
Insight into the synthesis of N-methylated polypeptides†
Christian
Muhl‡
a,
Lydia
Zengerling‡
a,
Jonathan
Groß
a,
Paul
Eckhardt
a,
Till
Opatz
 a,
Pol
Besenius
*a and
Matthias
Barz
*ab
a,
Pol
Besenius
*a and
Matthias
Barz
*ab
aDepartment of Chemistry, Johannes Gutenberg University Mainz, 55099 Mainz, Germany. E-mail: besenius@uni-mainz.de; barz@uni-mainz.de
bLeiden Academic Center for Drug Research (LACDR), Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: m.barz@lacdr.leidenuniv.nl
First published on 6th October 2020
Abstract
The ring-opening polymerization (ROP) of N-carboxy anhydrides (NCAs) is mostly divided into two classes: NCAs of α-substituted amino acids and N-methylated NCAs of α-unsubstituted glycine derivatives (NNCAs). The use of both monomer types offers different mechanistic features and results in a multitude of functional materials. To combine these properties, the synthesis and ROP of α-substituted and N-methylated NCAs (αNNCAs) of several amino acids were investigated. The current study provides insight into the influence of polymerization conditions and the limitations caused by the enhanced steric demand of the amino acid NCA monomers and their N-methylated derivatives. Namely, the effects of solvent polarity (DMF and DCM) and steric demand of the initiator by using neopentyl amine (NPA) and n-butyl amine (nBu) were studied. Analysis by HFIP-GPC and MALDI-ToF MS reveals that the polymerization and the resulting polymers are tremendously affected by the steric demand of both the initiators and the monomers, while electronic effects seem to have only minor influences. The experimental results are further compared with computational studies, based on coupled cluster (CC) calculations, which underline that electronic effects are of lower importance than steric constraints for the ROP of αNNCAs. Moreover, poly(N-methyl-L-methionine) forms helical secondary structures in solution. Therefore, this work combines mechanistic studies of the ROP of αNNCAs with initial studies on the solution properties of these polypeptides.
Introduction
Amino acids and peptides can be modified synthetically in a versatile manner to fulfill a desired function. Besides the incorporation of D-amino acids, cyclic amino acids or dehydroamino acids, the N-alkylation of amino acids plays an important role in modulating the solubility, metabolic stability and biological activity.1,2 The simplest form, N-methylation, prevents intramolecular hydrogen bonds and causes changes of conformation, polarity and steric demand. Furthermore, the lipophilicity,3 membrane permeability4 and metabolic stability5 are increased and the proteolytic stability6 is improved. All these properties make N-methylated amino acids interesting building blocks for the synthesis of polypeptides to obtain new biopolymers.Currently, most of the ongoing research on polypeptides is either focused on the synthesis, characterization and polymerization of α-amino acids,7,8N-substituted glycine derivatives (polypeptoids)9,10 and block copolymers thereof (polypept(o)ids),11–15 but only a few publications have addressed the synthesis and properties of N-substituted α-amino acid containing polymers. An example for polypeptoids relies on the use of the naturally occurring N-methyl glycine, also known as sarcosine. The corresponding N-substituted N-carboxy anhydride (NNCA) can be polymerized by nucleophilic ring opening polymerization (ROP) for the synthesis of the highly water-soluble polysarcosine. Due to the absence of the acidic proton at the nitrogen, only the normal amine mechanism (NAM) can occur and thereby well-defined polymers with a low dispersity (Đ < 1.1) are obtained.16 The polymerization of NNCAs with longer alkyl chains proceeds slowly (in the order Sar ≫ EtGly > PrGly > nBuGly > iBuGly).10 The reason for lower polymerization rates is the steric hindrance for β-C branched NNCAs and the aggregation of the side chains for linear or γ-C branched NNCAs.17
One of the most studied α- and N-substituted polypeptides is poly(N-methyl-L-alanine), which was first synthesized by a ROP of the corresponding NNCA by Goodman and Fried in 1967.18 Using conformational energy calculations, CD and NMR techniques, it was shown that this structure adopts a right-handed helix with all-trans peptide bonds.19–21 This polymer could also be synthesized from an activated urethane derivate of N-methyl-L-alanine with in situ NCA formation by heating in N,N-dimethylacetamide (DMAc) to 60 °C in the presence of the initiator n-butyl amine. The latter method is a phosgene-free synthesis and benefits from the absence of HCl to achieve control over molecular weights through the feed ratio of the monomer to the initiator.22,23 Cosani et al. describe their unsuccessful attempts in the polymerization of N-benzylglycine, N-benzyl-L-alanine or N-methyl-L-phenylalanine NCAs. As an alternative route for the preparation of poly(N-methyl-γ-methyl-L-glutamate) and poly(N-methyl-γ-ethyl-L-glutamate), they tested different methylation methods on poly(γ-methyl-L-glutamate) or poly(γ-ethyl-L-glutamate).24 Like poly(N-methyl-L-alanine), both polymers were found to form a right-handed helix in a TFE solution of remarkably high stability, which retained their conformation even in pure TFA as observed by CD (circular dichroism) measurements.25 A special case is the amino acid proline, the only proteinogenic amino acid containing a secondary amine. Poly(L-proline) (PLP) can form a right-handed PLP I helix (cis) in aliphatic alcohols and a left-handed PLP II helix (trans) in water and organic acids.26 The synthesis of well-defined PLP was challenging due to the impurity of the monomer and termination reactions in the polymerization. Highly pure NCA monomers can be synthesized from N-(Boc)-protected L-proline and were polymerized efficiently using dimethylamine as an initiator.27
In this work we describe the synthesis of different α-substituted and N-methylated NCAs (αNNCAs) with various side chains. Three synthetic strategies are widely used to prepare N-methylated amino acids: reductive amination, N-methylation by alkylation and a reductive ring opening of 5-oxazolidinones with a triethylsilane/TFA mixture.28,29 The latter method introduced by Freidinger et al. proceeds without racemization and was therefore chosen to synthesize NMMet and NMLeu in the current work.30 The polymerization of these αNNCAs strongly depends on the steric demand of the side chain and the resulting electronic structure of the applied monomers. Various conditions for the ROP of the prepared αNNCAs were investigated to study the influence of initiators with different steric demands and solvents with varying polarities. The resulting polymers were analyzed via GPC and MALDI-ToF measurements to obtain a deeper insight into the mechanistic pathway of the polymerization. Furthermore, the impact on secondary structure formation was investigated via CD spectroscopy and the results were compared with previous studies on these rarely described poly(N-alkylated α-substituted amino acids).
Experimental part
Schlenk techniques were used for the reactions with air- and moisture-sensitive reagents or intermediates, performed under an argon atmosphere, using laboratory glassware, and dried under high vacuum at 120 °C with a hot air gun. All the used solvents and reagents were purchased from ACROS Organics (Thermo Scientific GmbH, Nidderau), Alfer Aesar GmbH & Co. KG (Karlsruhe), Carbolution Chemicals GmbH (Saarbrücken), Iris Biotech GmbH (Markredwitz), Merck KGaA (Darmstadt), Sigma-Aldrich Chemie GmbH (Taufkirchen) and TCI Deutschland GmbH (Eschborn). Prior to use, water was demineralized, using PURELAB® flex by Elga. Solvents used for air- or moisture-sensitive reactions were bought anhydrous. DCM was dried using a solvent purification system. All the qualitative thin layer chromatography (TLC) analyses were carried out using silica coated aluminum sheets (60 Å, F254) purchased from MACHEREY-NAGEL GmbH & Co. KG (Düren). The detection of the analytes was performed using UV light (λ = 254 nm) and the detection reagent ninhydrin or KMnO4. Flash chromatography (FC) was performed by using silica gel with an average grain size of 15–40 μm, purchased from ACROS Organics™. Purification via medium pressure liquid chromatography (MPLC) was performed on a Sepacore® Easy Purification System (BÜCHI Labortechnik AG) equipped with a UV-Photometer C-640 (BÜCHI) and a Fraction Collector C-660 (BÜCHI). All runs were performed on a CHROMABOND Flash RS 120 C18 column (MACHEREY-NAGEL GmbH & Co. KG). Gel permeation chromatography (GPC) was performed with hexafluoroisopropanol (HFIP) containing 3 g L−1 potassium trifluoroacetate (KTFA) as an eluent at 40 °C. The column material was modified silica gel (PFG columns, particle size: 7 μm, porosity: 100 Å and 4000 Å), purchased from PSS Polymer Standards Service GmbH. For calibration, polymethyl methacrylate (PMMA, Polymer Standards Services GmbH) was used and toluene was used as the internal standard. A refractive index detector (G1362A RID) and a UV/VIS detector (at 230 nm, Jasco UV-2075 Plus) were used for polymer detection. All mass spectra were recorded on an electrospray ionization spectrometer (ESI) QT of Ultima. MALDI-ToF mass spectra were recorded using a Bruker rapifleX MALDI-ToF mass spectrometer equipped with a 337 nm N2 laser. Acceleration of the ions was performed with pulsed ion extraction (PIE, Bruker) at a voltage of 20 kV. The analyzer was operated in the reflection mode and the ions were detected using a microchannel plate detector. The mass spectra were processed using the X-TOF 5.1.0 software (Bruker, Billerica, MA, USA). Sample preparation was performed using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as the matrix, sodium trifluoroacetate as the cationizing salt, and dichloromethane as the solvent. All NMR spectra were recorded on a BRUKER Avance II 400 spectrometer and the measurements were carried out in deuterated solvents (CDCl3, DMSO-d6, and D2O). All the recorded NMR spectra were analyzed using MestReNova v12.0.0. CD spectroscopy was performed on a Jasco J-815 spectrometer at 20 °C. The spectra were analyzed using Spectra Manager 2.0. All spectra were recorded using a quartz cell with a path length of 1 mm at a concentration of c = 0.25 g L−1 polymer in HFIP. θMR was calculated from the following equation:with Mrepeating unit = 147.1 g mol−1, cM = 0.25 g L−1 and l = 0.1 cm. All measurements of optical rotation were carried out with a PerkinElmer 241 polarimeter at 589 nm. A quartz glass cuvette with a path length of 10 cm was used.
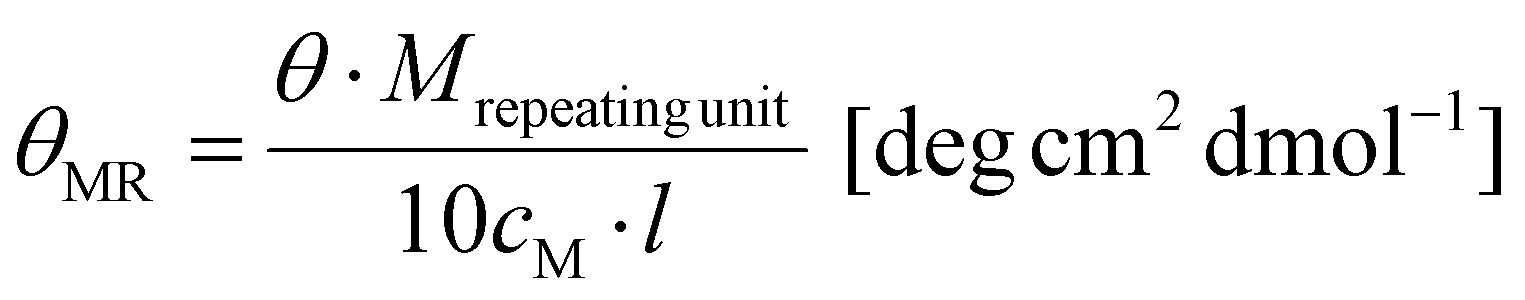
Synthesis of N-methyl-L-methionine NCA (NMMet-NCA)
For the synthesis of NMMet-NCA, 2.0 g (10.2 mmol, 1 eq.) of the corresponding amino acid was suspended in 150 mL of freshly distilled, anhydrous THF. Diphosgene (0.97 mL, 8 mmol, 0.8 eq.) was added at room temperature at once to the stirred suspension. After 15 min, the turbid solution became clear and was further stirred at room temperature for 30 min, before N2 was bubbled through the solution for 10 min. The solution was cooled with an ice bath and dry TEA (7.1 mL, 51.1 mmol, 5.1 eq.) was added slowly while precipitation occurred (TEA·HCl). After filtration under a N2 atmosphere, THF was removed under high vacuum and the residue was dissolved again in a minimum amount of dry ethyl acetate. The ethyl acetate solution was added to a mixture of dry n-hexane and diethyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and stored at −20 °C overnight. This resulted in the crystallization of the corresponding NCA, which was filtered under a N2 atmosphere and dried to yield 1.4 g (7.3 mmol, 72%). Crystal structure: CCDC 1976455;†1H-NMR (400 MHz, CDCl3) δ [ppm] = 4.29 (dd, 3J = 3.6, 3.2 Hz, 1H, –CH–), 3.00 (s, 3H, –NC
:1) and stored at −20 °C overnight. This resulted in the crystallization of the corresponding NCA, which was filtered under a N2 atmosphere and dried to yield 1.4 g (7.3 mmol, 72%). Crystal structure: CCDC 1976455;†1H-NMR (400 MHz, CDCl3) δ [ppm] = 4.29 (dd, 3J = 3.6, 3.2 Hz, 1H, –CH–), 3.00 (s, 3H, –NC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 2.67–2.51 (m, 2H, –C2SCH3), 2.32–2.23 (m, 1H, –C2CH2SCH3), 2.18–2.13 (m, 1H, –C2CH2SCH3), 2.11 (s, 3H, –SCH3); 13C-NMR (101 MHz, CDCl3): δ [ppm] = 168.64 (–COO), 152.13 (–CNOO), 59.82 (–CHN–), 28.86 (–N
3), 2.67–2.51 (m, 2H, –C2SCH3), 2.32–2.23 (m, 1H, –C2CH2SCH3), 2.18–2.13 (m, 1H, –C2CH2SCH3), 2.11 (s, 3H, –SCH3); 13C-NMR (101 MHz, CDCl3): δ [ppm] = 168.64 (–COO), 152.13 (–CNOO), 59.82 (–CHN–), 28.86 (–N![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 28.43 (–SCH2), 27.94 (–SCH2H2–), 15.56 (–SCH3).
H3), 28.43 (–SCH2), 27.94 (–SCH2H2–), 15.56 (–SCH3).
Synthesis of N-methyl-DL-methionine NCA (DL-NMMet-NCA)
The synthesis of DL-NMMet-NCA is analogous to the synthesis of NMMet-NCA. After the crystallization of the NCA, the solution was decanted in order to separate the crystallized NCA from the residual solvent. The NCA could not be collected via filtration, since the melting point of the crystals was below room temperature (20 °C). After decanting, the crystals melted and the resulting colorless and clear oil (yield: 64 mg, 0.3 mmol, 67%) was dried under reduced pressure to remove trace solvents. The oil was then characterized and used for polymerization. 1H-NMR (400 MHz, CDCl3) δ [ppm] = 4.29 (dd, 3J = 3.6, 3.2 Hz, 1H, –CH–), 3.00 (s, 3H, –NCH3), 2.67–2.51 (m, 2H, –C2SCH3), 2.32–2.23 (m, 1H, –C2CH2SCH3), 2.18–2.13 (m, 1H, –C2CH2SCH3), 2.11 (s, 3H, –SCH3).
Synthesis of N-methyl-DL-alanine NCA (NMAla-NCA)
N-Methyl-DL-alanine (1.70 g, 16.5 mmol, 1 eq.) was suspended in 100 mL of anhydrous THF. After the addition of diphosgene (1.59 mL, 13.2 mmol, 0.8 eq.), the suspension was stirred at room temperature for 1.5 hours until it became clear. N2 was bubbled through the solution for 15 min and THF was removed in vacuo. The residue was dissolved again in 20 mL of anhydrous THF and added to 200 mL of n-hexane. After storage at −20 °C overnight, NMAla-NCA (1.10 g, 8.2 mmol, 51%) crystallized and was collected via filtration under a N2 atmosphere and dried. Crystal structure: CCDC 1976456;†1H-NMR (400 MHz, CDCl3) δ [ppm] = 4.19 (q, 3J = 7.0 Hz, 1H, –CH–), 2.98 (s, 3H, –NCH3), 1.54 (d, 3J = 7.0 Hz, 3H, –CH3); 13C-NMR (101 MHz, CDCl3): δ [ppm] = 169.42 (–COO), 151.73 (–CNOO), 57.00 (–CHN–), 28.37 (–NH3), 15.19 (–CHH3).
Synthesis of N-methyl-L-leucine NCA (NMLeu-NCA)
N-Methyl-L-leucine (0.60 g, 3.7 mmol, 1 eq.) was suspended in 75 mL of anhydrous THF. After the addition of 1.9 mL phosgene in toluene (20% phosgene in toluene, 4 mmol, 1.1 eq.), the suspension was stirred at room temperature for 15 min until it became clear. Due to the incomplete reaction (reaction control was performed via1H-NMR), TEA (1.1 mL, 8 mmol, 2 eq.) was added to the cooled solution in order to convert N-carbamoyl chloride to the corresponding NCA. The formed precipitate (TEA·HCl) was filtered under a N2 atmosphere and THF was removed under reduced pressure from the clear, colorless solution. The residual oil could neither be crystallized nor precipitated and therefore the colorless oil was characterized and used for polymerization (NMLeu-NCA: 0.30 g, 1.7 mmol, 46%). 1H-NMR (400 MHz, CDCl3) δ [ppm] = 4.14 (t, 3J = 5.6 Hz, 1H, –CH–), 2.99 (s, 3H, –NCH3), 1.93 (dsep, 3J = 6.6, 1.2 Hz, 1H, –C(CH3)2), 1.78 (dd, 3J = 5.7, 2.2 Hz, 2H, –C2CH(CH3)2), 0.98 (dd, 3J = 6.7, 1.1 Hz, 6H, –CH(C3)2); 13C-NMR (101 MHz, CDCl3): δ [ppm] = 168.66 (–COO), 151.94 (–CNOO), 59.47 (–CHN–), 37.86 (–H2CH(CH3)2), 28.56 (–NH3), 24.00 (–CH2H(CH3)2), 22.86 (–CH(H3)2), 21.98 (–CH(H3)2).
Ring-opening polymerization of αNNCAs
In the following, the procedure is described exemplarily for the polymerization of NMAla-NCA, but it can be extended to all other αNNCAs.NMAla-NCA (78.9 mg, 0.61 mmol, 50 eq.) was transferred into a Schlenk tube under a dry nitrogen counter flow. Dry DMF (0.9 mL) and absolute n-butylamine (1.21 μL, 0.89 mg, 0.01 mmol, 1 eq.) in dry DMF were added using a stock solution. A steady flow of dry nitrogen was sustained during the polymerization, preventing any impurities from entering the Schlenk tube, while ensuring the escape of the produced CO2. The progress of the polymerization was monitored via IR spectroscopy by the decreasing intensities of the NCA-associated carbonyl peaks at 1858 and 1788 cm−1. Samples were taken using a nitrogen flushed syringe through a septum. If no conversion was observed after 16 h, equimolar amounts (relative to the initiator) of triethyl amine (TEA) in DMF were added, using a stock solution. The polymer precipitated after complete conversion in a mixture of cold diethyl ether and n-hexane (2:1). The suspension was centrifuged (3000 rpm, 10 min, 0–5 °C) and decanted. This procedure was repeated three times yielding poly(N-methyl-DL-alanine) (45 mg, 71%) as a colorless solid.
Results and discussion
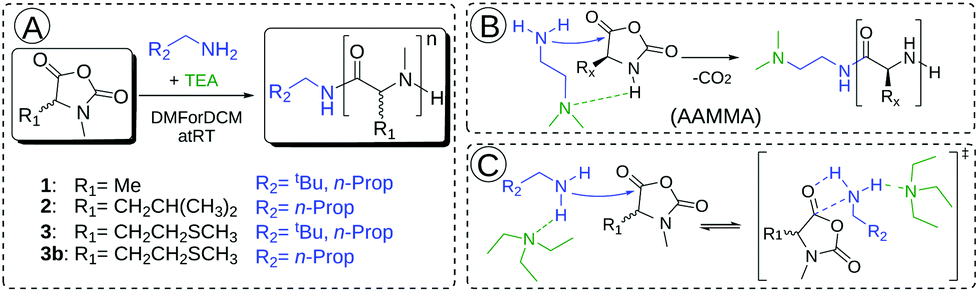
The synthesis of αNNCAs starts with the synthesis of the corresponding amino acids. N-methyl-L-methionine and N-methyl-L-leucine were prepared via a reductive ring-opening reaction using the corresponding Fmoc-protected 5-oxazolidinone according to literature procedures30,31 and in the last step the Fmoc-protecing group was removed. N-Methyl-DL-alanine was synthesized starting from 2-bromopropanoic acid and methylamine.32 The synthesis of α-substituted and N-methylated N-carboxy anhydrides (αNNCAs) was performed by suspending the corresponding N-methylated amino acids in dry THF and adding diphosgene at room temperature (Fig. 1A). After the suspension became clear, it was cooled down with an ice bath and anhydrous TEA was added slowly to precipitate the formed HCl, followed by filtration. In the case of N-methyl-L-leucine (NMLeu-NCA), addition of TEA was essential for ring closure, due to the reduced reactivity of the intermediate N-carbamoyl chloride. | ||
| Fig. 1 Synthetic route for the preparation of α-substituted and N-methylated N-carboxy anhydrides (structures 1, 2, 3 and 3b) using the Fuchs–Farthing method33,34 (A). 1H-NMR spectra of N-methyl-DL-alanine NCA (NMAla-NCA) (B), N-methyl-L-leucine NCA (NMLeu-NCA) (C) and N-methyl-L-methionine NCA (NMMet-NCA) (D) in CDCl3. Crystal structures via X-ray diffraction of NMAla-NCA and NMMet-NCA are illustrated in the corresponding NMR spectra. | ||
THF was removed under reduced pressure and the crude product was dissolved in a minimum amount of dry ethyl acetate. Addition of an excess amount of the n-hexane/diethyl ether mixture (1:1 by volume) caused the precipitation of αNNCAs, which could be collected by filtration under an inert atmosphere. The dried αNNCAs were characterized via1H-NMR spectroscopy (Fig. 1B–D). In addition, a small amount of each αNNCA was crystallized for characterization by X-ray diffraction. Crystal structures were obtained for all samples except for the N-methyl-L-leucine NCA, for which we were unable to obtain suitable crystals. In the next step, the ring-opening polymerization of the prepared αNNCAs (Fig. 2A) was carried out under various conditions, which are presented in Table 1.
 | ||
| Fig. 2 Polymerization of αNNCAs 1, 2, 3 and 3b in DMF (DCM) using different initiators (n-butyl amine and neopentyl amine) (A). Proposed accelerated amine mechanism through monomer activation (AAMMA) (B). Proposed mechanism for initiator activation caused by the added tertiary amine (TEA) (C).35 | ||
| Sample | Solvent | Initiator | M/I | Added base |
|---|---|---|---|---|
| NMAla-P1 | DMF | NPA | 25 | — |
| NMAla-P2 | DMF | NPA | 50 | — |
| NMAla-P3 | DMF | nBu | 50 | — |
| NMLeu-P1 | DMF | nBu | 50 | TEA |
| NMMet-P1 | DMF | NPA | 50 | DIPEA |
| NMMet-P2 | DMF | NPA + DABCO | 25 | — |
| NMMet-P3 | DMF | nBu | 25 | TEA |
| NMMet-P4 | DCM | NPA | 50 | TEA |
| DL-NMMet-P1 | DMF | nBu | 25 | TEA |
Monomer conversion could be monitored by FT-IR spectroscopy, since the intensities of the carbonyl vibration bands at 1786 and 1854 cm−1 are directly related to the remaining NCA concentration. Only the ROP of the N-methyl-DL-alanine NCA (NMAla-NCA) could be carried out to full conversion without the addition of a tertiary amine, whereas the polymerization of N-methyl-(D)L-methionine (NMMet-NCA, DL-NMMet-NCA) and NMLeu-NCA required equimolar amounts of a base (relative to the initiator) until initiation could be detected and reached full conversion after several days.
Complete conversion was confirmed by FT-IR spectroscopy and is exemplarily shown with NMAla-P1 in Fig. S1.† Due to this observation, the possible protonation of the initiator by residual HCl was tested by the reaction with silver nitrate solution (AgNO3). Therefore, all the used monomers, initiators and solvents were treated separately in relevant concentrations with AgNO3 solution. Since all samples showed no precipitation of AgCl, the residual HCl content could be neglected. Recently, Hadjichristidis and co-workers proposed the accelerated amine mechanism through monomer activation (AAMMA) for the ROP of NCAs, which is a combination of the normal amine mechanism (NAM) and the activated monomer mechanism (AMM) (Fig. 2B). They observed increased reaction kinetics by the use of initiators having primary and tertiary amines.35 However, monomer activation like in AMM or AAMMA is not possible for αNNCAs, due to N-methylation. Therefore, it is most likely that addition of a tertiary amine enhances only the reactivity of the attacking nucleophile due to coordination effects (Fig. 2C).36 In this regard, a possible reduced reactivity caused by the steric hindrance of the initiator was investigated in more detail. Both neopentyl amine and the less bulky n-butyl amine showed no reaction with the N-methylated NCAs of (D)L-methionine and L-leucine, unless equimolar amounts of TEA, DIPEA or DABCO were added to the polymerization system.
However, steric demand in general seems to be a critical factor for the ROP of αNNCAs, since NMAla-NCA could be initiated by both NPA and nBu, without the need for tertiary amine addition. Therefore, polymerization was carried out under various conditions and the resulting polymers were analyzed via HFIP-GPC (Fig. 3). The HFIP-GPC traces of NMAla-P1 to P3 showed an overall elution volume that is higher than expected when compared to NPA initiated polysarcosine (pSar) with a degree of polymerization (DP) of 50. Due to the same monomer to initiator ratios (M/I = 50) used for the polymerizations of NMAla-NCA and Sar-NCA together with comparable solubility in HFIP, the elution volumes were expected to be similar. Therefore, the discrepancy between elugrams is caused by differences in the DP of the samples NMAla-P1 to P3.
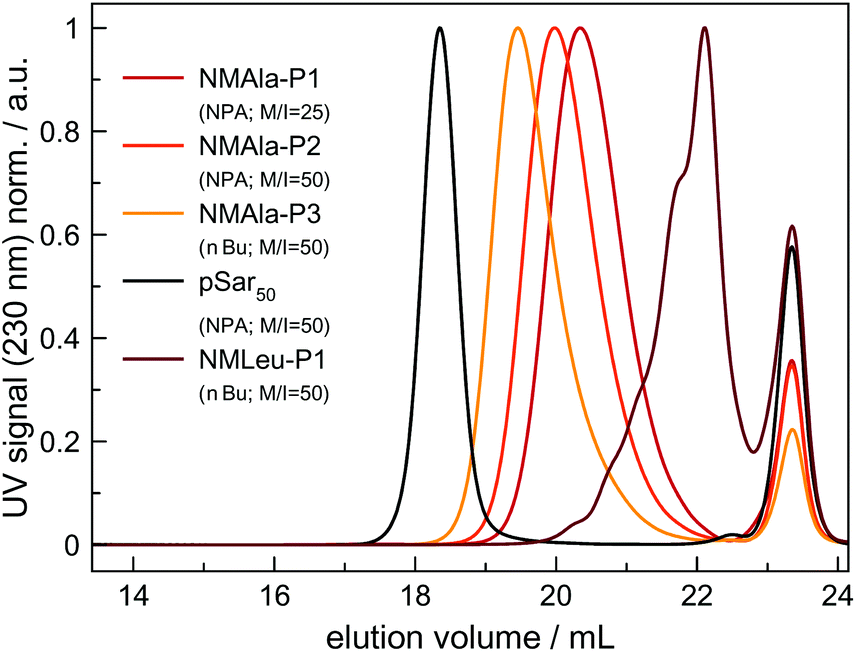 | ||
| Fig. 3 HFIP-GPC elugrams of NMAla-P1 to P3, NMLeu-P1 and polysarcosine for reference. For all the samples, toluene was used as the internal standard (peak at 23.4 mL) and polymerizations were carried out in anhydrous DMF. | ||
This means that a methyl group in the α-position is already decreasing the polymerization efficiency if a methyl substituent on the N atom is also present. In addition, the use of the less bulky n-butyl amine as the initiator leads to an increased degree of polymerization compared to initiation by neopentyl amine, when using similar M/I ratios. However, the steric demand of the side chain in the α-position has a stronger impact on the polymerization compared to the initiator itself. The GPC analysis of NMLeu-P1 leads to the suggestion that the polymerization of NMLeu-NCA is tremendously hampered and only results in the production of low molecular weight oligomers. The multiple shoulders at lower elution volumes with decreasing intensities can be explained by the presence of different oligomers. The GPC results underline the reduced reactivity of the β-C branched NMLeu-NCA towards a nucleophilic attack of a primary amine. For further analysis, the samples NMAla-P1 and P3 were characterized via MALDI-ToF mass spectrometry (Fig. 4). The average DPs were determined relative to the most intensive peak and resulted in Xn,MALDI(NMAla-P1) = 16 and Xn,MALDI(NMAla-P3) = 29. Both spectra showed additional polymer distributions caused by water initiation, which is especially pronounced in the spectrum of NMAla-P1. In this case, initiation by traces of water seems to be more effective than initiation by neopentyl amine, given the higher intensity of water-initiation associated peaks compared to peaks resulting from NPA-initiation. If the less bulky n-butyl amine is used as an initiator, the H2O-initiation related peaks decrease in intensity. These effects can also be observed in the NMR spectra of NMAla-P1 (Fig. S2†) and NMAla-P3 (Fig. S3†). The different amounts of the water initiated polymer lead to a higher discrepancy between the used M/I ratio and the DP determined via end group analysis by NMR at NMAla-P1 and a less pronounced discrepancy at NMAla-P3. The discrepancy is due to a reduction in the intensity for the initiator NMR signal compared to the polymer backbone NMR signal, caused by water initiation.
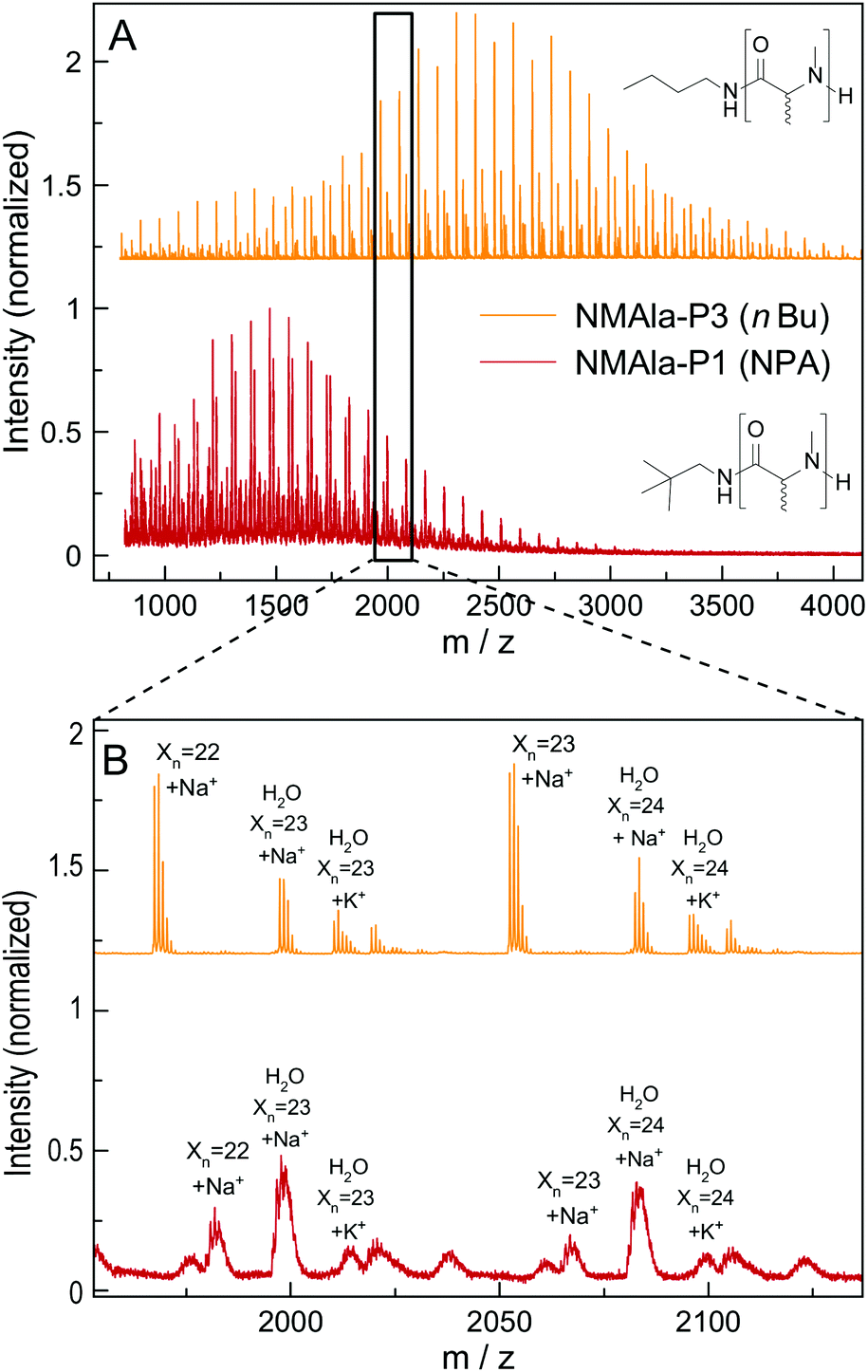 | ||
| Fig. 4 Full MALDI-ToF spectra of NMAla-P1 (red) and NMAla-P3 (orange) (A) and the corresponding magnifications (B) with assignment of the individual peaks. | ||
MALDI-ToF analysis thus clearly reveals that NMAla-NCA polymerization can be initiated by a primary amine, but the polymerization still suffers from the enhanced steric demand of the NCA monomer. The use of less bulky initiators helps to improve the initiation step, but does not seem to overcome the reduced reactivity towards a nucleophilic attack due to the high steric demand of the monomer. Our findings regarding the preparation of poly(N-methyl-DL-alanine) are in line with the results reported by Endo et al.23 They also obtained polymers with lower DPs as calculated from the initiator to monomer ratio, although they used a different approach for the preparation of the polypeptoids. In their protocol, the NCA was formed in situ, which can also contribute to the observed reduced degrees of polymerization, because impurities are not removed and could thus interfere with the polymerization.
To get a deeper insight into how the side chain influences the polymerization rate, the ROP of NMMet-NCA was performed under various conditions. The resulting polymers were characterized by HFIP-GPC (see Fig. 5). In all cases, the HFIP-GPC elugrams show the same trend as already observed for the NMAla-based experiments. A comparison of the N-methylated derivatives with the unmethylated derivatives of poly-L-methionine reveals that all polymerizations resulted in lower molecular weights as expected based on the monomer to initiator ratio. In analogy to the ROP of NMAla-NCA, the use of the less bulky initiator n-butyl amine slightly improved the obtained degrees of polymerization, but in contrast the addition of a tertiary amine was needed for the efficient initiation of NMMet-NCA. Note that the polymerization of NMMet-NCA proceeds significantly better than the ROP of NMLeu-NCA (see Fig. 3, red-brown curve), even though the side chains at the α-position are of comparable steric demand.
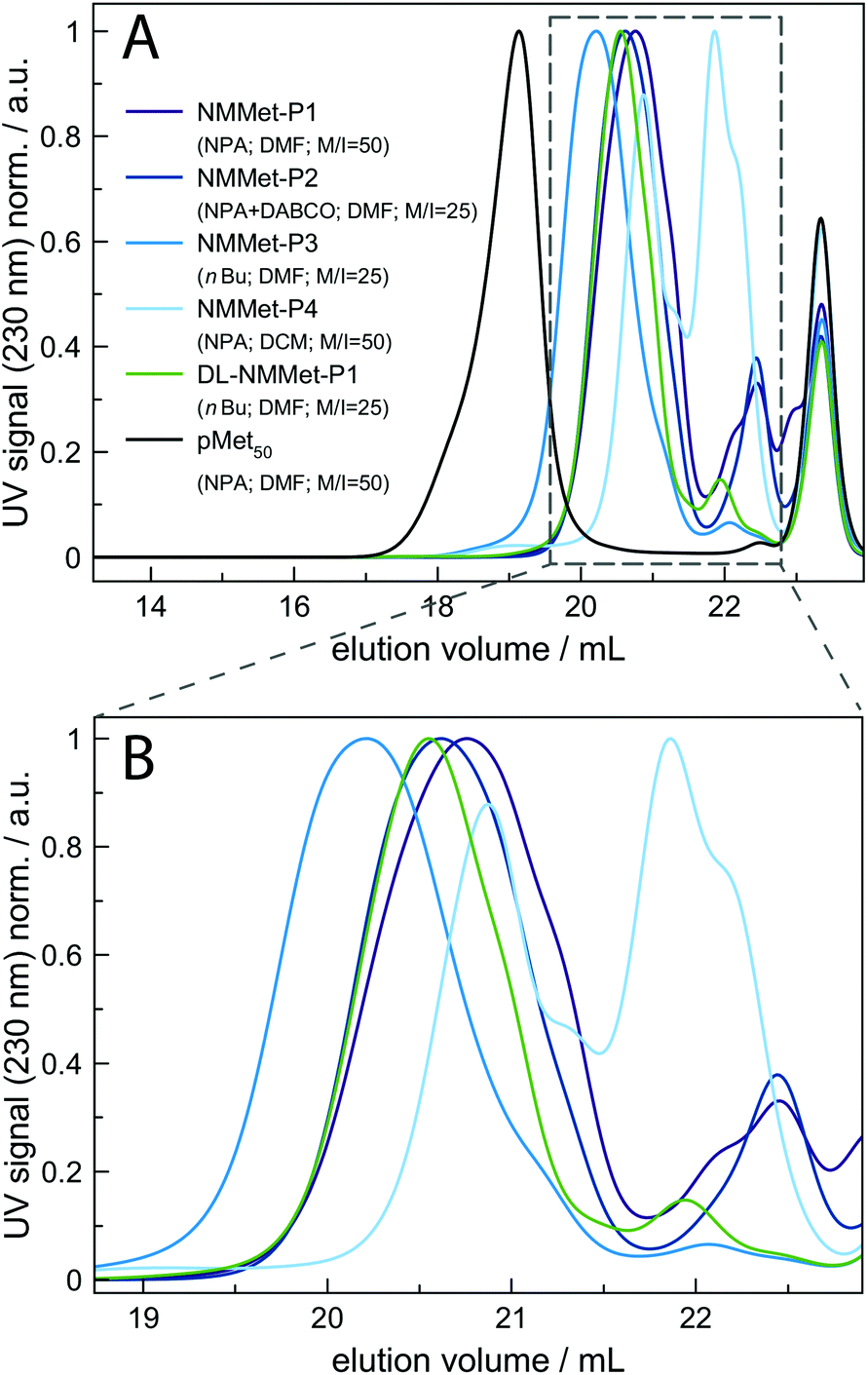 | ||
| Fig. 5 HFIP-GPC elugrams of NMMet-P1 to P4, DL-NMMet-P1 and poly-L-methionine for reference (A). For a better overview of the NMMet samples, the elution volume between 19 and 23 mL is magnified (B). For all the samples, toluene was used as the internal standard (peak at 23.4 mL). Bottom: Full MALDI-ToF spectra of NMMet-P1 (lower, purple), NMMet-P2 (middle, dark blue) and NMMet-P3 (upper, light blue) (A) and the corresponding magnifications (B) with assignment of the individual peaks. | ||
Very recently, Ling and co-workers have shown in a detailed DFT study that linear and γ-C branched N-substituents lead to similar reaction barriers, regarding the calculated Gibbs free energies for nucleophile addition.17 Given the observed differences in polymerization efficiency between NMLeu-NCA and NMMet-NCA, we conclude that branching in the side chains of α-substituted and N-methylated αNNCAs decreases the carbonyl reactivity much more compared to branching in the aliphatic side chains of N-alkylated and α-unsubstituted NCAs.
For additional insights, the samples NMMet-P1 to NMMet-P3 were analyzed by MALDI-ToF and the comparison of all three is shown in Fig. 6. In the mass spectra of NMMet-P1 and P2, most peaks are related to initiation by water. This trend could already be observed in the MALDI-ToF spectrum of NMAla-P1 (Fig. 4) and shows again that the steric demand of NPA presumably causes inefficient initiation. In comparison, initiation by the less bulky n-butyl amine yields polymers with a higher molecular mass, detectable by MALDI-ToF (Fig. 6) and HFIP-GPC (Fig. 5). The DP assignment of NMMet-P3 was performed using the most intense, initiator related peak and resulted in Xn,MALDI (NMMet-P3) = 8. This value has to be taken with caution though, since the mass spectrum is biased towards lower m/z ratios, due to a considerable mass discrimination effect.37
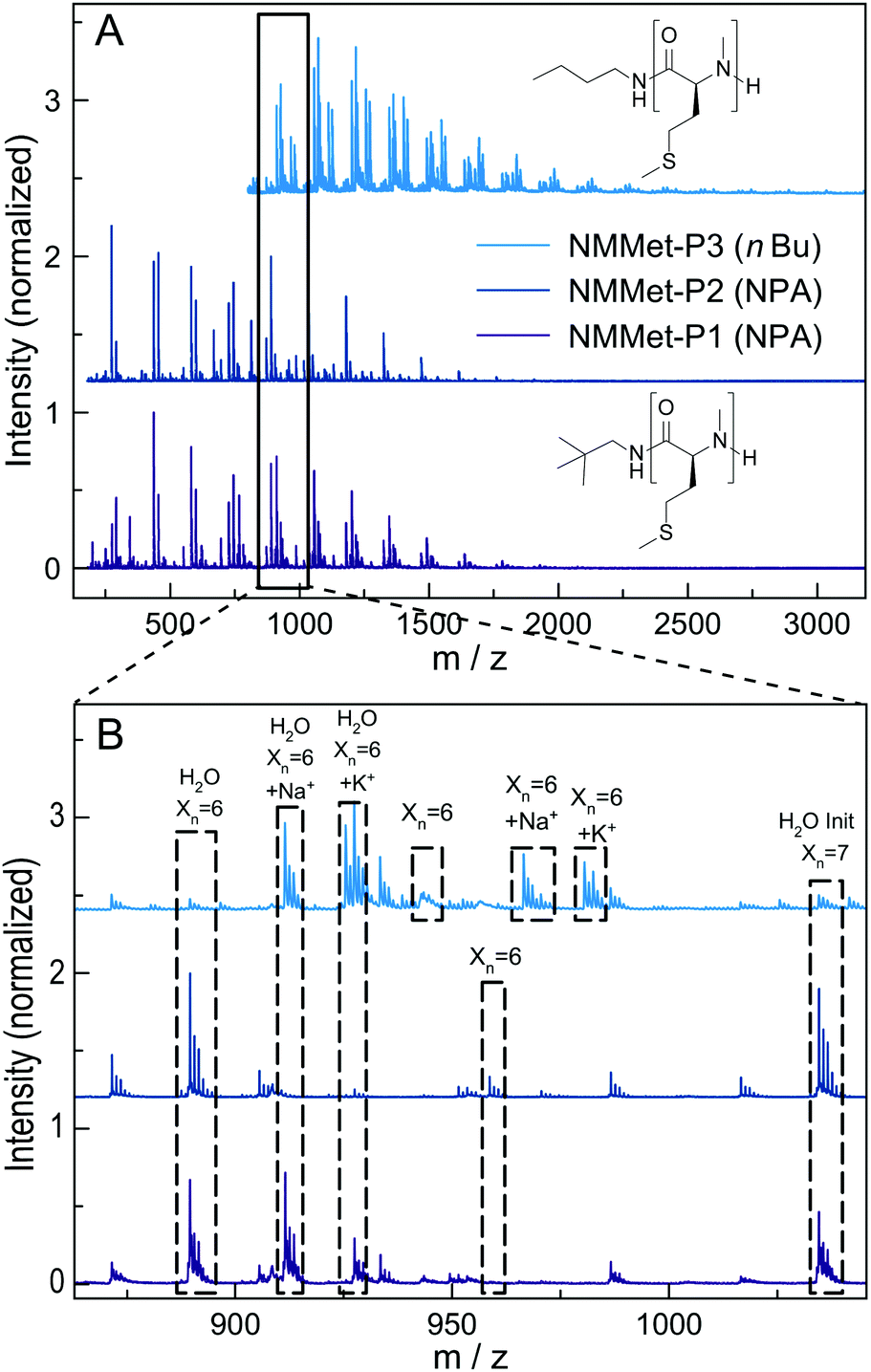 | ||
| Fig. 6 Full MALDI-ToF spectra of NMMet-P1 (lower, purple), NMMet-P2 (middle, dark blue) and NMMet-P3 (upper, light blue) (A) and the corresponding magnifications (B) with assignment of the individual peaks. | ||
To further support our findings and validate our explanation, coupled cluster (CC) calculations of the LUMO (lowest unoccupied molecular orbital) energies at the DLPNO-CCSD(T) level of theory with the cc-pVDZ basis set were performed to check for a possible electronic influence of the N-methyl substituents but the results were inconclusive. The CC method was chosen over DFT because the former allows a comparison of the calculated single point energies of different molecules and also delivers highly accurate results.38,39 In Fig. 7 the calculated frontier orbitals of NMMet-NCA, Met-NCA and ethylamine are illustrated and also the relative energy of the individual orbitals is provided.
 | ||
| Fig. 7 Frontier orbitals of NMMet-NCA (LUMO), Met-NCA (LUMO) and ethylamine (HOMO) and the corresponding relative energies, calculated using coupled clusters (CC: DLPNO-CCSD(T)/cc-pVDZ). | ||
Based on the Klopman–Salem theory, an interaction is favored if the related orbitals are similar in geometry and energy level.40,41 Based on the differences in frontier orbital energy levels, a higher reactivity would be expected in the case of NMMet-NCA with a primary amine, since the LUMO of NMMet-NCA has a smaller energy difference to the HOMO of ethyl amine (|ΔE| = 14.10 eV) compared to the LUMO of Met-NCAQ7(|ΔE| = 14.31 eV. However, this trend disagrees with the observed reactivities of the NCAs. Since the reduced reactivity does not seem to be a consequence of energetic properties, we conclude that the increased steric hindrance dominates and is the decisive factor for the decreased polymerization kinetics upon the N-methylation of α-substituted NCAs. The results presented so far, generated by experimental observations and GPC and MALDI-ToF analyses, showed decreased polymerization efficiency and lower degrees of polymerizations with increasing steric demand of the initiator and the substituents in the α-position of the applied NCA monomer. The reduced reactivity is also reflected in the need for the addition of a base (TEA) in order to start and promote the polymerization. These findings can be related exclusively to an enhanced steric hindrance, since an energetic contribution could be excluded by the comparison of the involved frontier orbitals, which were calculated using CC.
Besides differences in the polymerization rates, we also expected differences in the solution properties between polypeptides and poly(N-methylated α-amino acids), because N-methylation is known to suppress the formation of secondary structures of polypeptides due to the lack of intramolecular hydrogen bonding. However, hydrophobic side chains are still present and can contribute to interactions of the individual chain segments and domains. The use of enantiopure monomers may therefore lead to secondary structure formation, which can be seen in the CD spectrum of NMMet-P3 (Fig. 8).
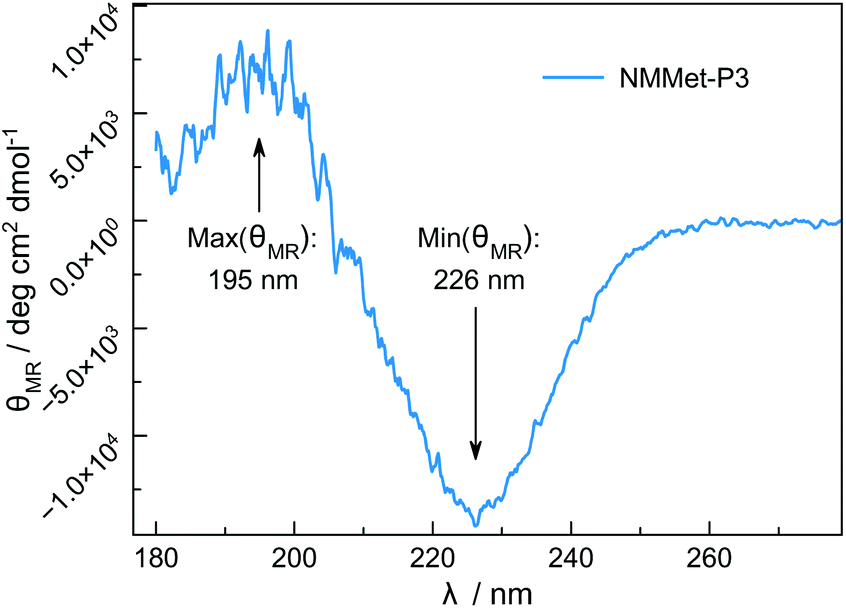 | ||
| Fig. 8 CD spectrum of NMMet-P3 in HFIP (c = 0.25 g L−1). | ||
The CD pattern of NMMet-P3 in HFIP shows a broad symmetric negative band at 226 nm and a positive band at 195 nm. The results have strong similarities to previous studies by Cosani et al. where they observed similar values (min(θMR) = 228 nm; max(θMR) = 196 nm) for poly(N-methyl-γ-methyl-L-glutamate) and poly(N-methyl-γ-ethyl-L-glutamate) in TFE.25 Along with conformational studies on poly(N-methyl-L-alanine), carried out by Goodman et al., the authors proposed a right-handed helix as the secondary structure.18 Despite a small blue shift of 1–2 nm for both bands (positive and negative), our observations match the results obtained by Cosani et al. Therefore, it is most likely that poly(N-methyl-L-methionine) also forms a helical secondary structure. Since N-methylation entails the loss of H-bond formation, the resulting structure has an increased hydrophobic character. The use of polar solvents, like DMF, could in consequence cause the formation of hydrophobic domains, due to insufficient solubilization of the polymer chains. In this regard, the polymerization of NMMet-NCA was further carried out in DCM to examine the influence of the solvent on the reaction properties. As can be seen in Fig. 4 (turquoise curve), the use of a less polar solvent, compared to DMF, has no benefit on the resulting polymerization and instead causes a bimodal distribution in HFIP-GPC.
Conclusions
In the present work, the influence of N-methylation on the polymerization properties of α-substituted N-carboxy anhydrides (NCAs) was investigated. To this end, different α-substituted and N-methylated NCAs (αNNCAs) were prepared and polymerized under various conditions. Next to the increasingly popular NCA polymerization of N-methylated glycine (sarcosine), and few reports using N-methylated alanine, we were intrigued that a strategy to polymerize αNNCAs had not been reported. We were able to demonstrate that the steric demand of both the initiator and side chain of the monomer has a major impact on the achievable degree of polymerization of the resulting polymer. The steric hindrance of the side chain in the α-position of the monomer is recognized as the major contribution to the low reactivity towards a nucleophilic attack by an amine. This strongly influences the initiation step of the polymerization and achievable chain lengths. Using the less bulky n-butyl amine compared to neopentyl amine resulted in more efficient initiation and propagation. Polymerization also benefitted from a heteroatom with free electron pairs in the side chain of the αNNCA, as shown with N-methyl-L-methionine. On the other hand, solvent polarity did not seem to influence the polymerization of αNNCAs. Moreover, the synthesized poly(N-methyl-α-amino acids) tend to form helical secondary structures in solution, which is in agreement with previously reported investigations using poly(N-methyl-L-alanine).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank O. Stach for his help with the synthesis of N-methyl-L-methionine, Dr D. Schollmeyer for X-ray crystal structure analysis and S. Türk for MALDI-ToF MS measurements. Parts of this research were conducted using the supercomputer Mogon and/or advisory services offered by Johannes Gutenberg University Mainz (hpc.uni-mainz.de), which is a member of the AHRP (Alliance for High Performance Computing in Rhineland Palatinate, http://www.ahrp.info) and the Gauss Alliance e.V. The authors gratefully acknowledge the computing time granted on the supercomputer Mogon at Johannes Gutenberg University Mainz (hpc.uni-mainz.de). L. Z. acknowledges the financial support from the Evonik Foundation (Werner Schwarze Scholarship). C. M., P. B., T. O., and M. B. would like to thank the German Research Foundation (DFG) for financial support (CRC 1066-2).Notes and references
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS.
- A. Sharma, A. Kumar, S. A. H. Abdel Monaim, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Biopolymers, 2018, 109, e23110 CrossRef.
- E. Biron, J. Chatterjee, O. Ovadia, D. Langenegger, J. Brueggen, D. Hoyer, H. A. Schmid, R. Jelinek, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2008, 47, 2595–2599 CrossRef CAS.
- P. P. Bose, U. Chatterjee, I. Hubatsch, P. Artursson, T. Govender, H. G. Kruger, M. Bergh, J. Johansson and P. I. Arvidsson, Bioorg. Med. Chem., 2010, 18, 5896–5902 CrossRef CAS.
- Q. G. Dong, Y. Zhang, M. S. Wang, J. Feng, H. H. Zhang, Y. G. Wu, T. J. Gu, X. H. Yu, C. L. Jiang, Y. Chen, W. Li and W. Kong, Amino Acids, 2012, 43, 2431–2441 CrossRef CAS.
- S. Sagan, P. Karoyan, O. Lequin, G. Chassaing and S. Lavielle, Curr. Med. Chem., 2012, 11, 2799–2822 CrossRef.
- Z. Song, Z. Tan and J. Cheng, Macromolecules, 2019, 52, 8521–8539 CrossRef CAS.
- A. Rasines Mazo, S. Allison-Logan, F. Karimi, N. J. A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC.
- B. A. Chan, S. Xuan, A. Li, J. M. Simpson, G. L. Sternhagen, T. Yu, O. A. Darvish, N. Jiang and D. Zhang, Biopolymers, 2017, 1–25 Search PubMed.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS.
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS.
- R. Luxenhofer, C. Fetsch and A. Grossmann, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2731–2752 CrossRef CAS.
- K. Klinker and M. Barz, Macromol. Rapid Commun., 2015, 36, 1943–1957 CrossRef CAS.
- Z. Song, Z. Han, S. Lv, C. Chen, L. Chen, L. Yin and J. Cheng, Chem. Soc. Rev., 2017, 46, 6570–6599 RSC.
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC.
- M. Sisido, Y. Imanishi and T. Higashimura, Makromol. Chem., 1977, 178, 3107–3114 CrossRef CAS.
- T. Bai and J. Ling, Biopolymers, 2019, 110, e23261 CrossRef.
- M. Goodman and M. Fried, J. Am. Chem. Soc., 1967, 89, 1264–1267 CrossRef CAS.
- J. E. Markt and M. Goodman, Biopolymers, 1967, 5, 809–814 CrossRef.
- M. Goodman, F. Chen and F. R. Prince, Biopolymers, 1973, 12, 2549–2561 CrossRef CAS.
- A. M. Liquori and P. De Santis, Biopolymers, 1967, 5, 815–820 CrossRef CAS.
- S. Yamada, K. Koga, A. Sudo, M. Goto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3726–3731 CrossRef CAS.
- Y. Shiraki, S. Yamada and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1674–1679 CrossRef CAS.
- A. Cosani, M. Palumbo, M. Terbojevich and E. Peggion, Macromolecules, 1978, 11, 1041–1045 CrossRef CAS.
- A. Cosani, M. Terbojevich, M. Palumbo, E. Peggion and M. Goodman, Macromolecules, 1979, 12, 875–877 CrossRef CAS.
- I. Z. Steinberg, W. F. Harrington, A. Berger, M. Sela and E. Katchalski, J. Am. Chem. Soc., 1960, 82, 5263–5279 CrossRef CAS.
- M. Gkikas, H. Iatrou, N. S. Thomaidis, P. Alexandridis and N. Hadjichristidis, Biomacromolecules, 2011, 12, 2396–2406 CrossRef CAS.
- L. Aurelio, R. T. C. Brownlee and A. B. Hughes, Chem. Rev., 2004, 104, 5823–5846 CrossRef CAS.
- M. L. Di Gioia, A. Leggio, F. Malagrinò, E. Romio, C. Siciliano and A. Liguori, Mini-Rev. Med. Chem., 2016, 16, 683–690 CrossRef CAS.
- R. M. Freidinger, J. S. Hinkle, D. S. Perlow and B. H. Arison, J. Org. Chem., 1983, 48, 77–81 CrossRef CAS.
- L. Aurelio, J. S. Box, R. T. C. Brownlee, A. B. Hughes and M. M. Sleebs, J. Org. Chem., 2003, 68, 2652–2667 CrossRef CAS.
- J. Etxabe, J. Izquierdo, A. Landa, M. Oiarbide and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 6883–6886 CrossRef CAS.
- A. C. Farthing, J. Chem. Soc., 1950, 3213–3217 RSC.
- C. J. Brown, D. Coleman and A. C. Farthing, Nature, 1949, 163, 834–835 CrossRef CAS.
- W. Zhao, Y. Gnanou and N. Hadjichristidis, Chem. Commun., 2015, 51, 3663–3666 RSC.
- J. Liu and J. Ling, J. Phys. Chem. A, 2015, 119, 7070–7074 CrossRef CAS.
- K. Martin, J. Spickermann, H. J. Räder, K. Müllen and J. Grotemeyer, Rapid Commun. Mass Spectrom., 1996, 10, 1471–1474 CrossRef CAS.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef.
- D. G. Liakos, Y. Guo and F. Neese, J. Phys. Chem. A, 2020, 124, 90–100 CrossRef CAS.
- L. Salem, J. Am. Chem. Soc., 1968, 90, 543–552 CrossRef CAS.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1976455 and 1976456. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py01055c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |