
Hybrid PEDOT/MnOx nanostructured electrocatalysts for oxygen reduction†
Julian A.
Vigil
 ,
Timothy N.
Lambert
*,
Maria
Kelly
and
Ruby
Aidun
,
Timothy N.
Lambert
*,
Maria
Kelly
and
Ruby
Aidun
Department of Materials, Devices & Energy Technologies, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA. E-mail: tnlambe@sandia.gov; Tel: +1 505 284 6967
First published on 5th May 2017
Abstract
A series of hybrid poly(3,4-ethylenedioxythiophene)/manganese oxide (PEDOT/MnOx) thin films have been prepared via a stepwise approach: electrodeposition of PEDOT, followed by formation of MnOx particles by a spontaneous redox reaction between PEDOT and KMnO4. Electrocatalytic characterization of the PEDOT/MnOx thin films demonstrates high activity toward the oxygen reduction reaction (ORR), with a shift in intrinsic ORR onset and half-wave potentials by ca. 0.2 V to lower overpotential relative to the PEDOT thin film. The most active PEDOT/MnOx thin film electrocatalyst, P-MnOx-20, demonstrates superior activity relative to the commercial 20% Pt/C catalyst in the half-wave region of the ORR potential window at equal mass loading, with a half-wave potential of 0.83 V (20% Pt/C, 0.81 V) and charge transfer resistance of 479 Ω (20% Pt/C, 862 Ω). The P-MnOx-20 film also demonstrates preference to a pseudo-four electron ORR pathway (n = 3.8) and high specific ORR activity, when considered on both a total mass (−96 mA mgtotal−1; 20% Pt/C: −108 mA mgtotal−1) and metal (or metal oxide) mass basis (−296 mA mgMnOx−1; 20% Pt/C: −540 mA mgPt−1). The P-MnOx-20 film has been identified as the most active PEDOT/ceramic composite electrocatalyst reported to date, which is rationalized by the high surface concentration of Mn(III), strong electronic coupling between PEDOT and MnOx, as well as a high active site density and efficiency achieved by the stepwise electrodeposition-redox approach.
Introduction
Electrochemical energy conversion and storage devices with high specific energy and efficiency are needed to stabilize an increasingly renewable electricity grid and enable electromobility (e.g., electric vehicles).1 Among these, fuel cells and metal–air batteries are promising candidates for the future of power generation and energy storage, respectively. The performance of both of these classes of devices, however, is limited by the sluggish kinetics of the oxygen reduction reaction (ORR) at the air cathode. Furthermore, the overpotential to drive the four electron/four proton transfer to surface oxygen species is in excess of 0.2 V, a significant source of efficiency loss in these devices.2,3 Platinum is one of the most active surfaces for facilitating the ORR at the solid–liquid–gas interface, and supported Pt catalysts are the leading choice for electrode catalyst layers. Besides its prohibitive cost, Pt can suffer from deactivation by particle coalescence, CO poisoning and competing alcohol oxidation reactions upon fuel crossover.4,5 In order to effectively replace Pt in these energy conversion and storage devices, new electrocatalytic materials are being pursued with a focus on raw material abundance, overall performance/stability and nanostructuring for improved active site density.6–10The rate determining step (RDS) of the ORR in both acidic and alkaline environments is acknowledged as the electron transfer to molecular oxygen, or equivalently, the displacement of the surface hydroxide.6,11 At low pH, the electron is transferred to chemisorbed O2 in the Inner Helmholtz Plane (IHP). The competing Outer Helmholtz Plane (OHP) electron transfer to solvated O2 tends to dominate at high pH, due to (1) stable surface OH coverage, and (2) reduction in overpotential for superoxide radical formation in the OHP.11–13 Although this phenomenon translates to lower alkaline ORR overpotential and relative independence to the catalyst surface thermodynamics (“nonspecificity”11–13), the OHP electron transfer favors the undesirable 2 e− ORR pathway to form HO2−. Active sites that take advantage of this alkaline RDS “nonspecificity” while maintaining 4 e− pathway selectivity are required to realize the stability benefits of operating in alkaline conditions. Catalyst materials from abundant sources with competitive activity against Pt have been discovered, returning interest to the alkaline fuel cell.13 Briefly, transition metal–nitrogen–carbon (M–N–C) materials (e.g., FeN4/C12 and NPC-Co4514), metal oxides (e.g., LaNiO3 and LaMnO3,15 NixCo3−xO4,8,16 MnO2 and Mn2O36,9,17–19), heteroatom-doped carbon (e.g., BNC-220 and S-graphene-105021), and conductive polymers (e.g., PEDOT and PANI22–24) have all demonstrated high ORR activity in alkaline environments.13
The high conductivity, stability, and electroactivity of intrinsically conductive polymers (ICPs) makes them compelling candidates for functional electrochemical and photo(electro)chemical materials. The conjugated backbone of the ICP not only provides rapid electron transfer as a purely conductive support, but results in a lower bandgap relative to conventional polymers, suggesting the ability to tune electronics for electron transfer (electrocatalysis) and light absorption (photocatalysis/photosensing).23,24 The ORR activity of poly(3,4-ethylenedioxythiophene) (PEDOT) was considered first by Khomenko et al. on a chemically polymerized PEDOT electrode (with PTFE binder).23 The authors rationalized the lack of observed ORR activity for PEDOT (relative to polythiophene and polymethylthiophene) by evidence that PEDOT is fully un-doped at the highly reducing ORR potentials. In contrast, the discovery of Pt-comparable ORR activity from a vapor polymerized PEDOT air electrode by Winther-Jensen et al. prompted renewed interest in PEDOT as an electrocatalytic material.22 The vapor polymerized PEDOT film reached a stable oxidation state only in the presence of air and remained more conductive by orders of magnitude relative to the film in the absence of air. It was later determined that the vapor polymerization method provides an inherent advantage for the ORR pathway on the PEDOT surface, observing a transition from 2 e− to 4 e− pathway at −0.45 V vs. Ag/AgCl25 (ca. 0.54 V vs. RHE), while electropolymerized PEDOT operated by a 2 e− pathway across the full ORR potential range.25,26
Jiang and co-workers have reported a series of PEDOT based catalysts: self-assembled PEDOT structures with ORR activity from pH 1–13,27 PEDOT doped with hemin for the incorporation of the active FeN4 center,28 and related S-, N-, Fe-doped C foams via pyrolysis of PEDOT:PSS for alkaline ORR.29 PEDOT:PSS anchored on rGO has also demonstrated enhanced ORR activity, proposed to originate from a conformational change of PEDOT and electronic interactions with rGO.30 Most relevant to the study reported here, PEDOT/ceramic composites have been reported with ORR activity: CoMn2O4–PEDOT31 and PEDOT/MnO2.32 PEDOT/MnO2/10AA carbon paper air electrodes were prepared by hydrothermal synthesis of α- and β-MnO2, followed by the direct polymerization of EDOT on the MnO2via chemical vapor deposition (CVD).32 The nanostructure of the hybrid electrode was not characterized, but hybridization resulted in an increase in ORR current (at −0.7 V vs. SCE) of 15.53 mA cm−2 for the more active phase α-MnO2/10AA to 30.91 mA cm−2 for PEDOT/α-MnO2/10AA.32 A composite CoMn2O4–PEDOT material was prepared by conversion of α-MnO2 nanorods to spinel CoMn2O4 nanoparticles, followed by chemical polymerization of EDOT in the presence of colloidal CoMn2O4 nanoparticles.31 The CoMn2O4–PEDOT electrocatalyst exhibits a preference for the 4 e− ORR pathway and half-wave potential only 50 mV more negative than the commercial Pt/C catalyst.31
Despite these developments in PEDOT-based electrocatalysts, none of these materials/composites have approached Pt-like ORR onset potential or specific mass activity;7 however, we recently reported on the co-electrodeposition of a MnOx/PEDOT composite thin film with ORR activity rivaling that of commercial 20% Pt/C.10 The unprecedented synergism between MnOx and PEDOT resulted in a >0.2 V shift in intrinsic ORR potential to lower overpotential while electrochemical impedance spectroscopy (EIS) indicated a low ORR charge transfer resistance (RCT). Thus, the co-electrodeposition approach was proposed to afford intimate contact and increased interfacial area between PEDOT and MnOx, providing facile electron transfer and enhancing the electronic coupling effect responsible for the lower ORR overpotential.
Here, we report on a series of hybrid PEDOT/MnOx thin film electrocatalysts derived from a stepwise process: PEDOT is electrodeposited from an organic solvent, and then decorated with MnOx particles via spontaneous redox with KMnO4 on the film surface. We provide chemical and electrochemical characterization of the hybrid PEDOT/MnOx films using scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), cyclic voltammetry (CV) and a quartz crystal microbalance (QCM). The electrocatalytic properties of the films for ORR in alkaline electrolyte have also been determined using a rotating disk electrode (RDE) with CV, linear scanning voltammetry (LSV), and EIS methods, and compared to commercial benchmark 20% Pt/C. Finally, the present work is compared to results on PEDOT–ceramic hybrid electrocatalysts in order to analyze property-activity relations to serve as a guide for future catalyst design in composite polymer–ceramic systems.
Experimental
Materials
Acetonitrile (99.8%, anhydrous), LiClO4 (>95%, ACS grade), KMnO4 (99+%, ACS grade), Nafion solution (5 wt% in alcohol) and 3,4-ethylenedioxythiophene (EDOT) were obtained from Sigma-Aldrich. Commercial benchmark electrocatalyst powder 20% Pt/C was obtained from E-Tek. All chemicals and materials were used as-received without purification or modification.Preparation of PEDOT and PEDOT/MnOx thin films
An EDOT acetonitrile solution containing 0.05 M EDOT monomer and 0.1 M LiClO4 was galvanostatically electropolymerized at a current density of 1 mA cm−2. The three-electrode cell consisted of a Pt foil counter electrode, Ag/Ag+ reference electrode and glassy carbon (GC) working electrode. The PEDOT film was deposited on the GC surface of two types of working electrodes: planar, silica-supported GC electrodes (A = 0.1963 cm2) were utilized for characterization, while RDE GC working electrodes (A = 0.0707 cm2, Bioanalytical Systems, Inc.) were utilized for electrochemical studies. Films were deposited for 20 s at 1 mA cm−2. The working electrode was then removed from the acetonitrile solution and allowed to soak in DI H2O to remove excess solvent, monomer and electrolyte from the film. The resulting blue PEDOT film was either used as-prepared (denoted PEDOT), or proceeded to soak in a 0.01 M KMnO4 solution to achieve the formation of MnOx on the film surface: a static soak in the KMnO4 solution for 10, 20 or 30 min resulted in hybrid PEDOT/MnOx films, denoted P-MnOx-10, P-MnOx-20, and P-MnOx-30, respectively. The hybrid films were then allowed to soak in DI H2O for 30 min to remove excess KMnO4 and dry under ambient conditions. No further preparation (e.g., binder application) was necessary before electrochemical investigation of the thin catalyst films.Results and discussion
Hybrid PEDOT/MnOx thin film electrocatalysts: electrodeposited PEDOT and spontaneous formation of MnOx
The preparation of hybrid PEDOT/MnOx thin film electrocatalysts is described fully in the Experimental Section, and depicted in (Scheme 1). Briefly, EDOT was electropolymerized from acetonitrile solvent containing 0.1 M LiClO4, at a constant current density of 1 mA cm−2 for 20 s (Scheme 1, 1).33 The PEDOT film was grown directly on a GC working electrode designed for the RDE cell. This PEDOT film was immersed in a bath of 0.01 M KMnO4 for varying times (10, 20, 30 min) to induce MnOx particle formation via spontaneous reduction of Mn(VII) (Scheme 1, 2).34–36 A SEM image and the corresponding energy-dispersive spectroscopy (EDS) element maps of the as-prepared P-MnOx-20 film are shown in Fig. 1a. C, O, and S appear to be equally abundant across the surface. Mn is present across the whole surface (consistent with nanoparticle growth),36 but also apparently concentrated on the PEDOT outgrowths of the film. Fig. 1a and b, as well as cross-sectional analysis in Fig. S1 (ESI†), show the irregular nature of the film growth. The bulk of the film ranges in thickness from 15–70 nm, with the larger extending features reaching as much as ca. 370 nm from the surface of the GC electrode. The irregular film is rationalized by the mechanism proposed by Aaron et al., where PEDOT chains are expected to polymerize and grow parallel to the electrode surface in organic solvent, and are thus less likely to form the close-packed, uniform thickness films that are characteristic of the micellar electropolymerization.10,33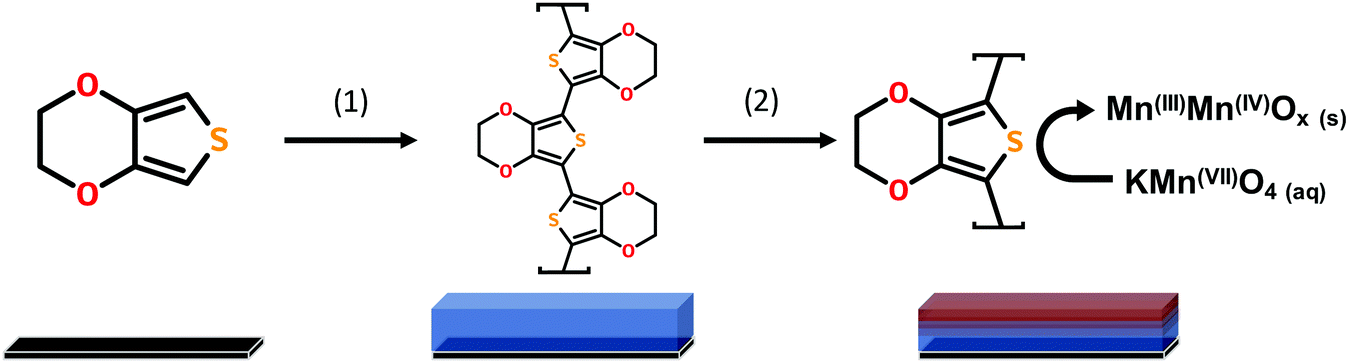 | ||
| Scheme 1 (1) Electropolymerization of EDOT (acetonitrile, 0.1 M LiClO4, 0.05 M EDOT, 1 mA cm−2); (2) spontaneous formation of MnOx particles on the PEDOT surface (0.01 M KMnO4, 10–30 min). | ||
 | ||
| Fig. 1 (a) Low-magnification SEM image and corresponding EDS C, O, S and Mn elemental maps of a P-MnOx-20 film; (b) high-magnification SEM image of a P-MnOx-20 film; (c) FTIR spectra of PEDOT/MnOx hybrid thin films after PEDOT background subtraction (arrow denotes 1044 cm−1 S(O) stretching mode of interest). | ||
KMnO4 is a common reagent known to react with alkenes, and has been successful in preparing MnOx particles in situ on conductive and high surface area carbons, such as acetylene black and carbon nanotubes.34,35 Similarly, Liu et al. recently adapted this methodology for conductive polymers, reporting redox-induced formation of MnO2 on PEDOT nanowire templates for supercapacitor arrays using KMnO4.36 Following MnO4− exposure, the authors showed that there is no measureable oxidative damage to the carbon backbone of PEDOT, resolved a small chemical shift in the S 2p binding energy via XPS, and showed the appearance of a S(O) stretching mode in the FTIR spectra. Thus, the authors’ suggested mechanism for the spontaneous growth of MnOx particles on a PEDOT surface is the oxidation of the thiophene sulfur atom to sulfoxide and/or sulfone, as Mn(VII) is reduced to Mn(IV).36 This hypothesis was further supported by DFT studies showing significantly favored orientation and charge transfer between the EDOT sulfur atom and the Mn atom in two phases of crystalline MnO2.32 FTIR spectra of the PEDOT/MnOx films prepared here are shown in Fig. 1c, and the presence of surface sulfoxide species via increased absorbance at the characteristic frequency of 1044 cm−1 (denoted by an arrow) after exposure to MnO4− (and PEDOT background subtraction)36,37 is consistent with the previously suggested growth mechanism.36
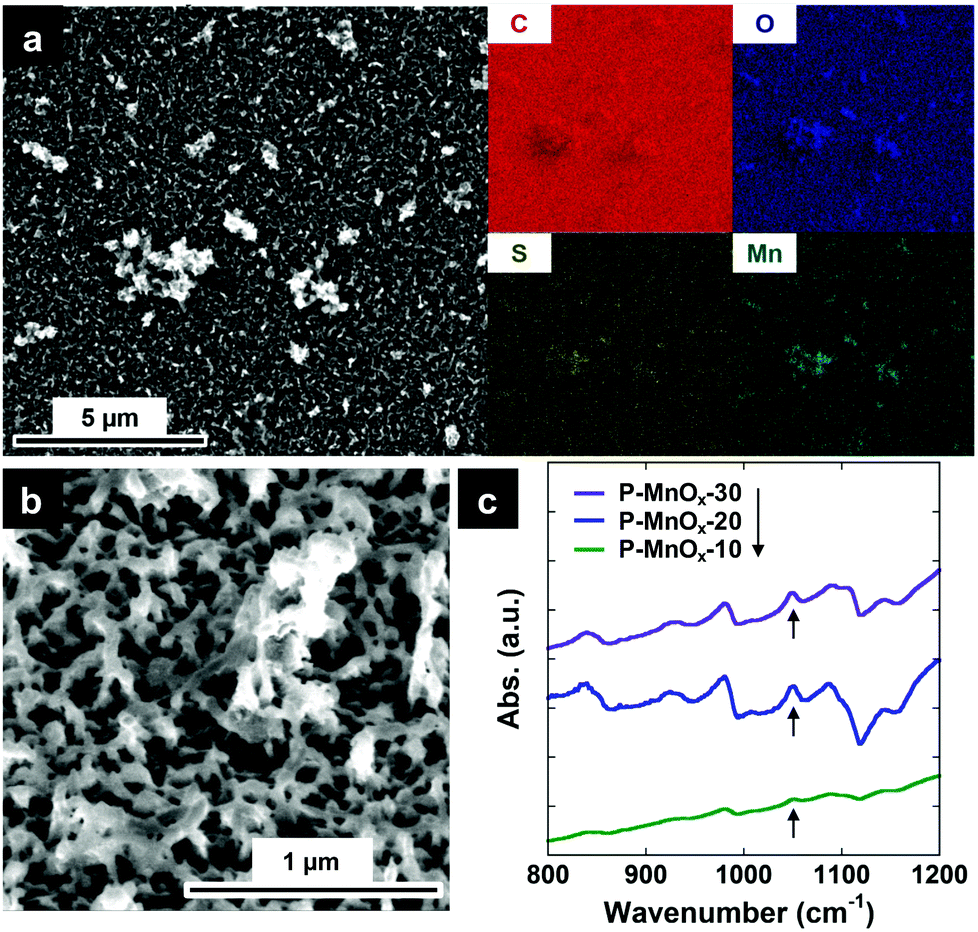
QCM measurements were used to understand the dynamics of the electrodeposition and to quantify the mass of PEDOT and subsequent MnOx on the PEDOT surface. Fig. 2a shows the mass change (calculated by the Sauerbrey equation) on a Ti/Pt crystal working electrode during the electropolymerization of PEDOT. The average mass loading of the PEDOT film after 20 s of electropolymerization is 20.8 ± 1.8 μg cm−2. Next, a PEDOT/MnOx film was prepared on the crystal identically to the method on a GC electrode: a 20 s PEDOT film was electropolymerized on the crystal, rinsed in DI H2O and transferred to a 0.01 M KMnO4 solution to monitor the uptake of MnOx by formation directly on the PEDOT film (Fig. 2b). 10, 20, and 30 min were chosen as the KMnO4 immersion times for the PEDOT films to assess the effects of changing MnOx formation time on film properties. Average MnOx mass loading of the hybrid films are 7.62 ± 1.28 μg cm−2, 9.94 ± 1.50 μg cm−2 and 11.51 ± 1.70 μg cm−2 for P-MnOx-10, P-MnOx-20 and P-MnOx-30, respectively. Thus, the average total mass loading for P-MnOx-10, P-MnOx-20 and P-MnOx-30 are 28.28 ± 3.13 μg cm−2, 30.6 ± 3.35 μg cm−2 and 32.17 ± 3.55 μg cm−2, respectively.
 | ||
| Fig. 2 (a) Electrode mass change during a typical galvanostatic electropolymerization of EDOT on a Ti/Pt QCM crystal; (b) electrode mass change during MnOx uptake on a 20 s (20.8 ± 1.8 μg cm−2) PEDOT film on the QCM crystal, when immersed in aqueous 0.01 M KMnO4. | ||
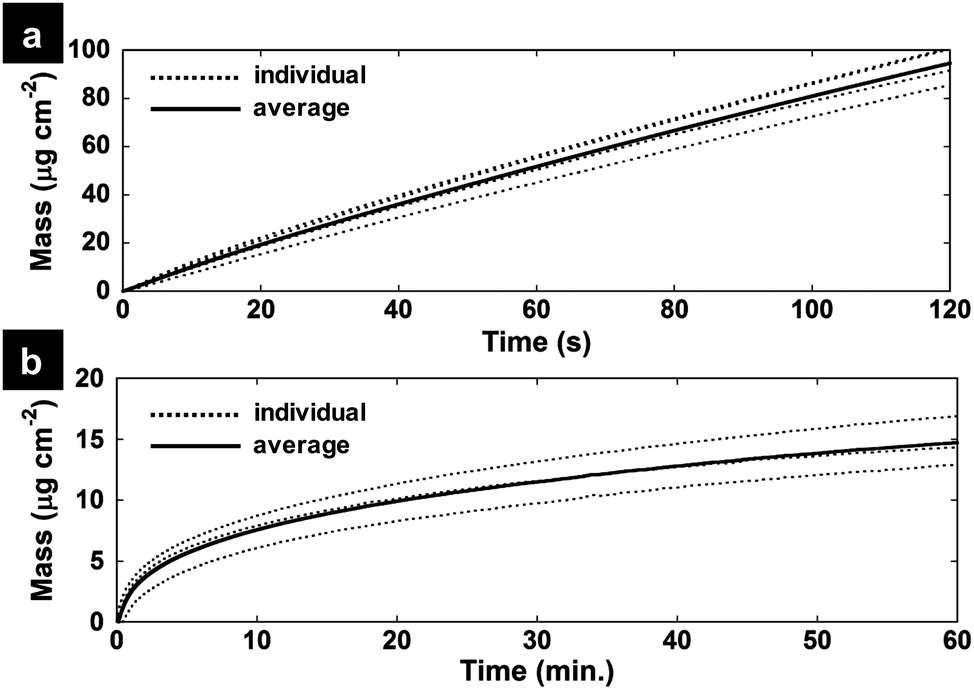
XPS survey spectra of the PEDOT, P-MnOx-10, P-MnOx-20, and P-MnOx-30 films are presented in Fig. 3a. The spectra indicate the presence of Mn (49 eV, 3p; 82–90 eV, 3s; 640–655 eV, 2p), S (163 eV, 2p; 228 eV, 2s), C (285 eV, 1s) and O (530 eV, 1s) for all PEDOT/MnOx films. The observation of Cl (207 eV, 2p) in the PEDOT only film suggests that the PEDOT is originally doped with ClO4− ions, which are removed (to the limit of detection) from the film upon reaction with aqueous MnO4− for as little as 10 min. Representative high resolution XPS spectra in the Mn 3s binding energy region are shown in Fig. 3b. Six measurements were taken in different locations on each film to analyze the characteristic 7S–5S multiplet peak splitting, which is strongly affected by Mn valence. This binding energy difference, ΔE(Mn 3s), can be used to determine relative concentrations of Mn(III) and Mn(IV) on the surface available for catalysis, according to known ΔE(Mn 3s) values for Mn(III)2O3 (5.4 eV) and Mn(IV)O2 (4.5 eV).38 Average experimental ΔE(Mn 3s) values as a function of MnOx formation time are shown in Fig. 3c, and the value is very close among the three films: all approximately within the standard error, the ΔE(Mn 3s) values are 4.95 ± 0.03 eV (P-MnOx-20) < 5.01 ± 0.06 eV (P-MnOx-30) < 5.05 ± 0.02 eV (P-MnOx-10). The values indicate a large concentration of Mn(III) on the surface for all three films.38 The electron transfer to O2 in the RDS of ORR occurs through the Mn(III)/Mn(IV) couple; thus, the Mn(III)/Mn(IV) ratio has been identified as a key activity descriptor for manganese oxide ORR electrocatalysts by our group and others.6,9,39 This result also shows that the spontaneous reduction of Mn(VII) on a PEDOT film results in a significantly reduced, mixed-valent manganese oxide (Mn(III)xMn(IV)yOz), as opposed to MnO2 (ΔE(Mn 3s) = 4.5 eV) as suggested by Liu et al.36
 | ||
| Fig. 3 (a) XPS survey spectra of the PEDOT and PEDOT/MnOx thin films on glassy carbon; (b) representative high resolution XPS spectra of the Mn 3s binding energy region; (c) Mn 3s multiplet splitting energy, ΔE(Mn 3s), as a function of the MnOx formation time for the PEDOT/MnOx thin films. | ||
Electrochemical properties of the PEDOT and PEDOT/MnOx films were determined in a three-electrode cell with a rotating glassy carbon working electrode, Ag/AgCl reference electrode (reported vs. RHE), and Pt counter electrode in 0.1 M KOH electrolyte. CV scans at a rate of 50 mV s−1 in the non-Faradaic potential window of 0.95–1.35 V are shown in Fig. 4a. The capacity of the electrode does not increase significantly upon formation of the PEDOT/MnOx hybrid (as compared to the 4-fold increase in capacitive current observed upon conversion of PEDOT nanowires to MnO2-NP/PEDOT nanowires by Liu et al.36), with integral charge of 244 μC (PEDOT), 354 μC (P-MnOx-10), 250 μC (P-MnOx-20), and 289 μC (P-MnOx-30) (Fig. 1a). Rigorous determination of the electrochemically active surface area (ECSA) or real surface area (RSA) requires experimentally determined double layer capacitance (CDL), and precise values of specific or reference capacitance, respectively. Because the specific and reference capacitance values are not known for the hybrid nanostructured film of PEDOT/MnOx, we have chosen to report the double-layer capacitance (CDL) as an approximation of the surface area accessible to the electrolyte and thus available for catalysis. CDL is proposed to provide a satisfactory comparison between PEDOT/MnOx films, as the PEDOT mass is the same for all films and the MnOx mass varies approximately within a standard deviation between the three samples. Fig. 4b shows the results of the repeated CV scans in a 0.1 V non-Faradaic potential window (all curves provided in Fig. S2, ESI†) at increasing scan rates. CDL values calculated from the slope of the correlation between scan rate and current near the switching potential demonstrate similar surface area for all films and reinforce the trends in charge capacity (Fig. 4a): 3.9 mF cm−2 (PEDOT) < 4.3 mF cm−2 (P-MnOx-20) < 4.9 mF cm−2 (P-MnOx-30) < 6.2 mF cm−2 (P-MnOx-10).
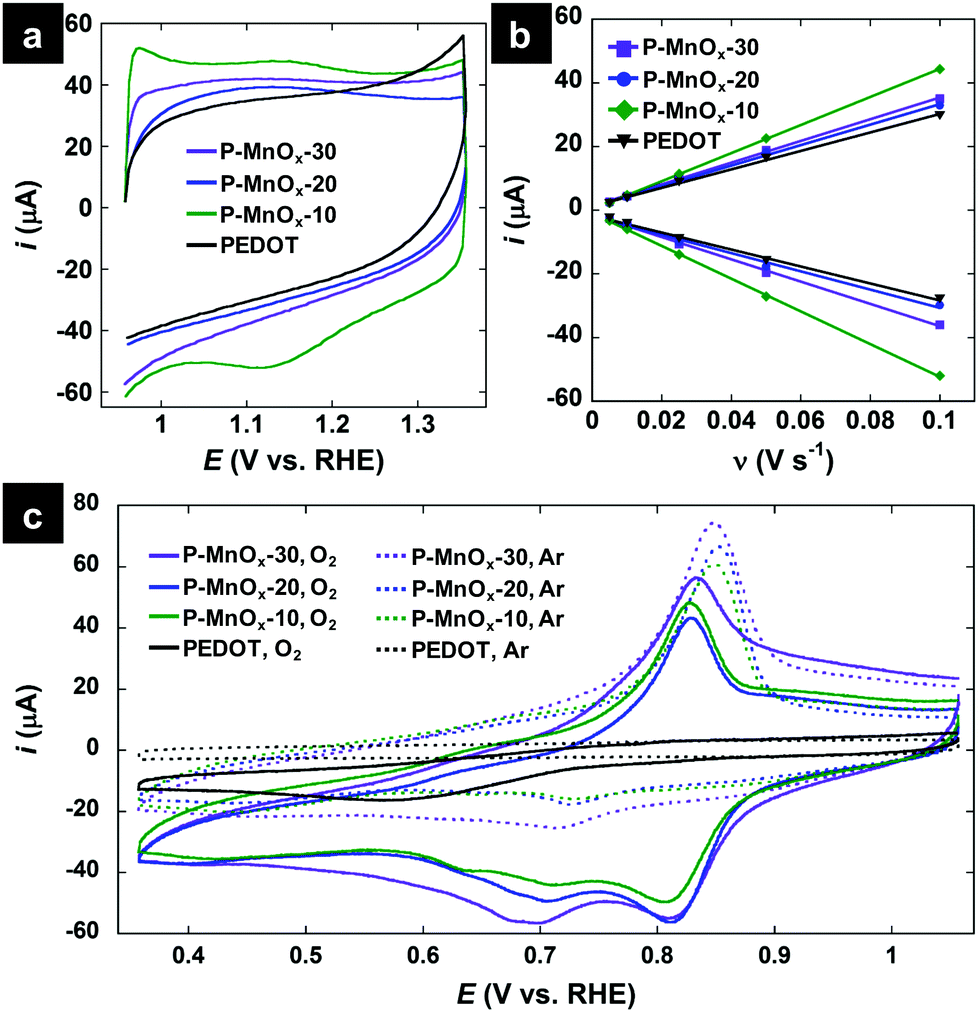 | ||
| Fig. 4 (a) CV scans at 50 mV s−1 in Ar-saturated 0.1 M KOH electrolyte; (b) current-scan rate plots to determine double-layer capacitance, where each point represents the near-switching potential (1.05 V vs. RHE) current of a CV curve at a discrete scan rate in Ar-saturated 0.1 M KOH electrolyte (all CV scans provided in Fig. S2, ESI†); (c) CV scans in the ORR potential window, in O2-free electrolyte (dotted curves, Ar-saturated 0.1 M KOH) and O2-saturated electrolyte (solid curves, O2-saturated 0.1 M KOH). | ||
CV scans in the ORR potential window in Ar-saturated and O2-saturated electrolyte are provided in Fig. 4c. The scans in O2-free electrolyte (dashed lines) show no redox activity from PEDOT and a sharp oxidation wave and weak reduction wave centered at ca. 0.77 V for the PEDOT/MnOx films, which can be attributed to the Mn(IV)/Mn(III) redox transitions on the surface.39 In O2-saturated electrolyte, a strong reduction wave is evident for the PEDOT film, signaling its ORR activity, however at a high overpotential (0.57 V). The O2 reduction wave for the PEDOT/MnOx films is shifted by more than 0.2 V to lower overpotential relative to PEDOT. The ORR peak potentials increase (improve) on the order of: PEDOT (0.57 V) < P-MnOx-10 (0.805 V) ∼ P-MnOx-30 (0.805 V) < P-MnOx-20 (0.81 V). The significant reduction in overpotential demonstrates the synergistic activity between MnOx and PEDOT, similar to that observed for the previously reported co-electrodeposited PEDOT/MnOx hybrid film.10 The stepwise synthetic method of MnOx formation directly on a PEDOT film precludes the evaluation of a MnOx control electrocatalyst on a GC electrode; however, the ORR peak potential for other amorphous MnOx catalysts are negative relative to the PEDOT/MnOx films,40,41 indicating there is beneficial electronic interaction between MnOx and PEDOT that facilitates the ORR at lower overpotential.10
Hydrodynamic LSV methods were employed to further investigate the ORR activity and determine the ORR electron transfer number (n) and kinetic ORR rate constant (k) values by Koutecky–Levich (K–L) analysis. Fig. 5a shows single LSV curves at 1600 RPM for the PEDOT, P-MnOx-10, P-MnOx-20 and P-MnOx-30 films, as well as a commercial benchmark, 20% Pt supported on Vulcan XC-72 (20% Pt/C). The ORR onset potential exhibits a similar positive shift upon the hybridization of MnOx with PEDOT, as seen using CV (Fig. 4c), from 0.68 V (PEDOT) to ca. 0.87 V (P-MnOx-10, P-MnOx-20, P-MnOx-30). The onset potential of 20% Pt/C is 0.89 V. Half-wave potentials for the five catalysts increase in the order of 0.57 V (PEDOT) < 0.81 V (20% Pt/C) < 0.82 V (P-MnOx-30) < 0.83 V (P-MnOx-10, P-MnOx-20). Diffusion-limiting ORR current is maximized with the P-MnOx-20 film, achieving −2.93 mA cm−2, vs. −1.07 mA cm−2, −2.74 mA cm−2 and −2.85 mA cm−2 for PEDOT, P-MnOx-10 and P-MnOx-30, respectively. It is worth noting that P-MnOx-20 demonstrates the highest diffusion-limiting current despite having the smallest charge capacity and CDL among the PEDOT/MnOx films (Fig. 4a and b). 20% Pt/C achieves diffusion-limiting current of −3.27 mA cm−2. Fig. 5b shows the LSV curves of the P-MnOx-20 and commercial 20% Pt/C catalysts normalized to specific catalyst mass, rather than the geometric area (Fig. 5a), in order to analyze the activity on an approximate ‘per-active-site’ basis. The P-MnOx-20 film outperforms 20% Pt/C at low overpotential (>0.7 V) when considered on a total catalyst mass basis, while the 20% Pt/C is clearly superior when normalized to metal (or metal oxide) mass basis. The diffusion-limiting specific mass activity (js) of 20% Pt/C is −108 mA mgtotal−1 (−540 mA mgPt−1), compared to −96 mA mgtotal−1 (−296 mA mgMnOx−1) for P-MnOx-20.
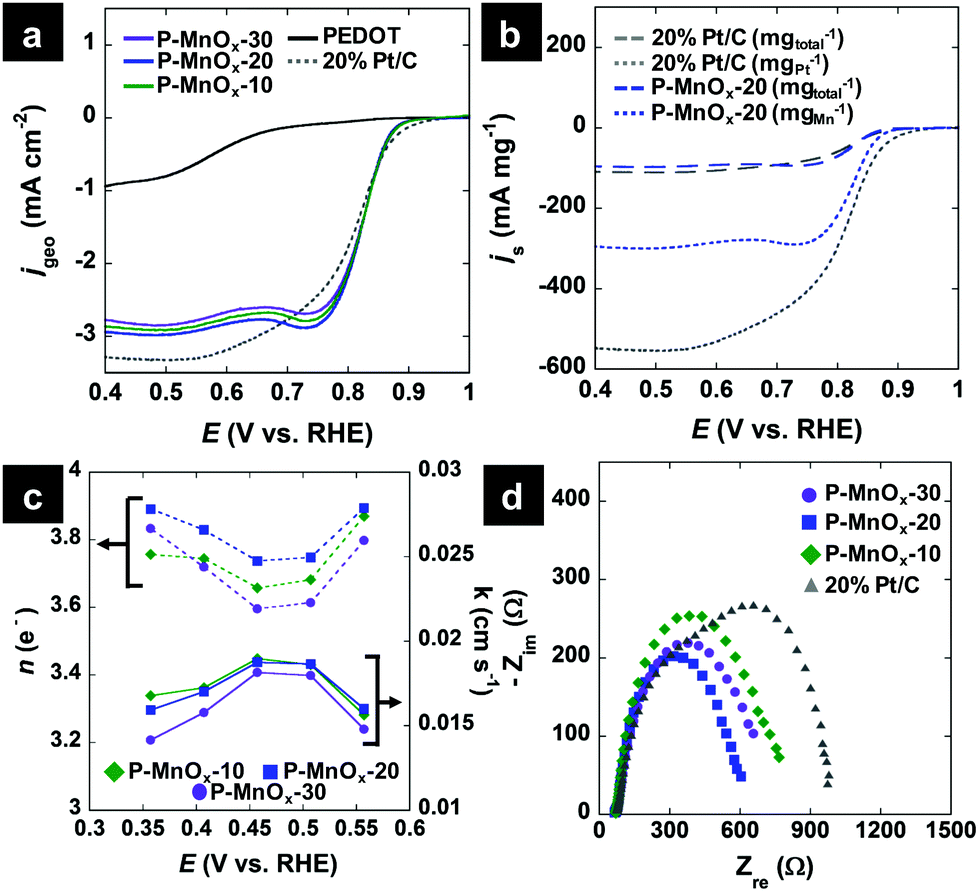 | ||
| Fig. 5 (a) Background-subtracted ORR geometric current density LSV curves at 1600 RPM in 0.1 M KOH electrolyte; (b) background-subtracted ORR specific mass activity LSV curves at 1600 RPM in 0.1 M KOH electrolyte; (c) K–L analysis, calculated n (dotted lines) and k (solid lines) metric values; (d) potentiostatic EIS Nyquist plots at the ORR half-wave potential in 0.1 M KOH electrolyte. | ||
Because of the similar activity in the onset and half-wave regions of the LSV curves for the PEDOT/MnOx films, K–L analysis and EIS were employed to determine the ORR pathway, kinetic constant, and charge transfer characteristics (All LSV curves are included in Fig. S3, ESI†). Fig. 5c shows the n and k values calculated from the slope and intercept, respectively, of the K–L plots, as a function of potential. Due to known issues in assumptions employed in the K–L analysis (particularly in alkaline electrolyte),42n was sometimes found to exceed the theoretical limit of 4 e− and is thus only considered in the range of 0.35–0.55 V. The average n values indicate a preference for the pseudo-four electron pathway for all PEDOT/MnOx catalysts, increasing in the order of: n = 3.7 (P-MnOx-10, P-MnOx-30) < 3.8 (P-MnOx-20). The pseudo-four electron pathway refers to the ORR pathway where O2 is first reduced electrochemically by two electrons to form a HO2− intermediate, followed by either (1) two-electron electrochemical reduction of HO2−, or (2) chemical disproportionation of HO2−, leading to regeneration and subsequent two-electron electrochemical reduction of O2. The competing electrochemical reduction and disproportionation of HO2− are evidenced by the high activity of Mn(III)/Mn(IV) oxides toward both processes.43k values show similarity in the intrinsic rate for the three PEDOT/MnOx catalysts, which could be expected from the identical slope in the kinetic-limited region of the LSV curves (Fig. 5a). Average k values show the following trend: 0.0175 cm s−1 (P-MnOx-10) > 0.0173 cm s−1 (P-MnOx-20) > 0.0162 cm s−1 (P-MnOx-30). Based on the ΔE(Mn 3s) analysis via XPS (Fig. 3c), P-MnOx-10 could be expected to exhibit the highest ORR rate, with (possibly) the highest concentration of surface Mn(III).6,9 However, it is likely that the Mn(III) surface concentration is similar between the three films, as ΔE(Mn 3s) values of both P-MnOx-10 and P-MnOx-20 are approximately within experimental standard deviation of P-MnOx-30. Optimal ORR charge transfer characteristics were determined by modelling EIS spectra to the equivalent Randles circuit. Fig. 5d shows the Nyquist plot spectra of the potentiostatic EIS experiments at 0.82 V with 0.01 V RMS perturbations. The real-axis intercepts demonstrate that P-MnOx-20 achieves the lowest ORR charge transfer resistance (RCT) in the half-wave region: 479 Ω (P-MnOx-20) < 545 Ω (P-MnOx-30) < 608 Ω (P-MnOx-10) < 862 Ω (20% Pt/C). Due to mechanical separation of the thin 30 μg cm−2 P-MnOx films from the electrode surface during long-term stability testing, the loading was increased to 80 μg cm−2 to assess the stability and MeOH tolerance of P-MnOx-20. The chronopotentiometric response is shown in Fig. S4 (ESI†) and demonstrates the stability of P-MnOx-20 at −1 mA cm−2 for three hours, including one hour in the presence of MeOH. The potential decreases only 15 mV over the three-hour period. The high stability rules out any near-term detrimental, irreversible MnOx reduction or competing MeOH oxidation.
Property-activity relations of PEDOT/MnOx electrocatalysts
The limited reports on PEDOT/ceramic composite electrocatalysts can be considered in two categories, PEDOT-modified metal oxide nanostructures31,32 and strongly coupled PEDOT-amorphous manganese oxide hybrid nanostructures10 (including this work). The high ORR overpotential and low mass activity of the CoMn2O4–PEDOT31 and PEDOT/α-MnO2/10AA32 composites (relative to the PEDOT/MnOx electrocatalysts reported here and in previous work10) is likely a synthetic limitation of the ceramic–polymer interfacial area, as the hybridization does not appear to significantly reduce intrinsic ORR overpotentials (onset, half-wave) of the ceramic. In contrast, the synergistic activity of hybrids derived from co-electrodeposition10 and redox-induced formation approaches are proposed to result in a nanostructure with strong electronic coupling and improved active site density by maximizing interfacial area between PEDOT and ceramic materials.We first reported a MnOx/PEDOT electrocatalyst prepared by an anodic, potentiostatic co-electrodeposition of MnOx and PEDOT from an aqueous micellar solution.10 The co-electrodeposition resulted in a uniformly distributed thin film of polymer and ceramic: uniform ca. 110 nm thickness (Fig. S5, ESI†), >50% MnOx content suggested by QCM studies, and nanoporosity on the order of ca. 10–50 nm.10 The MnOx/PEDOT exhibited a pseudo-four electron ORR pathway, half-wave potential 40 mV positive relative to 20% Pt/C, diffusion-limited mass activity of 40 mA mg−1, and Tafel slope (39 mV dec−1) lower than 20% Pt/C (Table 1).10 Here, the reported PEDOT/MnOx electrocatalysts were prepared via anodic, galvanostatic electrodeposition of PEDOT from organic solvent, followed by formation of MnOx particles via spontaneous reduction of aqueous MnO4− (Scheme 1). The route presented here results in a significantly different nanostructure to the co-electrodeposited films, with irregular thickness from ca. 20–370 nm, larger apparent porosity on the scale of ca. 50–200 nm, and <50% MnOx content (Fig. S5, ESI†). The P-MnOx-20 film showed similar high activity toward the ORR, with an n value of 3.8, half-wave potential 10 mV positive of 20% Pt/C, and RCT and Tafel slope (41 mV dec−1) lower than 20% Pt/C (Table 1 and Fig. S6, ESI†). The mass-transport corrected Tafel plots are also provided in Fig. S6 (ESI†) and confirm the low slope for P-MnOx-20. The similar n and Tafel slope values between co-electrodeposited MnOx/PEDOT and P-MnOx-20 suggest the same pathway/mechanism of ORR is taking place on the two hybrid surfaces. Furthermore, the Mn(III)/Mn(IV) ratio is a crucial indicator of ORR activity on manganese oxide surfaces,6,9 and the characteristic XPS Mn 3s multiplet splitting, ΔE(Mn 3s), suggest a very similar Mn(III)/Mn(IV) ratio on the surface of the co-electrodeposited MnOx/PEDOT film (4.98 ± 0.06 eV) as the P-MnOx-20 film (4.95 ± 0.03 eV) to facilitate the ORR (Fig. S5, ESI†). The diffusion-limited mass activity at 500 RPM for P-MnOx-20 is provided in Table 1 in order to compare to the electrodeposited PEDOT/MnOx10 catalyst that was reported at 400 RPM. The mass activity is higher for the P-MnOx-20 than the co-electrodeposited MnOx/PEDOT despite lower relative MnOx content and lower mass loading. For an ORR activity comparison of P-MnOx-20 to other PEDOT-based ORR electrocatalysts, the reader is referred to our previous articles7,10 for activity comparison tables.
| Catalyst | Ref. | Mass loading (μg cm−2) | Electrolyte/rotation rate | n (e−) | E half-wave (V vs. RHE) | Diffusion-limited mass activity (mA mg−1) | Tafel slope (mV dec−1) | EIS RCT (Ω) |
|---|---|---|---|---|---|---|---|---|
| P-MnOx-20 | This work | 30 | 0.1 M KOH/500 RPM | 3.8 | 0.83 | −65.3 | 41 | 479 |
| MnOx/PEDOT | 10 | 40 | 1 M KOH/400 RPM | 3.9 | 0.83 | −40.2 | 39 | 361 |
Thus, not only is the stepwise electropolymerization/spontaneous MnOx formation approach reported here potentially more versatile in nature, but it also results in a film with higher mass activity and catalyst (MnOx) efficiency than the co-electrodeposited PEDOT/MnOx. This result is proposed to originate from the MnOx residing exclusively on the surface of the PEDOT film in the P-MnOx-20 catalyst. In the co-electrodeposited approach, it is likely that potential MnOx electrocatalytic sites are obscured by PEDOT or internal to the larger MnOx platelets and not accessible. Thus, the stepwise approach of MnOx formation on a PEDOT film by spontaneous redox has proven an effective method for improved ORR activity via increased active site density and efficiency and represents an advance in PEDOT-based alkaline electrocatalysis for the ORR.
Conclusions
Hybrid PEDOT/MnOx thin film electrocatalysts were prepared by galvanostatic electrodeposition of PEDOT in organic solvent, followed by formation of MnOx particles on the surface via spontaneous reduction of aqueous MnO4−. High-resolution XPS studies in the Mn 3s binding energy region indicate a significant presence of Mn(III), in addition to the expected Mn(IV), on the surface of the electrode, with average ΔE(Mn 3s) multiplet splitting energy values of 5.05 eV (P-MnOx-10), 4.95 eV (P-MnOx-20), and 5.01 eV (P-MnOx-30). The electrocatalytic activity of the PEDOT/MnOx hybrid thin films towards the ORR was determined by CV and RDE LSV methods. The results indicate a strong electronic coupling between PEDOT and MnOx to facilitate the ORR at low overpotential, with a positive shift of >0.2 V in CV peak potential and LSV half-wave potential from PEDOT to P-MnOx-10, P-MnOx-20, and P-MnOx-30. Although all composite catalysts films show high activity at low overpotential, the P-MnOx-20 film exhibited the highest overall ORR activity, with onset and half-wave potentials of 0.87 and 0.83 V, respectively, n value of 3.8, and half-wave RCT of 479 Ω. Comparatively, commercial 20% Pt/C exhibited onset and half-wave ORR potentials of 0.89 and 0.81 V, respectively, n value of 3.9, and half-wave RCT of 862 Ω. Besides a more positive half-wave potential and lower RCT than 20% Pt/C, the P-MnOx-20 film also demonstrated high mass activity when considered on both a total mass (−96 mA mgtotal−1; 20% Pt/C: −108 mA mgtotal−1) and metal (or metal oxide) mass basis (−296 mA mgMnOx−1; 20% Pt/C: −540 mA mgPt−1). The reported P-MnOx-20 thin film was demonstrated to be the most active PEDOT–ceramic hybrid electrocatalyst reported so far for alkaline ORR. The origin of activity is attributed to strong electronic coupling between PEDOT and MnOx, high concentration of surface Mn(III), and the increase in active site density and efficiency realized by the spontaneous redox formation approach.Acknowledgements
Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy*s National Nuclear Security Administration under contract DE-NA0003525. Drs Michael Brumbach (XPS) and Erik Spoerke (IR) are thanked for their technical assistance and access to instrumentation.Notes and references
- O. Gröger, H. A. Gasteiger and J.-P. Suchsland, J. Electrochem. Soc., 2015, 162, A2605–A2622 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Larminie and A. Dicks, in Fuel Cell Systems Explained, John Wiley & Sons, Ltd, 2013, pp. 25–43 DOI:10.1002/9781118878330.ch2.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- R. J. Stanis, T. N. Lambert and M. A. Yaklin, Energy Fuels, 2010, 24, 3125–3129 CrossRef CAS.
- D. J. Davis, T. N. Lambert, J. A. Vigil, M. A. Rodriguez, M. T. Brumbach, E. N. Coker and S. J. Limmer, J. Phys. Chem. C, 2014, 118, 17342–17350 CAS.
- T. N. Lambert and J. A. Vigil, ECS Trans., 2016, 75, 965–970 CrossRef.
- T. N. Lambert, J. A. Vigil, S. E. White, D. J. Davis, S. J. Limmer, P. D. Burton, E. N. Coker, T. E. Beechem and M. T. Brumbach, Chem. Commun., 2015, 51, 9511–9514 RSC.
- T. N. Lambert, J. A. Vigil, S. E. White, C. J. Delker, D. J. Davis, M. Kelly, M. T. Brumbach, M. A. Rodriguez and B. S. Swartzentruber, J. Phys. Chem. C, 2017, 121, 2789–2797 CAS.
- J. A. Vigil, T. N. Lambert and K. Eldred, ACS Appl. Mater. Interfaces, 2015, 7, 22745–22750 CAS.
- N. Ramaswamy and S. Mukerjee, Adv. Phys. Chem., 2012, 2012, 491604 Search PubMed.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- Q. He and E. J. Cairns, J. Electrochem. Soc., 2015, 162, F1504–F1539 CrossRef CAS.
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293–301 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- C. Jin, F. Lu, X. Cao, Z. Yang and R. Yang, J. Mater. Chem. A, 2013, 1, 12170–12177 CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- T. N. Lambert, D. J. Davis, W. Lu, S. J. Limmer, P. G. Kotula, A. Thuli, M. Hungate, G. Ruan, Z. Jin and J. M. Tour, Chem. Commun., 2012, 48, 7931–7933 RSC.
- C. Jiang, Z. Guo, Y. Zhu, H. Liu, M. Wan and L. Jiang, ChemSusChem, 2015, 8, 158–163 CrossRef CAS PubMed.
- Y. Gong, H. Fei, X. Zou, W. Zhou, S. Yang, G. Ye, Z. Liu, Z. Peng, J. Lou, R. Vajtai, B. I. Yakobson, J. M. Tour and P. M. Ajayan, Chem. Mater., 2015, 27, 1181–1186 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. A. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- B. Winther-Jensen, O. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Science, 2008, 321, 671–674 CrossRef CAS PubMed.
- V. G. Khomenko, V. Z. Barsukov and A. S. Katashinskii, Electrochim. Acta, 2005, 50, 1675–1683 CrossRef CAS.
- Q. Zhou and G. Shi, J. Am. Chem. Soc., 2016, 138, 2868–2876 CrossRef CAS PubMed.
- R. Kerr, C. Pozo-Gonzalo, M. Forsyth and B. Winther-Jensen, ECS Electrochem. Lett., 2013, 2, F29–F31 CrossRef CAS.
- J. Ma, T. Xue, Z. Zou, B. Wang and M. Luo, Int. J. Electrochem. Sci., 2015, 10, 4562–4570 CAS.
- Z. Guo, Y. Qiao, H. Liu, C. Ding, Y. Zhu, M. Wan and L. Jiang, J. Mater. Chem., 2012, 22, 17153–17158 RSC.
- Z. Guo, H. Liu, C. Jiang, Y. Zhu, M. Wan, L. Dai and L. Jiang, Small, 2014, 10, 2087–2095 CrossRef CAS PubMed.
- Z. Guo, C. Jiang, C. Teng, G. Ren, Y. Zhu and L. Jiang, ACS Appl. Mater. Interfaces, 2014, 6, 21454–21460 CAS.
- M. Zhang, W. Yuan, B. Yao, C. Li and G. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 3587–3593 CAS.
- A. D. Chowdhury, N. Agnihotri, P. Sen and A. De, Electrochim. Acta, 2014, 118, 81–87 CrossRef CAS.
- Y.-L. Kuo, C.-C. Wu, W.-S. Chang, C.-R. Yang and H.-L. Chou, Electrochim. Acta, 2015, 176, 1324–1331 CrossRef CAS.
- N. Sakmeche, S. Aeiyach, J.-J. Aaron, M. Jouini, J. C. Lacroix and P.-C. Lacaze, Langmuir, 1999, 15, 2566–2574 CrossRef CAS.
- S.-B. Ma, K.-Y. Ahn, E.-S. Lee, K.-H. Oh and K.-B. Kim, Carbon, 2007, 45, 375–382 CrossRef CAS.
- S.-B. Ma, Y.-H. Lee, K.-Y. Ahn, C.-M. Kim, K.-H. Oh and K.-B. Kim, J. Electrochem. Soc., 2006, 153, C27–C32 CrossRef CAS.
- R. Liu, J. Duay and S. B. Lee, ACS Nano, 2010, 4, 4299–4307 CrossRef CAS PubMed.
- W. R. Fawcett and A. A. Kloss, J. Phys. Chem., 1996, 100, 2019–2024 CrossRef CAS.
- A. J. Nelson, J. G. Reynolds and J. W. Roos, J. Vac. Sci. Technol., A, 2000, 18, 1072–1076 CAS.
- A. S. Ryabova, F. S. Napolskiy, T. Poux, S. Y. Istomin, A. Bonnefont, D. M. Antipin, A. Y. Baranchikov, E. E. Levin, A. M. Abakumov, G. Kéranguéven, E. V. Antipov, G. A. Tsirlina and E. R. Savinova, Electrochim. Acta, 2016, 187, 161–172 CrossRef CAS.
- J. Yang and J. J. Xu, Electrochem. Commun., 2003, 5, 306–311 CrossRef CAS.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- A. S. Ryabova, A. Bonnefont, P. Zagrebin, T. Poux, R. Paria Sena, J. Hadermann, A. M. Abakumov, G. Kéranguéven, S. Y. Istomin, E. V. Antipov, G. A. Tsirlina and E. R. Savinova, ChemElectroChem, 2016, 3, 1667–1677 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00147a |
| This journal is © the Partner Organisations 2017 |