
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protodefluorinated Selectfluor® heteroaggregate photoinduces direct C(sp3)–H fluorinations without photocatalyst†
Shahboz
Yakubov
a and
Joshua P.
Barham
 *ab
*ab
aFakultät für Chemie und Pharmazie, Universität Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany. E-mail: Joshua-Philip.Barham@chemie.uni-regensburg.de
bDepartment of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
First published on 19th February 2025
Abstract
Herein, we uncover a hitherto hidden role of H-TEDA(BF4)2 – a cheap, stable, recoverable by-product of radical C(sp3)–H fluorinations using Selectfluor®. This forms a photoactive, mixed heteroaggregate with Selectfluor® which underlies the reactivity of visible light photochemical fluorination reactions of unactivated C(sp3)–H bonds. Where previous reports claim to be ‘photocatalytic’, reactions work without photocatalyst when H-TEDA(BF4)2 is dosed in at the start. Our results demonstrate that ‘photocatalysts’ are only necessary to generate a sufficient amount of nascent H-TEDA(BF4)2, whose heteroaggregate with Selectfluor® takes over as the main photoactive species. Mechanistic studies suggest the formation of a heteroaggregate between H-TEDA(BF4)2 and Selectfluor® under photoirradiation, which generates TEDA˙2+. A salient feature of our H-TEDA(BF4)2-promoted method is its flexibility to use the C(sp3)–H precursor substrate as the limiting reactant, simplifying product isolations.
Introduction
Considering the ubiquity of C–H bonds in molecules and the attractive properties that derive from a C–H to C–F replacement, methods for direct fluorinations of unactivated C(sp3)–H bonds are undoubtedly powerful additions to a synthetic chemist's arsenal.1 Of these, remote photochemical C(sp3)–H fluorinations using Selectfluor® (SF) are particularly attractive for their mild reaction conditions. A whole host of reports emerged since 2014, employing various photosensitizers (typically, aryl ketones such as acetophenone, anthraquinone, benzil, etc.; but also, tetracyanobenzene and methyl 4-fluorobenzoate)2 in catalytic loadings and SF typically as the limiting reactant, to fluorinate C(sp3)–H containing substrates under photoirradiation conditions. In recent years, cleavable photosensitizing auxiliaries (benzoyl, phthalimido)2h,3 covalently tethered to the C(sp3)–H containing substrate have been successfully used to enhance efficiency and overcome shortcomings in the generality of prior methods. When ‘photocatalysis’ was invoked, proposed mechanisms varied from excited triplet states participating directly in hydrogen atom transfer (HAT) with the target C(sp3)–H bond, to energy transfer (EnT) to SF to generate the TEDA˙2+ as the HAT agent. In any case, ultimately SF is the fluorine source and protodefluorinated SF (H-TEDA(BF4)2) is the endogenous byproduct of all such fluorination reactions (Scheme 1A). Recently, we reported that adding exogenous H-TEDA(BF4)2 deaggregates SF, forming a reactive heteroaggregate H-TEDA(BF4)2⋯SF (hereafter termed ‘HA’) that promotes remote C(sp3)–H fluorinations to higher yields in shorter times.4 However, it was yet unclear how HA enhances the reaction rate. In the case of photochemical/photocatalytic methods, we observed an induction period that was dramatically shortened upon addition of exogenous H-TEDA(BF4)2. Thus, we postulated that (i) it is HA that is the actual major photoactive species, (ii) the ‘photocatalyst’ present in prior reports is merely an initiator, generating a small amount of H-TEDA(BF4)2 in order for HA to carry the reaction forward, (iii) the ‘photocatalyst’ after its necessary role in generating H-TEDA(BF4)2 then plays a disruptive role, screening light from HA. Were (i), (ii) and (iii) to be the case, one could add promotor H-TEDA(BF4)2 at the start of the reaction without any ‘photocatalyst’, achieving comparable or higher efficiencies than prior ‘photocatalyst’-present radical C(sp3)–H fluorinations. Herein, we disclose the photoactivity of HA and leverage it for a variety of C(sp3)–H fluorinations (Scheme 1B). Moreover, we show how other H-bond donating/ion-pairing additives play similar roles, although H-TEDA(BF4)2 is the most general promotor. We hypothesize that H-bond donating/ion-pairing additives: (1) activate the N+–F bond of SF, promoting (ii) fluorine atom transfer (FAT) to the incipient substrate-derived C-centered radical; (2) lead to a photoactive HA involving SF, that upon excitation forms an exciplex with SF. Where previous reports generally employ the C(sp3)–H substrate in excess (e.g. 1.5 equiv.), a salient feature of our discovery is its flexibility to use the C(sp3)–H substrate as the limiting reactant, which is particularly effective in cases involving few/a single inherent fluorination site/s (especially for complex molecule remote fluorinations). This increases utility, ease of work-up and product separation from unreacted precursor (otherwise needed in excess and left over). | ||
| Scheme 1 (A) H-TEDA(BF4)2 generated after photochemical (radical) C(sp3)–H fluorination reactions. (B) This work: SF/H-TEDA(BF4)2 heteroaggregate as an active species in fluorination reactions. | ||
Results and discussion
To test our hypothesis, we initially examined hexyl propionate (1a) as a benchmark substrate. This does not contain any arylcarbonyl group (eliminating the possibility of background self-fluorination2c). In the presence of 2.0 eq. of H-TEDA(BF4)2 and 1.0 eq. of SF, under 405 nm LED light, the reaction of 1a (1.5 eq.) proceeded successfully and provided an excellent yield of fluorinated product 2a (91%) in only 3 h (Table 1, entry 1). This is higher than in the presence of 'photocatalyst' methyl 4-fluorobenzoate and absence of added H-TEDA(BF4)2 (0% yield after 3 h) – which required 24 h to afford 73% yield (entry 2) – reflecting a dramatic rate enhancement effect resulting from the HA. We determined that inert atmosphere, light, and H-TEDA(BF4)2 were all crucial for this ‘photocatalyst’-free reaction to proceed successfully (see ESI† for full optimization details). By structural derivatization we found certain features of H-TEDA(BF4)2 – such as its free N+–H, the BF4 counterion and the Cl atom on the methylene substituent – were essential for reactivity (see ESI†). To investigate if the promotive effect of H-TEDA(BF4)2 was simply due to its Brønsted acidity, we examined whether other acids could serve as promoters and whether any trend existed with their pKa(H) values (see ESI† for full details).5 Pyridinium tetrafluoroborate (Py-H·BF4) and Boc-Val-OH provided similar yields to each other (70%, Table 1, entries 3 and 4). Imidazolium tetrafluoroborate (Imid-H·BF4) and TFA provided 62% and 63% yields, respectively (entries 5 and 6). Tartaric acid provided a comparable yield (92%) to H-TEDA(BF4)2 (entry 7), however, its promotive effect was not general across different case studies unlike H-TEDA(BF4)2 (see ESI†). In summary, no relation was found between the extent of promotion and Brønsted acidity.|
|
||
|---|---|---|
| Entry | Deviation from H-TEDA(BF4)2/pKaH5 | Yielda (%) |
| a Yield was determined by 19F NMR and pentafluorobenzene was used as an internal standard. MFB: methyl 4-fluorobenzoate. b Result from ref. 2h, after 24 h. | ||
| 1 | None/2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 4 |
91 |
| 2 | No H-TEDA(BF4)2/with MFB (1 mol%) | 0/73b |
| 2 | Py-H·BF4/3.44 |
70 |
| 3 | Boc-Val-OH/4.01 (predicted)5a,b |
70 |
| 4 | Imid-H·BF4/6.44 |
62 |
| 5 | TFA/3.44 |
63 |
| 6 | Tartaric acid/2.985c |
92 |

Having demonstrated the effectiveness of the direct C(sp3)–H fluorination of 1a using our H-TEDA(BF4)2 system, we further explored the scope of this method by fluorinating numerous organic molecules with one or multiple unactivated C(sp3)–H bonds in their structures (Scheme 2). This method allowed us to fluorinate a variety of substrates with different functional groups, achieving higher yields than previously reported in ‘photocatalytic’ reactions. Secondary and tertiary positions in alkanes were fluorinated, affording 2b, 2c and 2d in good to excellent (67–96%) yields. Terminal halogens were well tolerated, affording 2f, 2g and 2h, in yields of 78%, 72%, and 70%, respectively. Previously reported yields for 'photocatalytic' fluorinations of 2f and 2g were only 58%.2h Substrates 2g, 2h, 2k, 2l, 2m, 2n, 2r and 2s underwent only C(sp3)–H benzylic fluorination, while substrates 2i, 2t and 2u provided several fluorinated isomers - those with benzylic fluorinated positions being the major products - in good to excellent yields (68–91%). To push the limits of utility of the ‘photocatalyst’-free fluorination method, we applied it to the late-stage fluorination of complex molecules such as Sclareolide and a dehydrocholic acid derivative, achieving 2o and 2p in yields of 88% and 80%, respectively; higher than previously reported yields under ‘photocatalyst’-present conditions (80% and 56%, respectively).2c,h Additionally, hexane-2-one was fluorinated to give 2v in 92% yield, a marked improvement on the previously reported 60% under ‘photocatalyst’-present conditions.2b
 | ||
| Scheme 2 Scope of H-TEDA(BF4)2–promoted photochemical C(sp3)–H fluorination. NMR yields were determined by 19F NMR with pentafluorobenzene as an internal standard (IS). Isolated yields are in parenthesis. Yields in grey: previously reported 'photocatalytic' methods. a1.0 eq. substrate and 2.0 eq. SF. bThe major isomer is depicted, and the yield corresponds to the mixture of regioisomers. c1.0 eq. substrate and 3.0 eq. SF. dNo purification was required. e1.0 eq. substrate and 1.2 eq. SF. | ||
In previous ‘photocatalytic’ reports,2 as well as our previous photosensitization auxiliary method,2hSF was consistently used as the limiting reagent (1.0 eq. SF and 1.5 eq. C(sp3)–H substrate), with reactions typically taking 24 h. In most previous reports, when deploying equimolar amounts of SF:C(sp3)–H substrate or an excess of SF, reactions either proceeded in very low yields or not at all. A key advantage of the ‘photocatalyst’-free method is it can achieve high-yielding reactions with the C(sp3)–H substrate as the limiting reagent. This increases utility of the reaction, especially for complex C(sp3)–H precursors and increases isolated yields by simplifying purification (eliminating the need to separate the product from the remaining excess of starting material, which is often challenging for fluorinated products). For example, using SF as the limiting reactant and 1.5 eq. of 1p gave product 2p in a 63% yield. When 1p was used as the limiting reactant (1.0 eq. substrate, 2.0 eq. SF), the yield increased to 80%. The same trend was observed in the formation of products 2e, 2g, 2h, 2j, 2a, 2k, 2l, and 2o, when using their C(sp3)–H precursors as limiting reactants. However, for substrates with multiple potential fluorination sites, employing the substrate as the limiting reactant while using SF in excess often results in the formation of mixture of regioisomeric byproducts. Therefore, stoichiometric adjustments were made in such cases, with SF as the limiting reagent, to ensure selective product formation and ease of isolation.
Among the various radical fluorination strategies reported,6 several involve catalytic or metal-assisted approaches that typically require prolonged reaction times or high-energy irradiation. Although product 2m (an ibuprofen derivative) was achieved in the same yield as a previous ‘photocatalyst’-present report (64%),6a the previous conditions were more forcing (302 nm UV light for 24 h) than the milder conditions herein (405 nm, 3 h). Product 2x was afforded in a slightly lower yield (78%) than our previous report (82%);2h however, by using substrate 1x as the limiting reactant, we were able to obtain difluorinated product 2y from substrate 1x in a good yield (74%). Overall, Scheme 2's results demonstrate that conditions where HA is the only photoactive species facilitate reactions with moderate to good yield improvements and in shorter reaction times. Given the overall higher efficiency of the ‘photocatalyst’-free C(sp3)–H fluorination conditions, we wondered how amenable they were to scale-up. Pleasingly, gram-scale reactions of 6 substrates performed over 24 h achieved comparable ( 2k, 2a, 2o, 2x) or even higher yields (2p, 2y) than those obtained in small-scale (<0.5 mmol) reactions (Scheme 2). In every case, H-TEDA(BF4)2 was recovered quantitatively and reused without any loss in reaction efficiency.
Consistent with previous ‘photocatalyst’-present reports, the reaction of 1a performed in the presence of 1.5 eq. TEMPO failed to afford any fluorinated product (2a). LC-MS analysis detected masses corresponding to TEMPO-trapped 1a (= 4) and TEDA˙2+ (= 5) (Scheme 3). This suggests that TEDA˙2+ and the C(sp3)–H derived radical from 1a are competent intermediates. By extension, the initial mechanistic step involving the C(sp3)–H activation of the substrate could be in common with previous reports and not be altered by the presence of 2.0 eq. H-TEDA(BF4)2. Our focus turned to discerning the nature of the photoactive species and the fluorinating agent. The existence of HA was confirmed in our previous report,4 but its photophysical properties and photochemical reactivity were not examined. If irradiation of HA gave rise to N+–F bond homolysis (of SF within HA, or by EnT to ‘free’ SF), then irradiating a mixture of SF and H-TEDA(BF4)2 in absence of a C(sp3)–H substrate should afford TEDA˙2+ (6, Scheme 4A). Indeed, LC-MS of such a reaction in the presence of TEMPO (2.0 eq.) detected compound 5 (Scheme 4B). Moreover, in the presence of an established H-atom donor THF (10.0 eq.), full conversion of SF to H-TEDA(BF4)2 occurred (2.0 eq. to 3.0 eq.).
 | ||
| Scheme 3 Radical trapping reaction of 1a with TEMPO. | ||
 | ||
| Scheme 4 (A) Formation of TEDA˙2+. (B) Radical trapping of SF and H-TEDA(BF4)2 with TEMPO. (C) Reaction of SF and H-TEDA(BF4)2 with THF as a hydrogen donor. | ||
UV-vis spectroscopy of the reaction mixture was performed using TFA as an active additive (Fig. 1) due to the limited solubility of H-TEDA(BF4)2 in MeCN (see ESI for details†). The results revealed growth of a new absorption (λmax = 358 nm) tailing into 400 nm, notably higher than any individual reaction component and which maximized at 2.0–5.0 eq. of TFA. This was attributed to HA. Luminescence spectroscopy was conducted on SF and H-TEDA(BF4)2 separately, as well as on their mixtures in various ratios. Both SF and H-TEDA(BF4)2 underwent marginal slow photodecomposition over successive excitations for 5–20 min (Fig. 2A and B; see ESI† for photodecomposition rates), with their peak intensities decreasing by ∼10% after 20 min. The emission intensity of H-TEDA(BF4)2 (λmax = 452 nm) was twice as high as that of SF (λmax = 467 nm) at isoconcentration. Mixing SF with H-TEDA(BF4)2 in a 1:1 ratio to afford the HA gave rise to a single peak (λ = 463 nm), resembling the emission wavelength of SF alone (Fig. 2A and C) but also resembling the higher emission intensity (2.4× vs. SF alone) of H-TEDA(BF4)2. The intensity of HA decreased by ∼15% over a 15 min period of successive excitations, which was a faster decay than either of its constituent molecules alone. When H-TEDA(BF4)2 was mixed with excesses of SF (1:1 → 1:4 ratio) (Fig. 2D), this led to a strong bathochromic shift in the emission peak (λmax = 463 → 490 nm) together with a marked decrease in emission intensity. At a 1:4 ratio, the peak at λmax = 490 nm is attributed to an exciplex of HA with SF. Exciplexes between arylketone reagents and SF were previously reported.2c,d This exhibited a pronounced decrease in intensity upon repeated excitations (Fig. 2E), with the peak wavelength shifting back to values more closely resembling SF, which is of course present in excess. This suggests quenching of HA by ‘free’ SF, presumably by EnT, leading to N+–F homolysis. Conversely, mixing SF (10 mM) with increasing concentrations of H-TEDA(BF4)2 (10 → 40 mM) did not alter the wavelength but did increase emission intensity up to 2.7× (Fig. 2F), corresponding simply to the higher emission intensity of ‘free’ H-TEDA(BF4)2. Clearly, all SF is constrained within HA; no SF is available to form the exciplex.
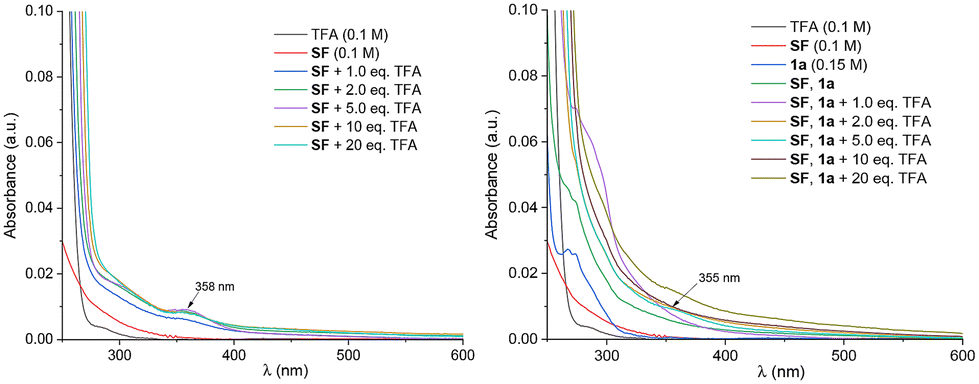 | ||
| Fig. 1 UV-visible spectroscopy of TFA, SF and their mixtures (left). UV-visible spectroscopy of TFA, 1a, SF and their mixtures (right). | ||
 | ||
| Fig. 2 Luminescence of SF and H-TEDA(BF4)2. (A) SF only (15 mM). (B) H-TEDA(BF4)2 only (15 mM). (C) SF and H-TEDA(BF4)2 in 1:1 ratio. (D) H-TEDA(BF4)2 (10 mM) with increasing concentrations of SF. (E) SF and H-TEDA(BF4)2 in a 4:1 ratio. (F) SF (10 mM) with increasing concentrations of H-TEDA(BF4)2. | ||
Based on our observations and consolidating knowledge from previous literature reports, we propose two mechanisms (Scheme 5), one involving a radical chain and one not.
 | ||
| Scheme 5 Proposed reaction mechanisms. | ||
Proposed mechanism 1
‘Photoactive HA-initiated HA-promoted-propagation chain’ – In this proposed mechanism (Scheme 5A), HA plays two roles, (1) a photoactive radical chain initiator; (2) a fluorine atom donor via a more reactive N+–F bond (vs. SF). Firstly, photoactive HA is formed immediately upon mixing of H-TEDA(BF4)2 with SF. Photoexcitation of HA affords *HA, which either (i) extrudes HF and forms two molecules of chain carrier TEDA˙2+, or (ii) forms an exciplex with SF, which reacts to regenerate HA and generate one molecule of TEDA˙2+. In the propagation cycle, TEDA˙2+ engages the C(sp3)–H substrate (1) in intermolecular HAT, affording an alkyl radical 1′ and regenerating H-TEDA(BF4)2. Then, the key role of HA comes into play as a more active F atom donor than SF in the FAT step. Owing to its more activated N+–F bond, 1′ reacts rapidly with HA in FAT, affording the fluorinated product 2. This enhanced reactivity minimizes opportunities for putative side reactions involving the alkyl radical (e.g. dimerization, disproportionation, HAT with substrate molecules, that would compromise the overall fluorinated product yield), leading to a more efficient and cleaner reaction when HA is present from the start. This could be one reason underlying enhanced reaction rates and overall yields in our HA-present 'photocatalyst'-free protocol, compared to previous ‘photocatalytic’ C(sp3)–H fluorinations. This mechanism is supported by emission spectroscopic detection of an exciplex (Fig. 2) and the detection of 5 upon irradiation of HA with TEMPO (Scheme 4B). Although this is inconsistent with the previously reported quantum yields of <0.15,2d,4 and the non-detection of HF/(F−) proposed to be generated in the initiation step, (i) a chain mechanism cannot be conclusively ruled out by low quantum yields, and (ii) formation of traces of HF that react with the glass vial cannot be ruled out.We deem a radical polar crossover-type SET between 1′ and HA/SF to be unlikely, as this would imply unstabilized 2° carbocationic intermediates, prone to Ritter-type reactivity7 with MeCN solvent (or with certain starting materials as Ritter-type nucleophiles). This is inconsistent with the observed efficiency and selectivity of the reaction (e.g. an excellent yield of 2u and the absence of Ritter-type products that we detected in studies of other reactions in our group7b,c).
Proposed mechanism 2
‘Photoactive HA asynchronous concerted σ-bond metathesis (non-chain)’ – In this proposed mechanism, photoactive HA is formed immediately upon mixing of H-TEDA(BF4)2 with SF. Photoexcitation of HA affords *HA, which acts like a previously reported EnT photosensitizer, forming an exciplex with the C(sp3)–H substrate.2c,d This undergoes asynchronous concerted σ-bond metathesis, initially forming 1′ and a non-free (stabilized) F˙, which rapidly combine within the aggregate/solvent cage to afford fluorinated product 2. Although F˙ is among the most reactive free radicals known, in this mechanism, 1′ and F˙ are generated in close proximity and the latter is still stabilized within HA so is not a ‘free’ radical. If 1′ was to diffuse away (TEMPO-1′ was detected), F˙ could engage the H-TEDA(BF4)2 – to which it is proximally formed – indiscriminately in HAT (at the N+–H bond, providing 6), shielding fresh molecules of 1. This non-chain mechanism would consist with previously reported quantum yields of <0.15,2d,4 and the photodecomposition of HA (Fig. 2C) upon successive excitations. The σ-bond metathesis being asynchronous (rather than synchronous concerted) is consistent with the TEMPO trapping experiment detecting both 4 and 5, implying a degree of escape of 1' from the aggregate/solvent cage.Conclusion
We report the discovery of a straightforward, scalable ‘photocatalyst’-free direct photochemical fluorination of unactivated C(sp3)–H bonds, which employs the natural reaction by-product H-TEDA(BF4)2 at the start of reactions to afford a photoactive species in situ. This results in moderate to good yield improvements over previous ‘photocatalyst’-present methods, dramatically increases reaction rates, and is conducive to scale-up (gram-scale). Importantly, it offers the flexibility to use the C(sp3)–H substrate as the limiting reactant, thereby increasing utility for complex molecule fluorinations under visible light irradiation, and simplifying work-up and purification. Mechanistic studies indicate the formation of a photoactive SF/H-TEDA(BF4)2 heteroaggregate (HA) that obviates the need for any exogenous photocatalyst. This rationalizes the common role of exogeneous ‘photocatalysts’ in previous studies: they are merely initiators needed to form HA, which is likely to be the primary photoactive species responsible for executing efficient photochemical radical C(sp3)–H fluorinations involving SF.Data availability
On behalf of all authors, I confirm that all the data pertaining to this article are available within the ESI† (experimental procedures, listed data, NMR spectra). No code or public datasets are relevant in this case. CCDC deposition numbers have been provided for the two X-ray diffraction (XRD) structures relating to this manuscript (see ESI†). Both CIF files and checkcif.pdf reports are provided for the XRD data.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. Y. is grateful for funding provided by the SynCat programme of the Elite Network of Bavaria. S. Y. and J. P. B. thank the Alexander von Humboldt Foundation for funding, provided within the framework of the Sofja Kovalevskaja Award endowed to J. P. B. by the German Federal Ministry of Education and Research. S. Y. is an associated member of the RTG 2620. J. P. B. is a member of DFG TRR 325 ‘Assembly Controlled Chemical Photocatalysis’ (444632635). We thank other members of the RTG and TRR for helpful discussions. J. P. B. is currently employed by the University of Strathclyde and is grateful for financial support.References
- (a) T. Liang, C. N. Neumann and T. Ritter, Introduction of Fluorine and Fluorine-Containing Functional Groups, Angew. Chem., Int. Ed., 2013, 52, 8214 ( Angew. Chem. , 2013 , 125 , 8372 ) CrossRef CAS PubMed; (b) A. Lin, C. B. Huehls and J. Yang, Recent advances in C–H fluorination, Org. Chem. Front., 2014, 1, 434 RSC; (c) P. A. Champagne, J. Desroches, J.-D. Hemel, M. Vandamme and J.-F. Paquin, Monofluorination of Organic Compounds: 10 Years of Innovation, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed; (d) B. Lantaño and A. Postigo, Radical fluorination reactions by thermal and photoinduced methods, Org. Biomol. Chem., 2017, 15, 9954 RSC; (e) R. Szpera, D. F. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, The Fluorination of C−H Bonds: Developments and Perspectives, Angew. Chem., Int. Ed., 2019, 58, 14824 CrossRef CAS PubMed; (f) S. Yakubov and J. P. Barham, Photosensitized direct C–H fluorination and trifluoromethylation in organic synthesis, Beilstein J. Org. Chem., 2020, 16, 2151 CrossRef CAS PubMed; (g) R.-B. Montserrat, C. C. Sazepin, J. C. Leung, T. Okbinoglu, P. Kennepohl, J. Paquin and G. M. Sammis, Fluorine Transfer to Alkyl Radicals, J. Am. Chem. Soc., 2012, 134, 4026 CrossRef PubMed; (h) F. Yin, Z. Wang, Z. Li and C. Li, Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (i) A. M. Hua, D. N. Mai, R. Martinez and R. D. Baxter, Radical C−H Fluorination Using Unprotected Amino Acids as Radical Precursors, Org. Lett., 2017, 19, 2949 CrossRef CAS PubMed; (j) A. M. Hua, S. L. Bidwell, S. I. Baker, H. P. Hratchian and R. D. Baxter, Experimental and Theoretical Evidence for Nitrogen–Fluorine Halogen Bonding in Silver-Initiated Radical Fluorinations, ACS Catal., 2019, 9, 3322 CrossRef CAS.
- (a) J. B. Xia, C. Zhu and C. Chen, Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef CAS PubMed; (b) J. B. Xia, C. Zhu and C. Chen, Visible light-promoted metal-free sp3-C–H fluorination, Chem. Commun., 2014, 50, 11701 RSC; (c) C. W. Kee, K. F. Chin, M. W. Wong and C. H. Tan, Selective fluorination of alkyl C–H bonds via photocatalysis, Chem. Commun., 2014, 5, 8211 RSC; (d) J. W. Kee, H. Shao, C. W. Kee, Y. Lu, H. S. Soo and C. H. Tan, Mechanistic insights for the photoredox organocatalytic fluorination of aliphatic carbons by anthraquinone using time-resolved and DFT studies, Catal. Sci. Technol., 2017, 7, 848 RSC; (e) S. Bloom, J. L. Knippel and T. Lectka, Direct monofluoromethylation of O-, S-, N-, and P-nucleophiles with PhSO(NTs)CH2F: the accelerating effect of α-fluorine substitution, Chem. Sci., 2014, 5, 1175 RSC; (f) D. D. Bume, C. R. Pitts, F. Ghorbani, S. A. Harry, J. N. Capilato, M. A. Siegler and T. Lectka, Ketones as directing groups in photocatalytic sp3 C–H fluorination, Chem. Sci., 2017, 8, 6918 RSC; (g) F. Ghorbani, S. A. Harry, J. N. Capilato, C. R. Pitts, J. Joram, G. N. Peters, J. D. Tovar, I. Smajlagic, M. A. Siegler, T. Dudding and T. Lectka, Carbonyl-Directed Aliphatic Fluorination: A Special Type of Hydrogen Atom Transfer Beats Out Norrish II, J. Am. Chem. Soc., 2020, 142, 14710 CrossRef CAS PubMed; (h) S. Yakubov, W. J. Stockerl, X. Tian, A. Shahin, M. J. P. Mandigma, R. M. Gschwind and J. P. Barham, Benzoates as photosensitization catalysts and auxiliaries in efficient, practical, light-powered direct C(sp3)–H fluorinations, Chem. Sci., 2022, 13, 14041 RSC; (i) C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Lectka, Direct, Catalytic Monofluorination of sp3 C–H Bonds: A Radical-Based Mechanism with Ionic Selectivity, J. Am. Chem. Soc., 2014, 136, 9780 CrossRef CAS PubMed.
- H. Egami, S. Masuda, Y. Kawato and Y. Hamashima, Photofluorination of Aliphatic C–H Bonds Promoted by the Phthalimide Group, Org. Lett., 2018, 20, 1367 CrossRef CAS PubMed.
- S. Yakubov, B. Dauth, W. J. Stockerl, W. M. da Silva, R. M. Gschwind and J. P. Barham, Protodefluorinated Selectfluor® Aggregatively Activates Selectfluor® for Efficient Radical C(sp3)−H Fluorination Reactions, ChemSusChem, 2024, 17, e202401057 CrossRef CAS PubMed.
- The pKa value of valine is known to be 2.32 according to this literature: (a) R. M. C. Dawson and W. H. Elliott, in Data for biochemical research, Oxford University Press, Oxford, 1959, pp. 200–205 Search PubMed. Only a predicted pKa value was found (4.01) for Boc-Val-OH: (b) Predicted NMR data calculated using Advanced Chemistry Development, Inc. (ACD/Labs) Software (© 1994–2024 ACD/Labs); ; (c) T. L. Edwards, V. L. Singleton and R. Boulton, Formation of Ethyl Esters of Tartaric Acid During Wine Aging: Chemical and Sensory Effects, Am. J. Enol. Vitic., 1985, 36, 118 CrossRef CAS.
- (a) S. Bloom, M. McCann and T. Lectka, Photocatalyzed Benzylic Fluorination: Shedding “Light” on the Involvement of Electron Transfer, Org. Lett., 2014, 16, 6338 CrossRef CAS PubMed; (b) Y. Amaoka, M. Nagatomo and M. Inoue, Metal-Free Fluorination of C(sp3)–H Bonds Using a Catalytic N-Oxyl Radical, Org. Lett., 2013, 15, 2160 CrossRef CAS PubMed; (c) Z. Li, L. Song and C. Li, Silver-Catalyzed Radical Aminofluorination of Unactivated Alkenes in Aqueous Media, J. Am. Chem. Soc., 2013, 135, 4640 CrossRef CAS PubMed; (d) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination, Angew. Chem., Int. Ed., 2012, 51, 10580 ( Angew. Chem. , 2012 , 124 , 10732 ) CrossRef CAS PubMed; (e) D. D. Bume, S. A. Harry, C. R. Pitts and T. Lectka, Sensitized Aliphatic Fluorination Directed by Terpenoidal Enones: A “Visible Light” Approach, J. Org. Chem., 2018, 83, 1565 CrossRef CAS PubMed; (f) D. D. Bume, C. R. Pitts, R. T. Jokhai and T. Lectka, Direct, visible light-sensitized benzylic C−H fluorination of peptides using dibenzosuberenone: selectivity for phenylalanine-like residues, Tetrahedron, 2016, 72, 6031 CrossRef CAS; (g) C. R. Pitts, D. D. Bume, S. A. Harry, M. A. Siegler and T. Lectka, Multiple Enone-Directed Reactivity Modes Lead to the Selective Photochemical Fluorination of Polycyclic Terpenoid Derivatives, J. Am. Chem. Soc., 2017, 139, 2208 CrossRef CAS PubMed; (h) J. N. Capilato, C. R. Pitts, R. Rowshanpour, T. Dudding and T. Lectka, Site-Selective Photochemical Fluorination of Ketals: Unanticipated Outcomes in Selectivity and Stability, J. Org. Chem., 2020, 85, 2855 CrossRef CAS PubMed; (i) A. Vasilopoulos, D. L. Golden, J. A. Buss and S. S. Stahl, Copper-Catalyzed C–H Fluorination/Functionalization Sequence Enabling Benzylic C–H Cross Coupling with Diverse Nucleophiles, Org. Lett., 2020, 22, 5753 CrossRef CAS PubMed; (j) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Oxidative aliphatic C-H fluorination with fluoride ion catalyzed by a manganese porphyrin, Science, 2012, 337, 1322 CrossRef CAS PubMed; (k) Y. Zhang, N. A. Fitzpatrick, M. Das, I. P. Bedre, H. G. Yayla, M. S. Lall and P. Z. Musacchio, A photoredox-catalyzed approach for formal hydride abstraction to enable Csp3−H functionalization with nucleophilic partners (F, C, O, N, and Br/Cl), Chem Catal., 2022, 2, 292 CrossRef CAS.
- (a) M. Lepori, I. Dey, C. Pratley and J. P. Barham, Merging New and Old Concepts: Tandem Oxidative Radical-Polar Crossover Ritter Amidation via Multicomponent Photo- and Electrochemical Processes, Eur. J. Org. Chem., 2024, 27, e202400840 CrossRef CAS; (b) S. Schmid, S. Wu, I. Dey, M. Domański, X. Tian and J. P. Barham, Photoelectrochemical Heterodifunctionalization of Olefins: Carboamidation Using Unactivated Hydrocarbons, ACS Catal., 2024, 14, 9648 CrossRef CAS; (c) M. Lepori, C. Pratley, I. Dey, V. Butera, V. Roider and J. P. Barham, Photocatalysis Enables Chemodivergent Radical Polar Crossover: Ritter-Type Amidation vs Heck-Type Olefin Carbofunctionalizations, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-cjjxv.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, spectral data and X-ray data. CCDC 2291634 and 2381200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qo00149h |
| This journal is © the Partner Organisations 2025 |