
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn improved digestion coil arrangement for high-pressure microwave-assisted flow digestion
Franz
Hallwirth
,
Herbert
Motter
and
Helmar
Wiltsche
 *
*
Graz University of Technology, Institute of Analytical Chemistry and Food Chemistry, Graz, Austria. E-mail: helmar.wiltsche@tugraz.at
First published on 6th October 2023
Abstract
A high pressure flow digestion system for microwave assisted sample digestion at a pressure of 40 bar and a digestion temperature of about 230 °C has been developed. The sample mineralization took place in a 2 mm inner diameter PFA digestion tube heated by microwave radiation in a pressurized microwave applicator. By employing computer simulation of the microwave field, the position and geometry of the coiled perfluoralkoxy (PFA) digestion tube was optimized. Thereby, a uniform absorption of the microwave radiation over the length of the digestion tube was attained, whereby the formation of hot- and cold spots was avoided. The high-pressure flow digestion system had a heated volume of 22 mL and was operated at 500 W microwave power and a carrier flow rate of 5 mL min−1. Digestion coil and connections were made of PFA allowing any kind of digestion acid mixture. Acid mixtures of nitric acid with hydrochloric and/or hydrofluoric acid were successfully used for sample digestion. The accuracy was evaluated with three certified reference materials (NIST SRM 1547 – peach leaves, IAEA-A-13 – animal blood and BCR-185R – bovine liver) digested with a mixture of 5 mL 6 mol L−1 HNO3 and 3 mol L−1 HCl. After 5 minutes sample mineralization in the high-pressure flow digestion system, the residual carbon concentration in these digests was <50 mg L−1. The agreement between the determined and the certified values ranged from 90–110% for Al, As, B, Ba, Ca, Cu, Fe, K, Mg, Mn, Na, Rb, Sr, and Zn using inductively coupled plasma optical emission spectrometry (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS) for analyte quantification. For Fe in SRM 1547 and Cd in BCR-185R inferior agreement of 86% and 121% with the certified values was encountered. The sample throughput of the fully automated system was 12 samples per hour.
Introduction
Sample digestion for element and trace element analysis by flame- or plasma-based techniques is time consuming and laborious but indispensable for most sample matrices prior to analysis. Incomplete sample digestion may result in undesirable spectral interference, carbon-based matrix effects1–6 as well as low analyte recoveries. Modern wet digestion systems such as microwave assisted high-pressure closed vessel batch digestion techniques have gained great popularity over the last few decades. The high-pressure up to 200 bar, reached by some systems, increases the boiling point of the digestion acid mixture and therefore allows for higher digestion temperatures up to 300 °C.7 This in turn improves the digestion capability of oxidizing mineral acids such as nitric acid and accelerates the sample decomposition. Nevertheless, all commercial closed vessel digestion systems require multiple time-consuming manual working steps. Opening and closing the single digestion vessel to weight in the sample material and adding the acid mixtures as well as the tedious cleaning steps between the digestion batches extend the whole digestion process. All these steps imply the permanent need of skilled staff and increase the potential risk of sample contamination or analyte losses.A promising alternative to circumvent the aforementioned drawbacks, is fully automated microwave heated high-pressure (>25 bar) flow digestion. Moreover, the potential for direct coupling of flow digestion systems to the analyte quantification by e.g. inductively coupled plasma mass spectrometry (ICP-MS) further decreases the manual workload.
The principles of flow-digestion for element analysis have been extensively described in the literature.8 With respect to heating the digestion acid/sample mixture by microwave radiation, two approaches have been reported: heating in a monomode cavity9–11 (focused microwave cavity) and heating in multimode cavities.12–17 These two cavities differ in their microwave field distribution. In mono-mode cavities, spatially well-defined regions of high and low E and H-field are present. By placing the digestion coil in a region of high field strength, rapid heating is ensured. However, two problematic aspects must be considered: firstly, the heated region within the cavity is spatially small (several centimeters) resulting in rapid heating and the formation of hot-spots that in turn might cause violent reaction of the digestion mixture. Secondly, only a small sample volume can be placed within this spatially limited region. Multimode cavities on the other side allow large sample volumes to be heated. However, the field distribution within the cavity is not defined and changes depending on volume, composition, temperature and geometrical location of the liquid phase. Consequently, the heating of the digestion mixture inside the digestion tube is characterized by zones of intense heating and zones of low field strength with low heating.
In this work we introduce a new generation of a microwave-assisted, high-pressure, flow digestion system. Several improvements over high-pressure flow digestion systems reported in literature16–19 were introduced: firstly, the microwave-heated volume of the digestion coil was nearly doubled from the previously used 13.5 mL to 22 mL.17 This allows for a longer residence time of the digestion mixture within the radiated/heated zone.16,17,19 Secondly, by extensively using microwave field simulation, a high-pressure microwave applicator was specifically designed for uniform power absorption along the digestion tube, essentially eliminating hot and cold spots. To the best of our knowledge, no microwave-assisted flow digestion system described in literature used a spatially optimized digestion coil for efficient and homogeneous microwave coupling. At the same time, the expensive and bulky modified commercial microwave digestion system previously used16,17 was replaced by a compact pressure-tight digestion vessel, specifically designed and built for the present flow digestion setup.
Experimental
Flow digestion setup
Fig. 1 shows a schematic of the high-pressure, continuous flow, pressure equilibrium flow digestion system. Diluted nitric acid 3% (v/v), used as carrier solution was constantly pumped through the system by an inert high-pressure pump (Fig. 1G, Azura P2.1S EPG90, Knauer, Germany).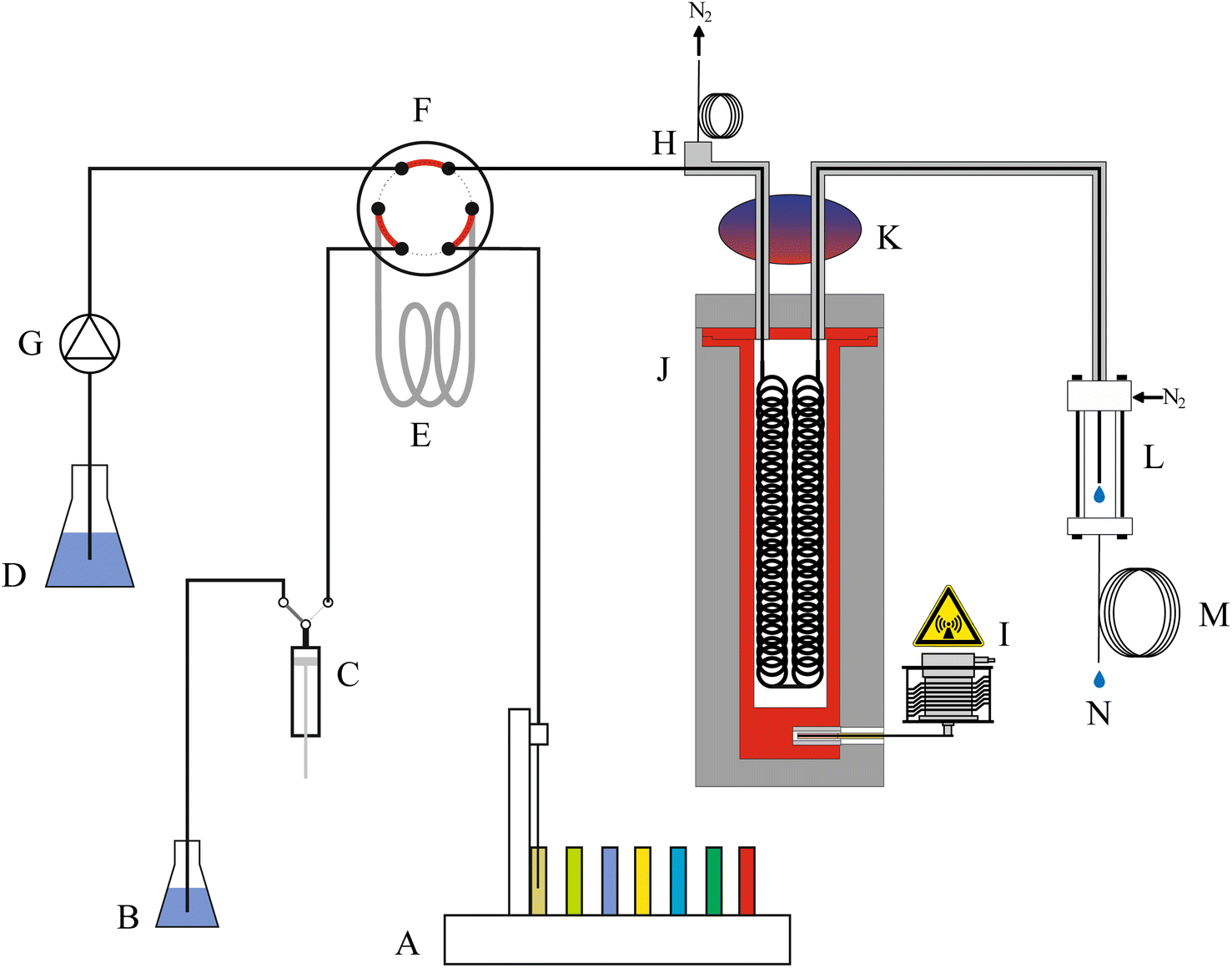 | ||
| Fig. 1 Schematic of the high-pressure, continuous flow, pressure equilibrium flow digestion system. The gray shaded components including the digestion vessel indicate the pressurized part. (A) Autosampler with sample, (B and D) nitric acid 3% (v/v) carrier solution, (C) precision dispenser, (E) sample loop, (F) six-port valve, (G) high pressure pump, (H) pressure interface including the nitrogen exit port, (I) magnetron, (J) high-pressure vessel, (K) cooler, (L) nitrogen inlet port/sample exit port, (M) pressure restriction loop, (N) digested sample. The counter-current nitrogen stream enters the digestion system at (L) and leaves the system at (H). | ||
Samples were either introduced as liquids or suspensions of fine grounded solids. These slurries of 1% (m/v) solid material in liquid phase could be handled by the system without the risk of clogging. Prior introduction in the flow digestion system, the slurries were stirred for 30 s by a polyetheretherketone (PEEK) paddle mixer to ensure a homogeneous slurry. The mixer was attached to the autosampler's (ASX-1400, Cetac, USA) sampling arm, next to the sampling capillary. Between the sampling steps the paddle was cleaned in a rinsing station with ultrapure water. The samples, supplied by the autosampler, were introduced into the high-pressure stream by means of a 12 mL PFA sample loop (Fig. 1E) connected to a wide-bore, polymer sealed 6-port valve (Fig. 1F, Azura VU 4.1 EWA01XA, Knauer, Germany). Using a precision dispenser (Fig. 1C, 1-Channel MultiDispenser, ProLiquid, Germany), varying sample volumes could be introduced into the flow digestion system. To avoid dilution of the digestion mixture by the carrier solution, each sample was embedded between two 2.5 mL segments of 6 mol L−1 nitric acid.
The digestion took place in a pressurized (40 bar) vessel (Fig. 1J) using the pressure equilibrium concept initially introduced by Knapp.18,19 Details of this arrangement are given below. After leaving the microwave irradiated zone the digested samples passed a cooling stage (Fig. 1K) and left the flow digestion setup through a restrictor capillary (Fig. 1M, 12 m PFA tube with 0.5 mm ID). The digests were collected in 50 mL sample tubes (Fig. 1N) for later analysis using the autosampler's second sampling needle.
A small but steady flow of nitrogen was introduced into the flow digestion system in a countercurrent direction to the sample stream (flow direction: Fig. 1L to H) to remove acid vapors originating from diffusion of steam through the PFA digestion tube. The applied nitrogen pressure thereby also defines the working pressure of the flow digestion system. A restrictor capillary at the nitrogen exit port (Fig. 1H) limits the nitrogen consumption to about 3 L min−1.
High-pressure microwave applicator
The design of the high-pressure microwave applicator aimed for three goals: firstly, the microwave absorption along the entire length of the digestion tube should be homogeneous. Thereby hot and cold-spots are avoided, allowing harsher reaction conditions and processing of slurries containing a larger fraction of solids. Secondly, a system pressure of 40 bar should be achieved. The boiling point of nitric acid at this pressure is about 230 °C. Thereby, efficient sample mineralization can be expected. Although, the pressure vessel was designed to withstand up to 80 bar, the flow digestion system is limited to 40 bar by the pressure-stability of the wide-bore 6-port valve for sample introduction (Fig. 1F), and the temperature-stability of the PFA tube which is about 260 °C even without additional mechanical stress. The third design goal was to limit the nitrogen-pressurized volume within the entire flow digestion system to below one liter in order to comply with European pressure vessel regulations.20Extensive microwave field simulations, calculated with HFSS Ansys Electromagnetic Suite (Version 15.0.0, Ansys, USA) resulted in a cylindrical waveguide with a diameter of 85 mm and circular loops of PFA tube (40 mm loop diameter) filled with acid/sample arranged within the waveguide as shown in Fig. 2b. The waveguide was constructed from a cylindrical pressure-tight aluminium body (height: 330 mm, ID 85 mm, OD: 125 mm) lined with PTFE to avoid corrosion and reduce the nitrogen-pressurized volume of the pressure vessel. The PFA digestion tube (ID 2 mm; OD 3 mm; 22 mL heated volume) was woven on two PTFE sheets (each 250 × 40 × 3 mm) using position holes drilled by a computer-numerically-controlled (CNC) milling machine. CNC machining was deemed necessary to attain the position of each loop of digestion tube as calculated by the microwave field simulation. The digestion tube assembly was attached to the lid of the pressure vessel as shown in Fig. 2a. This lid was equipped with a thin-walled PTFE protective cap to avoid corrosion of the lids body made of tool-steel. The pressure vessel and the lid were pressed together by a Tri-Clamp fitting, effectively sealing the pressurized part of the microwave applicator.
 | ||
| Fig. 2 Picture of the PFA digestion tube assembly retracted from the high-pressure microwave applicator (a) and side view of the computer simulation of the PFA digestion tube and the microwave antenna near the bottom of the applicator (b). Please note the different tilt angels of the PFA digestion-tube-loops in (b) that where optimized by microwave field simulation for similar power uptake for every loop. | ||
Microwave radiation was generated by an 850 W, 2.45 GHz magnetron (2M107A-795, National Electronics, Korea) powered by a CM 340 adjustable DC magnetron power supply (MKS instruments Inc, USA). The magnetron was mounted on a coaxial magnetron launcher (Model CMLD 1.1, S-Team, Slovakia). Via a 7/8′′ EIA to N adapter (LI-A78NFI, BroadcastStoreEurope, Denmark) and a low loss RG393 coaxial cable (Pasternack Enterprises, USA), the microwave radiation was transmitted to a λ/4 antenna mounted 25 mm off the bottom of the high-pressure microwave applicator.
Instrumentation
Analytes and residual carbon content (RCC) were quantified either by axially viewed inductively coupled plasma optical emission spectrometer (ICP-OES, Ciros Vision EOP, Spectro AMETEK, Germany) or ICP-MS (NexION 2000, PerkinElmer, USA). The following emission lines were used for ICP-OES: Al II 167.078 nm, As I 189.042 nm, B I 249.773 nm, Ba II 455.404 nm, C I 193.091 nm, Ca II 317.933 nm, Cd II 214.438 nm, Cu II 219.266 nm, K I 766.491 nm, Fe II 238.204 nm, Mg II 280.270 nm, Mn II 257.611 nm, Na I 588.995 nm, Pb II 220.353 nm, Sr II 407.771 nm, and Zn I 213.856 nm. Sc was used as internal standard in ICP-OES at a concentration of 1 mg L−1 employing the Sc II 361.384 nm emission line. Element concentrations determined by ICP-MS employed the following conditions: He collision mode: 43Ca, 48Ti, 52Cr, 56Fe, 60Ni; reaction cell with O2: As at m/z 91; no cell gas: 98Mo, 114Cd, 208Pb. 10 μg L−1 Ga, In and Ir were used as internal standards.References digestions of glucose, glycine, phenylalanine and nicotinic acid were performed by high-pressure flow digestion as well as by closed vessels batch digestion using two commercial microwave digestion systems (Multiwave 5000, Anton Paar GmbH, Austria and MARS 6, CEM Corporation, USA). For closed vessel batch digestion a temperature-controlled digestion program was used: within 30 minutes, the digestion acid was heated to 230 °C and thereafter, this temperature was maintained for additional 20 minutes.
Digestions of certified reference materials were performed by high-pressure flow digestion and by closed vessel batch digestion (Multiwave 3000, Anton Paar, Austria) employing the same temperature-controlled program as noted above. The same type of digestion vessels (HF100; ceramic support vessels with PFA liner; 40 bar maximum working pressure) were used with both, Multiwave 3000 and Multiwave 5000 (Anton Paar, Austria), whereby comparable digestion conditions were attained. iPrep (CEM Corporation, USA) digestion vessels were used with the MARS 6 batch digestion system.
Thermal images were recorded using an infrared thermal imager (TR120, Shenzhen Milesseey Technology Co., Ltd, China) using an emissivity coefficient of 0.92 (PTFE).
Reagents, samples and preparation of the slurries
All standard solutions and slurries were prepared using the high purity acids (HCl: RotipuranUltra, ROTH; Germany, HF: NORMATOM, VWR Chemicals, USA and HNO3: Fisher Scientific GmbH, Germany, purified by sub-boiling in a quartz still) and high purity water (18 MΩ cm, Barnstead Nanopure, Thermo Fisher Scientific, USA).Calibration solutions for element quantification were prepared by diluting a 100 mg L−1 multi element stock solution (Al, Ag, As, B, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, K, Li, Mg, Mn, Mo, Na, Ni, Pb, Sb, Se, Sr, Ti, Tl, V, Zn, Roth, Germany) with 3% (v/v) nitric acid. 1 g L−1 single element stock solutions of Sc, Ga, In and Ir (Roth, Germany) were used as internal standards after appropriate dilution. Potassiumhydrogenphthalate (pa, Merck, Germany) was used for preparing the calibration standards for RCC quantification by ICP-OES.
Digestion efficiency was evaluated by digesting solutions containing 10 g L−1 carbon prepared from analytical grade glucose, glycine, phenylalanine and nicotinic acid (all Merck, Germany) in 6 mol L−1 HNO3.
Three certified reference materials were investigated in this work: BCR-185R (Trace Elements in Lyophilized Bovine Liver; Institute for Reference Materials and Measurements, Belgium), IAEA A-13 (Trace and Minor Elements in Freeze Dried Animal Blood, International Atomic Energy Agency, Austria), and SRM 1547 (Peach Leaves, National Institute of Standards & Technology, USA).
Slurries of 1% (m/v) solids were prepared in 50 mL PP tubes (Sartstedt, Germany) by thoroughly mixing 0.5 g of the solid sample with about 10 mL water, adding concentrated acids and making up to volume with water for a final acid concentration of 6 mol L−1 nitric acid and 3 mol L−1 hydrochloric acid. The slurries were sonicated in an ultrasonic bath (Transsonic T 420, Elma, Germany) for 5 minutes before they were placed on the auto sampler of the flow digestion system. Thereby sample clotting was avoided. It is noteworthy, that the maximum particle diameter noted in the certificates of the solid CRMs is small (<125 μm for BCR-185R, <300 μm for IAEA A-15 and <75 μm for SRM 1547). For flow digestion, 5 mL slurry were embedded between two 2.5 mL segments of 6 mol L−1 HNO3 prior introduction into the microwave heated part. As noted above, slurries were automatically stirred for 30 s with a paddle mixer before the sample uptake by the high-pressure flow digestion system. Thereby segregation of solid particles and consequentially non-representative slurry sampling was avoided. The final volume of each sample after digestion was 50 mL.
The residual acid concentration in the digested samples was determined by manual titration with 0.10 mol L−1 NaOH (Roth, Germany) using phenolphthalein as indicator.
Results and discussion
Characterization of the high-pressure microwave applicator
In Fig. 3b the heating of the carrier solution by microwave radiation is illustrated. Contrary to conventional operation of the flow digestion system, no flow of the carrier solution was applied. Thereby the static heating pattern of the digestion coil can be visualized. Already 60 s of microwave radiation at 400 watts power sufficed to attain a homogeneous heating of the entire digestion coil, confirming the computer simulation data shown in Fig. 3c. It is important to note, that the used design aim for the digestion coil was uniform heating. This aim was achieved. It would also be possible to arrange the digestion coil in a way, that the microwave absorption is spatially different, e.g. that more energy is delivered to the beginning and the end of the digestion tube. Such arrangements are subject to further investigation.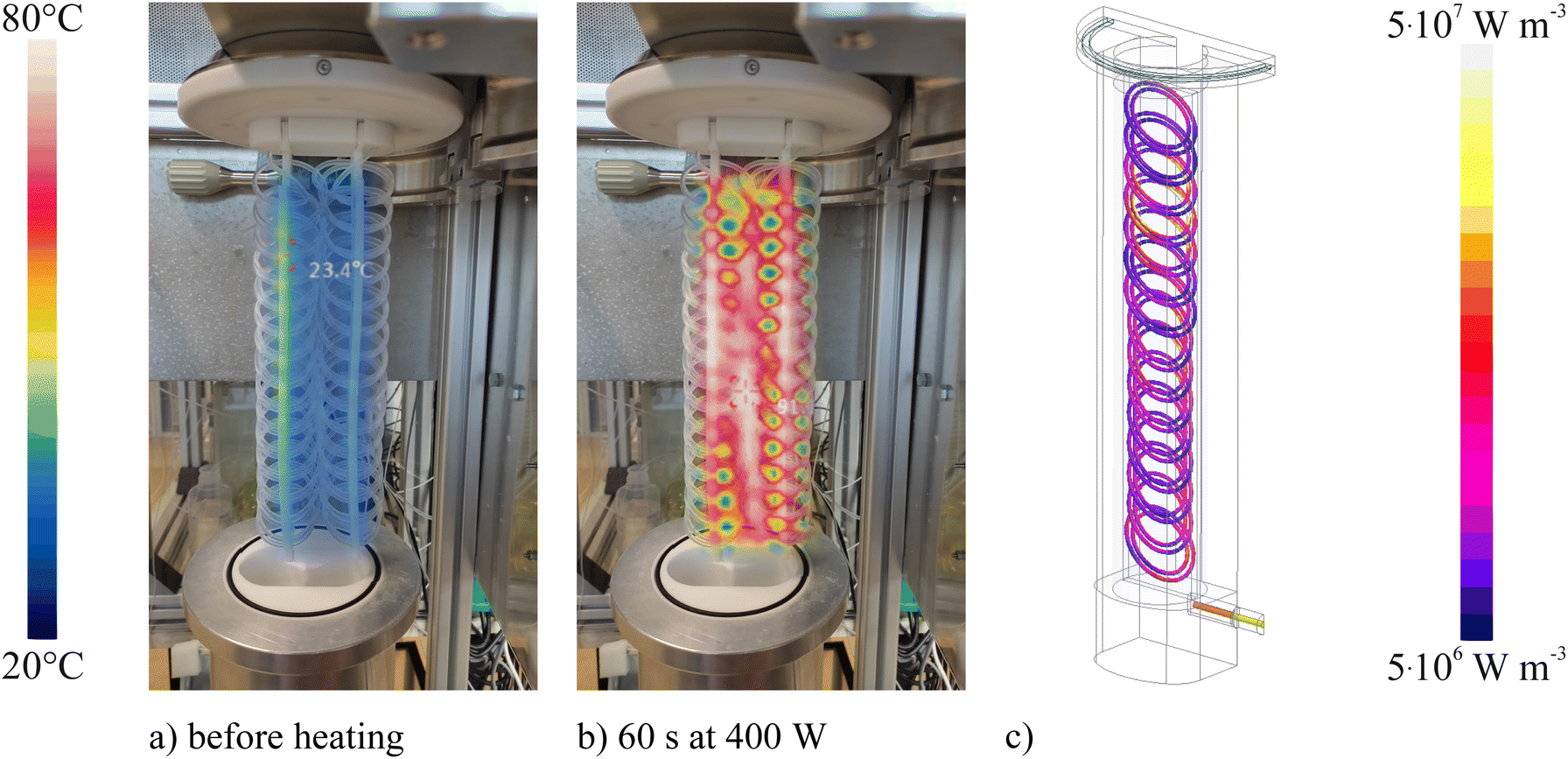 | ||
| Fig. 3 Image of the digestion coil overlaid by thermal images before (a) and after heating with 400 W for 60 s (b). The digestion coil was filled with 3% (v/v) nitric acid, though no flow was applied to identify the static heating pattern. The computer simulation of the volume loss density within the PFA digestion tube is shown in (c). Due to the symmetry of the digestion coil, only half of the microwave applicator was included in the simulation, whereby the simulation time could be reduced. | ||
The ratio between the vapor- and the liquid phase inside the digestion coil is a measure for characterizing the microwave heating.17 Small values (<5%) suggest low temperatures within the digestion coil due to poor microwave coupling or insufficient microwave power. High vapor-to-liquid phase ratios (>30%) indicate the formation of large volumes of vapor and steam. This in turn leads to a reduction in residence time within the heated zone due to premature sample ejection from the heated zone and consequentially ineffective sample mineralization. The initial system optimization showed, that for 40 bar system pressure, 5 mL min−1 carrier flow rate and 500 W microwave power the highest vapor-to-liquid phase ratio of 13 ± 1% was obtained. This value was deemed acceptable and comparable to that used in previous high-pressure flow digestion systems.17 An even higher vapor-to-liquid phase ratios would have required higher microwave power. However, in the present system, the microwave power level is limited by the RG393 coaxial cable. Though the microwave damping, that is equivalent to power loss, by this cable is lowest for all commercially available cable types, power levels exceeding 500 W resulted in an inacceptable heating of the coaxial cable. Up to 180 °C were measured during brief initial tests. For this reason, typically waveguides are used to transmit high-power microwave radiation instead of coaxial cables. However, during the initial verification of the reactor, a vector network analyzer (VNA) directly connected to the microwave applicator via a coaxial cable was used. To allow VNA diagnostic also in later stages of the applicator evaluation, the coaxial cable connection was retained. It is important to note, that there is no fundamental problem in converting the current microwave feed from coaxial cable to a waveguide-based one.
The digestion efficiency can be quantified by evaluating the RCC after digestion.21 Similar to previous works,16,17 we used glucose, glycine, phenylalanine and nicotinic acid as selected representative test substances for this evaluation. While glucose reacts violently with nitric acid once heated and thereby releases large amounts of CO2, glycine requires temperatures of about 240 °C to be digested appreciably.19 Nicotinic acid is very difficult do digest and phenylalanine even forms insoluble intermediate reaction products that require higher temperature to be solubilized again. Phenylalanine and nicotinic acid are known to be hard to digest because their molecular structure includes a phenyl ring.22,23
The digestion efficiencies expressed as RCC for the representative test substances are listed in Table 1, together with RCC values obtained by batch digestion at the same maximum operating pressure of 40 bar and the same acid concentration. Glucose was digested effectively by both, high-pressure flow digestion and batch digestion. For glycine, the flow digestion was inferior to batch digestion. While the latter achieved essentially complete digestion, in high-pressure flow digestion some 19% RCC remained. Though this value is comparable to previous reports for high-pressure flow digestion at 40 bar17 the residence time of the sample/acid mixture within the heated zone was significantly shorter than in batch digestion (4 minutes compared to 20 minutes + 30 minutes ramping up the temperature). Most likely, a prolonged residence time within the radiated zone would lead to a complete digestion of glycine as well. The results for phenylalanine and nicotinic acid proved to be comparable for both, batch and flow digestion. For phenylalanine a RCC of about 30% was observed. Compared to our previous high-pressure flow digestion systems17 this is an improvement by more than a factor of two. We attribute this to the improved heating in the microwave applicator. While our previous high pressure flow digestion systems employed multi-mode cavities, the current applicator provides uniform power absorption in a mono-mode cavity.
| Digestion procedure | Glucose RCC | Glycine RCC | Phenylalanine RCC | Nicotinic acid RCC |
|---|---|---|---|---|
| High-pressure flow digestion | <LOQ (0.5%) | 19 ± 5% | 28 ± 2% | 93 ± 11% |
| Batch digestion: Multiwave 5000, Anton Paar | <LOQ (0.5%) | <LOQ (0.5%) | 29 ± 7% | 97 ± 2% |
| Batch digestion: MARS 6, CEM | <LOQ (0.5%) | <LOQ (0.5%) | 27 ± 6% | 101 ± 8% |
Once more, nicotinic acid proved to be very difficult to digest. Neither batch nor flow digestion were able to reduce the RCC significantly. This is also in accordance with previous observations.17
Though most parts wetted by the sample/digestion acid mixture were constructed from high purity polymers such as PFA, stainless steel and aluminium were used for constructing the high-pressure autoclave. As steam and acid vapors slowly diffuse through PFA at elevated temperatures, all pressure-bearing metallic components are constantly flushed by a small, counter current flow of nitrogen. Following the pressure equilibrium concept,18,19 thereby the contamination of the sample with metals used as structural materials is avoided. To assess, that the counter-current flow of nitrogen actually avoids contamination, blank digestions with 5 mL of an acid mixture of 6 mol L−1 nitric acid, 3 mol L−1 hydrochloric acid and 1 mol L−1 hydrofluoric acid were performed. Subsequent analysis by ICP-MS revealed only small contamination of the blanks: 0.8 ± 0.1 μg L−1 Al, 0.9 ± 0.2 μg L−1 Cr, 3.4 ± 0.3 μg L−1 Fe, 0.7 ± 0.2 μg L−1 Mn, 0.9 ± 0.1 μg L−1 Ni, 0.8 ± 0.2 μg L−1 Ti (50 mL final volume, n = 4). These values are similar to the blank levels obtained with previous high-pressure flow digestion systems.17
Precision and accuracy evaluation
Three certified reference materials were used to assess the accuracy and precision of the high-pressure flow digestion. As the digestion acid mixture is important for some elements, also closed vessel batch digestions were performed for the CRMs. Thereby effects from different digestion systems can be identified even when unsatisfactory agreement of the quantified analyte content with the certified value is attained.In general, the results listed in Table 2 are in close agreement with the certified ones and comparable to those obtained closed vessel batch digestion. Even though aluminium and stainless steel components were used in the high-pressure flow digestion system, no significant contamination of the samples with Al, Cr, Fe or Mn was encountered, confirming the very low blanks obtained during blank digestions. It is important to note, that even though the residence time of the sample/acid mixture within the microwave heated part of the flow system was only 5 minutes, all CRM digests were clear and without any visual particles.
| Sample | Certified value (mg kg−1) | High-pressure flow digestion (mg kg−1) | Closed vessel batch digestion (mg kg−1) | |
|---|---|---|---|---|
| NIST SRM 1547(peach leaves) | Al | 248.9 ± 6.5 | 270 ± 20 | 240 ± 10 |
| B | 28.73 ± 0.81 | 31 ± 5 | 25 ± 2 | |
| Ba | 123.7 ± 5.5 | 120 ± 9 | 120 ± 4 | |
| Ca | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590 ± 160 590 ± 160 |
15300 ± 700 |
15400 ± 300 |
|
| Fe | 219.8 ± 6.8 | 190 ± 8 | 200 ± 10 | |
| K | 24330 ± 380 |
24700 ± 270 |
24600 ± 200 |
|
| Mg | 4320 ± 150 | 3890 ± 160 | 4230 ± 150 | |
| Mn | 97.8 ± 1.8 | 101 ± 5 | 91 ± 4 | |
| Sr | 53.0 ± 5.0 | 52 ± 2 | 56 ± 5 | |
| Zn | 17.97 ± 0.53 | 21 ± 2 | 17 ± 4 | |
| IAEA-A-13 (freeze dried animal blood) | Ca | 226–332 | 270 ± 80 | 250 ± 20 |
| Cu | 3.7–4.8 | 5.8 ± 0.3 | 2.8 ± 1 | |
| Fe | 2200–2500 | 2500 ± 200 | 2200 ± 100 | |
| K | 2100–2700 | 2500 ± 300 | 2400 ± 300 | |
| Mg | 81–139 | 85 ± 15 | 89 ± 6 | |
| Na | 11600–13500 |
10700 ± 1000 |
11000 ± 400 |
|
| Rb | 1.7–3.1 | 2.2 ± 0.1 | ND | |
| Zn | 12–14 | 12 ± 1 | 12 ± 2 | |
| SRM BCR 185R (bovine liver) | As | 0.033 ± 0.0029 | 0.026 ± 0.004 | ND |
| Cd | 0.544 ± 0.017 | 0.39 ± 0.02 | 0.50 ± 0.07 | |
| Cu | 277 ± 5 | 300 ± 20 | 250 ± 10 | |
| Mn | 11.07 ± 0.29 | 12 ± 1 | 10.0 ± 0.5 | |
| Zn | 138.6 ± 2.1 | 156 ± 15 | 137 ± 5 | |
While the general close agreement between the certified values and the values attained by flow digestion as sample preparation was attained, larger deviations (>10%) were encountered for Fe in SRM 1547 (peach leaves) and Cd in BCR 185R (bovine liver). In the first case, the Fe results for flow digestion as well as closed vessel batch digestion were similar, but lower than the certified ones. As the same digestion acid mixture was used in both cases, it seems reasonable to assume, that these systematic low results are connected to the acid mixture.16 Contrary to this, the low results for Cd using high-pressure flow digestion in BCR-185R (bovine liver) cannot be attributed to the digestion acid mixture, as acceptable values were obtained by closed vessel batch digestion. Though selective losses of Cd during the flow digestion seem unlikely, further investigations will be conducted.
The residual acid concentration after flow digestion was about 1.6 mol L−1 for all CRMs without significant differences among the reference materials. This indicates a large excess of digestion acid during digestion.
The residual carbon concentration in the digested CRMs was 29 ± 2 mg L−1, 38 ± 2 mg L−1, and 43 ± 2 mg L−1 for SRM 1547, IAEA-A-13, and BCR 185R, respectively. These values can be regarded negligible with respect to potential carbon-based matrix effects2 and underline the digestion effectivity of the presented high-pressure flow digestion system.
Blank digestions with 5 mL of an acid mixture of 6 mol L−1 nitric acid and 3 mol L−1 hydrochloric acid performed immediately after the CRM analysis showed no elevated blank levels for even for elements present in high concentration in the CRMs such as Ca, K or Na. This finding is not surprising, as the carrier solution is continuously pumped through the flow system, constantly cleaning all surfaces that come into contact with the sample and the digestion acid mixture.
Conclusion
A new microwave applicator for high-pressure flow digestion was developed and shown to evenly heat the sample/acid digestion mixture over the entire length of the digestion tube within the microwave field. The good agreement between computer simulation and actual heating pattern confirms the simulation. While this work aimed at a uniform heating of the digestion acid within the digestion tube, further research into different heating patterns is thereby possible. For instance, it seems interesting to investigate, if a gradual heating at the beginning of the digestion tube followed by an intense heating phase thereafter allows for higher concentrated slurries of very reactive samples or even samples containing more than 5% (v/v) ethanol.As all surfaces wetted by the sample were made of high purity fluoro-polymers, aggressive digestion acid mixtures containing HNO3, HCl and HF could be used without contaminating the digested sample with dissolved structural materials. Moreover, even prolonged operation of the system did not cause significant corrosion of the high-pressure flow system.
Another advantage of the presented flow digestion approach is the fully automated digestion of sample slurries without the risk of contamination due to various sample handling steps commonly necessary in closed vessel batch digestion.
The sample throughput was 12 samples per hour. It should be noted, that an increase of this sample throughput can be easily accomplished by having more than one digestion coil in the microwave applicator. Though this then also necessitates a change in the high pressure valve configuration, a sample throughput of 36 samples per hour for three digestion coils could be attained. Moreover, a direct coupling to the analyte quantification by ICP-MS is currently under investigation.
For a system pressure of 40 bar, the high pressure flow digestion system was able to attain RCC values for glucose and phenylalanine in line with closed vessel batch digestion. However, the mineralization of glycine was inferior to batch digestion. We attribute this to the limited residence time of the reaction mixture within the heated zone of the flow system. Nevertheless, this was deemed acceptable. The accuracy of the high-pressure flow digestion procedure was evaluated using three CRMs. The agreement between the determined and the certified values ranged from 90–110% for Al, As, B, Ba, Ca, Cu, Fe, K, Mg, Mn, Na, Rb, Sr, and Zn. For Fe in SRM 1547 and Cd in BCR-185R inferior agreement of 86% and 121% with the certified values was encountered.
Compared to other flow digestion systems reported in literature, the presented high-pressure flow digestion system has several advantages: firstly, the relatively high digestion temperature of about 230 °C results in effective sample mineralization and low RCC. This is in sharp contrast to continuous-,14,15,24–26 closed-,27,28 or stopped29 flow digestion systems at ambient pressure and consequently only about 120 °C digestion temperature. Another disadvantage of the aforementioned ambient pressure flow digestion system as well as of medium pressure flow digestion systems12,30 (2–20 bar system pressure) is the inherent limitation of the digestion acid concentration and maximum microwave power. Upon exceeding a system-specific limit of either of the two variables, excessive formation of CO2 or nitrous oxides prematurely eject the sample/acid mixture from the microwave heated zone resulting in poor digestion performance in terms of RCC. Due to the high system pressure of 40 bar used in the presented high-pressure flow digestion system, most of the gasses formed during the digestion remain dissolved in the liquid phase during the digestion step and are liberated thereafter during expansion in the restrictor capillary. Thereby, the flow pattern during the digestion is not disturbed and no premature ejection of the sample occurs.
Another advantage of the presented high pressure flow digestion system is the large (22 mL) heated volume in the pressurized microwave applicator. This represents an improvement over previous high-pressure flow digestion systems,16,17,19 particularly when considering the aforementioned homogeneous heating of the digestion acid by microwave radiation.
While from the digestion point of view the presented high pressure flow digestion system is superior to ambient pressure flow digestion systems, the main disadvantage is the system complexity. Moreover, particularly for manufacturing the high pressure reactor a workshop capable of machining metals and PTFE to tight tolerances is necessary.
Conflicts of interest
No conflicts to disclose.Acknowledgements
The authors acknowledge the support of Heimo Kotzian and the Anton Paar GmbH for the microwave field simulations.References
- J. Machát, V. Otruba and V. Kanický, Spectral and non-spectral interferences in the determination of selenium by inductively coupled plasma atomic emission spectrometry, J. Anal. At. Spectrom., 2002, 17, 1096–1102 RSC.
- H. Wiltsche, M. Winkler and P. Tirk, Matrix effects of carbon and bromine in inductively coupled plasma optical emission spectrometry, J. Anal. At. Spectrom., 2015, 30, 2223–2234 RSC.
- Y. Cheung, G. C. Y. Chan and G. M. Hieftje, Flagging matrix effects and system drift in organic-solvent-based analysis by axial-viewing inductively coupled plasma-atomic emission spectrometry, J. Anal. At. Spectrom., 2013, 28, 241–250 RSC.
- G. Grindlay, L. Gras, J. Mora and M. T. C. de Loos-Vollebregt, Carbon-related matrix effects in inductively coupled plasma atomic emission spectrometry, Spectrochim. Acta, Part B, 2008, 63B, 234–243 CrossRef CAS.
- G. Grindlay, L. Gras, J. Mora and M. T. C. de Loos-Vollebregt, Carbon-, sulfur-, and phosphorus-based charge transfer reactions in inductively coupled plasma–atomic emission spectrometry, Spectrochim. Acta, Part B, 2016, 115, 8–15 CrossRef CAS.
- G. Grindlay, J. Mora, M. de Loos-Vollebregt and F. Vanhaecke, A systematic study on the influence of carbon on the behavior of hard-to-ionize elements in inductively coupled plasma-mass spectrometry, Spectrochim. Acta, Part B, 2013, 86, 42–49 CrossRef CAS.
- C. B. Ojeda and F. S. Rojas, in Encyclopedia of Analytical Science, ed. P. Worsfold, C. Poole, A. Townshend and M. Miró, Academic Press, Oxford, 3rd edn, 2019, pp. 85–97 Search PubMed.
- H. Wiltsche and G. Knapp, in Microwave-Assisted Sample Preparation for Trace Element Determination, ed. E. M. M. Flores, Elsevier, Amsterdam, 2014, ch. 9 Search PubMed.
- L. J. M. Stewart and R. M. Barnes, Flow-through, microwave-heated digestion chamber for automated sample preparation prior to inductively coupled plasma spectrochemical analysis, Analyst, 1994, 119, 1003–1010 RSC.
- D. L. Tsalev, M. Sperling and B. Welz, On-line microwave sample pre-treatment for hydride generation and cold vapour atomic absorption spectrometry. Part 1. The manifold, Analyst, 1992, 117, 1729–1733 RSC.
- S. P. Fili, E. Oliveira and P. V. Oliveira, On-line digestion in a focused microwave-assisted oven for elements determination in orange juice by inductively coupled plasma optical emission spectrometry, J. Braz. Chem. Soc., 2003, 14, 435–441 CrossRef CAS.
- S. J. Haswell and D. Barclay, On-line microwave digestion of slurry samples with direct flame atomic absorption spectrometric elemental detection, Analyst, 1992, 117, 117–120 RSC.
- R. E. Sturgeon, S. N. Willie, B. A. Methven, J. W. H. Lam and H. Matusiewicz, Continuous-flow microwave-assisted digestion of environmental samples using atomic spectrometric detection, J. Anal. At. Spectrom., 1995, 10, 981–986 RSC.
- J. L. Burguera and M. Burguera, Determination of lead in biological materials by microwave-assisted mineralization and flow injection electrothermal atomic absorption spectrometry, J. Anal. At. Spectrom., 1993, 8, 235–241 RSC.
- M. A. Z. Arruda, M. Gallego and M. Valcarcel, Flow-through microwave digestion system for the determination of aluminium in shellfish by electrothermal atomic absorption spectrometry, J. Anal. At. Spectrom., 1995, 10, 501–504 RSC.
- H. Wiltsche, P. Tirk, H. Motter, M. Winkler and G. Knapp, A novel approach to high pressure flow digestion, J. Anal. At. Spectrom., 2014, 29, 272–279 RSC.
- T. Linhares Marques, H. Wiltsche, H. Motter, J. A. Nóbrega and G. Knapp, High pressure microwave-assisted flow digestion system using a large volume reactor-feasibility for further analysis by inductively coupled plasma-based techniques, J. Anal. At. Spectrom., 2015, 30, 1898–1905 RSC.
- G. Knapp, US Pat., 5672316, 1995.
- U. Pichler, A. Haase, G. Knapp and M. Michaelis, Microwave-Enhanced Flow System for High-Temperature Digestion of Resistant Organic Materials, Anal. Chem., 1999, 71, 4050–4055 CrossRef CAS.
- Directive 2014/29/EU, Safety of Simple Pressure Vessels in the EU, 2014 Search PubMed.
- C. A. Bizzi, M. F. Pedrotti, J. S. Silva, J. S. Barin, J. A. Nóbrega and E. M. M. Flores, Microwave-assisted digestion methods: towards greener approaches for plasma-based analytical techniques, J. Anal. At. Spectrom., 2017, 32, 1448–1466 RSC.
- K. W. Pratt, H. M. Kingston, W. A. MacCrehan and W. F. Koch, Voltammetric and liquid chromatographic identification of organic products of microwave-assisted wet ashing of biological samples, Anal. Chem., 1988, 60, 2024–2027 CrossRef CAS PubMed.
- M. M. Daniel, J. D. Batchelor, C. B. Rhoades Junior and T. J. Bradley, The Effect of Digestion Temperature on Matrix Decomposition Using a High Pressure Asher, At. Spectrosc., 1998, 19, 198–203 CAS.
- M. Burguera, J. L. Burguera and O. M. Alarcon, Flow injection and microwave-oven sample decomposition for determination of copper, zinc and iron in whole blood by atomic absorption spectrometry, Anal. Chim. Acta, 1986, 179, 351–357 CrossRef CAS.
- M. Burguera, J. L. Burguera, C. Rondon, C. Rivas, P. Carrero, M. Gallignani and M. R. Brunetto, In vivo sample uptake and on-line measurements of cobalt in whole blood by microwave-assisted mineralization and flow injection electrothermal atomic absorption spectrometry, J. Anal. At. Spectrom., 1995, 10, 343–347 RSC.
- S. Recknagel, P. Brätter, A. Tomiak and U. Rösick, Determination of selenium in blood serum by ICP-OES including an on-line wet digestion and Se-hydride formation procedure, Fresenius. J. Anal. Chem., 1993, 346, 833–836 CrossRef CAS.
- V. Carbonell, M. de la Guardia, A. Salvador, J. L. Burguera and M. Burguera, On-line microwave oven digestion flame atomic absorption analysis of solid samples, Anal. Chim. Acta, 1990, 238, 417–421 CrossRef CAS.
- V. Carbonell, A. Morales-Rubio, A. Salvador, M. de la Guardia, J. L. Burguera and M. Burguera, Atomic absorption spectrometric analysis of solids with on-line microwave-assisted digestion, J. Anal. At. Spectrom., 1992, 7, 1085–1089 RSC.
- E. R. Pereira-Filho and M. A. Z. Arruda, Mechanised flow system for on-line microwave digestion of food samples with off-line catalytic spectrophotometric determination of cobalt at ng l-1 levels, Analyst, 1999, 124, 1873–1877 RSC.
- H. Staufenberg, Vollautomatisches Durchfluß-Mikrowellenaufschlußsystem, LaborPraxis, 1993, vol. 11, pp. 58–59 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |