DOI:
10.1039/C9QM00586B
(Research Article)
Mater. Chem. Front., 2019,
3, 2746-2750
Utilizing the aggregation-induced emission phenomenon to visualize spontaneous molecular directed motion in the solid state†
Received
18th September 2019
, Accepted 10th October 2019
First published on 12th October 2019
Abstract
The real-time monitoring of spontaneous molecular directed motion is a highly important but very challenging task. In this work, a rod-like aggregation-induced emission (AIE) molecule was carefully designed and facilely synthesized. The AIE molecule, the salicylaldehyde 4-butoxyaniline Schiff base (SBA), exhibited a unique self-recovery property from a semi-ordered structure to an ordered structure along with significant fluorescence changes after grinding. The fluorescence changes were monitored to obtain important kinetic information regarding the spontaneous molecular directed motion process, including the kinetic order, rate constants, half-life, and apparent activation energy. Unlike instrumental analytical methods such as PXRD and AFM, which only give information on a stable state of samples, the proposed fluorescence method provides a new perspective for real-time visualization of spontaneous molecular directed motion in situ in the solid state.
Introduction
Spontaneous molecular directed motions induced by intermolecular interactions such as hydrogen bonding, π–π interactions, C–H⋯π interactions and hydrophobic interactions are an important driving force for molecular self-assembly, and are ubiquitous in various biochemical pathways such as DNA/RNA pairing, enzyme folding/unfolding, membrane transport and molecular recognition.1–6 Thus, the observation and measurement of spontaneous molecular directed motion are important for revealing life processes. However, due to the random nature of molecular motions, their characterization remains a challenging task.
In recent years, monitoring changes in fluorescence has been demonstrated to be an effective method to visualize various chemical and physical processes.7–9 For example, small molecule aggregation processes, the macromolecule cononsolvency effect, polymer glass transitions and gelation processes have been successfully visualized in real-time in situ by using different luminescent molecules.10–13 Recently, Prof. Tang and co-workers designed and synthesized a new aggregation-induced emission (AIE) molecule with self-recovery properties.14 Due to the intense and adjustable luminescence property of the AIE molecule in the solid state, the spontaneous molecular motion of the AIE molecule from the amorphous state to the crystal state induced by π–π interactions was visualized. However, the molecular motion is random and nondirected, making it difficult to fully understand and characterize it. Especially, kinetic information of the molecular motion was barely investigated. To date, the visualization of spontaneous molecular directed motion in the solid state is still not realized.
In order to visualize spontaneous molecular directed motions in the solid state, a new AIE molecule, the salicylaldehyde 4-butoxyaniline Schiff base (SBA), was deliberately prepared in this work. SBA was designed to consist of two parts, a salicylaldehyde aniline Schiff base part and a butoxy part, as illustrated in Scheme 1. The salicylaldehyde aniline Schiff base is a typical AIE group, which is expected to endow SBA with visible luminescence changes after stimulating with mechanical force such as grinding.15–18 The butoxy group has strong hydrophobic interactions, and can facilitate the self-recovery of SBA in the solid state.19–21 Moreover, inspired by the design strategy of rod-like liquid crystal molecules which can undergo directional alignment in the mesogenic state, the butoxy group was attached in the para-position of the N atom, resulting in a rod-like structure for SBA. This structural feature is expected to help SBA maintain a semi-ordered spatial arrangement even after grinding.
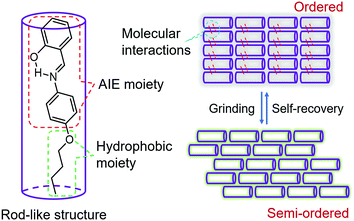 |
| | Scheme 1 Design strategy of SBA. | |
Results and discussion
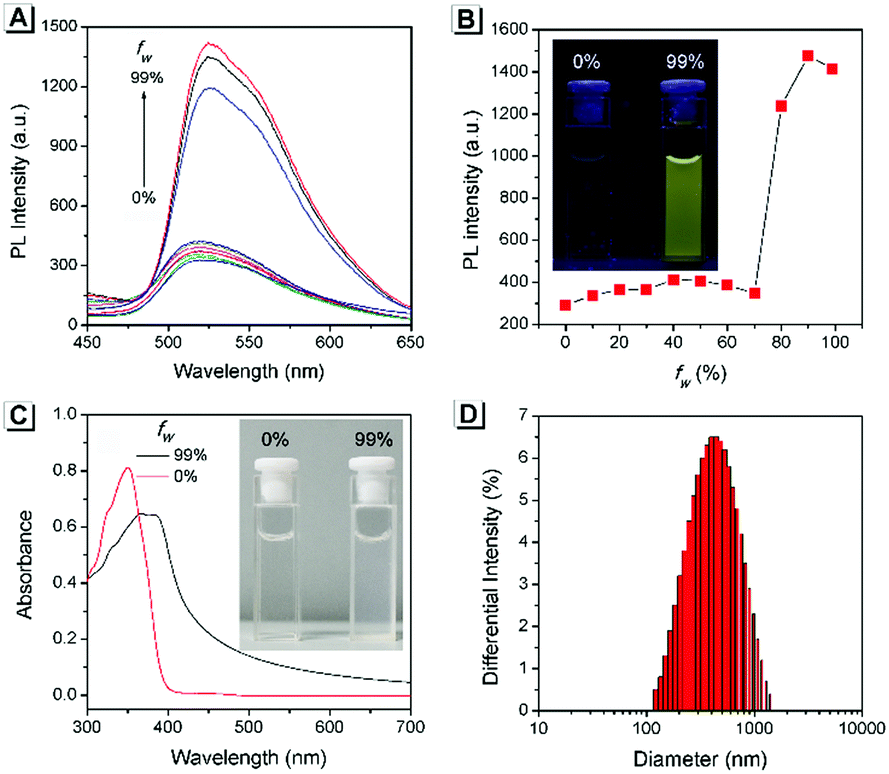
The AIE property of SBA was first investigated in EtOH/H2O mixtures with different water fractions (fw). As shown in Fig. 1A and B, SBA exhibited weak fluorescence in the good solvent of EtOH but intense fluorescence in the poor solvent of H2O, suggesting its AIE feature.22–25 The aggregation of SBA in H2O was confirmed by absorption spectra and dynamic light scattering (DLS). In EtOH, SBA exhibited flat baselines in the absorption spectrum over 400 nm. In contrast, a level-off tail in the absorption spectrum was observed in H2O, which could be attributed to the light scattering of the aggregated suspensions (Fig. 1C).26–28 Moreover, DLS results suggested that nano-particles with a diameter around 500 nm existed in H2O but no particles were observed in EtOH (Fig. 1D). The above results demonstrated that the intense fluorescence of SBA in aqueous solution originated from aggregation, i.e., AIE fluorescence.
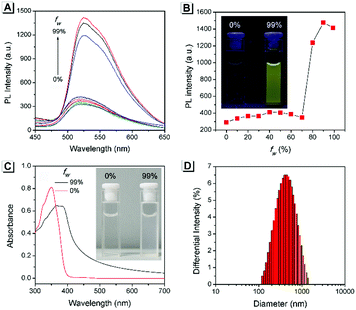 |
| | Fig. 1 (A) Fluorescence spectra of SBA in water/ethanol mixtures with different fw. (B) Fluorescence intensity of SBA at 525 nm as a function of fw. (C) Absorption spectra of SBA in water/ethanol mixtures with fw of 0% and 99%. (D) DLS result of SBA in water/ethanol mixtures with an fw of 99%. Inset: A photo of SBA in water/ethanol mixtures with fw of 0% and 99%; (B) under UV light irradiation and (C) under daylight. | |
The self-recovery property of SBA in the solid state can be visually observed through the change in its fluorescence. As shown in Fig. 2A, SBA exhibited almost no fluorescence in the solid state. After grinding, intense but transitory green emission around 525 nm was observed, which disappeared within 1 min at room temperature. Meanwhile, no obvious changes were observed in the UV-vis diffuse reflectance spectra, suggesting that no new substance was formed after grinding (Fig. 2B). The emission turn-on property by grinding and fast self-recovery was highly reversible. As shown in Fig. 2C, after repeatedly toggling between the ground and unground states (10 times), the fluorescence of SBA at 525 nm remained almost constant without degradation, indicating its good fatigue resistance.
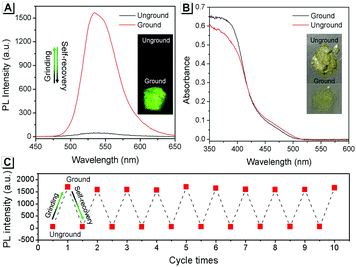 |
| | Fig. 2 (A) Fluorescence and (B) UV-vis diffuse reflectance spectra of SBA before and after grinding. (C) Fatigue resistance of the fluorescence intensity of SBA during 10 grinding/self-recovery cycles. All spectra were recorded at 0 °C. | |
To understand these phenomena, single-crystal X-ray diffraction (SCXRD) analysis was performed. The results confirmed that SBA was a rod-like molecule in the crystal, and the dihedral angle between two conjugate planes was 49.0° (Fig. 3A). The molecules were distributed in layers. Inside the layers, the molecules were arranged in “head-to-head” and “tail-to-tail” fashion (Fig. 3B and C). The minimum distance between a hydrogen atom and an adjacent benzene ring in another layer was about 2.9 Å, indicating that strong C–H⋯π interactions existed between the layers (generally, the necessary condition for C–H⋯π interactions is that the distance between the hydrogen atom and the adjacent benzene ring is smaller than 3.0 Å29–31). The existence of C–H⋯π interactions possibly facilitates the non-radiative transition of electrons in the excited state, which might be the primary reason for the fluorescence quenching in the crystal state. Under mechanical force, the characteristic stacked structure and C–H⋯π interactions were destroyed, resulting in intense green emission. This proposed mechanism for the fluorescence turn-on of SBA after grinding is shown in Scheme 1.
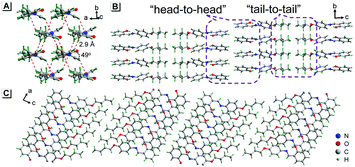 |
| | Fig. 3 Crystal structure of SBA viewed from different directions. | |
To provide support for this proposed mechanism, contrast experiments were carried out. Two control compounds were synthesized having shorter carbon chains compared to SBA, namely the salicylaldehyde 4-ethyoxyaniline Schiff base (SEA) and the salicylaldehyde 4-propoxyaniline Schiff base (SPA) (Scheme S1, ESI†). The crystal structures of SEA (Fig. S2, ESI†) and SPA (Fig. S3, ESI†) were very similar to that of SBA, and the SEA and SPA molecules were also arranged head-to-head and tail-to-tail in layers in their crystals, respectively. However, the fluorescence changes of SEA and SPA before and after grinding were quite different compared to SBA (Fig. 4A). For SEA, there was barely any change in emission after grinding. For SPA, only 1.5-fold emission enhancement was observed after grinding. For SBA, the emission enhancement after grinding was as high as 35-fold compared to SBA without grinding. These results confirmed that shorter carbon chains could not effectively insulate the salicylaldehyde aniline Schiff base moieties in adjacent molecules after grinding, resulting in weaker fluorescence changes (Fig. 4B).
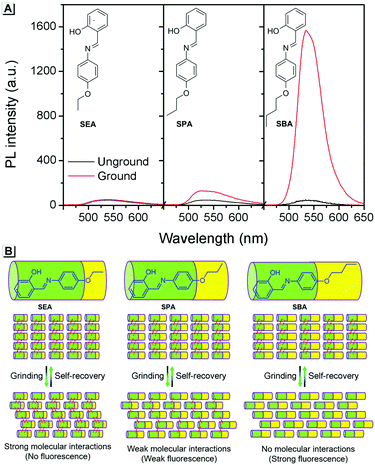 |
| | Fig. 4 (A) Fluorescence spectra of SEA, SPA and SBA before and after grinding. (B) Proposed mechanism for the fluorescence changes. | |
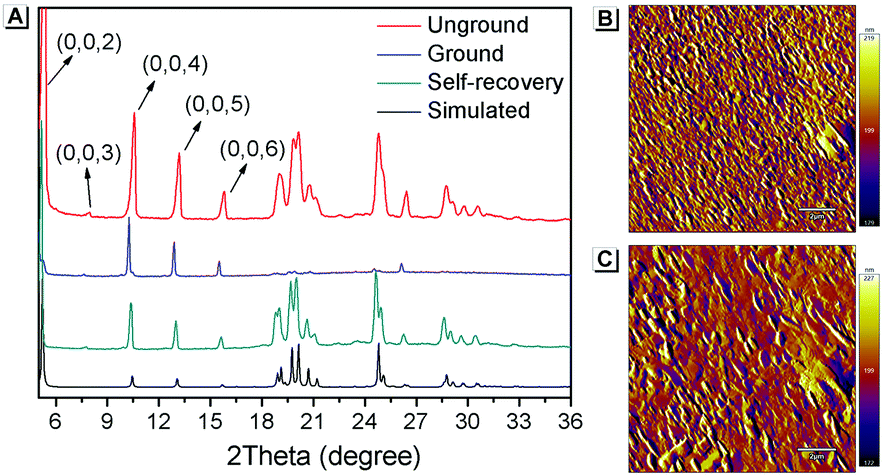
Furthermore, PXRD measurements were carried out at 0 °C to investigate the structural changes in SBA before and after grinding. At this temperature, the fluorescence emission of ground SBA is relatively stable (the temperature-dependent recovery rate will be discussed in detail later). As shown in Fig. 5A, the PXRD patterns of unground SBA fitted well with the simulated one, demonstrating its uniform crystal state. Surprisingly, although most of the diffraction peaks decreased and even disappeared after grinding, some peaks still remained, which indicated that SBA was not completely converted from the crystal to the amorphous form. After analysis and comparison, it was confirmed that these remaining diffraction peaks belonged to the (0, 0, n) crystal plane, indicating that the molecular arrangement was still ordered along the c-axis after grinding (i.e., a semi-ordered structure). More importantly, when the ground SBA was kept in the dark at room temperature, all the original diffraction peaks reappeared gradually, which suggested that the ordered crystal structure of SBA was recovered.32–34 This unique self-recovery process from semi-ordered structure to ordered structure along with fluorescence changes provides a visual prototype to understand spontaneous molecular directed motion in the solid state.
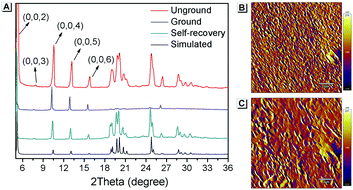 |
| | Fig. 5 (A) PXRD spectra of SBA in different states. AFM patterns of ground SBA (B) before and (C) after self-recovery. The scale bar is 2 μm. | |
Atomic force microscopy (AFM) was used to investigate the change in morphology of SBA in different states. As shown in Fig. 5B, splintery patches were observed in the luminescent state of SBA after grinding. After tuning back to the non-luminescent state, the splintery patches disappeared and larger patches were observed in the same area (Fig. 5C). The AFM patterns suggested that the morphology of SBA became more orderly after self-recovery in the dark, which was as expected.
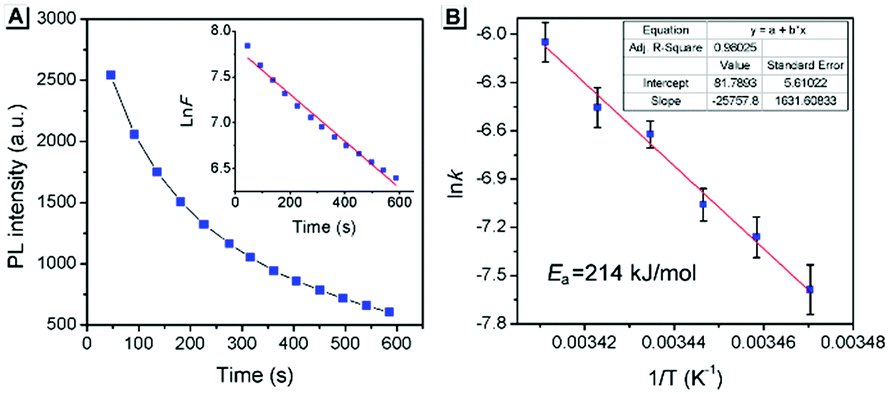
The above PXRD and AFM analyses provided useful information about the stable state of SBA, while real-time information for SBA can be obtained using the fluorescence method. Thus, time-dependent fluorescence spectra were recorded at different temperatures and kinetic analyses were carried out, which offered rich kinetic information about the spontaneous molecular directed motion. As shown in Fig. 6A, the luminescence intensity decay at 525 nm can be fitted well with first-order reaction kinetics (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) F is proportional to t, where t is time and F is the emission intensity at 525 nm), suggesting that the spontaneous molecular directed motion of ground SBA was a first-order dynamic process. The rate constant (k) and half-life of ground SBA (t1/2) were calculated according to the integral rate equation of a first-order reaction:
F is proportional to t, where t is time and F is the emission intensity at 525 nm), suggesting that the spontaneous molecular directed motion of ground SBA was a first-order dynamic process. The rate constant (k) and half-life of ground SBA (t1/2) were calculated according to the integral rate equation of a first-order reaction:
| | | −kt = lnF | (1) |
| | | t1/2 = ln2/k | (2) |
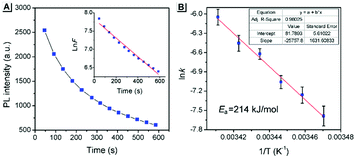 |
| | Fig. 6 (A) The thermal fading kinetics of ground SBA at 20 °C. Inset: The curves fitted with first-order reaction dynamics. (B) The rate constants obtained from different temperatures fitted with Arrhenius expressions. The inset shows the fitting parameters. | |
The calculated values of k and t1/2 from 15 °C to 20 °C are shown in Table S1 (ESI†). Based on these data, the apparent activation energy (Ea) of spontaneous molecular directed motion from the semi-ordered structure to the ordered structure was evaluated according to the Arrhenius expression:
| | | lnk = −Ea/RT + B | (3) |
where
T is the temperature,
R is the ideal gas constant and
B is a constant. As shown in
Fig. 6B, the apparent
Ea was calculated to be 214 kJ mol
−1, which is higher than most room-temperature spontaneous chemical reactions. These results indicate that intermolecular interactions provided a strong driving force for spontaneous molecular directed motion of
SBA, which can overcome the large activation barrier. Moreover, according to the Arrhenius expression, the large apparent
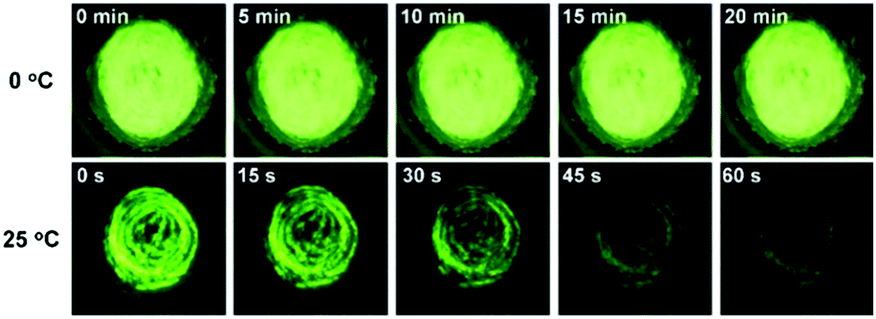
Ea suggests that the spontaneous molecular directed motion can be effectively controlled by temperature. As shown in
Fig. 7, the green emission of ground
SBA was quenched quickly at 25 °C, and it almost disappeared after keeping in the dark for 1 min. In contrast, the green emission of ground
SBA barely changed at 0 °C when it was kept in the dark for 20 min.
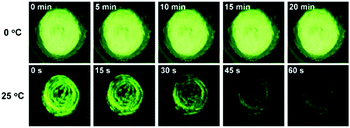 |
| | Fig. 7 Photo of ground SBA kept in the dark at 0 °C and 25 °C. | |
Furthermore, density functional theory (DFT) calculations and non-covalent interaction (NCI) analysis were performed to determine the binding energy of the molecules. The single crystal was used as the initial geometry for structural optimization. As shown in Fig. 8 and Fig. S4 (ESI†), the calculated binding energy was 52.7 kJ mol−1, which was mainly contributed by the C–H⋯π interactions between two adjacent molecules. In addition, it can be seen from the crystal structure (Fig. 3A) that each SBA molecule was surrounded by four other SBA molecules. Therefore, the total binding energy is 4 × 52.7 kJ mol−1, i.e., 211 kJ mol−1, which is in good agreement with the measured apparent Ea of spontaneous molecular directed motion from the semi-ordered structure to the ordered structure. Thus, it can be inferred that C–H⋯π interactions between adjacent molecules are the main driving force for spontaneous molecular directed motion.
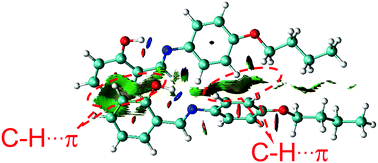 |
| | Fig. 8 The NCI analysis of SBA. | |
Conclusions
In this work, a rod-like AIE molecule of SBA was prepared, which exhibited unique self-recovery property with significant fluorescence changes after grinding in the solid state. PXRD measurements indicated that there was a directed motion of SBA molecules from a semi-ordered structure to an ordered structure. The fluorescence changes were monitored, which provided rich kinetic information regarding the spontaneous molecular directed motion process, including the kinetic order, rate constants, half-life and apparent activation energy. Furthermore, DFT calculations and NCI analysis were performed, which demonstrated that the C–H⋯π interactions between adjacent molecules were the main driving force for the spontaneous molecular directed motion of SBA. Unlike instrumental analytical methods such as PXRD and AFM, which only provide information about the stable state of samples, the proposed fluorescence method offers a new strategy for real-time visualization of spontaneous molecular directed motion in situ in the solid state.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21501150, 51502079, 21671174) and Zhongyuan thousand talents project.
Notes and references
- K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152–15160 CrossRef CAS PubMed.
- M. Harigai, M. Kataoka and Y. Imamoto, J. Am. Chem. Soc., 2006, 128, 10646–10647 CrossRef CAS PubMed.
- M. del Carmen Fernandez-Alonso, F. J. Canada, J. Jimenez-Barbero and G. Cuevas, J. Am. Chem. Soc., 2005, 127, 7379–7386 CrossRef PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- M. Minuth and C. Richert, Angew. Chem., Int. Ed., 2013, 52, 10874–10877 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2019, 14, 1677–1686 CrossRef CAS PubMed.
- N. Jiang, T. Shen, J. Z. Sun and B. Z. Tang, Sci. China Mater., 2019, 62, 1227–1235 CrossRef.
- H. Zhang, J. Z. Sun, J. Liu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Appl. Phys., 2019, 126, 050901 CrossRef.
- Y. Li, S. Liu, T. Han, H. Zhang, C. Chuah, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Mater. Chem. Front., 2019, 3, 2207–2220 RSC.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961 CrossRef.
- Z. Wang, J. Nie, W. Qin, Q. Hu and B. Z. Tang, Nat. Commun., 2016, 7, 12033 CrossRef PubMed.
- J. Xue, W. Bai, H. Duan, J. Nie, B. Du, J. Z. Sun and B. Z. Tang, Macromolecules, 2018, 51, 5762–5772 CrossRef CAS.
- Z. Qiu, E. K. K. Chu, M. Jiang, C. Gui, N. Xie, W. Qin, P. Alam, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2017, 50, 7620–7627 CrossRef CAS.
- P. Alam, N. L. C. Leung, Y. Cheng, H. Zhang, J. Liu, W. Wu, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 4536–4540 CrossRef CAS PubMed.
- W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163–2166 CrossRef CAS PubMed.
- P. Song, X. Chen, Y. Xiang, L. Huang, Z. Zhou, R. Wei and A. Tong, J. Mater. Chem., 2011, 21, 13470–13475 RSC.
- Q. Feng, Y. Li, L. Wang, C. Li, J. Wang, Y. Liu, K. Li and H. Hou, Chem. Commun., 2016, 52, 3123–3126 RSC.
- X. Zhang, J. Shi, G. Shen, F. Gou, J. Cheng, X. Zhou and H. Xiang, Mater. Chem. Front., 2017, 1, 1041–1050 RSC.
- X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS.
- S. Xue, X. Qiu, Q. Sun and W. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC.
- Y. Seong-Jun, C. Jong Won, G. Johannes, K. Kil Suk, C. Moon-Gun, K. Dongho and P. Soo Young, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. L. Wang, C. Fan, B. Xin, J. P. Zhang, T. Luo, Z. Q. Chen, Q. Y. Zhou, Q. Yu, X. N. Li, Z. L. Huang, C. Li, M. Q. Zhu and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1554–1562 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- K. Li, Y. Liu, Y. Li, Q. Feng, H. Hou and B. Z. Tang, Chem. Sci., 2017, 8, 7258–7267 RSC.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- Y. He, Y. Li, H. Su, Y. Si, Y. Liu, Q. Peng, J. He, H. Hou and K. Li, Mater. Chem. Front., 2019, 3, 50–56 RSC.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- C. Zhao, R. M. Parrish, M. D. Smith, P. J. Pellechia, C. D. Sherrill and K. D. Shimizu, J. Am. Chem. Soc., 2012, 134, 14306–14309 CrossRef CAS.
- O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on experimental procedures. A video for the self-recovery process of SBA. Additional experimental data, including crystal data, 1H-NMR spectra, 13C-NMR spectra, mass spectrometry, etc. CCDC 1941969–1941971. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00586b |
| ‡ J. Liu and C. Xing contributed equally. |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Qianqian
Deng
a,
Cuiping
Yang
a,
Qiuchen
Peng
ab,
Hongwei
Hou
a,
Qianqian
Deng
a,
Cuiping
Yang
a,
Qiuchen
Peng
ab,
Hongwei
Hou