
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation behavior of TiC and TiCN and their potential photocatalytic activity in a semi-oxidized state
Shiyun
Tang
a,
Guoqiang
Song
 *b,
Junjiang
Guo
a,
Claudia
Li
c,
Kang Hui
Lim
c,
Pinli
Diao
c,
Wenting
Chen
b,
Feiyang
Hu
d,
Jaka
Sunarso
e,
Hao
Tang
*b and
Sibudjing
Kawi
*c
*b,
Junjiang
Guo
a,
Claudia
Li
c,
Kang Hui
Lim
c,
Pinli
Diao
c,
Wenting
Chen
b,
Feiyang
Hu
d,
Jaka
Sunarso
e,
Hao
Tang
*b and
Sibudjing
Kawi
*c
aSchool of Chemical Engineering, Guizhou Institute of Technology, Guiyang 550003, China
bSchool of Intelligent Materials and New Energy, Institute of Carbon Neutral New Energy, Yuzhang Normal University, Nanchang 330103, China. E-mail: gq.song@git.edu.cn
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore, 117585, Singapore
dCollege of Chemistry and Materials, Jiangxi Agricultural University, No. 1101 Zhimin Avenue, Nanchang 330045, China
eResearch Centre for Sustainable Technologies, Faculty of Engineering, Computing and Science, Swinburne University of Technology, Jalan Simpang Tiga, Kuching, Sarawak 93350, Malaysia
First published on 29th June 2025
Abstract
The oxidation behavior of titanium carbide (TiC) and titanium carbonitride (TiCN) warrants significant attention. This study investigates the oxidation behavior of internally doped TiC and TiCN in ambient air by varying the oxidation temperature and explores their photocatalytic degradation activity. Macroscopic observations, combined with XRD and SEM analyses, reveal a pronounced color change during oxidation, with TiC exhibiting superior oxidation resistance compared to TiCN. TiC and TiCN were oxidized at 800 °C and 500 °C for 2 h, respectively, achieving substantial oxidation. During the oxidation process, TiC is directly oxidized to TiO2, while in TiCN, C is first replaced by O, followed by N. The final oxidation product for both materials is rutile TiO2. TG-DSC and TPO-IR analyses suggest that the C species in TiC is more complex than that in TiCN, attributed to the presence of C atoms with varying graphitization degrees embedded within the TiC lattice. In assessing photocatalytic degradation activity following semi-oxidization, TiC and TiCN exhibited optimal activity after oxidation for 2 h at approximately 400 °C and 500 °C, respectively, with residual rates of rhodamine B (RhB) at 3.85% and 9.37% after 240 min of degradation and adsorption. These results indicate that TiCN demonstrates superior adsorption and degradation capacities for RhB compared to oxidized TiC. Furthermore, UV-Vis DRS and XPS detection results confirm the formation of N and/or C inner-doped modified TiO2 in TiC and TiCN following appropriate semi-oxidization.
1 Introduction
Titanium carbide (TiC) and titanium carbonitride (TiCN) exhibit exceptional physical and chemical properties, including excellent hardness, elevated Young's modulus, significant electrical and thermal conductivity, high biocompatibility, elevated melting points and robust corrosion resistance.1–3 Therefore, TiC and TiCN are integral in the fabrication of wear-resistant coatings,3 organic crack and coke-resistant coatings,4 hard composites,5 bionic materials,6 and electrocatalysts,7,8 across various fields such as machinery, chemical engineering, microelectronics, and bioengineering. MoSi2 and its composites have been successfully prepared and reinforced with TiC, TiCN, and TiB2via hot-pressing, yielding a homogeneous microstructure,9 indicating that the incorporation of TiC, TiCN and TiB2 enhanced the mechanical strength and wear resistance of the resultant materials. Additionally, using TiC as a primary material, biocompatible nanostructured membranes could be developed.10 The Ti-based films demonstrated high corrosion resistance, bioactivity, biocompatibility, and non-toxicity, making them suitable for various medical applications, including orthopedic prostheses, connective tissue surgery, and dental implants. Recent advancements by Zhang et al. introduced novel flexible lithium-sulfur battery architecture that employs TiC nanofibers and in situ grown vertical graphene as the electrocatalytic anode.11 This innovation significantly enhances the performance of lithium–sulfur batteries, presenting a promising approach for achieving high energy density. Despite the critical role of oxidation in preserving the superior properties of these materials,12,13 comprehensive studies on the oxidation behavior of TiC and TiCN remain limited. Therefore, this study aims to systematically investigate the oxidation behavior of TiC and TiCN.The oxidation behavior of TiC has been studied extensively. Lavrenko et al.14 and Voitovich et al.15 proposed a detailed mechanism for the high-temperature oxidation of TiC, identifying four distinct stages between 1000 and 1200 °C: In Stage I, oxidation is influenced by the solubility of oxygen in the TiC lattice; Stages II and III are governed by the diffusion of oxygen within an inner oxide scale and titanium cations within an outer oxide scale, respectively; the final stage is controlled by reactions at the interface between the carbide and the inner scale. They further posited that TiC oxidation is influenced by the diffusion of oxygen as well as the concurrent diffusion of Ti and carbon (C) from the material into the scale. At lower temperatures (350–500 °C), Shamada et al.16 observed that the oxidation behavior of TiC also follows a four-step process: (I) fast step; (II) slow step; (III) fast step and (IV) slow step. Interestingly, they discovered two distinct activation energies in steps II–IV: 125–150 kJ mol−1 below approximately 420 °C and 42–71 kJ mol−1 above that temperature, with varying oxidation products at different stages and pressures. Gherrab et al.17 proposed that TiC samples with smaller particle sizes are more prone to oxidation, indicating that a reduction in particle size increases the oxidation ratio and lowers the oxidation exothermic peak temperature.18
The oxidation behavior of TiCN has also been similarly widely explored. Christoph et al.19 reported that the substitution of C in TiCN occurs more rapidly than that of other elements during oxidation, treating TiCN as an ideal TiC–TiN solid solution, the concepts of which are extensively covered in ref. 20. The oxidation of TiCN-based cermets is initiated at the particle's outer edge and gradually penetrates the core, with an upper oxidation temperature limit of approximately 1000 °C, yielding TiO2, N2 and CO as final products.21 Bi et al. investigated the structural properties and electronic structure of TiCN thin films, prepared by laser treatment in a controlled atmosphere, attributing oxygen resistance in TiCN to the binding strength between Ti 3d and C 2p orbitals.22 Moreover, comparisons of the oxidation performance of TiC and TiCN were conducted, and TiCN, TiC and TiN were assessed in a mixture of O2 and Ar at 800 °C.23 Their findings indicated that TiC had the strongest antioxidant activity, while TiN exhibited the weakest. They also noted that higher free C content in TiCN and TiC enhances oxidation resistance. Tang et al. prepared TiC and TiN coatings on the inner surface of SS304 pipe through chemical vapor deposition, observing that TiC begins reacting with oxygen at around 810 °C, producing CO2, which demonstrated greater oxidation resistance than TiN, which oxidized at 350 °C according to temperature-programmed oxidation experiments.24
Excessive oxidation of TiC and TiCN can lead to a rapid decline in their mechanical properties.25,26 However, controlled oxidation may yield beneficial effects.27,28 Some researchers have explored the semi-oxidation performance of TiC and TiCN within the ability to degrade pollutants using sunlight29,30 and ultraviolet (UV) light.31,32 Doping TiO2 with metal or non-metal ions, such as Fe,33 Cu,34 Pt,35 N,36 and C,37,38 can enhance its catalytic activity.39 Non-metal ion doping in the TiO2 photocatalyst has shown great industrial application prospects, as it not only enhances visible light responsiveness, but also preserves photocatalytic activity in the UV region.40,41 However, external doping introduces complexities and potential impurities.12,42
This study proposes an internal doping method to prepare non-metal doped TiO2, wherein TiC and TiCN are partially oxidized to produce semi-oxidized TiC (C-doped TiO2) and TiCN (C, N-codoped TiO2). This approach effectively mitigates the issues associated with impurity introduction and complex processing.43 Previous studies have demonstrated similar concepts, i.e., Tang et al. synthesized N-doped TiO2@SiO2 catalysts by partially oxidizing TiN@SiO2 composite aerogels,44 which exhibited excellent adsorption/photocatalytic degradation of rhodamine B (RhB) under visible light. Tayade et al. prepared N-doped anatase type TiO2 (N–TiO2) through hydrothermal treatment of TiN with H2O2.45 Their results indicated that the optimal N-doped TiO2 exhibited superior photocatalytic activity compared to P25 (TiO2 with an average particle size of 25 nm46) and N-P25. In addition, Hao et al. prepared C, N co-doped TiO2–TiC0.7N0.3 composite coatings on the surfaces of Al2O3 spheres by oxidizing TiC0.7N0.3 powder in air at 800 °C,47 yielding a composite coating that demonstrated enhanced photocatalytic performance under UV and visible light, degrading approximately 20% of methylene blue within 6 h. In summary, investigating the oxidation behavior of TiC and TiCN can facilitate the straightforward synthesis of non-metal doped TiO2 in a semi-oxidized state, thereby enhancing its potential applications in photocatalysis through this internal doping effect.
2 Experimental
TiC and TiCN powder samples, with purities of 99.5% and 99.0%, respectively, were selected for the oxidation experiment from Shanghai Yien Chemical Technology Co., Ltd. A multi-section precise temperature control tube furnace (Anhui Beiyike Equipment Technology Co. Ltd, China) was used as the heating apparatus. The samples were placed in a boat crucible and heated to the target temperature at a rate of 10 °C min−1 under an ambient atmosphere. Following thermal insulation and oxidation for 2 h, the furnace temperature was decreased to ambient temperature at a cooling rate of 10 °C min−1, producing the oxidized samples.The photocatalytic activity of the oxidized TiC and TiCN catalysts was evaluated by the degradation of RhB in aqueous solution under simulated solar irradiation with a 500 W xenon lamp (Beijing Precise Technology Co. Ltd, China). The lamp was positioned inside a cylindrical vessel and surrounded by a circulating water jacket for cooling. 200 mg of catalyst was suspended in 50 mL of an aqueous solution of 4 mg L−1 RhB. Prior to the experiment, the solution was ultrasonicated for 5 min at room temperature. Then, a 90-min dark reaction was conducted to achieve adsorption equilibrium, and a filter was used to eliminate UV light during the subsequent 150-min light reaction. The distance between the light source and the bottom of the solution was approximately 5 cm. At specified time intervals, 4 mL of the suspension was extracted for analysis and filtered. The catalyst was recovered by filtration and then subjected to calcination treatment in a muffle furnace. Specifically, calcination at 300 °C for 2 h effectively removed pollutants adsorbed on the catalyst surface, enabling regeneration of the photocatalyst. The regenerated catalyst was subsequently reused in fresh pollutant degradation experiments, with this cycle repeated to assess the catalyst's activity, stability and degradation efficiency changes across multiple cycles. Finally, the concentration of RhB was monitored using a UV-Visible spectrophotometer (UV-Vis, TU-1901, Beijing Puxi General Instrument Co., Ltd, China) at the characteristic absorption wavelength of 554 nm. The adsorption and photocatalytic degradation efficiency is expressed by the residual ratio, which is defined as follows:
| η = (Ct/C0) × 100% | (1) |
The microstructure and phase composition of the oxidized samples were characterized by scanning electron microscopy (SEM) (Nova Nano SEM 450, FEI company, USA), and X-ray diffraction (XRD) (Ultima IV rotary anode X-ray diffractometer, Rigaku Electric Co., Ltd) with Cu Ka (λ = 0.15406 nm), equipped with a graphite monochromator, Ni filter, voltage 40 kV, tube current 25 mA, scanning rate 0.03° s−1, and scanning range 2θ = 10°–80° with 0.03° of step size, respectively. Thermal analysis was carried out by thermogravimetric differential scanning calorimetry (TG-DSC) (ZH1450, Beijing Hengjiu Test Equipment Co., Ltd) and temperature programmed oxidation infrared analysis (TPO-IR) (Sichuan Science Instruments Co., Ltd). A detailed description of the TPO-IR device is provided in ref. 48. X-ray photoelectron spectroscopy (XPS) (Thermo Scientific k-alpha+, Thermofisher) was used for qualitative and quantitative analysis of oxidized samples. The optical characteristics were detected by UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) (Cary 5000, Agilent, USA).
3 Results and discussion
3.1 Comparison of the oxidation behavior of TiC and TiCN at different temperatures
The macroscopic changes in TiC and TiCN during oxidation were pronounced, particularly in terms of color. As illustrated in Fig. 1, the oxidation of TiC powder demonstrates a complex color evolution. Initially, the TiC powder, characterized by a lead gray color and metallic luster, transitions to dark brown following oxidation at 300 °C. A further increase in temperature to 350 °C results in an unexpected dark blue hue. This phenomenon occurs because elevated temperatures accelerate the growth of the oxide film on the TiC surface. As the film thickness increases, it triggers a thin-film interference effect, selectively enhancing wavelengths corresponding to deep blue and thus manifesting the observed hue. The metallic luster persists until the oxidation temperature reaches 500 °C, at which point significant oxidation occurs, and the sample nearly loses its metallic sheen, showing a dark brown earthy luster. At 800 °C, the color of TiC powder shifts to a distinct grayish-brown, indicating substantial oxidation, likely due to the formation of TiO2. The observation of the sample color at 950 °C suggests that TiC powder is not completely oxidized at this temperature. | ||
| Fig. 1 Photos of TiC samples after oxidation at different temperatures. | ||
The color changes of TiCN powder during oxidation within the 400 °C to 650 °C range are shown in Fig. 2. The TiCN powder retains its original carbon black color until the oxidation temperature exceeds 400 °C. Slightly above this temperature, the color lightens, reaching a light yellow at 450 °C. The appearance of light yellow may be attributable to the formation of TiN during the oxidation of TiCN, given that the original TiN is golden yellow.49 This suggests that C atoms are preferentially replaced by O atoms to form TiN during the oxidation of TiCN, followed by the continued replacement of N atoms by O atoms, ultimately resulting in the formation of TiO2. With the increase in oxidation temperature, the light yellow coloration of TiCN powder becomes more pronounced, particularly at 550 °C. Subsequently, as the oxidation deepens, the color of the sample lightens further, likely due to the gradual replacement of TiN by TiO2. Therefore, it is proposed that TiCN undergoes significant oxidation starting at 450 °C and reaches advanced oxidation by 550 °C.
 | ||
| Fig. 2 Photos of TiCN samples after oxidation at different temperatures. | ||
Fig. 3 presents the SEM images of TiC and TiCN samples after oxidation at various temperatures. In Fig. 3(a), the morphology of the original TiC particles exhibits relatively rough surfaces and sharp edges, with a discernible layered structure on the cross-section of the particles. This layered structure may indicate the orientation of the weak bonding forces within TiC. Following oxidation at 500 °C, as illustrated in Fig. 3(b), the edges of the TiC particles show clear signs of passivation, although the particle surfaces remain relatively flat. Fig. 3(c) and (d) reveal some cracks on the surface of TiC particles following oxidization at 700 and 800 °C, suggesting that the oxidation process has penetrated the TiC particles. In the case of TiCN, the original TiCN powder exhibits an irregular polyhedron morphology with well-defined edges (Fig. 3(e)). After oxidation at 400 °C, these edges become smoother, and slight cracks begin to appear on the particle surfaces (Fig. 3(f)). As oxidation proceeds to 500 °C (Fig. 3(g)), the cracks become more pronounced, and further oxidation at 600 °C leads to deeper penetration of cracks into the interior of the TiCN particles (Fig. 3(h)). These observations indicate that TiCN experiences significant oxidation at 600 °C, demonstrating a lower oxidation resistance compared to TiC.
 | ||
| Fig. 3 SEM images of samples after oxidation at different temperatures: TiC (a) original sample; (b) 500 °C; (c) 700 °C; (d) 800 °C and TiCN (e) original sample; (f) 400 °C; (g) 500 °C; (h) 600 °C. | ||
Fig. 4 presents the crystal phase structures of TiC and TiCN after oxidation at various temperatures, as analyzed by XRD. Both TiC and TiCN exhibit distinct diffraction peaks prior to and following oxidation. These peaks are relatively narrow and sharp, indicating a high degree of crystallinity. As illustrated in Fig. 4(a), TiC oxidized at temperatures below 500 °C maintains its crystal phase as a pure TiC crystal, characterized by a face-centered cubic (FCC) NaCl-type structure.50 The reflections at 2θ angles of 35.78°, 41.54°, 60.36°, 72.26°, and 76.02° correspond to (111), (200), (220), (311), and (222) crystal faces of the NaCl-type structure of TiC (JCPDS card number 38-1420), respectively. A minor presence of anatase TiO2 (JCPDS card No. 21-1272) was detected in the sample oxidized at 500 °C, albeit with low intensity, while TiC remains the predominant component, although its content decreases. Upon oxidation at 600 °C, a small amount of rutile TiO2 (JCPDS card number 21-1276) coexists with anatase TiO2. The anatase TiO2 is absent after oxidation at 700 °C, with rutile TiO2 content increasing, consistent with the known phase transformation of anatase TiO2 to rutile at this temperature. Despite a reduction in TiC content, its peak intensity remains significant, indicating that it continues to be the primary phase. However, the TiC signal weakens substantially due to increased oxidation, particularly at 800 °C, where TiC content drops sharply but does not vanish completely. This is consistent with the macro-level changes (color transformation) and micro-level findings (SEM results). In conclusion, TiC exhibits notable oxidation starting at 500 °C, and reaches a severe oxidation stage by 800 °C.
 | ||
| Fig. 4 X-ray diffraction pattern of samples after oxidation at different temperatures: TiC (a) and TiCN (b). | ||
Generally, the structural characterization of TiCN can be effectively described by the TiN structure,51 wherein N sites are occupied by C, resulting in either an FCC or a tetragonal structure.52Fig. 4(b) presents diffraction peaks at 2θ angles of 31.06°, 41.92°, 60.96°, 72.26°, and 76.92°, corresponding to the (111), (200), (220), (311), and (222) crystal facets of TiC0.5N0.5 (JCPDS card number 38-1489), thereby confirming the FCC structure of TiCN. As the oxidation temperature reaches 400 °C, the anatase phase of TiO2 becomes prominent, with the rutile phase beginning to appear. The proportions of both anatase and rutile TiO2 gradually increase with the rise in oxidation temperature, while the content of TiCN significantly decreases. Notably, TiCN appears to be completely transformed at 450 °C, with the TiN crystal phase (JCPDS card number 50-0681) observed at 2θ angles of 31.06° and 41.92°. This indicates that C atoms are preferentially replaced by O atoms during the oxidation of TiCN. A rapid increase in the content of rutile TiO2 is observed at 500 °C, suggesting substantial oxidation of TiCN at this temperature, which is consistent with macroscopic and microscopic observations. Analysis of the oxidation results reveals that the amount of anatase TiO2 produced from the oxidation of TiCN consistently exceeds that of rutile TiO2. Thus, it can be concluded that the oxidation resistance of TiC, which possesses a higher C content, is significantly superior to that of TiCN, corroborating findings reported by Tang24 and Ajikumar.53
TPO-IR analysis offers several advantages for the detection of carbon in solid minerals, including rapid analysis, low sample consumption, high accuracy, minimal interference, and elevated detection limits.48,54 The characteristic peak temperature (Tmax) of C species in TiC and TiCN samples during oxidation and decomposition into CO2 can be accurately determined using TPO-IR.55Fig. 5(a) reveals the TPO-IR curve for TiC, revealing five distinct Tmax values at 496 °C, 597 °C, 670 °C, 751 °C and 994 °C, respectively. The presence of multiple Tmax values suggests that the TiC structure contains C atoms with varying graphitization degrees, which have different temperatures for oxidative decomposition.48Fig. 5(b) displays the TPO-IR curve for TiCN, which shows a single sharp peak at a Tmax of 533 °C. This indicates the presence of only one C species in TiCN, likely connected by a Ti–C–N bond. Thus, it can be inferred that the TiC oxidation is most pronounced at 994 °C, whereas TiCN exhibits significant activity at 533 °C.
 | ||
| Fig. 5 TPO-IR curves of undoped samples TiC (a) and TiCN (b). | ||
Fig. 6 presents the TG and DSC curves of TiC and TiCN over a continuous temperature range of 50 to 1150 °C. In Fig. 6(a), TiC exhibits a continuous exothermic reaction from approximately 400 to 1150 °C, with peak exothermic activities observed at 484 °C, 781 °C, 900 °C, 973 °C, and 1070 °C, respectively. TG curves indicate a significant increase in the weight of TiC during the thermal oxidation process. Fig. 6(b) illustrates that TiCN also demonstrates exothermic behavior during thermal oxidation, occurring between approximately 400 °C and 800 °C, with peaks at 468 °C, 488 °C, and 658 °C. Based on the principle that C oxidizes first, it can be inferred that 468 °C is the oxidation temperature of C in TiCN, while 488 °C is the oxidation temperature of N. In general, rutile is the most stable form of TiO2. XRD test results indicate that at 600 °C, a substantial amount of anatase TiO2 is present, suggesting the limited oxidation capacity of TiCN at this temperature. It can therefore be speculated that the heat release observed at 658 °C corresponds to the phase transformation of TiO2 from anatase into rutile and the continued oxidation of N. This is consistent with the TG curve data, which show an increase in the mass of TiC and TiCN during oxidation, as the C and N atoms of TiC and TiCN are gradually replaced by O atoms, the relative atomic mass of which is greater than that of C and N.
 | ||
| Fig. 6 TG and DSC curves of TiC (a) and TiCN (b). | ||
3.2 Analysis of photocatalytic properties of semi-oxidized TiC and TiCN
Notably, RhB, an organic dye prevalent in aqueous environments, poses significant water pollution challenges.56,57 This study employs RhB as a model compound to evaluate the adsorption and photocatalytic activity of semi-oxidized TiC and TiCN. Fig. 7 shows the residual ratio of RhB following adsorption and photocatalysis with original and semi-oxidized TiC and TiCN. In Fig. 7(a), the dark reaction phase for TiC samples reaches adsorption equilibrium in approximately 30 min, with an average adsorption efficiency of about 10%. The residual ratio of RhB adsorbed and degraded by oxidized TiC is lower than that of original TiC. As oxidation temperatures increase (100 °C, 200 °C, 300 °C and 400 °C), the residual ratio of RhB significantly decreases to 70.15%, 48.68%, 40.18% and 9.37%, respectively, after 150 min of visible light irradiation. In contrast, at higher oxidation temperatures (500 °C, 600 °C, 700 °C, 800 °C and 900 °C), the residual ratio of RhB increased evidently to 15.48%, 32.09%, 38.55%, 61.82% and 76.23%, respectively, after 150 min of visible light irradiation, indicating a notable increase in residual dye.58 This indicates that only when TiC forms surface C-doped TiO2 at an appropriate oxidation temperature can it achieve the best photocatalytic degradation effect on RhB. Both insufficient and excessive oxidations lead to a decrease in its photocatalytic degradation performance. Fig. 7(b) indicates that the dark reaction phase for TiCN samples achieves adsorption equilibrium in approximately 15 min, with an average adsorption efficiency of about 20%. The adsorption and degradation efficiencies of oxidized TiCN on RhB improve to varying extents. At an oxidation temperature of 400 °C, the residual ratio of RhB is approximately 50.80% after 150 min of visible light irradiation. The adsorption and degradation capacity of TiCN increases significantly, with residual ratios of RhB as low as 3.85% at oxidation temperatures of 450 °C and 500 °C. However, as the oxidation temperature rises further to 550 °C and 600 °C, the residual ratio of RhB increases to 13.90% and 28.98%, respectively, after 150 min of light exposure.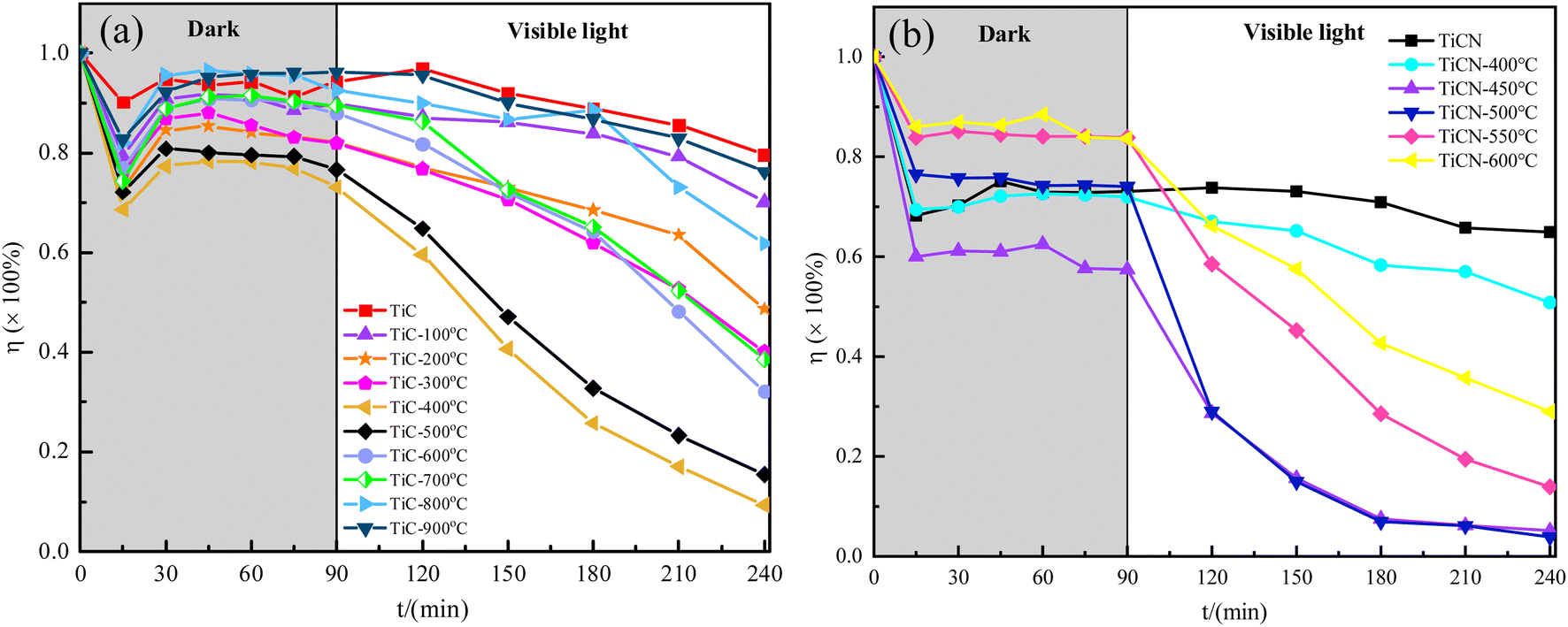 | ||
| Fig. 7 Residual ratio of RhB after adsorption and photocatalysis by the original and semi-oxidized TiC (a) and TiCN (b). | ||
To assess catalyst stability, two catalysts with optimal RhB degradation activity under visible light—TiC-400 °C and TiCN-500 °C—were selected for recovery and subjected to three rounds of cyclic degradation experiments, as shown in Fig. 8. Within the same cycle duration, which consists of a 90 min dark reaction followed by 150 min of visible light irradiation, and after three repeated uses, the residual ratios for TiC-400 °C were 9.97% in the first cycle test, 11.88% in the second, and 12.95% in the third. For TiCN-500 °C, the corresponding values were 5.37%, 5.88%, and 6.51% respectively. These results demonstrate that both catalysts exhibit excellent stability, with only a marginal decrease in activity after multiple cycles of use.
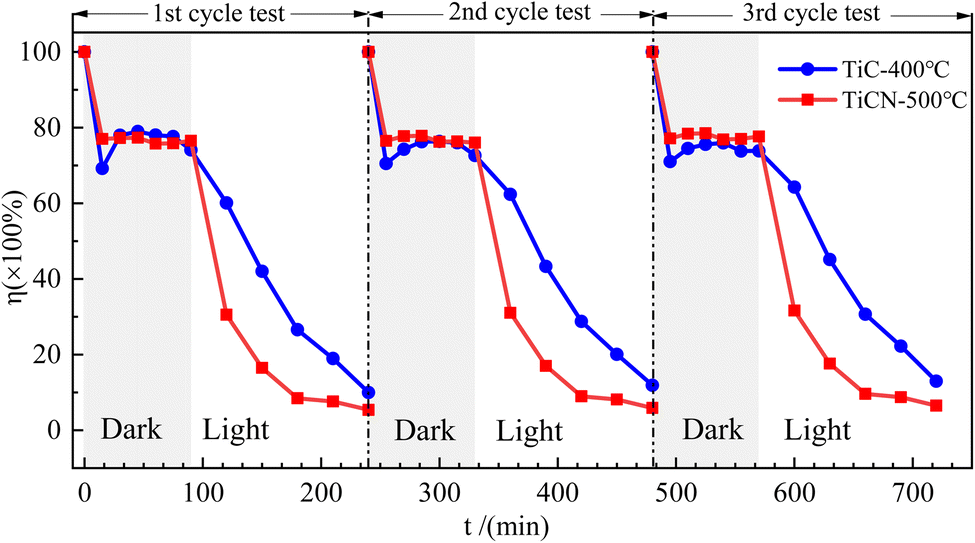 | ||
| Fig. 8 The results of cyclic recovery and degradation of RhB by TiC-400 °C and TiCN-500 °C catalysts. | ||
The experimental results indicate that oxidized TiC and TiCN exhibit superior photocatalytic activity compared to the original samples. By controlling the oxidation temperature, a specific concentration of C and/or N-doped semi-oxidized TiC and TiCN can be achieved, resulting in optimal photocatalytic performance. The optimal oxidation temperature for TiC is identified as 400 °C, while for TiCN, it ranges from 450 °C to 500 °C. Notably, TiCN oxidized at 500 °C demonstrates enhanced photocatalytic activity relative to TiC oxidized at 450 °C when evaluating only photocatalytic degradation activity. In addition, when considering the maximum adsorption and degradation efficiency, the residual ratio of RhB after adsorption and degradation by TiCN oxidized at 500 °C is 3.85%, which is 9.37% lower than that of RhB treated with TiC oxidized at 400 °C. This suggests that the adsorption and degradation capabilities of oxidized TiCN are superior to those of oxidized TiC. The comparison indicates that TiCN is doped with both C and N ions, while TiC is only doped with C ions. This difference in dopant ions may account for the observed variations in their adsorption and degradation capabilities.
The photocatalytic activity of TiC and TiCN in the visible light spectrum is attributed to the C/N-doped TiO2 formed in their semi-oxidized states. To elucidate the photocatalytic properties of C/N–TiO2 through this doping mechanism, UV-Vis DRS and XPS analyses were conducted on both the oxidized and non-oxidized samples.
Fig. 9 presents the UV-Vis DRS spectra of P25, TiC, TiCN, and their oxidized counterparts at varying temperatures. P25 exhibits absorption solely in the ultraviolet region (<400 nm), whereas the absorption spectra of oxidized TiC and TiCN extend into the visible region, displaying significant and broader absorption shoulders. This phenomenon can be attributed to the partial oxidation of TiC and TiCN, leading to the formation of C/N-doped TiO2. The energy levels associated with C and N introduce states within the band gap of TiO2, facilitating interactions with its conduction and valence bands.59 The electron transitions between these impurity energy levels and the respective bands result in a red shift of the absorption edge of TiO2.60 Consequently, the absorption threshold of TiO2 shifts from the ultraviolet to the visible region, indicating a reduction in the band gap.61,62 These findings are further corroborated by the comparative analysis of RhB degradation and UV-Vis spectra, confirming the enhanced photocatalytic activity of semi-oxidized TiC and TiCN.
 | ||
| Fig. 9 UV-Vis DRS spectra of TiC (a) and TiCN (b). | ||
XPS is effective for assessing the oxidation states of the surface layer of materials. Fig. 10 presents the XPS spectra of TiC and TiCN before and after oxidation. As depicted in Fig. 10(a), the Ti 2p spectrum of the TiC sample exhibits two distinct double-peak groups. The binding energies for one group are 461.18 eV (Ti 2p1/2) and 455.08 eV (Ti 2p3/2),63 which correspond to Ti 2p in TiC. The second group shows binding energies of 458.98 eV (Ti 2p1/2) and 464.68 eV (Ti 2p3/2), corresponding to Ti 2p in TiO2.64 Despite the minimal presence of a natural oxide layer on the surface of TiC, the XPS signals associated with TiO2 can be detected in the pristine TiC sample. Following oxidation at 500 °C, both TiC and TiO2 features are prominent in the XPS spectra, with a decrease in TiC content and a corresponding increase in TiO2 content relative to the original TiC. Most of the TiC is converted into TiO2 when the oxidation temperature reaches 800 °C, with the TiO2 bimodal Ti 2p peaks becoming highly pronounced and the TiC Ti 2p signals becoming significantly weaker. The C1s signal also exhibits notable changes, following a trend similar to that of Ti 2p. The C1s peak at a binding energy of 281.78 eV is attributed to TiC. As depicted in Fig. 10(b), the C1s signal in TiC is robust after oxidation at 500 °C, while it becomes nearly undetectable following oxidation at 800 °C, indicating complete oxidation of the TiC surface layer to TiO2 at this temperature. Collectively, these results show that the surface layer of TiC can effectively form an inner doped C–TiO2 phase after oxidation at 500 °C, whereas this formation does not occur at 800 °C. This behavior provides a compelling explanation for the observed experimental results pertaining to RhB degradation.
 | ||
| Fig. 10 XPS fitting curves of Ti (a), C (b) of TiC, and Ti (c), C (d) and N (e) of TiCN. | ||
The structure of TiCN is more complex than that of TiC. As a complex solid solution of TiC and TiN, TiCN includes Ti–N, Ti–C, and C–N bonds.65Fig. 10(c)–(e) depict the XPS Ti 2p, C 1s, and N 1s fitting curves of TiCN after oxidation at varying temperatures. The Ti 2p curve fitting distinguishes Ti from different sources, identifying three sets of Ti 2p double peaks.66 Specifically, peaks at 460.88 eV (Ti 2p1/2) and 455.18 eV (Ti 2p3/2) are a group of double peaks, corresponding to Ti–C bond in TiCN; peaks at 463.08 eV (Ti 2p1/2) and 456.68 eV (Ti 2p3/2) are derived from Ti–N bonds in TiCN; and peaks at 464.58 eV (Ti 2p1/2) and 458.48 eV (Ti 2p3/2) are derived from TiO2.67 As shown in Fig. 10(a), the Ti signal in TiCN is absent in the sample after oxidation at 500 °C. Combined with the observed changes in the signal peaks of C and N, it can be concluded that the Ti–N and O–Ti–O bonds persist at this temperature. These findings indicate that C and N are doped within TiCN following oxidation at 500 °C, which explains the enhanced photocatalytic activity of semi-oxidized TiCN compared to the original sample under visible light, further corroborating the above results of photocatalytic degradation of RhB.
In conclusion, TiC and TiCN are internally doped with C and/or N in a semi-oxidized state, with the doping levels modifiable through the degree of oxidation to optimize the photocatalytic activity. Fig. 11 illustrates the schematic representation of the internal doping effect of C and/or N resulting from the oxidation of TiC and TiCN surface layers. The oxidation process is initiated at the surface, leading to a gradual and uneven replacement of C and/or N in TiC and TiCN with O atoms, ultimately forming C and/or N-doped TiO2 exhibiting photocatalytic activity under visible light. The optimal doping oxidation temperatures for TiC and TiCN were determined to be 400 °C and 500 °C, respectively, with complete oxidation of their surface layers at 900 °C and 800 °C respectively.
 | ||
| Fig. 11 Schematic diagram of the C and/or N internal doping effect formed by oxidation of TiC and TiCN surface layers. | ||
4 Conclusions
In summary, this study systematically investigated the oxidation behavior of TiC and TiCN in ambient air at various temperatures and explored their photocatalytic degradation activity. The results showed that TiC has higher oxidation resistance than TiCN, and both materials can be significantly oxidized at 800 °C and 500 °C for 2 h, respectively. During oxidation, TiC is directly converted to TiO2, while TiCN undergoes a two-step process where C is replaced by O first, followed by N, with the final product being rutile TiO2. The analysis of oxidation behaviors indicated that the C species in TiC are more complex than those in TiCN. Notably, the optimal photocatalytic performance for TiC and TiCN was observed after oxidation at 400 °C and 500 °C for 2 h, respectively. Under these conditions, the residual rates of RhB degraded by oxidized TiC and TiCN were 9.37% and 3.85%, respectively, suggesting that oxidized TiCN has superior adsorption and degradation capabilities compared to oxidized TiC. The UV-Vis DRS and XPS results confirmed the formation of C/N-doped TiO2 in TiC and TiCN after appropriate semi-oxidation, which enhanced their photocatalytic activity, however, their performance in degrading other pollutants except RhB remains to be studied.Data availability
The data supporting this article have been all presented in the manuscript.Author contribution
Shiyun Tang: conceptualization, formal analysis, investigation, software, writing – original draft. Guoqiang Song: investigation, software, writing – review & editing, supervision. Junjiang Guo: methodology, software. Claudia Li: conceptualization, investigation, writing – review & editing. Kang Hui Lim: investigation, writing – review & editing. Pinli Diao: software, writing – original draft. Wenting Chen: software, writing – original draft. Feiyang Hu: methodology, software. Jaka Sunarso: writing – review & editing. Hao Tang: methodology, software, writing – original draft, supervision. Sibudjing Kawi: conceptualization, investigation, software, writing – review & editing, supervision.Conflicts of interest
We declare no competing financial interests or personal relationships that could influence this work and all the authors have read, reviewed and approved the manuscript for publication. The work described in the paper is original research that has not been published previously, and is not under consideration for publication elsewhere, in whole or in part.Acknowledgements
The research was supported by the Science and Technology Planning Project of Guizhou Province (Qiankehezhicheng [2021] Yiban 493 and Qiankehejichu-ZK [2023] Yiban 129), the National Natural Science Foundation of China (22062003), GIT Academic Seedling Training and Innovation Exploration Project (GZLGXM-19), the National Research Foundation, Singapore, A*STAR under its Low-Carbon Energy Research (LCER) Funding Initiative (FI) Project (U2102d2011, WBS: A-8000278-00-00), and the Singapore Ministry of Education Academic Research Fund (MOE AcRF) Tier 1 Project (WBS: A-0009184-01-00).References
- S. Zhou, W. Bi, J. Zhang, L. He, Y. Yu, M. Wang, X. Yu, Y. Xie and C. Wu, Adv. Mater., 2024, 36, e2400808 CrossRef PubMed.
- S. Jena, A. Madhuri, M. Gupta and B. P. Swain, J. Alloy. Compd., 2024, 175217 CrossRef CAS.
- S. A. Klymenko, S. V. Lytovchenko, S. A. Klymenko, A. S. Manokhin, M. Y. Kopeykina, A. O. Chumak and Y. O. Melnychuk, J. Superhard Mater., 2024, 46, 204–211 CrossRef.
- N. Czaplicka, A. Rogala and I. Wysocka, Int. J. Mol. Sci., 2021, 22, 12337 CrossRef CAS PubMed.
- A. Saurabh, C. M. Meghana, P. K. Singh and P. C. Verma, Mater. Today: Proc., 2022, 56, 412–419 CAS.
- J. Sahoo, S. Sarkhel, N. Mukherjee and A. Jaiswal, ACS Omega, 2022, 7, 45962–45980 CrossRef CAS PubMed.
- H. S. Gujral, G. Singh, J. H. Yang, C. I. Sathish, J. Yi, A. Karakoti, M. Fawaz, K. Ramadass, H. Ala'A and X. Yu, Carbon, 2022, 195, 9–18 CrossRef CAS.
- A. Jangam, S. Das, N. Dewangan, P. Hongmanorom, W. M. Hui and S. Kawi, Catal. Today, 2020, 358, 3–29 CrossRef CAS.
- H. Mao, Y. Zhang, J. Wang, K. Cui, H. Liu and J. Yang, Coatings, 2022, 12, 801 CrossRef CAS.
- D. Li, D. Dai, G. Xiong, S. Lan and C. Zhang, Adv. Sci., 2023, 10, 2300658 CrossRef CAS.
- Y. Zhang, P. Zhang, S. Zhang, Z. Wang, N. Li, S. R. P. Silva and G. Shao, InfoMat, 2021, 3, 790–803 CrossRef CAS.
- X. Luo, W. Li, L. Yuan, G. Xie and Y. Su, Nano Trends, 2024, 8, 100061 CrossRef.
- H. Li, X. Zhao, G. Xie and Y. Su, Appl. Phys. Lett., 2025, 126, 053504 CrossRef CAS.
- V. A. Lavrenko, L. A. Glebov, A. P. Pomitkin, V. G. Chuprina and T. G. Protsenko, Oxid. Met., 1975, 9, 171–179 CrossRef CAS.
- V. B. Voitovich, High Temp. Mater. Process., 1997, 16, 243–254 CAS.
- S. Shimada and M. Kozeki, J. Mater. Sci., 1992, 27, 1869–1875 CrossRef CAS.
- M. Gherrab, V. Garnier, S. Gavarini, N. Millard-Pinard and S. Cardinal, Int. J. Refract. Hard Met., 2013, 41, 590–596 CrossRef CAS.
- Y. Wang, H. Yang and X. Chen, ACS Appl. Electron. Mater., 2025, 7, 2910–2918 CrossRef CAS.
- C. Kainz, N. Schalk, C. Saringer and C. Czettl, Surf. Coat. Technol., 2021, 406, 126633 CrossRef CAS.
- A. Saurabh, C. M. Meghana, P. K. Singh and P. C. Verma, Mater. Today: Proc., 2022, 56, 412–419 CAS.
- J. Dai, G. Xie, X. Huo, J. Li, S. Deng and Y. Su, Small, 2025, 21, 2407773 CrossRef CAS.
- J. He, Z. He, Y. Qin, H. Zhao and Y. Bi, J. Therm. Spray Technol., 2022, 31, 2280–2299 CrossRef CAS.
- O. I. Ali, I. G. Gyurika, T. Korim and M. Jakab, Process. Appl. Ceram., 2024, 18, 213–223 CrossRef CAS.
- S. Tang, N. Shi, J. Wang and A. Tang, Ceram. Int., 2017, 43, 3818–3823 CrossRef CAS.
- O. I. Ali and I. G. Gyurika, Arch. Adv. Eng. Sci., 2024, 1–12 Search PubMed.
- Y. Su, Y. Liu, W. Li, X. Xiao, C. Chen, H. Lu, Z. Yuan, H. Tai, Y. Jiang and J. Zou, Mater. Horiz., 2023, 10, 842–851 RSC.
- H. Li, Y. Ding, K. Luo, Q. Zhang, H. Yuan, S. Xu and M. Xu, iScience, 2025, 28, 111750 CrossRef CAS.
- J. Li, G. Xie, L. Dai, M. Yang and Y. Su, J. Compos. Sci., 2025, 9, 123 CrossRef CAS.
- Y. Zhou, W. Yang, L. Feng, J. Hong, M. Abbas and S. Kawi, Appl. Catal., B, 2025, 363, 124782 CrossRef CAS.
- J. Huang, G. Xie, X. Xu, Z. Geng and Y. Su, ACS Appl. Mater. Interfaces, 2024, 16, 58838–58847 CrossRef CAS.
- Z. Li, S. Wang, J. Wu and W. Zhou, Renewable Sustainable Energy Rev., 2022, 156, 111980 CrossRef.
- A. S. Nur, M. Sultana, A. Mondal, S. Islam, F. N. Robel, A. Islam and M. S. A. Sumi, J. Water Process. Eng., 2022, 47, 102728 CrossRef.
- M. Hasanuzzaman, M. Mokammel, M. J. Islam and S. Hashmi, Adv. Mater. Process. Technol., 2024, 10, 1320–1343 Search PubMed.
- A. Aljaafari, Curr. Nanosci., 2022, 18, 499–519 CrossRef CAS.
- I. Majeed, H. Ali, A. Idrees, A. Arif, W. Ashraf, S. Rasul, M. A. Khan, M. A. Nadeem and M. A. Nadeem, Energy Adv., 2022, 1, 842–867 RSC.
- N. R. Reddy, P. M. Reddy, N. Jyothi, A. S. Kumar, J. H. Jung and S. W. Joo, J. Alloy. Compd., 2023, 935, 167713 CrossRef CAS.
- A. Patidar, V. R. Dugyala, S. Chakma, M. N. Galodiya and A. S. Giri, Res. Chem. Intermed., 2024, 50, 1035–1063 CrossRef CAS.
- Y. Guo, T. Zhang, Z. Chen, C. Duan, C. Li, S. Kawi and Y. Li, J. Alloy. Compd., 2024, 1005, 175964 CrossRef CAS.
- S. Cheng, Z. Sun, K. H. Lim, T. Z. H. Gani, T. Zhang, Y. Wang, H. Yin, K. Liu, H. Guo and T. Du, Adv. Energy Mater., 2022, 12, 2200389 CrossRef CAS.
- S. Cheng, Z. Sun, K. H. Lim, K. Liu, A. A. Wibowo, T. Du, L. Liu, H. T. Nguyen, G. K. Li and Z. Yin, Appl. Catal., B, 2024, 343, 123583 CrossRef CAS.
- Y. Zeng, K. G. Haw, Y. Wang, S. Zhang, Z. Wang, Q. Zhong and S. Kawi, ChemCatChem, 2021, 13, 491–505 CrossRef CAS.
- J. Qu, T. Yang, P. Zhang, F. Yang, Y. Cai, X. Yang, C. M. Li and J. Hu, Appl. Catal., B, 2024, 348, 123827 CrossRef CAS.
- P. Wang, Y. Dai, Z. Song, Y. Wang, J. Wei, Z. Liu, Y. Ma, F. Yang, J. Qu and S. Liu, Chem. Eng. J., 2025, 511, 162084 CrossRef CAS.
- S. Tang, J. Ran, J. Guo and A. Tang, J. Chem., 2019, 2019, 8587949 Search PubMed.
- T. S. Natarajan, V. Mozhiarasi and R. J. Tayade, Photochem, 2021, 1, 371–410 CrossRef.
- A. Marucco, F. Catalano, I. Fenoglio, F. Turci, G. Martra and B. Fubini, Chem. Res. Toxicol., 2015, 28, 87–91 Search PubMed.
- L. Hao, Z. Wang, Y. Zheng, Q. Li, S. Guan, Q. Zhao, L. Cheng, Y. Lu and J. Liu, Appl. Surf. Sci., 2017, 391, 275–281 CrossRef CAS.
- Q. Xu, S. Tang, A. Tang and Y. Tian, J. Anal. Methods Chem., 2022, 2022, 3255760 Search PubMed.
- H. Nakajima, W. Phae-Ngam, S. Chaiyakun, V. Tapanwong, T. Wongpinij, T. Lertvanithphol and M. Horprathum, Appl. Surf. Sci., 2024, 673, 160900 CrossRef CAS.
- C. Y. Tsai and K. Mondal, Cryst. Res. Technol., 2021, 56, 2000231 CrossRef CAS.
- S. Jena, A. Madhuri, M. Gupta and B. P. Swain, J. Alloy. Compd., 2024, 175217 CrossRef CAS.
- F. Habibi and A. Samadi, Surf. Coat. Technol., 2024, 478, 130472 CrossRef CAS.
- P. K. Ajikumar, M. Kamruddin, T. R. Ravindran, S. Kalavathi and A. K. Tyagi, Ceram. Int., 2014, 40, 10523–10529 CrossRef CAS.
- C. Li, G. Song, K. H. Lim, F. Hu, J. Sunarso, N. Yang, M. S. Wong, S. Liu and S. Kawi, Adv. Compos. Hybrid Mater., 2024, 7, 99 CrossRef CAS.
- S. Das, K. H. Lim, T. Z. Gani, S. Aksari and S. Kawi, Appl. Catal., B, 2023, 323, 122141 CrossRef CAS.
- T. Zhang, T. Wang, F. Meng, M. Yang and S. Kawi, J. Mater. Chem. C, 2022, 10, 5400–5424 RSC.
- S. Qiu, Q. Li, X. Li, J. Ma, L. Wu, X. Xie, L. Wu, S. Askari, N. Dewangan and J. Ashok, Adv. Sustain. Syst., 2024, 8, 2300340 CrossRef CAS.
- T. Zhang, F. Meng, Y. Cheng, N. Dewangan, G. W. Ho and S. Kawi, Appl. Catal., B, 2021, 286, 119853 CrossRef CAS.
- Y. Zeng, Y. Wang, P. Hongmanorom, Z. Wang, S. Zhang, J. Chen, Q. Zhong and S. Kawi, Chem. Eng. J., 2021, 409, 128242 CrossRef CAS.
- S. Xu, Z. Zhang, H. Zhang, Z. Li, H. Peng, S. Kawi, A. Xie and P. Qiu, Chem. Eng. J., 2024, 158693 Search PubMed.
- V. R. Akshay, B. Arun, M. Mukesh, A. Chanda and M. Vasundhara, Vacuum, 2021, 184, 109955 CrossRef CAS.
- Y. Wang, T. Wang, H. Arandiyan, G. Song, H. Sun, Y. Sabri, C. Zhao, Z. Shao and S. Kawi, Adv. Mater., 2024, 2313378 CrossRef CAS PubMed.
- W. Xu, Y. Peng, D. Hu, I. Razanau, D. Gu and W. Xiao, ChemCatChem, 2024, 16, e202400873 CrossRef CAS.
- L. Wang, L. Wang, Y. Shi, B. Zhao, Z. Zhang, G. Ding and H. Zhang, Chemosphere, 2022, 306, 135628 CrossRef CAS PubMed.
- Q. Cao and D. Li, Solid State Commun., 2023, 361, 115079 CrossRef CAS.
- J. Tao, T. Luttrell and M. Batzill, Nat. Chem., 2011, 3, 296–300 CrossRef CAS PubMed.
- Y. Zhang, Z. Bao, R. Wang, Y. Su, Y. Wang, X. Cao, R. Hu, D. Sha, L. Pan and Z. Sun, Nanoscale, 2024, 16, 1751–1757 RSC.
| This journal is © The Royal Society of Chemistry 2025 |