
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllylic C(sp3)–H alkylation via synergistic organo- and photoredox catalyzed radical addition to imines†
Jiaqi
Jia
 ab,
Rajesh
Kancherla
b,
Magnus
Rueping
*ab and
Long
Huang
*a
ab,
Rajesh
Kancherla
b,
Magnus
Rueping
*ab and
Long
Huang
*a
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany. E-mail: magnus.rueping@rwth-aachen.de; long.huang@rwth-aachen.de
bKAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal, 23955-6900, Saudi Arabia
First published on 30th April 2020
Abstract
A new catalytic method for the direct alkylation of allylic C(sp3)–H bonds from unactivated alkenes via synergistic organo- and photoredox catalysis is described. The transformation achieves an efficient, redox-neutral synthesis of homoallylamines with broad functional group tolerance, under very mild reaction conditions. Mechanistic investigations indicate that the reaction proceeds through the N-centered radical intermediate which is generated by the allylic radical addition to the imine.
1. Introduction
The selective C–H bond functionalization has expanded rapidly over the past decade owing to its tremendous potential to streamline the synthesis of natural products, pharmaceuticals, agrochemicals and materials.1,2 Allylic C–H activations are of particular interest due to the synthetic versatility of the resulting alkenes and opportunities for further functionalization. Despite the recent progress in the direct oxidation,3 amination,4 carboxylation,5 alkylation6 and arylation7 of allylic C–H bonds, direct C(sp3)–C(sp3) bond formations remain a major challenge for synthetic organic chemistry.Homoallylic amines are versatile building blocks and precursors for the synthesis of bioactive molecules and functional materials.8a Thus, routes for their efficient preparation have received considerable interest (Scheme 1A and B).8 Although advances in conventional organic synthesis have been achieved, several issues need to be addressed. These include the frequent requirement of cryogenic conditions, multi-step syntheses of the organometallic reagents and the generation of stoichiometric amounts of by-products.9 Recent developments in visible light photocatalyzed reactions have enabled new synthetic pathways towards the functionalization of imines.10,11 Notably, the polarity-reversed allylation of imines with prefunctionalized alkenes has been reported.10e This reaction, however, requires stoichiometric amount of Hantzsch ester as electron/proton donor and activator. We recently questioned whether the direct coupling between an aminoalkyl moiety and an allylic C(sp3)–H bond of simple olefins could be feasible. This strategy is generically useful considering it can overcome the challenges of functional group compatibility associated with preparation of allylation precursors and the use of excess reductants. Therefore, it could offer a wealth of opportunities for elaboration of both complex molecules and feedstock olefins.
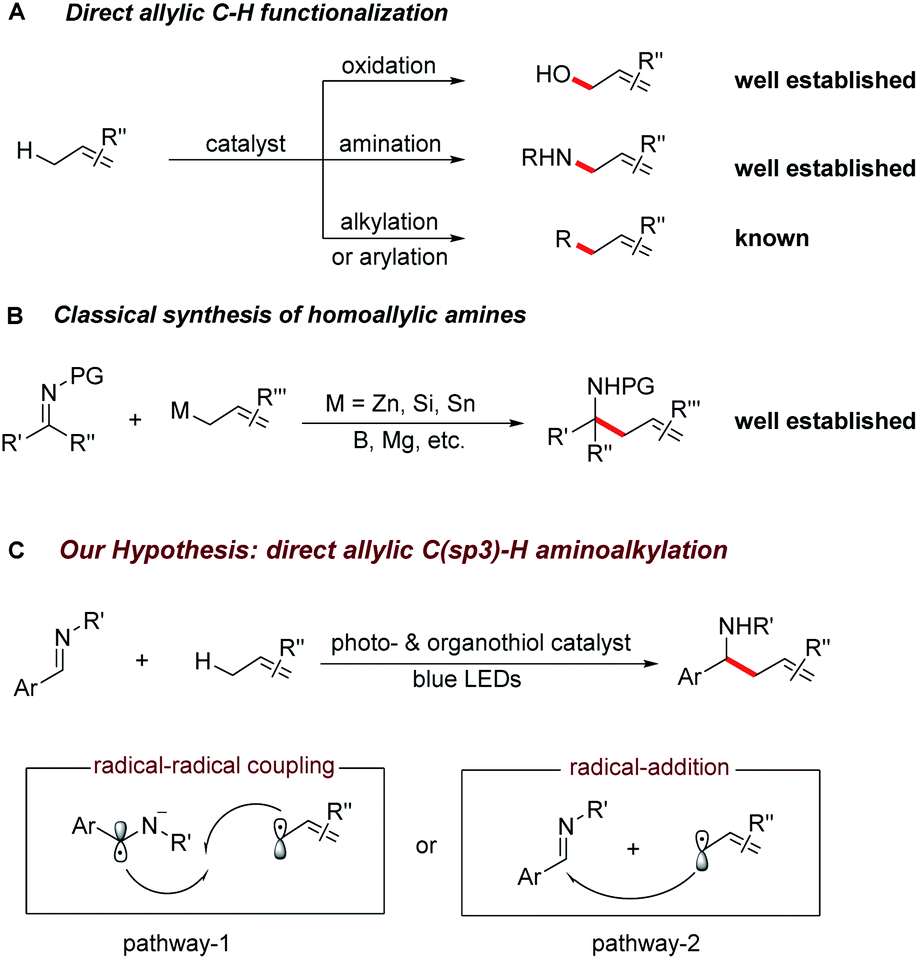 | ||
| Scheme 1 (A) The reaction development of direct allylic C–H activation of olefins. (B) Classical methods towards homoallylic amines synthesis. (C) Photoredox activation of allylic C(sp3)–H bonds for the homoallylic amines synthesis, initial proposed reaction pathways. | ||
In line with our recent reports on reductive Umpolung of imines10f,g as well as recent development on synergistic organo- and photoredox catalysis,12 we envisioned that upon visible light irradiation, the resulting oxidizing excited state of a photocatalyst PC(n)+* could be quenched by a thiol catalyst RSH through proton coupled electron transfer (PCET)13 in the presence of a basic additive14 to give the corresponding reduced PC(n−1)+ complex and a transient thiyl radical RS·.15 The generated electrophilic thiyl radical would then react with the alkene substrate (allylic C–H bond dissociation energy BDE is about 340 kJ mol−1) via hydrogen atom transfer to form an allylic radical along with the regeneration of thiol catalyst (S–H BDE is typically 365 kJ mol−1).16
Next, the oxidation of PC(n−1)+ species with imine ArCH = NR′ via single electron transfer would result in the formation of the aminoalkyl radical anion17 and the following radical–radical cross coupling18 with the allylic radical19 would result in the desired C–C bond formation (pathway 1). Alternatively, the allylic radical could add to the imine to form a N-centred radical, which is subsequently reduced by PC(n−1)+ to give the desired product (pathway 2). Herein, we report the successful development of a new protocol for the direct alkylation of allylic C(sp3)–H bonds via synergistic organo- and photoredox catalysis (Scheme 1C) and provide insight into the reaction mechanism of this efficient addition reaction.
2. Results and discussions
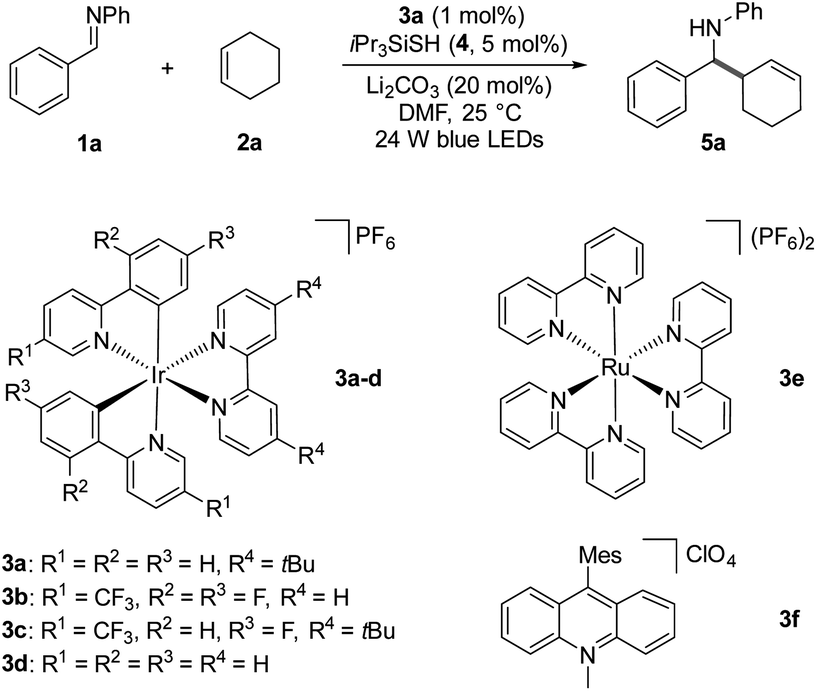
Based on our mechanistic hypothesis, we first evaluated the reaction conditions with imine 1a and cyclohexene 2a. To our delight, we found that the reaction of 1a with 2a (5 equiv) in the presence of [Ir(ppy)2(dtbbpy)]PF63a (ppy = 2,2′-bipyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine,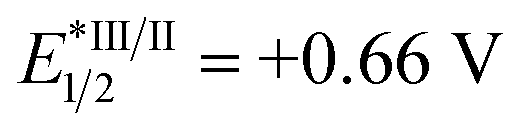 vs. SCE in MeCN)20 (1 mol%), organocatalyst 4 (5 mol%) and Li2CO3 (20 mol%) in DMF under irradiation with 24 W blue LEDs (λmax = 405 nm) afforded the desired homoallylic amine product 5a in 81% yield (Table 1, entry 1). Remarkably, we did not observe any reductive homocoupling product under these reaction conditions.10g Control experiments confirmed that the reaction did not proceed in the absence of the photocatalyst and light (entries 2 and 3). Moreover, only traces of the desired product were obtained in the absence of thiol 4, indicating the necessity of the hydrogen atom transfer (HAT) catalyst (entry 4). The use of other Ir-based photocatalysts gave 5a in lower yields (entries 5–7). In sharp contrast, only 5% of the desired coupling product was obtained when less reducing Ru(bpy)3(PF6)2 (EII/I1/2 = −1.35 V vs. SCE)21 was employed as photocatalyst (entry 8). The organic photocatalyst mesityl acridinium which is known as a weak reductant in its reduced form was next examined,22 but no product was observed (entry 9). Interestingly, a moderate yield was obtained in the absence of a basic additive (entry 10). In addition, we found that DMF was a suitable solvent while MeCN was inferior (entry 11). Notably, 34% of 5a was obtained when 2a was used as limiting reagent (entry 12). The use of other thiols failed to improve the yield (see ESI†).
vs. SCE in MeCN)20 (1 mol%), organocatalyst 4 (5 mol%) and Li2CO3 (20 mol%) in DMF under irradiation with 24 W blue LEDs (λmax = 405 nm) afforded the desired homoallylic amine product 5a in 81% yield (Table 1, entry 1). Remarkably, we did not observe any reductive homocoupling product under these reaction conditions.10g Control experiments confirmed that the reaction did not proceed in the absence of the photocatalyst and light (entries 2 and 3). Moreover, only traces of the desired product were obtained in the absence of thiol 4, indicating the necessity of the hydrogen atom transfer (HAT) catalyst (entry 4). The use of other Ir-based photocatalysts gave 5a in lower yields (entries 5–7). In sharp contrast, only 5% of the desired coupling product was obtained when less reducing Ru(bpy)3(PF6)2 (EII/I1/2 = −1.35 V vs. SCE)21 was employed as photocatalyst (entry 8). The organic photocatalyst mesityl acridinium which is known as a weak reductant in its reduced form was next examined,22 but no product was observed (entry 9). Interestingly, a moderate yield was obtained in the absence of a basic additive (entry 10). In addition, we found that DMF was a suitable solvent while MeCN was inferior (entry 11). Notably, 34% of 5a was obtained when 2a was used as limiting reagent (entry 12). The use of other thiols failed to improve the yield (see ESI†).
|
|
||
|---|---|---|
| Entrya | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), cyclohexene 2a (1.0 mmol), Li2CO3 (20 mol%), iPr3SiSH 4 (5 mol%), 3a–f (1 mol%), rt, solvent (2 mL), 24 W LEDs (450 nm), 24 h. b Yield of product after isolation. | ||
| 1 | None | 81 |
| 2 | No photocatalyst | 0 |
| 3 | No light | 0 |
| 4 | No thiol 4 | Trace |
| 5 | Photocatalyst 3b instead of 3a | 38 |
| 6 | Photocatalyst 3c instead of 3a | 58 |
| 7 | Photocatalyst 3d instead of 3a | 64 |
| 8 | Photocatalyst 3e instead of 3a | 5 |
| 9 | Photocatalyst 3f instead of 3a | 0 |
| 10 | No Li2CO3 | 55 |
| 11 | MeCN as solvent | 66 |
| 12 | 2a as limiting reagent | 34 |
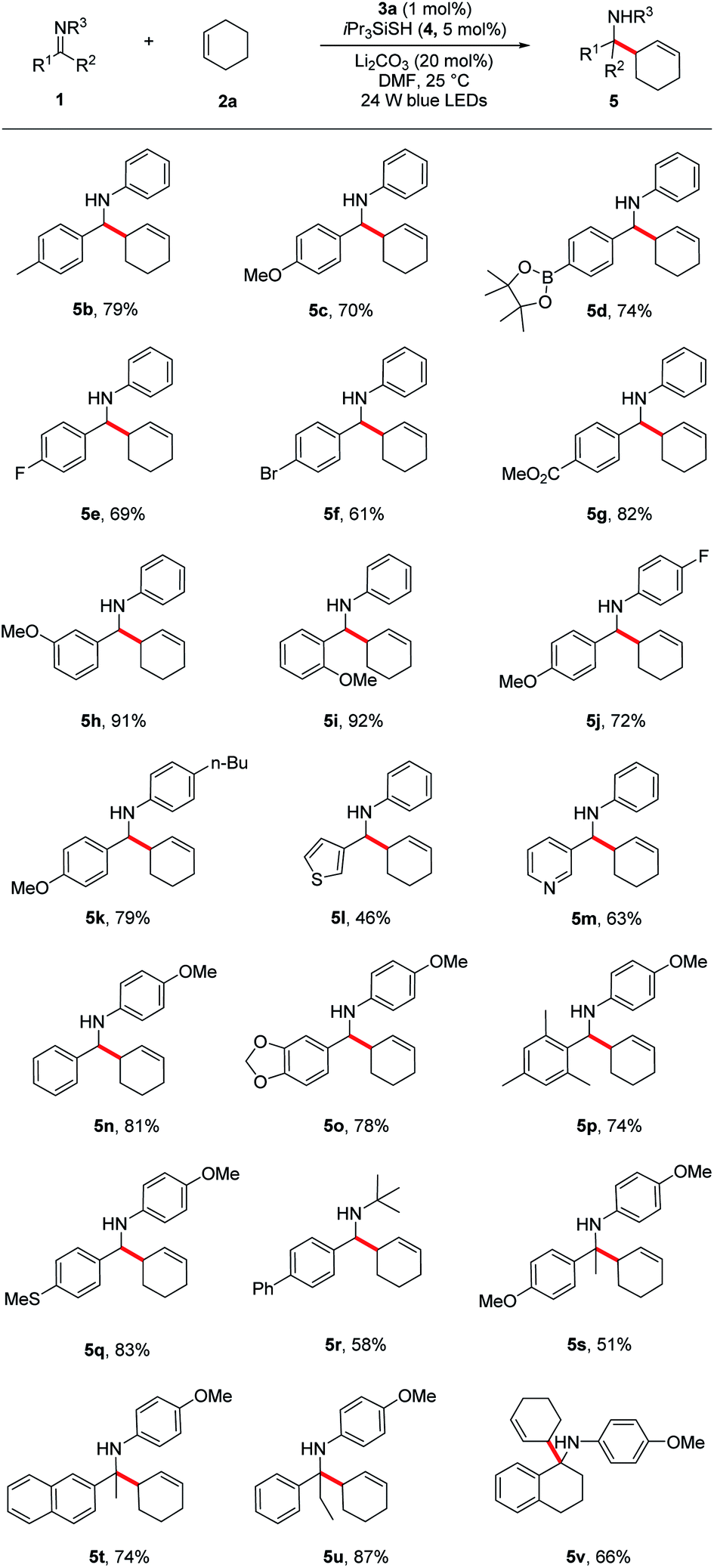
Under the optimized reaction conditions, we next screened various imines 1 in reaction with cyclohexene 2a. As shown in Table 2, a wide range of N-aryl imines with various substituents on the aryl ring underwent the aminoalkylation smoothly, affording the desired homoallylic amines 5b–k in good to excellent yields (61–92%) regardless of their electronic nature as well as substituent position. Notably, imines containing heterocyclic groups such as thiophene and pyridine were compatible with the reaction conditions and gave the corresponding products 5l and 5m in 46% and 63% yield, respectively. Moreover, the reaction proceeded in excellent yields for imines bearing the readily removable N-p-methoxyphenyl group (5n–q, 74–83%). In addition to N-aryl imines, imine 1r derived from an alkyl amine was also tolerated, albeit in moderate yield (5r, 58%).
a Reaction conditions: imine 1 (0.3 mmol), cyclohexene 2a (1.5 mmol), Li2CO3 (20 mol%), iPr3SiSH 4 (5 mol%), 3a (1 mol%), DMF (1.5 mL), 24 W LEDs (450 nm), rt, 24 h; yields after isolation; dr between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1.2:1. :1 to 1.2:1.
|
|---|
|
Encouraged by these results, we next evaluated more challenging ketimine substrates for the construction of all-carbon quaternary centers. Not surprisingly, aromatic ketimines were converted to the corresponding homoallylamines in good yields (5s–v, 51–87%). Next, we evaluated the scope of allylic radicals that can be generated with this protocol. Under the optimized conditions, a variety of cyclic and acyclic alkenes were successfully aminoalkylated using imine 1a, furnishing the corresponding products in good yields (Table 3). In addition to cyclohexene 2a described above, several other cyclic alkenes 2b–d (five, seven and eight membered derivatives) were tested and the corresponding homoallylic amines 6a–d were prepared in good to excellent yields under the reaction conditions (6a–d, 62–90%). An excellent regioselectivity was observed with 1-(tert-butyl)cyclohex-1-ene (2d), indicating that steric effects play a predominant role in determining the regioselectivity. For asymmetrically substituted alkene 2f which has two different C–H bonds at the allylic sites, isomers of aminoalkylation were observed. The selective formation of 6f and 6f′ suggests that the proton abstraction of allylic methine C–H bonds is favoured over primary C–H bonds due to the formation of more stabilized carbon centred radical. It is notable that unprotected allylic alcohol 2g was also aminoalkylated providing the corresponding product 6g in 51% yield.
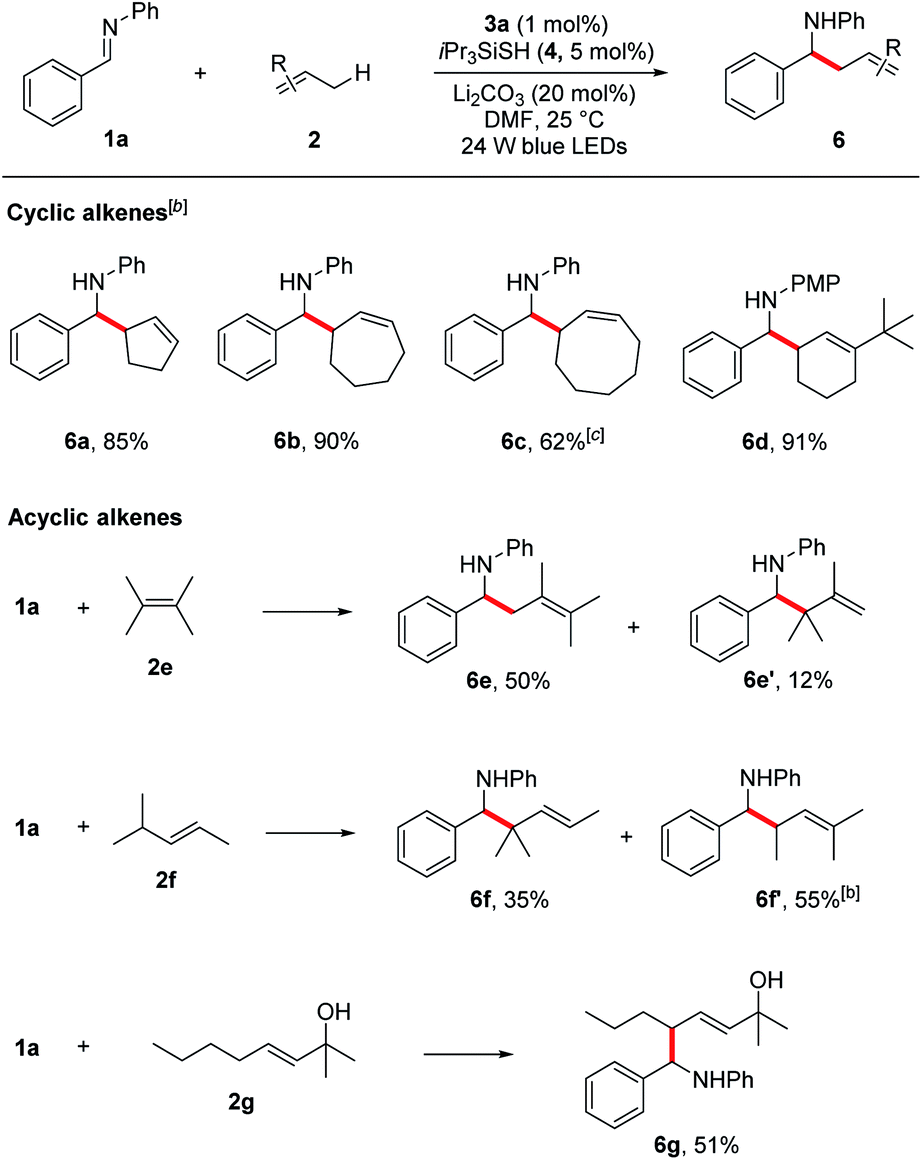
Owing to the wide applications of natural product modification in the fields of medicine, health science, pharmacy and biology, we subsequently focused on the aminoalkylation of several naturally occurring substrates as well as their derivatives (Fig. 1). Both regioisomers of pinenes could be converted to the homoallylic amines 6h and 6i with excellent regioselectivities at the methylene C–H bonds. This can be explained by the electrophilic thiol radical selectively abstracts the allylic C–H bond to form the more stabilized radical of pinenes. Meanwhile, the steric effect also plays a major part in determining the regioselectivity, the C–C bond formation selectively takes place in the sterically less-hindered allylic position. Steroid derivatives such as cholesterol and pregnenolone could also be functionalized under the photoredox conditions and the aminoalkylation took place selectively at the allylic position, affording the corresponding diastereomers 6j–l in good yields. In comparison with literature precedent,7c the low to moderate diastereoselectivities indicates that a possible different reaction pathway was involved for the current transformation.
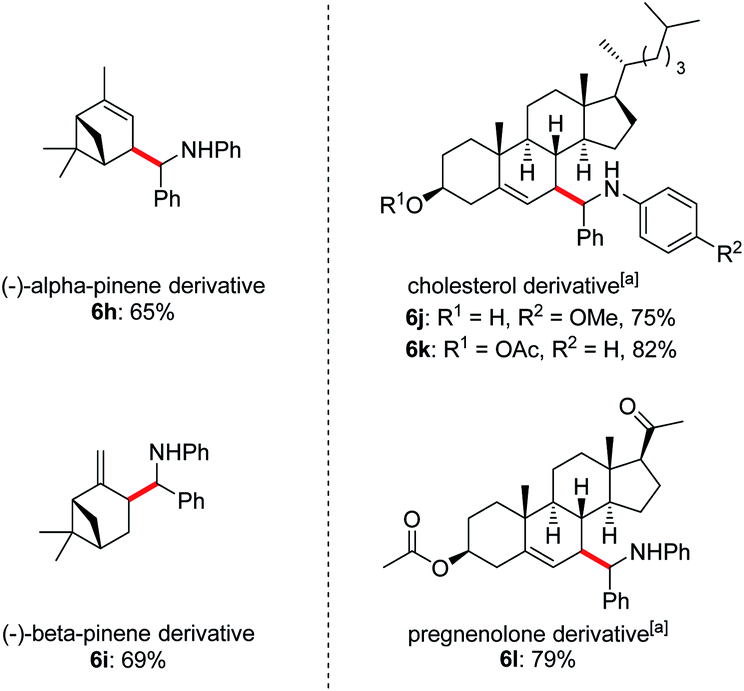 | ||
| Fig. 1 Late-stage diversification of complex molecules. Reaction conditions: imine 1 (0.2 mmol), alkene 2 (1.0 mmol), Li2CO3 (20 mol%), iPr3SiSH 4 (5 mol%), 3a (1 mol%), DMF (1.0 mL), 24 W LEDs (450 nm), rt, 24 h, for dr see ESI.†a2 (0.6 mmol), iPr3SiSH 4 (10 mol%), DMF (2 mL). b Isomers ratios: 6h (1.2:1:1:1), 6i (3:3:1:1), 6j (1.6:1.6:1:1), 6k (1.2:1:1:1), 6l (1.9:1.9:1). | ||
In order to gain some insight into the reaction mechanism, we first performed radical inhibition experiments. The reaction was greatly suppressed in the presence of various radical inhibitors, indicating the involvement of a radical process. It is noteworthy that radical initiators failed to give any product even at elevated temperature (see ESI†). Next, a series of experiments were conducted to better understand the nature of photoinduced single electron transfer step. The photocatalyst [Ir(ppy)2(dtbbpy)]PF63a on photoexcitation can result in a long-lived triplet excited state *3a (τ0 = 454.71 ± 0.30 ns in DMF, Fig. S1†), which can result in SET in the presence of suitable quencher. Therefore, Stern–Volmer quenching experiments were performed in order to confirm the quenching of 3a. Initially, steady-state fluorescence quenching of 3a using different concentrations of silanethiol 4 was carried out in the presence of Li2CO3, where a predominant quenching was observed with a linear correlation (Fig. 2a). Further, a linear correlation similar to steady-state experiments was also observed by time-resolved emission spectroscopy studies where excited-state life-time of *3a is quenched by different concentrations of 4 in the presence of Li2CO3 (Fig. 2a). Linear correlation by both the time-resolved and steady-state Stern–Volmer quenching experiments reveal the dynamic quenching of triplet excited-state of *3a by 4 in the presence of base. Next, a similar quenching experiments was carried out in the absence of base which also displayed considerable quenching of *3a (Fig. 2a). In-line with the observed result, a moderate yield of 55% of homoallylic amine 5a was obtained in the absence of a basic additive (Table 1, entry 10). Simultaneous steady-state Stern–Volmer quenching studies using 1a proved that there is no quenching of excited-state of *3a even in the absence and presence of base (Fig. 2b). In order to understand the electron transfer event better, the electron transfer rate constant kET was determined by time-resolved emission spectroscopy measurements. An electron transfer rate constant, kET = (2.22 ± 0.06) × 109 L mol−1 s−1 was obtained by plotting the difference between the observed rate constant (kobs) and rate constant without quencher (k0) versus different concentrations of 4 in presence of base which gave a linear fit (Fig. 2c and d).
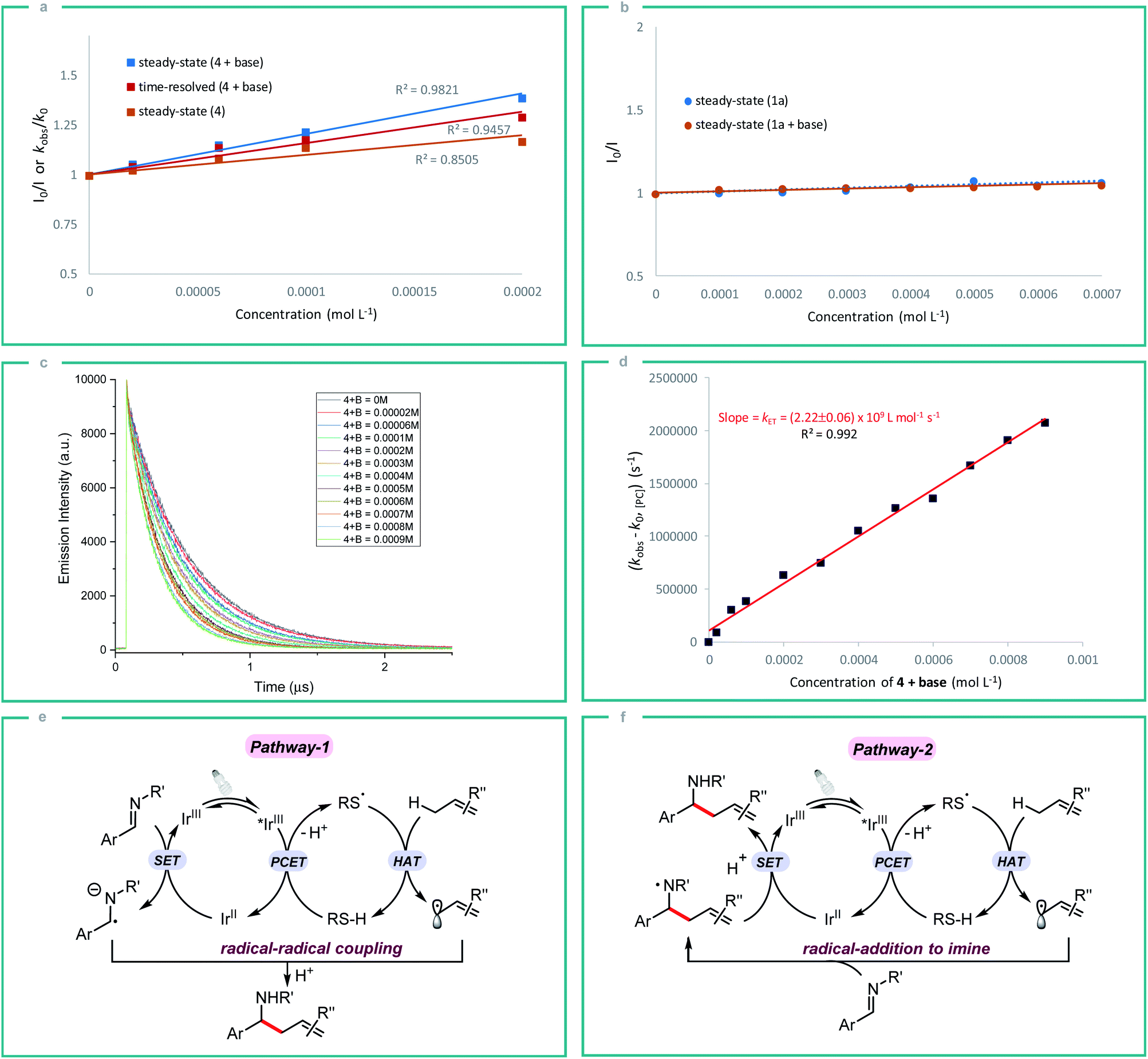 | ||
| Fig. 2 (a) Steady-state and time-resolved Stern–Volmer quenching of 3a with 4 and the mixture of 4 + base (Li2CO3); (b) Steady-state Stern–Volmer quenching experiment with 1a and the mixture of 1a + base (Li2CO3); (c) phosphorescence lifetimes of excited-state photocatalyst *3a (1 × 10−5 M) at different concentrations of quencher mixture 4 + base (B = Li2CO3). (d) Stern–Volmer analysis yielded a rate constant, kET, of 2.22 × 109 L mol−1 s−1 by the PCET between *3a and 4 in the presence of base; (e & f) proposed mechanistic pathways for aminoalkylation of allylic C(sp3)–H bonds. | ||
Initially, we assumed that the alkylation process may proceed through the radical–radical coupling pathway (Fig. 2e, pathway-1), where the allylic radical couples with the persistent radical anion intermediate that is generated by the SET reduction of imine. However, the lack of informative byproducts formation including reduced amine and vicinal diamine derived from the N-centered radical anion prompted us to think otherwise. And if such an intermediate is involved in the reaction process, there should be a yield dependence on the electronics of the imine. Based on our substrate scope both electron rich (1b) and electron deficient (1g) imines gave the expected product in good yield which indicates the possibility of an alternate pathway. Furthermore, the reduction potential of imines 1a and 1c (E1/2 = −1.91 and −2.01 V vs. SCE respectively)10c is very low compared to the redox potential of photocatalyst 3a (IrIII/IrII = −1.51 V) which does not favour the formation of radical anion intermediate. Nevertheless, both the imines 1a and 1c gave the expected product in good yield. All these observations strongly indicate that the reaction proceeds through the N-centered radical intermediate which is generated by the allylic radical addition to the imine. This N-centered radical subsequently gets reduced by IrII to give the expected product (Fig. 2f, pathway-2).23
In conclusion, we developed a new method for the efficient synthesis of homoallylic amines through the direct alkylation of allylic C(sp3)–H bonds of simple alkenes with imines by using the combination of photoredox and organocatalysis. The reaction tolerates a wide range of functional groups and proceeds under mild reaction conditions. The reactivity and utility of this protocol can be further emphasized in the late stage allylic C(sp3)–H bond functionalization of a range of complex natural products and derivatives. The strategy developed herein could be of relevance for organic synthesis as well as pharmaceutical development and may inspire the discovery other direct allylic C(sp3)–H functionalizations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/3754).References
- For selected reviews on C–H functionalization in the synthesis of complex molecules, see: (a) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (c) P. Tao and Y. Jia, Sci. China: Chem., 2016, 59, 1109–1125 CrossRef CAS.
- For reviews on photoredox catalysis in C–H functionalizations, see: (a) D. C. Fabry and M. Rueping, Acc. Chem. Res., 2016, 49, 1969–1979 CrossRef CAS PubMed; (b) X.-Q. Hu, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960–1962 CrossRef CAS PubMed; (c) S. Roslin and L. R. Odell, Eur. J. Org. Chem., 2017, 2017, 1993–2007 CrossRef CAS; (d) Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 28, 741–750 CrossRef CAS; (e) W.-J. Zhou, Y.-H. Zhang, Y.-Y. Gui, L. Sun and D.-G. Yu, Synthesis, 2018, 50, 3359–3378 CrossRef CAS; (f) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2019, 62, 24–57 CrossRef CAS.
- For selected examples of allylic C–H oxidation: (a) M. B. Andrus and Z. Zhou, J. Am. Chem. Soc., 2002, 124, 8806–8807 CrossRef CAS PubMed; (b) D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448–6451 CrossRef CAS PubMed.
- For reviews on allylic C–H aminaton: (a) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS PubMed; (b) R. L. Grange, E. A. Clizbe and P. A. Evans, Synthesis, 2016, 48, 2911–2968 CrossRef CAS.
- N. Ishida, Y. Masuda, S. Uemoto and M. Murakami, Chem.–Eur. J., 2016, 22, 6524–6527 CrossRef CAS PubMed.
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 56–57 CrossRef CAS PubMed; (c) J. M. Howell, W. Liu, A. J. Young and M. C. White, J. Am. Chem. Soc., 2014, 136, 5750–5754 CrossRef CAS PubMed; (d) R. Zhou, H. Liu, H. Tao, X. Yu and J. Wu, Chem. Sci., 2017, 8, 4654–4659 RSC.
- (a) T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118–3123 RSC; (b) M. Sekine, L. Ilies and E. Nakamura, Org. Lett., 2013, 15, 714–717 CrossRef CAS PubMed; (c) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef CAS PubMed; (d) L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed.
- (a) H. Ding and G. K. Friestad, Synthesis, 2005, 2005, 2815–2829 CrossRef; (b) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS PubMed; (c) R. Wang, Y. Luan and M. Ye, Chin. J. Chem., 2019, 37, 720–743 CrossRef CAS.
- (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef CAS; (b) R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS PubMed.
- For a recent review on photoredox catalyzed funtionalization of imines, see: (a) J. A. Leitch, T. Rossolini, T. Rogova, J. A. P. Maitland and D. J. Dixon, ACS Catal., 2020, 10, 2009–2025 CrossRef CAS . For selected examples, see: ; (b) T. Ju, Q. Fu, J.-H. Ye, Z. Zhang, L.-L. Liao, S.-S. Yan, X.-Y. Tian, S.-P. Luo, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2018, 57, 13897–13901 CrossRef CAS PubMed; (c) N. R. Patel, C. B. Kelly, A. P. Siegenfeld and G. A. Molander, ACS Catal., 2017, 7, 1766–1770 CrossRef CAS PubMed; (d) W.-J. Zhou, G.-M. Cao, G. Shen, X.-Y. Zhu, Y.-Y. Gui, J.-H. Ye, L. Sun, L.-L. Liao, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed; (e) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed; (f) E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776–6779 CrossRef CAS PubMed; (g) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed; (h) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed; (i) J. L. Jeffrey, F. R. Petronijević and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404–8407 CrossRef CAS PubMed; (j) D. Hager adn and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef PubMed; (k) For a general review on photocatalytic C-Heteroatom bond formation, see: A. Ruffoni, and D. Leonori, Photocatalytic Carbon-Heteroatom Bond Formation, Science of Synthesis, 2019, p. 517 Search PubMed.
- M. Hopfner, D. M. Wei, F. W. Heinemann and H. Kisch, Photochem. Photobiol. Sci., 2002, 1, 696–703 RSC.
- For recent examples on synergistic organo- and photoredox catalysis, see: (a) A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017, 9, 1073 CrossRef CAS PubMed; (b) A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS PubMed; (c) R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621–16625 CrossRef CAS PubMed; (d) T. Miura, Y. Funakoshi, J. Nakahashi, D. Moriyama and M. Murakami, Angew. Chem., Int. Ed., 2018, 57, 15455–15459 CrossRef CAS PubMed; (e) W. Xu, J. Ma, X.-A. Yuan, J. Dai, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 10357–10361 CrossRef CAS PubMed; (f) N. Zhou, X.-A. Yuan, Y. Zhao, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3990–3994 CrossRef CAS PubMed; (g) M. Das, M. D. Vu, Q. Zhang and X.-W. Liu, Chem. Sci., 2019, 10, 1687–1691 RSC; (h) Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed; (i) M. Zhang, X.-A. Yuan, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312–316 CrossRef CAS PubMed; (j) H. Jiang and A. Studer, Chem. - Eur. J., 2019, 25, 7105–7109 CrossRef CAS PubMed.
- For selected reviews on PCET, see: (a) R. Knowles and H. Yayla, Synlett, 2014, 25, 2819–2826 CrossRef; (b) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (c) D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed; (d) N. Hoffmann, Eur. J. Org. Chem., 2017, 2017, 1982–1992 CrossRef CAS.
- K. A. Ogawa and A. J. Boydston, Org. Lett., 2014, 16, 1928–1931 CrossRef CAS PubMed.
- For reviews on thiyl radicals, see: (a) F. Dénès, C. H. Schiesser and P. Renaud, Chem. Soc. Rev., 2013, 42, 7900–7942 RSC; (b) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- For reviews on persistence radical effect, see: (a) A. Studer, Chem. - Eur. J., 2001, 7, 1159–1164 CrossRef CAS PubMed; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed; (c) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC.
- (a) J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed; (b) H. Mitsunuma, S. Tanabe, H. Fuse, K. Ohkubo and M. Kanai, Chem. Sci., 2019, 10, 3459–3465 RSC.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- At the current stage, we can not rule out that the N-centered radical abstacts hydrogen atom from benzylic C–H bond to form a benzyl carbon radical, which can be further reduced to generate carbon anion then undergoes protonation to afford the product..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00819b |
| This journal is © The Royal Society of Chemistry 2020 |