
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompetitive reversed quartet mechanisms for photogenerated ground state electron spin polarization†
Martin L.
Kirk
 *abc,
David A.
Shultz
*d,
Patrick
Hewitt‡
d,
Anil Reddy
Marri
d and
Art
van der Est
*e
*abc,
David A.
Shultz
*d,
Patrick
Hewitt‡
d,
Anil Reddy
Marri
d and
Art
van der Est
*e
aDepartment of Chemistry and Chemical Biology, The University of New Mexico, MSC03 2060, 1 University of New Mexico, Albuquerque, NM 87131-0001, USA
bThe Center for High Technology Materials, The University of New Mexico, Albuquerque, New Mexico 87106, USA
cCenter for Quantum Information and Control (CQuIC), The University of New Mexico, Albuquerque, New Mexico 87131-0001, USA
dDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695-8204, USA
eDepartment of Chemistry, Brock University, St. Catharines, Ontario, Canada L2S 3A1. E-mail: mkirk@unm.edu; shultz@ncsu.edu; avanderest@brocku.ca
First published on 8th August 2023
Abstract
Photoinduced electron spin polarization (ESP) of a spin-½ organic radical (nitronyl nitroxide, NN) in a series of Pt(II) complexes comprised of 4,4′-di-tert-butyl-2,2′-bipyridine (bpy) and 3-tert-butylcatecholate (CAT) ligands, where the CAT ligand is substituted with (CH3)n-meta-phenyl-NN (bridge-NN) groups, is presented and discussed. We show the importance of attenuating the energy gap between localized NN radical and chromophoric excited states to control both the magnitude and sign of the optically-generated ESP, and to provide deeper insight into the details of the ESP mechanism. Understanding electronic structure contributions to optically generated ESP will enhance our ability to control the nature of prepared states for a variety of quantum information science applications, where strong ESP facilitates enhanced sensitivity and readout capabilities at low applied magnetic fields and higher temperatures.
Introduction
A critical advantage of using molecular spin systems for quantum information science (QIS) applications1–3 stems from the inherent ability to control the molecular electronic structure by synthetic design, with a particular emphasis on the optical and magnetic properties of these systems and how they can be further manipulated by external fields to perform various gate operations. Photoexcitation of closed-shell S = 0 donor–acceptor molecules creates a transient electron–hole pair. Here, the hole is created by removal of an electron from the HOMO level of the Donor, with the promoted electron being transferred to a LUMO orbital that is spatially localized on the acceptor. When these D–A molecules are covalently coupled to one or more persistent radicals, multiple pairwise excited state exchange interactions define the nature of the coupled spin system in the excited state. These exchange-coupled excited states can be exploited for the optical generation of well-defined initialized ground spin-states, which can display long-lived electron spin polarization (ESP: non-Boltzmann population of ms levels in ground- (GS) and excited states (ES))4–22 that allows for further spin manipulation and utilization as a quantum processor or information storage system.5Photogenerated multispin qubits have been shown to be important for the fast generation of ground state ESP. The fast relaxation from the ESP generating ES manifold ensures that the observed EPR response arises solely from the GS, without contributions from the spin polarized excited state(s). Thus, the lifetime of the GS ESP is dependent on the intrinsic T1/T2 of the spin carrying moiety, with long relaxation times enhancing the ability to use microwave pulses for further manipulation of the spin system and to perform gate operations that are critical to QIS applications. Strong spin polarization (e.g., ∼90%) can also be achieved in the absence of a photoprocess, but this requires ultra-low temperatures (0.2 K for g·β·H = 0.3 cm−1) and/or very high magnetic fields (7.5 K at 10 T). As a result, understanding the factors that lead to strong optically generated ground state ESP will allow for enhanced sensitivity and readout at lower applied magnetic fields and at higher temperatures.3,23
Experimental
Synthesis
See the ESI† for synthesis and spectroscopy details.Electronic absorption spectroscopy
Electronic absorption spectra of the complexes were collected using a UV-3600 Shimadzu UV-Vis-NIR spectrometer as methylene chloride solutions.Steady state- and transient EPR spectroscopies
Fluid solution EPR spectra for characterization purposes were recorded on a Bruker ELEXSYS E500 cw X-band spectrometer at NC State University. Samples for steady state and transient EPR (TREPR) measurements at Brock University were prepared by dissolving the solid Pt complexes in 2-methyl-THF (2-MTHF) to a concentration of ∼0.5 mM. The samples were placed in 4 mm O.D. quartz EPR tubes and degassed by repeated freeze–pump–thaw cycles. The frozen, degassed samples were then transferred without thawing from liquid nitrogen to the spectrometer cryostat, which was stabilized at 20 K. The steady state cw-EPR spectra were collected using field modulation detection with a modulation amplitude of 0.1 mT and a microwave power of 6.3 μW. TREPR time/field datasets were collected with direct detection at the same microwave power that was used for the steady state experiments. The TREPR samples were irradiated at 532 nm using 10 ns laser flashes from a frequency doubled NdYAG laser with a flash energy of ∼4 mJ. The modified Bruker EPR 200D-SRC X-band spectrometer used for the EPR experiments has been described in detail elsewhere.24Results and discussion
Mechanisms for ESP generation
Our approach to creating, controlling, and understanding the electronic origins of GS- and ES ESP, in addition to controlling the relevant photoprocesses that make this possible, utilizes bespoke ligand-to-ligand charge transfer (LL′CT) chromophores11,25–29 that have been appended with one or more stable organic radicals.6–11,25 These radical-elaborated (diimine)Pt(CAT-bridge-NN) (NN = nitronyl nitroxide; CAT = catecholate) complexes possess short-lived25–27 CAT → diimine LL′CT excited states that are comprised of three S = ½ spin centers, which are spatially localized on the CAT donor, the diimine acceptor, and the persistent NN radical,11 respectively (Fig. 1A).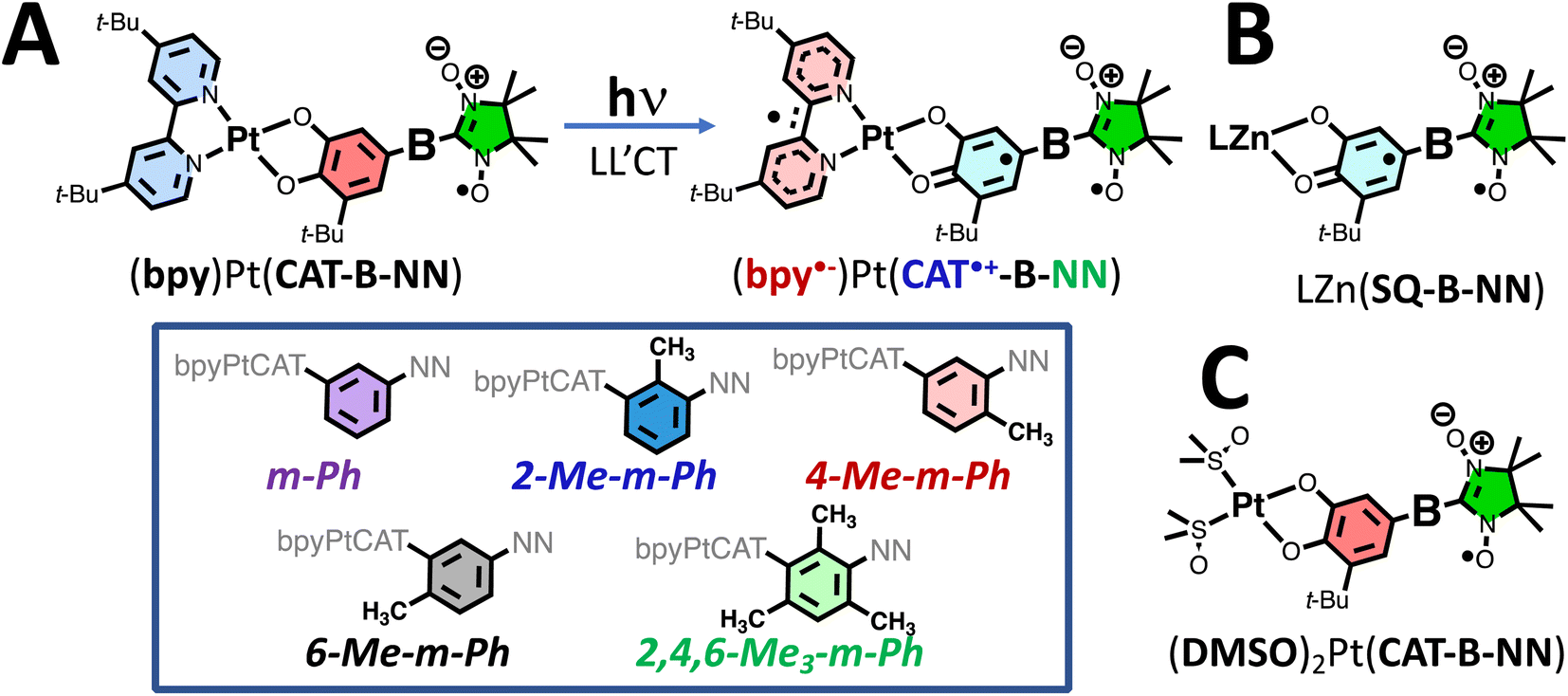 | ||
| Fig. 1 (A) Photoinduced ligand-to-ligand charge transfer (LL′CT, top). A valence bond description of the photoprocess shows the CAT → bpy CT oxidizing the CAT to the S = ½ CAT˙+ (i.e., a SQ) and simultaneously reducing the bpy to an S = ½ radical anion. Color-coded bridge units for the complexes detailed in this study are provided (inset, bottom). (B) Zn(II) semiquinone- (SQ) nitronyl nitroxide (NN) complexes that serve as ground state analogs of the excited state CAT˙+–NN˙ pairwise exchange interaction, JCAT˙+–NN. SQ–NN magnetic exchange in the ZnII complexes approximates excited state CAT˙+–NN exchange in the PtII complexes shown in (A). (C) Non ESP generating Pt(II) complexes that lack the LL′CT chromophore acceptor ligand (two DMSO ligands replace bpy), but retain the CAT-B-NN ligand. | ||
Pairwise excited state exchange interactions exist between the CAT˙+ (CAT˙+ = orthosemiquinone radical, SQ) and the diimine anion radical (i.e., Jbpy˙−–CAT˙+ when diimine = 4,4′-di-tert-butyl-2,2′-bipyridine, bpy) and between the CAT˙+ moiety and the localized persistent NN radical (i.e., JCAT˙+–NN; Fig. 1A and B). This results in a more complex and higher density manifold of excited states when compared to the open-shell singlet- and triplet states formed by photoexcitation of molecules that possess closed-shell singlet ground state configurations.28,30–32 For radical-elaborated (diimine)Pt(CAT-bridge-NN) complexes there are three low-lying excited states that differ by specific spin configurations: 2S1 (the spin singlet LL′CT chromophoric configuration S1 and NN doublet with Stot = ½), 2T1 (the spin triplet LL′CT chromophoric configuration T1 coupled to the NN doublet to give Stot = ½), and 4T1 (the spin triplet LL′CT chromophoric configuration coupled to the NN doublet to give Stot = 3/2; Fig. 2). As such, all three of these excited state potential energy surfaces (PESs) are expected to be highly nested but displaced from the GS PES. The judicious choice of the bridge fragment affects both the sign and magnitude of JCAT˙+–NN in these ES configurations, but has a negligible effect on Jbpy˙−–CAT˙+.25,27 Consequently, Jbpy˙−–CAT˙+ ensures that the chromophoric triplets in the 2T1 and 4T1 states are markedly lower in energy than the chromophoric singlet in the 2S1 state (i.e., E(2T1, 4T1) < E(2S1)).11
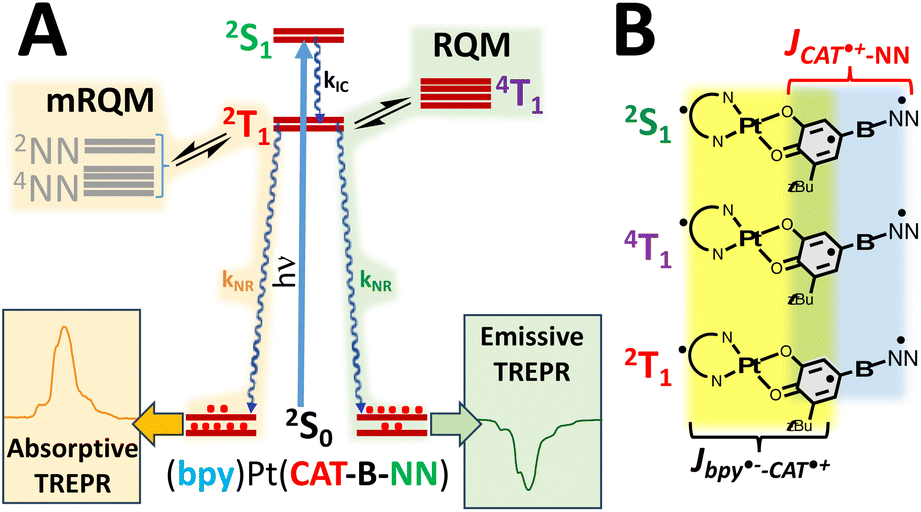 | ||
| Fig. 2 (A) Jablonski diagram relevant to the mRQM (orange, left pathway) and the RQM (green, right pathway) that results in enhanced absorptive- or emissive TREPR spectra, respectively. (B) Relative orientations of electron spins in the LL′CT manifold. Colored arrows depict relative orientations of bpy˙−, CAT˙+ and NN spins. | ||
The relative energy ordering of 2T1 and 4T1 states can be conveniently altered by the nature of the bridge connectivity (m-Ph vs. p-Ph coupling).7–11 For p-Ph linkages, we observe ferromagnetic JCAT˙+–NN, which leads to E(4T1) < E(2T1),7,8 while for m-Ph linkages the JCAT˙+–NN exchange coupling is antiferromagnetic and E(4T1) > E(2T1).9,10 Thus, our understanding of molecular design features in these systems, an a priori knowledge of Jbpy˙−–CAT˙+ from spectroscopy,11 and our understanding of ES JCAT˙+–NN exchange interactions based on ground state analog studies (Fig. 1B)33–39 leads to an accurate estimation of the relative energies of the 2S1, 2T1, and 4T1 LL′CT excited states in these systems.
In addition, the NN radical possesses both 2NN and 4NN excited states that are nearly isoenergetic with the LL′CT excited states, leading to additional control over the generation of ES ESP by the reversed quartet (RQM)40–42 and modified RQM (mRQM)10 mechanisms (vide infra). We can conveniently obtain the energy of the 2NN state from electronic absorption spectroscopy of the DMSO complexes shown in Fig. 1C, while the 4NN state energy has been suggested experimentally43 and evaluated computationally.10,36 The S = ½ ground state configuration, “localized” NN excited states (involved in the transitions shown in Fig. 3B; vide infra), 4NN, and LL'CT configurations and states are illustrated in Fig. S1.†
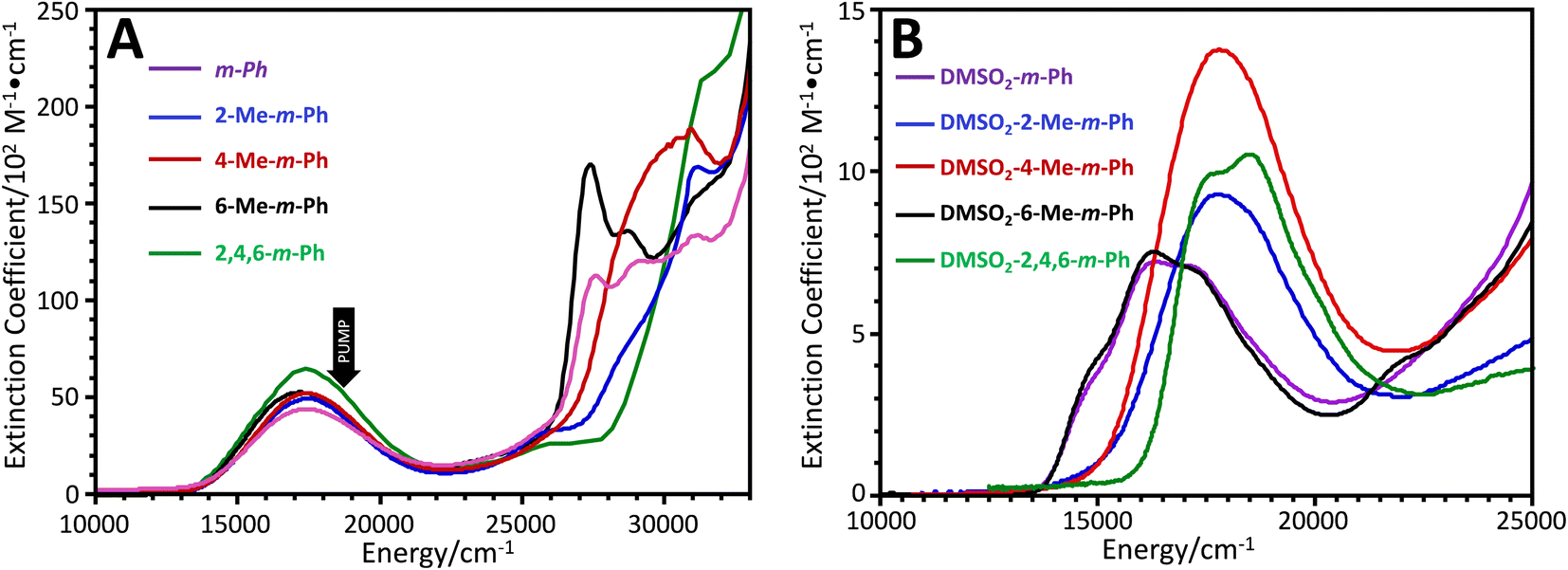 | ||
| Fig. 3 (A) Electronic absorption spectra of (bpy)Pt(CAT-B-NN) complexes (shown in Fig. 1A) as solutions in CH2Cl2 at 298 K. (B) Electronic absorption spectra of (DMSO)2Pt(CAT-B-NN) complexes (shown in Fig. 1C) as solutions in CH2Cl2 at 298 K. | ||
Optical excitation into the LL′CT band creates the initial 2S1 doublet excited state that rapidly undergoes exchange-mediated enhanced intersystem crossing to the 2T1 state (Fig. 2A).25 Molecular diffusion is precluded in low temperature optical glasses, and this eliminates molecule–molecule collision-based mechanisms for the generation of any ES ESP that could be transferred to the GS.44 However, both the RQM and mRQM require equilibration between different states to generate ES ESP that can subsequently be transferred to the electronic GS. Both of these mechanisms invoke equilibration amongst the ms levels of 2T1 and 4T1 (RQM) and the 2T1 and 2/4NN (mRQM) states by direct spin orbit coupling (SOC via zero-field splitting),40,41,45 spin-vibronic coupling,46 vibronic spin–orbit coupling,46 or vibronically-assisted intramolecular energy transfer when the energy gaps between these states are on the order of kBT. We note that both the RQM and mRQM can also be operative simultaneously. For strong chromophore-radical exchange, spin-vibronic mechanisms are expected to dominate,46 but the 2T1–4T1,2/4NN gaps can be attenuated by bond rotations that are facilitated by matrix effects in the optical glass to generate ESP. Fast relaxation25 of the spin-polarized 2T1 state by charge recombination results in spin-polarization transfer to the 2S0 ground state to afford GS ESP,19,21,42 which is conveniently detected by transient EPR (TREPR) spectroscopy.44
A hallmark of the RQM is the correlation of the absorptive- or emissive TREPR signal with the sign of the chromophore-radical exchange coupling parameter (JCR).40,41 However, our recent work has shown that when radical-elaborated (diimine)Pt(CAT-bridge-NN) excited states are similarly close in energy to 2T1, this results in ES equilibria and associated transitions between these states and the sign of the photoinduced TREPR does not necessarily correlate with the sign of JCR (JCR ≈ JCAT˙+–NN when Jbpy˙−–CAT˙+ ≫ JCAT˙+–NN). This realization resulted in our development of the modified reversed quartet mechanism (mRQM).7,8,10 Indeed, our choice of LL′CT chromophore and stable radical is based on these nearly isoenergetic LL′CT- and radical-centered ESs, leading to an expanded set of structural parameters that contribute to key design principles for controlling both the sign and magnitude of the photoinitiated ESP and its subsequent transfer to the GS, expanding the utility of fast photoinduced ESP for future QIS sensing applications and developing a greater understanding of coupled molecular qubit interactions.
Herein, we show how the RQM and mRQM become competitive mechanisms for the generation of either absorptive or emissive ES ESP. This occurs when the antiferromagnetic JCAT˙+–NN exchange interaction is modified by specific steric interactions between the stable NN radical and the bridging unit that couples the NN radical to the LL′CT chromophore.
Electronic absorption spectroscopy
Fig. 3A shows the electronic absorption spectra of the (bpy)Pt(CAT-B-NN) complexes used in this study. The bpy → CAT LL′CT band for each complex is present as an isolated band near 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1. Higher energy transitions near 28000 cm−1 are associated with the Ar–NN chromophore, and methyl substituent-dependent changes in transition energies and intensities are reminiscent of the para-Ph-Men-bridged complexes that we have studied previously.7,8 In addition, we have probed the effects of methyl groups on the bridge fragment in LZn(SQ-B-NN) complexes. These complexes are important since they serve as GS analogs of the excited state exchange interactions in (bpy)Pt(CAT-B-NN), and therefore allow for the ES JCAT˙+–NN exchange interaction to be estimated with precision.33,34
000 cm−1. Higher energy transitions near 28000 cm−1 are associated with the Ar–NN chromophore, and methyl substituent-dependent changes in transition energies and intensities are reminiscent of the para-Ph-Men-bridged complexes that we have studied previously.7,8 In addition, we have probed the effects of methyl groups on the bridge fragment in LZn(SQ-B-NN) complexes. These complexes are important since they serve as GS analogs of the excited state exchange interactions in (bpy)Pt(CAT-B-NN), and therefore allow for the ES JCAT˙+–NN exchange interaction to be estimated with precision.33,34
These prior studies on LZn(SQ-B-NN) complexes detailed how ring rotations lead to differential conjugation between the bridge, SQ, and NN groups, which result in inhibition of resonance33,34 and notable differences in both the SQ → NN intraligand charge transfer band intensity and energy. Similarly, methyl groups that disrupt bridge-NN conjugation in the complexes studied here (e.g., 2-Me-m-Ph, 4-Me-m-Ph, and 2,4,6-Me3-m-Ph) result in hypso- and hyperchromic shifts of the Ar–NN transition (Fig. 3B). In marked contrast, the LL′CT transitions for these complexes display only minor changes in bandshape/intensity as a function of the bridge substitution patterns, reflecting their dominant CAT → bpy LL′CT character. The most notable change in the LL′CT transition within this series of molecules is the hyperchromic shift of the LL′CT band in 2,4,6-Me3-m-Ph. This is expected to derive from an increase in the CAT → bpy dipole due to the attenuated CAT → bridge delocalization that results from the flanking methyl groups.
Fig. 3B shows the low-energy electronic absorption bands of the (DMSO)2Pt(CAT-B-NN) complexes, which lack the bpy acceptor ligand but retain the CAT-B-NN moiety. The electronic absorption spectra of these complexes are important for understanding the role of localized 2NN states (via vibronic coupling/intramolecular energy transfer) or 4NN (via spin-vibronic coupling/vibronic spin–orbit coupling)40,41,45,46 in the generation of ESP. The electronic absorption spectra of the (DMSO)2Pt(CAT-B-NN) complexes reveal that the weaker, localized configurationally mixed transitions that derive from NN(SOMO) → NN(LUMO) and NN(HOMO) → NN(SOMO) one-electron promotions, occur in the same energy region as the more intense LL′CT band.10,43,47 We note that the 2NN excited state formed by this localized NN-based transition is also present in the (bpy)Pt(CAT-B-NN) complexes, but is obscured by the LL′CT transition that possesses a markedly larger oscillator strength. Furthermore, spectroscopic-43 and computational studies10 suggest there are two NN excited states in this region, and our computations indicate that this second state may be a localized 4NN quartet state (see Fig. 2A).10
The methyl substituent-induced changes in the 28000 cm−1 transition discussed above are mirrored by changes in the localized NN-based transitions near 17000 cm−1, as shown in Fig. 3B. Specifically, methyl groups that disrupt NN-bridge conjugation result in ∼1000 to 2000 cm−1 hypsochromic shifts of this band. Thus, the presence of bridge methyl groups changes the energy gaps between 2/4NN-based excited states and the those of the LL′CT manifold, providing a mechanism for changing the sign and magnitude of the GS ESP by tuning the degree of LL′CT–2,4NN mixing and equilibria.
Ground state cw- and transient electron paramagnetic resonance spectroscopy
Fig. 4A displays the 20 K X-band field-modulation detected cw-EPR spectra of the five meta-phenylene-bridged (bpy)Pt(CAT-B-NN) complexes used in this study. These spectra derive from the S = ½ NN radical spin in the electronic ground state, where the system is at thermal equilibrium. The lineshapes of the spectra are characterized by anisotropic g- and 14N hyperfine tensors that are typical of NN radicals.48–50 The corresponding spin-polarized TREPR spectra measured with direct detection are presented in Fig. 4B. The TREPR spectra reveal only the transient spin polarized signals and are effectively light-minus-dark difference spectra. No TREPR signals were observed that could be assigned as originating from the high-spin 2,4T1 ESs or the localized 2,4NN radical states. Rather, the observed lineshapes are virtually identical to the thermal equilibrium cw-spectra shown in Fig. 4A (see Fig. 4C for the derivative TREPR signal of 2,4,6-Me3-m-Ph), and the signal-to-noise ratios of the TREPR spectra suggest that strong ground state ESP is generated in these systems. Notably, the lifetime of the (DMSO)2Pt(CAT-B-NN) complexes is ∼70 ± 5 ps (see ref. 25) and these molecules, which lack the bpy diamine acceptor moiety, do not display spin polarized TREPR spectra when they are irradiated at 532 nm in resonance with the localized 2NN band (see Fig. S2†). The ground state cw-spectra collected at thermal equilibrium are virtually identical to one another and have been normalized to the same maximum intensity. However, using this same normalization procedure, the relative intensities – and in one case (6-Me-m-Ph)9 the sign – of the TREPR spectra are markedly different. This strongly indicates that both the sign and magnitude of the GS ESP can be controlled by the magnitude of the excited state antiferromagnetic JCAT˙+–NN exchange interaction, in concert with the energy gaps between 2/4NN-based excited states and the those of the 2T1 LL′CT state. This observation is in marked contrast to the tenets of the RQM,40,41 where the sign of JCAT˙+–NN is expected to determine the sign of the GS ESP.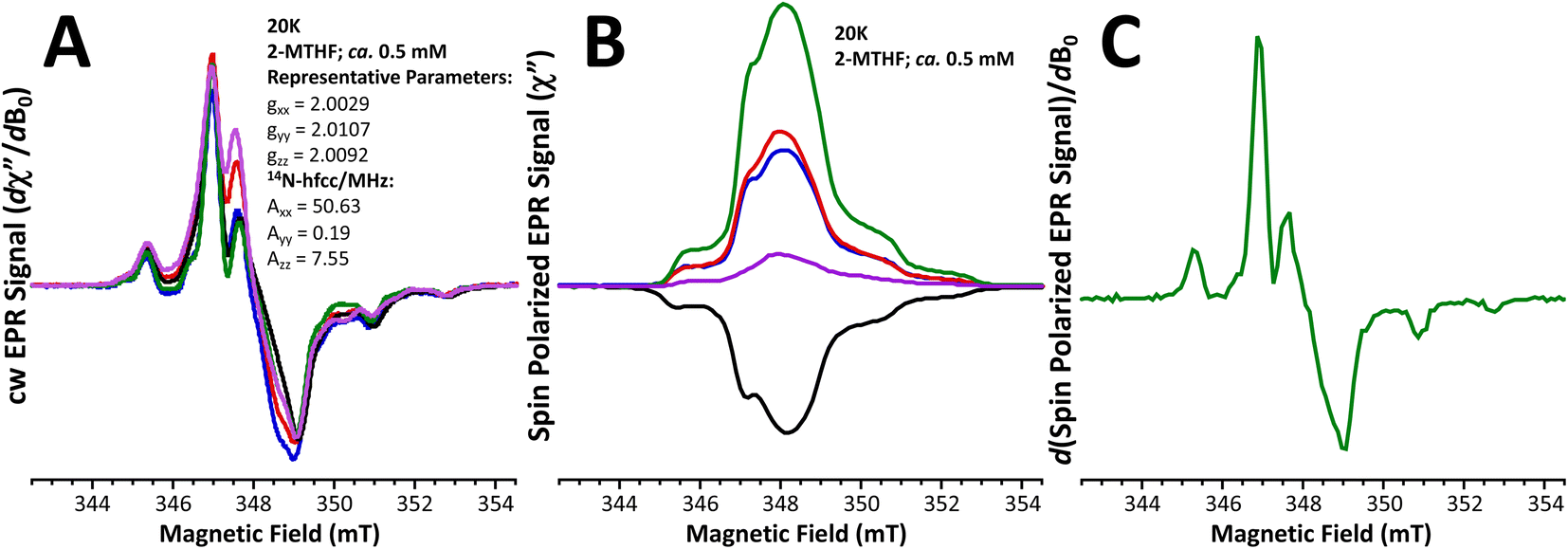 | ||
Fig. 4 Low-temperature X-band EPR spectra at thermal (Boltzmann) equilibrium and spin-polarized EPR spectra of (bpy)Pt(CAT-bridge-NN) complexes at 20 K in 2-MTHF: , ,  , ,  , ,  , ,  . (A) Field modulation detected cw-EPR spectra of the ground states in thermal equilibrium without light excitation. (B) Light-induced, spin-polarized TREPR spectra, measured with direct detection and extracted from the time/field datasets at 0.6 μs after the laser flash. Light excitation at 532 nm, concentrations ∼0.5 mM. C: First-derivative of TREPR spectrum of 2,4,6-Me3-m-Ph illustrating that TREPR signals in B are from ground states. . (A) Field modulation detected cw-EPR spectra of the ground states in thermal equilibrium without light excitation. (B) Light-induced, spin-polarized TREPR spectra, measured with direct detection and extracted from the time/field datasets at 0.6 μs after the laser flash. Light excitation at 532 nm, concentrations ∼0.5 mM. C: First-derivative of TREPR spectrum of 2,4,6-Me3-m-Ph illustrating that TREPR signals in B are from ground states. | ||
Molecular- and electronic structure contributions to electron spin polarization
Meta connectivity of CAT and NN moieties through a phenylene-ring bridge ensures antiferromagnetic exchange coupling (JCAT˙+–NN < 0) of CAT˙+ and NN spins in the LL′CT excited state. Although the number and position of methyl groups on the bridge modulates the magnitude of the antiferromagnetic CAT˙+–NN exchange, these steric constraints can't change the sign of JCAT˙+–NN.9 Thus, the 2T1 state is expected to lie ∼1.5JCAT˙+–NN below the 4T2 state for every complex in this series when Jbpy˙−–SQ ≫ JCAT˙+–NN.9,11 However, disruption of conjugation between the phenyl bridge and NN results in the energy of the 2/4NN excited states being shifted to higher energy by ∼1000 to 2000 cm−1 as is clearly observed in Fig. 3B. Note that the spectra in Fig. 3 were collected in the solution phase at room temperature, while the TREPR experiments are recorded at 20 K in 2-MTHF. Nevertheless, we have observed a strong correlation of solution phase conformations with those in molecular crystals.9,33,34 Steric interactions caused by methyl groups attached to a phenylene bridge in LZn-SQ-B-NN complexes (Fig. 1B) modulate fluid solution electronic absorption spectra consistent with the SQ-B and B-NN bond torsions that have been determined by X-ray crystallography.9,33,34 As a result of the bridge dependence on the 2/4NN energies and the magnitude of JCAT˙+–NN, either or both of the 2,4NN and 4T1 excited states may mix with the 2T1 excited state to generate ESP in the 2T1 state. The ESP generated in the 2T1 state is then subsequently transferred to the 2S0 ground state upon fast spin-allowed radiationless decay (kNR, Fig. 2A).Table 1 lists the primary steric interactions of the methyl group(s) – if present – for each complex as well as the sign of the observed TREPR signal. Four of the five complexes exhibit absorptive GS ESP consistent with the ESP arising from equilibration of the 2T1 state with lower-lying 2/4NN localized radical states as described previously.10 Notably, only 6-Me-m-Ph exhibits emissive ESP,9 and this is the only complex in which the methyl group is expected to cause a bond torsion of the CAT–Ph moiety, but with no torsion about the Ph–NN bond (as is the case for the LZn-SQ-6-Me-m-Ph-NN complex9). The observed emissive ESP for 6-Me-m-Ph is consistent with a small 2T1–4T1 gap by virtue of the weak JCAT˙+–NN exchange interaction,9 and a relatively larger 2T1–2,4NN gap due to unhindered conjugation of phenylene- and NN π-systems. As a result, the dominant ESP-generating interaction is between the 4T1 and the 2T2 states, leading to the observed emissive ESP in the electronic GS.





The complexes that possess methyl groups which disrupt phenylene–NN π-conjugation (2-Me-m-Ph, 4-Me-m-Ph, and 2,4,6-Me3-m-Ph), have higher-lying localized 2/4NN excited states compared to m-Ph and 6-Me-m-Ph, and this is observed in Fig. 3B. For 2-Me-m-Ph, 4-Me-m-Ph, and 2,4,6-Me3-m-Ph the observed TREPR signals are consistent with ESP originating from 2T1-2/4NN equilibration via the mRQM. Thus, the TREPR and electronic absorption spectra for the (bpy)Pt(CAT-B-NN) complexes presented here are consistent with the mechanistic trend for ESP generation that is illustrated in Fig. 5. While equilibration of 2T1 with both4T1 and 2/4NN is possible, only the dominant pathways leading to the observed TREPR signals are shown in Fig. 5.
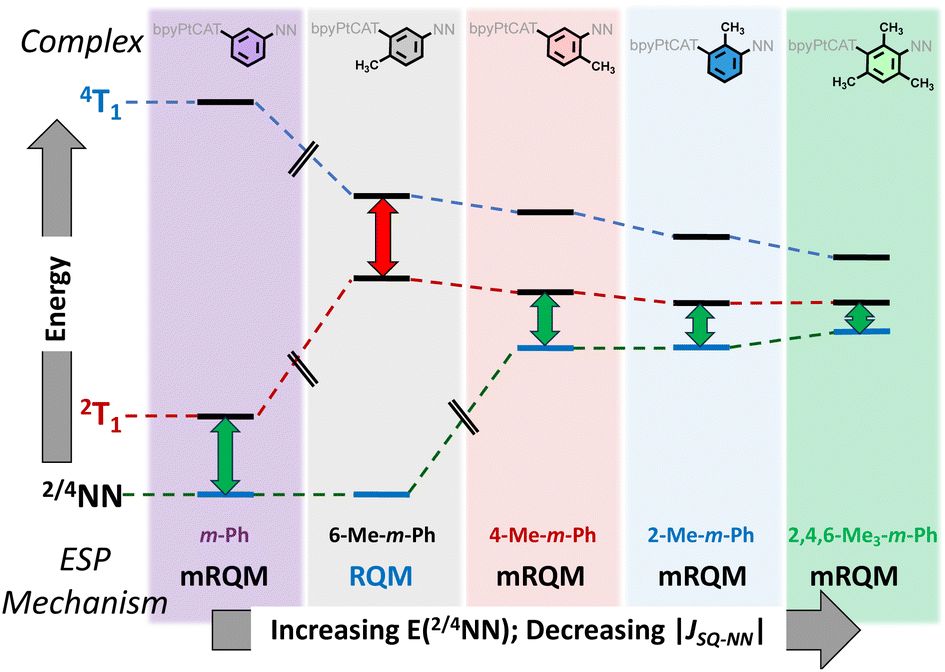 | ||
| Fig. 5 Qualitative illustration of the effects of specific positions of methyl groups on the m-Ph bridge unit for attenuation of JCAT˙+–NN (6-Me-m-Ph; based on exchange coupling),9 or an attenuation of JCAT˙+–NN with an increase in the energy of the NN excited states (4-Me-, 2-Me-, and 2,4,6-Me3-mPh) based on electronic absorption spectra, Fig. 3B. Energy gaps are illustrative only, and double headed arrows indicate the major ESP contribution. | ||
Conclusions
A series of (bpy)Pt(CAT-B-NN) complexes that possess a CAT → bpy LL′CT excited state, with three unpaired spins and variable ES antiferromagnetic CAT˙+–NN magnetic exchange coupling, has been studied with respect to ESP generation by the RQM and mRQM. Despite the antiferromagnetic JCAT˙+–NN exchange coupling present in every complex in this series, the photogenerated ground state ESP in these complexes varies from strong and emissive to strong and absorptive. Steric interactions that modulate JCAT˙+–NN and the energies of the NN-localized 2/4NN radical ESs result in competitive interactions between the 2T1 state and the 4T1 and 2,4NN states, and this ultimately determines the mechanism (RQM or mRQM) for generating the observed GS ESP. Our TREPR and electronic absorption spectral results suggest that for four of the five complexes, the variable-intensity absorptive ESP is derived from interactions between the chromophoric 2T1 and 4T1 states (RQM), while for 6-Me-m-Ph the emissive ESP is derived from interactions with the 2/4NN localized ESs (mRQM).The results presented here are notably different than those for isostructural (bpy)Pt(CAT-B-NN) complexes that possess methyl-substituted phenyl bridges with para-connectivity of CAT and NN groups.7,8 For those complexes, the para-substituted phenyl bridge enforces ferromagnetic CAT˙+–NN exchange coupling, which ensures that the energy of the 4T1 state always lies below the energy of the 2T1 state. Thus, we only observe absorptive TREPR signals with intensities that vary with the methyl substitution pattern on the phenyl bridges.7,8
In addition, the observation of only GS ESP in all of the (bpy)Pt(CAT-B-NN) complexes that we have studied thus far is markedly different from the majority of reports in which GS and ES ESP are observed in ground- and excited doublets and the excited state quartet.19,21,42 Another novel aspect of this study is that the meta-substituted phenyl bridges enable both absorptive and emissive ESP signals in the TREPR spectra of the GS only. This is due to ES relaxation to the electronic ground state being markedly faster than the time scale of the TREPR experiment. The inclusion of non-chromophore localized- and dark states in the excited state manifold, which are nearly degenerate with the 2S1, 2T1, and 4T1 states, creates new opportunities for the generation and control of both ground and excited state ESP that extends beyond the RQM. Here, we have shown the importance of attenuating the energy gap between localized NN excited states and the 2T1 state to control both the magnitude and sign of the optically generated ESP, and to provide deeper insight into the details of the mRQM. Understanding electronic structure contributions to optically generated ESP will contribute to enhancing our ability to control the nature of prepared states for a variety of QIS applications, with strong ESP leading to enhanced sensitivity and readout capabilities at low fields and higher temperatures.3,23
Data availability
ESI contains synthetic- and characterization details of all new complexes.Author contributions
M. L. K. and D. A. S. conceptualized and supervised the project; P. H. and A. R. M. synthesized and characterized all molecules and complexes; A.v.d.E. collected TREPR spectra, and M. L. K., D. A. S., and A.v.d.E. analyzed the TREPR data; M. L. K., D. A. S., P. H., A. R. M., and A.v.d.E. wrote the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M.L.K. and D.A.S. acknowledge financial support from DOE (DE-SC0020199). This work was also supported by an NSERC Discovery Grant (2015-04021 to A.v.d.E.). Mass spectrometry was performed by the Michigan State University MS facility.Notes and references
- D. W. Laorenza and D. E. Freedman, J. Am. Chem. Soc., 2022, 144, 21810–21825 CrossRef CAS PubMed.
- C. J. Yu, S. von Kugelgen, D. W. Laorenza and D. E. Freedman, ACS Cent. Sci., 2021, 7, 712–723 CrossRef CAS PubMed.
- M. R. Wasielewski, Phys. Today, 2023, 76, 28–34 CrossRef CAS.
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123, 1417–1551 CrossRef CAS PubMed.
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, A. R. Marri, P. Hewitt and A. van der Est, J. Am. Chem. Soc., 2022, 144, 21005–21009 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, P. Hewitt and A. van der Est, J. Am. Chem. Soc., 2022, 144, 12781–12788 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, P. Hewitt and A. van der Est, J. Phys. Chem. Lett., 2022, 13, 872–878 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, P. Hewitt, D. E. Stasiw, J. Chen and A. van der Est, Chem. Sci., 2021, 12, 13704–13710 RSC.
- M. L. Kirk, D. A. Shultz, J. Chen, P. Hewitt, D. Daley, S. Paudel and A. van Der Est, J. Am. Chem. Soc., 2021, 143, 10519–10523 CrossRef CAS PubMed.
- B. Stein, C. Tichnell, J. Chen, D. A. Shultz and M. L. Kirk, J. Am. Chem. Soc., 2018, 140, 2221–2228 CrossRef CAS PubMed.
- G. P. Wiederrecht, W. A. Svec and M. R. Wasielewski, J. Am. Chem. Soc., 1999, 121, 7726–7727 CrossRef CAS.
- M. T. Colvin, E. M. Giacobbe, B. Cohen, T. Miura, A. M. Scott and M. R. Wasielewski, J. Phys. Chem. A, 2010, 114, 1741–1748 CrossRef CAS PubMed.
- M. T. Colvin, R. Carmieli, T. Miura, S. Richert, D. M. Gardner, A. L. Smeigh, S. M. Dyar, S. M. Conron, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 5314–5325 CrossRef CAS PubMed.
- M. Zarea, M. A. Ratner and M. R. Wasielewski, J. Chem. Phys., 2015, 143, 054101 CrossRef PubMed.
- N. E. Horwitz, B. T. Phelan, J. N. Nelson, C. M. Mauck, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 4455–4463 CrossRef CAS PubMed.
- Y. H. Huang, M. D. Krzyaniak, R. M. Young and M. R. Wasielewski, Appl. Magn. Reson., 2022, 53, 949–961 CrossRef CAS.
- Y. H. Huang, M. D. Krzyaniak, R. M. Young and M. R. Wasielewski, Appl. Magn. Reson., 2022, 53, 949–961 CrossRef CAS.
- Y. F. Qiu, A. Equbal, C. J. Lin, Y. H. Huang, P. J. Brown, R. M. Young, M. D. Krzyaniak and M. R. Wasielewski, Angew. Chem., Int. Ed., 2023, 62, e202214668 CrossRef CAS PubMed.
- T. Quintes, M. Mayländer and S. Richert, Nat. Rev. Chem., 2023, 7, 75–90 CrossRef PubMed.
- Z. Wang, Y. Gao, M. Hussain, S. Kundu, V. Rane, M. Hayvali, E. A. Yildiz, J. Zhao, H. G. Yaglioglu, R. Das, L. Luo and J. Li, Chem.–Eur. J., 2018, 24, 18663–18675 CrossRef CAS PubMed.
- M. Mayländer, S. Chen, E. R. Lorenzo, M. R. Wasielewski and S. Richert, J. Am. Chem. Soc., 2021, 143, 7050–7058 CrossRef PubMed.
- M. Mayländer, P. Thielert, T. Quintes, A. Vargas Jentzsch and S. Richert, J. Am. Chem. Soc., 2023, 145, 14064–14069 CrossRef PubMed.
- W. Xu, P. Chitnis, A. Valieva, A. van der Est, Y. N. Pushkar, M. Krzystyniak, C. Teutloff, S. G. Zech, R. Bittl, D. Stehlik, B. Zybailov, G. Z. Shen and J. H. Golbeck, J. Biol. Chem., 2003, 278, 27864–27875 CrossRef CAS PubMed.
- C. R. Tichnell, D. R. Daley, B. W. Stein, D. A. Shultz, M. L. Kirk and E. O. Danilov, J. Am. Chem. Soc., 2019, 141, 3986–3992 CrossRef CAS PubMed.
- J. Yang, D. K. Kersi, C. P. Richers, L. J. Giles, R. Dangi, B. W. Stein, C. Feng, C. R. Tichnell, D. A. Shultz and M. L. Kirk, Inorg. Chem., 2018, 57, 13470–13476 CrossRef CAS PubMed.
- J. Yang, D. K. Kersi, L. J. Giles, B. W. Stein, C. Feng, C. R. Tichnell, D. A. Shultz and M. L. Kirk, Inorg. Chem., 2014, 53, 4791–4793 CrossRef CAS PubMed.
- J. A. Zuleta, J. M. Bevilacqua and R. Eisenberg, Coord. Chem. Rev., 1991, 111, 237–248 CrossRef CAS.
- J. Best, I. V. Sazanovich, H. Adams, R. D. Bennett, E. S. Davies, A. J. H. M. Meijer, M. Towrie, S. A. Tikhomirov, O. V. Bouganov, M. D. Ward and J. A. Weinstein, Inorg. Chem., 2010, 49, 10041 CrossRef CAS PubMed.
- W. Paw, S. D. Cummings, M. A. Mansour, W. B. Connick, D. K. Geiger and R. Eisenberg, Coord. Chem. Rev., 1998, 171, 125–150 CrossRef CAS.
- M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115–137 CrossRef CAS.
- S. D. Cummings and R. Eisenberg, in Progress in Inorganic Chemistry: Synthesis, Properties, and Applications, ed. E. I. Stiefel, 2004, vol. 52, pp. 315–367 Search PubMed.
- D. A. Shultz, M. L. Kirk, J. Zhang, D. E. Stasiw, G. Wang, J. Yang, D. Habel-Rodriguez, B. W. Stein and R. D. Sommer, J. Am. Chem. Soc., 2020, 142, 4916–4924 CrossRef CAS PubMed.
- D. E. Stasiw, J. Zhang, G. Wang, R. Dangi, B. W. Stein, D. A. Shultz, M. L. Kirk, L. Wojtas and R. D. Sommer, J. Am. Chem. Soc., 2015, 137, 9222–9225 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, D. E. Stasiw, G. F. Lewis, G. Wang, C. L. Brannen, R. D. Sommer and P. D. Boyle, J. Am. Chem. Soc., 2013, 135, 17144–17154 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, D. E. Stasiw, D. Habel-Rodriguez, B. Stein and P. D. Boyle, J. Am. Chem. Soc., 2013, 135, 14713–14725 CrossRef CAS PubMed.
- M. L. Kirk and D. A. Shultz, Coord. Chem. Rev., 2013, 257, 218–233 CrossRef CAS.
- M. L. Kirk, D. A. Shultz, E. C. Depperman, D. Habel-Rodriguez and R. D. Schmidt, J. Am. Chem. Soc., 2012, 134, 7812–7819 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, E. C. Depperman and C. L. Brannen, J. Am. Chem. Soc., 2007, 129, 1937–1943 CrossRef CAS PubMed.
- A. K. Tripathi, V. Rane, S. Kundu and R. Das, J. Chem. Phys., 2019, 151, 154305 CrossRef PubMed.
- V. Rozenshtein, A. Berg, E. Stavitski, H. Levanon, L. Franco and C. Corvaja, J. Phys. Chem. A, 2005, 109, 11144–11154 CrossRef CAS.
- E. M. Giacobbe, Q. X. Mi, M. T. Colvin, B. Cohen, C. Ramanan, A. M. Scott, S. Yeganeh, T. J. Marks, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 3700–3712 CrossRef CAS PubMed.
- G. Bussière, R. Beaulac, H. Bélisle, C. Lescop, D. Luneau, P. Rey and C. Reber, in Transition Metal and Rare Earth Compounds: Excited States, Transitions, Interactions III, ed. H. Yersin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 97–118, DOI:10.1007/b96861.
- M. D. E. Forbes, L. E. Jarocha, S.-Y. Sim and V. F. Tarasov, Adv. Phys. Org. Chem., 2013, 47, 1–83 CrossRef CAS.
- Y. Teki, Chem.–Eur. J., 2020, 26, 980–996 CrossRef CAS PubMed.
- J. Tatchen, N. Gilka and C. M. Marian, Phys. Chem. Chem. Phys., 2007, 9, 5209–5221 RSC.
- A. M. Putz, U. Schatzschneider and E. Rentschler, Phys. Chem. Chem. Phys., 2012, 14, 1649–1653 RSC.
- S. Hase, D. Shiomi, K. Sato and T. Takui, J. Mater. Chem., 2001, 11, 756–760 RSC.
- E. T. Chernick, Q. Mi, R. F. Kelley, E. A. Weiss, B. A. Jones, T. J. Marks, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2006, 128, 4356–4364 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, D. Habel-Rodriguez, R. D. Schmidt and U. Sullivan, J. Phys. Chem. B, 2010, 114, 14712–14716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Frozen solution EPR spectra are devoid of features characteristic of aggregation and are stable to extended irradiation at 532 nm. See Fig. S2 and S3 for details. See DOI: https://doi.org/10.1039/d3sc03049k |
| ‡ Present address: UES Inc., Dayton, OH 45432, USA. |
| This journal is © The Royal Society of Chemistry 2023 |