
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoupling the effects of hydrophilic and hydrophobic moieties at the neuron–nanofibre interface†
Adam D.
Martin
 *a,
Jonathan P.
Wojciechowski
b,
Eric Y.
Du
c,
Aditya
Rawal
d,
Holly
Stefen
a,
Carol G.
Au
a,
Liming
Hou
a,
Charles G.
Cranfield
e,
Thomas
Fath
a,
Lars M.
Ittner
a and
Pall
Thordarson
c
*a,
Jonathan P.
Wojciechowski
b,
Eric Y.
Du
c,
Aditya
Rawal
d,
Holly
Stefen
a,
Carol G.
Au
a,
Liming
Hou
a,
Charles G.
Cranfield
e,
Thomas
Fath
a,
Lars M.
Ittner
a and
Pall
Thordarson
c
aDementia Research Centre, Department of Biomedical Science, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, NSW 2109, Australia. E-mail: adam.martin@mq.edu.au
bDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK
cSchool of Chemistry, The Australian Centre for Nanomedicine, The ARC Centre of Excellence in Convergent Bio-Nano Science & Technology, University of New South Wales, Sydney, NSW 2052, Australia
dNMR Facility, Mark Wainwright Analytical Centre, The University of New South Wales, Sydney 2052, New South Wales, Australia
eSchool of Life Sciences, University of Technology Sydney, Ultimo, NSW 2007, Australia
First published on 18th December 2019
Abstract
Peptide-based nanofibres are a versatile class of tunable materials with applications in optoelectronics, sensing and tissue engineering. However, the understanding of the nanofibre surface at the molecular level is limited. Here, a series of homologous dilysine–diphenylalnine tetrapeptides were synthesised and shown to self-assemble into water-soluble nanofibres. Despite the peptide nanofibres displaying similar morphologies, as evaluated through atomic force microscopy and neutron scattering, significant differences were observed in their ability to support sensitive primary neurons. Contact angle and labelling experiments revealed that differential presentation of lysine moieties at the fibre surface did not affect neuronal viability; however the mobility of phenylalanine residues at the nanofibre surface, elucidated through solid- and gel-state NMR studies and confirmed through tethered bilayer lipid membrane experiments, was found to be the determining factor in governing the suitability of a given peptide as a scaffold for primary neurons. This work offers new insights into characterising and controlling the nanofibre surface at the molecular level.
Introduction
The extracellular matrix (ECM) is an entangled mesh network of fibres which provides important physical and chemical support for cells. Attempts to mimic the ECM have been the subject of intensive research for many years, with a number of elegant examples including polymer-based scaffolds,1–5 decellularised tissue,6–8 supramolecular materials,9–11 and more recently, multicomponent hydrogels.12–14 Each scaffold presents its own advantages, from the scalability of polymer synthesis, to the preservation of in vivo organisation for decellularised tissue, to the tunable and responsive nature of supramolecular interactions.One class of supramolecular scaffolds rapidly gaining popularity is that of peptide hydrogels. Due to their extraordinary diversity, there are many classes of peptides which can self-assemble into hydrogels. These include ionic, complementary peptide sequences such as the RADA and MAX sequences,15–17 or shorter peptides containing aromatic moieties at their N-terminus such as Fmoc or naphthalene which drive self-assembly.18–20 These shorter peptides are attractive targets for mimicking the ECM, due to their ease of synthesis and minimum gelation concentrations which are typically below 1% (w/v). However, one drawback associated with these scaffolds is that small changes in the peptide amino acid sequence can result in significant changes in peptide self-assembly and properties of the resultant hydrogels, including their ability to support cell growth. These changes include, but are not limited to, changes in N-terminal capping group, changes to amino acid chirality and changes to amino acid sequence.21,22
The alteration of amino acid sequence in self-assembling short peptides has previously been used to tune peptide pKa,23,24 enabling self-assembly under more physiologically relevant conditions. Other examples have incorporated the RGD sequence derived from fibronectin to enhance cell adhesion.25,26 Orthogonal functionalisation of hydrogels through click chemistry have enabled post-synthetic modifications to scaffolds.27,28 The ordering of amino acids in tripeptide hydrogels has been shown to affect the morphology of fibroblasts cultured upon these scaffolds.29 Recently we reported two tetrapeptides containing D-amino acids, which exhibited sequence dependent self-assembly, which could then be used to culture sensitive primary neurons on peptide coated coverslips.30 It should be noted that there is currently still no reported scaffold which can support primary neurons in 3D, owing in part to a lack of understanding surrounding the optimal cell–scaffold interface required for 3D encapsulation of primary neurons.
Expanding on our previous work raised the question of whether the amino acid sequence played a role in the ability of self-assembling peptides to support the growth of primary neurons. As such, a library of Fmoc-capped tetrapeptides was synthesised which bear two L-lysine residues and two L-phenylalanine residues in different positions (Fig. 1). After discovering significant differences in the ability of the substrates to support primary neuronal growth, we thoroughly examined the nanofibre surface of these six tetrapeptides, with differences suggested by zeta potential, contact angle, fluorescence labelling, solid and gel state NMR and electrical impedance spectroscopy experiments on tethered bilayer lipid membranes (tBLMs). This comprehensive characterisation of the nanofibre surface ultimately leads to an understanding of what constitutes a permissive surface for primary neurons, and the role that both hydrophilic and hydrophobic moieties play in the design of a suitable substrates.
 | ||
| Fig. 1 The six peptides used in this study, all bearing an Fmoc N-terminal capping group, two L-lysine and two L-phenylalanine residues. | ||
Results and discussion
Peptide library synthesis and morphological characterisation
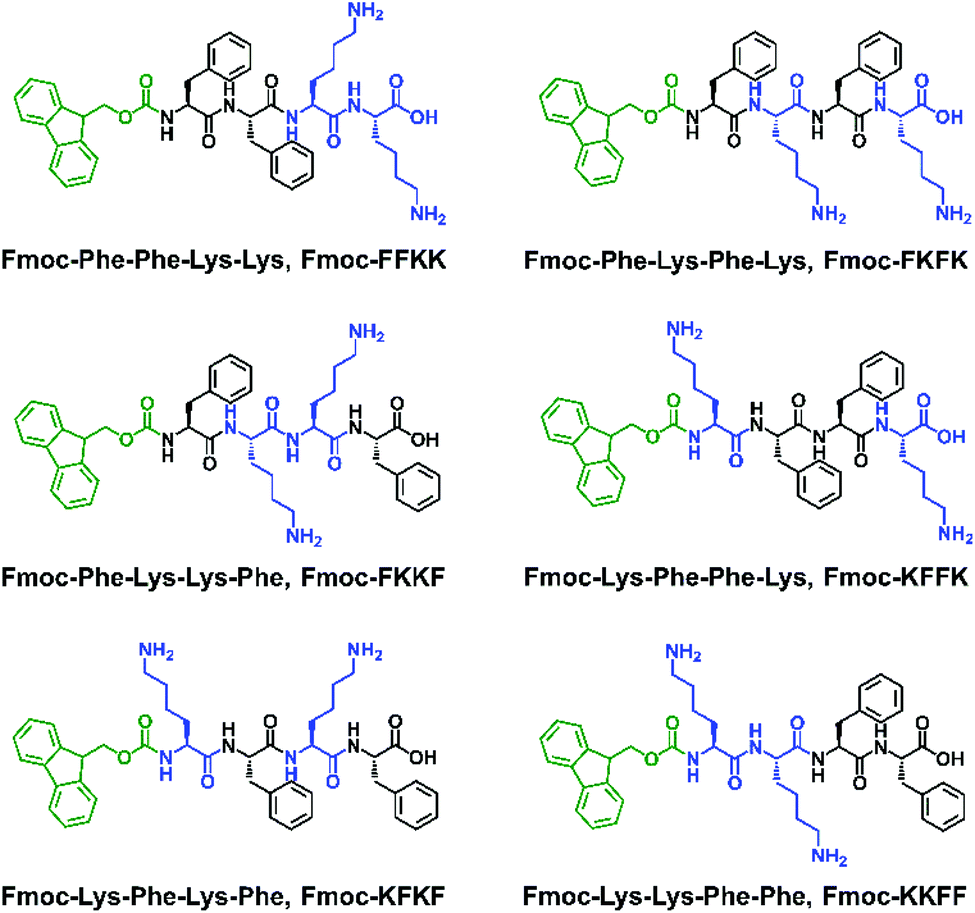
Fmoc-capped tetrapeptides were synthesised using solid phase peptide synthesis, followed by purification using semi-preparative HPLC and lyophilisation. SEM imaging (Fig. S3†) revealed that all tetrapeptides had already formed fibrous structures upon lyophilisation after HPLC purification, indicating their predisposition to self-assemble in an aqueous environment. Dissolution of the lyophilised peptides in water yielded solutions of nanofibres, as confirmed through viscosity measurements (Fig. S4†) and AFM imaging (Fig. 2a–f).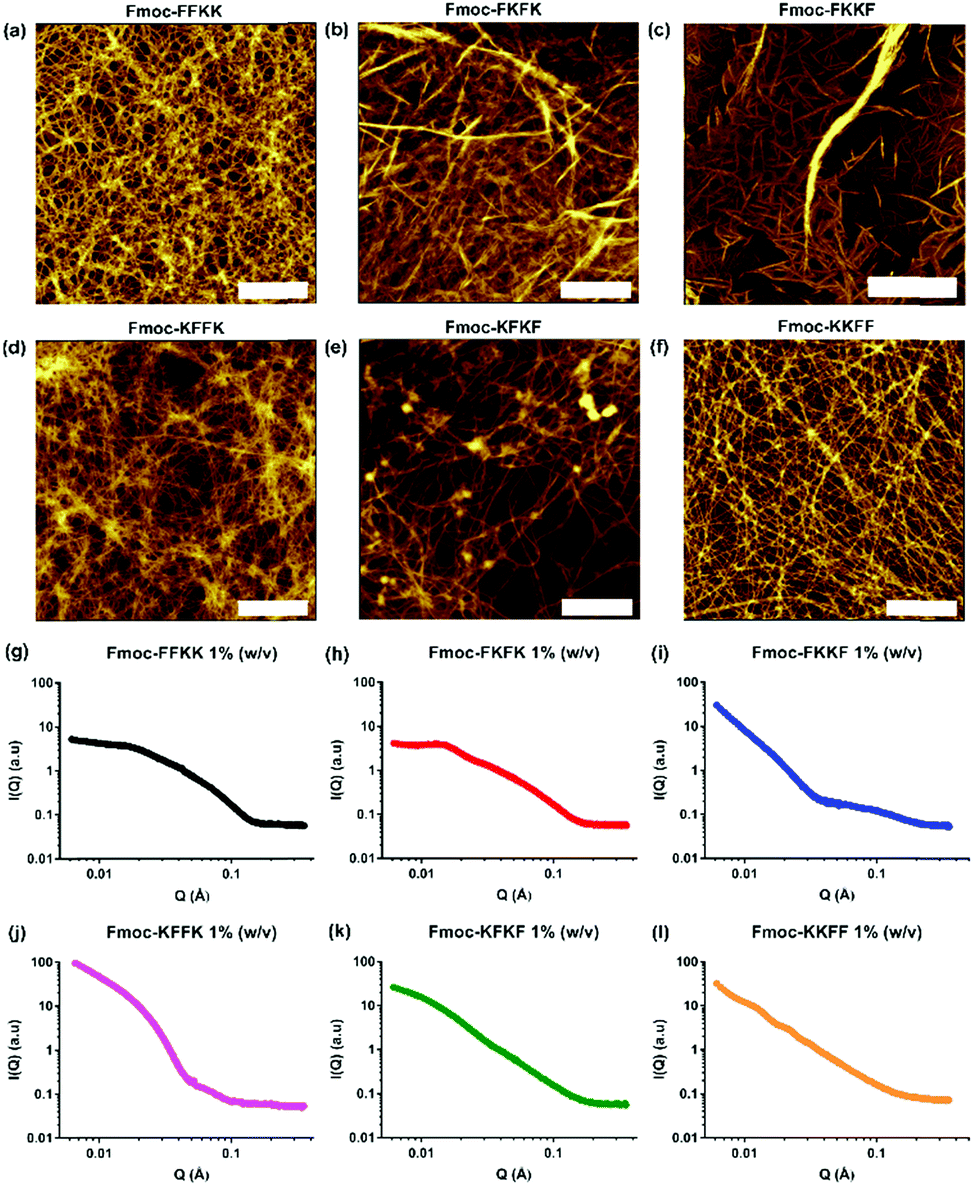 | ||
| Fig. 2 AFM images of tetrapeptides dissolved in water at 0.5% (w/v) and spread coated onto mica substrates, confirming that each of (a) Fmoc-FFKK, (b) Fmoc-FKFK, (c) Fmoc-FKKF, (d) Fmoc-KFFK, (e) Fmoc-KFKF and (f) Fmoc-KKFF self-assemble to give fibrous structures. Scale bar represents 1 μm. (g–l) Small angle neutron scattering measurements performed on tetrapeptides dissolved in D2O at 1% (w/v) confirm self-assembly into cylindrical architectures, however the differences in scattering patterns reveal each tetrapeptide possesses a unique fibre morphology in situ. | ||
For all tetrapeptides, similar nanofibre morphologies were observed; with fibre diameters ranging from 4.3–7.6 nm (diameters and associated standard deviations are given in Table S1†). All tetrapeptides had similar pKa values (Fig. S5†) and self-assembled into micrometre-long fibres, except for Fmoc-FKKF, where truncated, straight fibres were observed. The similar fibre diameters suggest a common self-assembly mechanism, with the driving force for this likely the interplay between the hydrophobic Fmoc capping group and phenylalanine residues, as previously described.31,32 It should be noted that differences in SEM and AFM fibre diameters are likely due to differences associated with sample preparation.33 Due to the pre-assembly of these tetrapeptides into nanofibres, we were unable to obtain monomers in order to study the exact self-assembly mechanism for each tetrapeptide. Utilisation of a solvent switch mechanism (i.e. dissolution of peptides in DMSO before dilution into water) would likely not be representative of their self-assembly in pure water, as has been shown for the related peptide Fmoc-FF.34
To confirm that the AFM images obtained were representative of the in situ nanofibre morphology, small angle neutron scattering was performed. Solutions of peptide nanofibres were prepared at 1% (w/v) and scattering patterns for all tetrapeptides were fitted using a flexible cylinder model due to its physical relevance to the nanofibre morphology as determined by AFM. Fmoc-FKKF was observed to contain two distinct scattering regimes, likely from fibrous structure (high q) and larger aggregates (low q), thus was modelled using a flexible cylinder model at high q and a fractal model at low q. Structure factor peaks can be observed for Fmoc-FKFK at approximately 0.015 Å−1 and Fmoc-KKFF at 0.012 and 0.02 Å−1, due to repulsion between positively charged fibres. For all peptides apart from Fmoc-FKKF, the Kuhn length obtained was much smaller than the cylinder length (i.e. distance between intersecting fibres), indicating a flexible fibre. This supports the AFM images. For Fmoc-FKKF, the Kuhn length and fibre length are similar, indicating straight fibres as observed by AFM. Full fitting parameter outputs (fibre length, radius, Kuhn length and χ2 values) and plotted fits are detailed in the ESI (Table S2 and Fig. S6†).
Suitability of tetrapeptide nanofibres as substrates for primary neuronal cultures
As all tetrapeptides self-assembled into nanofibres with relatively similar diameters and morphologies, their ability to act as cell scaffolds was evaluated. To create peptide nanofibre-coated coverslips, tetrapeptides were dissolved at several concentrations and incubated overnight with glass slides. This technique is widely used for molecules such as poly-D-lysine and polyethyleneimine (PEI) due to electrostatic attraction between the negatively charged glass coverslip and the positively charged molecule.38 Initially, a robust, immortalised cell line (HEK293T) was seeded atop peptide nanofibres at several concentrations and the viability of these cells was evaluated through AlamarBlue assays and immunostaining (Fig. S7†). No significant differences in viability were observed across each tetrapeptide at several concentrations.Next, the ability of these tetrapeptide nanofibres to act as a cell substrate was extended to more sensitive primary neuronal cells. Primary neurons are currently the most relevant in vitro model of the brain and represent an important tool for studying mechanisms in neurodegeneration and drug screening.35–37 We have recently shown the ability to support primary neurons on lysine-containing nanofibres.30 After coating coverslips with 0.5% (w/v) solutions of tetrapeptide nanofibres and aspiration of excess media, primary neurons were seeded atop peptide nanofibre coated coverslips and incubated for seven days in vitro (DIV), whereupon viability was assessed either through fixing and immunostaining (Fig. 3a–f), or a metabolic viability assay (Fig. 3g). Somewhat surprisingly, significant differences were observed in neuronal adhesion to substrates, in particular Fmoc-FKKF and Fmoc-KFFK. It is known that primary neurons are notoriously sensitive to their environment, with the interaction between neuron cell surface and substrate playing a crucial role in cell adherence and development.
 | ||
| Fig. 3 Evaluation of nanofibre surfaces for culturing sensitive primary hippocampal neurons. Glass coverslips were coated with a 0.5% (w/v) peptide nanofibre solution and primary neurons seeded atop these substrates. Primary neurons were fixed at DIV7 and stained for neuronal markers β3-tubulin (green) and MAP2 (red), alongside DAPI. Representative images for (a) Fmoc-FFKK, (b) Fmoc-FKFK, (c) Fmoc-FKKF, (d) Fmoc-KFFK, (e) Fmoc-KFKF and (f) Fmoc-KKFF are shown. Scale bar represents 100 μm. Cell viability was also quantified through an Alamar Blue assay for primary neurons (g). Poly-D-lysine coated glass were used as a positive viability control. The symbol * represents p < 0.05, **p < 0.01, with p values determined by a one-way ANVOA and Tukey's post-test. | ||
For primary neuronal cultures, subtle differences in the tetrapeptide surface can have an important effect on neuronal adhesion to nanofibre substrates. From the immunostaining and viability measurements, Fmoc-FKKF is significantly poorer (p < 0.01) at supporting primary neurons than all other substrates, with Fmoc-KFFK significantly worse than Fmoc-FKFK and Fmoc-KFKF. No significant differences in viability were observed between the other four tetrapeptides Fmoc-FFKK, Fmoc-FKFK, Fmoc-KFKF and Fmoc-KKFF. This suggests that the amino acid sequence plays an important role in the ability of these tetrapeptides to support sensitive primary neuronal cells.
Effect of secondary structure and hydrophilic residues on the nanofibre surface
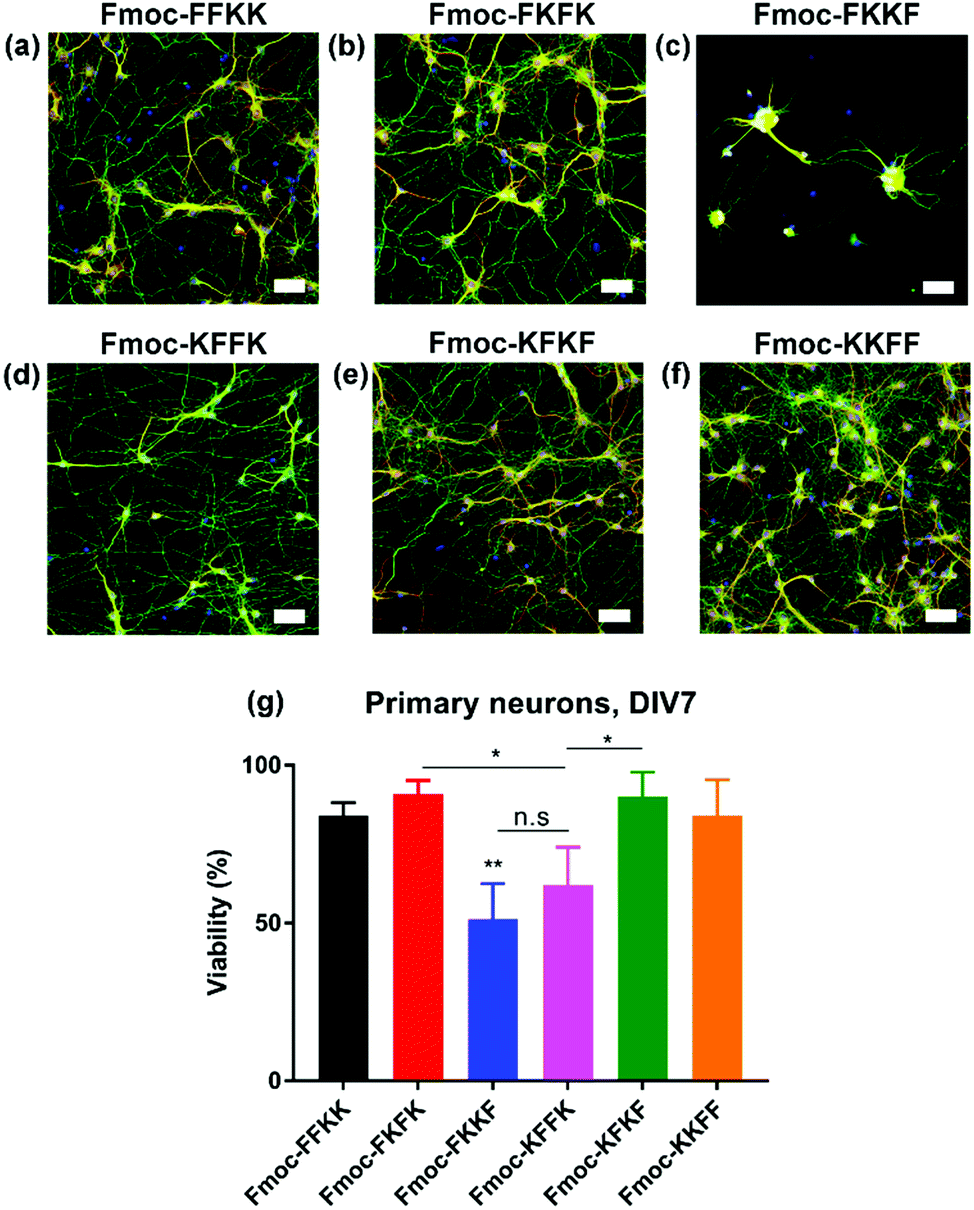
In order to determine whether the differences observed in neuronal adhesion were related to differences in the structure of the peptide nanofibres, the secondary structure of the nanofibres was analysed through circular dichroism. Here, a 0.5% (w/v) nanofibre solution was initially prepared and diluted to 0.025% (w/v) to obtain a concentration which was appropriate for measurements. It is evident that the majority of the tetrapeptides exist in a disordered, or random conformation, as evidenced by a lack of peaks below 240 nm (Fig. 4a). One exception to this is Fmoc-FKFK, which displays a positive peak at 220 nm which is consistent with a β-sheet structure. It should be noted that this differs from the CD spectra of previously reported lysine tetrapeptides Fmoc-FFKK and Fmoc-FKFK,30 owing to the incorporation of D-amino acids into these peptides, which likely affects their secondary structure. | ||
| Fig. 4 Effects of amino acid sequence on the secondary structure and surface of tetrapeptide nanofibres. (a) Circular dichroism measurements for tetrapeptide solutions diluted to 0.025% (w/v) show a disordered structure for all peptides, excepting Fmoc-FKFK which displays a classic β-sheet peak at 220 nm. (b) Zeta potential measurements performed at 0.5% (w/v) show differences in surface charge dependent on amino acid sequence. (c) Differences are also seen in contact angle measurements which approximate the hydrophobicity of a glass coverslip coated with a 0.5% (w/v) nanofibre solution. Control experiments using untreated coverslips and poly-D-lysine treated coverslips (the current gold standard for neuronal tissue culture) are also presented. Finally, (d) FITC labelling of lysine residues, quantified by analytical HPLC, confirms that the more hydrophobic surfaces obtained using Fmoc-FKFK is likely due to a lower amount of solvent-accessible lysines. | ||
As no significant differences were found in the secondary structure of the tetrapeptides (Fmoc-FKFK excepted), zeta potential measurements were performed in order to assess whether there may variability in the surface charge of the nanofibres, which in turn may affect their ability to support primary neurons. Zeta potential measurements on peptide solutions prepared at 0.5% (w/v) revealed differences in the surface charge of the self-assembled fibres (Fig. 4b). All zeta potential values recorded were highly positive, which is consistent with previous reports for lysine containing self-assembling peptides.39,40 The three peptides with the highest zeta potential were Fmoc-FFKK, Fmoc-FKFK and Fmoc-KFFK, with these values being at least 20 mV larger than those measured for Fmoc-FKKF, Fmoc-KFKF and Fmoc-KKFF. However, it should be noted that zeta potential assumes spherical particles, which is not the case for these highly anisotropic fibres. Nonetheless, this data provides a useful relative comparison, which suggests that the order of amino acids in the peptide affects the surface charge of the nanofibre.
To investigate whether the differences observed in zeta potential measurements were translated to the tetrapeptide coated coverslips used for culturing primary neurons, contact angle measurements were performed on glass coverslips coated with 0.5% (w/v) peptide solutions, identical to how the substrates for culturing primary neurons were prepared. From these measurements, it is evident that Fmoc-FKFK coated coverslips are significantly more hydrophobic than all other peptides (Fig. 4c and S8†), whereas Fmoc-FKKF and Fmoc-KFFK were the most hydrophilic of the tetrapeptides. All peptide-coated coverslips were significantly more hydrophobic than poly-D-lysine coated coverslips, which is the current gold standard for primary neuronal culture. Somewhat surprisingly, the tetrapeptide coated coverslips were also more hydrophobic than uncoated glass coverslips. As both poly-D-lysine and all peptides, excepting Fmoc-FKKF and to an extent Fmoc-KFFK, can be successfully used to culture primary neurons, this suggests that surface hydrophobicity does not play a major role in determining substrate suitability for primary neurons.
To determine whether the trends observed in the contact angle measurements were due to the presentation (or lack thereof) of lysine residues at the nanofibre surface, fluorescein isothiocyanate (FITC) was used to label any solvent accessible lysine residues at the surface of the nanofibres. FITC has previously been used to label proteins and peptides at lysine residues,41,42 therefore nanofibre solutions were prepared at 0.5% (w/v) before diluting 2× to reduce any aggregation-induced labelling artefacts. After overnight incubation of the nanofibre solutions with FITC, methanol was used to solvate the nanofibres into monomers and fibre labelling was quantified using analytical HPLC (Fig. 4d, S9 and S10†). Consistent with the contact angle measurements, Fmoc-FKFK had a significantly lower degree of labelling when compared with the other tetrapeptides. This may be due to the amplified β-sheet structure of Fmoc-FKFK relative to the other tetrapeptides. Fmoc-FFKK also trends towards a lower degree of labelling, however the difference is not significant relative to the other tetrapeptides. The fibre labelling suggests that the increased hydrophobicity of the Fmoc-FKFK nanofibre surface is due to decreased solvent accessibility of lysine residues, however this is insufficient to decrease the viability of neurons cultured atop these substrates. This can be rationalised by considering the size of the nanofibres (4–8 nm) versus the size of the neuronal cell body (approximately 20 μm) and that many nanofibres would simultaneously be in contact with the neuronal cell body, providing sufficient favourable electrostatic interactions for the adhesion of primary neurons to substrates.
The effect of hydrophobic moieties on the nanofibre surface
The characterisation of the tetrapeptide nanofibres thus far has focused on the hydrophilic portion of the nanofibres. Zeta potential identified differences in surface charge (however all nanofibres are still highly positively charged) and contact angle and fibre labelling measurements have given insight into the degree of solvent accessible, hydrophilic lysine residues at the surface of the nanofibre. However, these insights do not correspond to the differences in primary neuronal viability observed in Fig. 3. Therefore, it was theorised that the hydrophobic phenylalanine and Fmoc moieties may play an important role in determining substrate suitability for primary neurons. Indeed, it has been shown that the analogous, more hydrophobic peptides Fmoc-FF and Fmoc-GFF are cytotoxic towards HeLa cells and tumour spheroids, respectively, due to their interactions with the cell membrane.43,44With this in mind, solid state and gel state NMR was performed on the tetrapeptides to obtain conformational information, which can yield insights into the nature of the fibre surface. Solid state NMR has been used previously in combination with other techniques to determine the molecular association and surface chemistry of MAX1 and MAX8 peptides, however these studies required isotopic labelling.45,46 Using our unlabelled tetrapeptides complex spectra were obtained, however key differences in the solid state and gel state NMR can be identified. In the solid state 13C NMR (Fig. 4a), the peaks located from 155–165 ppm are representative of the carbamate carbon from the Fmoc moiety. For Fmoc-FFKK and Fmoc-FKFK, only one peak was present, whereas for the remaining tetrapeptides, two peaks were observed. This suggests that for Fmoc-FFKK and Fmoc-FKFK, only one conformation of the Fmoc group is present. A sharp set of peaks for Fmoc-FKFK between 50-60 ppm in the solid state 13C NMR suggests an ordered C-α region, confirming the enhanced β-sheet structure of this tetrapeptide.
Further evidence for the β-sheet structure of Fmoc-FKFK can be seen in the gel state NMR for Fmoc-FKFK, which shows two small peaks at approximately 5 and 5.3 ppm (Fig. 5b). The presence of these peaks have previously been shown to represent a β-sheet structure.47,48 In the aromatic region (7–8 ppm) of the gel state NMR, an increased number of peaks (8) are observed for Fmoc-FKKF and Fmoc-KFFK, suggesting that the aromatic regions of these peptides are conformationally flexible. This is likely the mechanism for the reduced viability of primary neurons cast upon nanofibre coatings of these two peptides, as the presence of aromatic moieties is known to be beneficial for both positively and negatively charged anti-bacterial peptides due to their ability to penetrate the cell membrane.49,50
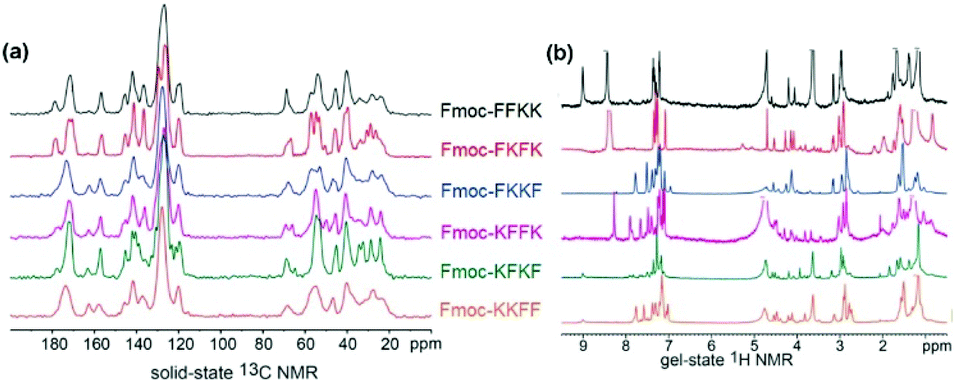 | ||
| Fig. 5 Examining tetrapeptide self-assembly through (a) solid state 13C NMR and (b) gel state NMR. Comparing the spectra reveals β-sheet formation for Fmoc-FKFK and increased aromatic flexibility for Fmoc-FKKF and Fmoc-KFFK. Solid state NMR measurements were conducted using lyophilised peptides and gel state NMR measurements were recorded at a concentration of 1% (w/v) in D2O, where gelation was triggered through the addition of deuterated PBS (pD 7.4). | ||
To verify this result, tBLMs consisting of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipids were treated with solutions of the tetrapeptide nanofibres at different concentrations and the AC swept frequency electrical impedances monitored over time to determine whether the tetrapeptides can disrupt this cell-mimetic membrane. This technique has previously been used to assess the activity of various bespoke and natural peptides on lipid bilayers.51–53 Upon addition of 1 and 10 μM solutions of tetrapeptide nanofibres, minimal disruption of the membrane bilayer was observed. However, upon treatment of the membranes with 100 μM nanofibre solutions, significant increases in membrane conductance can be seen for Fmoc-FKKF and Fmoc-KFFK (Fig. 6c and d), with a notable increase also for Fmoc-FFKK (Fig. 6a) and smaller increase for the other tetrapeptides. For clarity, a 100 μM solution corresponds to approximately 0.01% (w/v), however it is highly likely that the concentration of the peptide at the lipid membrane interface is far higher due to electrostatic interactions between the anionic lipid phosphates and the cationic peptide lysine residues. This suggests that at 100 μM, the nanofibres of Fmoc-FKKF and Fmoc-KFFK are strongly interacting with the lipid membranes. As determined in the NMR measurements, these membrane interactions are likely due to the increased conformational flexibility of the hydrophobic residues of Fmoc-FKKF and Fmoc-KFFK causing alterations in lipid bilayer packing, expanding intrinsic membrane pores, as described previously.51,53,54 This ability to disrupt the lipid membrane is likely the reason that decreased neuronal viability is seen on substrates of Fmoc-FKKF and Fmoc-KFFK.
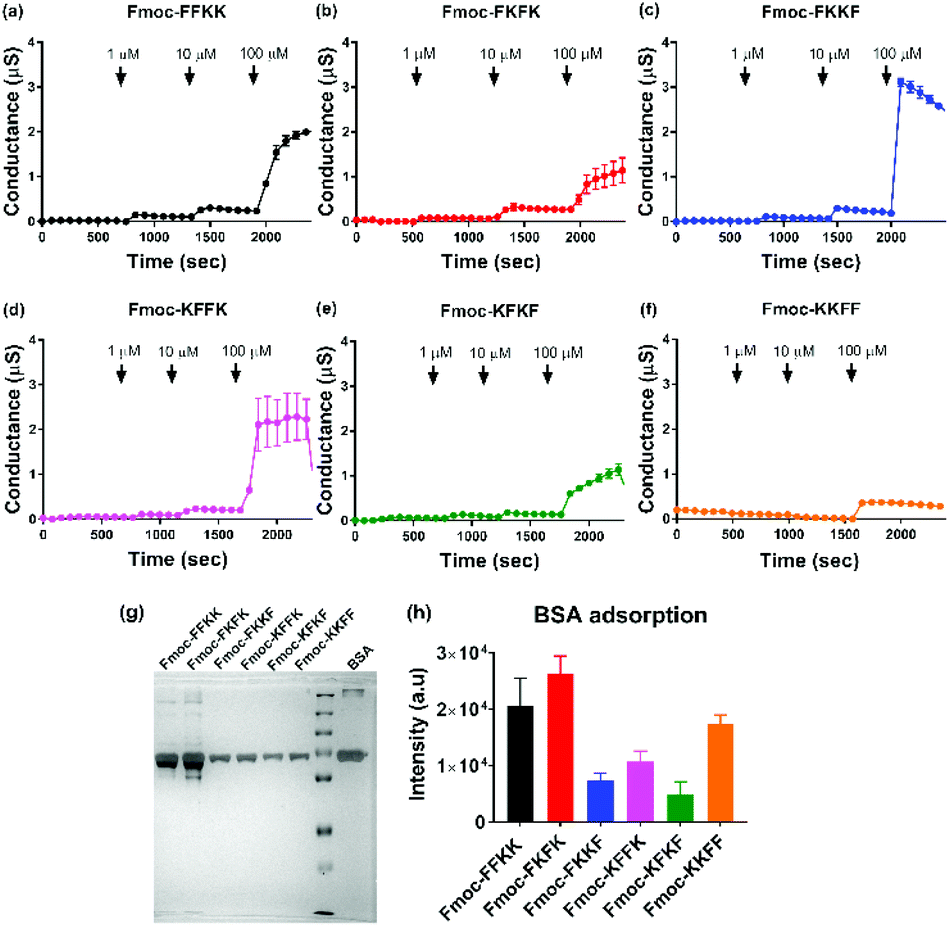 | ||
| Fig. 6 Evaluating the ability of tetrapeptide nanofibres to disrupt membranes and adsorb proteins. Electrical impedance spectroscopy was performed on sparsely tethered bilayer lipid membranes (tBLMs) consisting of 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipids, with changes in conductance observed upon treatment of the membranes with (a–f) peptide nanofibre solutions at different concentrations. (g) Representative silver stained SDS-PAGE gel and (h) associated quantification (n = 3, ±standard deviation) showing adsorption of serum proteins onto tetrapeptide nanofibres after incubation with cell culture medium containing 10% FBS. | ||
It has previously been shown that FKKF and KFFK peptide sequences (without the Fmoc group) are able to interact with membranes. The FKKF tetrapeptide sequence is highly conserved in the pIII protein coat of the fd filamentous bacteriophage, which is the protein responsible for the infectivity of the bacteriophage.55,56 Mutation of these residues has been shown to significantly decrease bacteriophage activity.57 The KFFK tetrapeptide sequence is mostly conserved across the phenol-soluble modulin alpha (PSMα) series of peptides, which are responsible for the virulence of S. aureus through the interaction with and lysis of red and white blood cell membranes.58–60 Therefore both of these tetrapeptide sequences are known to interact with membranes, a trait which is seemingly conserved when these sequences are capped with an Fmoc moiety and self-assemble into nanofibres.
Additionally, the disassembly of the peptide nanofibre coatings was assessed through analytical HPLC after incubation with cell culture media over five days. Analytical HPLC of the cell culture media after five days incubation revealed the presence of Fmoc-FKKF and Fmoc-KFFK for coverslips coated with these peptides (Fig. S11†), although at such low concentrations (<10 μM) that they are unlikely to have any significant effects on viability, which is supported by the tBLM measurements above, as no membrane disruption is observed at these concentrations. For the remaining four peptides, negligible amounts of peptides could be detected in the culture medium supernatant. This suggests that the mobility of hydrophobic residues on the nanofibre surface is likely the critical factor determining neuronal adhesion to our peptide substrates.
Finally, the effect of serum proteins on the ability of the tetrapeptide nanofibres to interact with the cell membrane was investigated. Higher levels of protein adsorption were observed on Fmoc-FFKK and Fmoc-FKFK substrates (Fig. 6g and h), suggesting an inverse correlation with solvent accessible lysines and protein adsorption. However, the levels of protein adsorption onto tetrapeptide nanofibres did not reflect the differences observed in neuronal cell viability. tBLM experiments were also performed using DMEM containing 10% FBS (Fig. S13†). Apart from Fmoc-KKFF, the results match those reported in Fig. 6, with Fmoc-FKKF and Fmoc-KFFK yielding large increases in membrane conductance, suggesting strong membrane interactions. Fmoc-KKFF aggregated upon dilution into DMEM containing 10% FBS, hence it is likely that the increased membrane conductance observed are due to the interaction of these aggregates with the membrane. For reference, no aggregation of any other peptides were observed upon dilution into DMEM/FBS, nor did Fmoc-KKFF aggregate when diluted into the buffer used for tBLM measurements in Fig. 6. Therefore, it is shown that even in complex cellular milieu, hydrophobic mobility at the nanofiber surface plays an integral role in determining whether nanofibers can interact with cell membranes, which in turn determines the suitability of these substrates for supporting sensitive cells such as neurons.
Conclusions
In conclusion, we have synthesised all possible permutations of tetrapeptides which bear two L-phenylalanines and two L-lysines, capped at their N-terminus with an Fmoc-moiety. Whilst all tetrapeptides self-assembled into nanofibres of similar diameters as determined by AFM and small angle neutron scattering, significant differences were observed in their ability to support sensitive primary neurons, with Fmoc-FKKF significantly worse than other substrates and Fmoc-KFFK showing decreased neuronal viability. A comprehensive investigation of the nanofibre surface was undertaken, with zeta potential, contact angle and nanofibre labelling experiments suggesting differences in the presentation of lysine groups at the surface of the nanofibres. However, this did not correlate with decreased neuronal viability, likely owing to the differences in scale between the nanofibres (4–8 nm) and the neuronal cell body (approximately 20 μm). In contrast, solid state and gel state NMR revealed that the aromatic moieties of Fmoc-FKKF and Fmoc-KFFK were much more conformationally flexible than the four other tetrapeptides, with the ability of these flexible aromatic residues to interact with the cell membrane confirmed through electrical impedance spectroscopy experiments using tethered bilayer lipid membranes, which showed strong interactions with Fmoc-FKKF and Fmoc-KFFK. This work decouples the differential effects of hydrophilic and hydrophobic moiety presentation at the surface of a peptide nanofibre, and their effects on cell viability. It is envisioned that these insights will assist in the rational design of new neuronal–scaffold interfaces for the 3D culturing of primary neurons, which could give insights into neurogenesis, ageing and disease mechanisms.Ethical statement
All experiments were performed in compliance with the Australian Code for the care and use of animals for scientific purposes (2013) and were approved by the Animal Ethics Committee of the University of New South Wales, Australia.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Mark Wainwright Analytical Centre (UNSW) for access to instruments and the Australian Nuclear Science and Technology Organization (ANSTO) for access to the QUOKKA beamline (proposal 6928). The authors received funding from the National Health and Medical Research Council (NHMRC) (1081916, 1132524) to LI, (1083209) to TF and the Australian Research Council (ARC) (CE140100036 and DP190101892) to PT, (DP106001664) to CGC and (DP170100781, DP170100843) to LI, (DP180101473) to TF. ADM is an ARC-NHMRC Dementia Research Development Fellow (1106751) and LI is a NHMRC Principal Research Fellow (1136241).Notes and references
- A. Lee, A. R. Hudson, D. J. Shiwarski, J. W. Tashman, T. J. Hinton, S. Yerneni, J. M. Bliley, P. G. Campbell and A. W. Feinberg, Science, 2019, 365, 482–487 CrossRef CAS PubMed.
- J. A. Shadish, G. M. Benuska and C. A. DeForest, Nat. Mater., 2019, 18, 1005–1014 CrossRef CAS.
- C. Yang, F. W. Del Rio, H. Ma, A. R. Killaars, L. P. Basta, K. A. Kyburz and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4439–4445 CrossRef PubMed.
- A. Cangialosi, C. K. Yoon, J. Liu, Q. Huang, J. Guo, T. D. Nguyen and D. H. Gracias, Science, 2017, 357, 1126–1130 CrossRef CAS PubMed.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- Y. Jin, J. S. Lee, J. Kim, S. Min, S. Wi, J. H. Yu, G. E. Chang, A. N. Cho, Y. Choi, D. H. Ahn, S. R. Cho, E. Cheong, Y. G. Kim, H. P. Kim, Y. Kim, D. S. Kim, H. W. Kim, Z. Quan, H. C. Kang and S. W. Cho, Nat. Biomed. Eng., 2018, 2, 522–539 CrossRef CAS PubMed.
- E. Bassat, Y. E. Mutlak, A. Genzelinakh, I. Y. Shadrin, K. B. Umansky, O. Yifa, D. Kain, D. Rajchman, J. Leach, D. R. Bassat, Y. Udi, R. Sarig, I. Sagi, J. F. Martin, N. Bursac, S. Cohen and E. Tzahor, Nature, 2017, 547, 179–184 CrossRef CAS PubMed.
- N. Noor, A. Shapira, R. Edri, I. Gal, L. Wertheim and T. Dvir, Adv. Sci., 2019, 6, 1900344 CrossRef PubMed.
- R. Freeman, M. Han, Z. Alvarez, J. A. Lewis, J. R. Wester, N. Stephanopolous, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808–813 CrossRef CAS PubMed.
- A. N. Moore and J. D. Hartgerink, Acc. Chem. Res., 2017, 50, 714–722 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 26, 9060–9093 CrossRef PubMed.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS PubMed.
- S. Lindsey, J. H. Piatt, P. Worthington, C. Sonmez, S. Satheye, J. P. Schneider, D. J. Pochans and S. A. Langhans, Biomacromol, 2015, 16, 2672–2683 CrossRef CAS PubMed.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- A. Lampel, R. V. Ulijn and T. Tuttle, Chem. Soc. Rev., 2018, 47, 3737–3758 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- A. L. Rodriguez, C. L. Parish, D. R. Nisbet and R. J. Williams, Soft Matter, 2013, 9, 3915–3919 RSC.
- M. He, J. Li, S. Tan, R. Wang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 18718–18721 CrossRef CAS PubMed.
- G. Cheng, V. Castaletto, R. R. Jones, C. J. Connon and I. W. Hamley, Soft Matter, 2011, 7, 1326–1333 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- C. A. DeForest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS PubMed.
- L. M. de Leon-Rodriguez, Y. Hemar, G. Mo, A. K. Mitra, J. Cornish and M. A. Brimble, Acta Biomater., 2017, 47, 40–49 CrossRef CAS PubMed.
- S. Marchesan, C. D. Easton, K. E. Styan, L. J. Waddington, F. Kushkaki, L. Goodall, K. M. McLean, J. S. Forsythe and P. G. Hartley, Nanoscale, 2014, 6, 5172–5180 RSC.
- A. D. Martin, S. W. Chua, C. G. Au, H. Stefen, M. Przybyla, Y. Lin, J. Bertz, P. Thordarson, T. Fath, Y. D. Ke and L. M. Ittner, ACS Appl. Mater. Interfaces, 2018, 10, 25127–25134 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castila, H. Su, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 11, 3531–3540 CrossRef PubMed.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Z. Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- M. Bi, A. Gladbach, J. van Ersel, A. Ittner, M. Przybyla, A. van Hummel, S. W. Chua, J. van der Hoven, W. S. Lee, J. Muller, J. Parmar, G. von Jonquieres, H. Stefen, E. Guccione, T. Fath, G. D. Housley, M. Klugmann, Y. D. Ke and L. M. Ittner, Nat. Commun., 2017, 8, 473 CrossRef PubMed.
- A. Ittner, S. W. Chua, J. Bertz, A. Volkerling, J. van der Hoven, A. Gladbach, M. Przybyla, M. Bi, A. van Hummel, C. H. Stevens, S. Ippati, L. S. Suh, A. Macmillan, G. Sutherland, J. J. Kril, A. P. G. Silva, J. P. McKay, A. Poljak, F. Delerue, Y. D. Ke and L. M. Ittner, Science, 2016, 354, 904–908 CrossRef CAS PubMed.
- I. Leshchyns’ka, H. T. Liew, C. Shepherd, G. M. Halliday, C. H. Stevens, Y. D. Ke, L. M. Ittner and V. Sytnyk, Nat. Commun., 2015, 6, 8836 CrossRef PubMed.
- D. Mazia, G. Schatten and W. Sale, J. Cell Biol., 1975, 66, 198–200 CrossRef CAS PubMed.
- S. Toksoz, R. Mammadov, A. B. Tekinay and M. O. Guler, J. Colloid Interface Sci., 2011, 356, 131–137 CrossRef CAS PubMed.
- Y. Sun, W. Li, X. Wu, N. Zhang, Y. Zhang, S. Ouyang, X. Song, X. Fang, R. Seeran, W. Xue, L. He and W. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 2348–2359 CrossRef CAS PubMed.
- B. Law, R. Weissleder and C. H. Tung, Biomacromolecules, 2006, 7, 1261–1265 CrossRef CAS PubMed.
- J. Kim, J. Yoon and R. C. Hayward, Nat. Mater., 2009, 9, 159–164 CrossRef PubMed.
- W. T. Truong, Y. Su, D. Gloria, F. Braet and P. Thordarson, Biomater. Sci., 2015, 3, 298–307 RSC.
- J. P. Wojciechowski, A. D. Martin, A. F. Mason, C. M. Fife, S. M. Sagnella, M. Kavallaris and P. Thordarson, ChemPlusChem, 2017, 82, 383–389 CrossRef CAS.
- K. Nagy-Smith, E. Moore, J. P. Schneider and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9816–9821 CrossRef CAS PubMed.
- K. Nagy-Smith, P. J. Beltramo, E. Moore, R. Tycko, E. M. Furst and J. P. Schneider, ACS Cent. Sci., 2017, 3, 586–597 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS PubMed.
- F. Avbelj, D. Kocjan and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17394–17397 CrossRef CAS PubMed.
- H. D. Glossop, G. Heruka De Zoysa, Y. Hemar, P. Cardoso, K. Wang, J. Lu, C. Valery and V. Sarojini, Biomacromolecules, 2019, 20, 2515–2529 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- T. Berry, D. Dutta, R. Chen, A. Leong, H. Wang, W. A. Donald, M. Parviz, B. Cornell, M. Wilcox, N. Kumar and C. G. Cranfield, Langmuir, 2018, 34, 11586–11592 CrossRef CAS PubMed.
- Z. Su, J. J. Leitch, R. J. Faragher, A. L. Schwan and J. Lipkowski, Electrochim. Acta, 2017, 243, 364–373 CrossRef CAS.
- C. G. Cranfield, S. T. Henriques, B. Martinac, P. Duckworth, D. J. Craik and B. Cornell, Langmuir, 2017, 33, 6630–6637 CrossRef CAS PubMed.
- A. Alghalayini, A. Garcia, T. Berry and C. G. Cranfield, Antibiotics, 2019, 8, 12 CrossRef CAS PubMed.
- F. M. Marassi and S. J. Opella, Protein Sci., 2003, 12, 403–411 CrossRef CAS PubMed.
- A. C. Zeri, M. F. Mesleh, A. A. Nevzorov and S. J. Opella, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6458–6463 CrossRef CAS PubMed.
- D. A. Marvin, R. D. Hale, C. Nave and M. Helmer Citterich, J. Mol. Biol., 1994, 235, 260–286 CrossRef CAS PubMed.
- Z. Yao, B. P. Cary, C. A. Bingman, C. Wang, D. F. Kreitler, K. A. Satyshur, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2019, 141, 7660–7664 CrossRef CAS PubMed.
- E. Tayeb-Fligelman, O. Tabachnikov, A. Moshe, O. Goldschmidt-Tran, M. R. Sawaya, N. Coquelle, J. P. Coquelle and M. Landau, Science, 2017, 355, 831–833 CrossRef PubMed.
- K. M. Towle, C. T. Lohans, M. Miskolzie, J. Z. Acedo, M. J. van Belkum and J. C. Vederas, Biochemistry, 2016, 55, 4798–4806 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05686f |
| This journal is © The Royal Society of Chemistry 2020 |