
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC(sp2)–H cyclobutylation of hydroxyarenes enabled by silver-π-acid catalysis: diastereocontrolled synthesis of 1,3-difunctionalized cyclobutanes†
Lei
Tang‡
 ,
Qi-Nan
Huang‡
,
Feng
Wu
,
Yuanjiu
Xiao
,
Jin-Lan
Zhou
,
Tong-Tong
Xu
,
Wen-Biao
Wu
*,
Shuanglin
Qu
* and
Jian-Jun
Feng
*
,
Qi-Nan
Huang‡
,
Feng
Wu
,
Yuanjiu
Xiao
,
Jin-Lan
Zhou
,
Tong-Tong
Xu
,
Wen-Biao
Wu
*,
Shuanglin
Qu
* and
Jian-Jun
Feng
*
State Key Laboratory of Chemo/Biosensing and Chemometrics, Advanced Catalytic Engineering Research Center of the Ministry of Education, College of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, P. R. China. E-mail: 1334154923@qq.com; squ@hnu.edu.cn; jianjunfeng@hnu.edu.cn
First published on 30th August 2023
Abstract
Ring-opening of bicyclo[1.1.0]butanes (BCBs) is emerging as a powerful strategy for 1,3-difunctionalized cyclobutane synthesis. However, reported radical strain-release reactions are typically plagued with diastereoselectivity issues. Herein, an atom-economic protocol for the highly chemo- and diastereoselective polar strain-release ring-opening of BCBs with hydroxyarenes catalyzed by a π-acid catalyst AgBF4 has been developed. The use of readily available starting materials, low catalyst loading, high selectivity (up to >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 d.r.), a broad substrate scope, ease of scale-up, and versatile functionalizations of the cyclobutane products make this approach very attractive for the synthesis of 1,1,3-trisubstituted cyclobutanes. Moreover, control experiments and theoretical calculations were performed to illustrate the reaction mechanism and selectivity.
:2 d.r.), a broad substrate scope, ease of scale-up, and versatile functionalizations of the cyclobutane products make this approach very attractive for the synthesis of 1,1,3-trisubstituted cyclobutanes. Moreover, control experiments and theoretical calculations were performed to illustrate the reaction mechanism and selectivity.
Introduction
Cyclobutanes represent important structural units in natural products and other biologically significant molecules.1 Moreover, the cyclobutane scaffold, especially the 1,3-difunctionalized cyclobutane skeleton, is often incorporated in drug design, such as PF-03654746,2 linsitinib,3 and TAK-828F4 (Scheme 1A). In these cases, a 1,3-substituted cyclobutane linker can act as an aryl isostere with reduced planarity; flexible ethyl- or propyl-linkers can also be replaced by conformationally restricted 1,3-disubstituted cyclobutanes to limit the number of possible conformations.1b Despite the importance of these cyclobutanes, catalytic methods for their synthesis remained relatively less explored in parallel with their homologues.5–7 Moreover, diastereocontrolled synthesis of 1,1,3-trisubstituted cyclobutanes featuring quaternary carbon stereocenters remains challenging.7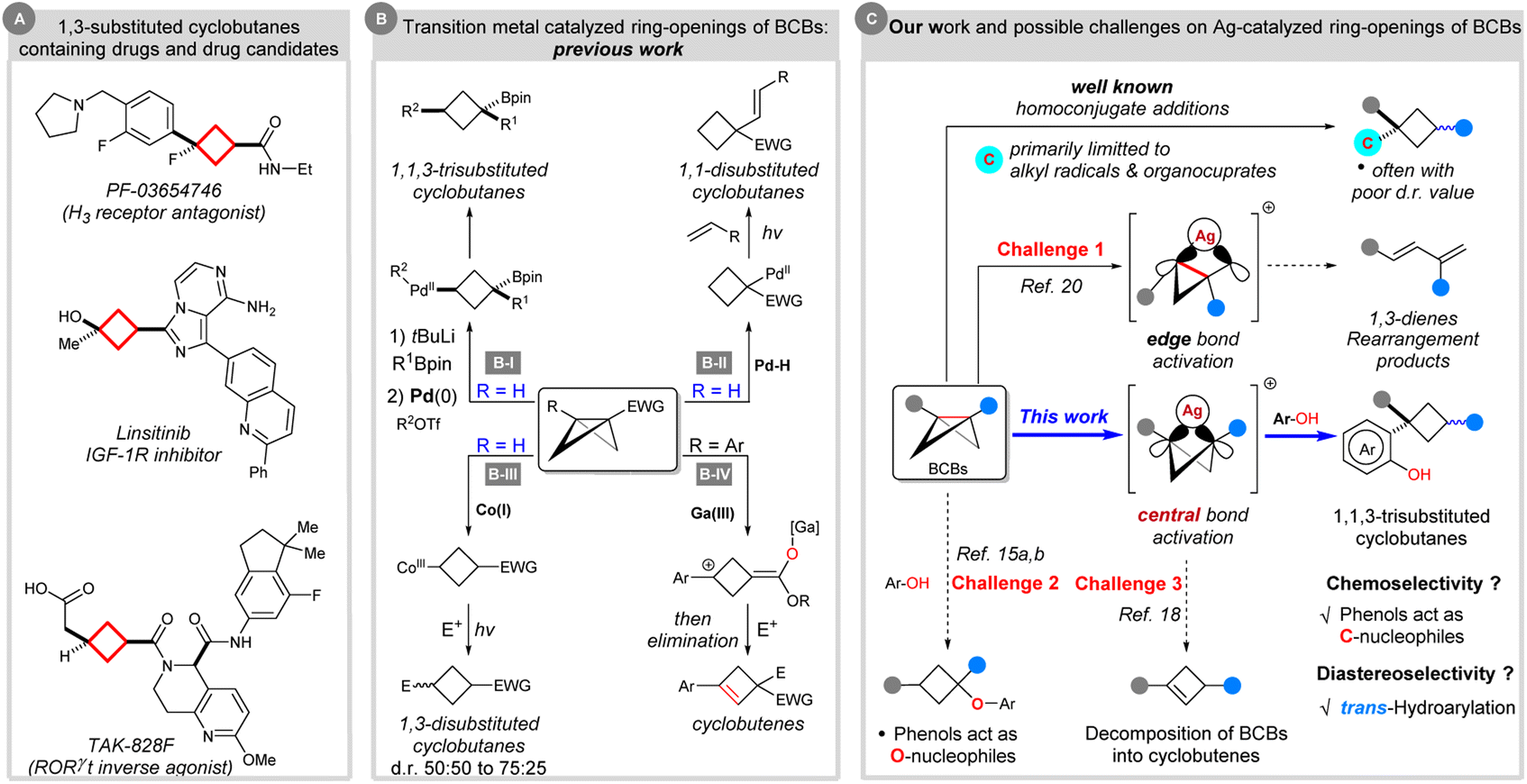 | ||
| Scheme 1 Transition metal catalyzed ring-opening reactions of BCBs for the synthesis of cyclobutane derivatives and their scientific context. | ||
In recent years, strain-release driven transformations have recaptured significant attention in synthetic organic chemistry,8 materials science,9 analytical chemistry10 and bioconjugation.11 As the smallest of fused carbocycles, bicyclo[1.1.0]butanes (BCBs) are highly strained (ring strain energy ∼ 66 kcal mol−1) yet bench-stable, synthetically versatile carbocycles.12 The release of ring tension embedded in BCBs, coupled with the π-type reactivity for the central C–C σ-bond, allows for the design or discovery of new reactions for the synthesis of ring systems.13 Among them, ring-opening reactions via homo- or heterolysis of the spring-loaded C–C bond represent powerful tools enabling quick and efficient access to multisubstituted cyclobutane derivatives. In this direction, there are six general strategies for intermolecular ring-opening reactions of BCBs: (1) radical strain-release reactions with radical nucleophiles. This strategy provides powerful methods for making mostly 1,3-disubstituted alkylated cyclobutanes, albeit mainly with poor diastereoselectivity (not shown).7e,14 (2) Polar strain-release reactions with 2-electron-based nucleophiles. The nucleophilic ring opening reactions of BCBs concerned mainly the addition of various heteroatom (O, N, P)-centred nucleophiles,15 such as Hoz's O-cyclobutylation,15a Aggarwal's α-selective ring-opening,15b Gaoni's azidation,15c Baran's amination,15d Wipf's hydrophosphination15e and others.15f By contrast, the successful use of carbon nucleophilic reagents in addition reactions to BCBs still lags behind and had been limited to strong nucleophiles like organocuprates.7a–d Once again, poor diastereoselectivity was detected in these examples (not shown). (3) Simultaneous activation of BCBs by nucleophiles and electrophiles. This method usually relies on the 1,2-migration process of BCB–boronate complexes, and functionalization by capture of an electrophile, thereby leading to 1,1,3-trisubstituted cyclobutane products with moderated to excellent diastereoselectivity (Scheme 1B-I).7f–h (4) Palladium hydride enabled hydroalkenylation of BCBs to afford 1,1-disubstituted cyclobutanes (Scheme 1B-II).16 (5) Polarity-reversal strategy. In 2020, Gryko's group disclosed elegant work on Umpolung BCB activation with Co(I) complexes. Co(I)-catalysis allowed the in situ formation of nucleophilic cyclobutyl radicals upon light-driven homolysis of the intermediate Co(III)–alkyl species. This can react with electrophiles to give 1,3-disubstituted cyclobutanes with up to a 75:25 d.r. value (Scheme 1B-III).17 Besides these, (6) oxygenophilic Lewis acid catalyzed ring-opening reactions of BCBs with electrophiles and final intramolecular E1 elimination giving rise to cyclobutene products (Scheme 1B-IV).18
Despite significant progress, the above strategies are typically plagued with diastereoselectivity issues. Among them, the known strategy to solve the diastereoselectivity issues and synthesize 1,1,3-trisubstituted cyclobutanes had been limited to palladium and oxygenophilic bismuth Lewis acid catalysis, which were developed by Aggarwal's group7f,h and Biju's group, respectively.19 Therefore, the development of novel transition metal catalyzed methodologies and exploration of further reaction pathways of BCBs is of great value to BCB chemistry.
In the 1970s, Paquette20a,b and others20c–e have shown that BCBs are capable of silver catalyzed rearrangements. Mechanistic studies suggested that the argento cationic intermediate formed by cleavage of an edge bond of BCBs could further undergo rearrangement to generate 1,3-dienes (Scheme 1C). On the basis of our experience in strained ring chemistry21 and in order to expand the library of known BCBs, we envisioned that such a carbophilic silver catalysis strategy would enable a different approach to access the cyclobutyl cations from direct activation of the central bond of BCBs. The capture of this intermediate with a naphthol (or phenol) would lead to the formation of the aimed 1,1,3-trisubstituted cyclobutane via Friedel–Crafts-type C-alkylation and protodemetalation. However, there are challenges associated with this hypothesis: (i) the issue of site-selectivity (C–C bond cleavage: edge bond versus central bond);20 (ii) the chemoselectivity issue (C- versus O-cyclobutylation);15a,b (iii) the competitive bicyclobutane-to-cyclobutene isomerization.18 Besides these, (iv) the other problem that needs to be solved is the control of the diastereoselectivity.
Results and discussion
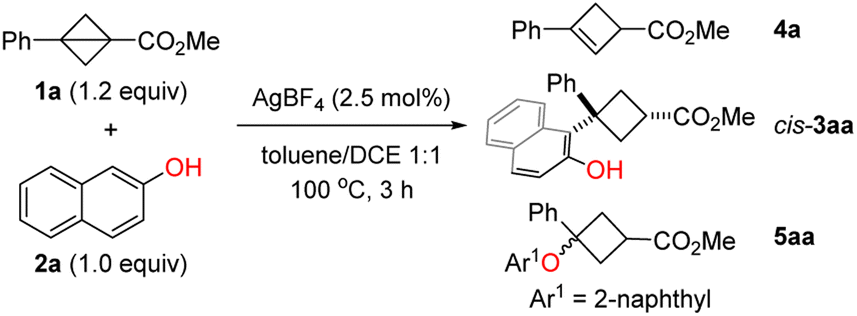
To test the hypothesis, we initiated our investigation from the reaction of BCB 1a and 2-naphthol (2a). After screening of various reaction parameters, we found that the desired C(sp2)–H cyclobutylation occurred with AgBF4 (2.5 mol%) as the catalyst in toluene/DCE (1:1) at 100 °C; cis-3aa was obtained in 85% NMR yield with a 95:5 d.r. value along with 11% NMR yield of 4a resulting from isomerization of 1a (Table 1, entry 1). Control experiments showed that both the amount and type of silver salt and the solvent are essential (entries 2–5). The reactions with commonly used Brønsted and oxygenophilic Lewis acid including TfOH, TsOH, Ga(OTf)3, Sc(OTf)3, Cu(OTf)2, and FeCl3 afforded desired products with poor yield and diastereoselectivity (entries 6–8; see the ESI† for the complete set of optimization data). Of note, when Zn(OTf)2 was employed, 63% NMR yield of O-nucleophilic ring-opening product 5aa was obtained as the major product (entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Variation | cis-3aayb (%) (d.r. = cis/trans) | 4a y (%) | 5aa y (%) |
|
a The reactions were performed with 1a (1.2 equiv.), 2a (1.0 equiv.) and AgBF4 (2.5 mol%) in toluene/1,2-dichloroethane(DCE) (1:1, v/v) at 100 °C for 3 h.
b NMR yield with CH2Br2 as an internal standard.
c
1a (1.1 equiv.) was used.
d
1a (1.1 equiv.), 2a (1.0 equiv.) and the catalyst (10 mol%) in toluene at 80 °C for 12 h.
|
||||
| 1 | None | 85 (95:5) |
11 | 0 |
| 2c | 10 mol% AgBF4 was used | 73 (94:6) |
14 | 0 |
| 3c | 10 mol% AgBF4 in toluene | 78 (91:9) |
17 | 0 |
| 4c | 10 mol% AgBF4 in DCE | 51 (94.5:5.5) |
9 | 0 |
| 5d | AgOTf instead of AgBF4 | 61 (83:17) |
18 | 0 |
| 6d | TfOH instead of AgBF4 | 0 | 0 | 0 |
| 7d | TsOH·H2O instead of AgBF4 | 75 (77:23) |
8 | 0 |
| 8d | Ga(OTf)3 instead of AgBF4 | 37 (77:23) |
0 | 0 |
| 9d | Zn(OTf)2 instead of AgBF4 | 9 (62:38) |
31 | 63 |
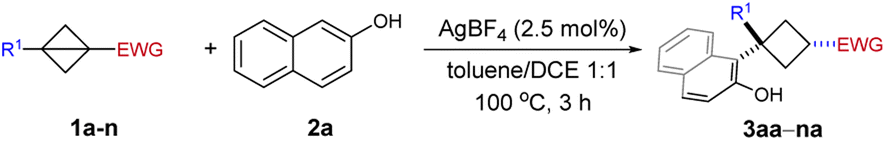
Under the optimized conditions, we next explored the substrate scope of BCBs as summarized in Table 2. We firstly examined the nature of the ester group and both alkyl (3aa–3ca, entries 1–3) and benzyl (entry 4, 3da) esters were obtained in good yield with good to excellent diastereoselectivity. The reaction of phenyl ester 1e was also successful yet with eroded diastereoselectivity (entry 5). Different from Biju's work, apart from BCB esters, 1,3-disubstituted bicyclobutanes bearing other electron-withdrawing groups such as BCB ketone 1f, Weinreb amide derived BCB 1g and sulfonyl BCB 1h provided the corresponding ring-opening products in acceptable yield with up to >98:2 d.r. (3fa–3ha, entries 6–8). The sulfonyl group is a localizing electron-withdrawing group, which stabilizes the negative charge by exerting mainly an inductive effect. By contrast, the carbonyl group is a charge delocalizing group.15a Notably, sulfonyl BCB is a suitable substrate in our silver-π-acid catalytic system. However, it is not compatible with Biju's oxygenophilic bismuth Lewis acid catalytic system.19 This result implies that the cationic Ag catalyst could preferably activate the bridging C–C bond in BCB without the need to coordinate to the ester oxygen. Subsequently, a variety of substituents at the aromatic ring of BCB esters have been examined. BCBs with substituents in the para- and meta-positions were compatible with our catalyst system and afforded the corresponding 1,1,3-trisubstituted cyclobutanes in good yield with up to > 98:2 d.r. (3ia–3na, entries 9–14). The replacement of methyl (1i) by a strongly electron-withdrawing CF3 group (1l) was an exception as the yield decreased from 76% for 3ia to 25% for 3la (entry 9 vs. entry 12). It is probably because the BCB containing an electron-deficient unit can't stabilize the in situ generated cyclobutyl cation.
|
|
||||
|---|---|---|---|---|
| Entry | R1 | EWG | Yieldb (%) | d.r.c |
|
a Unless otherwise noted, the reactions were performed with 1 (0.36 mmol), 2a (0.3 mmol) and AgBF4 (2.5 mol%) in toluene/1,2-dichloroethane(DCE) (1:1, v/v, 2 mL) at 100 °C for 3 h.
b Isolated yield of cis-3.
c Determined by 1H NMR spectroscopic analysis of the crude reaction product.
d Combined isolated yield of the diastereomers which cannot be separated by chromatography.
|
||||
| 1 | Ph | CO2Me | 80 (3aa) | 95:5 |
| 2 | Ph | CO2Et | 80 (3ba) | 93:7 |
| 3 | Ph | CO2iPr | 76 (3ca) | 90:10 |
| 4 | Ph | CO2Bn | 77 (3da) | 93:7 |
| 5 | Ph | CO2Ph | 64 (3ea) | 81:19 |
| 6 | Ph | C(O)(2-naphthyl) | 50 (3fa)d | 86:14 |
| 7 | Ph | C(O)NMe(OMe) | 80 (3ga) | >98:2 |
| 8 | Ph | SO2Ph | 56 (3ha)d | 67:33 |
| 9 | 4-MeC6H4 | CO2Me | 76 (3ia) | >98:2 |
| 10 | 4-CF3OC6H4 | CO2Me | 74 (3ja) | 92:8 |
| 11 | 4-FC6H4 | CO2Me | 80 (3ka) | >98:2 |
| 12 | 4-CF3C6H4 | CO2Me | 25 (3la) | >98:2 |
| 13 | 3-MeC6H4 | CO2Me | 75 (3ma) | >98:2 |
| 14 | 3-FC6H4 | CO2Me | 76 (3na)d | 80:20 |
We then examined the scope of naphthols and phenols (Scheme 2).This method is amenable to a series of 2-naphthols bearing different R3 substituents, including aryl (2b22 and 2f), halogen (2c, 2h and 2j), and propargyl (2g) groups at the C4–C7 positions of 2-naphthols, and led to the corresponding trisubstituted cyclobutanes with synthetically useful phenoxy functionalities, in moderate to excellent yields (43–92%) with up to >98:2 d.r. 1-Naphthols also furnished the corresponding product with good yield and excellent diastereoselectivity (2k–m). Relatively low yields and selectivities were observed with p-methoxy- and phenyl-substituted phenols (3an and 3ao), while 3,5-dimethylphenol (2p) afforded the corresponding product in a good yield and d.r. value.
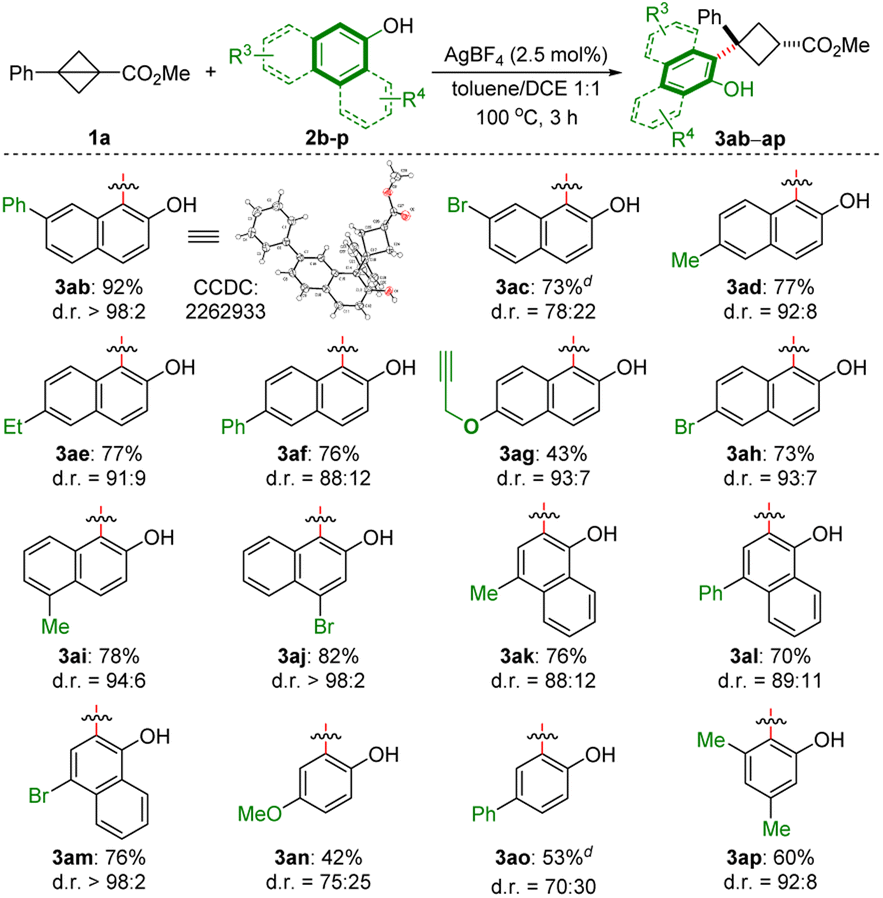 | ||
| Scheme 2 Survey of the scope of naphthols and phenols.a–d a The reactions were performed with 1a (0.36 mmol), 2b–p (0.3 mmol) and AgBF4 (2.5 mol%) in toluene/1,2-dichloroethane(DCE) (1:1, v/v, 2 mL) at 100 °C for 3 h. b Isolated yield of cis-3. c d.r. value was determined by 1H NMR spectroscopic analysis of the crude reaction product. dCombined isolated yield of the diastereomers which cannot be separated by chromatography. | ||
The reaction proved to be easily scalable and was performed on a preparative scale (1.0 mmol) without any loss in efficiency and selectivity, furnishing the product cis-3am in 81% yield with >98:2 d.r. (Scheme 3). The synthetic utility of the products was demonstrated by carrying out a series of functional group interconversions of the phenolic hydroxyl- and ester groups. On one hand, a number of different groups, including the phosphine group (7), H (8) and alkyl group (9), could be incorporated into the aromatic ring via cross-coupling after converting the phenoxy group into triflate 6. On the other hand, ester 3aa can undergo addition, hydrolysis and reduction reactions to give tertiary alcohol 9, carboxylic acid 10 and primary alcohol 12 respectively. Notably, 1-benzoxepin derivatives 11 and 13 featuring a bridged ring system can be synthesized through Keck macrolactonization and intramolecular Mitsunobu reactions respectively.
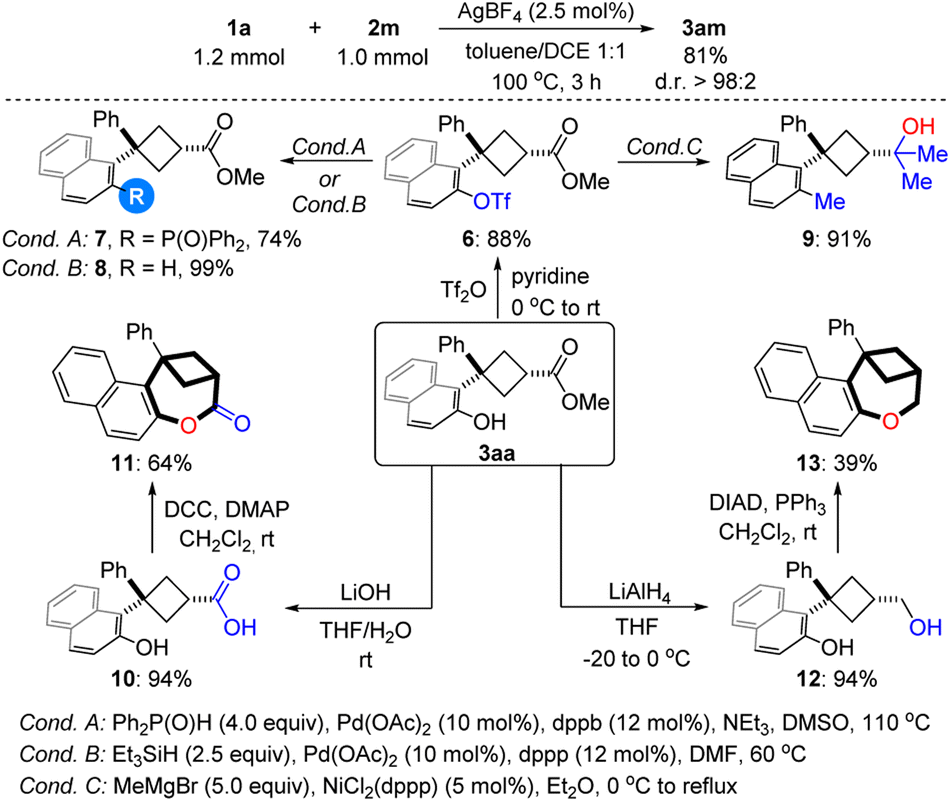 | ||
| Scheme 3 Scale-up synthesis and synthetic transformations. | ||
To interrogate the mechanism, a series of control experiments were conducted. The desired reaction did not occur when 2-methoxynaphthalene was employed (Scheme 4A). Moreover, the deuterium labeling experiment confirmed the critical role of the hydroxyl group of naphthol in those C(sp2)–H cyclobutylations (Scheme 4B). When 3aa with 75:25 d.r. was applied under the standard conditions, no change in the diastereoselectivity of 3aa was found (see, ESI†). This result suggests that high diastereoselectivity may not be obtained via an isomerization pathway (trans- to cis-3aa). The treatment of 4a with standard conditions gave 3aa in 22% NMR yield. However, cyclobutene 4a was far less reactive than bicyclobutane 1a (Scheme 4CversusTable 1 entry 2).
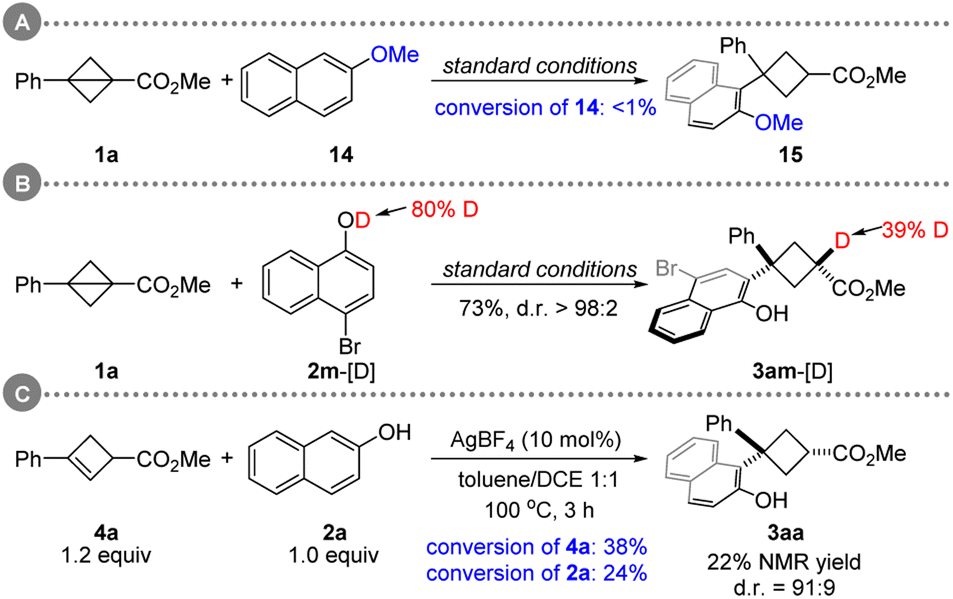 | ||
| Scheme 4 Mechanistic experiments. | ||
To further elucidate the mechanistic details of this reaction and to explain the observed stereoselectivity, density functional theory (DFT) calculations23 were carried out on the model reaction of BCB 1a and 2-naphthol (2a) promoted by the silver catalyst. On the basis of the control experiments and DFT calculations, a plausible catalytic cycle for this diastereoselective transformation is summarized in Scheme 5 (we have considered different possible reaction pathways, and only discuss the most favorable one here; for more details see the ESI†). The molecular orbital analysis of 1a reveals that the bridging C–C bond exhibits the characteristics of a π-bond (Fig. S1†). Thus, the cationic Ag catalyst (a typical π-acid) preferably activates the bridging C–C bond rather than the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, leading to the ring-opening of BCB and formation of the carbon cation intermediate A (Fig. S2†). Then, the nucleophilic attack of A by the π-bond of 2a forms a new C–C bond and affords the intermediate B. Next, another molecule of 2a enters into the reaction with its π-bond coordinating to the π-acidic Ag atom of B, followed by 1,3-migration of Ag, leading to a silver enolate intermediate C with a hydrogen bond between the hydroxyl group and the enolate carbon. Subsequently, the proton is facilely transferred from the hydroxyl group to the enolate carbon of the BCB moiety, releasing the naphthol silver salt D and giving the protonated intermediate E. The process from B to E can be viewed as the replacement of the Ag atom by the proton. This process would not change the stereochemistry of the BCB carbon, because the substrate 2a could only approach the BCB ring from the top direction by coordination with the Ag atom (for more energetic and geometric information see Fig. S3 and S4†). In addition, this is in agreement with the deuterium labeling experiment (Scheme 4B) that the proton in the product is from the hydroxyl group of 2a. Finally, the naphthol anion moiety of D abstracts the proton of E, producing the final major product cis-3aa and releasing F, in which the π-acidic Ag+ catalyst is coordinated by the π-bond of 2a.
O bond, leading to the ring-opening of BCB and formation of the carbon cation intermediate A (Fig. S2†). Then, the nucleophilic attack of A by the π-bond of 2a forms a new C–C bond and affords the intermediate B. Next, another molecule of 2a enters into the reaction with its π-bond coordinating to the π-acidic Ag atom of B, followed by 1,3-migration of Ag, leading to a silver enolate intermediate C with a hydrogen bond between the hydroxyl group and the enolate carbon. Subsequently, the proton is facilely transferred from the hydroxyl group to the enolate carbon of the BCB moiety, releasing the naphthol silver salt D and giving the protonated intermediate E. The process from B to E can be viewed as the replacement of the Ag atom by the proton. This process would not change the stereochemistry of the BCB carbon, because the substrate 2a could only approach the BCB ring from the top direction by coordination with the Ag atom (for more energetic and geometric information see Fig. S3 and S4†). In addition, this is in agreement with the deuterium labeling experiment (Scheme 4B) that the proton in the product is from the hydroxyl group of 2a. Finally, the naphthol anion moiety of D abstracts the proton of E, producing the final major product cis-3aa and releasing F, in which the π-acidic Ag+ catalyst is coordinated by the π-bond of 2a.
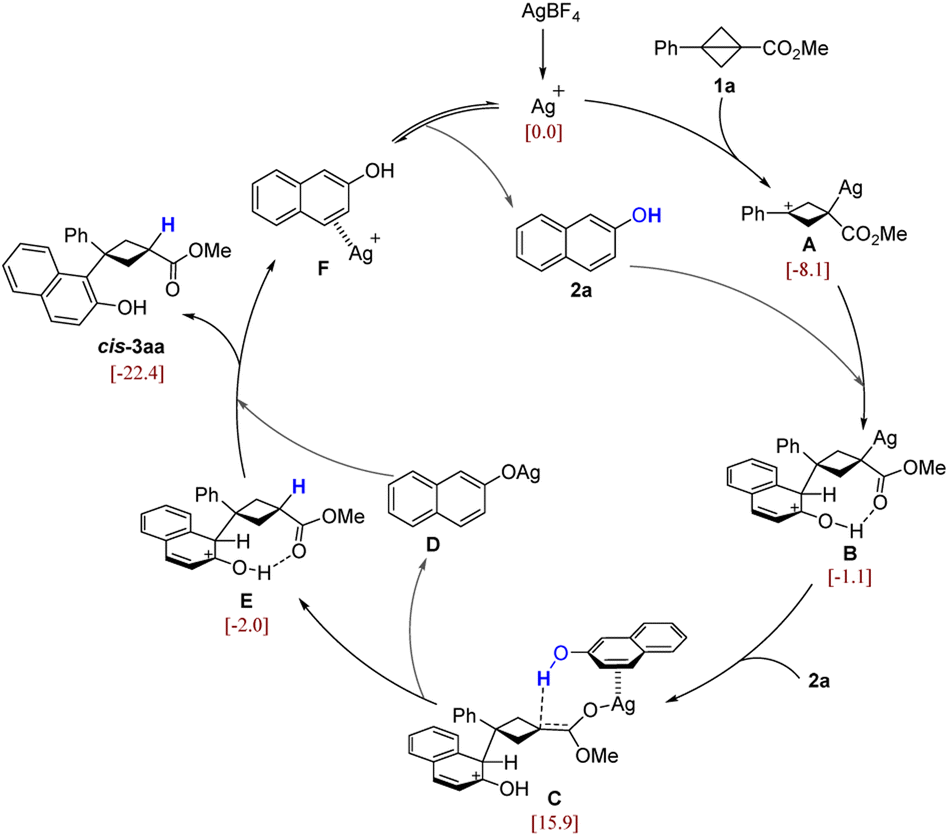 | ||
| Scheme 5 Proposed mechanism. The values in brackets are calculated relative Gibbs free energies (in kcal mol−1). | ||
The DFT studies show that the diastereoselectivity is determined by the nucleophilic attack step (A → B), where the nucleophile 2a approaches the carbocation A through either the top or the bottom directions, finally leading to isomers of trans- and cis-3aa, respectively. The transition states for these two nucleophilic attack modes are compared. As shown in Scheme 6, there is a hydrogen bond interaction in TS2-cis, which helps to stabilize this transition state. In contrast, it shows electrostatic repulsion between the acidic hydrogen and the positive Ag center in TS2-trans, which hinders this nucleophilic attack. Thus, TS2-cis is lower than TS2-trans by 3.3 kcal mol−1, which well agrees with the experiment that cis-3aa is the major product. It is of note that the nucleophilic attack could also occur by the oxygen atom of 2a. However, the calculations show that this O-nucleophilic attack is less favorable than both TS2-cis and TS2-trans (Fig. S3 and S5†). In addition, the reaction of cyclobutene 4a with 2a to form 3aa is also examined by DFT calculation, which is predicted to have a higher activation barrier (Fig. S6†), in agreement with lower yields (Scheme 4C).
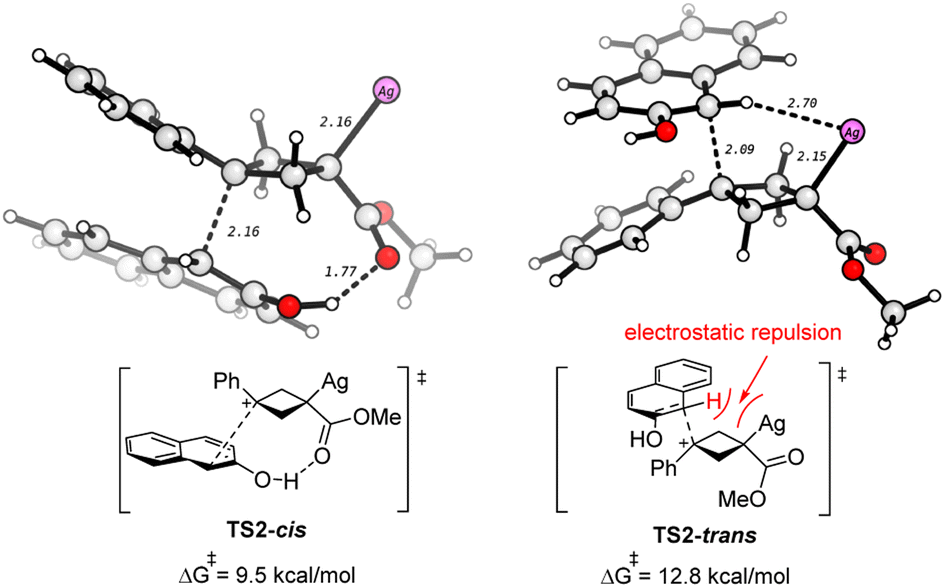 | ||
| Scheme 6 Comparison of the two transition states for the formation of cis- and trans-3aa. The selected bond distances are in Å. | ||
Conclusion
In summary, by taking advantage of hydroxyarenes as C-nucleophiles rather than O-nucleophiles in unusual silver catalyzed polar strain-release ring-opening of BCBs, an atom-economic and highly selective method (up to >98:2 d.r.) for the synthesis of 1,1,3-trisubstituted cyclobutanes was developed. The salient features of this transformation include readily available starting materials, low catalyst loading, wide functional-group compatibility, versatile functionalizations of the cyclobutane products and scalability. Notably, mechanistic experiments and DFT calculations were performed to gain insights into the reaction mechanism, which shows that the silver catalyst acts as a carbophilic π-acid rather than an oxygenophilic Lewis acid to effectively activate the BCB bridging C–C bond and promote the transformation. The diastereoselectivity is determined by hydrogen bond interaction and steric repulsions in the nucleophilic attack step. This reactivity mode may open opportunities for the development of other reaction processes.
Data availability
All detailed procedures, characterization data, NMR spectra and DFT studies are available in the ESI.†Author contributions
L. T., F. W., Y. X., J.-L., Z., and T.-T. X. performed the experiments and conducted the analytical characterization. Q.-N. H. and S. Q. executed the theoretical calculations. W.-B. W., S. Q. and J.-J. F. wrote the manuscript. J.-J. F. conceived the catalytic system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Fundamental Research Funds for the Central Universities and financial support from the Hunan Youth Talent (grant no. 2021RC3053).References
- (a) C. Hui, Y. Liu, M. Jiang and P. Wu, Cyclobutane-containing scaffolds in bioactive small molecules, Trends Chem., 2022, 4, 677 CrossRef CAS; (b) M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, Cyclobutanes in small-molecule drug candidates, ChemMedChem, 2022, 17, e202200020 CrossRef CAS PubMed.
- T. T. Wager, B. A. Pettersen, A. W. Schmidt, D. K. Spracklin, S. Mente, T. W. Butler, H. J. Howard, D. J. Lettiere, D. M. Rubitski, D. F. Wong, F. M. Nedza, F. R. Nelson, H. Rollema, J. W. Raggon, J. Aubrecht, J. K. Freeman, J. M. Marcek, J. Cianfrogna, K. W. Cook, L. C. James, L. A. Chatman, P. A. Iredale, M. J. Banker, M. L. Homiski, J. B. Munzner and R. Y. Chandrasekaran, Discovery of two clinical histamine H3 receptor antagonists: trans-N-ethyl-3-fluoro-3-[3-fluoro-4-(pyrrolidinylmethyl)-phenyl]cyclobutanecarboxamide (PF-03654746) and trans-3-fluoro-3-[3-fluoro-4-(pyrrolidin-1-ylmethyl)phenyl]-N-(2-methylpropyl)cyclobutanecarbox-amide (PF-03654764), J. Med. Chem., 2011, 54, 7602 CrossRef CAS PubMed.
- M. J. Mulvihill, A. Cooke, M. Rosenfeld-Franklin, E. Buck, K. Foreman, D. Landfair, M. O'Connor, C. Pirritt, Y. Sun, Y. Yao, L. D. Arnold, N. W. Gibson and Q.-S. Ji, Discovery of OSI-906 : a selective and orally efficacious dual inhibitor of the IGF-I receptor and insulin receptor, Future Med. Chem., 2009, 1, 1153 CrossRef CAS PubMed.
- K. Majima and M. Yamano, Diastereoselective synthesis of a cis-1,3-disubstituted cyclobutane carboxylic acid scaffold for TAK-828F, a potent retinoic acid receptor-related orphan receptor (ROR)-γt inverse agonist, J. Org. Chem., 2021, 86, 11464 CrossRef CAS PubMed.
- For reviews on synthesis of cyclobutanes, see: (a) M. Wang and P. Lu, Catalytic approaches to assemble cyclobutane motifs in natural product synthesis, Org. Chem. Front., 2018, 5, 254 RSC; (b) E. N. Hancock, J. M. Wahl and M. K. Brown, Recent advances in the synthesis of gem-dimethylcyclobutane natural products, Nat. Prod. Rep., 2019, 36, 1383 RSC; (c) K.-G. Wen, Y.-Y. Peng and X.-P. Zeng, Advances in the catalytic asymmetric synthesis of quaternary carbon containing cyclobutanes, Org. Chem. Front., 2020, 7, 2576 RSC; (d) J. Li, K. Gao, M. Bian and H. Ding, Recent advances in the total synthesis of cyclobutane-containing natural products, Org. Chem. Front., 2020, 7, 136 RSC; (e) J. Chen, Q. Zhu, H. Fang and P. Lu, Dancing on ropes-enantioselective functionalization of preformed four-membered carbocycles, Chin. J. Chem., 2022, 40, 1346 CrossRef CAS.
- For representative examples on synthesis of 1,3-difunctionalized cyclobutanes by cycloadditions, see: (a) J. M. Hoyt, V. A. Schmidt, A. M. Tondreau and P. J. Chirik, Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenes, Science, 2015, 349, 960 CrossRef CAS PubMed; (b) M. L. Conner and M. K. Brown, Synthesis of 1,3-Substituted Cyclobutanes by Allenoate-Alkene [2+2] Cycloaddition, J. Org. Chem., 2016, 81, 8050 CrossRef CAS PubMed; (c) J. M. Wahl, M. L. Conner and M. K. Brown, Allenoates in Enantioselective [2+2] Cycloadditions: From a Mechanistic Curiosity to a Stereospecific Transformation, J. Am. Chem. Soc., 2018, 140, 15943 CrossRef CAS PubMed; (d) G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Selective C-H Halogenation with a Highly Fluorinated Manganese Porphyrin, Angew. Chem., Int. Ed., 2018, 57, 1251 CrossRef CAS PubMed; (e) Z. Fan, D. A. Strassfeld, H. S. Park, K. Wu and J.-Q. Yu, Formal γ−C–H Functionalization of Cyclobutyl Ketones: Synthesis of cis-1,3-Difunctionalized Cyclobutanes, Angew. Chem., Int. Ed., 2023, 62, e202303948 CrossRef CAS PubMed; (f) C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Enantioselective Synthesis of 3-Substituted Cyclobutenes by Catalytic Conjugate Addition/Trapping Strategies, Angew. Chem., Int. Ed., 2020, 59, 2750 CrossRef CAS PubMed; (g) M. Yan, Q. Zhou and P. Lu, Collective synthesis of chiral tetrasubstituted cyclobutanes enabled by enantioconvergent Negishi cross-coupling of cyclobutenones, Angew. Chem., Int. Ed., 2023, 62, e202218008 CrossRef CAS PubMed.
- For synthesis of 1,1,3-trisubstituted cyclobutanes by ring opennings of BCBs, see: (a) Y. Gaoni, Conjugate addition of organocopper regents to 1-arylsulfonylbicyclobutanes synthesis of the racemic form of the six pheromone of the citrus mealybug, Planococcus citri(risso), Tetrahedron Lett., 1982, 23, 5215 CrossRef CAS; (b) R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, Enantioselective Synthesis of Cyclobutanes via Sequential Rh-catalyzed Bicyclobutanation/Cu-catalyzed Homoconjugate Addition, J. Am. Chem. Soc., 2013, 135, 9283 CrossRef CAS PubMed; (c) R. A. Panish, S. R. Chintala and J. M. Fox, A Mixed-Ligand Chiral Rhodium(II) Catalyst Enables the Enantioselective Total Synthesis of Piperarborenine B, Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters, Angew. Chem., Int. Ed., 2016, 55, 4983 CrossRef CAS PubMed; (d) Y. Gaoni, A. Tomazic and E. Potgieter, Stereochemistry of addition of organocopper reagents and of the hydride ion to 1-(arylsulfonyl)bicyclo[1.1.0]butanes, J. Org. Chem., 1985, 50, 2943 CrossRef CAS; (e) M. Silvi and V. K. Aggarwal, Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters, J. Am. Chem. Soc., 2019, 141, 9511 CrossRef CAS PubMed; (f) A. Fawcett, T. Biberger and V. K. Aggarwal, Carbopalladation of C–C σ-bonds enabled by strained boronate complexes, Nat. Chem., 2019, 11, 117 CrossRef CAS PubMed; (g) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, Difunctionalization of C−C σ-Bonds Enabled by the Reaction of Bicyclo[1.1.0]butyl Boronate Complexes with Electrophiles: Reaction Development, Scope, and Stereochemical Origins, J. Am. Chem. Soc., 2020, 142, 16766 CrossRef CAS PubMed; (h) B. Wölfl, N. Winter, J. Li, A. Noble and V. K. Aggarwal, Strain-Release Driven Epoxidation and Aziridination of Bicyclo[1.1.0]butanes via Palladium Catalyzed σ-Bond Nucleopalladation, Angew. Chem., Int. Ed., 2023, 62, e202217064 CrossRef PubMed; (i) F. W. Goetzke, A. M. L. Hell, L. van Dijk and S. P. Fletcher, A catalytic asymmetric cross-coupling approach to the synthesis of cyclobutanes, Nat. Chem., 2021, 13, 880 CrossRef CAS PubMed; (j) D. Egea-Arrebola, F. W. Goetzke and S. P. Fletcher, Rhodium-Catalyzed Asymmetric Arylation of Cyclobutenone Ketals, Angew. Chem., Int. Ed., 2023, 62, e202217381 CrossRef CAS PubMed; (k) M. Takatsuki, H. Aoyama, K. Murai, M. Arisawa and M. Sako, Heteroannulation of bicyclobutane derivatives via Au-catalyzed hydration to enol ethers and intramolecular cyclization giving spirocyclobutanes, Chem. Commun., 2023, 59, 7467 RSC.
- (a) M. Golfmann and J. C. L. Walker, Bicyclobutanes as unusual building blocks for complexity generation in organic synthesis, Commun. Chem., 2023, 6, 9 CrossRef CAS PubMed; (b) C. B. Kelly, J. A. Milligan, L. J. Tilley and T. M. Sodano, Bicyclobutanes: from curiosities to versatile reagents and covalent warheads, Chem. Sci., 2022, 13, 11721 RSC; (c) A. Fawcett, Recent advances in the chemistry of bicycle- and 1-azabicyclo[1.1.0]butanes, Pure Appl. Chem., 2020, 92, 751 CrossRef CAS; (d) M. A. A. Walczak, T. Krainz and P. Wipf, Ring-Strain-Enabled Reaction Discovery: New Heterocycles from Bicyclo[1.1.0]butanes, Acc. Chem. Res., 2015, 48, 1149 CrossRef CAS PubMed.
- (a) H. K. Hall Jr and A. B. Padias, Bicyclobutanes and Cyclobutenes: Unusual Carbocyclic Monomers, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 62 Search PubMed; (b) A. M. Dilmaç, E. Spuling, A. de Meijere and S. Bräse, Propellanes-From a Chemical Curiosity to “Explosive” Materials and Natural Products, Angew. Chem., Int. Ed., 2017, 56, 5684 CrossRef PubMed.
- A. Kaur, W. Lin, V. Dovhalyuk, L. Driutti, M. L. D. Martino, M. Vujasinovic, J.-M. Löhr, M. E. Sellin and D. Globisch, Chemoselective bicyclobutane-based mass spectrometric detection of biological thiols uncovers human and bacterial metabolites, Chem. Sci., 2023, 14, 5291 RSC.
- P. Zhang, R. Zhuang, X. Wang, H. Liu, J. Li, X. Su, X. Chen and X. Zhang, Highly Efficient and Stable Strain-Release Radioiodination for Thiol Chemoselective Bioconjugation, Bioconjugate Chem., 2018, 29, 467 CrossRef CAS PubMed.
- K. B. Wiberg, G. M. Lampman, R. P. Ciula, D. S. Connor, P. Schertler and J. Lavanish, Bicyclo[1.1.0]butane, Tetrahedron, 1965, 21, 2749 CrossRef CAS.
- (a) P. Wipf and M. A. A. Walczak, Pericyclic cascade reactions of (bicyclo-[1.1.0]butylmethyl)amines, Angew. Chem., Int. Ed., 2006, 45, 4172 CrossRef CAS PubMed; (b) M. A. A. Walczak and P. Wipf, Rhodium(I)-Catalyzed Cycloisomerizations of Bicyclobutanes, J. Am. Chem. Soc., 2008, 130, 6924 CrossRef CAS PubMed; (c) R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Intermolecular [2π+2σ]-photocycloaddition enabled by triplet energy transfer, Nature, 2022, 605, 477 Search PubMed; (d) Y. Liang, R. Kleinmans, C. G. Daniliuc and F. Glorius, Synthesis of polysubstituted 2-oxabicyclo[2.1.1]hexanes via visible-light-induced energy transfer, J. Am. Chem. Soc., 2022, 144, 20207 CrossRef CAS PubMed; (e) Y. Liang, F. Paulus, C. G. Daniliuc and F. Glorius, Catalytic formal [2π+2σ] cycloaddition of aldehydes with bicyclobutanes: expedient access to polysubstituted 2-oxabicyclo[2.1.1]hexanes, Angew. Chem., Int. Ed., 2023, 62, e202305043 CrossRef CAS PubMed; (f) R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, Strain-release [2π+2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes initiated by energy transfer, J. Am. Chem. Soc., 2022, 144, 7988 CrossRef CAS PubMed; (g) Y. Zheng, W. Huang, R. K. Dhungana, A. Granados, S. Keess, M. Makvandi and G. A. Molander, Photochemical intermolecular [3σ+2σ]-cycloaddition for the construction of aminobicyclo[3.1.1]heptanes, J. Am. Chem. Soc., 2022, 144, 23685 CrossRef CAS PubMed; (h) S. Agasti, F. Beltran, E. Pye, N. Kaltsoyannis, G. E. M. Crisenza and D. J. Procter, A catalytic alkene insertion approach to bicyclo[2.1.1]hexane bioisosteres, Nat. Chem., 2023, 15, 535 CrossRef CAS PubMed; (i) T. Yu, J. Yang, Z. Wang, Z. Ding, M. Xu, J. Wen, L. Xu and P. Li, Selective [2σ+2σ] cycloaddition enabled by boronyl radical catalysis: synthesis of highly substituted bicyclo[3.1.1]heptanes, J. Am. Chem. Soc., 2023, 145, 4304 CrossRef CAS PubMed; (j) M. Xu, Z. Wang, Z. Sun, Y. Ouyang, Z. Ding, T. Yu, L. Xu and P. Li, Diboron(4)-catalyzed remote [3+2] cycloaddition of cyclopropanes via dearomative/rearomative radical transmission through pyridine, Angew. Chem., Int. Ed., 2022, 61, e202214507 CrossRef CAS PubMed; (k) Y. Liu, S. Lin, Y. Li, J.-H. Xue, Q. Li and H. Wang, Pyridine-boryl radical-catalyzed [2π+2σ] cycloaddition of bicyclo[1.1.0]butanes with alkenes, ACS Catal., 2023, 13, 5096 CrossRef CAS.
- For addition of carbon-centered radicals, see: (a) X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, Ti-catalyzed radical alkylation of secondary and tertiary alkyl chlorides using Michael acceptors, J. Am. Chem. Soc., 2018, 140, 14836 CrossRef CAS PubMed; (b) G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J.-C. Cintrat, Photochemical strain-release-driven cyclobutylation of C(sp3)-centered radicals, Angew. Chem., Int. Ed., 2020, 59, 2618 CrossRef CAS PubMed; (c) C. J. Pratt, R. A. Aycock, M. D. King and N. T. Jui, Radical α-C-H cyclobutylation of aniline derivatives, Synlett, 2020, 31, 51 CrossRef CAS PubMed; (d) H. Gao, L. Guo, C. Shi, Y. Zhu, C. Yang and W. Xia, Transition metal-free radical α-oxy C-H cyclobutylation via photoinduced hydrogen atom transfer, Adv. Synth. Catal., 2022, 364, 2140 CrossRef CAS.
- (a) S. Hoz, C. Azran and A. Sella, Atomic motions and protonation stereochemistry in nucleophilic additions to bicyclobutanes, J. Am. Chem. Soc., 1996, 118, 5456 CrossRef CAS; (b) L. Guo, A. Noble and V. K. Aggarwal, α-Selective ring-opening reactions of bicyclo[1.1.0]butyl boronic ester with nucleophiles, Angew. Chem., Int. Ed., 2021, 60, 212 CrossRef CAS PubMed; (c) Y. Gaoni, Regiospecific additions of hydrazoic acid and benzylamine to 1-(arylsulfonyl)bicycle[1.1.0]butanes. application to the synthesis of cis and trans 2,7-methanoglutamic acids, Tetrahedron Lett., 1988, 29, 1591 CrossRef CAS; (d) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Strain-release amination, Science, 2016, 351, 241 CrossRef CAS PubMed; (e) J. A. Milligan, C. A. Busacca, C. H. Senanayake and P. Wipf, Hydrophosphination of bicyclo[1.1.0]butane-1-carbonitriles, Org. Lett., 2016, 18, 4300 CrossRef CAS PubMed; (f) Y. Gaoni, New bridgehead-substituted 1-(arylsulfonyl)bicycle[1.1.0]butanes and some novel addition reactions of the bicyclic system, Tetrahedron, 1989, 45, 2819 CrossRef CAS.
- Z. Zhang and V. Gevorgyan, Palladium hydride-enabled hydroalkenylation of strained molecules, J. Am. Chem. Soc., 2022, 144, 20875 CrossRef CAS PubMed.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, Polarity-reversal strategy for the functionalization of electrophilic strained molecules via light-driven cobalt catalysis, J. Am. Chem. Soc., 2020, 142, 5355 CrossRef CAS PubMed.
- K. Dhake, K. J. Woelk, J. Becica, A. Un, S. E. Jenny and D. C. Leitch, Beyond bioisosteres: divergent synthesis of azabicyclohexanes and cyclobutenyl amines from bicyclobutanes, Angew. Chem., Int. Ed., 2022, 61, e202204719 CrossRef CAS PubMed.
- During the preparation of our manuscript, an elegant study describing Bi(OTf)3-catalyzed ring-opening of BCBs with naphthols was reported: A. Guin, S. Bhattacharjee, M. S. Harariya and A. T. Biju, Lewis acid-catalyzed diastereoselective carbofunctionalization of bicyclobutanes employing naphthols, Chem. Sci., 2023, 14, 6585 RSC.
- (a) L. A. Paquette, R. P. Henzel and S. E. Wilson, The influence of structural features on the course of bicyclo[1.1.0]butane rearrangements catalysed by silver (I) ion, J. Am. Chem. Soc., 1972, 94, 7780 CrossRef CAS; (b) L. A. Paquette, S. E. Wilson and R. P. Henzel, Mechanistic aspects of the silver(I)-promoted rearrangements of tricyclo [4.1.0.0]heptane derivatives. Deuterium isotope effect studies and independent generation of argento carbonium ions, J. Am. Chem. Soc., 1972, 94, 7771 CrossRef CAS; (c) M. Sakai, H. H. Westberg, H. Yamaguchi and S. Masamune, Silver(I)-catalyzed rearrangement of bicyclobutanes. Some aspects of the mechanism II, J. Am. Chem. Soc., 1971, 93, 4611 CrossRef; (d) L. A. Paquette, Catalysis of strained σ-bond rearrangements by silver(I) ion, Acc. Chem. Res., 1971, 4, 280 CrossRef CAS; (e) K. C. Bishop III, Transition metal catalyzed rearrangements of small ring organic molecules, Chem. Rev., 1976, 76, 461 CrossRef; (f) R. K. Kumar and X. Bi, Catalytic σ-activation of carbon–carbon triple bonds: reactions of propargylic alcohols and alkynes, Chem. Commun., 2016, 52, 853 RSC; (g) M. Li, W. Wu and H. Jiang, Recent advances in silver-catalyzed transformations of electronically unbiased alkenes and alkynes, ChemCatChem, 2020, 12, 5034 CrossRef CAS; (h) Q.-Z. Zheng and N. Jiao, Ag-catalyzed C–H/C–C bond functionalization, Chem. Soc. Rev., 2016, 45, 4590 RSC; (i) J.-Y. Son, S. Aikonen, N. Morgan, A. S. Harmata, J. J. Sabatini, R. C. Sausa, E. F. C. Byrd, D. H. Ess, R. S. Paton and C. R. J. Stephenson, Exploring Cuneanes as Potential Benzene Isosteres and Energetic Materials: Scope and Mechanistic Investigations into Regioselective Rearrangements from Cubanes, J. Am. Chem. Soc., 2023, 145, 16355 CrossRef CAS PubMed; (j) E. Smith, K. D. Jones, L. O'Brien, S. P. Argent, C. Salome, Q. Lefebvre, A. Valery, M. Böcü, G. N. Newton and H. W. Lam, Silver(I)-Catalyzed Synthesis of Cuneanes from Cubanes and their Investigation as Isosteres, J. Am. Chem. Soc., 2023, 145, 16365 CrossRef CAS PubMed; (k) H. Takebe and S. Matsubara, Catalytic Asymmetric Synthesis of 2,6-Disubstituted Cuneanes through Enantioselective Constitutional Isomerization of 1,4-Disubstituted Cubanes, Eur. J. Org Chem., 2022, e202200567 CrossRef CAS.
- (a) T.-Y. Lin, C.-Z. Zhu, P. Zhang, Y. Wang, H.-H. Wu, J.-J. Feng and J. Zhang, Regiodivergent intermolecular [3+2] cycloadditions of vinyl aziridines and allenes: stereospecific synthesis of chiral pyrrolidines, Angew. Chem., Int. Ed., 2016, 55, 10844 CrossRef CAS PubMed; (b) C.-Z. Zhu, J.-J. Feng and J. Zhang, Rhodium(I)-catalyzed intermolecular aza-[4+3] cycloaddition of vinyl aziridines and dienes: atom-economical synthesis of enantiomerically enriched functionalized azepines, Angew. Chem., Int. Ed., 2017, 56, 1351 CrossRef CAS PubMed; (c) T.-Y. Lin, H.-H. Wu, J.-J. Feng and J. Zhang, Transfer of chirality in the rhodium-catalyzed chemoselective and regioselective allylic alkylation of hydroxyarenes with vinyl aziridines, Org. Lett., 2017, 19, 2897 CrossRef CAS PubMed.
- Deposition numbers 2262933 (for cis-3ab) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.†.
- DFT calculations were performed by using the Gaussian 16 package under the level of B3LYP-D3(BJ)/def2-TZVP-SMD(toluene)//B3LYP-D3(BJ)/6-31G(d,p) and SDD for the Ag atom. See the ESI† for computational details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization, and spectroscopic data. CCDC 2262933. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03258b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |