
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrthogonal coiled coils enable rapid covalent labelling of two distinct membrane proteins with peptide nucleic acid barcodes†
Georgina C.
Gavins
a,
Katharina
Gröger
a,
Marc
Reimann
 a,
Michael D.
Bartoschek
b,
Sebastian
Bultmann
b and
Oliver
Seitz
*a
a,
Michael D.
Bartoschek
b,
Sebastian
Bultmann
b and
Oliver
Seitz
*a
aInstitut für Chemie, Humboldt-Universität zu Berlin Brook-Taylor-Strasse 2, Berlin 12489, Germany. E-mail: oliver.seitz@chemie.hu-berlin.de
bCenter for Molecular Biosystems (BioSysM), Faculty of Biology, Ludwig-Maximilians-Universität München, Butenandtstr. 1, Munich 81377, Germany. E-mail: bultmann@bio.lmu.de
First published on 16th July 2021
Abstract
Templated chemistry offers the prospect of addressing specificity challenges occurring in bioconjugation reactions. Here, we show two peptide-templated amide-bond forming reactions that enable the concurrent labelling of two different membrane proteins with two different peptide nucleic acid (PNA) barcodes. The reaction system is based on the mutually selective coiled coil interaction between two thioester-linked PNA–peptide conjugates and two cysteine peptides serving as genetically encoded peptide tags. Orthogonal coiled coil templated covalent labelling is highly specific, quantitative and proceeds within a minute. To demonstrate the usefulness, we evaluated receptor internalisation of two membranous receptors EGFR (epidermal growth factor) and ErbB2 (epidermal growth factor receptor 2) by first staining PNA-tagged proteins with fluorophore–DNA conjugates and then erasing signals from non-internalized receptors via toehold-mediated strand displacement.
Introduction
Rapid and site-selective bioconjugation reactions are a key tool at the chemistry–biology interface to establish covalent bonds between proteins and chemically designed entities such as fluorescent labels, purification handles and small molecule drugs. A distinct challenge is to perform chemical bioconjugation reactions in or on living cells.1–7 Such reactions are useful for fluorescence labelling and imaging of proteins. Considering the naturally occurring functional group repertoire of biomolecules, it is difficult to site-specifically target a particular site or biomolecule without competing reactions at the multitude of other sites/biomolecules. One solution to the specificity problem is provided by using enzymes that guide the chemistry to peptide recognition tags or enable self-modification reactions at the enzyme's specific microenvironment.1–7 However, steric demand, the need for cofactors and carefully adjusted reaction conditions, or limited substrate scope can be reason for concern.8,9 By contrast, chemical bioconjugation methods potentially offer tolerance to a wider scope of reaction conditions and substrates. To enable site specific bioconjugation of a select protein, chemists introduced templates that recognize a particular arrangement of peptide side chains and orient functional groups in a way that facilitates a bimolecular reaction. Noteworthy examples are the bisarsenic bis-thiol adducts reacting with tetracysteine motifs10,11 and functionalized metal complexes12,13 guiding alkylation reactions to a Cys side chain in the vicinity of oligohistidine or oligoaspartate tags. We, and others, have previously introduced amide and thioether bond forming bioconjugation reactions templated by the formation of coiled coils.14–16The specificity demands are particularly high for orthogonal bioconjugation chemistries enabling the simultaneous modification of two different biomolecules on live cells. While orthogonal covalent labelling has been achieved with enzyme-based chemistries17–19 and through biosynthetic incorporation of unnatural amino acids,20 a chemical method remained elusive. Here, we report the first method to offer orthogonal, chemical bioconjugation on live cells using widely applicable peptide recognition tags. The method relies on coiled coil interactions pioneered by Matsuzaki21 for non-covalent labelling. Recently, Beatty used coiled coils for the imaging of two differently localized proteins via non-covalent labelling in fixed cells.22 For covalent modification in live cells, we designed a template system to guide an acylation reaction to N-terminal cysteine residues of genetically encoded peptide tags (Cys-Acc-POI in Scheme 1). Labelling agents (such as PNA-Don) recognize the targeted tag via formation of coiled coil complexes. This arranges the thioester in the immediate vicinity of the N-terminal cysteine and triggers a proximity-induced native chemical ligation-type transfer reaction.15,23,24 Previously, we have shown covalent labelling of single proteins with dyes and peptide nucleic acid (PNA)-based reporter groups.25PNA is a bio-stable oligonucleotide analogue acting as a unique barcode for the target protein, which can be addressed with complementary DNA, offering the application of DNA nanotechnology beyond typical fluorescence imaging.26–29 In this study, we introduce the simultaneous and orthogonal labelling of two membrane receptors: epidermal growth factor receptor (EGFR) and epidermal growth factor receptor 2 (ErbB2) on living Chinese Hamster Ovary (CHO) cells. We show that interfacing live cell protein labelling with DNA technology provides a direct read-out of EGFR/ErbB2 internalisation.
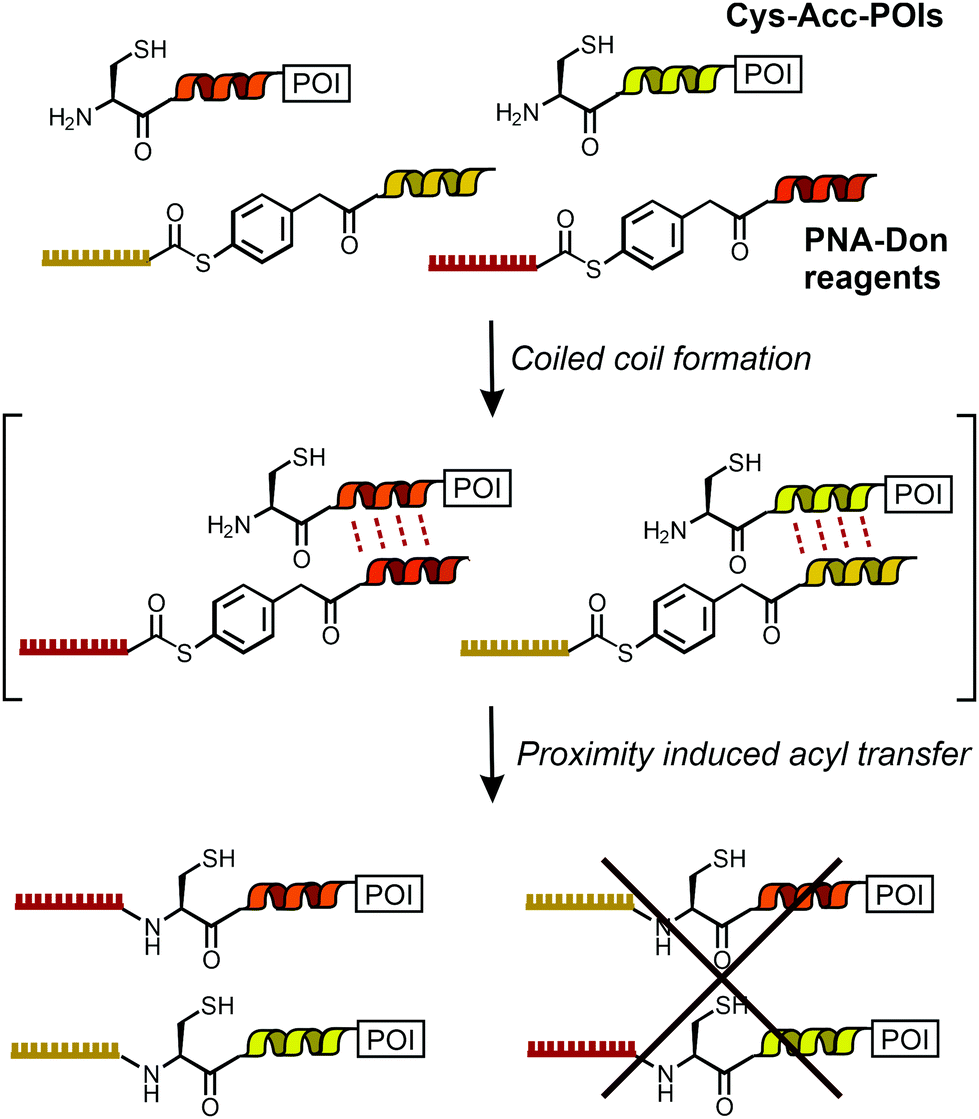 | ||
| Scheme 1 Dual labeling of Cys-Acc-tagged proteins with PNA templated by orthogonal coiled coils. | ||
Results and discussion
We utilized de novo designed parallel heterodimeric coiled coils (P1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P2 and P3:P4) developed by Gradišar and Jerala30 (Fig. 1A). With self-assembly of the P1:P2 and P3:P4 complexes specificity of the acyl transfer is controlled by templation. The labelling reagents PNA1-P2 and PNA3-P4 are comprised of donor peptides P2 and P4 joined to a PNAvia a thioester (Fig. 1B, for synthesis25 see Scheme S3-1, ESI†). To test the feasibility of orthogonal PNA transfer, the donor agent PNA1-P2 was added in excess to the acceptor peptide Cys-P1-TMR (200 nM); a likely scenario in live cell applications. Fluorescence-UPLC (Fl-UPLC) analysis showed that 84% of the acceptor carried the PNA tag after only 10 seconds (Fig. 1B, left). Within one minute labelling was complete. The PNA-acceptor acyl transfer product (P in Fig. 1A, see also Fig. S5, S4, ESI†) was mainly observed in the initial 10 seconds but with excess of thioester a second acyl transfer on the free cysteine led to a doubly PNA-acylated thioester (PP′).15 As previously reported for the K3/E3 coiled coil templated reaction,25 product P was reformed from PP′ upon addition of NaOH (Fig. S5-1 and 2, ESI†). If desired, PP′ can be converted to P in a templated reaction.25 For imaging applications this conversion is not necessary. Testing the second coiled coil template reaction revealed that transfer of PNA from PNA3-P4 onto Cys-P3-TMR proceeded equally rapidly (Fig. 1B right).
:P2 and P3:P4) developed by Gradišar and Jerala30 (Fig. 1A). With self-assembly of the P1:P2 and P3:P4 complexes specificity of the acyl transfer is controlled by templation. The labelling reagents PNA1-P2 and PNA3-P4 are comprised of donor peptides P2 and P4 joined to a PNAvia a thioester (Fig. 1B, for synthesis25 see Scheme S3-1, ESI†). To test the feasibility of orthogonal PNA transfer, the donor agent PNA1-P2 was added in excess to the acceptor peptide Cys-P1-TMR (200 nM); a likely scenario in live cell applications. Fluorescence-UPLC (Fl-UPLC) analysis showed that 84% of the acceptor carried the PNA tag after only 10 seconds (Fig. 1B, left). Within one minute labelling was complete. The PNA-acceptor acyl transfer product (P in Fig. 1A, see also Fig. S5, S4, ESI†) was mainly observed in the initial 10 seconds but with excess of thioester a second acyl transfer on the free cysteine led to a doubly PNA-acylated thioester (PP′).15 As previously reported for the K3/E3 coiled coil templated reaction,25 product P was reformed from PP′ upon addition of NaOH (Fig. S5-1 and 2, ESI†). If desired, PP′ can be converted to P in a templated reaction.25 For imaging applications this conversion is not necessary. Testing the second coiled coil template reaction revealed that transfer of PNA from PNA3-P4 onto Cys-P3-TMR proceeded equally rapidly (Fig. 1B right).
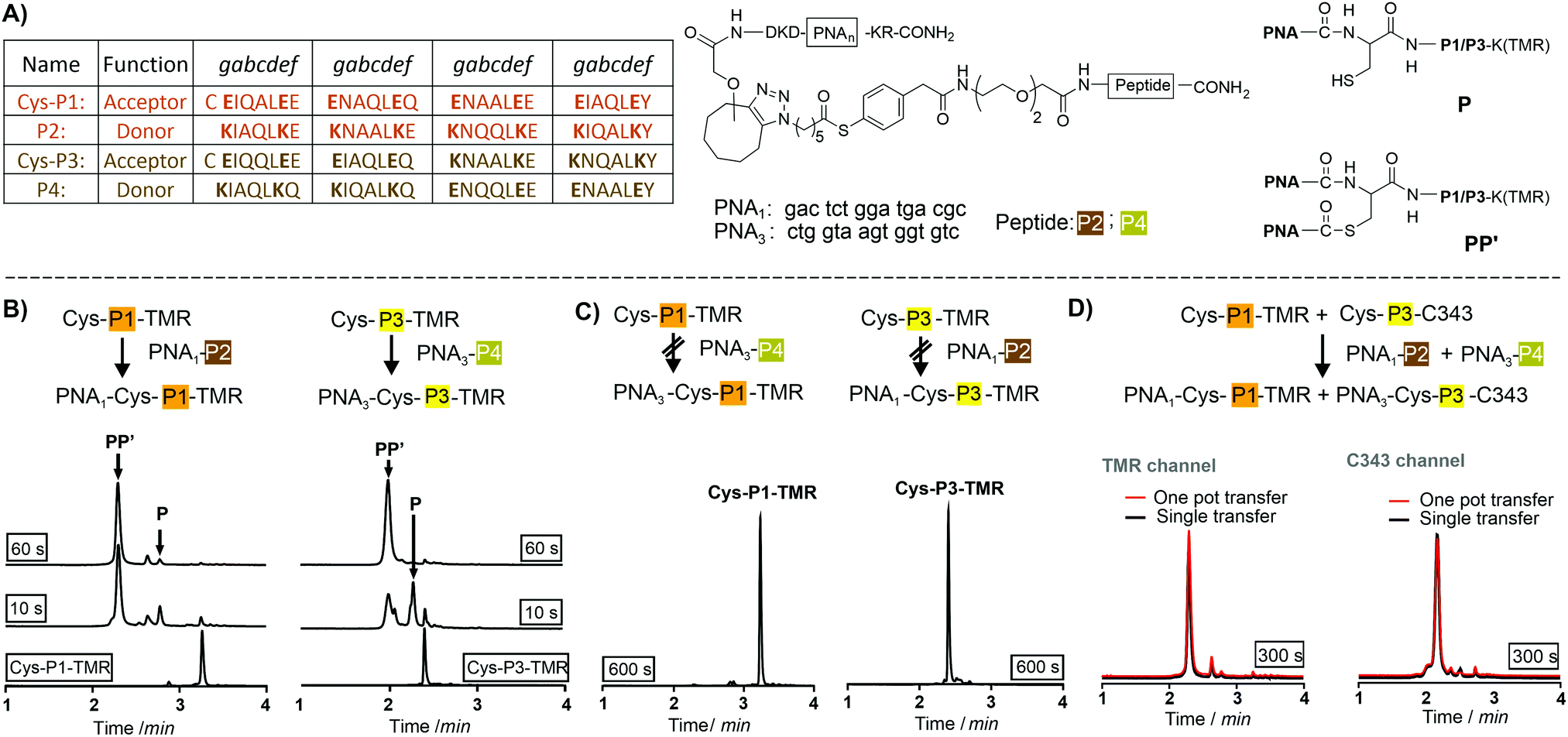 | ||
| Fig. 1 (A) Orthogonal coiled coils, PNA-labeling reagents and products formed in coil coil-templated PNA transfer. Fl-UPLC analysis of reactions involving (B) matched and (C) mismatched coiled coil peptides. Conditions: 200 nM Cys-P1-TMR or Cys-P3-TMR, 1200 nM PNA1-P2 or PNA3-P4 in 200 nM phosphate, 1 mM TCEP, 0.1% CHAPS, pH 7.2, 30 °C. (D) One-pot reactions of a Cys-P1-TMR/Cys-P3-C343/PNA1-P2/PNA3-P4-mixture (red traces) overlaid with Fl-UPLC traces for reactions involving Cys-P1-TMR + PNA1-P2 or Cys-P3-C343 + PNA3-P4 in separate pots (black traces). Detector settings: TMR, Ex 550 nm, Em 580 nm; C343, Ex 420 nm, Em 500 nm. | ||
The specificity was investigated by incubating mismatched peptides (PNA1-P2 with Cys-P3-TMR and PNA3-P4 with Cys-P1-TMR). No new peaks appeared after 10 minutes (Fig. 1C). Furthermore, all peptides were allowed to react in one pot. Cys-P3-C343 was used in place of the TMR labelled peptide to facilitate Fl-UPLC analysis. Cys-P3-C343 and Cys-P3-TMR had similar reactivity (Fig. S5-3, ESI†). The one pot dual label transfer reaction proceeded as quickly as reactions between individual coiled coil pairs (Fig. S5-7, ESI†). Fl-UPLC traces were superimposable suggesting that no new products were formed (Fig. 1D).
To realize live cell fluorescent labelling of the two cell surface proteins EGFR and ErbB2, we used three stable CHO cells enabling doxycycline inducible expression of Cys-P1-EGFR-eYFP, Cys-P3-ErbB2-eCFP, or both (Fig. S8-2, ESI†). Induced Cys-P1-EGFR-eYFP CHO cells were treated with PNA1-P2 or PNA3-P4 (100 nM), respectively, for 4 minutes. Subsequently, cells were incubated with fluorophore conjugated complementary oligonucleotides; Atto565-DNA1 for PNA1-P2 or Cy7-DNA3 for PNA3-P4. For the Cys-P1-EGFR-eYFP cell line, treatment with matched PNA1-P2 and Atto565-DNA1 stained membrane regions (Fig. 2A). Colocalization scatter plots showed that Atto565 signals were not found in pixels lacking an eYFP signal (Fig. S9-3, ESI†). No staining was observed with non-matching PNA-donor thioester and DNA-fluorophore. A similar set of experiments was performed with the Cys-P3-ErbB2-eCFP-CHO cells. Staining of membranous regions and colocalization with the eCFP signal succeeded when the dox induced cell were treated with matched PNA3-P4 and Cy7-DNA3 but not with PNA1-P2 and Atto565-DNA1 (compare Fig. 2B left and right). To verify that both the PNA conjugation and DNA hybridization steps were selective, every combination of PNA-Donor and fluorophore-DNA was tested on both Cys-P1-EGFR-eYFP and Cys-P3-ErbB2-eCFP cells (Fig. S9-1 and 2, ESI†). Labelling only occurred with the correct combination of peptides and complementary DNA.
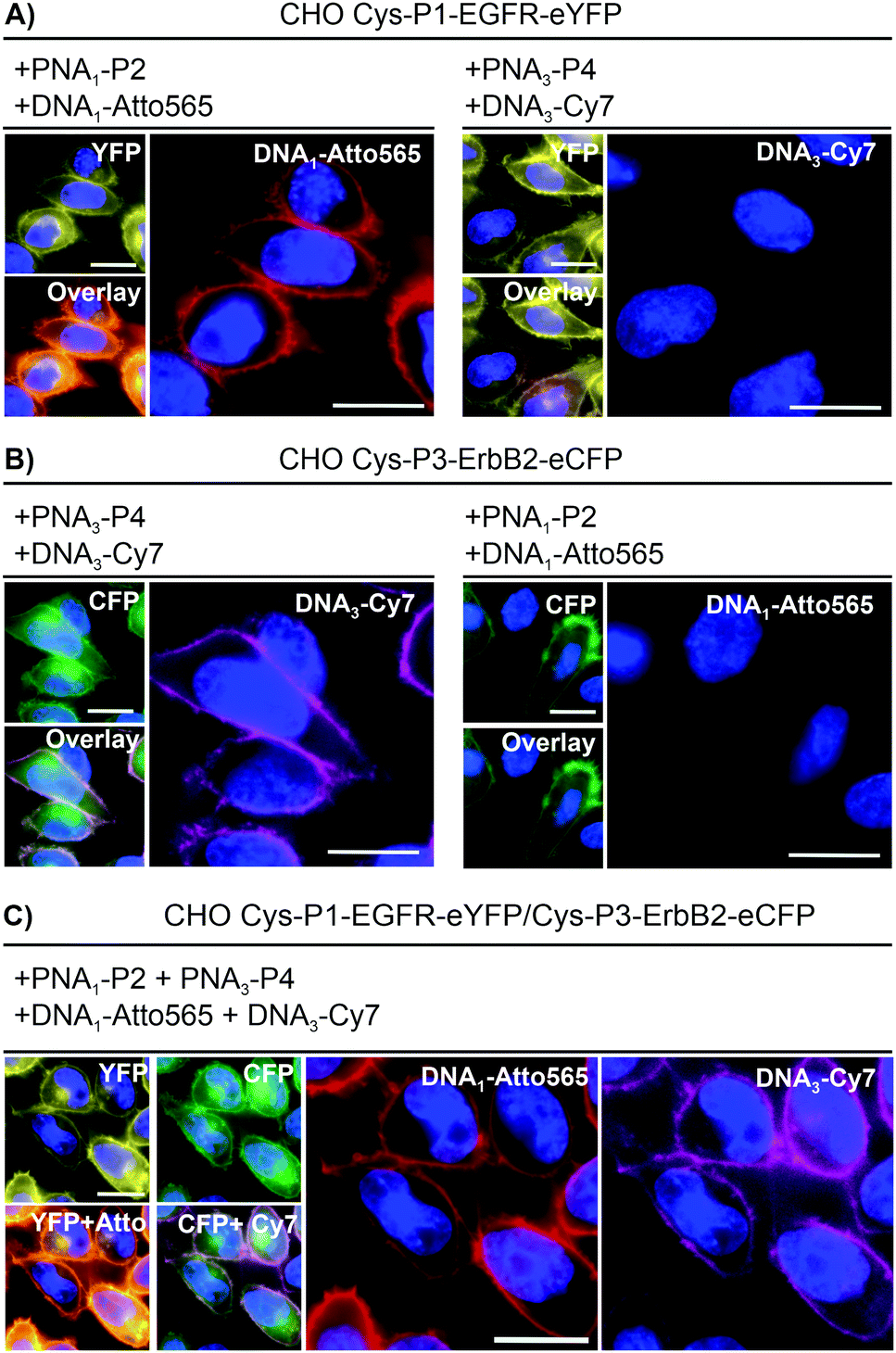 | ||
| Fig. 2 Dual color live cell labeling. After nuclear staining with Hoechst33342 (blue), (A) Cys-P1-EGFR-eYFP or (B) Cys-P3-ErbB2-eCFP expressing CHO cells were treated with matched (left) or mismatched (right) PNA labeling reagents/fluorophore-DNA. Conditions: 4 min with 100 nM PNA1-P2 (EGFR) or/and PNA3-P4 (ErbB2) in DPBS, washing with HBSS, 4 min hybridization with 200 nM Atto565-DNA1 (EGFR) or Cy7-DNA3 (ErbB2). (C) Simultaneous labeling of Cys-P1-EGFR-eYFP/Cys-P3-EGFR-eCFP cells. Scale bar = 10 μm. | ||
For one-pot dual colour labelling CHO cells expressing both Cys-P1-EGFR-eYFP and Cys-P3-ErbB2-eCFP were submitted to the conditions used for single colour labelling, only with addition of both thioesters, PNA1-P2 and PNA3-P4, together followed by hybridization of both oligonucleotides together (Fig. 2C). As before, Atto565 and Cy7 signals marked membrane regions.
To assess the usefulness of our dual colour labelling methodology, we analysed the concurrent internalisation of EGFR and ErbB2 (Fig. 3). These two members of the epidermal growth factor receptor family are critically involved in cancer genesis, and their trafficking is well studied.31,32 Of note, there are no known ligands for ErbB2, which renders classical ligand-based assays unsuitable. Cells were incubated with the PNA1-P2/PNA3-P4 mixture. For increasing brightness and enabling erasure of staining, PNA-labelled cells were treated with DNA complexes comprised of a 57/59 nt long DNA adaptor carrying two imaging strands (Atto565- or Dy751-DNA conjugate) with a 21/22mer portion complementary to the adaptor strand, and an 8mer overhang (Fig. 3A). This serves as a toehold which enables removal of the imaging strand by treatment with a fully complementary ‘eraser DNA’ utilizing a process known as toehold-mediated strand displacement. After staining with the DNA complexes, cells were incubated with either EGF (100 nM), geldanamycin (GA, 3 μM) or just media. Prior to analysis, erasure DNA was added for 8 minutes. Endocytosed receptors are protected from the eraser DNA and any remaining fluorescent signal must be due to internalisation. Therefore, quantification of the fluorescence provides a direct read-out of internalisation. Control experiments confirmed that imager DNA did not stain cells unselectively (Fig. S9-5, ESI†). Cells that were incubated for 20 min with cell medium only showed weak signals within intracellular locations (Fig. 3B, right) indicative of basal internalisation after serum starvation.33 Treatments with EGF and GA revealed the different internalisation behaviour. After 20 min EGF stimulation, EGFR internalisation increased moderately (≈53%) over basal levels, whilst ErbB2 internalisation remained unchanged (Fig. 3B middle and Fig. 3C). Previous measurements had shown that PNA-labelled EGFR undergoes autophosphorylation and internalizes upon EGF activation.25 The rather weak internalisation of EGFR observed here is probably due to coexpression with ErbB2, which has been reported to impair EGF stimulated endocytosis of EGFR.34 Both receptors responded more strongly to 20 min treatment with GA. Of note, GA increased EGFR internalisation by a 2.9-fold compared to 1.4-fold for ErbB2. The modest internalisation of ErbB2 is in agreement with a previous study, which showed that the surface levels of ErbB2 expressed on HEK293 cells were only moderately affected during 1 h of GA stimulation.35
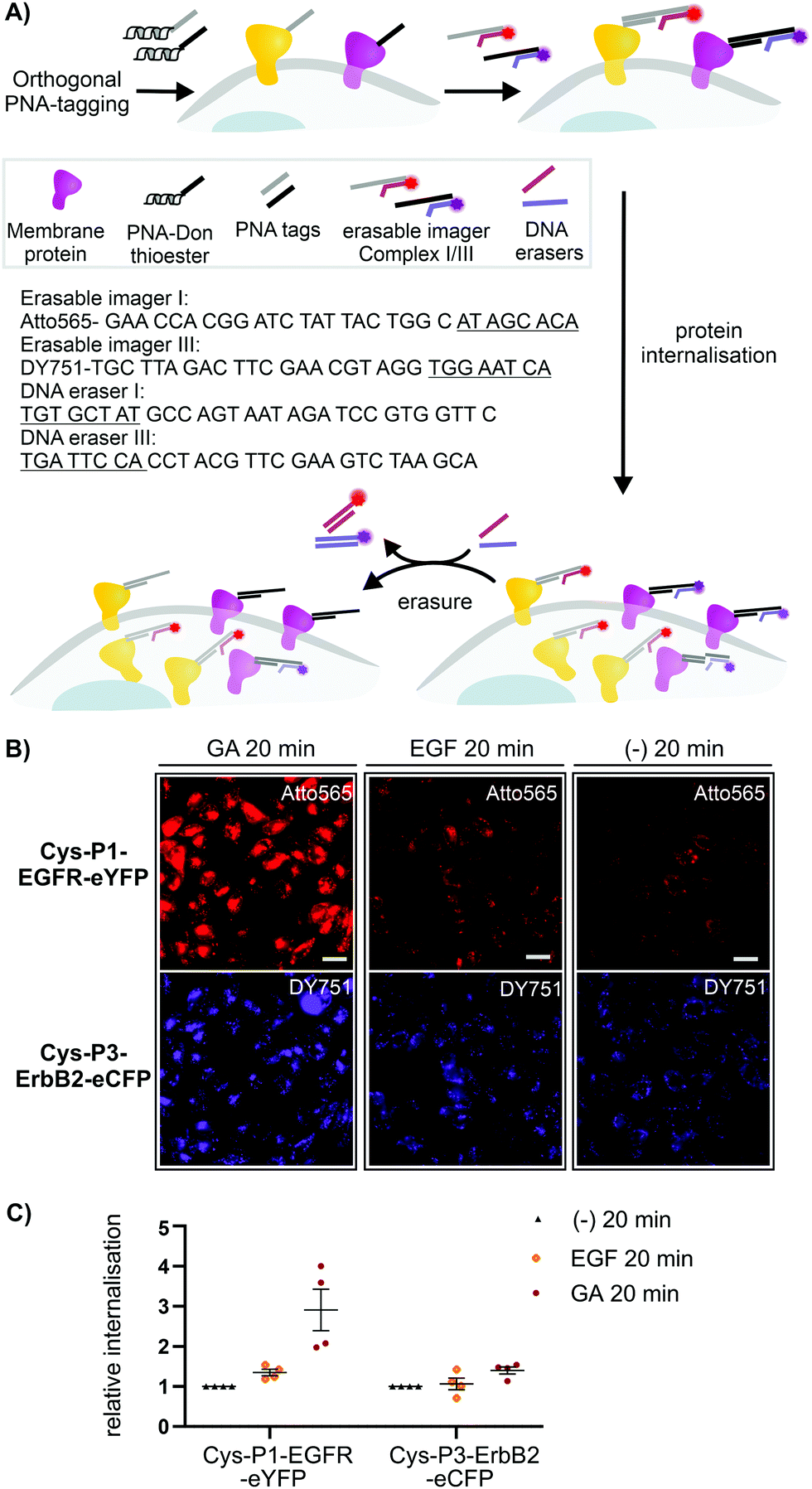 | ||
| Fig. 3 (A) Dual receptor internalization assay. (B) Fluorescent microscopy imaging of internalized EGFR and ErbB2 in CHO cells. Conditions: see ESI.† (C) Quantitative analysis of internalization. Data is presented as internalization relative to the negative control (−) from four independent replicates (n = 100 cells per condition per experiment). Sequences of imager strands and eraser strands are given in 5′ → 3′ direction. Toehold-related sequences are underlined. For other sequences see Table S6-1 (ESI†). | ||
Conclusions
Our data on simultaneous dual-colour labelling of EGFR and ErbB2 on CHO cells shows that coiled coil templation of two parallel acyl transfer reactions is rapid (4 min), bioorthogonal and compatible with cell culture conditions. We used parallel dual labelling to install two different PNA barcodes on EGFR and ErbB2, which enable selective fluorescence staining via hybridization with commercially available oligonucleotide–fluorophore conjugates. This barcoding method also unlocks the potential for interfacing DNA nanotechnologies, such as toehold-mediated strand displacement, which we used to facilitate the analysis of receptor internalisation by removing signals from non-internalized receptors. As a result, the signal obtained by a direct read-out positively correlates with the amount of internalized receptor. The internalisation assay proceeds with live cells and requires only 20 min. Here, we present orthogonal labelling of two proteins. Moreover, six coiled coil pairs from the same de novo designed set have already been shown to be orthogonally functional in mammalian cells by Lebar et al,36 where their interactions were used to simultaneously control protein localization. We believe that using orthogonal coiled coils to template covalent bond forming reactions on live cells is a useful new application and suggests intriguing possibilities for rapid oligonucleotide barcoding of multiple surface proteins on live cells.Author contributions
G. G. planned and performed experiments and drafted the manuscript. K. G. and M. R. did orienting studies on orthogonal coiled coil-templated reactions. M. D. B. and S. B. constructed the stable CHO cell lines. O. S. conceived the study and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Deutsche Forschungsgemeinschaft (SPP1623, Se 819/17-2). M. D. B. is a fellow of the International Max Planck Research School for Molecular Life Sciences (IMPRS-LS).References
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- C. Jing and V. W. Cornish, Acc. Chem. Res., 2011, 44, 784–792 CrossRef CAS PubMed.
- L. Xue, I. A. Karpenko, J. Hiblot and K. Johnsson, Nat. Chem. Biol., 2015, 11, 917–923 CrossRef CAS PubMed.
- X. Chen and Y.-W. Wu, Org. Biomol. Chem., 2016, 14, 5417–5439 RSC.
- J. Lotze, U. Reinhardt, O. Seitz and A. G. Beck-Sickinger, Mol. BioSyst., 2016, 12, 1731–1745 RSC.
- A. F. L. Schneider and C. P. R. Hackenberger, Curr. Opin. Biotechnol, 2017, 48, 61–68 CrossRef CAS PubMed.
- P. Wolf, G. Gavins, A. G. Beck-Sickinger and O. Seitz, ChemBioChem, 2021, 22, 1717–1732 CrossRef CAS.
- I. C. Vreja, I. Nikić, F. Göttfert, M. Bates, K. Kröhnert, T. F. Outeiro, S. W. Hell, E. A. Lemke and S. O. Rizzoli, ACS Nano, 2015, 9, 11034–11041 CrossRef CAS PubMed.
- H. Gelman, A. J. Wirth and M. Gruebele, Biochemistry, 2016, 55, 1968–1976 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063–6076 CrossRef CAS PubMed.
- H. Nonaka, S.-h. Fujishima, S.-h. Uchinomiya, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 9301–9309 CrossRef CAS PubMed.
- I. Takahira, H. Fuchida, S. Tabata, N. Shindo, S. Uchinomiya, I. Hamachi and A. Ojida, Bioorg. Med. Chem. Lett., 2014, 24, 2855–2858 CrossRef CAS.
- J. Wang, Y. Yu and J. Xia, Bioconjugate Chem., 2014, 25, 178–187 CrossRef CAS.
- U. Reinhardt, J. Lotze, S. Zernia, K. Mörl, A. G. Beck-Sickinger and O. Seitz, Angew. Chem., Int. Ed., 2014, 53, 10237–10241 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, K. Morl, A. G. Beck-Sickinger and O. Seitz, Bioconjugate Chem., 2015, 26, 2106–2117 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- I. Nikić, T. Plass, O. Schraidt, J. Szymański, J. A. G. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef PubMed.
- Y. Yano, A. Yano, S. Oishi, Y. Sugimoto, G. Tsujimoto, N. Fujii and K. Matsuzaki, ACS Chem. Biol., 2008, 3, 341–345 CrossRef CAS PubMed.
- J. K. Doh, S. J. Tobin and K. E. Beatty, Biochemistry, 2020, 59, 3051–3059 CrossRef CAS PubMed.
- U. Reinhardt, J. Lotze, K. Mörl, A. G. Beck-Sickinger and O. Seitz, Bioconjugate Chem., 2015, 26, 2106–2117 CrossRef CAS PubMed.
- J. Lotze, P. Wolf, U. Reinhardt, O. Seitz, K. Mörl and A. G. Beck-Sickinger, ACS Chem. Biol., 2018, 13, 618–627 CrossRef CAS PubMed.
- G. C. Gavins, K. Gröger, M. D. Bartoschek, P. Wolf, A. G. Beck-Sickinger, S. Bultmann and O. Seitz, Nat. Chem., 2021, 13, 15–23 CrossRef CAS PubMed.
- A. Zieba, F. Ponten, M. Uhlén and U. Landegren, Mod. Pathol., 2018, 31, 253–263 CrossRef CAS PubMed.
- S. B. Yeldell and O. Seitz, Chem. Soc. Rev., 2020, 49, 6848–6865 RSC.
- R. Jungmann, M. S. Avendaño, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313–318 CrossRef CAS PubMed.
- G. Liszczak and T. W. Muir, Angew. Chem., Int. Ed., 2019, 58, 4144–4162 CrossRef CAS PubMed.
- H. Gradišar and R. Jerala, J. Pept. Sci., 2011, 17, 100–106 CrossRef PubMed.
- V. Bertelsen and E. Stang, Membranes, 2014, 4, 424–446 CrossRef PubMed.
- A. Tomas, C. E. Futter and E. R. Eden, Trends Cell Biol., 2014, 24, 26–34 CrossRef CAS PubMed.
- X. Tan, P. F. Lambert, A. C. Rapraeger and R. A. Anderson, Trends Cell Biol., 2016, 26, 352–366 CrossRef CAS PubMed.
- B. S. Hendriks, L. K. Opresko, H. S. Wiley and D. Lauffenburger, J. Biol. Chem., 2003, 278, 23343–23351 CrossRef CAS PubMed.
- J. C. Stüber, F. Kast and A. Plückthun, ACS Chem. Biol., 2019, 14, 1154–1163 CrossRef PubMed.
- T. Lebar, D. Lainšček, E. Merljak, J. Aupič and R. Jerala, Nat. Chem. Biol., 2020, 16, 513–519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and supporting data. See DOI: 10.1039/d1cb00126d |
| This journal is © The Royal Society of Chemistry 2021 |