
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of α-substituted cyclic boronates via titanium-catalyzed cyclization of vinyl boronates with dihaloalkanes†
Ximei
Tian
ab and
Lipeng
Wu
 *ac
*ac
aState Key Laboratory of Low Carbon Catalysis and Carbon Dioxide Utilization, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou, 730000, P. R. China. E-mail: lipengwu@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
cCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, P. R. China
First published on 12th March 2025
Abstract
Cyclic boronates are versatile synthons for organic synthesis and for introducing ring systems into bioactive molecules. Existing synthetic methods have narrow substrate scope and the synthesis of α-substituted cyclic boronates is still elusive. Furthermore, no general method for synthesizing cyclic boronates with different ring sizes and hetero atom containing rings is available. Herein, we present a new and general synthetic method for synthesizing α-substituted cyclic boronates. Our approach has the advantage of using earth-abundant Ti as the catalyst and readily available dihaloalkanes, such as dichloromethane, as the reactant. Cyclic boronates that are otherwise difficult to access, such as α-substituted cyclic boronates with three-, four-, five-, and six-membered rings, heteroatom-containing rings, and cyclic boronates with spiro rings, are readily obtained.
Alkyl boronates are synthetically valuable compounds in organic chemistry,1–7 and they also find applications in medicinal chemistry, an example is bortezomib, which is an approved drug for treating relapsed multiple myeloma and mantle cell lymphoma.8,9 Therefore, significant efforts have been devoted to developing new synthetic methodologies for their preparation. Among them, the synthesis of cyclic boronates has attracted particular attention in recent years. Cyclic boronates are versatile synthons for functionalizing ring systems and introducing them into molecular architectures. The latter is a pivotal strategy in drug design and biological studies,10–12 which can improve the lipophilicity13 and metabolic stability of compounds,14 and provide entropically favorable binding via conformational constraint of drug molecules (Fig. 1A).15,16 In this respect, methodologies such as C–H boration of cycloalkanes,17–21 hydroboration of cyclopropenes,22–24 reaction of allylic carbonates, phosphonates, or homoallylic sulfonates with diboron,25–27 cyclopropanation of alkenyl boronates with ethyl diazoacetate,28 decarboxylative borylation of N-hydroxyphthalimide esters of cyclic carboxylic acids,29,30 borylation of benzyltrimethylammonium with cyclic rings,31 reaction of lithiated 1,1-diborylalkanes with α-halo small rings,32,33 and several other recent advances34–40 have been developed that essentially extended the availability of cyclic boronates (Fig. 1B). However, despite these achievements, several limitations remain: (1) many methods depend on existing ring systems, which limit the availability of the starting material; (2) methods that do not rely on existing ring systems are generally limited to the synthesis of three-membered ring boronates, and the synthesis of boronates with other four-, five-, or six-membered rings or spiro rings remains elusive; and (3) no general method for the synthesis of α-substituted cyclic boronates, which contain a quaternary carbon center, is available.12,32,33,37–39,41–43 Noticed that, it is very challenging to obtain α-substituted cyclic boronates using most of the developed methods such as C–H boration of cyclopropanes and hydroboration of cyclopropenes (Fig. 1B). Therefore, the development of novel and versatile synthetic methodologies is essential.
 | ||
| Fig. 1 (A) Representative drug molecules containing ring systems; (B) recently developed methods for the construction of cyclic boronates and their limitations; (C) titanium-catalyzed cyclization of vinyl boronates with dihaloalkanes for the synthesis of structurally diverse cyclic boronates. | ||
Recently, transition-metal-catalyzed reductive cyclopropanation of alkenes with gem-dihaloalkanes or its analogs (Simmons–Smith type reaction) was developed as an efficient method for constructing the cyclopropane motif.44–54 Their application for the synthesis of α-substituted cyclopropyl boronates were also studied.55,56 We also noticed that an excess amount of early-transition metal titanium(II) complexes or sub-stoichiometric titanium(III) complexes could be used to promote the cyclopropanation of gem-dihalides or thioacetals with alkenes or allylic alcohols.57–61 Taken together, and in line with our interest in exploring titanium as a potential catalyst for various synthetic valuable transformations,62–64 herein, we report the success of our investigation of titanium as a catalyst for the reductive cyclization of α-substituted vinyl boronates for the synthesis of structurally diverse cyclic boronates. Our system features the advantages of using readily available and inexpensive titanium as the catalyst and dichloromethane or other dihaloalkanes as the reactant. As a result, α-substituted cyclic boronates with three-, four-, five-, and six-membered rings, including heteroatom-containing rings, which are otherwise difficult to obtain, were readily produced. In addition, cyclic boronates with spiro rings were successfully accessed using this method (Fig. 1C).
To start the investigation, 1-phenyl vinyl boronate (1a, 0.2 mmol) was reacted with dichloromethane (2a, 1.0 mmol) using Mn (2 equiv.) as the reductant in THF (1 mL) at 80 °C (Table 1). Among the titanium complexes investigated, Cp2TiCl2 was the most efficient catalyst, giving the target product 3a in 35% yield (Table 1, entries 1–5). The addition of LiCl improved the yield of 3a to 48% (entries 6–8). The low yield of 3a was mainly due to the low conversion; thus, the yield of 3a was further improved by enhancing the amount of Mn and dichloromethane (entries 9–12). Under these conditions, an 80% yield of 3a was obtained at 80 °C; enhancing the temperature to 120 °C only slightly improved the yield of 3a to 87% (entry 13). Despite the minimally improved yield, to ensure good reproducibility and generality, we conducted the subsequent substrate scope studies at 120 °C.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Ti] | Additives | x | y | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.2 mmol), Mn (x equiv.), 2a (y equiv.), cat. (10 mol%), additive (1 equiv.), THF (1 mL) in 15 mL pressure tube, stirring, 80 °C, 20 h. b Yields were determined by GC with dodecane as internal standard. c Reaction was performed at 120 °C. | |||||
| 1 |

|
— | 2 | 5 | 8 |
| 2 | InTiCl3 | — | 2 | 5 | 14 |
| 3 | CpTiCl3 | — | 2 | 5 | 16 |
| 4 | Cp*TiCl3 | — | 2 | 5 | 17 |
| 5 | Cp2TiCl2 | — | 2 | 5 | 35 |
| 6 | Cp2TiCl2 | TMS-Cl | 2 | 5 | 23 |
| 7 | Cp2TiCl2 | MgCl2 | 2 | 5 | 40 |
| 8 | Cp2TiCl2 | LiCl | 2 | 5 | 48 |
| 9 | Cp2TiCl2 | LiCl | 4 | 5 | 53 |
| 10 | Cp2TiCl2 | LiCl | 5 | 5 | 62 |
| 11 | Cp2TiCl2 | LiCl | 5 | 8 | 72 |
| 12 | Cp2TiCl2 | LiCl | 5 | 10 | 80 |
| 13c | Cp2TiCl2 | LiCl | 5 | 10 | 87 |

Various 1-substituted vinyl boronates 1 were then reacted with dichloromethane 2a to produce the corresponding α-substituted cyclopropyl boronates 3 (Scheme 1). Vinyl boronates substituted with electron-donating groups such as –Me, –Et, –tBu, –Ph, –MeO reacted well to give the products 3a–g in 42–79% isolated yields. Interestingly, halide groups on the phenyl ring, such as –Cl, –Br, and –CF3, remained intact under the reaction conditions to give 3h–j (58–75% yields). In addition, heteroatom-containing groups such as –TMS, –Me2N, and morpholine had no deleterious effects on the results (3k–m, 66–76% yields). Functional groups such as ester and nitrile were also suitable, giving the corresponding products 3n and 3o in moderate yields of 58% and 68%, respectively. Vinyl boronates with multi-substituted and fused benzene rings, including 1,3-benzodioxole, 2,3-dihydrobenzo[b][1,4]dioxine, and dibenzothiophene, reacted well to give the corresponding cyclic boronates 3q–t in 58–78% yields. Interestingly, when deuterated dichloromethane (2b) was used, the corresponding d2-labeled cyclic boronate 3u was obtained. The presence of the gem-dimethyl group is believed to improve the potency or eliminate metabolic liabilities of biologically active compounds.47,65 We then successfully applied 2,2-dichloropropane (2c) for the dimethylcyclopropanation with vinyl boronates, and product 3v was obtained in good yield. Unfortunately, the current system does not work for aliphatic vinyl boronates (Scheme S1†).
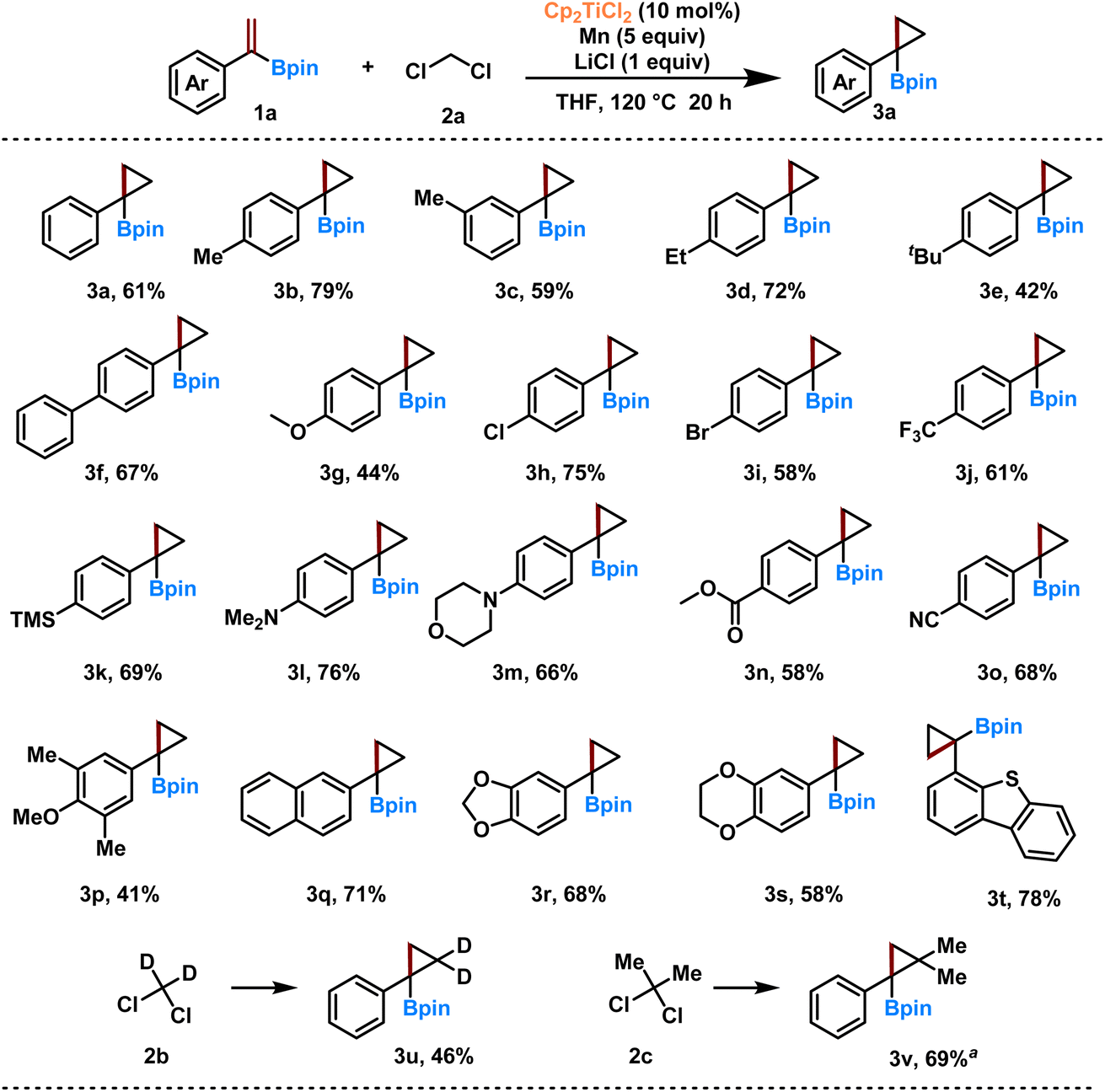 | ||
| Scheme 1 Substrate scope for the Ti-catalyzed synthesis of α-substituted cyclopropyl boronates-reaction conditions: 1 (0.2 mmol), 2 (10 equiv.), Cp2TiCl2 (10 mol%), Mn (5 equiv.), LiCl (1 equiv.), THF (1 mL), 15 mL pressure tube, 120 °C, 20 h; isolated yields; a2 equivalent of 2c. | ||
The developed method was then applied to the synthesis of cyclic boronates, which are not easily accessible by other methods (Scheme 2). We were pleased that when using 1,3-dichloropropane (2d) as the reactant, α-substituted cyclopentyl boronates 4a–d could be obtained. More interestingly, using bis(chloromethyl)sulfane (2e) as the dichloroalkane partner, products 5a–f with S-containing 5-membered rings were produced. To our knowledge, cyclic boronates like 5 have never been successfully synthesized. Then, 1,4-dichlorobutane (2f) was also successfully applied for the cyclization reaction, and α-substituted cyclohexyl boronate 6a was obtained.
 | ||
| Scheme 2 Substrate scope for the Ti-catalyzed synthesis of α-substituted cyclic boronates-reaction conditions: avinyl boronate (0.2 mmol), dichloroalkane (2 equiv.), Cp*TiCl3 (10 mol%), Mn (5 equiv.), LiCl (1 equiv.), THF (1 mL), 15 mL pressure tube, 80 °C, 20 h, isolated yields; bInTiCl3 (10 mol%), 120 °C; cvinyl boronate (0.2 mmol), bis(bromomethyl)benzene (2 equiv.), 1 equiv. AlCl3 in 1 mL DMA, 40 °C for 20 h, isolated yields; d40 mol% TiCl4, 60 °C, isolated yields after oxidation. | ||
To synthesize spirocyclic boronates, 1,1-dichloro-4,4-dimethylcyclohexane (2g) was reacted with vinyl boronate 1a, and we found that the target spirocyclic boronate 7a was produced in moderate yield. Inspired by this result, 4,4-dichlorotetrahydro-2H-thiopyran (2h) and 4,4-dichlorotetrahydro-2H-pyran (2i) were subjected to the reaction with vinyl boronates substituted with various functional groups, and the corresponding spirocyclic boronates 8a and 9a–h were obtained.
To our knowledge, no other methods have been reported for the synthesis of boronates such as 7–9. We further extended our methodology to synthesize more complex compounds and biomolecule-derived spirocyclic boronates 9i–l, including fenchol and menthol-derived molecules. Finally, spirocyclic boronate 10a, with a nitrogen-containing ring, was obtained in 63% yield by reacting ethyl 4,4-dichloropiperidine-1-carboxylate with 1a.
We also examined the use of 1,2-bis(bromomethyl)benzene (2j) as the dihaloalkane partner, the reaction of which with vinyl boronates would produce the corresponding benzene-fused cyclic boronates 11. Surprisingly, 2j reacted readily with vinyl boronates in the presence of Mn, even without a Ti catalyst, giving 11a in 35% GC yield. This might be due to the high reactivity of the benzylic bromide moiety, which generates a benzylic radical just in the presence of Mn. Adding 1 equiv. of a Lewis acid AlCl3, which is believed to facilitate the radical addition, increased the yield of 11a to 75% GC yields. Nevertheless, various substituted vinyl boronates reacted well, including –Cl, –COOMe, and –CN substituted derivatives (11a–h). Finally, efforts toward the synthesis of cyclobutyl boronate were conducted using 1,2-dichloroethane, and cyclobutyl boronate 12a was obtained in 34% yield under the optimized conditions using TiCl4.
Having generated a range of α-substituted cyclic boronates, the robustness of our method for gram-scale synthesis was assessed and it was found that boronates 3a and 3h were produced in 62% and 76% isolated yields on a 5 mmol scale (Scheme 3). The synthetic utility of these compounds was demonstrated in a series of downstream transformations. First, olefination of 3h with vinyl magnesium bromide produced terminal alkene 13a in 41% yield. Treating 3h with furan-2-yllithium followed by NBS afforded the arylated product 13b in 68% yield. The aryl chloride moiety could also be further borylated to produce bis-boronates 13c with one alkyl and aryl boronate functionality. The oxidation and Suzuki–Miyaura coupling of 3a were also performed, giving 1-phenylcyclopropanol 13d and the corresponding arylation product 13e. The arylation of 3a with a tethered free hydroxyl group was also achieved using a photo/Ni dual catalysis, the product of which could be further derivatized with 1-adamantanecarboxylic acid and 4-((6-(acryloyloxy)hexyl)oxy)benzoic acid to produce the corresponding products 13g and 13h.
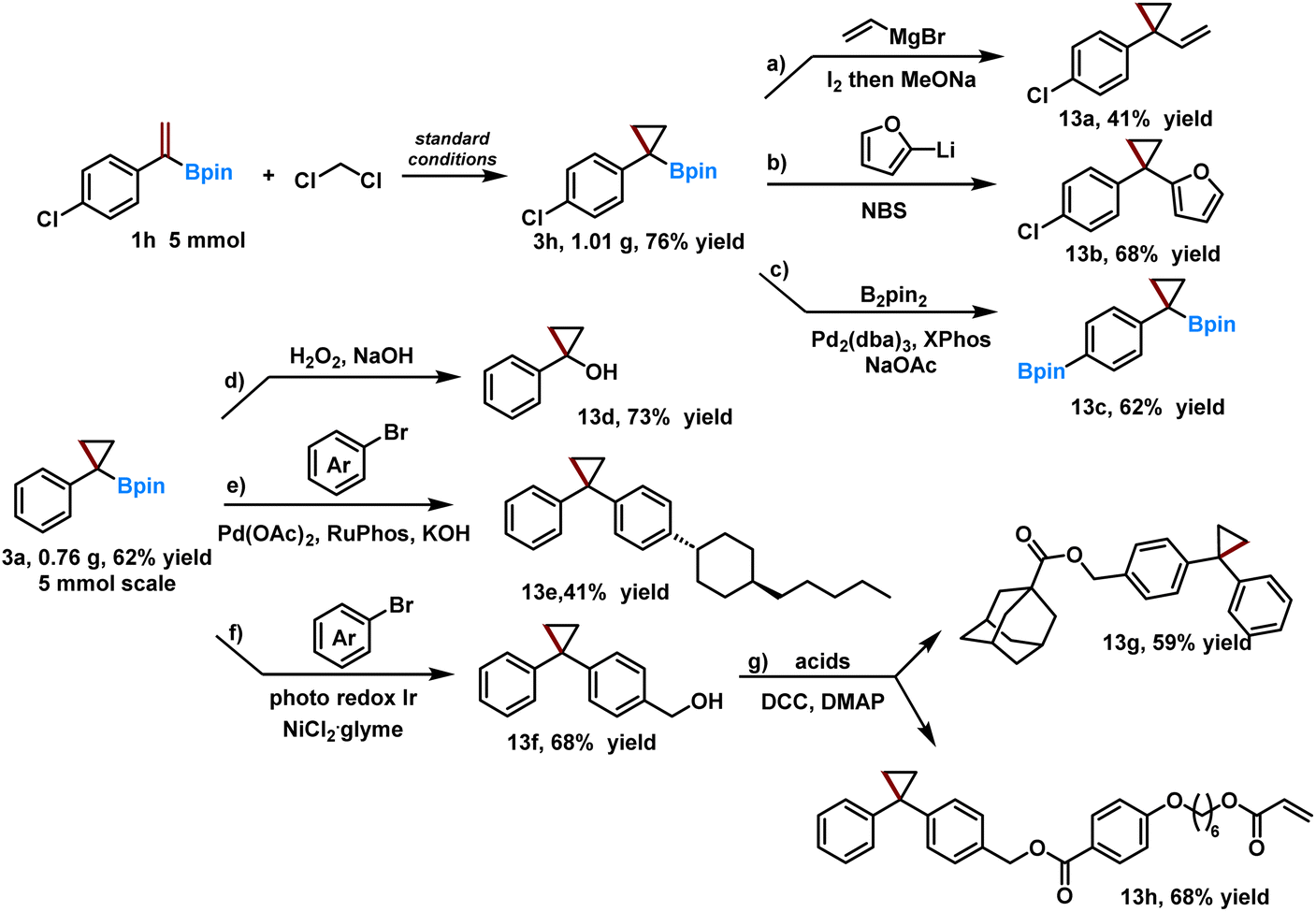 | ||
| Scheme 3 Gram-scale synthesis and synthetic utilization. (a) 0.5 mmol 3h, 2 mmol vinyl magnesium bromide (1.0 M in THF), 2 mmol I2, 4 mmol NaOMe; (b) 0.5 mmol 3h, 0.66 mmol furan, 0.66 mmol nBuLi (1.6 M in THF), 0.6 mmol NBS; (c) 0.5 mmol 3h, 0.51 mmol B2pin2, 1 mol% Pd2(dba)3, XPhos 2 mol%, 0.6 mmol NaOAc; (d) 0.5 mmol 3a, NaOH (2 M aq., 1 mL), H2O2 (30% aq., 0.5 mL); (e) 0.5 mmol 3a, 10 mol% Pd(OAc)2, 20 mol% RuPhos, 2 mmol KOH, 1 mmol aryl bromide; (f) 1.0 mmol 3a, 0.5 mmol aryl bromide, 1 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, 0.75 mmol morpholine, 5 mol% NiCl2 glyme and 5 mol% dtbbpy; (g) 0.5 mmol corresponding acids, 0.75 mmol 13f, 10 mol% DMAP, 0.55 mmol DCC. For detailed conditions, please refer to the ESI.† | ||
Several control experiments were then conducted to shed light on the potential reaction mechanism. Initially, we carried out radical inhibitor experiments by adding the radical scavengers 2,2,6,6-tetramethylpiperidinyl-1-oxide (TEMPO) or 9,10-dihydroanthracene (DHA) to the standard reaction (Scheme 4). We found that the inclusion of TEMPO (2 equiv.) shut down the reaction completely (Scheme 4a) and that adding increasing amounts of DHA led to a gradual reduction in the yield of 3a (Scheme 4b). These results indicate that a radical species might be involved in the reaction. Further support for this was obtained from radical clock experiments in which the ring-opening product 14b was obtained in 42% yield when (1-cyclopropylvinyl)benzene (14a) was allowed to react with dichloromethane (Fig. S1 and 2†). The production of 14b provided additional support for a radical reaction pathway and indicated the stepwise activation of the two –Cl moieties from dichloromethane. Moreover, the mass fragment corresponding to a possible α-boryl alkyl titanium intermediate 14c was observed by HRMS in the stoichiometric reaction after reaction for 1.5 h.
 | ||
| Scheme 4 Mechanistic studies: (a and b) radical inhibitor experiments by adding TEMPO or DHA to the standard catalytic reaction; (c) radical clock experiment using 14a as the substrate; (d) HRMS spectrum shows the formation of a key intermediate. | ||
We proposed the following reaction mechanism based on the above experimental results and literature reports (Fig. 2). First, Cp2TiIIICl is generated from the reduction of Cp2TiIVCl2 by Mn. The former is well known as a single-electron transfer (SET) agent66–71 that can activate the dihaloalkane species to form the monochloroalkyl radical species A. Radical addition to the vinyl boronates produces radical species B in which the boryl group can stabilize the neighboring carbon radical. Radical capture by Cp2TiIIICl then generates the key α-boryl alkyl titanium intermediate C (m/z fragment observed by HRMS), which undergoes reductive ring closure to the cyclic boronates with the regeneration of Cp2TiIVCl2.60
 | ||
| Fig. 2 Proposed catalytic cycle. | ||
A new synthetic method for the general synthesis of α-substituted cyclic boronates has been developed and is presented herein. This method has the advantages of using earth-abundant Ti as the catalyst and readily available dihaloalkanes, such as dichloromethane, as the reactant. As a result, α-substituted cyclic boronates with three-, four-, five-, and six-membered rings and heteroatom-containing rings, which are difficult to access using other methods, were readily obtained. Moreover, cyclic boronates with spiro rings were also gained. Studies on the mechanism indicate a Ti-catalyzed stepwise reductive cyclization pathway.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. Wu, X. Tian conceived the project and designed the experiments. X. Tian performed the experiments and analyzed the data. L. Wu, X. Tian wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22271295), Gansu Provincial Natural Science Foundation Key Project (23JRRA606), Major Program of the Lanzhou Institute of Chemical Physics, CAS (No. ZYFZFX-9), State Key Laboratory Program of the Lanzhou Institute of Chemical Physics (CHGZ-202208), CAS for generous financial support.Notes and references
- A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed.
- A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed.
- Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3160–3161 CrossRef CAS PubMed.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107–19110 Search PubMed.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Science, 2020, 368, 736–741 Search PubMed.
- M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910–6911 Search PubMed.
- L. J. Milo, J. H. Lai, W. Wu, Y. Liu, H. Maw, Y. Li, Z. Jin, Y. Shu, S. E. Poplawski, Y. Wu, D. G. Sanford, J. L. Sudmeier and W. W. Bachovchin, J. Med. Chem., 2011, 54, 4365–4377 Search PubMed.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 Search PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 Search PubMed.
- J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037–8047 Search PubMed.
- K.-L. Yu, N. Sin, R. L. Civiello, X. A. Wang, K. D. Combrink, H. B. Gulgeze, B. L. Venables, J. J. K. Wright, R. A. Dalterio, L. Zadjura, A. Marino, S. Dando, C. D'Arienzo, K. F. Kadow, C. W. Cianci, Z. Li, J. Clarke, E. V. Genovesi, I. Medina, L. Lamb, R. J. Colonno, Z. Yang, M. Krystal and N. A. Meanwell, Bioorg. Med. Chem. Lett., 2007, 17, 895–901 Search PubMed.
- K. Futatsugi, D. W. Kung, S. T. M. Orr, S. Cabral, D. Hepworth, G. Aspnes, S. Bader, J. Bian, M. Boehm, P. A. Carpino, S. B. Coffey, M. S. Dowling, M. Herr, W. Jiao, S. Y. Lavergne, Q. Li, R. W. Clark, D. M. Erion, K. Kou, K. Lee, B. A. Pabst, S. M. Perez, J. Purkal, C. C. Jorgensen, T. C. Goosen, J. R. Gosset, M. Niosi, J. C. Pettersen, J. A. Pfefferkorn, K. Ahn and B. Goodwin, J. Med. Chem., 2015, 58, 7173–7185 CrossRef CAS PubMed.
- J. P. Davidson, O. Lubman, T. Rose, G. Waksman and S. F. Martin, J. Am. Chem. Soc., 2002, 124, 205–215 Search PubMed.
- M. R. Harris, H. M. Wisniewska, W. Jiao, X. Wang and J. N. Bradow, Org. Lett., 2018, 20, 2867–2871 Search PubMed.
- C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 3375–3378 CrossRef CAS PubMed.
- Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599–10604 Search PubMed.
- Y. Shi, Y. Yang and S. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201463 CrossRef CAS PubMed.
- Q. Gao and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218025 CrossRef CAS PubMed.
- T. Xie, L. Chen, Z. Shen and S. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300199 CrossRef CAS PubMed.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed.
- B. Tian, Q. Liu, X. F. Tong, P. Tian and G. Q. Lin, Org. Chem. Front., 2014, 1, 1116–1122 Search PubMed.
- A. Parra, L. Amenos, M. Guisan-Ceinos, A. Lopez, J. L. Garcia Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef CAS PubMed.
- H. Ito, Y. Kosaka, K. Nonoyama, Y. Sasaki and M. Sawamura, Angew. Chem., Int. Ed., 2008, 47, 7424–7427 CrossRef CAS PubMed.
- H. Ito, T. Toyoda and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 5990–5992 CrossRef CAS PubMed.
- C. Zhong, S. Kunii, Y. Kosaka, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 11440–11442 CrossRef CAS PubMed.
- J. Carreras, A. Caballero and P. J. Perez, Angew. Chem., Int. Ed., 2018, 57, 2334–2338 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 Search PubMed.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 Search PubMed.
- J. Hu, H. Sun, W. Cai, X. Pu, Y. Zhang and Z. Shi, J. Org. Chem., 2016, 81, 14–24 Search PubMed.
- T. R. McDonald, J. A. Turner, A. L. Gabbey, P. Balasubramanian and S. A. L. Rousseaux, Org. Lett., 2024, 26, 3822–3827 CrossRef CAS PubMed.
- T. R. McDonald and S. A. L. Rousseaux, Chem. Sci., 2023, 14, 963–969 RSC.
- J. Hu, M. Tang, J. Wang, Z. Wu, A. Friedrich and T. B. Marder, Angew. Chem., Int. Ed., 2023, 62, e202305175 CrossRef CAS PubMed.
- F.-P. Wu, X. Luo, U. Radius, T. B. Marder and X.-F. Wu, J. Am. Chem. Soc., 2020, 142, 14074–14079 Search PubMed.
- C. B. Kelly, L. Thai-Savard, J. Hu, T. B. Marder, G. A. Molander and A. B. Charette, ChemCatChem, 2024, 16, e202400110 CrossRef CAS.
- M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS PubMed.
- Y.-W. Sun, J.-H. Zhao, X.-Y. Yan, C.-L. Ji, H. Feng and D.-W. Gao, Nat. Commun., 2024, 15, 10810 Search PubMed.
- R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 6525–6528 CrossRef CAS PubMed.
- K. Prysiazhniuk, O. Polishchuk, S. Shulha, K. Gudzikevych, O. P. Datsenko, V. Kubyshkin and P. K. Mykhailiuk, Chem. Sci., 2024, 15, 3249–3254 RSC.
- L. Thai-Savard, M. Sayes, J. Perreault-Dufour, G. Hong, L. A. Wells, M. C. Kozlowski and A. B. Charette, J. Org. Chem., 2023, 88, 1515–1521 CrossRef CAS PubMed.
- X. Y. Chen, F. C. Gao, P. F. Ning, Y. Wei and K. Hong, Angew. Chem., Int. Ed., 2023, 62, e202302638 Search PubMed.
- T. Fang, P. Zhang and C. Liu, Angew. Chem., Int. Ed., 2024, 63, e202415301 Search PubMed.
- H. Kanai and N. Hiraki, Chem. Lett., 2006, 8, 761–762 Search PubMed.
- Y. Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171–3175 Search PubMed.
- S. Pal, Y. Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686–11689 Search PubMed.
- J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902–13906 Search PubMed.
- J. Werth and C. Uyeda, Chem. Sci., 2018, 9, 1604–1609 RSC.
- H. Ikeda, K. Nishi, H. Tsurugi and K. Mashima, Chem. Sci., 2020, 11, 3604–3609 Search PubMed.
- C. L. Ji, J. Han, T. R. Li, C. G. Zhao, C. J. Zhu and J. Xie, Nat. Catal., 2022, 5, 1098–1109 CrossRef CAS.
- K. E. Berger, R. J. Martinez, J. Zhou and C. Uyeda, J. Am. Chem. Soc., 2023, 145, 9441–9447 Search PubMed.
- M. Liu, N. Le and C. Uyeda, Angew. Chem., Int. Ed., 2023, 62, e202308913 Search PubMed.
- D. T. Ngo, J. J. A. Garwood and D. A. Nagib, J. Am. Chem. Soc., 2024, 146, 24009–24015 Search PubMed.
- S. Ni, D. Spinnato and J. Cornella, J. Am. Chem. Soc., 2024, 146, 22140–22144 CrossRef CAS PubMed.
- Á. Gutiérrez-Bonet, S. Popov, M. H. Emmert, J. M. E. Hughes, A. F. Nolting, S. Ruccolo and Y. Wang, Org. Lett., 2022, 24, 3455–3460 Search PubMed.
- Y. Q. Tian, Y. K. Zhang, J. P. Niu and C. Zhang, ChemCatChem, 2024, 16, e202400972 CrossRef CAS.
- T. Fujiwara, M. Odaira and T. Takeda, Tetrahedron Lett., 2001, 42, 3369–3372 CrossRef CAS.
- T. Takeda, S. Kuroi, K. Yanai and A. Tsubouchi, Tetrahedron Lett., 2002, 43, 5641–5644 Search PubMed.
- T. Takeda, K. Arai, H. Shimokawa and A. Tsubouchi, Tetrahedron Lett., 2005, 46, 775–778 CrossRef CAS.
- T. Takeda, Bull. Chem. Soc. Jpn., 2005, 78, 195–217 Search PubMed.
- M. J. Duran-Pena, J. M. Botubol-Ares, J. R. Hanson, R. Hernandez-Galan and I. G. Collado, Org. Biomol. Chem., 2015, 13, 6325–6332 Search PubMed.
- X. Wang, P. Cui, C. Xia and L. Wu, Angew. Chem., Int. Ed., 2021, 60, 12298–12303 Search PubMed.
- B. Han, C. Ren, M. Jiang and L. Wu, Angew. Chem., Int. Ed., 2022, 61, e202209232 Search PubMed.
- B. Han, C. Ren and L. Wu, Organometallics, 2023, 42, 1248–1253 CrossRef CAS.
- T. T. Talele, J. Med. Chem., 2018, 61, 2166–2210 CrossRef CAS PubMed.
- M. Castro Rodríguez, I. Rodríguez García, R. N. Rodríguez Maecker, L. Pozo Morales, J. E. Oltra and A. Rosales Martínez, Org. Process Res. Dev., 2017, 21, 911–923 Search PubMed.
- A. R. Martínez, L. P. Morales, E. D. Ojeda, M. C. Rodríguez and I. Rodríguez-García, J. Org. Chem., 2021, 86, 1311–1329 Search PubMed.
- T. McCallum, X. Wu and S. Lin, J. Org. Chem., 2019, 84, 14369–14380 Search PubMed.
- T. Hilche, P. H. Reinsberg, S. Klare, T. Liedtke, L. Schafer and A. Gansäuer, Chem.–Eur. J., 2021, 27, 4903–4912 Search PubMed.
- T. Hilche, S. L. Younas, A. Gansäuer and J. Streuff, ChemCatChem, 2022, 14, e202200530 CrossRef CAS.
- A. Gansäuer, J. Org. Chem., 1998, 63, 2070–2071 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ESI Tables 1–6, ESI Fig. 1–3, ESI Scheme 1, products characterizations. See DOI: https://doi.org/10.1039/d5sc01132a |
| This journal is © The Royal Society of Chemistry 2025 |