
Immune-modulatory alginate protects mesenchymal stem cells for sustained delivery of reparative factors to ischemic myocardium†
Ravi K.
Ghanta‡
*a,
Samira
Aghlara-Fotovat‡
b,
Aarthi
Pugazenthi
a,
Christopher T.
Ryan
a,
Vivek P.
Singh
a,
Megumi
Mathison
a,
Maria I.
Jarvis
b,
Sudip
Mukherjee
b,
Andrea
Hernandez
b and
Omid
Veiseh
 *b
*b
aMichael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX, USA. E-mail: Ravi.Ghanta@bcm.edu; Tel: +1 713 873 3435
bDepartment of Bioengineering, Rice University, Houston, TX, USA. E-mail: omid.veiseh@rice.edu; Tel: +1 713 348 3082
First published on 11th August 2020
Abstract
Paracrine factors secreted by mesenchymal stem cells (MSCs) have been previously shown to improve cardiac function following acute myocardial infarction (MI). However, cell therapy activates the innate immune response, leading to the rapid elimination of transplanted cells and only short-term therapeutic delivery. Herein, we describe a new strategy to deliver sustained paracrine-mediated MSC therapy to ischemic myocardium. Using an immune evasive, small molecule modified alginate, we encapsulated rat MSC cells in a core–shell hydrogel capsule and implanted them in the pericardial sac of post-MI rats. Encapsulated cells allowed diffusion of reparative paracrine factors at levels similar to non-encapsulated cells in vitro. Encapsulation enabled sustained cell survival with localization over the heart for 2 weeks. The effect of the experimental group on ventricular function and fibrosis was compared with blank (cell free) capsules and unencapsulated MSCs injected into infarcted myocardium. MSC capsules improved post-MI ventricular function ∼2.5× greater than MSC injection. After 4 weeks, post-MI fibrosis was reduced ∼2/3 with MSC capsules, but unchanged with MSC injection. MSC encapsulation with alginate core–shell capsules sustains cell survival and potentiates efficacy of therapy.
1. Introduction
Preservation and restoration of ventricular function are the primary goals for heart failure therapies. Cellular therapy, the introduction of autologous or allogenic cells into ischemic or infarcted myocardium, has produced encouraging pre-clinical improvements in ventricular function.1,2 However, clinical studies have yielded modest and inconsistent results, questioning therapeutic utility.3,4 Although it was initially believed that cell therapy would improve heart function by transplanted cell differentiation into cardiomyocytes, the primary therapeutic benefit is now believed to be paracrine-mediated myocardial repair.5,6 Mesenchymal stem cells (MSCs) release potent angiogenic, mitogenic, and anti-fibrotic factors and are available through allogenic sources for practical clinical application.7 Consequently, MSCs have been studied in randomized clinical trials for the treatment of myocardial ischemia with noted improvements in ventricular function and remodeling associated with heart failure.8,9The optimal delivery method for cellular therapy to the heart remains undetermined, with intravenous injection, catheter-directed intracoronary delivery and direct intramyocardial injection as techniques utilized in most prior studies.10 Pericardial delivery of cell therapy, through percutaneous pericardial injection or deposition during cardiac surgery, represents a promising method to deliver reparative proteins to the heart.11–13 However, as fewer than 1% of cells survive hours after implantation, poor cell survival represents a continuing limitation, regardless of cell type or delivery strategy.14–19 Epicardial placement removes the transplanted cells from the hostile ischemic and inflamed environment of a myocardial infarction (MI), but may still permit paracrine mediated repair.
Strategies to improve cell survival beyond a few days are paramount to improve reproducibility and efficacy of therapy for successful clinical translation.20 Encapsulation of cells with biomaterials holds promise to improve cell viability, with prior groups utilizing alginate, polyethylene glycol, and hyaluronic acid to fabricate cellular gels or patches.21–23 We have previously developed a novel immune modulatory alginate (triazole-(triazole-thiomorpholine dioxide [TMTD] alginate) for cell encapsulation which significantly reduces foreign body response and pericapsular fibrotic overgrowth while permitting diffusion of nutrients and cytokines.24,25 The thiomorpholine functional group preferentially localizes to the capsule surface and significantly reduces macrophage adherence and subsequent activation.26 We have previously demonstrated preserved survival and function of TMTD alginate-encapsulated islet cells for over 6 months in the peritoneal cavity of immune competent rodents and non-human primates.24,25,27
We hypothesize that TMTD alginate can be utilized to create MSC capsules that can be implanted in the pericardial sac, with sustained cell survival and delivery of reparative paracrine factors to improve ventricular function. In this study, we fabricated TMTD alginate-MSC capsules and verified sustained paracrine factor production in vitro. We then demonstrated sustained survival and pericardial localization in vivo. Finally, we evaluated efficacy in a clinically relevant post-infarct rat model. Our studies suggest that our immune modulatory alginate encapsulation platform can be used in the pericardial space to provide enhanced therapeutic efficacy to the heart.
2. Materials and methods
2.1. Study overview and experimental design
In this study, we first designed and fabricated TMTD alginate 1.5 mm microcapsules with 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MSCs per capsules. Viability and elution of paracrine growth factors (Vascular Endothelial Growth Factor [VEGF]; adrenomedulin [ADM]; and Hepatocyte Growth Factor [HGF]) from MSC cell capsules were verified in vitro over 14 days using enzyme-linked immune-sorbent assays (ELISA) (Cat# ERVEGFA, ThermoFisher Scientific, Cat# 50-150-1296, Biomatik Corporation, Cat# EKU10058, Biomatik Corporation). MSC capsules were then implanted in the pericardium of Sprague Dawley rats (n = 3) overlying the right and left ventricles. Capsule localization was verified with In Vivo Imaging System (IVIS) and retrieved capsules were verified for viability after 14 days. Finally, MSC capsules were implanted in rats following acute MI by ligation of the left anterior descending (LAD) coronary artery. Efficacy of MSC capsules was assessed by echocardiography and histology over 28 days after MI and compared against blank capsule controls and MSC intramyocardial injection (n = 4–5 per group). For this study, all rats were male Sprague Dawley (175–199 g, 44–48 days, Envigo). Rat MSCs were utilized for in vitro characterization and in vivo efficacy after MI.
000 MSCs per capsules. Viability and elution of paracrine growth factors (Vascular Endothelial Growth Factor [VEGF]; adrenomedulin [ADM]; and Hepatocyte Growth Factor [HGF]) from MSC cell capsules were verified in vitro over 14 days using enzyme-linked immune-sorbent assays (ELISA) (Cat# ERVEGFA, ThermoFisher Scientific, Cat# 50-150-1296, Biomatik Corporation, Cat# EKU10058, Biomatik Corporation). MSC capsules were then implanted in the pericardium of Sprague Dawley rats (n = 3) overlying the right and left ventricles. Capsule localization was verified with In Vivo Imaging System (IVIS) and retrieved capsules were verified for viability after 14 days. Finally, MSC capsules were implanted in rats following acute MI by ligation of the left anterior descending (LAD) coronary artery. Efficacy of MSC capsules was assessed by echocardiography and histology over 28 days after MI and compared against blank capsule controls and MSC intramyocardial injection (n = 4–5 per group). For this study, all rats were male Sprague Dawley (175–199 g, 44–48 days, Envigo). Rat MSCs were utilized for in vitro characterization and in vivo efficacy after MI.
2.2. Materials
MSCs were purchased from AS One International, Inc. (Cat# RASMX-01001) Green-Fluorescent Protein (GFP) expressing MSCs (AS One International, Inc. Cat# #RASMX-01101) were also utilized for validation of sustained survival 14 days after implantation. In addition, we engineered wild type rat MSCs with Firefly Luciferase (F-LUC) using lenti-viral transfection (VectorBuilder) for IVIS image localization. Cell culture reagents were purchased from Thermo-Fisher Scientific. SLG20 alginate was purchased from Pronova. Syringe pumps, co-axial needles, and voltage generators were purchased from Harvard Apparatus, Ramé-Hart, and Gamma High Voltage. D-Luciferin was purchased from PerkinElmer. Calcein AM and calcein AM violet live cell stains were purchased from Thermo-Fisher Scientific (Cat# C3099 & C34858). Ethidium homodimer-1 was purchased from Thermo-Fisher Scientific (Cat# E1169). VEGF, HGF, and ADM ELISA kits were purchased from Fisher, and Biomatik Corporation. Unless otherwise noted, all chemicals are from Sigma Aldrich, USA and used without any further purification. MSC's were cultured in Dulbecco's Modified Eagle Medium (DMEM/F-12), supplemented with 10% embryonic stem cell fetal bovine serum, and 1% anti–anti. Cells were maintained at 37 °C in a 5% CO2 atmosphere.Sprague Dawley rats (175–199 g, 44–48 days) were purchased from Envigo and maintained under standard housing conditions. All animals were treated and cared for in compliance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication 86-23, revised 1996). The protocol was approved by the Institutional Animal Care and Use Committees.
2.3. Synthesis of TMTD
4-Propargylthiomorpholine 1,1-dioxide (1 eq.) was added to a 250 mL round bottom flask and dissolved in methanol:water mixture (5:1). Consequently, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (0.25 eq.), triethylamine (0.25 eq.), and copper iodide (0.1 eq.) was added. The reaction mixture was purged with argon for 15 min and cooled to 0 °C following which 11-azido-3,6,9-trioxaundecan-1-amine (1 eq., 6.30 g, 28.86 mmol) was added. The reaction mixture was stirred at room temperature for 15 min and afterward heated to 55 °C for overnight. The reactions was cooled to room temperature and filtered through Celite to remove any insoluble portion. The filtrate was dried using rotavap under reduced pressure with silica. The crude reaction was then purified by liquid chromatography with dichloromethane: ultra (22% MeOH in DCM with 3% NH4OH) mixture 0% to 40% on a 120 g ISCO silica column and further characterized with ESI mass and NMR mass spectroscopy.
NMR: 1H (600 MHz; CDCl3): 2.87 (2H, s, NH2–CH2–CH2–O), 3.05 (8H, m, N–CH2–CH2–S), 3.51 (2H, t, J = 5.2 Hz, NH2–CH2), 3.61 (8H, m, PEG) 3.81 (2H, s, thiomorpholine–CH2–triazole), 3.89 (2H, t, J = 5.2 Hz, N–CH2–CH2–O), 4.55 (t, 2H, J = 5.2, N–CH2–CH2–O), 7.69 (1H, s, triazole).
13C (600 MHz; CDCl3): 41.74 (NH2–CH2), 50.42 (N–CH2), 50.56 (N–CH2 thiomorpholine) 51.52 (S–CH2 thiomorpholine) 52.2 (thiomorpholine–CH2–triazole), 69.54–73.14 (m, PEG), 124.10 (CH triazole), 143.32 (C triazole).
ESI MS: [M + H+] = 392.1939.
2.4. Synthesis of TMTD modified UPVLVG alginate
In a round bottom flask, 2 g (1 eq.) of UPVLVG (BP-1903-04; Novamatrix) was dissolved in water (75 mL). Then TMTD small molecule (3.99 g, 10.20 mmol, 1 eq.) was dissolved in water under vortexing. The pH of TMTD solution is adjusted to 7.4 using HCl. The TMTD aqueous solution is then slowly added to the UPVLVG solution under stirring condition. Subsequently a solution of (4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride) (DMTMM, 0.5 eq.) was added dropwise to the mixture of UPVLVG and TMTD. The reaction was heated to 55 °C and stirred for overnight. The solution was filtered through a cyano-silica gel and dialyzed in a 40 cm long 10–12 K pretreated dialysis tubing in a beaker using saline (2 days) and Milli-Q water (3 days). The dialyzed solution was frozen in −80 °C and lyophilized until dry.NMR: 1H (600 MHz; D2O): 3.07 (4H, s, N–CH2–CH2–S), 3.17–3.40 (m, alginate protons), 3.46 (4H, s, N–CH2–CH2–S), 3.50–3.70 (16H, m, ethoxy), 3.7–5.2 (m, alginate protons), 8.08 (1H, s, triazole).
Elemental: C: 35.67%, H: 4.34%, N: 5.08%, O: 33.50%. By elemental analysis data there is 16.0% modification of the starting alginate.
2.5. Preparation of alginates for encapsulation
SLG20 alginate, used for the core of the core–shell hydrogels, was purchased from PRONOVA. It was dissolved in 0.8% saline at 1.4% weight per volume. SLG100 was prepared at 3% weight per volume in 0.8% saline. It was then blended with TMTD small molecule alginate prepared in section 2.4 above, using a 70:30 ratio of TMTD:SLG100.
2.6. Cell preparation
In order to suspend cells in alginate, cells that had reached 70–90% confluency were removed of supernatant and washed with 5 ml's of DPBS. Washing solution was aspirated and followed by 5 ml's Trpysin EDTA to disperse the cell layer. Cells were incubated for 3 minutes, then observed under a microscope to ensure detachment. 7 mL's of complete growth medium were added to the flask to stop the EDTA reaction. The cell suspension was transferred to a 50 ml conical tube and centrifuged for 5 minutes at 250g. The supernatant was aspirated, and the pellet was broken. The cells were washed 3 times with Ca2+ free Krebs solution and then centrifuged again. After centrifugation, the buffer was aspirated and the desired volume of SLG20 alginate was added to the cells and pulled up by a 5 ml luerlock syringe.2.7. Core–shell capsule synthesis
Following alginate and cell preparation, hydrogels were synthesized using a two fluid co-axial electrostatic spraying device which consisted of two syringe pumps and a voltage generator. Each of the pumps held a syringe, one containing SLG20 alginate with the cell suspension, and the other containing modified TMTD/SLG100 alginate. The syringes were connected using a co-axial needle that was suspended over a 1:4 barium chloride:mannitol crosslinking bath. The bath was supplemented with 0.65% Tween20. The pumps were set to run at 5 ml per hour and 6 ml per hour for the core and shell respectively. Capsule size was maintained using a voltage of ∼5.6 kV. Capsules incubated in the crosslinking bath for 15 minutes and were subsequently washed three times with HEPES buffer, followed by three times with complete media. Capsules were aliquoted into 1.5 mLs and stored at room temperature overnight. Prior to implant, samples were washed with 0.9% sterile saline. Unmodified control capsules were made using SLG20 alginate in the core and shell.
2.8. Viability testing
Following encapsulation, capsules were distributed into a 96 well plate at a density of 1 capsule per well. On days 1, 7, 14, and 28 capsules were washed with Ca+ KREBS and stained using 50 μl of live/dead stock solution containing 20 μl of 2 μM calcein AM and 40 μl of 4 μM EthD-1 in 8 mL DPBS. The capsules were incubated for 20 minutes at 37 °C following which, capsules were washed with PBS and fixed using 4% PFA for imaging. Capsules were imaged under a fluorescence microscope at 4× magnification, in the TexRed, GFP, and Transmitted channels. Images were processed in Adobe Photoshop.2.9. Enzyme-linked immunosorbent assay
Cells from each group were grown up into T-150 flasks in DMEM media with 10% embryonic stem cell-FBS, 1% anti–anti. At confluency, culture media was aspirated and 10 ml TrypLE was added. Cells were incubated for 3 minutes at 37 °C in a 5% CO2 humidified atmosphere. Once the cell layer was dispersed, 10 ml's complete media was added to stop the reaction. The cell suspension was transferred to a 50 ml conical tube and centrifuged at 250g for 5 minutes. The supernatant was aspirated and cells were re-suspended in 1 ml complete media. 10 μl of the cell suspension was mixed with 10 μl of trypanblue, and cell concentration was counted using a Countess™ hemocytometer. The volume required to achieve a concentration of 10000 cells per well was calculated based on the concentration of live cells in the sample. That volume was transferred into 8 wells of a 96 well plate. Of the remaining cell suspension volume, 10 million cells were resuspended in 1 ml alginate to be synthesized into core shell capsules as described below. Once fabricated, capsules were plated in 8 wells of a 96 well plate at a density of 1 capsule per well. All samples were incubated at 37 °C in a 5% CO2 humidified atmosphere. Media was changed regularly. At 24 hours and 30 days after plating, supernatant was collected from each well and frozen at −20 °C. ELISA's for VEGFA, HGF, and adrenomedullin were purchased from Thermo Scientific. Frozen samples were thawed and run on the kits according to protocols provided. All samples were run with an n = 5.
2.10. Capsule implantation & experiment MI model
Animals were anesthetized with inhalational isoflurane in 100% O2 (5.0% induction; 2.5% maintenance). Endotracheal intubation was performed and the animals ventilated with positive-pressure ventilation. A left lateral thoracotomy was performed. The pericardial sac was identified and a small portion opened sharply exposing the left ventricle at the intended LAD ligation site. For animals that underwent MI, the LAD was identified and ligated with a 7-0 poly-propylene suture (Ethicon, Somerville, New Jersey) 1–2 mm from its origin.28 Visual confirmation of infarction was noted by edema and discoloration of the anterior LV wall. MSC or blank cell capsules were then deposited on a sterile surgical towel and then deposited in the pericardial space on the epicardial surface of the right and left ventricle with surgical forceps. For each animal 1.5 mL of capsules were deposited. For animals in the MSC injection group, 0.2 cc (4 × 106 cells per rat) were injected into the left ventricle myocardium in the infarct zone using a 27 gauge syringe. Cells were injected obliquely and the needle was not withdrawn for 10 seconds to minimize chance for cell leakage. The chest was then sutured closed in layers and the animals were extubated and allowed to recover.2.11. In vivo imaging system imaging (IVIS)
In vivo MSC capsule localization was performed with IVIS (IVIS Lumina II, PerkinElmer) using engineered F-LUC + MSC cell capsules. Rats were anesthetized using 3.5% isoflurane and 1.5 L min−1 oxygen in an induction chamber. Once fully sedated, the chest was shaved and an intra-peritoneal injection of 100 mg kg−1 of 15 mg ml−1D-luciferin in saline. The rat was then imaged at 20, 40, and 60 minutes in field of view in D, binning set to 8, with an F stop set to 2 and manual subject height set to 1.70 cm. The exposure times selected were 60, 1, 5, and 15 seconds.2.12. Echocardiography
Echocardiography was performed under light anesthesia with 3% isoflurane using a FujiFilm Visualsonics Vevo 2100 Imaging System (VisualSonics, Canada). Echocardiography was conducted 3 days prior to surgery, 4 days after, and 4 weeks after infarct induction and capsule implant. Parasternal short-axis images were obtained at the level of papillary muscles and imaged using motion mode (M-Mode). Images were then analyzed by a blinded investigator. The formula (LVEDV − LVESV/LVEDV) × 100% was used to calculate the ejection fraction (EF). The change in EF from post-infarct to study termination was calculated as: [(EF at day 28 after surgery) − (EF at day 3 after surgery)]/(EF at day 3 after surgery).2.13. Capsule retrieval and histologic examination
At experiment termination, rats were anesthetized, and their chests were opened. The heart pericardial sac and both pleural spaces were widely opened. Capsules were identified and explanted. Retrieved capsules were rinsed in saline and examined under fluorescence microscopy. Images were processed in Adobe Photoshop (Adobe Inc., San Jose, California). For post-infarct subjects, animals were exsanguinated under deep anesthesia by an incision made in the inferior vena cava while beating hearts were perfused with 10 mL's of phosphate buffered saline (PBS pH 7.2) and fixed in a 4% paraformaldehyde solution. Each excised heart was cut transversally, and two slices (2–3 mm) were obtained, each above or below the transverse centerline of the infarct region. Tissues were then embedded in paraffin and cut into twelve 5 μm thick sections at 500 μm intervals. Six sections per animal was mounted on the glass slide and stained with Masson-trichrome where fibrotic area stains blue and non-fibrotic region stains red. Cytation 5 cell imaging multi-mode reader (BioTek instruments) at 10× magnification was used to obtain digital photographs of histological sections. All sections were quantified using ImageJ software version 1.52 (National Institutes of Health, Bethesda, Maryland). The percent of the LV wall area with fibrosis was calculated using: (total blue pixels from all sections)/(total blue plus red pixels from all sections) × 100.2.14. Statistical analysis
Results were plotted as mean ± SEM. ELISA data samples passed normality test with an alpha = 0.05 using a Shapiro Wilk test. ELISAs statistics were run using a two tailed unpaired t-test with welches correction with an f test to compare variances. For statistical analysis of efficacy studies including % LVEF and delta LVEF a generalized estimating equation (GEE) was utilized to confirm change in EF over the three timepoints for each group and a, 2 way ANOVA with Holm–Sidak's multiple comparisons test was used. For statistical analysis of histology quantification, ordinary one-way ANOVA and Holm–Sidak's multiple comparisons test was used.3. Results and discussion
3.1. Fabrication and characterization of encapsulated MSCs
Immune-evasive alginate TMTD was prepared as previously described (Fig. 1a and b).25,27 NMR and Mass spec show successful synthesis of TMTD alginate (methods 2.3–2.4). Core–shell hydrogel micro capsules were fabricated using a biocompatible alginate polymer through electrostatic spraying (Fig. 1c). TMTD alginate was dissolved in 0.8% saline and subsequently blended with SLG100. This modified alginate was used as the shell. SLG20 alginate was also dissolved in 0.8% saline and used for the core. Using a syringe pump and co-axial needle, spherical gels were formed and dropped into a 100 mM barium chloride bath and ionically crosslinked. Capsules maintained an approximate diameter of 1.5 mm (Fig. 2a). The hydrogels can be placed in the pericardial sac and used in the acute myocardial infarct space (Fig. 1d).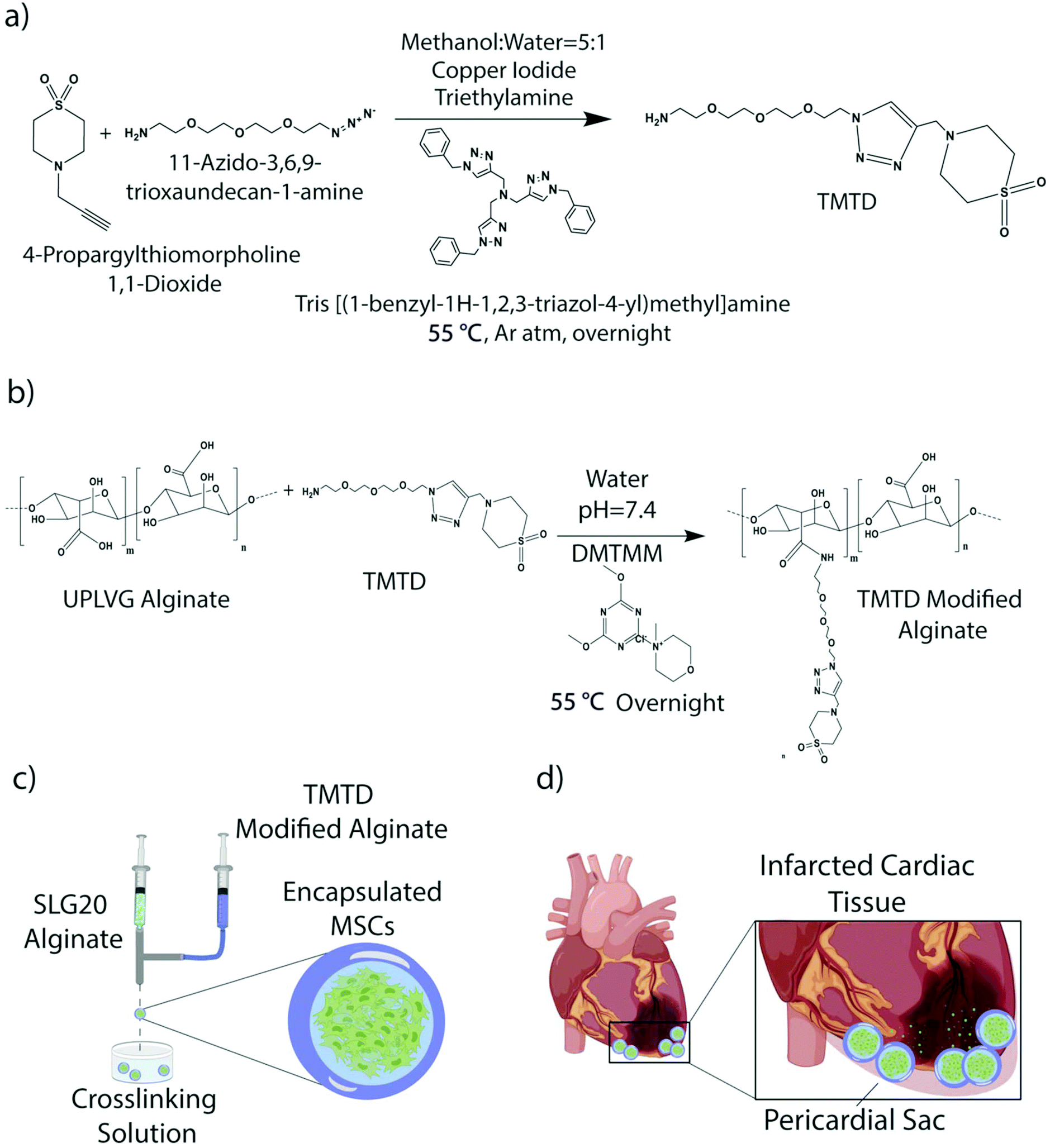 | ||
| Fig. 1 (a) Chemical schematic of TMTD small molecule synthesis and (b) TMTD small molecule modified alginate. (c) Schematic of electrostatic spraying mechanism used for encapsulation of MSCs into core shell capsules and (d) application of the platform in an acute myocardial infarct model. | ||
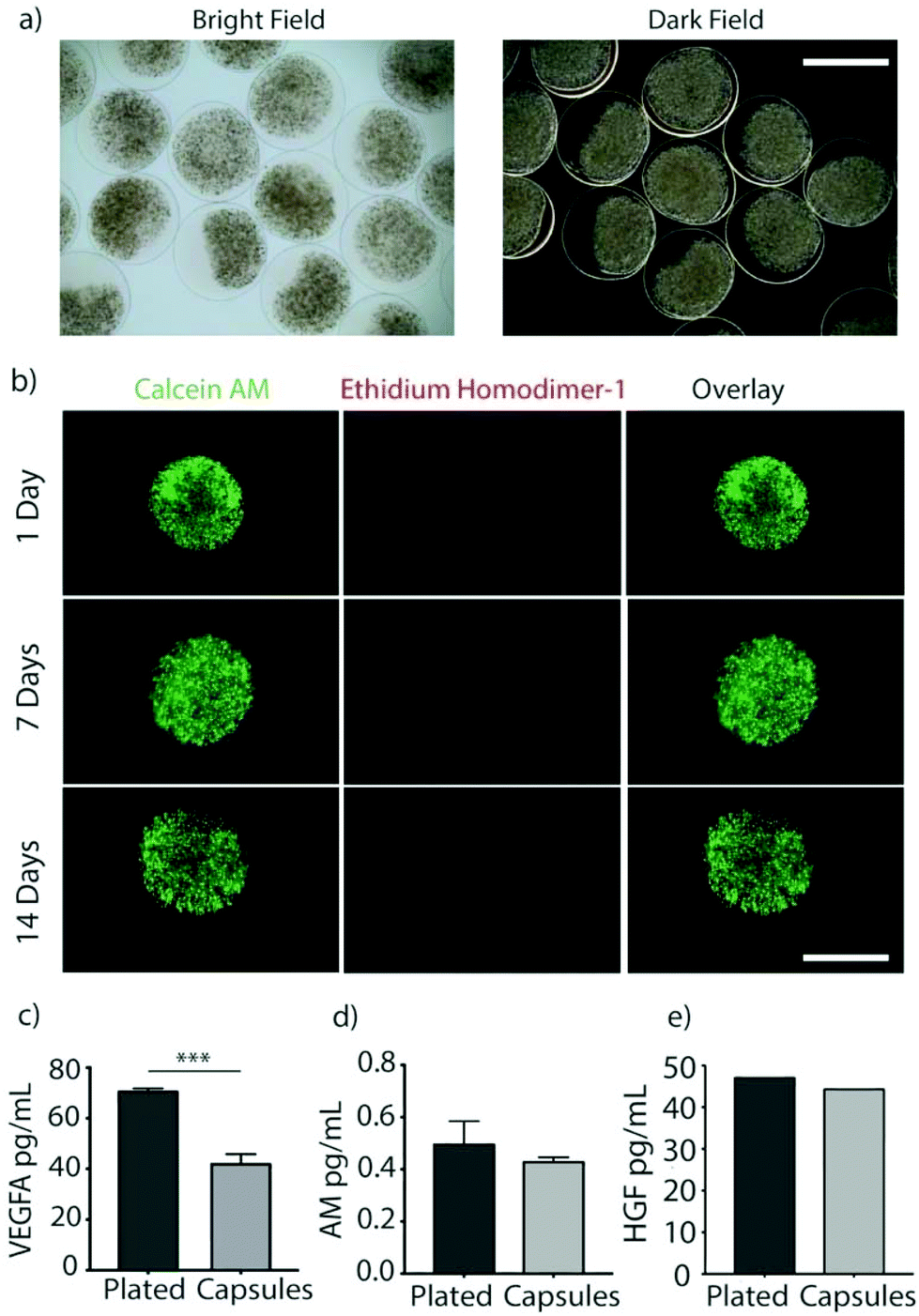 | ||
| Fig. 2 (a) Representative bright and dark field images of encapsulated MSCs showing the transparent shell, scale bar is 2000 μm. (b) Representative images of encapsulated MSCs stained with calcein AM (live) and ethidium homodimer (dead) over the span of 2 weeks shows cells remain viable over time, scale bar is 1000 μm. (c, d, & e) Concentrations of secreted paracrine factors measured in supernatant after 24 hours. Groups compare plated (un-encapsulated) and TMTD-encapsulated MSCs. (VEGFA; p = 0.0009, AM; p = 0.1156, HGF; p = 0.7058) (n = 5). | ||
Rat MSCs were encapsulated in the alginate hydrogels at a concentration of 10 × 106 cells per mL of SLG20 (∼40000 cells per capsule). The formed hydrogel is porous and allows for the inward diffusion of nutrients to enable cell viability, as well as outward diffusion of soluble factors secreted by MSCs. In vitro, encapsulated MSCs were maintained in complete DMEM and viability was assessed via live/dead staining. MSCs exhibited high viability in vitro over the course of two weeks (Fig. 2b). The amount of VEGF released by encapsulated MSCs over a 24-hour period was lower than non-encapsulated plated cells (70.85 ± 0.89 v 42.25 ± 3.55; p < 0.01), likely due to the negative charge of the alginate surface slowing diffusion of the positively charged VEGF molecule (Fig. 2c). However, a substantial amount of VEGF was still able to diffuse out of the hydrogel spheres.27 In contrast, no difference was observed between encapsulated MSCs and non-encapsulated plated cells in the release of ADM (0.49 ± 0.086 vs. 0.43 ± 0.015; p = 0.12) or HGF (47.24 ± 0.002 vs. 44.48 ± 0.002; p = 0.71) over 24 hours (Fig. 2d and e).
While it was initially assumed that the benefit of cell therapy resulted from trans-differentiation of transplanted cells into functioning cardiomyocytes, extensive subsequent work has shown that few cells do trans-differentiate and insufficient to account for the structural and functional improvements in cardiac function.29,30 The observed therapeutic benefits have been attributed to the release of a variety of cytokines and growth factors, including angiogenic VEGF, cardioprotective HGF, and anti-fibrotic ADM.31,32 This has led to a shift from a trans-differentiation mechanism to a paracrine mediated mechanism as the principal therapeutic benefit of cell therapy.6
While the diffusion of a positively charged molecule such as VEGF could potentially be impaired by the negative charge of the alginate surface, a substantial amount of VEGF was still able to diffuse out of the hydrogel spheres.27 Similarly, in vitro elution of HGF, and AM were measured and comparable to non-encapsulated cells (Fig. 2c–e). Thus, above results demonstrating that elution of paracrine factors was not meaningfully impaired by encapsulation, meet a critical prerequisite for therapeutic effects in this application.
3.2. in vivo biocompatibility, viability and localization of alginate MSC capsules in non-ischemic rats
MSCs encapsulated in TMTD modified alginate were implanted in the pericardial space on the epicardial surface of non-ischemic rats to validate in vivo viability and capsule localization. Photographs were collected during and 2 weeks following implantation of the capsules on the epicardial surface of the ventricles within the pericardial space via a left thoracotomy (Fig. 3a–c). Capsules demonstrated sufficient physical integrity for handling with surgical forceps for positioning. F-Luc MSCs were implanted in a rat and tracked through a longitudinal imaging study using a luminescence based in vivo imaging system (IVIS) for 14 days (Fig. 3d). MSC capsules remained viable and localized in the left lower chest overlying the heart. In a separate group of non-ischemic rats, GFP + MSC capsules were implanted in the pericardial space and retrieved after 14 days. Explanted retrieved capsules were translucent, with minimal peri-capsular tissue overgrowth. Viability was confirmed using live/dead staining (Fig. 3e). In contrast, explanted unmodified control capsules had extensive pericapsular formation and fibrotic overgrowth after 2 weeks (ESI Fig. 1†), which would block sustained delivery of paracrine factors and resulted in encapsulated cell death. For this reason, we did not include an unmodified capsule group in our experimental cohorts.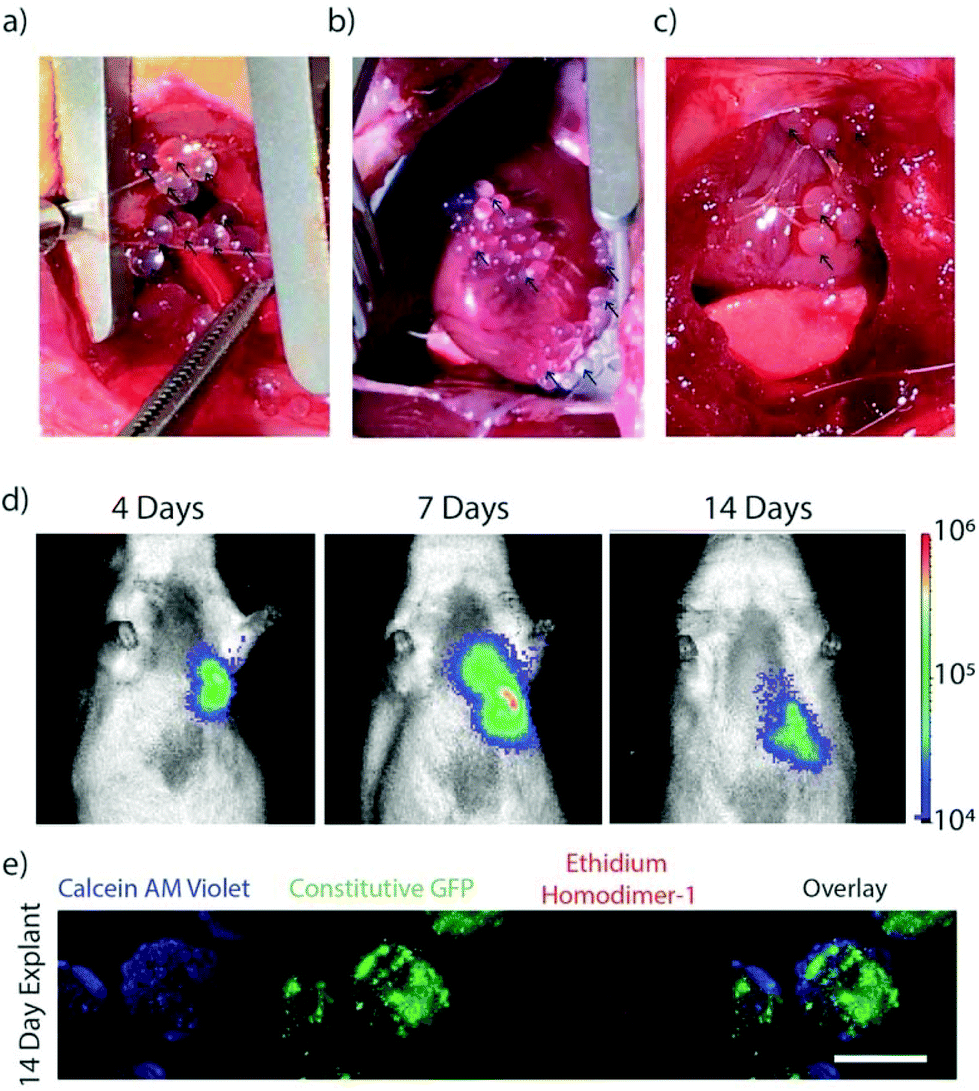 | ||
| Fig. 3 (a & b) Representative photos of encapsulated MSC's placed in the pericardial sac at the time of implant and (c) at two-week explant. In all photos, black arrows point to capsules. (d) IVIS luminescent images of encapsulated MSCs expressing firefly luciferase imaged at 4, 7, and 14 days post implant. Color scale min = 1.00 × 104 max = 1.00 × 106 (radiance p sec−1 cm−2 sr−1). (e) Representative images of encapsulated MSCs explanted from the pericardial sac after 2 weeks, stained with calcein AM (live) and ethidium homodimer (dead), scale bar is 1000 μm (e). | ||
Stem cell therapy for heart failure has demonstrated promise, however poor transplanted cell retention remains a critical limitation.1,3 A variety of natural and synthetic biomaterials have been utilized to enhance stem cell survival and engraftment.33–36 Alginate is a versatile hydrogel for biomedical application and cell encapsulation. The core shell design of the capsules enables diffusion of nutrients, yet blocks host antibodies and immune cells, to sustain cell survival.
These findings demonstrate that fabricated TMTD-alginate MSC capsules placed in the post-MI pericardial space maintain high cell viability over 14 days in vitro (Fig. 2a) and in vivo (Fig. 3d and e) with minimal peri-capsular overgrowth. In contrast, prior studies with hydrogels or epicardial patches demonstrated 80% major cell loss within 6 days in vitro and nearly 100% loss within 7 days in vivo.21,36,37
3.3. Therapeutic efficacy of encapsulated MSCs in ischemic rats
Encapsulated MSCs were implanted following acute MI and compared to blank capsule control and MSC intramyocardial injection over 28 days with serial echocardiography (Fig. 4a). In all subjects, the anterior left ventricular wall became hypokinetic and demonstrated a wall motion abnormality (Fig. 4b). Furthermore, EF decreased with infarction in all rats (Fig. 4c). At 28 days, blank capsule (control) rats demonstrated no change in EF (43 ± 3.24 vs. 45 ± 2.96; p = 0.6914). MSC injection rats demonstrated improvement in EF (51 vs. 60; p = 0.0092). MSC capsules demonstrated improvement in EF (46 vs. 64; p = 0.0046) and a greater degree of improvement compared to MSC injection (41% vs. 16%; p < 0.05) (Fig. 4d).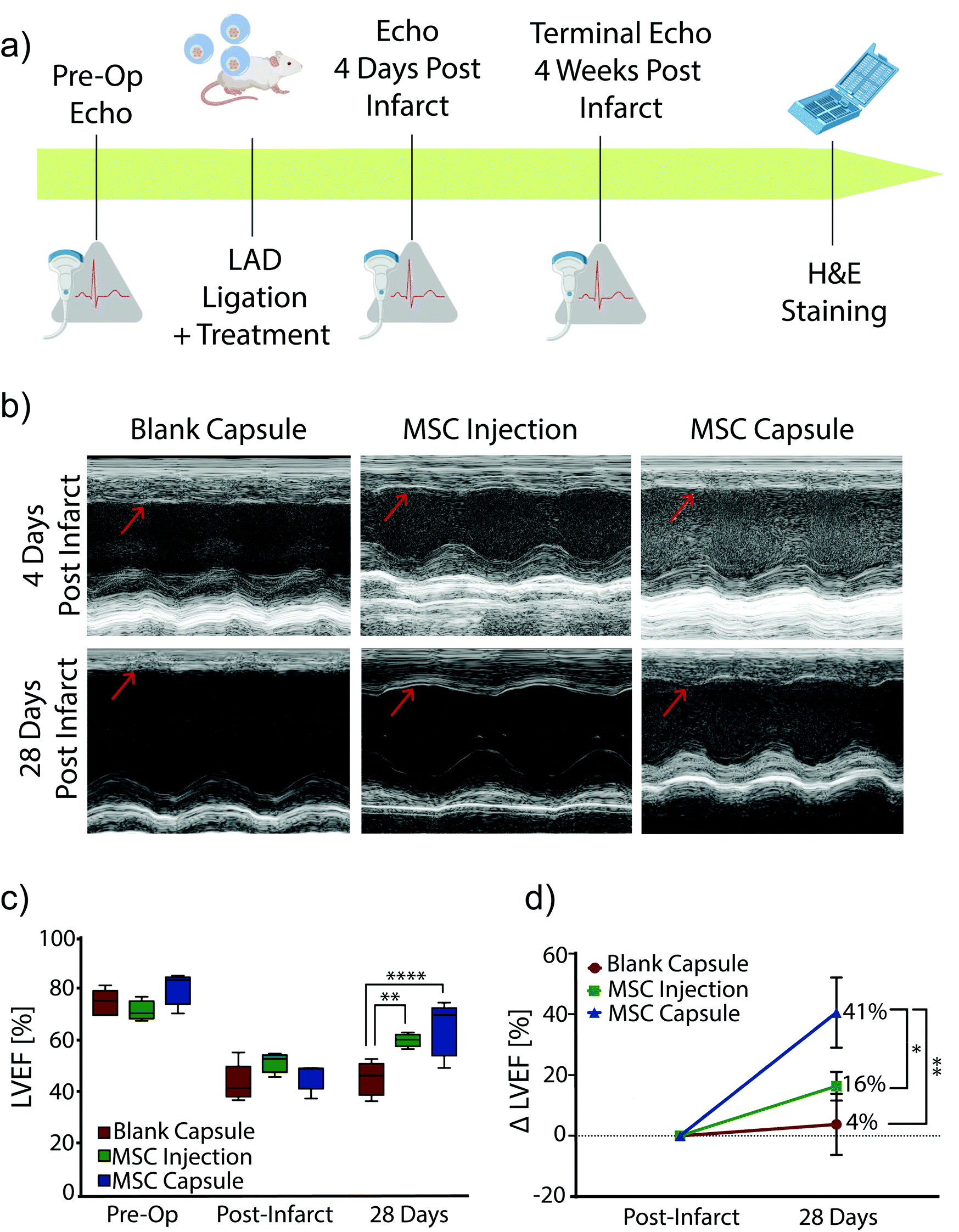 | ||
| Fig. 4 (a) Schematic of the study timeline and endpoint. (b) Representative M-mode echocardiograph images of the papillary muscle 4 days post infarct and 28 days post infarct (n = 4–5). Red arrows identify the left ventricular wall. (c) Left ventricular ejection fraction (LVEF) measurements 1 day prior to infarct, 4 days after, and 28 days (terminal) after acute myocardial infarction (n = 4–5). (d) Delta LVEF plotted to show the improvement in ejection fraction from 4 days post MI to 28 days post MI (terminal) for each group (n = 4–5). % Change was calculated using the formula [(LVEF 28 day (terminal) − LVEF 4-day post-MI)/LVEF 4-day post-MI] × 100. All data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
Explanted hearts were sectioned and stained with Masson's trichrome in order to assess fibrosis (Fig. 5a). Rats in the blank capsule (control) and MSC injection groups demonstrated fibrosis of 9% ± 2% and 11% ± 2% of the left ventricle respectively (p = 0.40). Rats in the MSC capsule group had decreased fibrosis (3% ± 0.5%) compared to blank (p = 0.021) and MSC injection groups (p = 0.0092) (Fig. 5b).
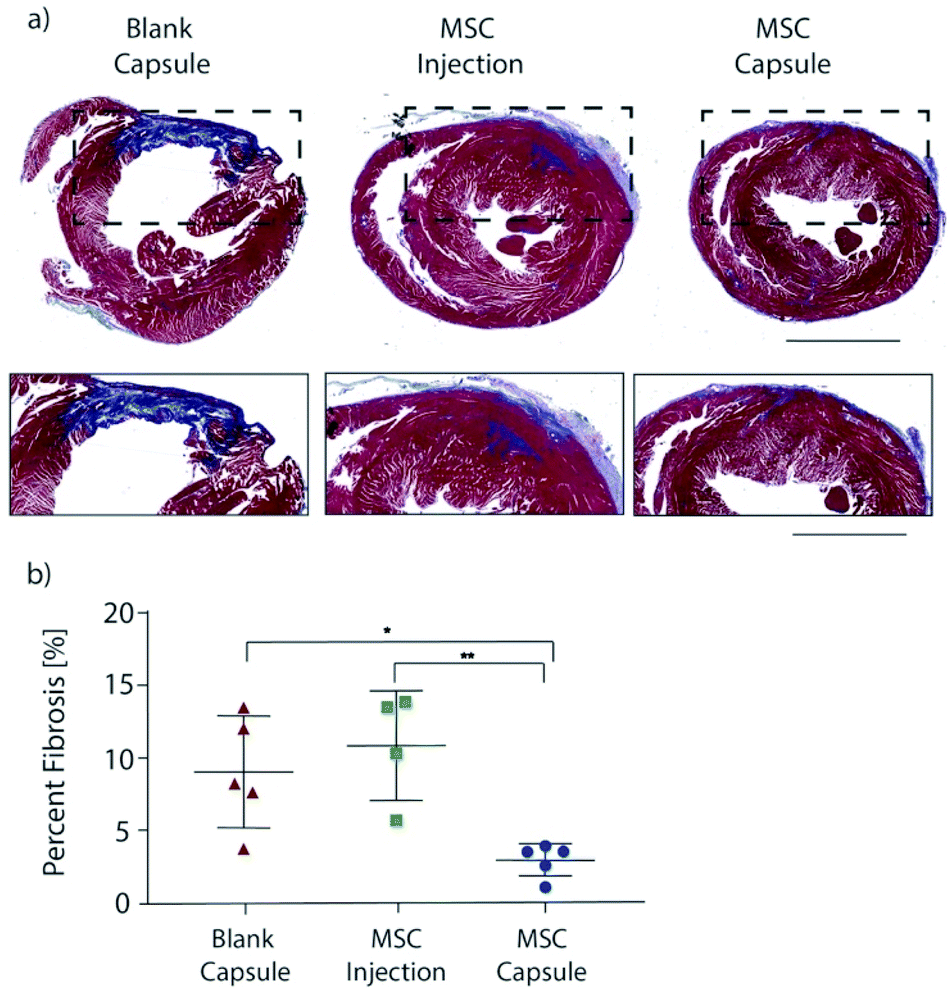 | ||
| Fig. 5 (a) Representative Masson's trichrome-stained myocardial sections 28 days after myocardial infarction and treatment administration. Blue depicts scar tissue, red illustrates viable myocardium. Magnified images of the infarcted region are portrayed below, scale bar is 1250 μm. (b) Quantitative analysis of the % fibrosis 28 days post-MI. All data are presented as the mean ± SEM. Ordinary one-way ANOVA and Holm–Sidak's multiple comparisons test was used. *P < 0.05, **P < 0.01. | ||
Thus, MSC encapsulation with TMTD-alginate led to a greater therapeutic benefit after MI when compared to blank capsules (negative control) and direct intramyocardial MSC injection (positive control) over 28 days. Both MSC injection and capsules improved LV EF, however MSC capsules led to a greater improvement in LV EF (41% vs. 16%). There was no change in post-MI fibrosis with MSC injection relative to controls, while in MSC capsule animals fibrosis was reduced to 1/3 of that seen in controls. This therapeutic advantage is likely secondary to prolonged cell survival, which enables sustained delivery of reparative paracrine factors.
The optimal route for cell therapy (intravenous, intra-coronary, direct intramyocardial, or pericardial) has not been determined. Both acute MI and chronic ischemia are associated with inflammation, which may exacerbate cell death and blunt therapeutic benefits of cell therapy.38 The hostile environment of the myocardium with inflammation and hypoxia may ultimately limit intramyocardial delivery. Pericardial delivery allows for therapeutics to be adjacent to the injured tissue without directly encountering the activated inflammatory cells. This route is especially appealing for delivery of reparative proteins and factors provided sufficient diffusion occurs. Lovich and colleagues demonstrated that epicardial delivery leads to rapid diffusion of drugs throughout the myocardium, with 500× increase in potency compared to systemic delivery.39,40 Whyte and colleagues recently reported the utility of pericardial MSC delivery to improve cardiac function over a 4 weeks period.13 This innovative strategy, however, required weekly cellular injection into the epicardial reservoir to achieve efficacy. Use of MSC capsules allows for sustained reparative factor delivery without repeated cell administration.
This strategy also has significant potential for clinical translation. Alginate biomaterials are highly biocompatible and have been injected in the myocardium of heart failure patients in a randomized clinical trial, with favorable safety.41 Alginate cell capsules can be delivered to the epicardium via transcatheter pericardial installation as demonstrated in a pilot porcine study (ESI Fig. 2†), catheter instillation or following cardiac surgery.11 Furthermore, this platform can be utilized with alternative cell types or engineered cells designed for targeted delivery of specific reparative factors.26
4. Conclusions
In summary, we have demonstrated fabrication and in vivo therapeutic application of TMTD-alginate MSC capsules for improvement of ventricular functioning and remodelling in a post-MI rat model. MSC capsules demonstrated sustained cell survival and superior therapeutic efficacy to MSC injection. Combined, our results demonstrate that this approach could facilitate treatment of MI. Furthermore, this biomaterial cell therapy strategy can be utilized to deliver a range of therapeutic factors to the heart.Conflicts of interest
O.V. is co-founder and holds equity in Sigilon Therapeutics. Entity, which hold intellectual property rights related to the materials used in this study.Acknowledgements
This work was supported by the American Association of Thoracic Surgery Research Award and the Baylor College of Medicine Cardiovascular Research Institute. The Cancer Prevention Research Institute of Texas (Grant ID, RR160047), the National Institutes of Health (NIH Grant 1R01DK120459) Research Supplements to Promote Diversity in Health-Related Research (A. N.), and the Rice University Academy Fellowship (M. J.). This work was also partially supported by the funding from the Emerson Collective to A. J. and W. P. CTR is supported by the National Institutes of Health/National Heart, Lung, and Blood Institute Research Training Program in Cardiovascular Surgery (T32 HL139430). We would also like to acknowledge Nicholas Stevens for his assistance with this project.Notes and references
- E. Braunwald, Circ. Res., 2018, 123, 132–137 CrossRef CAS PubMed.
- M. N. Banerjee, R. Bolli and J. M. Hare, Circ. Res., 2018, 123, 266–287 CrossRef CAS PubMed.
- G. Curfman, J. Am. Med. Assoc., 2019, 321, 1186–1187 CrossRef PubMed.
- R. Madonna, L. W. Van Laake, S. M. Davidson, F. B. Engel, D. J. Hausenloy, S. Lecour, J. Leor, C. Perrino, R. Schulz, K. Ytrehus, U. Landmesser, C. L. Mummery, S. Janssens, J. Willerson, T. Eschenhagen, P. Ferdinandy and J. P. Sluijter, Eur. Heart J., 2016, 37, 1789–1798 CrossRef PubMed.
- T. Yoshioka, N. Ageyama, H. Shibata, T. Yasu, Y. Misawa, K. Takeuchi, K. Matsui, K. Yamamoto, K. Terao, K. Shimada, U. Ikeda, K. Ozawa and Y. Hanazono, Stem Cells, 2005, 23, 355–364 CrossRef CAS.
- M. Gnecchi, H. He, O. D. Liang, L. G. Melo, F. Morello, H. Mu, N. Noiseux, L. Zhang, R. E. Pratt, J. S. Ingwall and V. J. Dzau, Nat. Med., 2005, 11, 367–368 CrossRef CAS PubMed.
- T. Schinkothe, W. Bloch and A. Schmidt, Stem Cells Dev., 2008, 17, 199–206 CrossRef CAS PubMed.
- J. M. Hare, J. E. Fishman, G. Gerstenblith, D. L. DiFede Velazquez, J. P. Zambrano, V. Y. Suncion, M. Tracy, E. Ghersin, P. V. Johnston, J. A. Brinker, E. Breton, J. Davis-Sproul, I. H. Schulman, J. Byrnes, A. M. Mendizabal, M. H. Lowery, D. Rouy, P. Altman, C. Wong Po Foo, P. Ruiz, A. Amador, J. Da Silva, I. K. McNiece, A. W. Heldman, R. George and A. Lardo, J. Am. Med. Assoc., 2012, 308, 2369–2379 CrossRef CAS PubMed.
- A. B. Mathiasen, A. A. Qayyum, E. Jorgensen, S. Helqvist, A. Fischer-Nielsen, K. F. Kofoed, M. Haack-Sorensen, A. Ekblond and J. Kastrup, Eur. Heart J., 2015, 36, 1744–1753 CrossRef CAS PubMed.
- Z. Liu, R. Mikrani, H. M. Zubair, A. Taleb, M. Naveed, M. Baig, Q. Zhang, C. Li, M. Habib, X. Cui, K. R. Sembatya, H. Lei and X. Zhou, Eur. J. Pharmacol., 2020, 876, 173049 CrossRef CAS PubMed.
- D. Ladage, I. C. Turnbull, K. Ishikawa, Y. Takewa, K. Rapti, C. Morel, I. Karakikes, L. Hadri, J. Muller-Ehmsen, K. D. Costa, R. J. Hajjar and Y. Kawase, Gene Ther., 2011, 18, 979–985 CrossRef CAS PubMed.
- J. Zhang, Z. Wu, Z. Fan, Z. Qin, Y. Wang, J. Chen, M. Wu, Y. Chen, C. Wu and J. Wang, J. Physiol., 2018, 596, 2037–2054 CrossRef CAS PubMed.
- W. Whyte, E. T. Roche, C. E. Varela, K. Mendez, S. Islam, H. O'Neill, F. Weafer, R. N. Shirazi, J. C. Weaver, N. V. Vasilyev, P. E. McHugh, B. Murphy, G. P. Duffy, C. J. Walsh and D. J. Mooney, Nat. Biomed. Eng., 2018, 2, 416–428 CrossRef CAS PubMed.
- Y. Tokita, X. L. Tang, Q. Li, M. Wysoczynski, K. U. Hong, S. Nakamura, W. J. Wu, W. Xie, D. Li, G. Hunt, Q. Ou, H. Stowers and R. Bolli, Circ. Res., 2016, 119, 635–651 CrossRef CAS.
- K. U. Hong, Q. H. Li, Y. Guo, N. S. Patton, A. Moktar, A. Bhatnagar and R. Bolli, Basic Res. Cardiol., 2013, 108, 346 CrossRef PubMed.
- M. Hofmann, K. C. Wollert, G. P. Meyer, A. Menke, L. Arseniev, B. Hertenstein, A. Ganser, W. H. Knapp and H. Drexler, Circulation, 2005, 111, 2198–2202 CrossRef.
- S. H. Li, T. Y. Lai, Z. Sun, M. Han, E. Moriyama, B. Wilson, S. Fazel, R. D. Weisel, T. Yau, J. C. Wu and R. K. Li, J. Thorac. Cardiovasc. Surg., 2009, 137, 1225–1233 CrossRef.
- M. Gyongyosi, J. Blanco, T. Marian, L. Tron, O. Petnehazy, Z. Petrasi, R. Hemetsberger, J. Rodriguez, G. Font, I. J. Pavo, I. Kertesz, L. Balkay, N. Pavo, A. Posa, M. Emri, L. Galuska, D. L. Kraitchman, J. Wojta, K. Huber and D. Glogar, Circ. Cardiovasc. Imaging, 2008, 1, 94–103 CrossRef PubMed.
- L. M. McGinley, J. McMahon, A. Stocca, A. Duffy, A. Flynn, D. O'Toole and T. O'Brien, Hum. Gene Ther., 2013, 24, 840–851 CrossRef CAS PubMed.
- O. Veiseh and A. J. Vegas, Adv. Drug Delivery Rev., 2019, 144, 148–161 CrossRef CAS PubMed.
- R. D. Levit, N. Landazuri, E. A. Phelps, M. E. Brown, A. J. Garcia, M. E. Davis, G. Joseph, R. Long, S. A. Safley, J. D. Suever, A. N. Lyle, C. J. Weber and W. R. Taylor, J. Am. Heart Assoc., 2013, 2, e000367 Search PubMed.
- E. A. Phelps, N. Landazuri, P. M. Thule, W. R. Taylor and A. J. Garcia, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3323–3328 CrossRef CAS PubMed.
- N. H. Chi, M. C. Yang, T. W. Chung, J. Y. Chen, N. K. Chou and S. S. Wang, Biomaterials, 2012, 33, 5541–5551 CrossRef CAS PubMed.
- O. Veiseh, J. C. Doloff, M. Ma, A. J. Vegas, H. H. Tam, A. R. Bader, J. Li, E. Langan, J. Wyckoff, W. S. Loo, S. Jhunjhunwala, A. Chiu, S. Siebert, K. Tang, J. Hollister-Lock, S. Aresta-Dasilva, M. Bochenek, J. Mendoza-Elias, Y. Wang, M. Qi, D. M. Lavin, M. Chen, N. Dholakia, R. Thakrar, I. Lacik, G. C. Weir, J. Oberholzer, D. L. Greiner, R. Langer and D. G. Anderson, Nat. Mater., 2015, 14, 643–651 CrossRef CAS PubMed.
- A. J. Vegas, O. Veiseh, M. Gurtler, J. R. Millman, F. W. Pagliuca, A. R. Bader, J. C. Doloff, J. Li, M. Chen, K. Olejnik, H. H. Tam, S. Jhunjhunwala, E. Langan, S. Aresta-Dasilva, S. Gandham, J. J. McGarrigle, M. A. Bochenek, J. Hollister-Lock, J. Oberholzer, D. L. Greiner, G. C. Weir, D. A. Melton, R. Langer and D. G. Anderson, Nat. Med., 2016, 22, 306–311 CrossRef CAS PubMed.
- A. J. Vegas, O. Veiseh, J. C. Doloff, M. Ma, H. H. Tam, K. Bratlie, J. Li, A. R. Bader, E. Langan, K. Olejnik, P. Fenton, J. W. Kang, J. Hollister-Locke, M. A. Bochenek, A. Chiu, S. Siebert, K. Tang, S. Jhunjhunwala, S. Aresta-Dasilva, N. Dholakia, R. Thakrar, T. Vietti, M. Chen, J. Cohen, K. Siniakowicz, M. Qi, J. McGarrigle, S. Lyle, D. M. Harlan, D. L. Greiner, J. Oberholzer, G. C. Weir, R. Langer and D. G. Anderson, Nat. Biotechnol., 2016, 34, 345–352 CrossRef CAS PubMed.
- M. A. Bochenek, O. Veiseh, A. J. Vegas, J. J. McGarrigle, M. Qi, E. Marchese, M. Omami, J. C. Doloff, J. Mendoza-Elias, M. Nourmohammadzadeh, A. Khan, C. C. Yeh, Y. Xing, D. Isa, S. Ghani, J. Li, C. Landry, A. R. Bader, K. Olejnik, M. Chen, J. Hollister-Lock, Y. Wang, D. L. Greiner, G. C. Weir, B. L. Strand, A. M. A. Rokstad, I. Lacik, R. Langer, D. G. Anderson and J. Oberholzer, Nat. Biomed. Eng., 2018, 2, 810–821 CrossRef CAS PubMed.
- M. Mathison, R. P. Gersch, A. Nasser, S. Lilo, M. Korman, M. Fourman, N. Hackett, K. Shroyer, J. Yang, Y. Ma, R. G. Crystal and T. K. Rosengart, J. Am. Heart Assoc., 2012, 1, e005652 Search PubMed.
- C. E. Murry, M. H. Soonpaa, H. Reinecke, H. Nakajima, H. O. Nakajima, M. Rubart, K. B. Pasumarthi, J. I. Virag, S. H. Bartelmez, V. Poppa, G. Bradford, J. D. Dowell, D. A. Williams and L. J. Field, Nature, 2004, 428, 664–668 CrossRef CAS PubMed.
- X. L. Tang, Q. Li, G. Rokosh, S. K. Sanganalmath, N. Chen, Q. Ou, H. Stowers, G. Hunt and R. Bolli, Circ. Res., 2016, 118, 1091–1105 CrossRef CAS PubMed.
- N. Nagaya, K. Kangawa, T. Itoh, T. Iwase, S. Murakami, Y. Miyahara, T. Fujii, M. Uematsu, H. Ohgushi, M. Yamagishi, T. Tokudome, H. Mori, K. Miyatake and S. Kitamura, Circulation, 2005, 112, 1128–1135 CrossRef PubMed.
- C. Toma, M. F. Pittenger, K. S. Cahill, B. J. Byrne and P. D. Kessler, Circulation, 2002, 105, 93–98 CrossRef PubMed.
- A. D. Celiz, J. G. Smith, R. Langer, D. G. Anderson, D. A. Winkler, D. A. Barrett, M. C. Davies, L. E. Young, C. Denning and M. R. Alexander, Nat. Mater., 2014, 13, 570–579 CrossRef CAS PubMed.
- V. F. Segers and R. T. Lee, Circ. Res., 2011, 109, 910–922 CrossRef CAS PubMed.
- M. Habib, K. Shapira-Schweitzer, O. Caspi, A. Gepstein, G. Arbel, D. Aronson, D. Seliktar and L. Gepstein, Biomaterials, 2011, 32, 7514–7523 CrossRef CAS PubMed.
- E. T. Roche, C. L. Hastings, S. A. Lewin, D. Shvartsman, Y. Brudno, N. V. Vasilyev, F. J. O'Brien, C. J. Walsh, G. P. Duffy and D. J. Mooney, Biomaterials, 2014, 35, 6850–6858 CrossRef CAS PubMed.
- Z. Liu, H. Wang, Y. Wang, Q. Lin, A. Yao, F. Cao, D. Li, J. Zhou, C. Duan, Z. Du, Y. Wang and C. Wang, Biomaterials, 2012, 33, 3093–3106 CrossRef CAS PubMed.
- R. J. Vagnozzi, M. Maillet, M. A. Sargent, H. Khalil, A. K. Z. Johansen, J. A. Schwanekamp, A. J. York, V. Huang, M. Nahrendorf, S. Sadayappan and J. D. Molkentin, Nature, 2020, 577, 405–409 CrossRef CAS.
- M. A. Lovich, A. E. Wei, M. Y. Maslov, P. I. Wu and E. R. Edelman, J. Pharm. Sci., 2011, 100, 4993–5006 CrossRef CAS PubMed.
- M. Y. Maslov, E. R. Edelman, M. J. Pezone, A. E. Wei, M. G. Wakim, M. R. Murray, H. Tsukada, I. S. Gerogiannis, A. Groothuis and M. A. Lovich, J. Controlled Release, 2014, 194, 257–265 CrossRef CAS PubMed.
- H. N. Sabbah, M. Wang, R. C. Gupta, S. Rastogi, I. Ilsar, M. S. Sabbah, S. Kohli, S. Helgerson and R. J. Lee, JACC Heart Failure, 2013, 1, 252–258 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00855a |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |