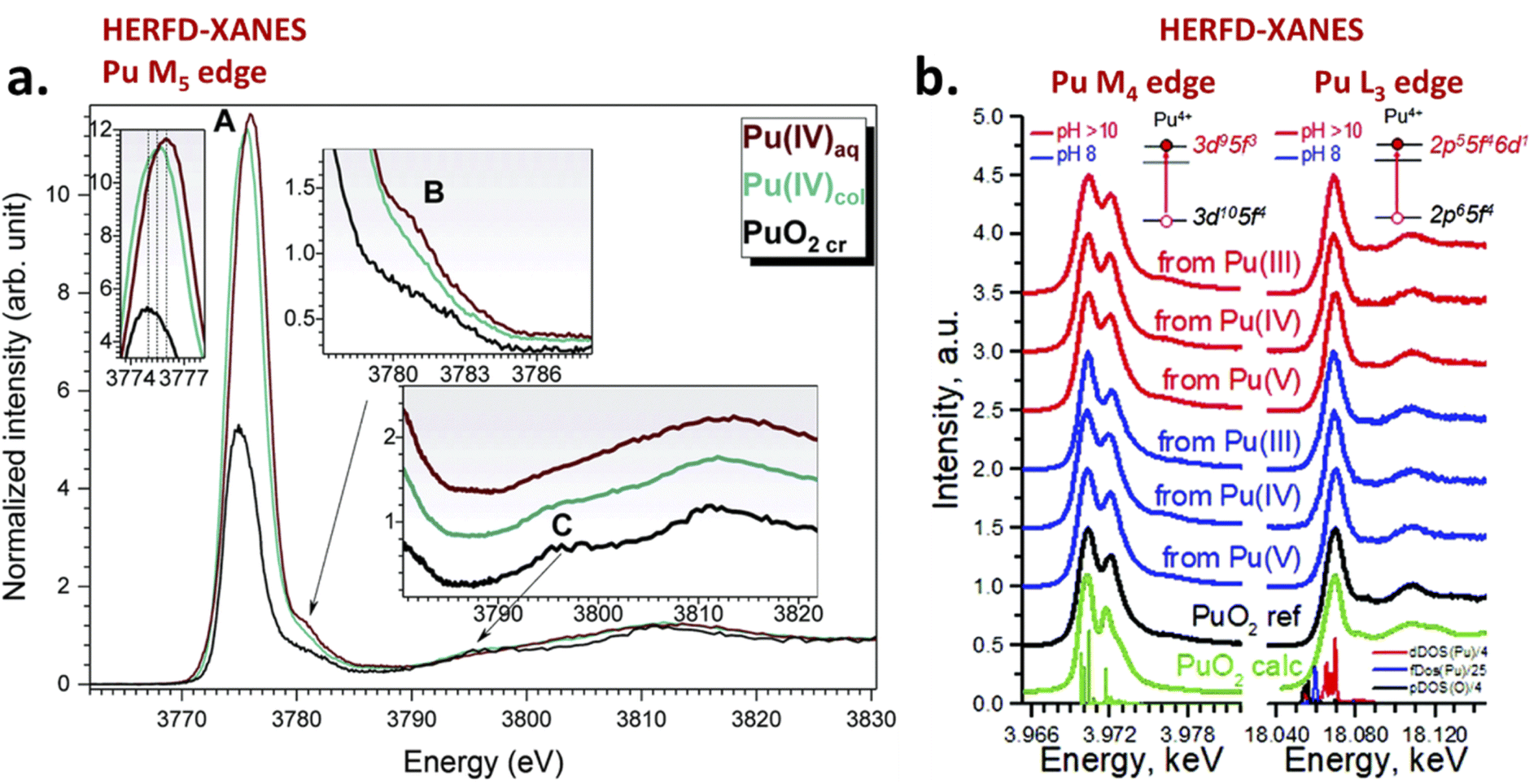 | ||
| Fig. 19 (a) HERFD-XANES spectra acquired at the Pu M5 edge for aqueous Pu(IV) containing 6% Pu(VI), bulk PuO2 and Pu(IV) colloids. Adapted from Vitova et al.124 with permission from the Royal Society of Chemistry, copyright 2018. (b) HERFD-XANES spectra acquired at the Pu M4 edge for PuO2 nanoparticles prepared by the addition of ammonia to Pu(III–V) aqueous solutions. Adapted from Gerber et al.111 with permission from the Royal Society of Chemistry, copyright 2020. | ||
The Pu M4 and L3 edges HERFD-XANES spectra acquired on PuO2 nanoparticles prepared by the addition of ammonia to Pu(III), Pu(IV) and Pu(V) solutions recently ruled out the potential contribution of other oxidation states to these nanoparticle structures.111 In agreement with computational approaches, the L3 edge HERFD-XANES spectra acquired on six samples of PuO2 nanoparticles confirmed the strong predominance of the Pu tetravalent state and did not evidence any convincing variation when compared with a bulk reference (Fig. 19b). Thus, the observations agreed with the PuO2 structure interpretations extracted from XRD, HR-TEM and HEXS techniques. Note that a slight difference in the post-edge region (18.09 keV) was observed and attributed to a nanoparticle surface effect due to the sensitivity of the region for decreases in CN in agreement with analogous investigations carried out on ThO2 nanoparticles.204 Interestingly, the authors also mentioned the absence of variations in their repeated measurements after 4 months (samples encapsulated as wet pastes in sample holders) which is particularly interesting when compared with other actinide oxides, for instance UO2 which has been demonstrated to partially oxidize and grow in size.203 Probing the Pu M4 edge with the HERFD-XANES technique allowed the authors to go deeper into the comparison by examining the 5f crystal field splitting with an Anderson impurity model (SIAM, Fig. 19b). In addition to the white line shape and position, similar spectral features were indeed observed between the nanoparticles and the bulk PuO2. This particularly includes the observation of a shoulder at high energy which agreed with the theoretical calculations thus discarding the potential contribution of other Pu oxidation states to the nanoparticle structure.111,114,207 The technique also allowed the discrimination of the additional contribution of Pu(III) and Pu(VI) aqueous ions in colloidal dispersions of PuO2 nanoparticles prepared at low pH.113
Recently, it was described the transformation of Pu(VI) into PuO2 nanoparticles in the presence of an excess of ammonia (pH = 11). During the process, Pu M4 edge measurements evidenced the formation of a metastable Pu(V) intermediate similar to NH4PuO2CO3.114 Kinetic measurements supported by HERFD confirmed the two-step process for the formation of the nanoparticles (Pu(VI) → Pu(V) → Pu(IV)) and the exclusive presence of Pu(IV) – and the absence of Pu(V) – in the finally formed PuO2 nanoparticles of ca. 2 nm (Fig. 20).114,207
 | ||
| Fig. 20 (a) Formation kinetics of the transformation of aqueous Pu(VI) into PuO2 nanoparticles through a metastable Pu(V) intermediate (conditions: 6 × 10−5 M of Pu in excess of ammonia, pH 11) with crystal structures of the formed phases (inset). (b) Corresponding HERFD XANES spectra acquired at the Pu M4 edge for the intermediate Pu(V) phase and the final PuO2 nanoparticles compared with references. Reproduced from ref. 114 with permission from Wiley-VCH Verlag GmbH & Co., copyright 2019. | ||
6. Going beyond the fundamental properties of PuO2 nanoparticles
The size vs. property relationship of PuO2 nanoparticles appears less investigated in comparison to studies related to their synthesis approach and fundamental electronic and structural characterizations. In contrast to bulk PuO2 which is known to be a hard, dense and chemically poorly reactive material generally showing an olive green color, interesting features have been nevertheless mentioned in the literature for the nanosized analogues. Popa and Walter indicated that PuO2 nanoparticles prepared under hot compressed water conditions have a lighter green color.71,118 By contrast, the PEG/ammonia route used for the preparation of PuO2 nanoparticles116 leads to the observation of a very smooth black color powder which contrasts with conventional observations made for bulk PuO2 (Fig. 21). Recently, PuO2 nanoparticles obtained by auto-radiolytic decomposition of Pu oxalate compounds were found to exhibit a variety of colors despite evidence of similar preparation and storage conditions for the precursors (room temperature in air) and similar properties for the obtained nanocrystals (FTIR, XRD, UV-vis spectroscopy).20,120 It is noteworthy that the authors reported the formation of PuO2 nanocrystals from both Pu(III) and Pu(IV) oxalates. The few examples related to the variability of powder colors strongly suggest that nanosizing PuO2 causes a significant impact on the electronic transitions responsible for optical absorption which is most probably related to surface characteristics. It is also interesting to note that unusual colors (dull yellow, buff, kaki, black…) have been reported for bulk PuO2 depending on the synthesis route, chemical purity, particle size and stoichiometry.45,208 Reflectance spectroscopy might be used to report these interesting observations and further advances are expected in the domain. | ||
| Fig. 21 Pictures from PuO2 nanoparticles prepared by (a) firing at 485 °C of precursors obtained by the PEG/ammonia route116 (personal picture); (b and c) decomposition of Pu(IV) oxalates by auto-radiolysis over 14.5 months;120 (d and e) decomposition of Pu(III) oxalates by auto-radiolysis over 16 and 15 months, respectively.120 Figures (b–e) were reproduced from ref. 120 with permission from John Wiley and Sons, copyright 2021. | ||
Through magnetization experiments, the nanosize-induced property has been demonstrated for PuO2 nanoparticles. Bulk PuO2 exhibits a temperature independent paramagnetic behavior and is characterized by the absence of magnetic moments despite the electronic configuration of Pu(IV).117,209 Magnetization experiments carried out on a mixture of PuO2 nanodots and nanorods interestingly demonstrated a magnetic susceptibility value M/H(300 K) = χDC(300 K) = 0.835 × 10−3 emu mol−1, corresponding to 1.05 × 10−8 m3 mol−1 after conversion. This value appears in between the values reported for Pu(III) and Pu(V) at 0.47 × 10−8 m3 mol−1 and 1.63 × 10−8 m3 mol−1, respectively, but more interestingly it is constrained between the values reported for PuO2 and Pu(IV), being 2.84 × 10−8 m3 mol−1 for the latter.210–212 Such an intermediate behaviour suggests a possible influence of the nanoparticle surface if a crystalline and diamagnetic PuO2 core is considered.
The evaluation of the catalytic properties of actinide oxides is a growing topic and was recently discussed in the comprehensive review of Leduc et al.10 Although plutonium appears neglected for obvious reasons, very interesting results have been obtained with nanosized uranium and thorium oxides suggesting promising developments. The selective and reversible extraction of Pu(IV) colloids from the soluble ionic species was carried out in a mixture composed of n-octanol and trichloroacetic acid.86 This interesting approach allowed pointing out the complexity of the surface reactivity of Pu nanoparticles raising questions about their behavior in migration schemes. Meanwhile, the strong adsorption of Pu(IV) intrinsic colloids onto materials acquiring a negative surface charge when contacted with water (glass, paper, silica…) has been reported.45 With regard to the formation of the nanoparticles, Powell et al. evidenced a striking difference when Pu(IV) solution was brought in contact with goethite or quartz with the observation of 2–5 nm Pu4O7 and PuO2 nanoparticles at the surface of the respective materials. The difference noticed in the case of goethite was explained by the epitaxial growth of the nanoparticles as a result of the similarity of the lattice parameters.55
Bulk PuO2 is a highly refractory material requiring drastic conditions to be dissolved (boiling nitric media, HF, O3, etc.).45,213–215 By comparison, easier dissolution and an increased dissolution rate have been reported in 8 M HNO3 at 80 °C for PuO2 nanomaterials prepared by hydrothermal synthesis.71 Whereas high fired PuO2 did not show significant behavior in sonicated water, the ultrasound treatment of nanostructured PuO2 in pure water and an Ar/CO atmosphere was found to yield Pu(IV) intrinsic colloids.94,96 This behavior was attributed to the improved reactivity of nanostructured PuO2 in comparison to the bulk, but also to redox mechanisms fostered for such nanoscale oxides. Such an approach opens interesting perspectives for the preparation of PuO2 nanoparticles with specific properties: core–shell nanoparticles, functionalization, dissolution, etc. For instance, recent results reported the significant sonochemical dissolution of nanoscale ThO2 often used as a PuO2 simulant in dilute 0.5 M H2SO4 (20 kHz, 20 °C) in contrast with the known strong refractory behaviour of bulk ThO2. The mechanism was evidenced to combine the physical effects of ultrasound, in situ generation of H2O2 and complexation properties of sulfates which further enabled the accumulation of a neo-formed thorium peroxo sulfate with 1D morphology.216,217 The transposition of this work to nanoscale PuO2 is an on-going study that demonstrated interesting preliminary results.123 Moreover, the enhanced reactivity of AnO2 nanoparticles is particularly interesting since the experimental conditions used for the preparation of several nanostructured PuO2 samples are similar to the ones used for its industrial preparation dedicated to MOX nuclear fuel.116,121 The potential observation of production specification differences (e.g. density, sintering properties, microstructure, etc.) might be explained by the contribution of nanoscale objects.
7. Conclusion and outlook
This review summarizes the recent progress in the synthesis, characterization and multi-scale structural properties of PuO2 nanoparticles. Beyond the interest related to the fundamental knowledge of nanoscale PuO2 (and more generally, AnO2), this overview demonstrates that these nanomaterials constitute a promising topic in nuclear chemistry due to their potential implications for industrial and technological purposes. Their potential role in radioactivity migration in the geosphere represents another important question focusing on the past nuclear responsibility and future energy-related activities. Safety surface and underground storage, corrosion of the spent nuclear fuel, preparation of reference materials for safeguards and forensics, behavior of nuclear fuel in operation or in the case of accidents, or decommissioning of polluted sites are some of the global challenges requiring a thorough characterization of PuO2 nanoparticles. Nuclearized and newly developed or improved analytical techniques, in combination with the available access to large instruments for hot samples, highly contributed to achieving new insights about the piece of the chemistry puzzle addressed by the PuO2 nanoparticles.The preparation of PuO2 nanoparticles with a controlled and reproducible size is of paramount interest for potential applications and laboratory screening. The observed size is very often in the 1–5 nm range which appears particularly interesting when focusing the size vs. structural properties relationship. Recent results demonstrate that PuO2 particles in the 1–10 nm size range show the most significant effect on the modification of their local and electronic structures. Larger particles generally correspond to bulk PuO2 to a greater extent, although their surface chemistry may imply specific reactivity in agreement with what was observed with other actinide oxides in heterogeneous catalysis, for instance. Nevertheless, accessing PuO2 nanoparticles with controlled size and morphology appears challenging. The preparation of well-defined mixed oxide nanoparticles and their nuclear technological options also represent an interesting and relevant question deserving answers. The highly debated and intriguing issue related to the crystalline nature and the potential presence of higher valence oxidation states in the PuO2 nanoparticles has been fixed. The development of nuclearized analytical techniques and the increased sensitivity of spectroscopy approaches have significantly contributed to the state-of-the art knowledge related to the electronic structure and oxidation state of the PuO2 nanoparticles. Further democratization of these recent approaches (including for instance SAXS, HERFD-XANES, O–K edge spectroscopy) and their relevant combination appear essential for the careful and thorough characterization of the physico-chemical properties of the PuO2 nanoparticles.
More generally, PuO2 nanoparticles are defined as crystalline particles in agreement with the PuO2 bulk fluorite FCC structure. However, recent studies confirm a size-dependent structural disorder definitely related to the nanoparticle surface but also to a gradient of Pu–Pu bond length variation within the core by analogy with smaller clusters exhibiting some structural similarities. Further insights are still expected in solution about the formation mechanism of the nanoparticles and the potential contribution of smaller cluster species. Similarly, understanding the nature of the nanoparticle surface and its interaction with the surroundings (adsorption of species, interaction with other particles, dissolution, aging processes, etc.) is challenging. The radiolysis effect of plutonium isotopes on the local structure of the nanoparticles, on their stability, on their surface and the surrounding and interface chemistry is also a fair question. Finally, the related behaviour of hot particles with the biosphere including marine species appears essential.
Conflicts of interest
There are no conflicts to declare.References
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826–859 CrossRef CAS PubMed.
- H. E. Rudel, M. K. M. Lane, C. L. Muhich and J. B. Zimmerman, ACS Nano, 2020, 14, 16472–16501 CrossRef CAS PubMed.
- M. S. Chavali and M. P. Nikolova, SN Appl. Sci., 2019, 1, 607 CrossRef CAS.
- S. Rajeshkumar and P. Naik, Biotechnol. Rep., 2018, 17, 1–5 CrossRef CAS PubMed.
- M. Fernandez-Garcia, A. Martinez-Arias, J. C. Hanson and J. A. Rodriguez, Chem. Rev., 2004, 104, 4063–4104 CrossRef CAS PubMed.
- T.-D. Nguyen, Nanoscale, 2013, 5, 9455–9482 RSC.
- Z. Wu, S. Yang and W. Wu, Nanoscale, 2016, 8, 1237–1259 RSC.
- E. Villarreal, G. G. Li, Q. Zhang, X. Fu and H. Wang, Nano Lett., 2017, 17, 4443–4452 CrossRef CAS PubMed.
- J. Leduc, M. Frank, L. Jürgensen, D. Graf, A. Raauf and S. Mathur, ACS Catal., 2019, 9, 4719–4741 CrossRef CAS.
- E. A. Goodilin, P. S. Weiss and Y. Gogotsi, ACS Nano, 2019, 13, 10879–10886 CrossRef CAS PubMed.
- S. N. Kalmykov and M. A. Denecke, Actinide Nanoparticle Research, Springer, Berlin, Heidelberg, 1st edn, 2011 Search PubMed.
- L. M. Moreau, A. Herve, M. D. Straub, D. R. Russo, R. J. Abergel, S. Alayoglu, J. Arnold, A. Braun, G. J. P. Deblonde, Y. Liu, T. D. Lohrey, D. T. Olive, Y. Qiao, J. A. Rees, D. K. Shuh, S. J. Teat, C. H. Booth and S. G. Minasian, Chem. Sci., 2020, 11, 4648–4668 RSC.
- O. Walter, K. Popa and O. D. Blanco, Open Chem., 2016, 14, 170–174 CAS.
- R. M. Pallares and R. J. Abergel, Nanoscale, 2020, 12, 1339–1348 RSC.
- G. E. Sigmon and A. E. Hixon, Chem.–Eur. J., 2019, 25, 2463–2466 CrossRef CAS PubMed.
- H. Geckeis, M. Zavarin, B. Salbu, O. C. Lind and L. Skipperud, in Plutonium Handbook, D. L.Clark, D. A.Geeson and R. J. Hanrahan Jr, La Grange Park, IL, USA, 2019, ch 25, pp. 1979–2118 Search PubMed.
- C. R. Armstrong, H. A. Brant, P. R. Nuessle, G. Hall and J. R. Cadieux, Sci. Rep., 2016, 6, 21512 CrossRef CAS PubMed.
- G. X. Wang, E. R. Batista and P. Yang, Appl. Sci., 2020, 10, 4655 CrossRef CAS.
- The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. Edelstein and J. Fuger, Springer, Netherlands, 2011 Search PubMed.
- T. A. Lennox, D. N. Millington and R. E. Sunderland, Prog. Nucl. Energy, 2007, 49, 589–596 CrossRef CAS.
- K. V. Vrinda Devi, J. Ramkumar, K. Biju and D. B. Sathe, J. Nucl. Mater., 2019, 518, 129–139 CrossRef CAS.
- H. Wu, Y. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522–16523 CrossRef CAS PubMed.
- V. F. Petrunin, Phys. Procedia, 2015, 72, 536–539 CrossRef CAS.
- W.-Q. Shi, L.-Y. Yuan, Z.-J. Li, J.-H. Lan, Y.-L. Zhao and Z.-F. Chai, Radiochim. Acta, 2012, 100, 727–736 CrossRef CAS.
- G. Kauric, O. Walter, A. Beck, B. Schacherl, O. Dieste Blanco, J. F. Vigier, E. Zuleger, T. Vitova and K. Popa, Mater. Today Adv., 2020, 8, 100105 CrossRef.
- L. R. Khanal, J. A. Sundararajan and Y. Qiang, Energy Technol., 2020, 8, 1901070 CrossRef.
- T. Wiss, V. V. Rondinella, R. J. M. Konings, D. Staicu, D. Papaioannou, S. Bremier, P. Poml, O. Benes, J. Y. Colle, P. Van Uffelen, A. Schubert, F. Cappia, M. Marchetti, D. Pizzocri, F. Jatuff, W. Goll, T. Sonoda, A. Sasahara, S. Kitajima and M. Kinoshita, Radiochim. Acta, 2017, 105, 893–906 CrossRef CAS.
- V. V. Rondinella and T. Wiss, Mater. Today, 2010, 13, 24–32 CrossRef CAS.
- Y. Torres, C. Garcia-Ostos, C. Arevalo, F. J. Gotor, J. J. Pavon, P. Trueba and J. A. Rodriguez-Ortiz, J. Nucl. Sci. Technol., 2017, 54, 167–173 CrossRef CAS.
- J. Spino and D. Papaioannou, J. Nucl. Mater., 2008, 372, 416–420 CrossRef CAS.
- T. Ozawa, Y. Ikusawa, T. Abe and K. Maeda, La Grange Park, Orlando, FL, 2004 Search PubMed.
- V. Tyrpekl, J. F. Vigier, D. Manara, T. Wiss, O. D. Blanco and J. Somers, J. Nucl. Mater., 2015, 460, 200–208 CrossRef CAS.
- R. Jovani-Abril, M. Gibilaro, A. Janssen, R. Eloirdi, J. Somers, J. Spino and R. Malmbeck, J. Nucl. Mater., 2016, 477, 298–304 CrossRef CAS.
- Q. Yan, Y. W. Mao, X. S. Zhou, J. H. Liang, M. Y. Ye and S. M. Peng, J. Nucl. Mater., 2018, 512, 417–422 CrossRef CAS.
- J. Spino, H. Santa Cruz, R. Jovani-Abril, R. Birtcher and C. Ferrero, J. Nucl. Mater., 2012, 422, 27–44 CrossRef CAS.
- J. Spino, D. Papaioannou, I. Ray and D. Baron, 2002.
- A. Brunstad, Ind. Eng. Chem., 1959, 51, 38–40 CrossRef.
- A. Y. Romanchuk, I. E. Vlasova and S. N. Kalmykov, Front. Chem., 2020, 8, 630 CrossRef CAS PubMed.
- C. Walther and M. A. Denecke, Chem. Rev., 2013, 113, 995–1015 CrossRef CAS PubMed.
- A. Y. Romanchuk, S. N. Kalmykov, A. B. Kersting and M. Zavarin, Russ. Chem. Rev., 2016, 85, 995–1010 CrossRef CAS.
- A. B. Kersting, Inorg. Chem., 2013, 52, 3533–3546 CrossRef CAS PubMed.
- A. P. Novikov, S. N. Kalmykov, S. Utsunomiya, R. C. Ewing, F. Horreard, A. Merkulov, S. B. Clark, V. V. Tkachev and B. F. Myasoedov, Science, 2006, 314, 638–641 CrossRef CAS PubMed.
- P. H. Santschi, K. A. Roberts and L. Guo, Environ. Sci. Technol., 2002, 36, 3711–3719 CrossRef CAS PubMed.
- D. Clark, S. Hecker, G. Jarvinen and M. Neu, in The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. Edelstein and J. Fuger, Springer Netherlands, 2011, ch. 7, pp. 813–1264, DOI:10.1007/978-94-007-0211-0_7.
- C. Walther, J. Rothe, B. Brendebach, M. Fuss, M. Altmaier, C. M. Marquardt, S. Buechner, H. R. Cho, J. I. Yun and A. Seibert, Radiochim. Acta, 2009, 97, 199–207 CrossRef CAS.
- A. Safonov, E. Lavrinovich, A. Emel'yanov, K. Boldyrev, V. Kuryakov, N. Rodygina, E. Zakharova and A. Novikov, Sci. Rep., 2022, 12, 4557 CrossRef CAS PubMed.
- A. Wolfsberg, Z. Dai, L. Zhu, P. Reimus, T. Xiao and D. Ware, Environ. Sci. Technol., 2017, 51, 5582–5590 CrossRef CAS PubMed.
- M. A. Denecke, N. Bryan, S. Kalmykov, K. Morris and F. Quinto, in Experimental and Theoretical Approaches to Actinide Chemistry, 2018, ch. 8, pp. 378–444, DOI:10.1002/9781119115557.
- V. Neck, M. Altmaier, A. Seibert, J. I. Yun, C. M. Marquardt and T. Fanghanel, Radiochim. Acta, 2007, 95, 193–207 CrossRef CAS.
- W. Runde and M. P. Neu, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Netherlands, Dordrecht, 2011, pp. 3475–3593, DOI:10.1007/978-94-007-0211-0_32.
- A. B. Kersting, D. W. Efurd, D. L. Finnegan, D. J. Rokop, D. K. Smith and J. L. Thompson, Nature, 1999, 397, 56–59 CrossRef CAS.
- H. Zanker and C. Hennig, J. Contam. Hydrol., 2014, 157, 87–105 CrossRef PubMed.
- A. Y. Romanchuk, S. N. Kalmykov, A. V. Egorov, Y. V. Zubavichus, A. A. Shiryaev, O. N. Batuk, S. D. Conradson, D. A. Pankratov and I. A. Presnyakov, Geochim. Cosmochim. Acta, 2013, 121, 29–40 CrossRef CAS.
- B. A. Powell, Z. R. Dai, M. Zavarin, P. H. Zhao and A. B. Kersting, Environ. Sci. Technol., 2011, 45, 2698–2703 CrossRef CAS PubMed.
- M. Schmidt, S. S. Lee, R. E. Wilson, K. E. Knope, F. Bellucci, P. J. Eng, J. E. Stubbs, L. Soderholm and P. Fenter, Environ. Sci. Technol., 2013, 47, 14178–14184 CrossRef CAS PubMed.
- R. Kirsch, D. Fellhauer, M. Altmaier, V. Neck, A. Rossberg, T. Fanghänel, L. Charlet and A. C. Scheinost, Environ. Sci. Technol., 2011, 45, 7267–7274 CrossRef CAS PubMed.
- E. Balboni, K. F. Smith, L. M. Moreau, T. T. Li, M. Maloubier, C. H. Booth, A. B. Kersting and M. Zavarin, ACS Earth Space Chem., 2020, 4, 1993–2006 CrossRef CAS.
- A. E. Hixon and B. A. Powell, Environ. Sci. Technol., 2014, 48, 9255–9262 CrossRef CAS PubMed.
- A. E. Hixon and B. A. Powell, Environ. Sci.: Process. Impacts, 2018, 20, 1306–1322 CAS.
- K. F. Smith, K. Morris, G. T. W. Law, E. H. Winstanley, F. R. Livens, J. S. Weatherill, L. G. Abrahamsen-Mills, N. D. Bryan, J. F. W. Mosselmans, G. Cibin, S. Parry, R. Blackham, K. A. Law and S. Shaw, ACS Earth Space Chem., 2019, 3, 2437–2442 CrossRef CAS PubMed.
- E. I. Hamilton, Mar. Pollut. Bull., 1985, 16, 305–309 CrossRef.
- E. I. Hamilton, Mineral. Mag., 1985, 49, 177–194 CrossRef CAS.
- P. J. Kershaw, D. C. Denoon and D. S. Woodhead, J. Environ. Radioact., 1999, 44, 191–221 CrossRef CAS.
- G. J. Hancock, S. G. Tims, L. K. Fifield and I. T. Webster, in Stratigraphical Basis for the Anthropocene, ed. C. N. Waters, J. A. Zalasiewicz, M. Williams, M. Ellis and A. M. Snelling, Geological Society Publishing House, Bath, 2014, vol. 395, pp. 265–281 Search PubMed.
- M. R. Beccia, P. L. Solari, M. Monfort, C. Moulin and C. Den Auwer, New J. Chem., 2018, 42, 7582–7591 RSC.
- T. Dumas, M. Guigue, P. Moisy, R. Colina-Ruiz, J. Mustre de Leon, M. Matara-Aho, P. L. Solari, M. Monfort, C. Moulin, M. R. Beccia and C. D. Auwer, ChemistrySelect, 2018, 3, 2021–2024 CrossRef CAS.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- O. L. Gaskova, Geochem. Int., 2006, 44, 501–507 CrossRef.
- N. P. Laverov, V. I. Velichkin, B. I. Omel'yanenko and S. V. Yudintsev, Geology of Ore Deposits, 2003, vol. 45, pp. 1–18 Search PubMed.
- K. Popa and O. Walter, in Comprehensive Nuclear Materials, ed. R. J. M. Konings and R. E. Stoller, Elsevier, Oxford, 2020, second edn, pp. 579–592, DOI:10.1016/B978-0-12-803581-8.11780-1.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS PubMed.
- M. Altmaier, X. Gaona and T. Fanghanel, Chem. Rev., 2013, 113, 901–943 CrossRef CAS PubMed.
- International Atomic Energy Agency, Radioactive Particles in the Environment: Sources, Particle Characterization and Analytical Techniques, IAEA-TECDOC-1663, IAEA, Vienna, 2011 Search PubMed.
- J. Jeevanandam, A. Barhoum, Y. S. Chan, A. Dufresne and M. K. Danquah, Beilstein J. Nanotechnol., 2018, 9, 1050–1074 CrossRef CAS PubMed.
- V. Gubala, L. J. Johnston, Z. Liu, H. Krug, C. J. Moore, C. K. Ober, M. Schwenk and M. Vert, Pure Appl. Chem., 2018, 90, 1283–1324 CrossRef CAS.
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS PubMed.
- H. Gleiter, Acta Mater., 2000, 48, 1–29 CrossRef CAS.
- D. H. Everett, Pure Appl. Chem., 1972, 31, 577–638 CrossRef.
- F. A. Khan, in Applications of Nanomaterials in Human Health, ed. F. A. Khan, Springer Singapore, Singapore, 2020, pp. 1–13, DOI:10.1007/978-981-15-4802-4_1.
- The transuranium elements: research papers. 1. Papers 1.1 to 6.39, ed. G. T. Seaborg, J. J. Katz and W. M. Manning, N.Y.: McGraw-Hill, USA, 1949 Search PubMed.
- C. Walther, H. R. Cho, C. M. Marquardt, V. Neck, A. Seibert, J. I. Yun and T. Fanghanel, Radiochim. Acta, 2007, 95, 7–16 CrossRef CAS.
- J. Rothe, C. Walther, M. A. Denecke and T. Fanghanel, Inorg. Chem., 2004, 43, 4708–4718 CrossRef CAS PubMed.
- V. Neck and J. I. Kim, Radiochim. Acta, 2001, 89, 1–16 CrossRef CAS.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef CAS PubMed.
- R. E. Wilson, S. Skanthakumar and L. Soderholm, Angew. Chem., Int. Ed., 2011, 50, 11234–11237 CrossRef CAS PubMed.
- H. Nitsche, Chem. Rev., 2013, 113, 855–857 CrossRef CAS PubMed.
- K. E. Knope and L. Soderholm, Inorg. Chem., 2013, 52, 6770–6772 CrossRef CAS PubMed.
- C. Tamain, T. Dumas, D. Guillaumont, C. Hennig and P. Guilbaud, Eur. J. Inorg. Chem., 2016, 2016, 3536–3540 CrossRef CAS.
- C. Tamain, T. Dumas, C. Hennig and P. Guilbaud, Chem.–Eur. J., 2017, 23, 6864–6875 CrossRef CAS PubMed.
- K. E. Knope, S. Skanthakumar and L. Soderholm, Inorg. Chem., 2015, 54, 10192–10196 CrossRef CAS PubMed.
- D. W. Wester, Inorg. Chem., 1982, 21, 3382–3385 CrossRef CAS.
- D. W. Ockenden and G. A. Welch, J. Chem. Soc., 1956, 3358–3363, 10.1039/jr9560003358.
- C. Micheau, M. Virot, S. Dourdain, T. Dumas, D. Menut, P. L. Solari, L. Venault, O. Diat, P. Moisy and S. I. Nikitenko, Environ. Sci.: Nano, 2020, 7, 2252–2266 RSC.
- D. Rai and J. L. Swanson, Nucl. Technol., 1981, 54, 107–112 CrossRef CAS.
- E. Dalodière, M. Virot, V. Morosini, T. Chave, T. Dumas, C. Hennig, T. Wiss, O. Dieste Blanco, D. K. Shuh, T. Tyliszcak, L. Venault, P. Moisy and S. I. Nikitenko, Sci. Rep., 2017, 7:43514, 1–10 Search PubMed.
- E. Dalodiere, PhD thesis, Université de Montpellier, 2017.
- M. H. Lloyd and R. G. Haire, Nucl. Appl., 1968, 5, 114–122 CAS.
- M. H. Lloyd and R. G. Haire, Radiochim. Acta, 1978, 25, 139–148 CrossRef CAS.
- P. Thiyagarajan, H. Diamond, L. Soderholm, E. P. Horwitz, L. M. Toth and L. K. Felker, Inorg. Chem., 1990, 29, 1902–1907 CrossRef CAS.
- P. Zhao, M. Zavarin, Z. Dai and A. B. Kersting, Appl. Geochem., 2020, 122, 104725 CrossRef CAS.
- R. G. Haire, M. H. Lloyd, M. L. Beasley and W. O. Milligan, J. Electron Microsc., 1971, 20, 8–16 Search PubMed.
- A. I. Abdel-Fattah, D. X. Zhou, H. Boukhalfa, S. Tarimala, S. D. Ware and A. A. Keller, Environ. Sci. Technol., 2013, 47, 5626–5634 CrossRef CAS PubMed.
- J. I. Kim and B. Kanellakopulos, Radiochim. Acta, 1989, 48, 145–150 CrossRef CAS.
- L. M. Toth and H. A. Friedman, J. Inorg. Nucl. Chem., 1978, 40, 807–810 CrossRef CAS.
- M. H. Lloyd and O. K. Tallent, Nucl. Technol., 1973, 18, 205–215 CrossRef CAS.
- C. Maillard and J. M. Adnet, Radiochim. Acta, 2001, 89, 485–490 CrossRef CAS.
- T. W. Newton, D. E. Hobart and P. D. Palmer, Radiochim. Acta, 1986, 39, 139–147 CrossRef CAS.
- A. K. Pikaev, V. P. Shilov and A. V. Gogolev, Russ. Chem. Rev., 1997, 66, 763–788 CrossRef.
- I. R. Triay, R. S. Rundberg, A. J. Mitchell, M. A. Ott, D. E. Hobart, P. D. Palmer, T. W. Newton and J. L. Thompson, Size Determinations of Plutonium Colloids Using Autocorrelation Photon Spectroscopy, Los Alamos National Lab. (LANL), Los Alamos, NM (United States), 1989 Search PubMed.
- E. Gerber, A. Y. Romanchuk, I. Pidchenko, L. Amidani, A. Rossberg, C. Hennig, G. B. M. Vaughan, A. Trigub, T. Egorova, S. Bauters, T. Plakhova, M. Hunault, S. Weiss, S. M. Butorin, A. C. Scheinost, S. N. Kalmykov and K. O. Kvashnina, Nanoscale, 2020, 12, 18039–18048 RSC.
- A. Y. Romanchuk, T. V. Plakhova, A. V. Egorov, T. B. Egorova, P. V. Dorovatovskii, Y. V. Zubavichus, A. A. Shiryaev and S. N. Kalmykov, Dalton Trans., 2018, 47, 11239–11244 RSC.
- E. Gerber, A. Y. Romanchuk, S. Weiss, A. Kuzenkova, M. O. J. Y. Hunault, S. Bauters, A. Egorov, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Environ. Sci.: Nano, 2022, 9, 1509–1518 RSC.
- K. O. Kvashnina, A. Y. Romanchuk, I. Pidchenko, L. Amidani, E. Gerber, A. Trigub, A. Rossberg, S. Weiss, K. Popa, O. Walter, R. Caciuffo, A. C. Scheinost, S. M. Butorin and S. N. Kalmykov, Angew. Chem., Int. Ed., 2019, 58, 17558–17562 CrossRef CAS PubMed.
- C. Ekberg, K. Larsson, G. Skarnemark, A. Odegaard-Jensen and I. Persson, Dalton Trans., 2013, 42, 2035–2040 RSC.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, E. Dalodiere, O. D. Blanco, T. Wiss, X. Le Goff, M. Odorico, D. Prieur, A. Rossberg, L. Venault, N. Dacheux, P. Moisy and S. I. Nikitenko, Nanoscale Adv., 2020, 2, 214–224 RSC.
- D. Hudry, C. Apostolidis, O. Walter, A. Janssen, D. Manara, J. C. Griveau, E. Colineau, T. Vitova, T. Prussmann, D. Wang, C. Kubel and D. Meyer, Chem.–Eur. J., 2014, 20, 10431–10438 CrossRef CAS PubMed.
- K. Popa, O. Walter, O. D. Blanco, A. Guiot, D. Bouexiere, J. Y. Colle, L. Martel, M. Naji and D. Manara, CrystEngComm, 2018, 20, 4614–4622 RSC.
- L. Balice, D. Bouexiere, M. Cologna, A. Cambriani, J. F. Vigier, E. De Bona, G. D. Soraru, C. Kubel, O. Walter and K. Popa, J. Nucl. Mater., 2018, 498, 307–313 CrossRef CAS.
- J. F. Corbey, L. E. Sweet, S. I. Sinkov, D. D. Reilly, C. M. Parker, J. M. Lonergan and T. J. Johnson, Eur. J. Inorg. Chem., 2021, 2021, 3277–3291 CrossRef CAS.
- R. M. Orr, H. E. Sims and R. J. Taylor, J. Nucl. Mater., 2015, 465, 756–773 CrossRef CAS.
- V. Tyrpekl, M. Belis, T. Wangle, J. Vleugels and M. Verwerft, J. Nucl. Mater., 2017, 493, 255–263 CrossRef CAS.
- L. Bonato, PhD thesis, Université de Montpellier, 2020.
- T. Vitova, I. Pidchenko, D. Fellhauer, T. Pruessmann, S. Bahl, K. Dardenne, T. Yokosawa, B. Schimmelpfennig, M. Altmaier, M. Denecke, J. Rothe and H. Geckeis, Chem. Commun., 2018, 54, 12824–12827 RSC.
- I. R. Triay, D. E. Hobart, A. J. Mitchell, T. W. Newton, M. A. Ott, P. D. Palmer, R. S. Rundberg and J. L. Thompson, Radiochim. Acta, 1991, 52–3, 127–131 CrossRef.
- D. Bouexiere, K. Popa, O. Walter and M. Cologna, RSC Adv., 2019, 9, 6542–6547 RSC.
- L. J. Cash, M. D. Hoover, R. A. Guilmette, P. N. Breysse and L. Bertelli, Radiat. Prot. Dosim., 2016, 173, 318–324 Search PubMed.
- H. Smith, G. N. Stradling, B. W. Loveless and G. J. Ham, Health Phys., 1977, 33, 539–551 CrossRef CAS.
- R. J. Batrice, J. N. Wacker and K. E. Knope, in Experimental and Theoretical Approaches to Actinide Chemistry, ed. J. K. Gibson and W. A. Jong, 2018, ch. 2, pp. 53–127, DOI:10.1002/9781119115557.
- D. E. Hobart, D. E. Morris, P. D. Palmer and T. W. Newton, Presented in Part at the Conference: Nuclear Waste Isolation in the Unsaturated Zone: FOCUS `89, Las Vegas, NV (United States), 18-21 Sep 1989; Other Information: PBD: 1989, United States, 1989 Search PubMed.
- M. Altmaier, X. Gaona, D. Fellhauer, D. L. Clark, W. H. Runde and D. E. Hobart, in Plutonium Handbook, ed. D. L. Clark, D. A. Geeson and R. J. Hanrahan Jr, American Nuclear Society, La Grange Park, IL, USA, 2019 Search PubMed.
- L. Medyk, D. Manara, J. Y. Colle, D. Bouexiere, J. F. Vigier, L. Marchetti, P. Simon and P. Martin, J. Nucl. Mater., 2020, 541, 152439 CrossRef CAS.
- F. Cappia, D. Hudry, E. Courtois, A. Janssen, L. Luzzi, R. J. M. Konings and D. Manara, Mater. Res. Express, 2014, 1, 025034 CrossRef CAS.
- C. Jegou, M. Gennisson, S. Peuget, L. Desgranges, G. Guimbretiere, M. Magnin, Z. Talip and P. Simon, J. Nucl. Mater., 2015, 458, 343–349 CrossRef CAS.
- Z. Talip, S. Peuget, M. Magnin, M. Tribet, C. Valot, R. Vauchy and C. Jegou, J. Nucl. Mater., 2018, 499, 88–97 CrossRef CAS.
- T. Shinonaga, D. Donohue, H. Aigner, S. Burger, D. Klose, T. Karkela, R. Zilliacus, A. Auvinen, O. Marie and F. Pointurier, Anal. Chem., 2012, 84, 2638–2646 CrossRef CAS PubMed.
- E. Villa-Aleman, N. J. Bridges, T. C. Shehee and A. L. Houk, J. Nucl. Mater., 2019, 515, 140–149 CrossRef CAS.
- M. J. Sarsfield, R. J. Taylor, C. Puxley and H. M. Steele, J. Nucl. Mater., 2012, 427, 333–342 CrossRef CAS.
- B. L. Scott, A. L. Pugmire, J. T. Stritzinger, D. K. Veirs, L. E. Wolfsberg and M. P. Wilkerson, J. Nucl. Mater., 2019, 521, 155–160 CrossRef CAS.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- L. Amidani, G. B. M. Vaughan, T. V. Plakhova, A. Y. Romanchuk, E. Gerber, R. Svetogorov, S. Weiss, Y. Joly, S. N. Kalmykov and K. O. Kvashnina, Chem.–Eur. J., 2021, 27, 252–263 CrossRef CAS PubMed.
- S. J. L. Billinge and I. Levin, Science, 2007, 316, 561 CrossRef CAS PubMed.
- J. Blazek and E. P. Gilbert, Carbohydr. Polym., 2011, 85, 281–293 CrossRef CAS.
- D. Honecker, M. Bersweiler, S. Erokhin, D. Berkov, K. Chesnel, D. A. Venero, A. Qdemat, S. Disch, J. K. Jochum, A. Michels and P. Bender, Nanoscale Adv., 2022, 4, 1026–1059 RSC.
- L. M. Toth, H. A. Friedman and M. M. Osborne, J. Inorg. Nucl. Chem., 1981, 43, 2929–2934 CrossRef CAS.
- T. Dumas, M. Virot, D. Menut, C. Tamain, C. Micheau, S. Dourdain and O. Diat, J. Synchrotron Radiat., 2022, 29, 30–36 CrossRef CAS PubMed.
- M. Dembowski, C. A. Colla, P. Yu, J. Qiu, J. E. S. Szymanowski, W. H. Casey and P. C. Burns, Inorg. Chem., 2017, 56, 9602–9608 CrossRef CAS PubMed.
- M. Nyman, Coord. Chem. Rev., 2017, 352, 461–472 CrossRef CAS.
- B. H. Zhai, Q. Tian, N. Li, M. H. Yan and M. J. Henderson, J. Synchrotron Radiat., 2022, 29, 281–287 CrossRef CAS PubMed.
- L. Kumar, P. Kumar, A. Narayan and M. Kar, Int. Nano Lett., 2013, 3, 8 CrossRef.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114, 12909–12919 CrossRef CAS.
- S. Skanthakumar, D. Gorman-Lewis, A. Locock, M. H. Chiang, M. P. Jensen, P. C. Burns, J. Fein, C. D. Jonah, K. Attenkofer and L. Soderholm, Boston, MA, 2003.
- W.-Q. Shi, L.-Y. Yuan, C.-Z. Wang, L. Wang, L. Mei, C.-L. Xiao, L. Zhang, Z.-J. Li, Y.-L. Zhao and Z.-F. Chai, Adv. Mater., 2014, 26, 7807–7848 CrossRef CAS PubMed.
- W. D. Neilson, H. Steele, N. Kaltsoyannis and S. T. Murphy, Phys. Chem. Chem. Phys., 2022, 24, 8245–8250 RSC.
- W. J. Huang, R. Sun, J. Tao, L. D. Menard, R. G. Nuzzo and J. M. Zuo, Nat. Mater., 2008, 7, 308–313 CrossRef CAS PubMed.
- A. Kuzmin and J. Chaboy, IUCrJ, 2014, 1, 571–589 CrossRef CAS PubMed.
- D. Vollath, F. D. Fischer and D. Holec, Beilstein J. Nanotechnol., 2018, 9, 2265–2276 CrossRef CAS PubMed.
- D. Nafday, S. Sarkar, P. Ayyub and T. Saha-Dasgupta, ACS Nano, 2018, 12, 7246–7252 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- P. P. Rodenbough, C. Zheng, Y. Liu, C. Hui, Y. Xia, Z. Ran, Y. Hu and S.-W. Chan, J. Am. Ceram. Soc., 2017, 100, 384–392 CrossRef CAS.
- V. Swamy, D. Menzies, B. C. Muddle, A. Kuznetsov, L. S. Dubrovinsky, Q. Dai and V. Dmitriev, Appl. Phys. Lett., 2006, 88, 243103 CrossRef.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- X. Gao, I. Di Bernardo, P. Kreider, T. Tran-Phu, X. B. Cai, N. Wang, Y. Zhu, M. B. Venkataraman, J. Lipton-Duffin, A. Bayon, W. Lipinski and A. Tricoli, ACS Catal., 2019, 9, 9880–9890 CrossRef CAS.
- P. M. Diehm, P. Agoston and K. Albe, ChemPhysChem, 2012, 13, 2443–2454 CrossRef CAS PubMed.
- T. V. Plakhova, A. Y. Romanchuk, D. V. Likhosherstova, A. E. Baranchikov, P. V. Dorovatovskii, R. D. Svetogorov, T. B. Shatalova, T. B. Egorova, A. L. Trigub, K. O. Kvashnina, V. K. Ivanov and S. N. Kalmykov, J. Phys. Chem. C, 2019, 123, 23167–23176 CrossRef CAS.
- F. Zhang, S. W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127–129 CrossRef CAS.
- D. Prieur, W. Bonani, K. Popa, O. Walter, K. W. Kriegsman, M. H. Engelhard, X. F. Guo, R. Eloirdi, T. Gouder, A. Beck, T. Vitova, A. C. Scheinost, K. Kvashnina and P. Martin, Inorg. Chem., 2020, 59, 5760–5767 CrossRef CAS PubMed.
- T. V. Plakhova, A. Y. Romanchuk, S. M. Butorin, A. D. Konyukhova, A. V. Egorov, A. A. Shiryaev, A. E. Baranchikov, P. V. Dorovatovskii, T. Huthwelker, E. Gerber, S. Bauters, M. M. Sozarukova, A. C. Scheinost, V. K. Ivanov, S. N. Kalmykov and K. O. Kvashnina, Nanoscale, 2019, 11, 18142–18149 RSC.
- A. J. Garcia-Adeva, D. R. Conradson, P. Villella and S. D. Conradson, J. Phys. Chem. B, 2003, 107, 6704–6716 CrossRef CAS.
- M. Auffan, J. Rose, C. Chanéac, J.-P. Jolivet, A. Masion, M. R. Wiesner and J.-Y. Bottero, in Nanoethics and Nanotoxicology, ed. P. Houdy, M. Lahmani and F. Marano, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 269–290, DOI:10.1007/978-3-642-20177-6_12.
- T. Belin, N. Millot, F. Villiéras, O. Bertrand and J. P. Bellat, J. Phys. Chem. B, 2004, 108, 5333–5340 CrossRef CAS.
- S. Moxon, A. R. Symington, J. S. Tse, J. Dawson, J. M. Flitcroft, S. C. Parker, D. J. Cooke, R. M. Harker and M. Molinari, Phys. Chem. Chem. Phys., 2020, 22, 7728–7737 RSC.
- B. E. Tegner, M. Molinari, A. Kerridge, S. C. Parker and N. Kaltsoyannis, J. Phys. Chem. C, 2017, 121, 1675–1682 CrossRef CAS.
- L. Zhang, B. Sun, Q. Zhang, H. Liu, K. Liu and H. Song, Appl. Surf. Sci., 2021, 537, 147882 CrossRef CAS.
- J. M. Haschke, T. H. Allen and L. A. Morales, Science, 2000, 287, 285–287 CrossRef CAS PubMed.
- T. Gouder, A. B. Shick and F. Huber, Top. Catal., 2013, 56, 1112–1120 CrossRef CAS.
- A. Seibert, T. Gouder and F. Huber, Radiochim. Acta, 2010, 98, 647–657 CrossRef CAS.
- M. Kato, A. Komeno, H. Uno, H. Sugata, N. Nakae, K. Konashi and M. Kashimura, J. Nucl. Mater., 2009, 393, 134–140 CrossRef CAS.
- R. P. Turcotte and T. D. Chikalla, in Defects and Transport in Oxides, ed. M. S. Seltzer and R. I. Jaffee, Springer US, Boston, MA, 1974, pp. 159–175, DOI:10.1007/978-1-4615-8723-1_8.
- E. Villa-Aleman, A. L. Houk, N. J. Bridges and T. C. Shehee, J. Raman Spectrosc., 2019, 50, 899–901 CrossRef CAS.
- J. F. Park, C. D. L., C. D. K., O. R. J. and S. V. H., U.S. Deparmtent of Commerce, National Technical Information Service, 1973..
- G. M. Kanapilly, O. G. Raabe and H. A. Boyd, Proc. 3rd Int. Congr. Int. Radiat. Protection Ass., 1974, USAEC, Oak Ridge, pp. 1237–1242 Search PubMed.
- R. L. Fleischer and O. G. Raabe, Health Phys., 1977, 32, 253–257 CrossRef CAS PubMed.
- R. L. Fleischer, Health Phys., 1975, 29, 69–73 CrossRef CAS PubMed.
- H. E. Sims, K. J. Webb, J. Brown, D. Morris and R. J. Taylor, J. Nucl. Mater., 2013, 437, 359–364 CrossRef CAS.
- W. D. Neilson, H. Steele and S. T. Murphy, J. Phys. Chem. C, 2021, 125, 15560–15568 CrossRef CAS PubMed.
- R. C. Belin, P. M. Martin, J. Lechelle, M. Reynaud and A. C. Scheinost, Inorg. Chem., 2013, 52, 2966–2972 CrossRef CAS PubMed.
- B. R. Scott, in Encyclopedia of Toxicology, ed. P. Wexler, Academic Press, Oxford, 3rd edn, 2014, pp. 29–43, DOI:10.1016/B978-0-12-386454-3.00057-9.
- C. Madic, Science, 2000, 287, 243–244 CrossRef CAS.
- Chemistry of the Elements, ed. N. N. Greenwood and A. Earnshaw, Butterworth-Heinemann, Oxford, second edn, 1997, pp. 1250–1284, DOI:10.1016/B978-0-7506-3365-9.50037-7.
- G. C. Allen, P. A. Tempest and F. G. A. Stone, Proc. R. Soc. London, Ser. A, 1986, 406, 325–344 CAS.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. Den Auwer, F. J. Espinosa-Faller, P. L. Gordon, N. J. Hess, R. Hess, D. W. Keogh, L. A. Morales, M. P. Neu, W. Runde, C. D. Tait, D. K. Veirs and P. M. Villella, Inorg. Chem., 2003, 42, 3715–3717 CrossRef CAS PubMed.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, L. A. Morales, M. P. Neu, P. Paviet-Hartmann, W. Runde, C. D. Tait, D. K. Veirs and P. M. Villella, J. Am. Chem. Soc., 2004, 126, 13443–13458 CrossRef CAS PubMed.
- M. K. Richmann, D. T. Reed, A. J. Kropf, S. B. Aase and M. A. Lewis, J. Nucl. Mater., 2001, 297, 303–312 CrossRef CAS.
- C. Hennig, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035120 CrossRef.
- P. Martin, S. Grandjean, M. Ripert, M. Freyss, P. Blanc and T. Petit, J. Nucl. Mater., 2003, 320, 138–141 CrossRef CAS.
- S. Calvin, XAFS for Everyone, CRC Press, 2013 Search PubMed.
- R. Schmitt, A. Nenning, O. Kraynis, R. Korobko, A. I. Frenkel, I. Lubomirsky, S. M. Haile and J. L. M. Rupp, Chem. Soc. Rev., 2020, 49, 554–592 RSC.
- S. D. Conradson, K. D. Abney, B. D. Begg, E. D. Brady, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, G. H. Lander, A. J. Lupinetti, L. A. Morales, M. P. Neu, P. D. Palmer, P. Paviet-Hartmann, S. D. Reilly, W. H. Runde, C. D. Tait, D. K. Veirs and F. Wastin, Inorg. Chem., 2004, 43, 116–131 CrossRef CAS PubMed.
- S. D. Conradson, Appl. Spectrosc., 1998, 52, 252A–279A CrossRef CAS.
- A. Romanchuk, A. Trigub, T. Plakhova, A. Kuzenkova, R. Svetogorov, K. Kvashnina and S. Kalmykov, J. Synchrotron Radiat., 2022, 29, 288–294 CrossRef CAS PubMed.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- E. Gerber, A. Y. Romanchuk, S. Weiss, S. Bauters, B. Schacherl, T. Vitova, R. Hübner, S. Shams Aldin Azzam, D. Detollenaere, D. Banerjee, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Inorg. Chem. Front., 2021, 8, 1102–1110 RSC.
- L. Amidani, T. V. Plakhova, A. Y. Romanchuk, E. Gerber, S. Weiss, A. Efimenko, C. J. Sahle, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Phys. Chem. Chem. Phys., 2019, 21, 10635–10643 RSC.
- S. D. Conradson, I. Al Mahamid, D. L. Clark, N. J. Hess, E. A. Hudson, M. P. Neu, P. D. Palmer, W. H. Runde and C. D. Tait, Polyhedron, 1998, 17, 599–602 CrossRef CAS.
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 253002 CrossRef CAS PubMed.
- K. O. Kvashnina and S. M. Butorin, Chem. Commun., 2022, 58, 327–342 RSC.
- M. Paffett, 2004, LALP-04-060.
- G. Raphael and R. Lallement, Solid State Commun., 1968, 6, 383–385 CrossRef CAS.
- E. Dalodière, M. Virot, T. Dumas, D. Guillaumont, M. C. Illy, C. Berthon, L. Guerin, A. Rossberg, L. Venault, P. Moisy and S. I. Nikitenko, Inorg. Chem. Front., 2018, 5, 100–111 RSC.
- M. Autillo, L. Guerin, H. Bolvin, P. Moisy and C. Berthon, Phys. Chem. Chem. Phys., 2016, 18, 6515–6525 RSC.
- M. Autillo, L. Guerin, D. Guillaumont, P. Moisy, H. Bolvin and C. Berthon, Inorg. Chem., 2016, 55, 12149–12157 CrossRef CAS PubMed.
- X. Beaudoux, M. Virot, T. Chave, G. Leturcq, G. Jouan, L. Venault, P. Moisy and S. I. Nikitenko, Dalton Trans., 2016, 45, 8802–8815 RSC.
- J. L. Ryan and L. A. Bray, Abstr. Pap. Am. Chem. Soc., 1979, 14 CAS.
- M. E. Woods, P. D. Benny, A. J. Unger, S. Phongikaroon and K. G. Myhre, J. Radioanal. Nucl. Chem., 2021, 327, 991–995 CrossRef CAS.
- L. Bonato, M. Virot, X. Le Goff, P. Moisy and S. I. Nikitenko, Ultrason. Sonochem., 2020, 69, 105235 CrossRef CAS PubMed.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, P. Lecante, D. Prieur, X. Le Goff, C. Hennig, N. Dacheux, P. Moisy and S. I. Nikitenko, Chem.–Eur. J., 2019, 25, 9580–9585 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |