
Time-dependent covalent network formation in extrudable hydrogels†
 a
and
Alshakim
Nelson
*a
a
and
Alshakim
Nelson
*a
Abstract
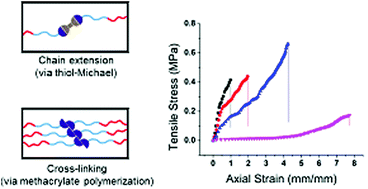
The fabrication of hydrogel materials has gained increased attention for a broad range of biomedical and biotechnological applications. However, one longstanding challenge in the field is to develop hydrogels that can be easily processed into the desired form factor, while achieving the necessary final physical and biochemical properties. Herein, we report a shear-thinning hydrogel ink that can be photo-cured to create a stretchable, suturable hydrogel whose polymer network is formed via the combination of thiol-Michael addition and radical polymerization. A shear-thinning hydrogel based on bis-methacrylated Pluronic® F-127 was modified with varying equivalents of 2,2′-(ethylenedioxy)diethanethiol (EDT) as an additive. We observed that aging the hydrogel over time prior to extrusion allowed the relatively slow thiol-Michael addition to occur (between thiol and methacrylate) prior to UV initiated photopolymerization of the methacrylates. The viscoelastic properties of these hydrogels could be tuned based on the amount of EDT added, and the aging time of the hydrogel formulation. The changes to the physical properties of the hydrogels were attributed to the increased chain length between network junctions that resulted from the thiol-Michael addition reactions. The optimized hydrogel composition was then extruded from a coaxial nozzle to produce hydrogel tubes that, after curing, were resistant to tearing and were suturable. These extrudable synthetic hydrogels with tunable viscoelastic properties are promising for tissue engineering applications and as surgical training models for human vasculature.
- This article is part of the themed collection: Polymer Chemistry Pioneering Investigators 2021
 Please wait while we load your content...
Please wait while we load your content...

