
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirectional molecular sliding movement in peptide hydrogels accelerates cell proliferation†
Shuxin
Song
a,
Jingyu
Wang
*b,
Zhifei
Cheng
a,
Zhimou
Yang
 c,
Linqi
Shi
a and
Zhilin
Yu
*a
c,
Linqi
Shi
a and
Zhilin
Yu
*a
aKey Laboratory of Functional Polymer Materials, Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: yzh026@nankai.edu.cn
bSchool of Biomedical Engineering and Technology, Tianjin Medical University, Tianjin 300070, China. E-mail: wangjy_026@tmu.edu.cn
cState Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials, Ministry of Education, College of Life Sciences, Nankai University, Tianjin 300071, China
First published on 16th December 2019
Abstract
Adjusting the mechanical cues generated in cellular microenvironments is important for manipulating cell behaviour. Here we report on mechanically dynamic hydrogels undergoing directional domain sliding motion and investigate the effect of the well-defined mechanical motion on accelerating cell proliferation. The mechanically dynamic hydrogels were prepared via self-assembly of an amphiphilic peptide consisting of two alternating polar and nonpolar domains cross-linked by disulfide bonds at a nonsymmetrical position. The cross-linked peptide assembled into entangled nanofibers driven by the hydrophobic collapse involving a partial-length sequence due to the covalent constraint. Reduction of the disulfide bonds led to formation of non-equilibrated peptide bilayers, which underwent directional domain sliding motion along each promoted by the thermodynamically favourable transition from the partial to full hydrophobic collapse. The mechanical cues resulting from the directional domain sliding motion within the mechanically dynamic hydrogels accelerated cell proliferation when incubating cells on the hydrogel, compared to the thermodynamically static counterparts, via a mechanotransduction mechanism as supported by the facilitated translocation of yes-associated proteins into the nucleus of the cells. Our finding demonstrates the great potential of mechanically dynamic hydrogels as new-generation biomimetic extracellular matrices in tissue engineering and regeneration.
Introduction
Mechanical cues surrounding cells play critical roles in modulating cell behaviour and tissue generation.1–4 This natural phenomenon has inspired design and creation of biomimetic extracellular matrices (ECMs) exhibiting specific mechanical cues for efficient manipulation of cell adhesion, spreading, proliferation, and differentiation.5–9 Conventionally, the mechanical cues within artificial ECMs are associated with external physical forces or the intrinsic mechanical properties of soft materials.10–16 On the one hand, the external physical force can be applied from different sources with variable strengths to stimulate the mechanotransduction pathway of cells.17–19 On the other hand, a considerable number of biocompatible hydrogels with tuneable interacting strengths with cells via incorporating cell-binding epitopes into hydrogels or controlling the mechanical properties of hydrogels have been developed and broadly utilized as artificial ECMs.20–34 However, while the former approach suffers from the less precise and inefficient transduction of mechanical signals to in vivo cells, the mechanical signals generated by the latter strategy exhibit limited dynamic features to actively stimulate cells. To address these challenges, alternative to the above two approaches, creation of hydrogels undergoing internal mechanical movements as artificial matrices allows for generation of intrinsic mechanical cues and thereby directly and efficiently manipulating cell behaviour.35–41 In addition, precise control over the mechanical movements within dynamic hydrogels potentially leads to the mechanical cues with specific intensity and life-time, which is beneficial for different cellular processes with distinct requirements. Despite the progress achieved in soft materials possessing unique mechanical cues, precise control over the mechanical movements within biocompatible hydrogels beyond the dynamic motions to generate mechanical cues remains challenging.Self-assembly of peptides into well-defined nanostructures has been considered as an efficient strategy to create advanced soft materials with great potential in tissue regeneration and disease diagnosis and treatment.42–57 We have demonstrated that rationally establishing structural complementarity at the interacting interface between peptides allows for dictating their self-assembly and precisely controlling their organization within assemblies via covalent and/or noncovalent bonding strategies.57 Conventionally, the association of peptides at their desirable interacting interface during their self-assembly is thermodynamically favourable.58 This implies that incorporation of stimulus-responsive groups into peptides potentially leads to stimulus-dependent interacting interfaces between peptides, thus promoting the transition between different organizing fashions of peptides and creating hydrogels undergoing thermodynamically favourable well-defined mechanical movements upon exposure to stimuli. On the basis of these considerations, herein we report on mechanically dynamic hydrogels consisting of amphiphilic peptide bilayers undergoing reduction-responsive directional domain sliding motion and investigate the effect of the resulting sliding motion on cell proliferation.
To create the mechanically dynamic (MD) hydrogels, we designed an amphiphilic peptide VECSS consisting of two alternating hydrophilic (glutamic acid) and hydrophobic (valine) domains cross-linked by disulfide bonds at a non-central position (Fig. 1). Based on the previous reports, alternating polar and nonpolar peptides conventionally assemble into nanofibrils driven by the hydrophobic collapse into antiparallel β-sheet bilayers.59–62 Therefore, we hypothesize that the covalent constraint arising from the disulfide bonds leads to formation of the hydrophobic interfaces between peptide VECSS bilayers consisting of only a part of the residues within the sequences, accompanied by two disordered segments protruding along the bilayers. Reduction of the disulfide bonds eliminates the covalent constraint between the two domains, resulting in formation of non-equilibrated VECSH bilayers with the hydrophobic interfaces involving only partial-length sequences. The hydrophobicity of the protruding segments promotes formation of the thermodynamically favourable hydrophobic collapses involving the full-length sequences, thus leading to the directional domain sliding movement along each other within the non-equilibrated VECSH bilayers (Fig. 1). Hence, self-assembly of VECSS into nanofibers and reduction of the disulfide bonds potentially allow for creation of the mechanically dynamic hydrogels undergoing well-defined internal domain sliding motions, thus generating mechanical signals that affect cell behaviour. In this study, we synthesized and characterized the self-assembly of peptides VECSS and VECSH, and prepared the hydrogels VECSS, VECSH, and VECSH-MDvia adding calcium chloride. We eventually investigated the domain sliding movement within the mechanically dynamic hydrogel VECSH-MD and cultured different cells on the resulting hydrogels. There results allow us to elucidate the role of the directional domain sliding motion within the mechanically dynamic hydrogels in accelerating cell proliferation via a mechanotransduction mechanism.
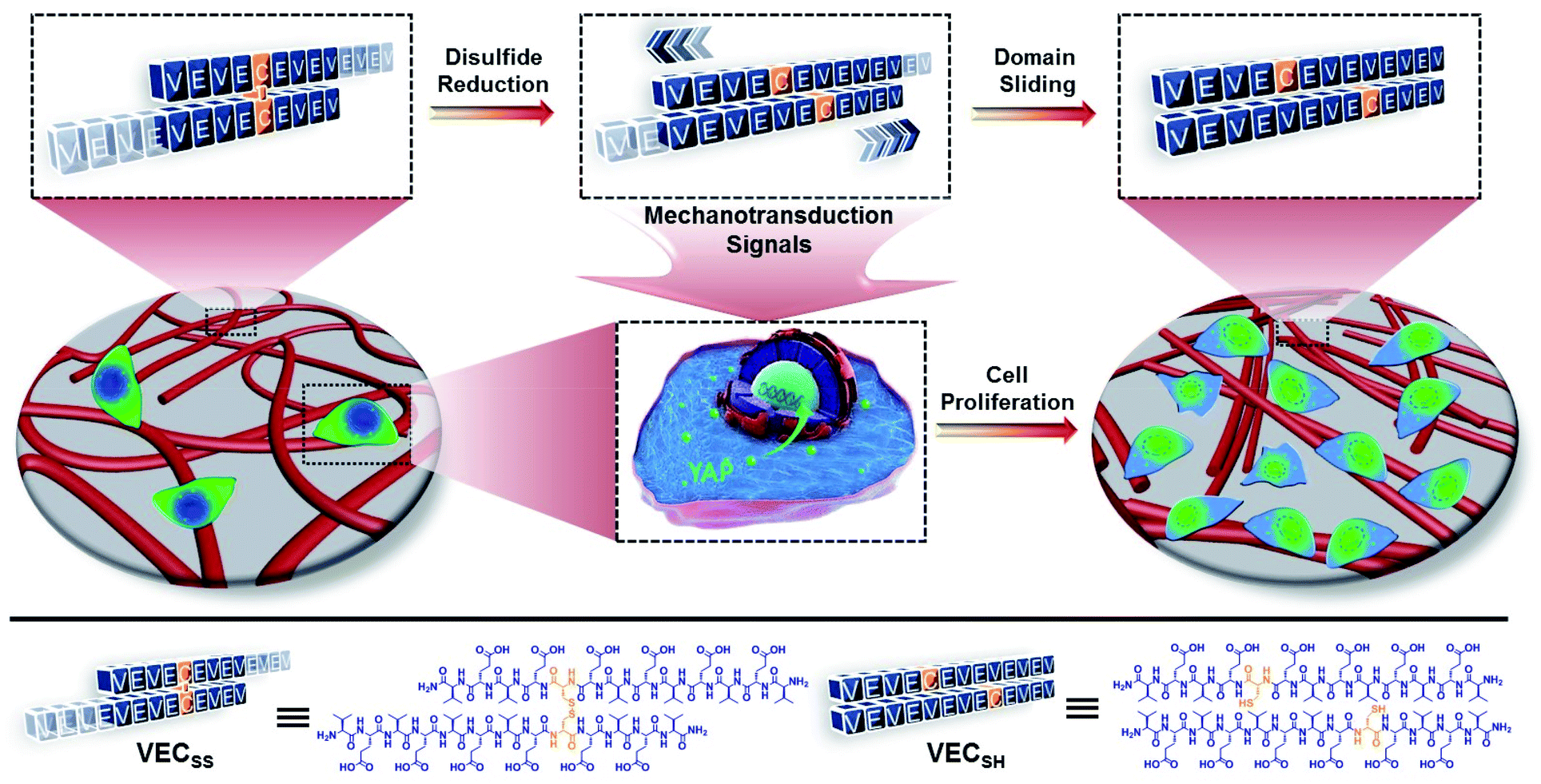 | ||
| Fig. 1 Schematic representation of mechanically dynamic hydrogels with well-defined mechanical motion prepared via hydrogelation of the peptide VECSS consisting of two alternating hydrophobic and hydrophilic domains cross-linked by a disulfide bond and reduction of the disulfide bonded VECSS into non-equilibrated VECSH bilayers, thus promoting the directional domain sliding motion along each other. The underlying directional mechanical motion accelerates cell proliferation when culturing cells on the hydrogels via a mechanotransduction mechanism involving the translocation of yes-associated protein (YAP) into the nucleus. | ||
Results and discussion
Self-assembly of peptides
To confirm the hypothesis of the antiparallel organization of the peptides, we characterized the secondary structures formed by peptides VECSS and VECSH using circular dichroism (CD) and Fourier transform infrared spectroscopy (FTIR) (Fig. 2A and B). The CD spectrum of the peptide VECSS showed the negative and positive exciton coupling peaks at 213 and 190 nm, indicative of the formation of β-sheets for the peptide VECSS.63 Addition of a reductive agent tris(2-carboxyethyl)phosphine (TCEP) to VECSS led to reduction of disulfide bonds and quantitative conversion of the peptide VECSS to VECSH, which was verified by mass spectroscopy and liquid chromatography (Fig. S4†). The negative and positive maximal CD peaks at 213 and 190 nm were maintained in the spectrum of the peptide VECSH, suggesting the formation of β-sheets for the peptide VECSH as well. In addition, under the identical mass concentration, increase of both the negative and positive maximal CD intensities of the peptide VECSH compared with those of VECSS implies improved β-sheet population.64 This improvement is potentially attributed to the transition from the partial to the full hydrophobic collapse caused by the peptide VECSS or VECSH, respectively, as a result of release of the covalent constraint within VECSH. The secondary structures formed by peptides VECSS and VECSH were confirmed by FTIR studies (Fig. 2B). FTIR spectra of both peptides VECSS and VECSH displayed two transmittance peaks at 1624 cm−1 and 1692 cm−1, therefore indicating formation of antiparallel β-sheets by peptides VECSS and VECSH.65 CD and FTIR studies demonstrate that antiparallel β-sheets are formed by peptides VECSS and VECSH, which is the basis for our hypothesis of occurrence of the hydrophobic collapse transition upon release of the disulfide bond constraint.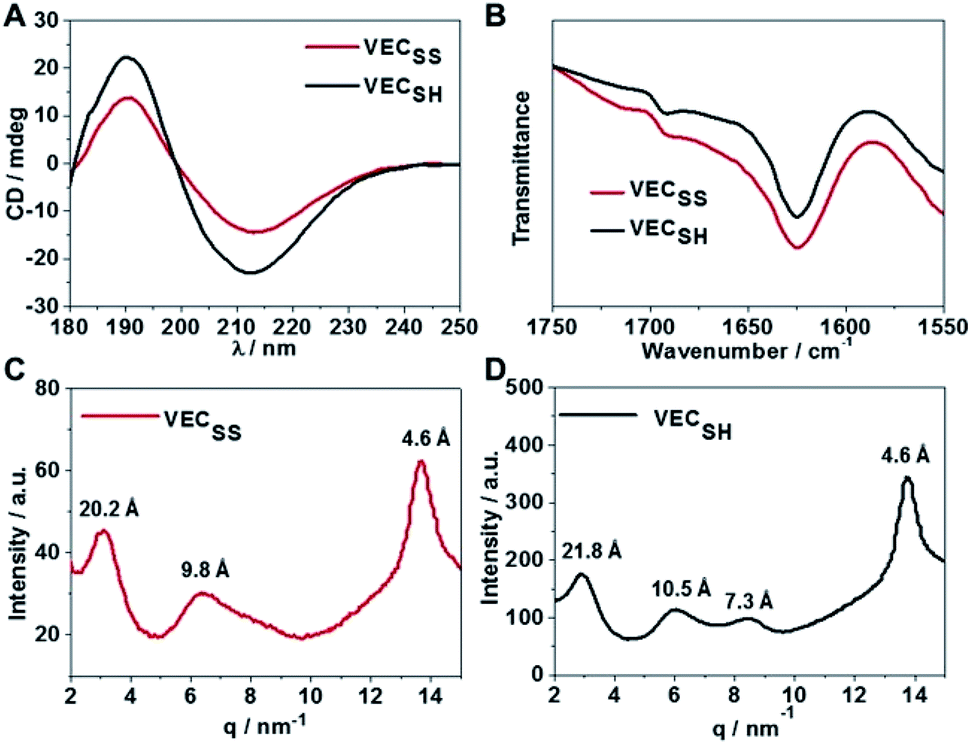 | ||
| Fig. 2 (A) Circular dichroism (CD) spectra of peptides VECSS and VECSH at an identical mass concentration diluting from the solution aged for 2 days. (B) Fourier transform infrared spectroscopy (FTIR) spectra of peptides VECSS and VECSH in the aged solutions. Wide-angle X-ray scattering (WAXS) profiles of the powder of peptides VECSS (C) and VECSH (D) obtained by lyophilizing the aged solutions. | ||
We investigated the internal structural features of the assemblies formed by peptides VECSS and VECSH using the wide-angle X-ray scattering method (WAXS) (Fig. 2C and D). WAXS spectra showed two Bragg reflections at 20.3 and 4.6 Å for the peptide VECSS and two reflections at 21.8 and 4.6 Å for the peptide VECSH, corresponding to the width of the monomers and the periodic spacings of the β-sheet secondary structures.66–69 The smaller width of the VECSS monomers compared with that of the VECSH dimers might be attributed to the covalent constraint arising from disulfide bonds, thus indicating that the peptide VECSS is organized in a direction perpendicular to the long axis of nanofibers, in which the two domains within the peptide VECSS localize in different faces within the bilayers. In addition, while a Bragg reflection at 9.8 Å associated with the second order reflection of the width of the dimers was observed for the peptide VECSS, two reflections at 10.5 and 7.3 Å associated with the second or third order of the width of the dimers were detected for the peptide VECSH. WAXS studies clearly reveal the formation of the β-sheets by both peptides VECSS and VECSH with a conventional organizing fashion of alternating hydrophobic and hydrophilic peptides.
We subsequently characterized the morphology of the resulting nanostructures formed from peptides VECSS and VECSH by transmission electron microscopy (TEM) and atomic force microscopy (AFM) (Fig. 3A–D). TEM images showed that the peptide VECSS assembled into flexible nanofibers entangled with each other, whereas bundled stiff nanofibers were formed by the peptide VECSH. The width of the resulting flexible and rigid nanofibers was estimated to be 4.47 or 5.93 nm, respectively (Fig. S11†). The width of the nanofibers formed by the peptide VECSH is comparable with the length of the fully stretched peptide backbone (Fig. S14†), suggesting the full collapse of the hydrophobic interface of full-length sequences to form the antiparallel β-sheets. However, the width of the nanofibers formed by the peptide VECSS is comparable to the length of approximately 8-residue segments, which is consistent with the length of the constrained hydrophobic interfaces within the peptide VECSS (Fig. S13†). This result indicates that only partial-length sequences within the constrained hydrophobic interfaces participate in the formation of antiparallel β-sheets, accompanied by two protruding segments adopting a flexible conformation along the nanofibers. This interpretation is consistent with the difference between the CD intensity of peptides VECSS and VECSH, in which a lower intensity of the β-sheet CD signals for the peptide VECSS compared to VECSH was observed.
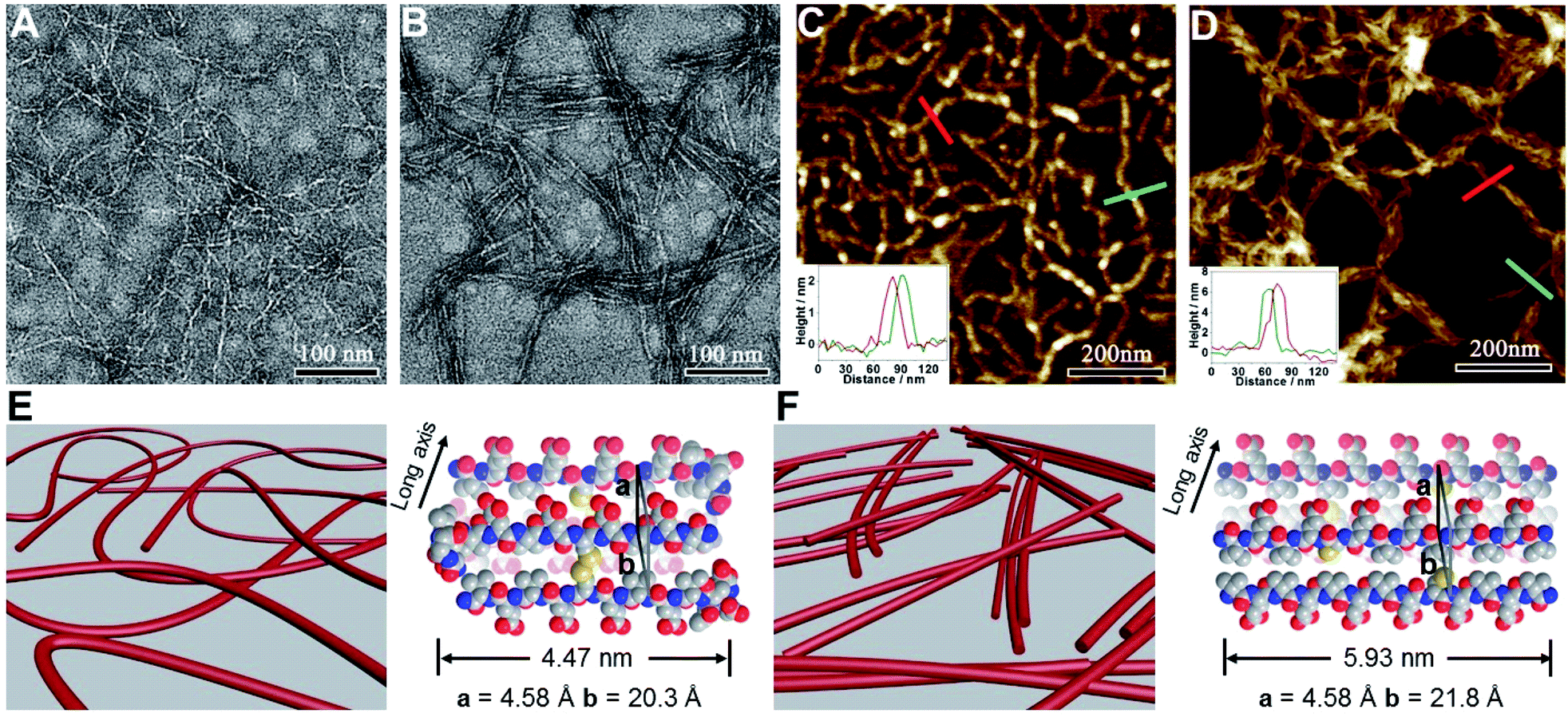 | ||
| Fig. 3 (A–D) TEM and AFM images of the nanofibers formed by peptides VECSS (A and C) and VECSH (B and D) prepared from the aged solutions. TEM samples were stained with uranyl acetate (2%). The insets in (C) and (D) indicate the height profiles of the selected nanostructures. (E and F) Graphical representation of the resulting nanofibers formed from VECSS (E) and VECSH (F), in which the organizing models of peptides VECSS and VECSH of the monomers within the nanofibers are shown. | ||
The morphology of the assemblies of peptides VECSS and VECSH was confirmed by AFM (Fig. 3C and D), in which flexible and bundled nanofibers with a height of 2.1 and 6.2 nm were observed for peptides VECSS and VECSH, respectively. The height of the VECSS nanofibers suggests formation of the bilayer structures consistent with the behaviour of alternating hydrophilic and hydrophobic peptides. However, the height of the VECSH nanofibers implies that the nanofiber bundles consist of approximately three individual nanofibers. Combining the morphological and conformational studies, the underlying mechanism for the self-assembly of peptides VECSS and VECSH into flexible or stiff nanofibers is illustrated (Fig. 3E and F). Based on the formation of antiparallel β-sheets, the partial hydrophobic collapse involving approximately 8-residue-length segments promotes the self-assembly of the peptide VECSS into flexible nanofibers appended with protruding disordered segments along the nanofibers, whereas stiff nanofibers are formed by VECSH driven by the full hydrophobic collapse involving the full-length sequences. This mechanism potentially allows for the hydrophobic collapse transition upon reduction of the disulfide bonds within the peptide VECSS.
Preparation and characterization of hydrogels
To test the possibility of creation of peptide hydrogels, we carried out the rheological studies of peptides VECSS and VECSH. We found that addition of 7.5 equivalents of calcium chloride into the aged VECSS70 or annealed VECSH solution resulted in rapid hydrogelation caused by the Ca2+–COOH coordination, leading to the formation of static hydrogels VECSS and VECSH (Fig. 4A). The storage modulus of the resulting hydrogels VECSS and VECSH was estimated to be 2569 and 3308 Pa, respectively (Fig. 4B). Compared to hydrogel VECSH, decrease of the rigidity of hydrogel VECSS is attributed to the partial hydrophobic collapse only involving the eight residues localizing within the constrained hydrophobic interface. This finding is consistent with the relationship between the rigidity of the hydrogels formed by alternating amphipathic peptides and their sequence length.61 Therefore, our rheological results suggest that the static hydrogels VECSS and VECSH consist of peptide bilayers with the partial or full hydrophobic collapse, respectively. This inspires us to create a mechanically dynamic (MD) hydrogel VECSH-MD undergoing the partial → full hydrophobic collapse transition. The hydrogel VECSH-MD was created via initially reducing the disulfide bonds within equilibrated VECSS bilayers in solution to produce non-equilibrated VECSH bilayers, and immediately adding CaCl2 for hydrogelation (Fig. 4A). Time-dependent rheological studies determined the initial and final storage modulus of the resulting hydrogel VECSH-MD to be 2520 and 3319 Pa (Fig. 4C), which are close to the moduli of the static hydrogels VECSS and VECSH, respectively. These results indicate the transition from the partial to full hydrophobic collapse for the non-equilibrated VECSH bilayers within the hydrogel VECSH-MD caused by the release of covalent constraint upon reduction of the disulfide bonds, thus leading to a well-defined directional domain sliding movement within the hydrogel VECSH-MD.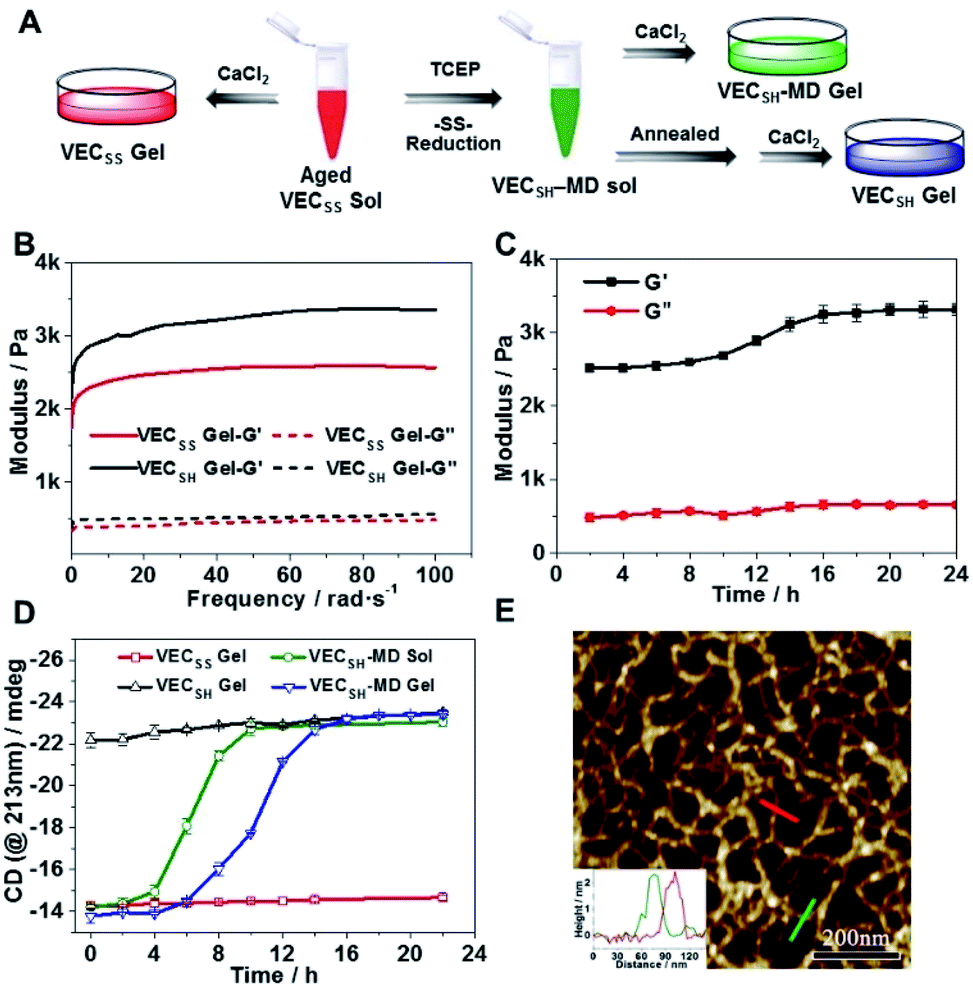 | ||
| Fig. 4 (A) Graphical illustration of the preparation of static hydrogels VECSS and VECSH, and mechanically dynamic hydrogels VECSH-MD. (B) Mechanical properties of static hydrogels VECSS and VECSH upon addition of CaCl2 into the aged or annealed solution. (C) Time-dependent mechanical properties of the mechanically dynamic hydrogel VECSH-MD. (D) Time-dependent intensity of the CD signals (@213 nm) of static hydrogels VECSS and VECSH, mechanically dynamic hydrogels VECSH-MD, and the solution VECSH-MD. (E) AFM image of non-equilibrated VECSH-MD assemblies prepared from the hydrogel VECSH-MD aged for 2 h, in which the inset shows the height profiles of selected nanofibers. | ||
We further investigated the underlying mechanism for the partial → full hydrophobic collapse transition within the hydrogel VECSH-MD. Due to the strong correlation between the hydrophobic collapse fashion and the β-sheet population, recording the intensity of the β-sheet CD signals of the hydrogels allows us to monitor the hydrophobic collapse transition of the domains upon reduction of disulfide bonds. CD spectra of the hydrogel VECSH-MD showed a gradual increase of the β-sheet signal intensity in the time range between 4 and 14 h and a subsequent plateau value close to that of the hydrogel VECSH, demonstrating occurrence of the partial → full hydrophobic collapse transition for non-equilibrated VECSH bilayers within the hydrogel VECSH-MD (Fig. 4D and S17†). Both time-dependent rheological and CD studies suggest formation of the equilibrated VESSH bilayers in the hydrogel VECSH-MD once the hydrophobic collapse transition is complete. In contrast, CD spectra of the static hydrogels VECSS and VECSH displayed constant β-sheet intensities, indicating no change of the hydrophobic collapse within the two equilibrated peptide bilayers. In principle, the transition of the hydrophobic collapse of the non-equilibrated VECSH bilayers could proceed via two plausible pathways: (1) the dynamic dissociation–reassociation of the peptide VECSH; (2) the directional domain sliding motion for the non-equilibrated VECSH bilayers. It is reasonable that dissociation of the domains will induce initial decrease of the β-sheet CD intensity of the hydrogel VECSH-MD. However, this is in contrast to the observed constant β-sheet CD intensity of the hydrogel VECSH-MD at the beginning stage in the time-dependent CD studies. Hence, our results exclude the dynamic dissociation–reassociation process during the hydrophobic collapse transition within the hydrogel VECSH-MD, thus suggesting a directional domain sliding mechanism for the hydrophobic collapse transition within MD hydrogels (Fig. 4D). Gradual increase of the intensity of the β-sheet signals was also observed in the non-equilibrated VECSH solution (Fig. S18 and S19†), despite a relatively rapid process compared to the hydrogel VECSH-MD. This might be attributed to the trapping of the sliding motion within hydrogels, resulting in a long period to reach the thermodynamic equilibrium. These results demonstrate successful creation of mechanically dynamic hydrogels via reduction of the disulfide bonds within equilibrated VECSS and thereby leading to non-equilibrated VECSH assemblies, which undergo the directional domain sliding motion promoted by the thermodynamically favourable hydrophobic collapse transition.
To confirm the mechanism of the directional domain sliding movement for the hydrophobic collapse transition within the mechanically dynamic hydrogel VECSH-MD, we carried out an AFM experiment to estimate the morphology of the nanostructures in the early stage of the hydrogel VECSH-MD (Fig. 4E). The AFM image clearly showed formation of tangled nanofibers by the non-equilibrated VECSH bilayers with a height of 2.2 nm, which is close to that of the nanofibers formed by the peptide VECSS. This result demonstrates that the organizing fashion of the non-equilibrated peptide VECSH bilayers is maintained similar to that of equilibrated peptide VECSS bilayers. This is consistent with the CD result of the comparable intensity of β-sheet signals of the non-equilibrated peptide VECSH bilayers and equilibrated peptide VECSS bilayers. Therefore, the AFM study confirmed the directional domain sliding mechanism for the hydrophobic collapse transition within the non-equilibrated VECSH bilayers, thus implying creation of the mechanically dynamic hydrogel VECSH-MD. The underlying mechanical movement within the hydrogel VECSH-MD is initiated by reduction of the disulfide bonds, leading to the metastable VECSH bilayers stabilized by the hydrophobic interactions involving only 8-residue-length segments. The hydrophobicity of the two protruding segments allows for thermodynamically favourable hydrophobic interactions involving the full-length sequences, thus promoting the directional domain sliding movement within the non-equilibrated peptide VECSH bilayers to form the equilibrated VECSH bilayers through several intermediates involving sequences in variable lengths (Fig. 5).
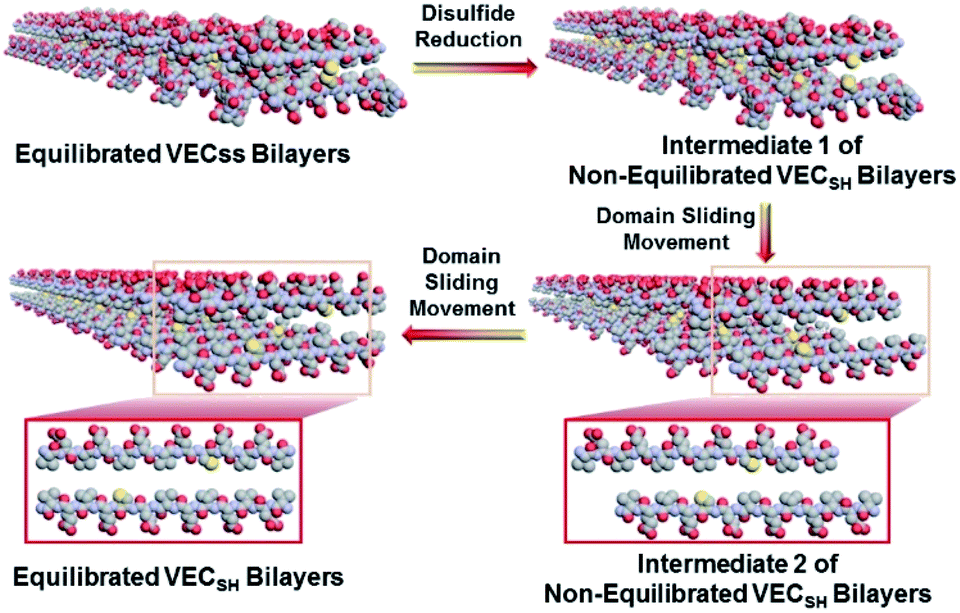 | ||
| Fig. 5 Schematic representation of the mechanically dynamic process within the mechanically dynamic hydrogel VECSH-MD upon reduction of disulfide bonds and the resulting directional sliding movement promoted by the thermodynamically favourable hydrophobic collapse transition. The included molecular graphics are produced only for illustration. | ||
Cell proliferation on hydrogels
On the basis of the preparation and characterization of the mechanically dynamic hydrogel VECSH-MD, we cultured both human hepatocyte LO2 cells and rat aortic vascular smooth muscle A10 cells on the mechanically dynamic hydrogels to investigate the effect of the directional domain sliding motion on the cell proliferation (Fig. 6). Meanwhile, the static hydrogels VECSS and VECSH prepared from the equilibrated solution were also utilized as the culturing scaffolds to confirm the effect of molecular sliding motion on cell proliferation. To ensure the exposure of the cells to the domain sliding motion promoted by the hydrophobic collapse transition, cells were cultured on the hydrogels for various time points ranging from 4 to 48 hours, which cover the transition of the hydrophobic collapse of the non-equilibrated VECSH bilayers. CCK-8 assay revealed that culturing human hepatocyte LO2 cells on the three hydrogels for 4 h resulted in an almost same cell viability for the three hydrogels, indicating the identical cell adhesion of the three hydrogels. When the culturing time is prolonged to 48 h, the cells incubated on the hydrogel VECSH-MD exhibited the highest viability among the three hydrogels (Fig. 7).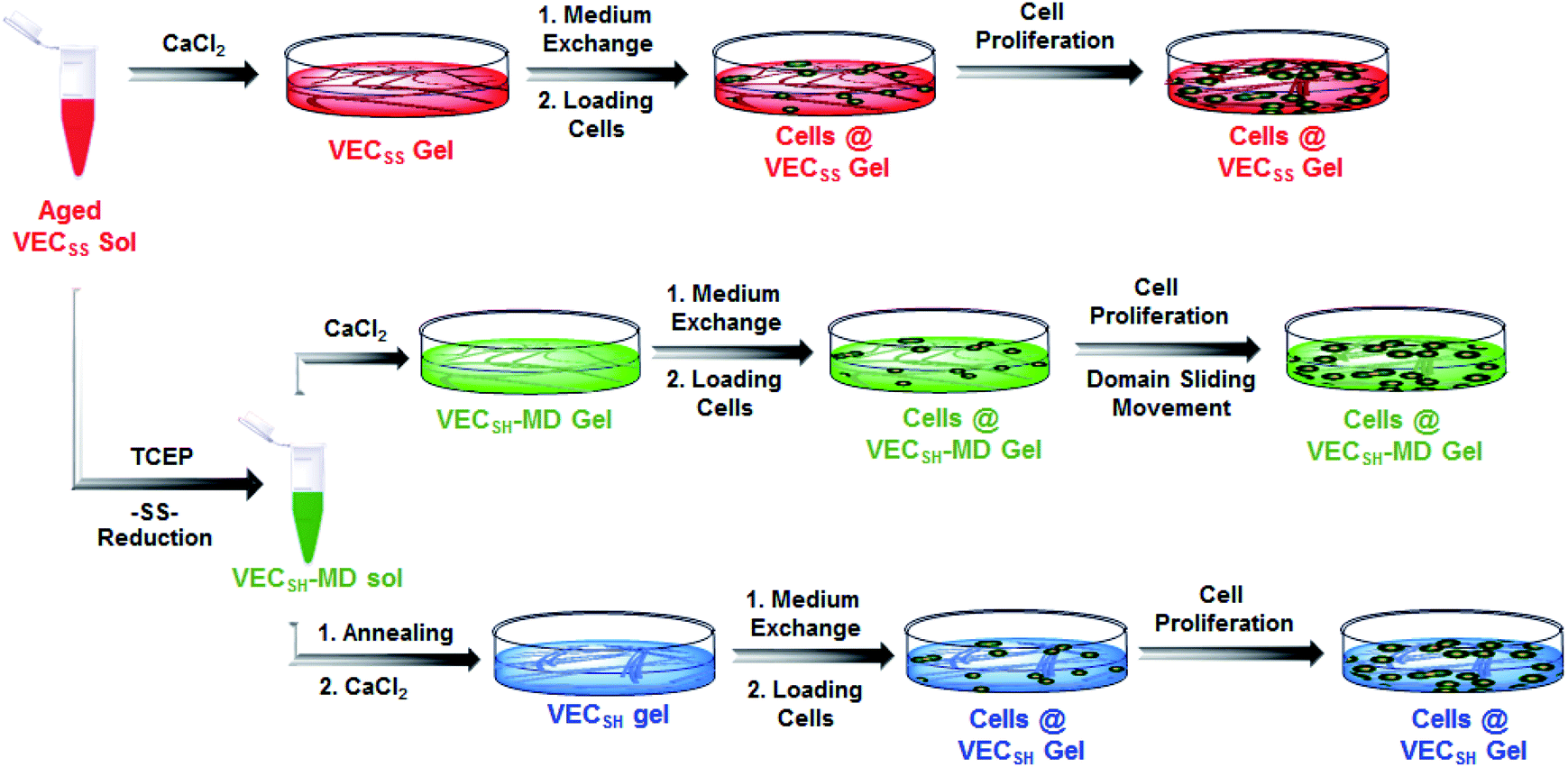 | ||
| Fig. 6 Graphical illustration of the preparation of the hydrogels and cell culture on the surface of static hydrogels VECSS and VECSH, as well as the mechanically dynamic hydrogel VECSH-MD. | ||
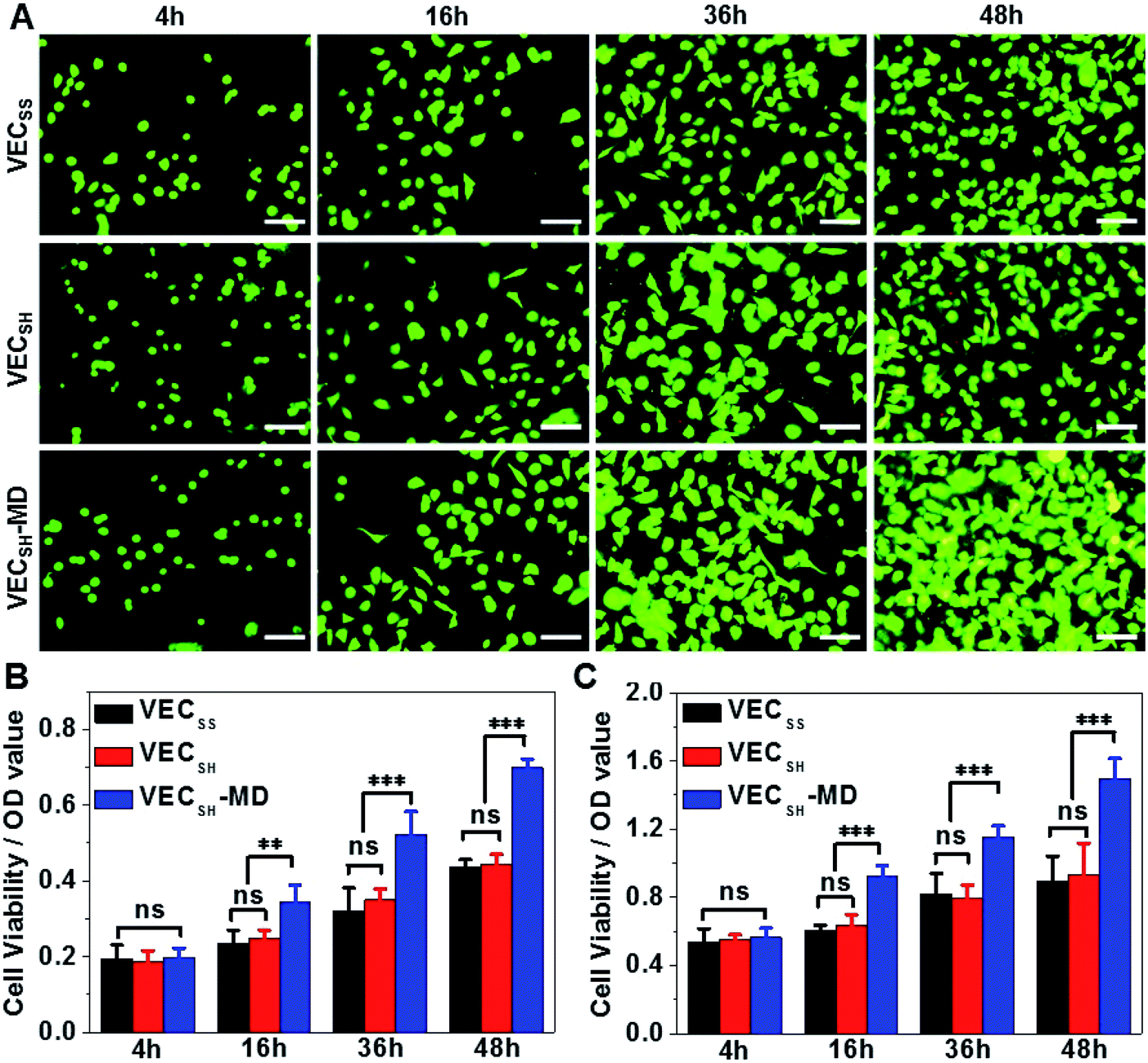 | ||
| Fig. 7 (A) Representative fluorescence images of calcein AM-stained (A) human hepatocyte LO2 cells cultured on the hydrogel VECSH-MD for 4, 16, 36 or 48 h. Scale bar: 50 μm. Cell viability of LO2 (B) and A10 (C) cells cultured on static hydrogels VECSS and VECSH and mechanically dynamic hydrogels VECSH-MD for 4, 16, 36 or 48 h. *, **, ***: suggestive of significant differences (ANOVA: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). ns: indicative of no significant differences. | ||
Considering the fact that the assemblies within the hydrogel VECSH-MD are the intermediates for the thermodynamically stable nanostructures within the static hydrogels VECSS and VECSH, the directional domain sliding movement with the hydrogel VECSH-MD gives rise to the stiffness of the hydrogel ranging between those of static hydrogels VECSS and VECSH as revealed by rheological studies. In addition, the directional domain sliding movement within the hydrogel VECSH-MD potentially generates mechanical forces. Both the underlying processes might affect the mechanical cues generated in the mechanically dynamic hydrogel VECSH-MD. It is worth noting that the equilibrated state of the hydrogel VECSH-MD consists of identical structural components (equilibrated VESSH bilayers) to those of the hydrogel VECSH. Hence we attributed the accelerated cell proliferation to the mechanical cues resulting from the directional domain sliding motion within the mechanical dynamic hydrogels. To elucidate the effect of the directional domain sliding motion on cell proliferation, we also cultured rat aortic vascular smooth muscle A10 cells on the three hydrogels (Fig. 7C and S20†). Analogous to the LO2 cells, the viability of A10 cells incubated on the hydrogel VECSH-MD for 48 h is the highest one among the three matrices. This result confirms the pervasive effect of the domain sliding motion within the hydrogel VECSH-MD on the acceleration of cell proliferation. Considering the absence of specific binding epitopes within the mechanically dynamic hydrogels to cells, it is reasonable that the transduction of mechanical signals from hydrogels to incubated cells is independent of the types of cells. Hence, we believe that the acceleration of cell proliferation by the mechanically dynamic hydrogels could be applied to many other types of cells. Thus our findings demonstrate the great potential of mechanically dynamic hydrogels as artificial scaffolds to accelerate cell proliferation.
To confirm the results of cell proliferation cultured on peptide hydrogels, we also carried out DNA analysis by estimating the percentage of 5-ethynyl-2′-deoxyuridine (EdU)-incorporating DNA in LO2 and A10 cells (Fig. 8 and S21†). LO2 and A10 cells were cultured on the hydrogel VECSH-MD and the static hydrogels for various time points in the presence of EdU, which is a thymidine derivative and can be incorporated into the cellular nucleus during DNA replication.71 We found that the percentage of LO2 and A10 cells containing EdU residue cultured on the three hydrogels for 4 h is identical and pretty low due to the suppressed cell proliferation. Culturing both LO2 and A10 cells on the hydrogels longer than 16 h exhibited an increase of the percentage of the cells containing EdU, indicating the cell proliferation. The percentage of the cells containing EdU was estimated to be 53.5 ± 6.2% and 53.0 ± 3.9% for LO2 and A10 cells, respectively, when incubating the cells on the hydrogel VECSH-MD for 48 h (Fig. 8B and C). This value is much higher than that of cells cultured on the static hydrogels VECSS and VECSH. The results of the EdU assays indicate the accelerated cell proliferation when culturing on the hydrogel VECSH-MD compared to the two static hydrogels, thus demonstrating the effect of directional domain sliding motion within hydrogels on accelerating cell proliferation.
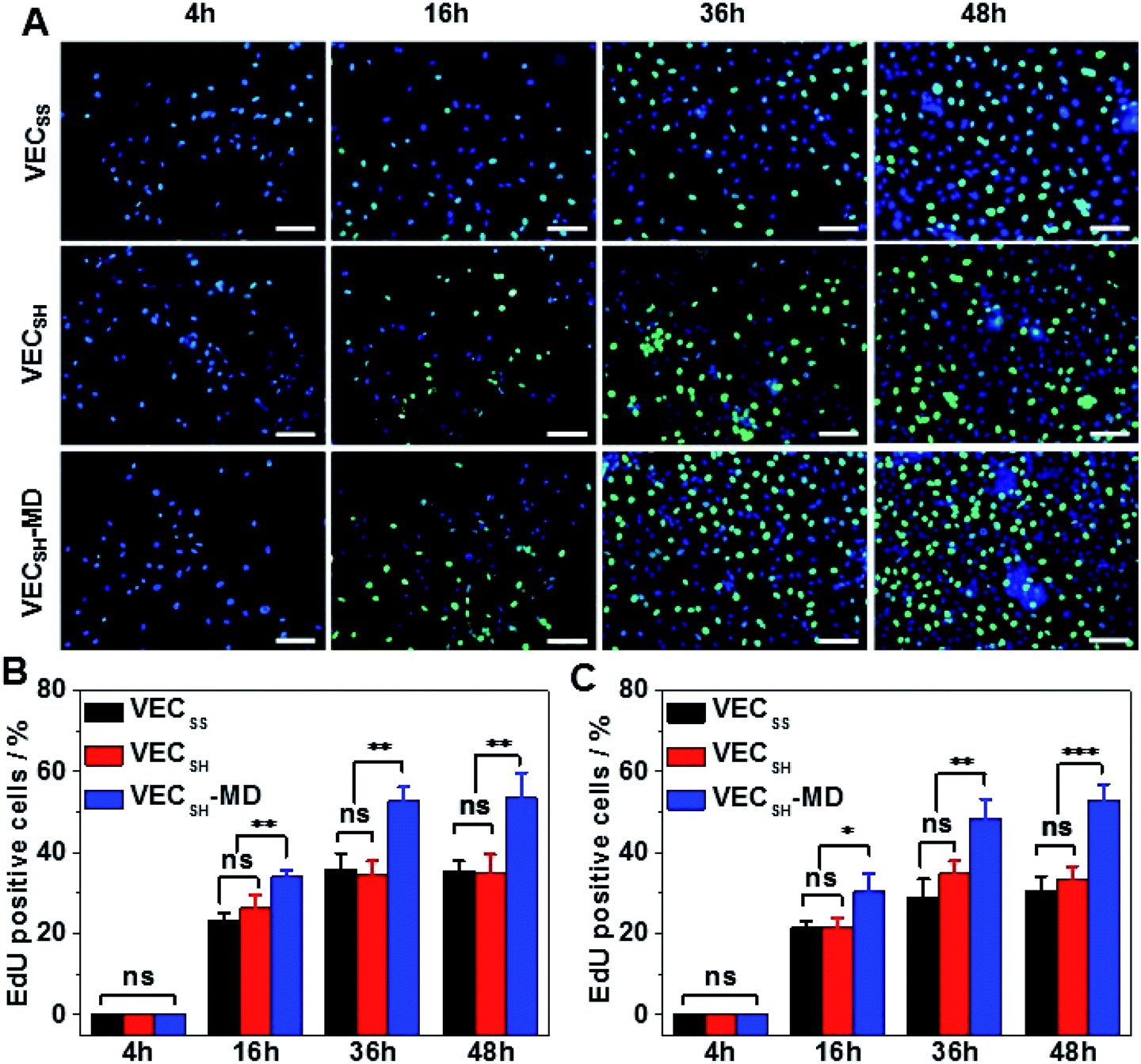 | ||
| Fig. 8 (A) Representative fluorescence images of 4′,6-diamidino-2-phenylindole (DAPI) and Alexa Fluor 488 co-stained human hepatocyte LO2 cells cultured on the hydrogel VECSH-MD for 4, 16, 36, or 48 h. Blue: DAPI; green: Alexa Fluor 488; cyan: merged DAPI and Alexa Fluor 488. Scale bar: 50 μm. The percentage of the EdU-incorporated cells of LO2 (B) and A10 (C) cells cultured on hydrogels VECSS, VECSH, and VECSH-MD for 4, 16, 36 or 48 h. *, **, ***: suggestive of significant differences (ANOVA: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). ns: indicative of no significant differences. | ||
Mechanotransduction signals in cells
To further gain insight into the mechanism of the accelerated cell proliferation induced by the mechanically dynamic hydrogels, we estimated the activity of the Hippo signalling pathway in human hepatocyte LO2 cells cultured on the static hydrogels VECSS and VECSH and the mechanically dynamic VECSH-MD by monitoring the translocation of the downstream effector yes-associated protein (YAP) in YAP-GFP overexpression (OE) LO2 cells (Fig. 9). The Hippo signalling pathway is a conventional mechanotransduction pathway regulating cell proliferation, apoptosis, and self-renewal associated with mechanical cues,72 in which a kinase cascade containing the phosphorylation of the downstream transcription co-activator YAP protein takes place.73 Previous studies have demonstrated that the mechanical cues perceived by cells promote the nuclear translocation of unphosphorylated YAP for downstream gene activation.74 Hence, monitoring the distribution of YAP in the cytoplasm and nucleus allows us to estimate the mechanical cues generated in the mechanically dynamic hydrogels and confirm the mechanotransduction pathway for the accelerated cell proliferation induced by the directional domain sliding motion.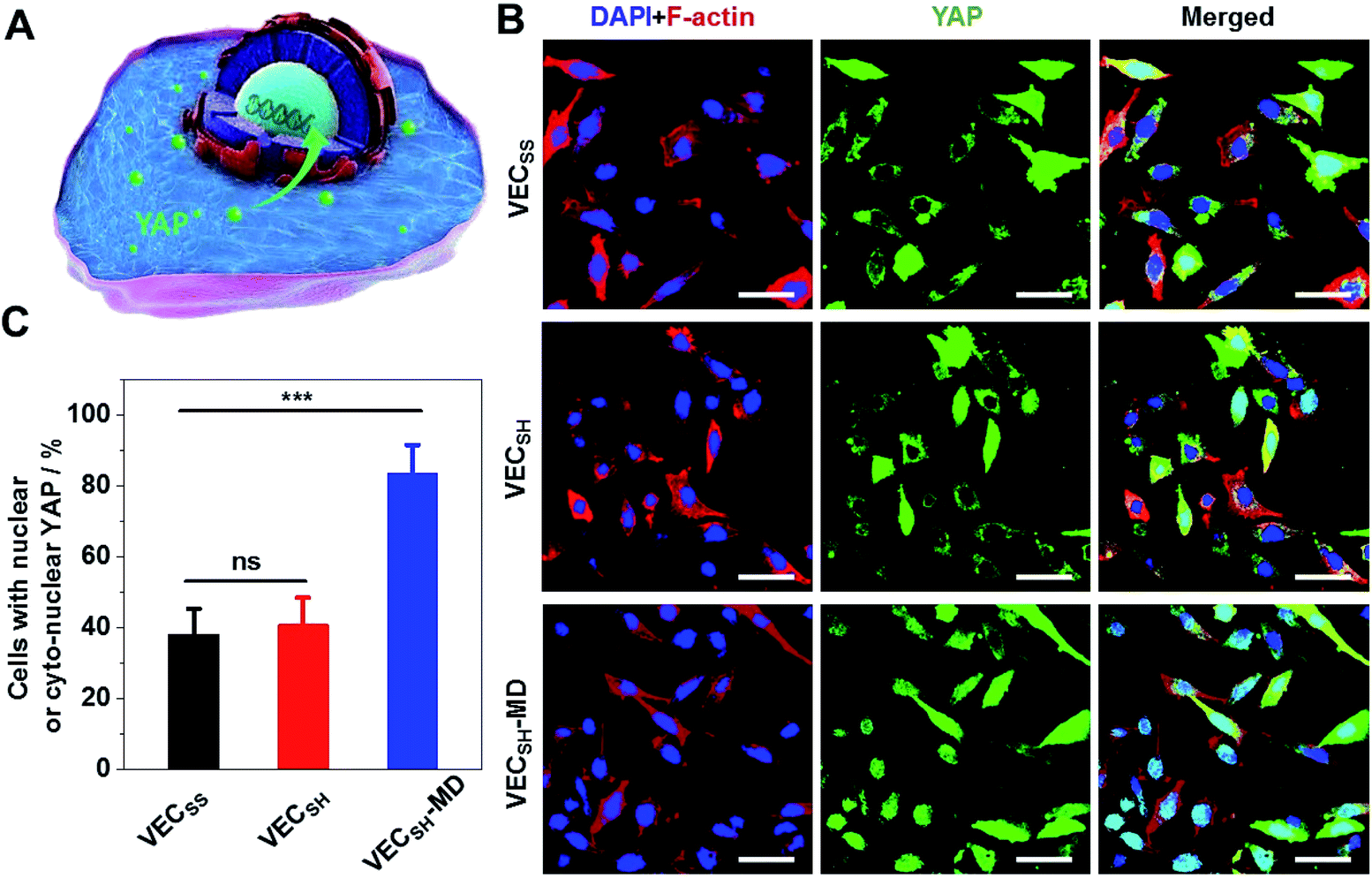 | ||
| Fig. 9 (A) Schematic representation of the mechanotransduction Hippo pathway involving the translocation of YAP protein into the nucleus in cells. (B) CLSM images of the YAP-GFP OE LO2 cells cultured on static hydrogels VECSS and VECSH, as well as the mechanically dynamic hydrogel VECSH-MD for 36 h. Blue: DAPI; red: Alexa Fluor 647-phalloidin; green: GFP labeled YAP. Scale bar: 50 μm. (C) Statistical analysis of the percentage of the cells containing nuclear or cyto-nuclear YAP cultured on different hydrogels. ***: suggestive of significant differences (ANOVA: ***p ≤ 0.001). ns: indicative of no significant differences. | ||
Confocal laser scanning microscopy (CLSM) images showed that the YAP-GFP OE LO2 cells cultured on the static hydrogels VECSS and VECSH and the mechanically dynamic hydrogel VECSH-MD for 36 h contained the YAP localized in the cytoplasm, nucleus or cyto-nucleus (Fig. S22†), implying participation of the mechanotransduction pathway in cell proliferation (Fig. 9B). In particular, the population of the cells with nuclear or cyto-nuclear YAP cultured on the hydrogel VECSH-MD was improved compared with that cultured on static hydrogels VECSS and VECSH. Statistical analysis of the cells cultured on different hydrogels revealed that the percentage of the cells with nuclear or cyto-nuclear YAP was estimated to be 38.3%, 40.9%, and 83.7% for the hydrogels VECSS, VECSH, and VECSH-MD, respectively (Fig. 9C). The slight difference between the distribution of the YAP protein in cells cultured on the static hydrogels VECSS and VECSH might be attributed to the distinct hydrogel stiffness. In contrast, the significantly improved translocation of the YAP into nucleus in cells cultured on the hydrogel VECSH-MD compared to VECSS and VECSH is most likely caused by the directional domain sliding movement within the hydrogels. The YAP-GFP localization assays demonstrate that the cells can sense the mechanical cues generated by the mechanical movements within the mechanically dynamic hydrogel VECSH-MD. It also supports a potential mechanotransduction mechanism for the accelerated cell proliferation of cells induced by the directional domain sliding movement within the hydrogels. Hence our results clearly demonstrate the implication of the mechanically dynamic hydrogels as cellular matrices for accelerating cell proliferation, thus providing an alternative material for tissue engineering and regeneration.
Conclusions
In summary, we have created mechanically dynamic hydrogels undergoing well-defined mechanical movement by designing and synthesizing an amphiphilic peptide consisting of two alternating hydrophilic and hydrophobic sequences cross-linked by disulfide bonds at a non-symmetrical position. Due to the covalent constraint resulting from the disulfide bonds, the amphiphilic peptides formed the bilayers organized in an antiparallel orientation and the flexible nanofibers stabilized by the hydrophobic interactions involving only partial-length sequences, accompanied by two disordered segments protruding along the assemblies. Reduction of the disulfide bonds between the two domains within the hydrogels formed by the amphiphilic peptides leads to the transition from a partial to full hydrophobic collapse and thus promoting the directional sliding motion for the two domains along each other, thus creating mechanically dynamic hydrogels. The well-defined directional mechanical motion at the molecular level within the hydrogels accelerates proliferation of both human hepatocyte LO2 cells and rat aortic vascular smooth muscle A10 cells when culturing cells on the hydrogels, compared to static counterparts, through a mechanotransduction mechanism. Our results demonstrate the great potential of mechanically dynamic hydrogels as a new category of biomimetic extracellular matrices in tissue engineering and regeneration.Experimental
General materials
Rink amide 4-methyl-benzhydrylamine (MBHA) resin (loading density: 0.436 mmol g−1) and all Fmoc-protected amino acids used in solid-phase peptide synthesis were obtained from Bide Pharmatech Co., Ltd (China). Hydrogen peroxide (30% H2O2) and tris(2-carboxyethyl) phosphine hydrochloride (TCEP·HCl) were purchased from J&K Scientific Ltd. Organic solvents for peptide synthesis and purification were provided by Tianjin Concord Technology Co., Ltd. Water used in all experiments was purified using Arium Pro Ultrapure water systems (Sartorius, 18.2 MΩ).Peptide synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:3:2 for 3 h. After cleavage, the filtered solution was concentrated by rotary evaporation, and the crude peptide was precipitated from cold diethyl ether (−20 °C). The crude peptide was then purified using a preparative reverse phase HPLC setup equipped with a Durashell C18 column (C18, 10 μm, 150 Å, 30 × 150 mm). A gradient from “5% acetonitrile + 95% water” to “95% acetonitrile + 5% water” was used as the eluent at a flow rate of 10 mL min−1.
:5:3:2 for 3 h. After cleavage, the filtered solution was concentrated by rotary evaporation, and the crude peptide was precipitated from cold diethyl ether (−20 °C). The crude peptide was then purified using a preparative reverse phase HPLC setup equipped with a Durashell C18 column (C18, 10 μm, 150 Å, 30 × 150 mm). A gradient from “5% acetonitrile + 95% water” to “95% acetonitrile + 5% water” was used as the eluent at a flow rate of 10 mL min−1.
Hydrogel preparation
Cell culture on hydrogels
Peptides were sterilized upon exposure to UV light for 60 min prior to usage. The solution of VECSS (2 wt%) was prepared by dissolving the peptide VECSS in ammonia water (pH = 10) as a clear solution and adjusting the solution pH to 5.4 by using diluted HCl solution. The VECSS solution was aged for 2 days prior to the preparation of the static hydrogel VECSS promoted by addition of 7.5 equivalents of CaCl2. The aged VECSS solution was mixed with 8 equivalents of TCEP to reduce the disulfide bonds within 2 h. Annealing the resulting solution led to the equilibrated peptide VECSH solution, which was used to prepare the static hydrogel VECSH by adding 7.5 equivalents of CaCl2. On the other hand, the aged VECSS solution was converted to the non-equilibrated peptide VECSH within 2 h after adding 8 equivalents of TCEP, to which 7.5 equivalents of CaCl2 were immediately added to prepare the mechanically dynamic hydrogel VECSH-MD. It is worth noting that in each well of 384-well plates, 20 μL of corresponding peptide solutions was added and mixed with 1 μL CaCl2 (2 M) solution to prepare the hydrogels. After incubating at room temperature for 30 min, the hydrogels in 384-well plates were centrifuged at 2000 rpm for 20 min and washed with 60 μL of Dulbecco's Modified Eagle Medium (DMEM) until the colour of the medium was maintained. In the cases of the solutions treated with TCEP, the centrifugation–washing process was repeated until no TCEP was detected to avoid cytotoxicity.YAP-GFP OE LO2 cell culture on the hydrogel VECSH-MD
To prepare the YAP-GFP overexpression (OE) LO2 cells, a mixture of pEGFP-YAP plasmid (500 ng) and P3000 (1 μL) was dissolved in Opti-MEM medium (25 μL), and lipofectamine 3000 (0.75 μL) was diluted in Opti-MEM medium (25 μL). The diluted plasmid solution was mixed with the diluted lipofectamine 3000 reagent and incubated for 15 min. Subsequently, the original LO2 cells were incubated with the plasmid–lipid complex for 24 h, leading to the YAP-GFP OE LO2 cells used in the following cell culturing experiments. The protocol for culturing the YAP-GFP OE LO2 cells on static hydrogels VECSS and VECSH and the mechanically dynamic VECSH-MD for 36 h is identical to that for the general cells as described in the above section.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21774065, 31600768, 51933006, and 21620102005) and the Fundamental Research Funds for the Central Universities (Nankai University, ZB19100123 and 63186058).Notes and references
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- A.-S. Smith, Nat. Phys., 2010, 6, 726–729 Search PubMed.
- S. V. Plotnikov, A. M. Pasapera, B. Sabass and C. M. Waterman, Cell, 2012, 151, 1513–1527 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- J. C. Friedland, M. H. Lee and D. Boettiger, Science, 2009, 323, 642–644 CrossRef CAS PubMed.
- L. Yuguo, G. Shiva, L. Jonathan and S. Tatiana, Biomaterials, 2011, 32, 39–47 CrossRef PubMed.
- C. Yang, M. W. Tibbitt, L. Basta and K. S. Anseth, Nat. Mater., 2014, 13, 645–652 CrossRef CAS PubMed.
- M. Uroz, A. Garcia-Puig, I. Tekeli, A. Elosegui-Artola, J. F. Abenza, A. Marín-Llauradó, S. Pujals, V. Conte, L. Albertazzi, P. Roca-Cusachs, Á. Raya and X. Trepat, Nat. Mater., 2019, 18, 1015–1023 CrossRef CAS PubMed.
- B. D. Hoffman, C. Grashoff and M. A. Schwartz, Nature, 2011, 475, 316–323 CrossRef CAS PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- L. Montero de Espinosa, W. Meesorn, D. Moatsou and C. Weder, Chem. Rev., 2017, 117, 12851–12892 CrossRef CAS PubMed.
- K. Uto, J. H. Tsui, C. A. DeForest and D.-H. Kim, Prog. Polym. Sci., 2017, 65, 53–82 CrossRef CAS PubMed.
- K. H. Vining and D. J. Mooney, Nat. Rev. Mol. Cell Biol., 2017, 18, 728–742 CrossRef CAS PubMed.
- N. Kong, Q. Peng and H. Li, Adv. Funct. Mater., 2014, 24, 7310–7317 CrossRef CAS.
- J. Zhang, C. Cheng, J. L. Cuellar-Camacho, M. Li, Y. Xia, W. Li and R. Haag, Adv. Funct. Mater., 2018, 28, 1804773 CrossRef.
- K. A. Günay, T. L. Ceccato, J. S. Silver, K. L. Bannister, O. J. Bednarski, L. A. Leinwand and K. S. Anseth, Angew. Chem., Int. Ed., 2019, 58, 9912–9916 CrossRef PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Macromolecules, 2004, 37, 7331–7337 CrossRef CAS.
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS PubMed.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- M. Zhou, A. Smith, A. K. Das, N. Hodson, R. Collins, R. Ulijn and J. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- L. E. O'leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- S. Sur, J. B. Matson, M. J. Webber, C. J. Newcomb and S. I. Stupp, ACS Nano, 2012, 6, 10776–10785 CrossRef CAS PubMed.
- V. Hernandez-Gordillo and J. Chmielewski, Biomaterials, 2014, 35, 7363–7373 CrossRef CAS PubMed.
- P. W. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS PubMed.
- J. Shi, G. Fichman and J. P. Schneider, Angew. Chem., Int. Ed., 2018, 57, 11188–11192 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. Watanabe, T. Ooya, K.-H. Nitta, K. D. Park, Y. H. Kim and N. Yui, Biomaterials, 2002, 23, 4041–4048 CrossRef CAS PubMed.
- J.-H. Seo, S. Kakinoki, Y. Inoue, T. Yamaoka, K. Ishihara and N. Yui, Soft Matter, 2012, 8, 5477–5485 RSC.
- J. H. Seo, S. Kakinoki, T. Yamaoka and N. Yui, Adv. Healthcare Mater., 2015, 4, 215–222 CrossRef CAS PubMed.
- X. Tong and F. Yang, Adv. Mater., 2016, 28, 7257–7263 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H.-p. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–336 CrossRef CAS PubMed.
- A. Cipitria and M. Salmeron-Sanchez, Adv. Healthcare Mater., 2017, 6, 1700052 CrossRef PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- J. H. Collier, J. S. Rudra, J. Z. Gasiorowski and J. P. Jung, Chem. Soc. Rev., 2010, 39, 3413–3424 RSC.
- X. Zhao, F. Pan, H. Xu, M. Yaseen, H. Shan, C. A. Hauser, S. Zhang and J. R. Lu, Chem. Soc. Rev., 2010, 39, 3480–3498 RSC.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- C. Ren, J. Zhang, M. Chen and Z. Yang, Chem. Soc. Rev., 2014, 43, 7257–7266 RSC.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- X. Q. Dou and C. L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- A. N. Moore and J. D. Hartgerink, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS PubMed.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- P. Zhang, Y. Cui, C. F. Anderson, C. Zhang, Y. Li, R. Wang and H. Cui, Chem. Soc. Rev., 2018, 47, 3490–3529 RSC.
- G. B. Qi, Y. J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 CrossRef PubMed.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- S. Lou, X. Wang, Z. Yu and L. Shi, Adv. Sci., 2019, 6, 1802043 CrossRef PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- S. Zhang, Biotechnol. Adv., 2002, 20, 321–339 CrossRef CAS PubMed.
- C. J. Bowerman and B. L. Nilsson, Pept. Sci., 2012, 98, 169–184 CrossRef CAS PubMed.
- M. R. Caplan, E. M. Schwartzfarb, S. Zhang, R. D. Kamm and D. A. Lauffenburger, Biomaterials, 2002, 23, 219–227 CrossRef CAS PubMed.
- A. S. Carlini, R. Gaetani, R. L. Braden, C. Luo, K. L. Christman and N. C. Gianneschi, Nat. Commun., 2019, 10, 1735 CrossRef PubMed.
- Z. Yu, F. Tantakitti, T. Yu, L. C. Palmer, G. C. Schatz and S. I. Stupp, Science, 2016, 351, 497–502 CrossRef CAS PubMed.
- D. G. Osterman and E. Kaiser, J. Cell. Biochem., 1985, 29, 57–72 CrossRef CAS PubMed.
- Y. Zou, Y. Li, W. Hao, X. Hu and G. Ma, J. Phys. Chem. B, 2013, 117, 4003–4013 CrossRef CAS PubMed.
- M. S. Lamm, K. Rajagopal, J. P. Schneider and D. J. Pochan, J. Am. Chem. Soc., 2005, 127, 16692–16700 CrossRef CAS PubMed.
- R. P. Nagarkar, R. A. Hule, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2008, 130, 4466–4474 CrossRef CAS PubMed.
- H. Cui, A. G. Cheetham, E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 12461–12468 CrossRef CAS PubMed.
- Z. Yu, F. Tantakitti, L. C. Palmer and S. I. Stupp, Nano Lett., 2016, 16, 6967–6974 CrossRef CAS PubMed.
- Annealing the solution of VECSS solution led to oxidation of disulfide bonds potentially due to the trace amount of H2O2 remained from the thiol cross-linking reactions. Hence an aging process for the solution of peptide VECSS was carried out for thermodynamic equilibration.
- F. Chehrehasa, A. C. Meedeniya, P. Dwyer, G. Abrahamsen and A. Mackay-Sim, J. Neurosci., 2009, 177, 122–130 CAS.
- J. Huang, S. Wu, J. Barrera, K. Matthews and D. Pan, Cell, 2005, 122, 421–434 CrossRef CAS PubMed.
- G. Halder, S. Dupont and S. Piccolo, Nat. Rev. Mol. Cell Biol., 2012, 13, 591–600 CrossRef CAS PubMed.
- S. Chakraborty, K. Njah, A. V. Pobbati, Y. B. Lim, A. Raju, M. Lakshmanan, V. Tergaonkar, C. T. Lim and W. Hong, Cell Rep., 2017, 18, 2464–2479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05808g |
| This journal is © The Royal Society of Chemistry 2020 |