
Dually-dynamic covalent tetraPEG hydrogels end-linked with boronate ester and acylhydrazone groups
Charalambos
Michael
a,
Demetris E.
Apostolides
a,
Costas S.
Patrickios
 *a and
Takamasa
Sakai
b
*a and
Takamasa
Sakai
b
aDepartment of Chemistry, University of Cyprus, 1 University Avenue, Aglanjia, 2109 Nicosia, Cyprus. E-mail: cmicha16@ucy.ac.cy; apostolides.demetris@ucy.ac.cy
bDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: costasp@ucy.ac.cy; sakai@tetrapod.t.u-tokyo.ac.jp
First published on 25th July 2022
Abstract
Well-defined dually dynamic hydrogels were prepared by end-linking four-armed poly(ethylene glycol) stars (tetraPEG stars) through two different types of dynamic covalent cross-links, boronates and acylhydrazones, leading to robust, self-healable materials. This required the prior end-functionalization of tetraPEG stars, originally bearing four hydroxyl terminal groups, with glucoronate, acylhydrazide and benzaldehyde groups, resulting in three differently end-functional star polymers. A first type of dually dynamic hydrogel resulted from the combination of the first two differently end-functionalized tetraPEG stars, cross-linked by 4-formylphenyl boronic acid, a small molecule bearing both an aldehyde and a boronic acid group, respectively complementary to the acylhydrazide and glucoronate end-groups of the two above-mentioned tetraPEG stars. For comparison, a singly-dynamic hydrogel cross-linked with only acylhydrazone groups was also prepared, as well as a double-like hydrogel combining the constituents of both of the above-mentioned hydrogels. All three types of hydrogels were prepared at three different pH values, 8.5, 10.5 and 12.5, leading to a total number of nine samples. All nine samples were investigated for their self-healing, mechanical, viscoelastic and aqueous swelling/degradation properties. This study sets the basis for the development of well-defined polymeric dynamic covalent hydrogels where their self-healing and stability can be readily tuned.
Introduction
Polymer networks (thermosets and elastomers) and gels (organogels and hydrogels) represent an important part, ∼25%, of the annual global polymer production of more than 300 million tons,1 and have a similar environmental footprint to linear and other non-network commercial polymers, with the extra burden of their cross-linked structure which even further delays their disintegration in nature. Alleviating the problem of environmental pollution arising from polymer networks and gels can be accomplished by extending the service life of objects made of these materials via rendering them self-repairable, on the one hand, and, on the other hand, by accelerating their degradability once disposed. Both of these targets can be attained by the implementation of dynamic covalent bonds2–4 as cross-links5,6 in these materials. Although covalent in nature, dynamic covalent bonds break and reform, i.e., they display a dynamic equilibrium involving partner exchange, leading to self-healing in the bulk material, or to disintegration when the system is diluted, e.g., under conditions of rain.Several choices are available for such dynamic covalent bonds, including boronate esters exchanging at and above the pKa of the corresponding boronic acid, imines (hydrazones and oximes) exchanging under acidic conditions, Diels-Alder adducts and nitroxides exchanging thermally, and disulfides exchanging under reduction-oxidation conditions.7 Boronate esters represent an attractive choice, characterized by their extreme reversibility, but limited by their consequent reduced stability.8 On the other hand, acylhydrazones possess high hydrolytic stability but lower reversibility, particularly the aryl-substituted ones.9 Combination of the two types of dynamic covalent groups, acylhydrazones and boronates, as cross-links in the same polymer network, is expected to endow the material with a superior spectrum of properties, and, in particular, good reversibility with sufficient stability, leading to self-repairable and strong polymer networks. Furthermore, fine-tuning the extent of each property, reversibility vs. stability, according to the demands of the application at hand, can be readily accomplished by adjusting the percentage of each of the two kinds of bonds.
Conventional polymeric hydrogels are infamously fragile due to their pronounced spatial inhomogeneity arising from the uneven distribution of cross-links, a result of free radical cross-linking copolymerization usually employed for their preparation.10 Many successful efforts have been undertaken during the past 20 years to mechanically reinforce polymer hydrogels, including interpenetration (double-networks),11 utilization of mechanical (rather than chemical) cross-links (slide-ring gels),12 doping with inorganic nanoparticles (nanocomposite gels),13 and well-defined gels (tetraPEG gels) based on four-armed star poly(ethylene glycol)s (tetraPEG stars) interconnected at the tips of their arms (end-linked).14 However, only the last of these four approaches really addresses the problem of eliminating network heterogeneity, and is, consequently, the most general and promising.
Hydrogels of well-defined structure, cross-linked with a mixture of acylhydrazone and boronate dynamic covalent bonds will be readily self-healable and robust materials. This is the aim undertaken in this study. In particular, herein we employ tetraPEG stars of molar mass of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (“TP10k”) which are appropriately end-functionalized so that they bear different types of terminal groups. Appropriate combination of the various kinds star polymers leads to the formation of hydrogels bearing both boronate and acylhydrazone cross-links. Although tetraPEG gels separately end-linked with boronate15,16 or acylhydrazone17 groups have already been reported in the literature, the simultaneous utilization of both types of cross-links in the same material has not yet been documented. On the other hand, some hydrogels18 and organogels19 have appeared in the literature, with the simultaneous utilization of both acylhydrazone and boronate cross-links; however, the gels in these reports were randomly cross-linked. The dually dynamic tetraPEG hydrogels prepared in this investigation are characterized in terms of their self-healing, mechanical, viscoelastic and aqueous swelling/degradation properties, the last of which (swelling and degradation) are rarely studied for dynamic covalent hydrogels.
000 g mol−1 (“TP10k”) which are appropriately end-functionalized so that they bear different types of terminal groups. Appropriate combination of the various kinds star polymers leads to the formation of hydrogels bearing both boronate and acylhydrazone cross-links. Although tetraPEG gels separately end-linked with boronate15,16 or acylhydrazone17 groups have already been reported in the literature, the simultaneous utilization of both types of cross-links in the same material has not yet been documented. On the other hand, some hydrogels18 and organogels19 have appeared in the literature, with the simultaneous utilization of both acylhydrazone and boronate cross-links; however, the gels in these reports were randomly cross-linked. The dually dynamic tetraPEG hydrogels prepared in this investigation are characterized in terms of their self-healing, mechanical, viscoelastic and aqueous swelling/degradation properties, the last of which (swelling and degradation) are rarely studied for dynamic covalent hydrogels.
Thus, the synthetic novelty in this investigation is the preparation of well-defined hydrogels based on a mixture of a number of differently end-functionalized tetraPEG stars. These novel hydrogel materials would have a broad application potential, appropriate for cases when robust hydrogels are required, and covering a wide spectrum of needs in terms of the relative character in self-repairability vs. stability, which can be regulated in a straightforward manner, namely, through the fraction of each kind of cross-link in the system.
Experimental section
Materials
Diethyl ether (purity ≥99%), 4-hydroxybenzhydrazide (≥97%), benzaldehyde (≥99%), 4-formylphenylboronic acid (≥95%), D-(+)-gluconic acid δ-lactone (≥99%), 4-hydroxybenzaldehyde (98%), methanesulfonyl chloride (≥99.7%), potassium carbonate (≥99%), hydrazine hydrate (80% w/w solution in water), dialysis tubing composed of benzoylated cellulose with molecular weight cut-off of 3000 g mol−1 (average flat width 32 mm), methanol (99.8%), sodium carbonate (≥99%), sodium hydrogen carbonate (≥99.7%), sodium phosphate monobasic monohydrate (≥98%), sodium phosphate dibasic (≥99%), glacial acetic acid, potassium phosphate tribasic (≥98%), sodium hydroxide (≥98%) and 80% w/w aqueous ammonia solution were purchased from Sigma-Aldrich-Merck, Germany. Tetrahydroxy-terminated four-arm star poly(ethylene glycol) of Mn = 10000 g mol−1 (TP10k-OH) was purchased from NOF Corporation, Tokyo, Japan. Dichloromethane (DCM, 99%) was purchased from Labscan, Ireland. N,N-dimethylformamide (DMF, 99.8%) and triethylamine (TEA, ≥99%) were purchased from Scharlau, Spain. Dimethylsulfoxide (DMSO, ≥99%) was purchased from Fisher, UK. Deuterated chloroform (CDCl3, 99.8%) and deuterated dimethylosulfoxide (d6-DMSO, 99.8%) were purchased from Merck, Germany. Aqueous buffer solutions (100 mM) at pH 8.5, 10.5 and 12.5 were prepared from sodium phosphate dibasic/sodium phosphate monobasic monohydrate, sodium carbonate/sodium hydrogen carbonate, and potassium phosphate tribasic, respectively.
Synthesis of benzaldehyde-protected hydrazide
For this synthesis, 3.1 g (29.9 mmol) benzaldehyde, 3.5 g (23 mmol) 4-hydroxybenzhydrazide and 60 mL DMF were first placed into a round bottomed flask. Subsequently, the mixture was refluxed at 80 °C for 24 h. Next, after most of the DMF was removed under reduced pressure, the reaction mixture was precipitated in chloroform. The precipitate obtained was dried in a vacuum oven at 60 °C for 5 h to give the product as a white powder.Modification of TP10k-OH
Preparation of the “hydrazone” cross-linked hydrogels
These dynamic covalent hydrogels were formed by mixing aqueous solutions of TP10k-Bz and TP10k-Hz at their stoichiometric ratio. The synthesis was performed in three different buffers (100 mM) of pH values equal to 8.5, 10.5 and 12.5, and at a polymer concentration of 15% w/v each. A typical experimental procedure is given below. First, 0.034 g (3.22 μmol) TP10k-Hz and 0.0335 g (3.22 μmol) TP10k-Bz were dissolved separately in 225 μL buffer each. Afterwards, the two aqueous solutions were mixed and vortexed at room temperature. The hydrogels were left for seven days to be formed completely.Preparation of the “boronate” cross-linked hydrogels
This category of dynamic covalent hydrogels was formed by mixing solutions of TP10k-Gluco, 4-formylphenylboronic acid (4-FPBA) and TP10k-Hz at their stoichiometric ratio. The synthesis took place in three different buffers (100 mM) of pH values equal to 8.5, 10.5 and 12.5, and at a polymer concentration of 15% w/v each. A typical experimental procedure is given below. Initially, 0.034 g (3.17 μmol) of TP10k-Gluco and 0.0019 g (12.7 μmol) of 4-FPBA were dissolved together in 225 μL of aqueous buffer solution of the chosen pH. Then, 0.0334 g (3.17 μmol) of TP10k-Hz was dissolved separately in another 225 μL of aqueous buffer solution of the same pH as above. Subsequently, these two solutions were mixed and vortexed at room temperature. The hydrogels were left for seven days to be formed completely.Preparation of the “double” hydrogels
These dynamic covalent hydrogels were formed by mixing solutions of TP10k-Gluco, 4-formylphenylboronic acid (4-FPBA), TP10k-Hz and TP10k-Bz at their stoichiometric ratio. The synthesis was performed in three different buffers (100 mM) of pH values equal to 8.5, 10.5 and 12.5, and at a polymer concentration of 30% w/v each. A typical experimental procedure is given below. Firstly, 0.034 g (3.17 μmol) of TP10k-Gluco and 0.0019 g (12.7 μmol) of 4-FPBA were dissolved together in 150 μL of buffer. Then, 0.0674 g (6.34 μmol) TP10k-Hz and 0.0335 g (3.17 μmol) TP10k-Bz were dissolved separately in additional 150 μL of buffer each, and, subsequently, the three aqueous solutions were mixed and vortexed at room temperature. The hydrogels were left for seven days to be formed completely.Aqueous degrees of swelling of the hydrogels
The degrees of swelling of the three types of hydrogels, (“boronate” cross-linked, “hydrazone” cross-linked and “double” hydrogels) were measured in aqueous buffered solutions. Each type of hydrogel was formed in aqueous buffer solutions of three different pH values, 8.5, 10.5 and 12.5, and, later on, transferred into a vial with 10 mL of aqueous buffer solution (100 mM) of a pH value the same as that of hydrogel formation. Every hydrogel was left in the buffer to equilibrate for twenty-five days. The status of the hydrogel was observed (whether fully dissolved or not), and the swollen mass of the hydrogels was determined gravimetrically.Characterization with 1H NMR spectroscopy
1H NMR spectroscopy was performed on a 500 MHz Avance Bruker spectrometer, equipped with an Ultrashield magnet, to confirm the structures (quantitative end-functionalization reactions) of the tetraPEG star building blocks of the hydrogels (TP10k-Gluco, TP10k-Bz and TP10k-Hz) and their precursors (TP10k-Ms, TP10k-NH2 and TP10k-HzProt). The 1H NMR spectra were recorded in the deuterated solvents CDCl3 and d6-DMSO. The 1H NMR spectra of the “boronate” cross-linked hydrogel in D2O buffered (100 mM) at pD = 8.5, three and seven days after mixing its components, and that of the in situ formed adduct between TP10k-Gluco and FPBA at the same pD were also recorded.Rheology
Viscoelastic characterization of the hydrogels at their as-prepared state was performed on a Discovery HR2 rheometer from Thermal Analysis Instruments (TA), operating in parallel plate geometry, with an upper plate of 20 mm in diameter and a temperature-controlled (Peltier) lower plate maintained at 20 °C. The rheometer was operated in the oscillation frequency mode, with angular frequency values from 0.1 to 100 rad s−1, 1% strain, and an approximate plate separation of 3 mm. The rheological measurements provided the frequency dependence of the storage (G′) and loss (G′′) shear moduli of the “hydrazone” cross-linked, “boronate” cross-linked, and “double” hydrogels, prepared at a total solids concentration of 15, 15 and 30% w/v, respectively, each in aqueous buffer solutions of pH 8.5, 10.5 and 12.5. Rheological strain sweep experiments were also conducted on selected samples (“boronate” and “double” hydrogels formed at pH = 8.5) in order to more quantitatively probe their self-healing capability. In these experiments, the strain sweep first indicated the critical strain at which the sample fails. Next, a strain 20 to 50% higher than the critical was instantaneously applied to the sample, which was subsequently allowed to recover. Finally, the extent to which the sample regained its original storage shear modulus was calculated.Mechanical testing
An Instron 5944 mechanical testing system (Instron, Norwood, MA) was used to characterize the compressive mechanical properties of the hydrogels at their as-prepared state. The samples were cylindrical with a diameter of 9.5 mm and a height of 8.1 mm, and were compressed at a constant speed of 1.37 mm min−1. “Boronate”, “hydrazone” and “double” hydrogels with a total solids concentration of 15, 15, 30% w/v, respectively, were prepared, each at three different pH values of 8.5, 10.5 and 12.5. Attempted tension experiments on the same mechanical testing system failed due to sample slippage from the instrument's grasps. However, rough tension experiments were performed on selected samples (“boronate” and “double” formed at pH = 10.5) by holding the samples using our fingers and pulling apart, which allowed an estimation of the strain at break. These semi-quantitative experiments were performed both on virgin and self-healed (cut and pushed against each other for 24 h) samples, which also allowed an estimation of the self-healing efficiency.Self-healing
Self-repair experiments were performed on “boronate” cross-linked, “hydrazone” cross-linked, and “double” hydrogels formed at pH 8.5, 10.5 and 12.5, with a total solids concentration of 15, 15, 30% w/v, respectively. All the different types of hydrogels were prepared at the three pHs, one sample with and another without the presence of dye: rhodamine B, methylene blue, riboflavin, cresol purple, or one of two different universal pH indicators. After seven days, both the dyed and non-dyed hydrogels were cut in half, resulting in two pieces each. Subsequently, a piece of the colored and a piece of the non-dyed hydrogel were pressed together along their cut surfaces and left for 8 h in a closed container.Results and discussion
End-functionalized TetraPEG stars
The preparation of the designed hydrogels required the syntheses of three differently end-functionalized final tetraPEG stars, whose syntheses involved the prior preparation of three intermediates. The three final tetraPEG stars were the ones end-functionalized with glucoronate (“TP10k-Gluco”), benzaldehyde (“TP10k-Bz”) and acylhydrazide (“TP10k-Hz”) groups, whereas the intermediate products were tetraPEG stars end-functionalized with mesylate (“TP10k-Ms”), primary amine (“TP10k-NH2”), and benzaldehyde-protected acylhydrazide (“TP10k-ProtHz”) groups. All synthetic procedures are illustrated in Fig. 1, where the employed reaction conditions, solvents, and reagents with their excesses are also provided. It must be pointed out that all end-functionalizations were complete (100%, confirmed using 1H NMR spectroscopy), in order to secure as high cross-linking conversion in the hydrogels as possible, whereas the lower-than-perfect recovery yields, ranging between 78 and 97%, were due to material losses during the various purification procedures (extraction, precipitation, dialysis). Fig. 1 also illustrates schematics of the three differently end-functionalized final tetraPEG stars, TP10k-Gluco, TP10k-Bz and TP10k-Hz, plus the chemical structure and schematic of 4-formylphenyl boronic acid (FPBA), a small molecule subsequently used for the hydrogel synthesis. | ||
| Fig. 1 Reaction schemes followed for the preparation of the three differently end-functionalized tetraPEGs, as well as their schematic representations plus that of FPBA. | ||
The 1H NMR spectra of TP10k-Bz and TP10k-Hz, as well as their intermediates, were provided in our previous work.17 Thus, here we only give the 1H NMR spectra (Fig. 2) of TP10k-Gluco and its intermediates, synthesized following the procedure described by Yesilyurt et al.15
 | ||
| Fig. 2 1H NMR spectra of (I) the starting compound TP10k-OH, (II) the purified mesylate-terminated tetraPEG (TP10k-mesyl), (III) the purified amine-terminated tetraPEG (TP10k-NH2), and (IV) the final targeted product of diol-terminated tetraPEG (TP10k-Gluco). | ||
Mesylate-terminated tetraPEG (TP10k-mesyl)
The synthesis of TP10k-mesyl was perfomed by the mesylation of TP10k-OH, resulting in the activation of the four terminal hydroxyl groups of the tetraPEG star with mesyl groups. The final product was isolated in 78% yield. Its structure and purity were confirmed by 1H NMR spectroscopy. Fig. 2(II) shows the 1H NMR spectrum of the synthesized and purified TP10k-mesyl. The spectrum presented the full conversion of the hydroxyl groups (1H NMR spectrum of starting material given in Fig. 2(I)) to mesyl groups, due to the appearance of the peak e at 3.1 ppm corresponding to the methyl protons of the mesyl group.Amine-terminated tetraPEG (TP10k-NH2)
The synthesis of TP10k-amine was achieved by the nucleophilic attack of ammonia to the mesyl terminal groups of TP10k-mesyl, resulting in their conversion to primary amine terminal groups. The final product was isolated in 96% yield, while its structure and purity were confirmed using 1H NMR spectroscopy. Fig. 2(III) shows the 1H NMR spectrum of the purified TP10k-NH2. The spectrum shows that the mesyl groups were completely replaced by primary amine groups, evidenced by the disappearance of the methyl protons of TP10k-mesyl at 3.1 ppm and the appearance of peak c at 2.7 ppm, corresponding to the methylene group next to the terminal amine.Diol-terminated tetraPEG (TP10k-Gluco)
The synthesis of TP10k-Gluco was performed by the nucleophilic attack of the primary amine groups of TP10k-NH2 onto the glucurolactone ring, leading to ring opening and incorporation of one glucuronate group at each of the terminal amine groups of TP10k-NH2. The final product was isolated in 97% yield, and its structure and purity were confirmed using 1H NMR spectroscopy. Fig. 2(IV) shows the 1H NMR spectrum of the purified TP10k-Gluco. The spectrum indicates that the amine groups reacted completely with the lactone, manifested by the appearance of the amide bond peak d at 7.7 ppm, and the peaks of the opened glucurolactone, i, h, g, k, j, e and f, but also by the disappearance of the methylene peak at 2.7 ppm.Hydrogel types
Fig. 3 illustrates schematically the three hydrogel types and the strategy they were prepared from the combination of the various building blocks. These building blocks include the three differently end-functionalized tetraPEG stars, TP10k-Gluco, TP10k-Bz and TP10k-Hz, as well as FPBA. Our most novel hydrogel type, henceforth to be called “boronate”, was a dually dynamic hydrogel resulting from the indirect end-linking of the glucoronate and acylhydrazide end-functionalized tetraPEG stars through FPBA whose aldehyde and boronic acid groups are reactive to acylhydrazide and glucoronate groups, respectively. In addition to that, and for comparison, a dynamic hydrogel cross-linked only via acylhydrazone groups was also prepared (to be called “hydrazone” hydrogel),17 as well as a double-like hydrogel combining the constituents of both of the above-mentioned hydrogels (“double” hydrogel). Each of the three hydrogels was formed in aqueous buffers at three different pHs, 8.5, 10.5 and 12.5.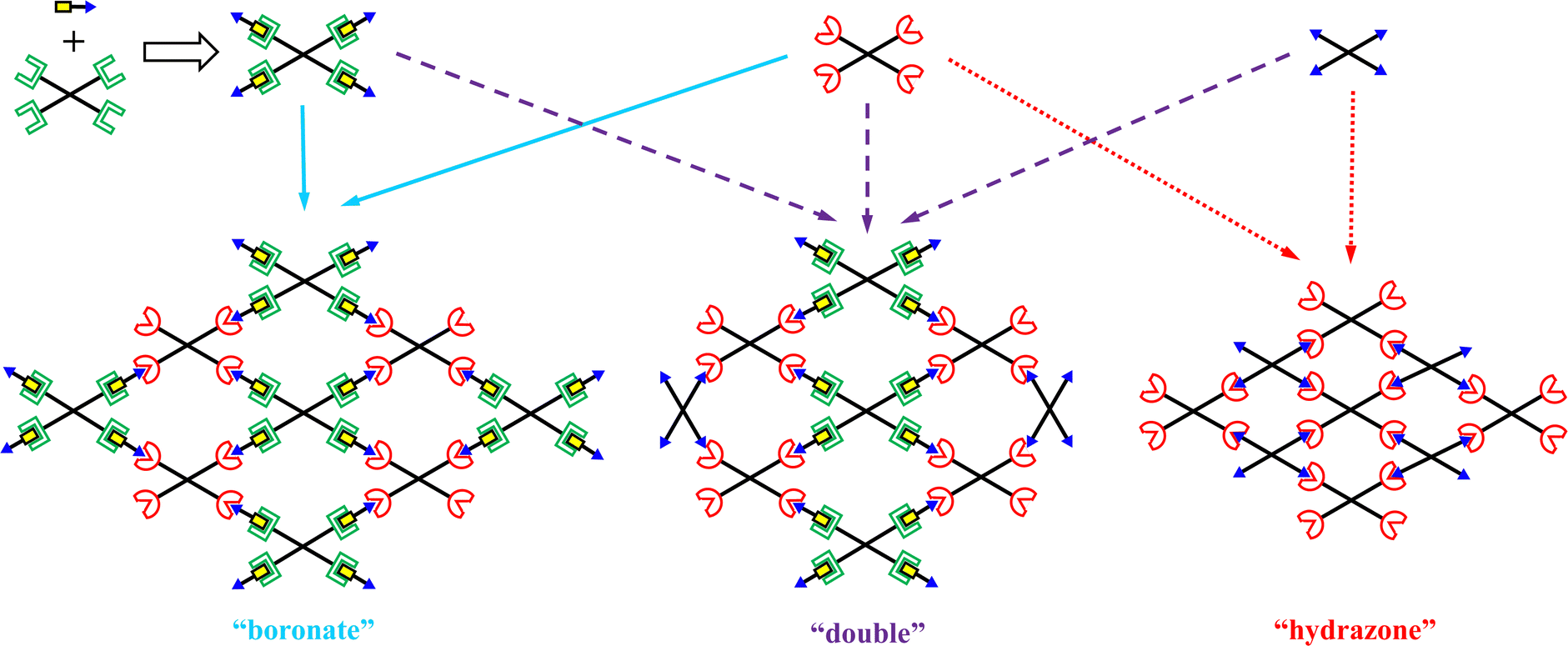 | ||
| Fig. 3 Preparation of the three types of hydrogels from the four available building blocks. | ||
Structure of boronate hydrogel
Next, we provide 1H NMR spectroscopy evidence for the structure of the “boronate” hydrogel prepared in two consecutive steps (see Fig. 3). We were able to record a 1H NMR spectrum in D2O buffered (100 mM) at pD = 8.5 not only for the product of the first step which is soluble, but also for the product of the second step which is the hydrogel. We would like to point out that NMR spectra of polymer networks, even in the swollen state, are very rare in the literature, due to peak broadening arising from low chain mobility in the semi-solid gel state. Here, the soft nature of the system apparently allowed the recording of the high resolution proton NMR spectrum, shown in Fig. 4.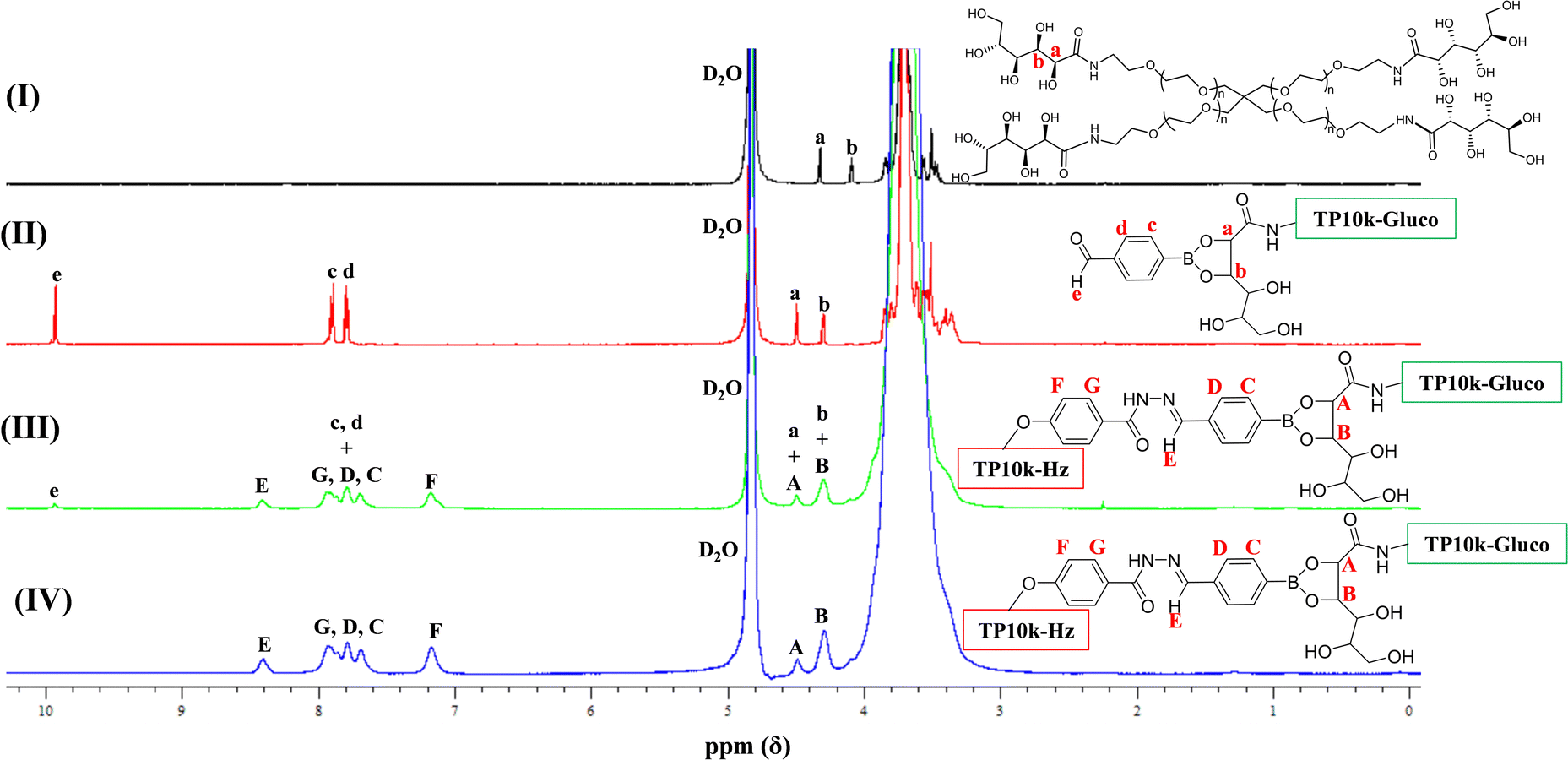 | ||
| Fig. 4 1H NMR spectra in D2O buffered at pD = 8.5 of (I) the starting TP10k-Gluco, (II) the fully-formed (within 1 h) adduct of TP10k-Gluco with a stoichiometric amount of FPBA, and (III and IV) the “boronate” hydrogel formed from the reaction of the TP10k-Gluco-FPBA adduct with a stoichiometric amount of TP10k-Hz after three days (III) and after seven (IV). | ||
The 1H NMR spectrum in part (II) of the figure clearly shows the complete conversion of the starting TP10k-Gluco (its 1H NMR spectrum is shown in part (I) of the figure) to its adduct with FPBA after 1 h of reaction (with a stoichiometric amount of FPBA), as evidenced by the downfield shift of the signals from the methine protons a and b, and the appearance of aromatic protons c, d and e at the correct positions and with the correct integrals in the spectrum. Parts (III) and (IV) of the figure display the 1H NMR spectra for the “boronate” hydrogel recorded three and seven days, respectively, after the addition of a stoichiometric amount of TP10k-Hz to the previously mentioned adduct. While near-complete conversion can already be detected within three days of the reaction, as evidenced from the large reduction in the signal from the benzaldehyde proton e at 9.8 ppm, complete conversion is achieved in seven days, when proton e totally disappears from the spectrum in part (IV).
Apparent degrees of swelling and degradation of the hydrogels
The degrees of swelling of hydrogels represent a very important thermodynamic property and easy to measure, usually gravimetrically.20–22 However, many studies do not report it, as they focus on the (bio)applications of the as-prepared hydrogel samples. Furthermore, regarding the “boronate”-cross-linked hydrogels, these would totally degrade upon their equilibration with excess water,15 due to the very fast dynamics of the boronate bond. Still, it would be very useful to obtain the time of complete gel dissolution. Thus, in this first part of hydrogel characterization, we study the swelling behavior of all gel samples, to obtain the relevant swelling properties and dissolution times. Fig. 5 displays the temporal evolution of the apparent aqueous degrees of swelling for the three hydrogel types, each at the three different pH values. The nine swelling profiles can be grouped into two types, according to their shape: six of them, which form a maximum, and three of them that tend to level off. The latter type corresponds to the most stable samples, which are the “hydrazone” hydrogels at the two lower pH values, 8.5 and 10.5, plus the “double” hydrogel at pH 8.5, whereas the former type corresponds to the rest of the samples. The maximum swelling value in the former type is created by the tendency of the hydrogel to absorb more solvent from the as-prepared state in order to attain equilibrium swelling leading to an increase in the degree of swelling at the beginning of the process, on the one hand, and, on the other hand, the eventual degradation of the sample arising from the dynamic nature of the cross-links in combination with the presence of a large excess of solvent, ultimately leading to the complete dissolution of the hydrogel.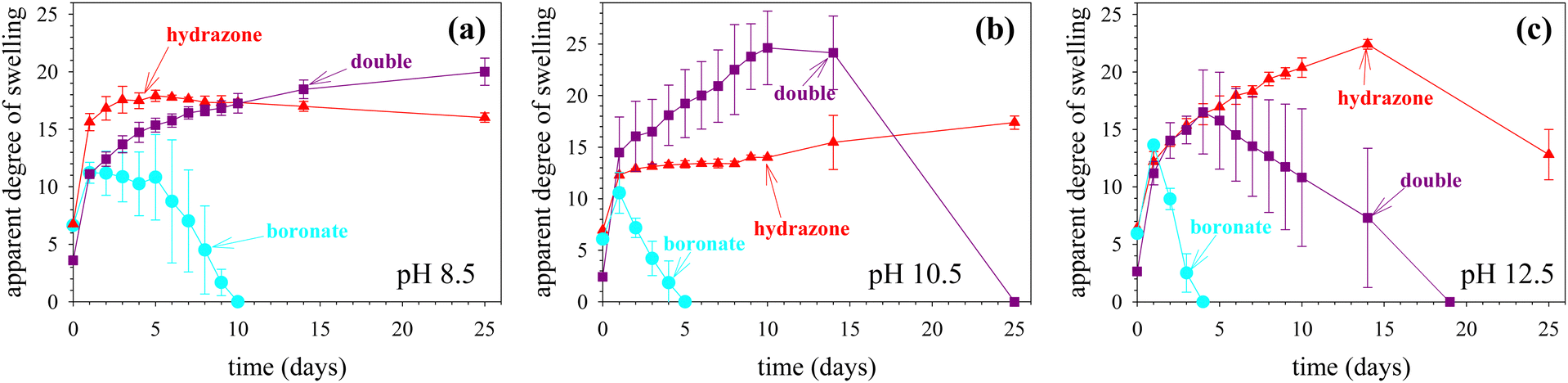 | ||
| Fig. 5 Temporal evolution of the apparent aqueous degrees of swelling of the three types of hydrogels, each prepared at three different pH values, (a) 8.5, (b) 10.5 and (c) 12.5, and equilibrated in an aqueous buffer of pH the same as that at its preparation. | ||
The time of complete gel dissolution for the six curves (also presenting a maximum) was taken from Fig. 5 and is listed in Table 1. For the three remaining samples, no gel dissolution was observed within the timespan of the observation of ∼50 days. In the same table, we also list the value of the maximum apparent degree of swelling observed. For the two samples not presenting a clear swelling maximum, the apparent swelling degree on the last day of the measurement is taken instead. For each hydrogel type, the hydrogel dissolution times in Table 1 decrease as the pH increases. This is due to the increased instability of both the acylhydrazone23 and boronate24 bonds as the pH increases above 8.5. At a given pH value, the dissolution times for the “hydrazone” cross-linked hydrogels are much longer than the corresponding values for the “boronate”-cross-linked hydrogels, consistent with the higher stability of the hydrazone bond, whereas the dissolution times for the “double” hydrogels are expectedly intermediate between those of the two single counterparts.
| Dissolution time (days) | Maximum degree of swelling | |||||
|---|---|---|---|---|---|---|
| pH | Boronate | Double | Hydrazone | Boronate | Double | Hydrazone |
| 8.5 | 10 | >50 | >50 | 11.2 ± 0.9 | 20.0 ± 1.2 | 17.9 ± 0.5 |
| 10.5 | 5 | 25 | >50 | 10.6 ± 2.0 | 24.6 ± 3.6 | 17.4 ± 0.6 |
| 12.5 | 4 | 19 | 35 | 13.6 ± 0.4 | 16.3 ± 3.7 | 22.4 ± 0.4 |
The values of the maximum apparent degrees of swelling are also listed in Table 1 and their relative values can be explained using similar arguments to those used for the dissolution times. The “boronate”-cross-linked gels present the lowest values of maximum apparent degrees of swelling, ∼11–14, because these samples degrade fast, faster than the rate of solvent absorption, thereby not being capable of attaining equilibrium swelling; an estimation of the equilibrium degree of swelling may be ∼17–18, i.e., the maximum apparent degrees of swelling of the non-degrading “hydrazone”-cross-linked hydrogels, discussed next. At the other extreme, the “hydrazone”-cross-linked gels present high maximum apparent degrees of swelling because they do not degrade at all or they degrade very slowly. Finally, the “double” hydrogels, containing both types of dynamic covalent cross-links, display the highest values of maximum degrees of swelling at two out of the three pH values of the study, 8.5 and 10.5, probably a result of the slow degradation, where the partially degraded gels can absorb more solvent due to their decreasing cross-linking density.
Self-healing capabilities of the hydrogels
As mentioned in the introduction, one of the most important properties of hydrogels cross-linked with dynamic covalent bonds is their self-healing capability, thereby extending their service life and reducing environmental impact from early disposal. To this end, all nine hydrogels, arising from the three hydrogel types, each prepared at three different pH values, were tested for their ability to self-mend. For each hydrogel, two samples were employed, one colored with the addition of a small amount of a dye to the reaction mixture of the polymer solution before gel formation. Each sample was subsequently cut in half, and two halves, one from each of the two different samples, were brought together and slightly compressed against each other by placing them in a glass vial with the same dimensions as that where gel preparation was performed. The results of these experiments are shown in Fig. 6 below, showing pictures of the two cut samples for each hydrogel before and after the attempted self-healing procedure. | ||
| Fig. 6 Self-healing tests performed on the three hydrogel types, each prepared at three different pH values, 8.5, 10.5 and 12.5, without the addition of catalyst. | ||
It can be seen from the figure that all “boronate”-cross-linked hydrogels self-healed. This can be attributed to the high reversibility of the boronic ester bond at the three pH values employed, as all three were above the pKa value of the phenylboronic acid of 7.6.25 In contrast, none of the “hydrazone”-cross-linked hydrogels could self-heal due to the low reversibility of the hydrazone bond in this mildly alkaline to alkaline pH range used.17 On the other hand, all the “double” hydrogels exhibited self-healing capability because of the presence of the boronate reversible cross-links as well. More quantitative self-healing tests were also performed on selected samples, as these are described in the end of the two following sections of the manuscript, “Viscoelastic Properties” and “Compressive Mechanical Properties”.
Viscoelastic properties of the hydrogels
The study of the viscoelastic properties of dynamic hydrogels is very important for understanding their reversibility and self-healing behavior. We studied these properties for all nine hydrogels using dynamic rheology and measuring the shear storage modulus, G′, and the shear loss modulus, G′′, as a function of the angular frequency, ω. Fig. 7 plots the values of the two shear moduli, G′ and G′′, against angular frequency, ω. With the exception of the “boronate” and “double” hydrogels at pH 8.5, the remaining seven hydrogels exhibit G′ values which are frequency independent, consistent with a totally elastic behavior within the employed angular frequency range. In contrast, the two boronate-containing hydrogels (“boronate” and the “double” hydrogels) at pH 8.5 display G′ values which increase with ω in the lower ω region, levelling off in the higher ω range. This is consistent with a hybrid behavior, with more viscous characteristics at low frequencies, and more elastic nature at higher frequencies. | ||
| Fig. 7 Viscoelastic properties of the three hydrogel types, (a) “boronate”, (b) “double” and (c) “hydrazone”, each prepared at three different pH values, 8.5, 10.5 and 12.5, studied by dynamic rheology. It is noted that G′′ values lower more than 100 compared to the corresponding G′ values (mainly the case in part (c) of the figure) are of limited accuracy and should, therefore, be considered as rough estimations of G′′. | ||
In contrast to the two types of frequency-dependence of the G′ values, three types of frequency-dependence are observed with the G′′ values. The first type concerns the three “hydrazone”-cross-linked hydrogels which present almost constant G′′ values (i.e., frequency-independent), indicating little or no viscous behavior, which is also consistent with the trends in their corresponding G′ values discussed in the previous paragraph above. The second type concerns the other two hydrogels, “boronate” and “double”, at the two higher pHs, 10.5 and 12.5, in which G′′ appears to decrease linearly as the frequency increases in the double-logarithmic plots. This suggests an increasingly viscous behavior as frequency is reduced, revealing that the samples display reversible characteristics at longer timescales, matching the time required for the dynamic cross-link to open up. However, as it will be explained in the following, the behavior in this region is still dominated by elastic rather than viscous character. Finally, the third type concerns the “boronate” and “double” hydrogels at pH 8.5, whose G′′ values exhibit a maximum with frequency. The frequency value at which G′′ is maximized, ωmax, delineates the regions of mainly viscous (for ω < ωmax) and mainly elastic (for ω > ωmax) behavior. This hybrid viscous/elastic behavior shown by the G′′ vs. ω profiles of these two hydrogels is also consistent with their G′ vs. ω profiles discussed in the previous paragraph.
Importantly, the G′ vs. ω curves intersect (or “cross with”; that is the reason why ωmax is frequently called ωcrossing) the G′′ vs. ω curves at (or near) ωmax, which is the characteristic relaxation frequency, the inverse of which can give the relaxation time. It is noteworthy that the second type of G′′ vs. ω profile is expected to also intersect the corresponding G′ vs. ω profile at a frequency lower than those studied herein. In fact, such an extrapolation (called superposition) can be employed to determine the projected intersection point and the relaxation frequency ωmax. For the two boronate-containing hydrogels at pH 8.5, the intersection of the curves of the two moduli takes place within the studied frequency range. The determined ωmax values are 2.31 and 0.339 rad s−1, corresponding to relaxation times, τ (=2π/ωmax), equal to 2.72 and 18.52 s, for the “boronate”-cross-linked and “double” hydrogels, respectively. The longer relaxation time for the “double” hydrogel compared to the “boronate” can be attributed to the higher polymer concentration and the co-presence of the more stable acylhydrazone cross-links in the “double” hydrogel.
As the G′ values in Fig. 7 are either frequency-independent or level off at a sufficiently high frequency, one may extract from the figure these high-frequency (“plateau”) shear storage modulus values and compare them with the theoretical ones. This is done in Table 2 which lists both the experimental, at the three hydrogel formation pH values, for all three network types, i.e., the two “single” (“boronate” and “hydrazone”) and the “double” networks, and the corresponding theoretical ones calculated from the phantom network model.26 Expectedly, the experimental G′ plateau values are lower (by a factor of 2.5–3.5, for most cases) than the corresponding experimental values. This difference can be attributed to incomplete or/and reversible end-linking reactions, and secondary or higher loop formation. The fair proximity of the experimental “plateau” values to the theoretical ones may suggest that the G′ plateau values from rheology are fairly consistent with the network (expected) structure.
| Experimental shear storage modulus, G′ (kPa) | Theoretical shear storage modulus, G′ (kPa) | |||
|---|---|---|---|---|
| Network type | pH = 8.5 | pH = 10.5 | pH = 12.5 | |
| “boronate” | 15 | 7.1 | 11 | 37 |
| “hydrazone” | 3.2 | 9.6 | 14 | 37 |
| “double” | 34 | 30 | 29 | 73 |
Rheology may also be utilized to further explore the self-healing capability of the hydrogels. To this end, we performed large amplitude oscillatory shear tests (“strain sweeps”) on the two self-mendable network types, the “boronate” and the “double”, focusing our attention on those prepared at pH = 8.5, where these materials have the most liquid-like behavior, so as to better show their self-repair, and also identify any differences between the two network types regarding this self-healing behavior. The results collected from these strain sweeps and subsequent hydrogel destruction followed by self-repair are plotted in Fig. 8.
 | ||
| Fig. 8 Strain sweep (parts (a) and (c)) and destruction followed by self-repair (parts (b) and (d)) rheological experiments for the “boronate” (parts (a) and (b)) and “double” (parts (c) and (d)) hydrogels, both prepared at pH 8.5. | ||
In these experiments, systematic strain sweeps were first performed to determine the critical strains at which the hydrogels would start failing (“flowing”; parts (a) and (c) of the figure, for the “boronate” and “double” hydrogels, respectively; “flow” occurs when G′′ becomes greater than G′). These experiments indicated that the less stiff “boronate” hydrogel required the higher critical strain of ∼400% to start failing, as compared to 33% for the stiffer “double” hydrogel. Knowing these critical strain values, we then equilibrated both hydrogels at the low oscillatory strain, γ, of 1%, and, subsequently, subjected each hydrogel to a step increase in strain, reaching a value of at least 20% higher than the corresponding critical strain, causing the destruction and “flow” of each hydrogel (parts (b) and (d) of the figure, for the “boronate” and “double” hydrogels, respectively). After that, we returned the strain to the low value of 1%, and waited for the recovery of each hydrogel. Both hydrogels fully recovered their initial elastic shear modulus values, with the recovery time for the stiffer “double” hydrogel being expectedly shorter, at ∼3 min, than that of the less stiff “boronate” hydrogel whose full recovery time was ∼9 min.
Compressive mechanical properties of the hydrogels
The mechanical properties of hydrogels are very important for their efficient use in various biomedical or technological applications. Although most hydrogels are infamously fragile, usually due to spatial inhomogeneities arising from uneven distribution of cross-links within the hydrogel volume,10 relatively recent research14 has shown that preparing hydrogels from large, well-defined building blocks, such as star polymers, and then end-linking them, can lead to mechanically robust materials, even when highly swollen in water.27 This was the reason why we chose tetraPEG stars as building blocks for our hydrogels in the present work. Fig. 9 and 10 show representative stress–strain curves in compression of the nine hydrogel samples, and the mechanical properties calculated from these curves, respectively. These mechanical properties are the elastic modulus, E, the stress at break, σmax, and the strain at break, εmax.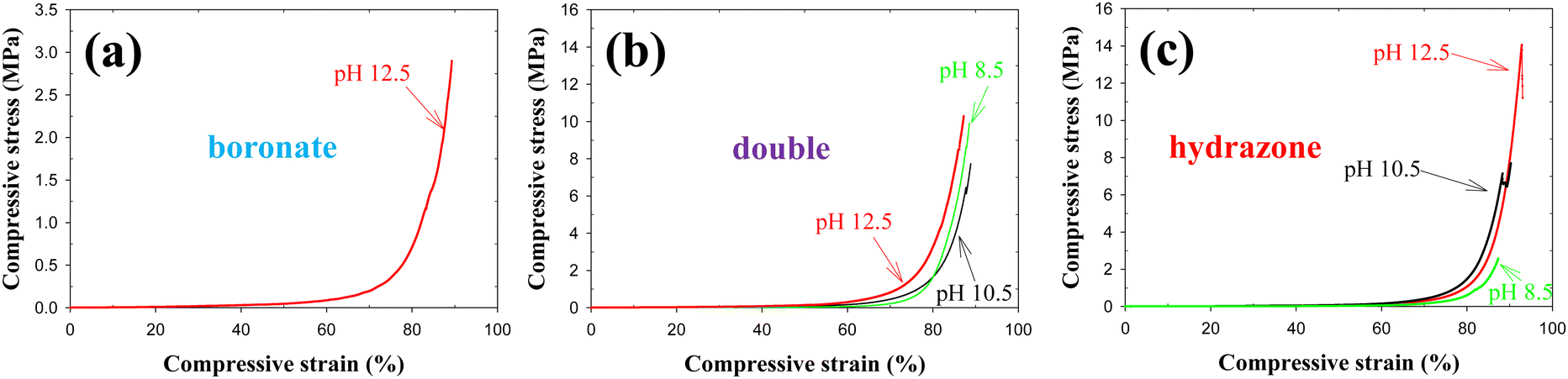 | ||
| Fig. 9 Representative stress–strain curves in compression for the three hydrogel types, “boronate”, “double” and “hydrazone”, prepared at the three different pH values, 8.5, 10.5 and 12.5. | ||
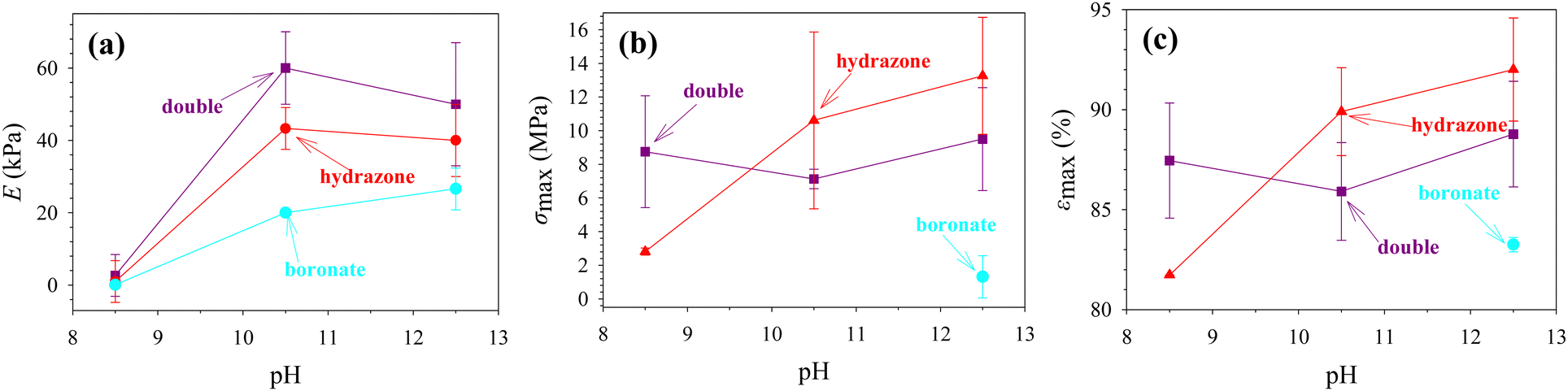 | ||
| Fig. 10 Compressive mechanical properties, (a) elastic modulus, E, (b) stress at break, σmax, and (c) strain at break, εmax, of the three hydrogel types, boronate, hydrazone and double, each prepared at three different pH values, 8.5, 10.5 and 12.5. | ||
At all three pHs, the values of the elastic moduli, E, decrease in the order: “double” > “hydrazone” > “boronate”. The highest values exhibited by the “double” hydrogels are due to the fact that we chose that each “double” hydrogel contains the same concentration of boronate and hydrazone cross-links as in the respective “boronate” and “hydrazone” hydrogels. This resulted in twice as high overall cross-link concentration in the “double” hydrogel relative to the two above-mentioned hydrogels (and also twice as high polymer concentration). The lower values of elastic modulus displayed by the “boronate”-cross-linked hydrogels as compared to the “hydrazone”-cross-linked hydrogels can be attributed to the greater reversibility of the boronate bond than the hydrazone bond.
The elastic modulus of the “boronate” hydrogels gradually increases with the pH. This is because the boronate bond becomes less reversible as the pH value rises above the pKa value of the boronic acid of 7.6.25 On the other hand, the elastic modulus of the “double” hydrogel presents a maximum with respect to pH (although the error bars of the measurements are relatively large especially at the two highest pH values). The low value of the elastic modulus of the “double” hydrogel at pH 8.5 can be attributed to the boronate bonds which become more reversible at this pH, whereas the relatively low value of the elastic modulus at pH 12.5 is due to the reversibility increase of the hydrazone bond, which is also the reason why the “hydrazone” hydrogel also exhibits a slight decrease in its elastic modulus upon the increase in pH from 10.5 to 12.5. Interestingly, one may observe in the figure an approximately additive relationship between the elastic moduli of the “double” hydrogel with the “boronate” and “hydrazone” constituent hydrogels because the “double” hydrogel is essentially the product of “adding” the two of them together. Finally, upon comparing the values of the elastic moduli, E, plotted in Fig. 10(a) with the values of the plateau shear storage moduli, G′, listed in Table 2, one may conclude that the well-known relationship E ∼ 3 G′ approximately holds, with the exception of all three samples prepared at pH 8.5 which are “too liquid”. The agreement is particularly good for the “boronate” cross-linked samples prepared at pHs 10.5 and 12.5, and satisfactory for the four other samples prepared at these two pHs, particularly if one takes into account the sizable error bars in Fig. 10(a).
Next, we discuss together the stress and strain at break of all samples. We must first point out that it was not possible to determine the values of the stress and strain at break for the “boronate”-cross-linked hydrogels at pH 8.5 and 10.5 because those samples did not fracture but rather deformed plastically. This was not the case for the “boronate”-cross-linked hydrogel at pH 12.5, which did present fracture. The former hydrogels do not fracture because of the high reversibility of the boronate bonds at pH 8.5 and 10.5, and in which the boronate cross-links exchange very fast, with exchange/relaxation times of 2.72 and 314 s, well within the timeframe of the compressive experiment, thereby immediately mending any incurring hydrogel damage in the course of the compression. The stress at break for the “boronate”-cross-linked hydrogel at pH 12.5 was lower than the corresponding values for the two other hydrogels at the same pH because of their decreased reversibility leading to hydrogels with more permanent cross-links. The stress at break values for the “double” hydrogel are pH-independent, within experimental error, and these are lower than those for the “hydrazone”-cross-linked hydrogels (at the two higher pHs), which can be attributed to the contribution of the boronate bonds to the dynamics of the double hydrogel. Nonetheless, all the hydrogels are very robust, most of them displaying stress at break values within the range between 7 and 13 MPa. Finally, the trends in the strain at break behavior of the three hydrogel types versus pH are identical to those observed in the stress at break, and, therefore, similar explanations may apply.
It is to be noted that we unsuccessfully attempted several times to perform tension experiments on the hydrogels using the same mechanical testing instrument, which were always hampered by sample slippage from the grasps of the instrument. Thus, we tried to perform tension experiments in a “more qualitative” (“semi-quantitative”) way, in the sense that we held the samples using our fingers and pulled them to verify their extensibility and approximately determine their tensile strain at break, missing to determine their tensile stress at break and tensile elastic modulus. These “semi-quantitative” experiments were performed both on virgin and self-healed samples, focusing on the two self-healable samples prepared at pH = 10.5, choosing them over the ones at pH = 8.5 which are “too liquid”, and also preferring them to those at pH 12.5 where self-healing capability may be reduced. Our results are summarized in Table 3.
| Tensile strain at break, εmax (%) | Self-healing efficiency based on tensile strain at break (%) | ||
|---|---|---|---|
| Network type | Virgin | Healed | |
| “boronate” | 390 | 220 | 55 |
| “double” | 300 | 120 | 40 |
The results in the table above show that virgin samples from both network types displayed good extension at break, being able to be elongated about 4 (the “boronate”) and 3 (the “double”) times their initial length before failure. The less stiff “boronate” network expectedly presented the higher εmax value. Furthermore, cut samples from both network types could self-heal upon pressing the two cut surfaces against each other for 24 h. The self-healed sample of the less stiff “boronate” gel again attained a higher strain at break (220%) in comparison with the stiffer “double” counterpart (120%), with the former network type also exhibiting a higher self-healing efficiency (55%) than the latter (40%). We did not attempt to determine the self-healing efficiency via mechanical testing in compression, because we anticipated a reduced accuracy in compressive mode where the sample is self-supported, thereby making any defects or cracks more difficult to appear.
Conclusions
The “boronate”-linked hydrogels developed in this study represent the first example of well-defined dually-dynamic tetraPEG gels. These hydrogels exhibit moderate aqueous stability, disintegrating within 4–10 days, depending on the pH, display excellent self-healing capability due to their short bond lifetimes, and show mechanical properties which improve only at the highest investigated pH of 12.5. On the other hand, the “hydrazone”-linked hydrogels, also investigated herein, exhibit excellent aqueous stability, poor self-healing capability, and very good mechanical properties. The “double” hydrogels expectedly display properties intermediate between the two above-mentioned hydrogels, as they are their hybrids. In particular, these hybrid hydrogels present good aqueous stability (compared to the moderate stability of the “boronate”), good self-healing capability (compared to non-self-healable “hydrazone”), and good mechanical properties. This study opens the way for the macromolecular engineering of more hybrid materials, where the ratio of the “boronate” and “hydrazone” components is changed, so as to optimize the property of interest, whether aqueous stability, self-healing capability, and mechanical properties, while keeping the remaining properties at acceptable levels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Regional Development Fund and the Republic of Cyprus are acknowledged for co-funding this research work through The Research and Innovation Foundation of Cyprus (Project Number: Excellence/0918/0325). The same organizations are also thanked for funding projects ANAVATHMISI/0609/12 and NEKYP/0308/02, which enabled the purchase of the rheometer and the NMR spectrometer (500 MHz), respectively, at the University of Cyprus, used in this investigation. Finally, we thank our colleague Prof. E. Leontidis of the Chemistry Department at the University of Cyprus for kindly providing access to his rheometer used in our experiments.References
- C. K. Varnava and C. S. Patrickios, Polymer networks one hundred years after the macromolecular hypothesis: A tutorial review, Polymer, 2021, 215, 1223322 CrossRef.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic covalent chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- J.-M. Lehn, Dynamers: Dynamic molecular and supramolecular polymers, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- J.-M. Lehn, From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Self-healing gels based on constitutional dynamic chemistry and their potential applications, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S. Y. An, D. Arunbabu, S. M. Noh, Y. K. Song and J. K. Oh, Recent strategies to develop self-healable crosslinked polymeric networks, Chem. Commun., 2015, 51, 13058–13070 RSC.
- A. W. Jackson and D. A. Fulton, Making polymeric nanoparticles stimuli-responsive with dynamic covalent bonds, Polym. Chem., 2013, 4, 31–45 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Synthesis and applications of boronic acid-containing polymers: From materials to medicine, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- D. E. Apostolides and C. S. Patrickios, Dynamic covalent polymer hydrogels and organogels cross-linked through acylhydrazone bonds: Synthesis, characterization, and applications, Polym. Int., 2018, 67, 627–649 CrossRef CAS.
- M. Shibayama, Spatial inhomogeneity and dynamic fluctuations of polymer gels, Macromol. Chem. Phys., 1998, 199, 1–30 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double-network hydrogels with extremely high mechanical strength, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Y. Okumura and K. Ito, The polyrotaxane gel: A topological gel by figure-of-eight cross-links, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Nanocomposite hydrogels: A unique organic–inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U.-I. Chung, Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Injectable self-healing glucose-responsive hydrogels with pH-regulated mechanical properties, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- V. Yesilyurt, A. M. Ayoob, E. A. Appel, J. T. Borenstein, R. Langer and D. G. Anderson, Mixed reversible covalent crosslink kinetics enable precise, hierarchical mechanical tuning of hydrogel networks, Adv. Mater., 2017, 29, 1605947 CrossRef PubMed.
- D. E. Apostolides, T. Sakai and C. S. Patrickios, Dynamic covalent star poly(ethylene glycol) model hydrogels: A new platform for mechanically robust, multifunctional materials, Macromolecules, 2017, 50, 2155–2164 CrossRef CAS.
- Y. Liu, Y. Liu, Q. Wang, Y. Han, H. Chen and Y. Tan, Doubly dynamic hydrogel formed by combining boronate ester and acylhydrazone bonds, Polymers, 2020, 12, 487 CrossRef CAS PubMed.
- S. Ren, P. Sun, A. Wu, N. Sun, L. Sun, B. Dong and L. Zheng, Ultra-fast self-healing PVA organogels based on dynamic covalent chemistry for dye selective adsorption, New J. Chem., 2019, 43, 7701–7707 RSC.
- D. E. Apostolides and C. S. Patrickios, Model dynamic covalent organogels based on end-linked three-armed oligo(ethylene glycol) star macromonomers, J. Polym. Sci., 2021, 59, 2309–2323 CrossRef CAS.
- N. R. Richbourg and N. A. Peppas, The swollen polymer network hypothesis: Quantitative models of hydrogel swelling, stiffness, and solute transport, Prog. Polym. Sci., 2020, 105, 101243 CrossRef CAS.
- S. P. O. Danielsen, H. K. Beech, S. Wang, B. M. El-Zaatari, X. Wang, L. Sapir, T. Ouchi, Z. Wang, P. N. Johnson, Y. Hu, D. J. Lundberg, G. Stoychev, S. L. Craig, J. A. Johnson, J. A. Kalow, B. D. Olsen and M. Rubinstein, Molecular characterization of polymer networks, Chem. Rev., 2021, 121(8), 5042–5092 CrossRef CAS PubMed.
- J. M. Sayer and W. P. Jencks, General base catalysis of thiosemicarbazone formation, J. Am. Chem. Soc., 1969, 91(23), 6353–6361 CrossRef CAS.
- D. Wu, W. Wang, D. Diaz-Dussan, Y.-Y. Peng, Y. Chen, R. Narain and D. G. Hall, In situ forming, dual-crosslink network, self-healing hydrogel enabled by a bioorthogonal nopoldiol–benzoxaborolate click reaction with a wide pH range, Chem. Mater., 2019, 31(11), 4092–4102 CrossRef CAS.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, The relationship among pKa, pH, and binding constants in the interactions between boronic acids and diols - it is not as simple as it appears, Tetrahedron, 2004, 60(49), 11205–11209 CrossRef CAS.
- M. Zhong, R. Wang, K. Kawamoto, B. D. Olsen and J. A. Johnson, Quantifying the impact of molecular defects on polymer network elasticity, Science, 2016, 353(6305), 1264–1268 CrossRef CAS PubMed.
- S. Nakagawa and N. Yoshie, Star polymer networks: A toolbox for cross-linked polymers with controlled structure, Polym. Chem., 2022, 13(15), 2074–2107 RSC.
| This journal is © The Royal Society of Chemistry 2022 |