DOI:
10.1039/C8QM00544C
(Research Article)
Mater. Chem. Front., 2019,
3, 1052-1061
An AIE fluorescent switch with multi-stimuli responsive properties and applications for quantitatively detecting pH value, sulfite anion and hydrostatic pressure†
Received
24th October 2018
, Accepted 7th January 2019
First published on 10th January 2019
Abstract
Stimuli-responsive fluorescent materials which can change their optical signal outputs upon changes in ambient physical or chemical parameters exhibit great application potential for comprehensive environmental monitoring. In this article, a new aggregation-induced emission fluorescent switch, 1',3′,3′-trimethyl-5′-(4-(1,2,2-triphenylvinyl)phenyl)spiro[chromene-2,2′-indoline]-6-carbonitrile (TPE-Sp-CN), was synthesized and applied in the quantitative detection of three parameters, namely pH value, sulfite anion and hydrostatic pressure, with different molecular states. According to the analysis of the optical spectra, TPE-Sp-CN can distinguish water pH values from 0 to 4, the detection limit of sulfite anion is 1.04 ppm, and the pressure sensing range is 1 atm to 6 GPa with the sensing sensitivity of a 9.876 nm GPa−1 shift in the PL spectrum. Furthermore, TPE-Sp-CN can quantitatively sense all three parameters with good linear relationships, which is rarely reported. Also, it is promising to use TPE-Sp-CN as an indicator to monitor the environment or disasters that are related to the above parameters, such as harmful geological activities.
1. Introduction
Stimuli-responsive fluorescent materials can change their chemical structures or packing modes upon environmental changes and provide different optical outputs. These materials have been widely investigated recently because of their extensive applications in the detection of chemicals1–4 and physical parameters,5,6 such as gases,7,8 hazardous ions,9–11 explosives,12–14 pH value,15,16 radio irradiation,17,18 temperature,19–22 and pressure.23–26 Although they possess high sensitivity, most stimuli-responsive materials can only detect one type of stimulus.27,28 In order to realize multi-parameter detection in a complicated environment, multi-stimuli responsive materials are usually fabricated by integration of several different stimuli-responsive moieties into one molecule or by tagging them to polymers, which require tedious and costly construction processes.29–32
Fluorescent molecular switches have received great attention as multi-stimuli-responsive materials.33–36 By manipulating its molecular structures, it is possible for a single molecule to respond to multiple parameters.37,38 It is obvious that the more “states” a molecular switch possesses, the more parameters it can detect.39,40 However, it is very challenging to achieve maximum numbers of readily distinguishable “states” for a small molecular switch because a very delicate design is required to avoid domination of one state over the others,41 and it is even more difficult to apply every state of the molecule to quantitatively sense chemical or physical parameters.
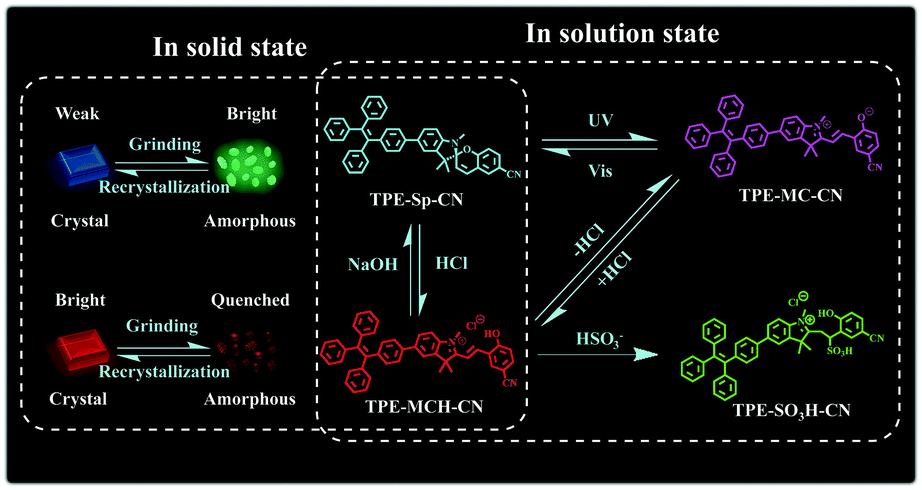
It is a feasible method to construct fluorescent molecular switches by chemically connecting fluorophores with molecular switches.42–46 Although they have been used in the field of multi-parameter sensing, fluorescent molecular switches with limited state numbers and qualitative detection still cannot fulfill the demands of comprehensive analysis of complex environments. In this article, we synthesized a new fluorescent switch based on tetraphenylethylene (TPE)-modified cyano-spiropyran (TPE-Sp-CN). Switches between multi-states have been realized with this extremely small molecule in both solution and solid states without the notorious aggregation-caused quenching effect (Scheme 1). In solution, four states of TPE-Sp-CN (ring-closed form), TPE-MCH-CN (protonated ring-open form), TPE-MC-CN (de-protonated ring-open form) and TPE-SO3H-CN (sulfite addition ring-open form) were observed, corresponding to different acidic, basic and ionic environments. In the solid state, four states of crystalline ring-open and ring-closed forms and amorphous ring-open and ring-closed forms were obtained by crystallization and grinding. Also, quantitative detection of multiple parameters of pH value, sulfite anion and hydrostatic pressure has been achieved with the different states of the molecules. Therefore, this work makes full use of every fragment of the molecule to push the limits of molecular structural modulation in sensing and provides a new material for multi-parameter sensing and detection. This is promising for comprehensive analysis of complicated environments, such as monitoring underground water systems to estimate the activity level of geological activities which are related to the three parameters.47–49
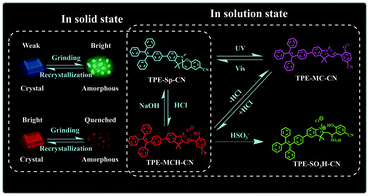 |
| | Scheme 1 Illustration of the switching states of TPE-Sp-CN with different stimuli. | |
2. Results and discussion
2.1. Detection of pH value
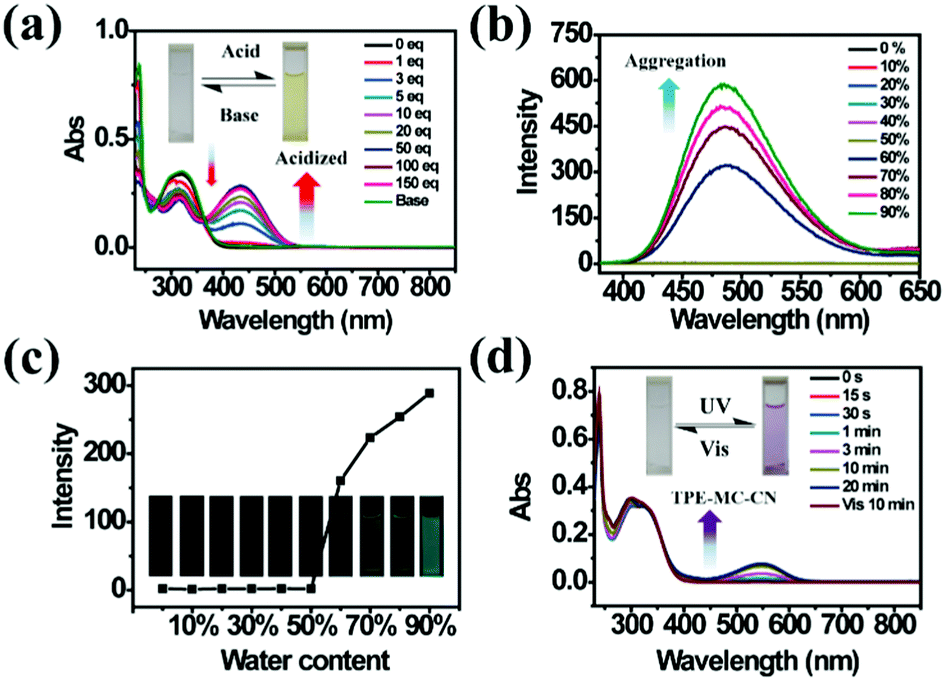
As a famous molecular switch, spiropyran (SP) has been researched for decades due to its reversible isomerization between the SP and MC (merocyanine)/MCH (protonated merocyanine) forms upon UV-Vis or acid–base stimulation. Here, TPE-Sp-CN also shows an acid response in solution (Fig. 1a). Upon gradual addition of hydrochloric acid to a TPE-Sp-CN acetonitrile solution (1 × 10−5 M), a transition from TPE-Sp-CN (abs: 238 nm and 320 nm) to TPE-MCH-CN (abs: 436 nm) occurred, and 50 equivalents of HCl were sufficient to achieve complete structural transition. Meanwhile, TPE-MCH-CN was deprotonated and returned to TPE-Sp-CN when sufficient NaOH was added. Then, we explored whether this acid–base response can be applied in pH sensing.
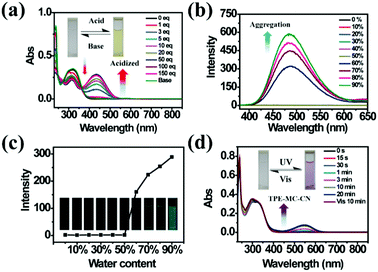 |
| | Fig. 1 (a) Absorption spectra of TPE-Sp-CN in acetonitrile solution (1 × 10−5 M) before and after addition of different equivalents of HCl followed by treatment with sufficient NaOH. (b) PL spectra of TPE-Sp-CN (1 × 10−5 M) in mixtures with different volume fractions of MeCN/H2O, and (c) corresponding plot of the fluorescence intensity changes with water content at 485 nm. (d) Absorption spectra of TPE-Sp-CN in methanol/acetonitrile (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixed solution with different exposure times to 254 nm hand-held UV light followed by visible light. Insets: Pictures of the corresponding TPE-Sp-CN solutions. :1) mixed solution with different exposure times to 254 nm hand-held UV light followed by visible light. Insets: Pictures of the corresponding TPE-Sp-CN solutions. | |
In order to determine the appropriate sensing conditions for pH values (such as the volume fractions of solvent and water), AIE experiments with TPE-Sp-CN and TPE-MCH-CN were performed in an acetonitrile/water system. Due to the non-irradiation transition of TPE, neither TPE-Sp-CN nor TPE-MCH-CN is emissive in acetonitrile solution (Fig. S1, ESI†). For TPE-Sp-CN, aggregates began to form at 60% water content accompanied with red shifts of the absorption spectra (Fig. S2, ESI†), and an emission appeared at 485 nm (Fig. 1b). Meanwhile, the intensity of the bluish green emission increased with increasing water fraction (Fig. 1c). For TPE-MCH-CN (formed by HCl treatment), we found that the water content and HCl equivalents have a synergetic influence on the structures of the molecules and their aggregation states. With the addition of 500 equivalents of HCl, less than 80% water content did not induce any aggregation of the molecules (Fig. S3a, ESI†). After the water content exceeded 80%, TPE-MCH-CN began to aggregate; however, no fluorescence appeared (Fig. S3b, ESI†). The quenching of fluorescence is suggested to be caused by the formation of strong dipole–dipole and π–π interactions after aggregation. Strong intermolecular interactions can be deduced from the bathochromic shift of the UV-Vis spectra from 436 nm to 483 nm.50–53 When the acid was decreased to 80 equivalents, aggregates formed when the water percentage exceeded 80%, with appearance of an emission at 675 nm. The UV-Vis spectra revealed that aggregates of the de-protonated product of TPE-MC-CN (abs: 596 nm) also formed in addition to the TPE-MCH-CN aggregates (abs: 483 nm), and the emission arose from the TPE-MC-CN aggregates (Fig. 3d and Fig. S3c, ESI†). When the acid was decreased to 20 equivalents, the de-protonated product of TPE-MC-CN (abs: 551 nm) was generated in solution when the water percentage only reached 20%, with a color change from yellow to purple (Fig. S3e and S4, ESI†). The formation of TPE-MC-CN was also confirmed by the absorption spectra of the photochromic process of TPE-Sp-CN (Fig. 1d). When the water content increased from 20% to 70%, the absorption showed solvatochromism from 551 nm to 536 nm without aggregation. When the water content was greater than 80%, TPE-MC-CN began to aggregate, and the emission peak red-shifted from 640 nm to 675 nm (Fig. S3f, ESI†). Therefore, it can be seen that hydrophobic TPE-Sp-CN tends to aggregate in water, whereas the organic salt TPE-MCH-CN has better solubility; also, the aggregation of TPE-MCH-CN has no emissive output (Fig. 2a).
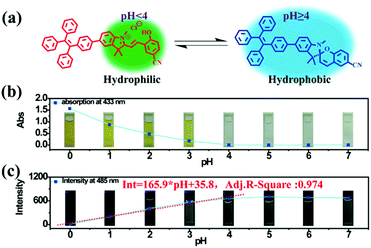 |
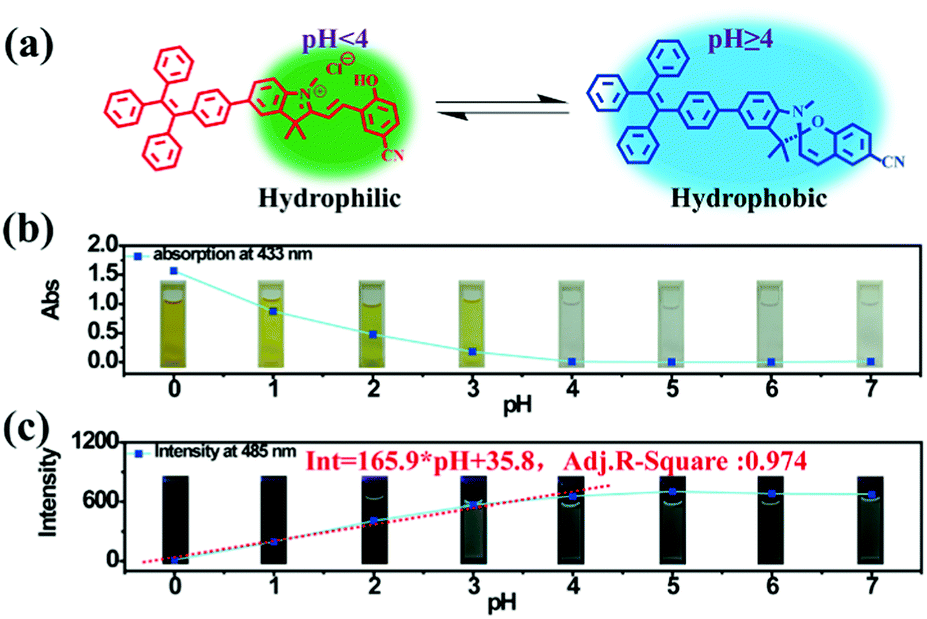
| | Fig. 2 (a) Illustration of switching between the ring-closed and protonated ring-open forms of TPE-Sp-CN at different pH values. (b) The plot of the absorption at 433 nm of the TPE-Sp-CN solutions (1 mL, 1 × 10−4 M) treated with 0.2 mL water samples with different pH values (0 to 7). (c) The plot of the emission intensity of TPE-Sp-CN in acid solutions (VMeCN:Vacid water = 1:9) against pH (0 to 7). | |
Based on the above experiments, a pH sensing process for water with different pH values was conducted as follows: 0.2 mL water sample was added to 1 mL TPE-Sp-CN acetonitrile solution with sufficient equilibration, and the corresponding absorption spectra were recorded; then, water was added to increase the water content to 90% and induce aggregation, and the corresponding PL spectra were recorded. As depicted in Fig. 2b, after equilibration was achieved, the solutions with different pH values showed different absorptions in the visible light range which can be distinguished by the naked eye. The pH (value < 4) of the solutions could be firstly judged by the absorption at 433 nm, and a lower pH corresponded to a higher absorption (Fig. S5, ESI†). In the second stage, after the water content reached 90%, fluorescence from aggregates appeared. In low pH conditions, TPE-MCH-CN aggregates formed with no emission (Fig. S6, ESI†). In higher pH conditions, the emission of TPE-Sp-CN aggregates at 485 nm gradually appeared and increased. The de-protonated ring-open form (TPE-MC-CN) also appeared, with an emission of 650 nm, due to a water-induced ring-opening process. The PL intensity of 485 nm is plotted against pH in Fig. 2c. For a solution with pH 0, the emissive intensity approached zero; with increasing pH, the intensity gradually enhanced and reached a platform at pH 4. The relationship between emission intensity and pH fulfills the linear equation:
| | | Int = 165.9 × pH + 35.8 | (1) |
The adj.
R-square value of the equation is 0.974. Therefore, the pH of acidic solutions can be detected by
TPE-Sp-CN with a good linear relationship.
2.2. Sulfite anion detection
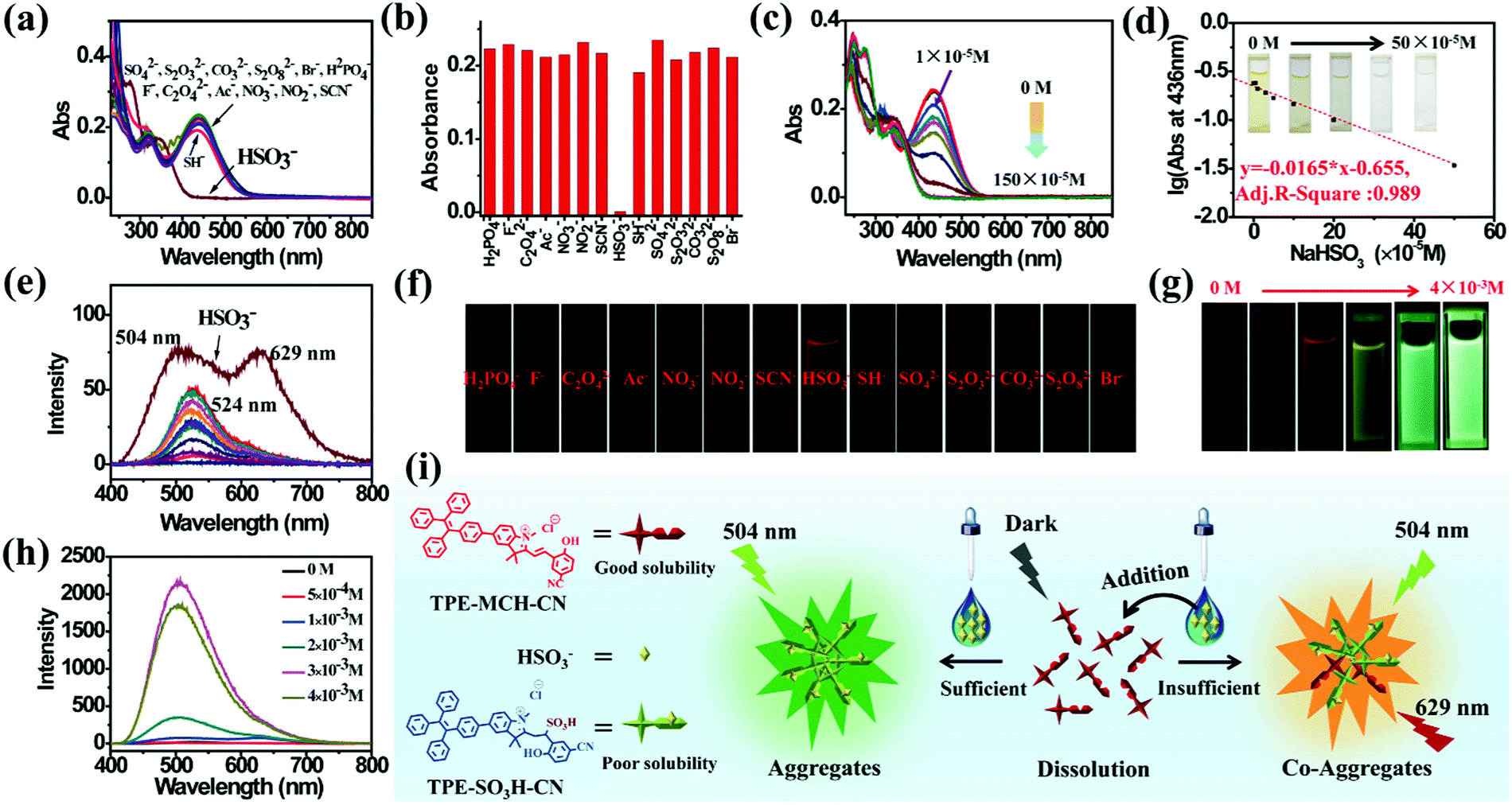
TPE-Sp-CN also exhibited the ability to monitor the sulfite concentration of a water system because the double bond of cyanine is reactive upon selective addition of nucleophilic reagents.54,55 Fourteen common anions, H2PO4−, F−, C2O42−, Ac−, NO3−, NO2−, CO32−, Br−, SH−, HSO3−, SCN−, SO42−, S2O32−, and S2O82−, were tested, and the corresponding absorption spectra were recorded (Fig. 3a). In the presence of 100 equivalents of these anions, only HSO3− led to decoloration of the TPE-MCH-CN solution (1 × 10−5 M) with disappearance of the absorption peak at 436 nm (Fig. 3b). This indicated that HSO3− broke conjugated structure of TPE-MCH-CN in addition reaction, and the additive product TPE-SO3H-CN formed finally (Scheme 1 and Fig. S7, ESI†). The MS results (m/z: 715.2588) verified that the addition reaction occurred (Fig. S8, ESI†), and the 1H NMR spectra indicated that the reaction proceeds by 1,2-addition rather than 1,4-addition (Fig. S9, ESI†). For TPE-MCH-CN, the proton signal at δ 4.12 (H1′) of the nitrogen-methyl group shifted to δ 4.14 (H1′′) after reaction with HSO3−, while the proton signal of the double methyl groups H2′ (δ 1.73) split into two signals, H2′′ (δ 1.61 and 1.55). Also, the proton signal of the nitrogen methyl group for neutral TPE-Sp-CN is δ 2.68. This indicates that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond was not destroyed in the addition reaction and that HSO3− was added to the Cβ position, as depicted in Fig. S9 (ESI†). After the reaction mechanism was confirmed, the detection limit was measured and was found to be 1 × 10−5 M (1.04 ppm, 104 × 10−5 g L−1) (Fig. 3c and d). In the low concentration range (0 to 50 × 10−5 M), the HSO3− concentration and logarithm of absorption at 436 nm fulfill the equation:
N double bond was not destroyed in the addition reaction and that HSO3− was added to the Cβ position, as depicted in Fig. S9 (ESI†). After the reaction mechanism was confirmed, the detection limit was measured and was found to be 1 × 10−5 M (1.04 ppm, 104 × 10−5 g L−1) (Fig. 3c and d). In the low concentration range (0 to 50 × 10−5 M), the HSO3− concentration and logarithm of absorption at 436 nm fulfill the equation:| | | y = −0.0165 × x − 0.655 | (2) |
in which “y” represents the logarithm of absorption at 436 nm and “x” refers to the HSO3− concentration; also, the adj. R-square value is 0.989. Thus, a low HSO3− concentration can be quantitatively sensed. Because TPE-SO3H-CN did not aggregate at relatively low concentrations (1 × 10−5 M), no fluorescence signal was observed after the addition reaction (Fig. S10, ESI†).
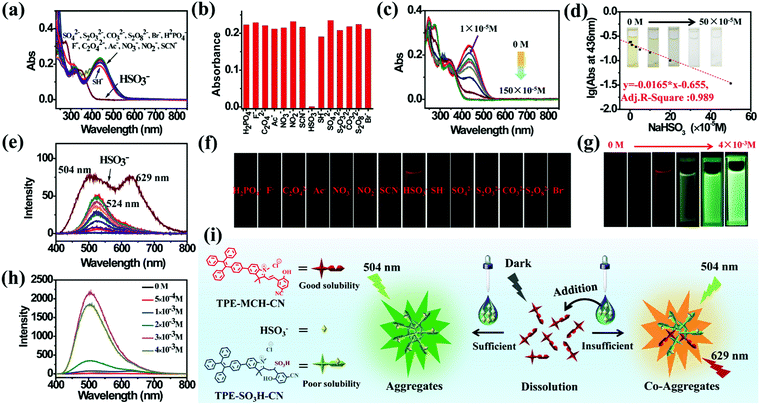 |
| | Fig. 3 (a) Absorption spectra of the mixed solutions of TPE-MCH-CN (1 × 10−5 M, 5 × 10−3 M HCl acidized) with fourteen anions (1 × 10−3 M) and (b) the corresponding absorbances at 436 nm. (c) Absorption spectra and (d) the corresponding absorbances at 436 nm of TPE-MCH-CN (1 × 10−5 M) with different equivalents of HSO3− (Vacetonitrile:Vwater = 3:7). (e) PL spectra and (f) corresponding fluorescence images of the mixed solutions of TPE-Sp-CN with fourteen anions. (g) Fluorescence images and (h) corresponding PL spectra of TPE-MCH-CN (1 × 10−4 M) with different HSO3− concentrations (Vacetonitrile:Vwater = 4:6). (i) Illustration of the emission mechanism in the course of HSO3− detection. | |
Different results were observed at a higher concentration of TPE-MCH-CN solution (1 × 10−4 M). Among the fourteen anions (1 × 10−3 M), HSO3− still decolored the dark yellow solution quickly (Fig. S11, ESI†). It is surprising that a fluorescent signal of the solution could be detected with a double emission of 504 nm and 629 nm after the reaction reached the equilibrium state (Fig. 3e). The orange-red fluorescence could be observed by the naked eye under 365 nm UV light (Fig. 3f). The double emission peaks are believed to arise from co-aggregates of TPE-SO3H-CN and TPE-MCH-CN. TPE-MCH-CN aggregates were proved to be non-emissive in the aggregation state because of the existence of strong π–π stacking and dipolar interactions within the aggregates (Fig. S3b, ESI†). However, when TPE-MCH-CN was dispersed at the molecular level in PMMA at a low concentration (weight ratio: 1/100), the red emission from separated molecules could be observed (Fig. S12, ESI†). In the same way, if unreacted TPE-MCH-CN co-aggregated with TPE-SO3H-CN, the strong interaction between TPE-MCH-CN would be interrupted by TPE-SO3H-CN; therefore, the red emission of TPE-MCH-CN would be observed. Meanwhile, at this high concentration, TPE-SO3H-CN, which has stronger intermolecular interactions, such as hydrogen bonds and ionic bonds, tends to aggregate more readily than TPE-MCH-CN. The emission peak at 504 nm should be derived from the AIE emission from the TPE-SO3H-CN aggregates. When different concentrations of HSO3−, ranging from 5 × 10−4 M to 4 × 10−3 M, were added to the detection solution, TPE-MCH-CN was gradually converted to TPE-SO3H-CN with decreasing absorption at 436 nm (Fig. S13, ESI†). Orange fluorescence appeared when the concentration of HSO3− reached 1 × 10−3 M, and it changed from orange (629 nm) to bright green (504 nm) with further increasing concentration of HSO3− (Fig. 3g and h). When the concentration of HSO3− was higher than 3 × 10−3 M, all the TPE-MCH-CN was consumed, and only bright green emission at 504 nm was observed. An illustration of the mechanism of the change in emission is depicted in Fig. 3i. Thus, due to the AIE output of TPE-SO3H-CN at high concentration, HSO3− can also be detected according to the emission spectra or even by the naked eye. In contrast, only very weak emission at 524 nm from a few TPE-Sp-CN aggregates (formed by water-induced de-protonation and isomerization) was detected for the other anion-treated solutions (Fig. 3e). This is because some of the emission from the TPE-Sp-CN aggregates was absorbed by unreacted TPE-MCH-CN, and only the emissions at longer wavelengths were detected; this resulted in an emission peak at 524 nm (Fig. S14, ESI†).
It is worthwhile to mention that two control molecules (SPTPE and TPE-Sp) with different linked structures of TPE and Sp could not distinguish HSO3− from other anions very well under the same conditions (Fig. S15 and S16, ESI†). This indicates that the appropriate molecular structure and the deliberately modified cyano group, which has an electron withdrawing effect, play important roles in the activation of the double bond and the detection of HSO3−. Meanwhile, the highly acidic detection conditions are also important; we excluded the addition reaction with other reactive anions of CN−, S2− OH−, etc. Thus, through the corresponding absorption and PL spectra, HSO3− can be detected by acidic TPE-MCH-CN solution.
2.3. Hydrostatic pressure sensing
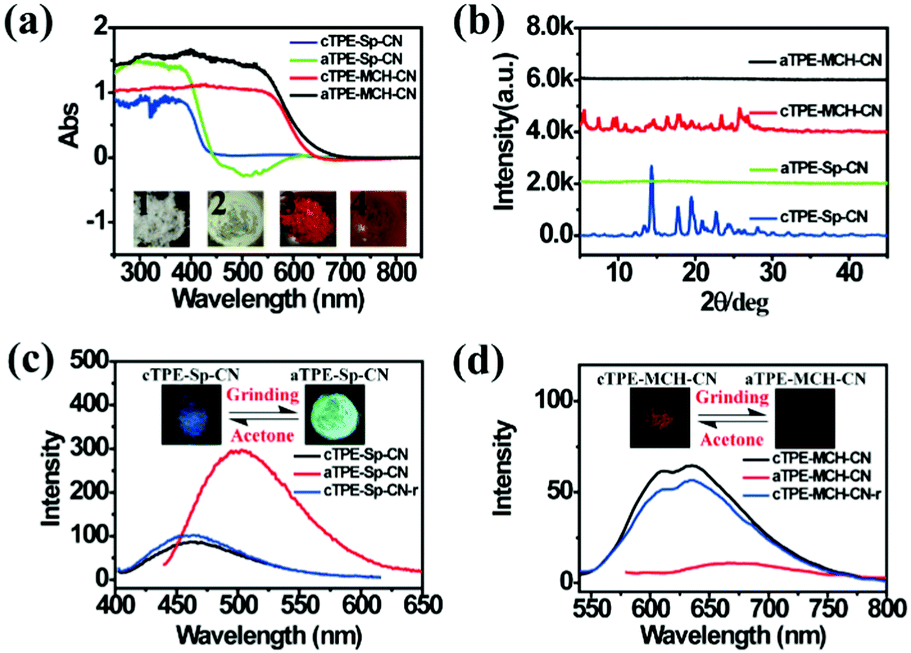
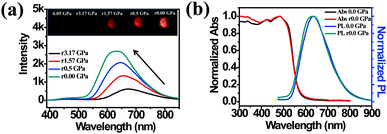
High pressure sensing has attracted great research interest56,57 because pressure is an important parameter in geo-exploration, industrial production and daily life. In order to study the pressure sensing properties of TPE-Sp-CN, we firstly investigated the mechanochromic properties of TPE-Sp-CN. TPE-Sp-CN crystals (cTPE-Sp-CN) were obtained by crystallization in acetone/hexane mixed solution. The white powder cTPE-Sp-CN, which has almost no absorption in the visible light range, turned pale green after grinding. The band-edge of the absorption spectrum red-shifted nearly 27 nm from 425 nm to 452 nm (Fig. 4a). Also, the XRD pattern (Fig. 4b) indicated that cTPE-Sp-CN had changed into amorphous TPE-Sp-CN (aTPE-Sp-CN), with characteristic peaks weakening and disappearing.58 Meanwhile, the emission of the powder also showed an abrupt change. The weak blue emission (465 nm) of cTPE-Sp-CN changed to bright green emission (502 nm) after the powder was converted to aTPE-Sp-CN (Fig. 4c). It is notable that the quantum yield of aTPE-Sp-CN (34.62%) is surprisingly higher (12.54 times) than that of cTPE-Sp-CN (2.76%), whereas the lifetimes of cTPE-Sp-CN and aTPE-Sp-CN showed no dramatic change (Fig. S17, ESI†). Therefore, after cTPE-Sp-CN was ground, the irradiation rate of the powder increased significantly (Table S1, ESI†). Although TPE-MCH-CN has the same molecular framework as TPE-Sp-CN, it exhibited an opposite changing trend of fluorescence intensity. TPE-MCH-CN crystals (cTPE-MCH-CN) were obtained by treating solid TPE-Sp-CN with an HCl/acetone mixture. After cTPE-MCH-CN was ground, the band-edge gas of the absorption spectrum bathochromically shifted nearly 60 nm, from 640 nm to 700 nm (Fig. 4a). Despite experiencing the same phase transition from the crystalline state to the amorphous state, the red emission at 636 nm of cTPE-MCH-CN was eventually quenched rather than becoming brighter (Fig. 4d). Amorphous TPE-MCH-CN (aTPE-MCH-CN) finally formed, and the quantum yield of cTPE-MCH-CN decreased from 4.52% to 0.01%. The amorphous states of both TPE-Sp-CN and TPE-MCH-CN can be recovered to crystalline states by solvent fuming with good reversibility.
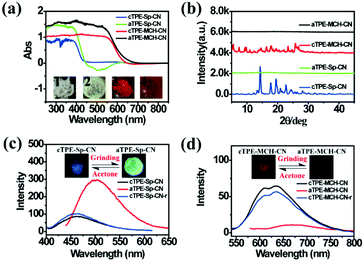 |
| | Fig. 4 (a) Absorption spectra and (b) XRD patterns of cTPE-Sp-CN, aTPE-Sp-CN, cTPE-MCH-CN and aTPE-MCH-CN. Inset: Images of 1: cTPE-Sp-CN, 2: aTPE-Sp-CN, 3: cTPE-MCH-CN and 4: aTPE-MCH-CN in ambient light. (c) and (d) Changes in the PL spectra during the cTPE-Sp-CN to aTPE-Sp-CN and cTPE-MCH-CN to aTPE-MCH-CN transitions, where the insets show fluorescence images under excitation at 365 nm; cTPE-Sp-CN-r and cTPE-MCH-CN-r represent the crystal states recovered by acetone gas. | |
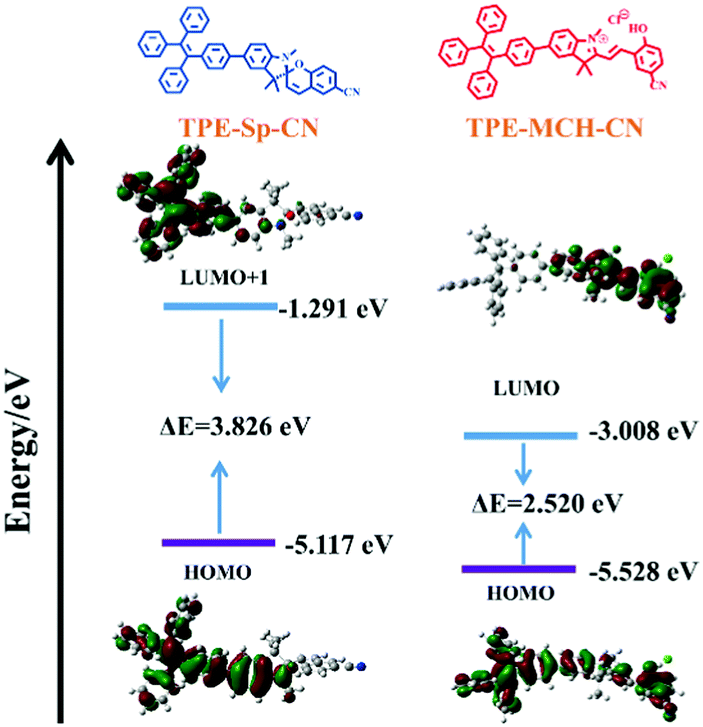
Theoretical calculations were performed to disclose the different emission mechanisms of TPE-Sp-CN and TPE-MCH-CN by B3LYP/6-31G(d) with Gaussian 09 (Fig. 5).59 The results indicate that the blue fluorescence of TPE-Sp-CN arises from localized excited (LE) emission of the HOMO to LUMO+1 transition with a band gap of 3.826 eV, whereas the red fluorescence of TPE-MCH-CN arises from an intramolecular charge transfer (ICT) transition with a smaller band gap of 2.520 eV. Based on the studies of the emission mechanism, both cTPE-Sp-CN and cTPE-MCH-CN show potential to be used as pressure sensors, which further encouraged us to explore their hydrostatic pressure-sensing properties.
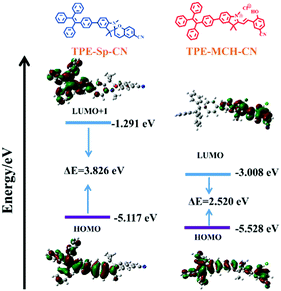 |
| | Fig. 5 The orbital amplitude plots and corresponding energy levels of optimized TPE-Sp-CN and optimized TPE-MCH-CN. | |
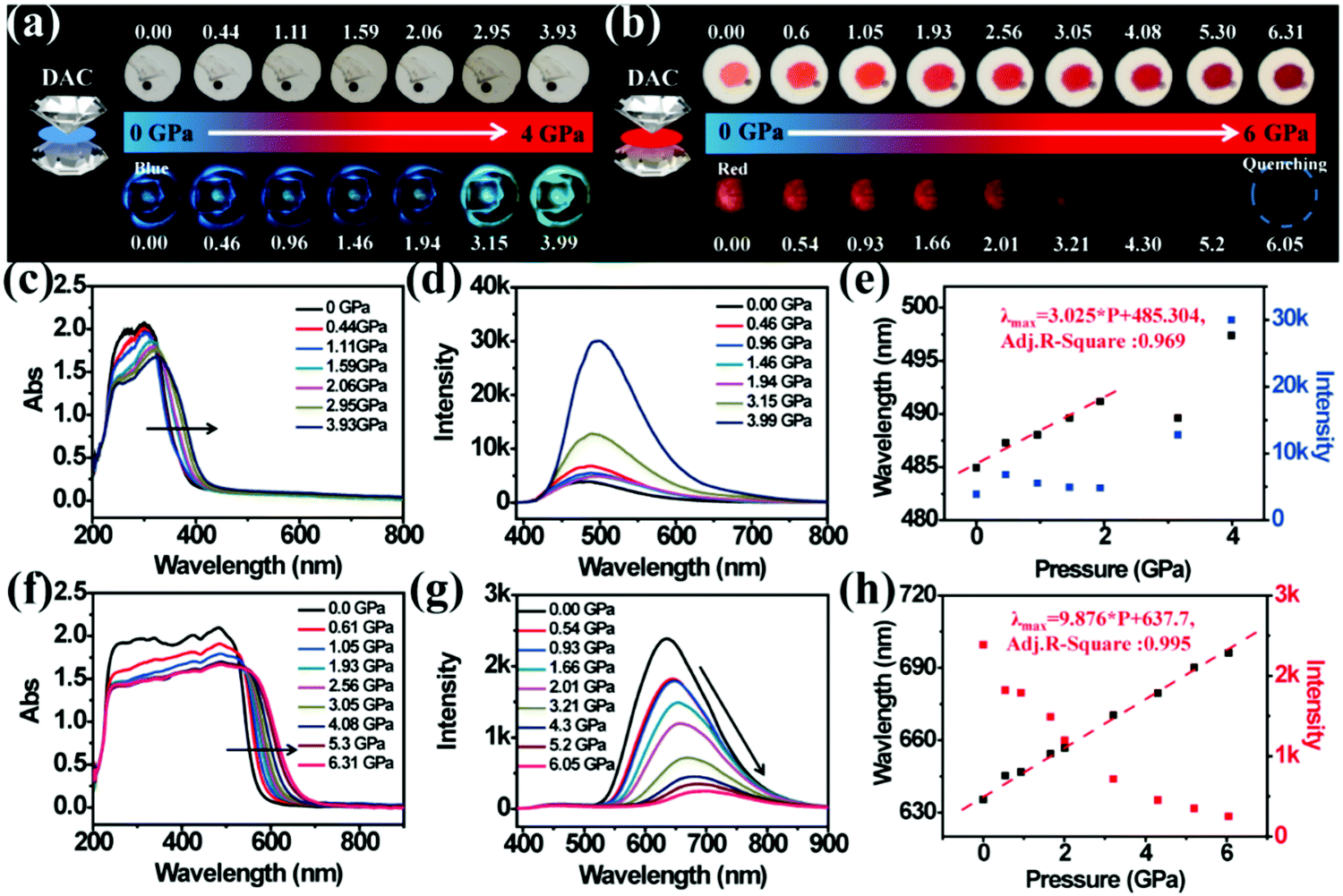
In order to investigate the relationship between structure and optical properties in hydrostatic pressure conditions, a single crystal of TPE-Sp-CN (CCDC 1821887†), which was cultivated from slow evaporation of its DCM/hexane solution, was placed in a DAC container for high pressure measurements with silicon oil as the pressure transmitting medium. The colour of the single crystal in ambient light showed no obvious change as the pressure increased from 0 GPa to 3.93 GPa (Fig. 6a), and the band edge of the absorption red-shifted from 400 nm to 420 nm (Fig. 6c). The emission of the single crystal exhibited a red shift from weak blue (485 nm) to cyan (497 nm) (Fig. 6a and d). In order to investigate the profound relationship between pressure and emission, the emission wavelength peaks were plotted against hydrostatic pressure (Fig. 6e). When the hydrostatic pressure was between 0 GPa and 1.94 GPa, the pressure and emission wavelength were linearly correlated and fit the linear equation:
in which “
λmax” represents the emission peak position, “
P” refers to the hydrostatic pressure, and “
a” and “
b” are the intercept and slope of the linear equation. The slope and intercept are 3.025 and 485.304, respectively, and the adj.
R-square value is 0.9694. However, as the pressure further increased, the linear relationship became inapplicable. The emission hypochromically shifted from 491 nm (1.94 GPa) to 489 nm (3.15 GPa), then red-shifted to 497 nm at 3.99 GPa. The corresponding emissive intensity exhibits more complex fluctuation with pressure; therefore, it is not suitable to utilize the emission intensity to sense pressure (
Fig. 6e). The configuration and conformation of
TPE-Sp-CN and its packing mode in a single crystal were investigated in order to interpret the emission wavelength changes. This single crystal of
TPE-Sp-CN belongs to the monoclinic crystal system (Fig. S18, ESI
†); there are four
TPE-Sp-CN molecules and four dichloromethane molecules in each crystal cell. The TPE moiety directs to the inside of the crystal cell, whereas the cyano group directs to the outside of the crystal cell. No π–π interactions were found between
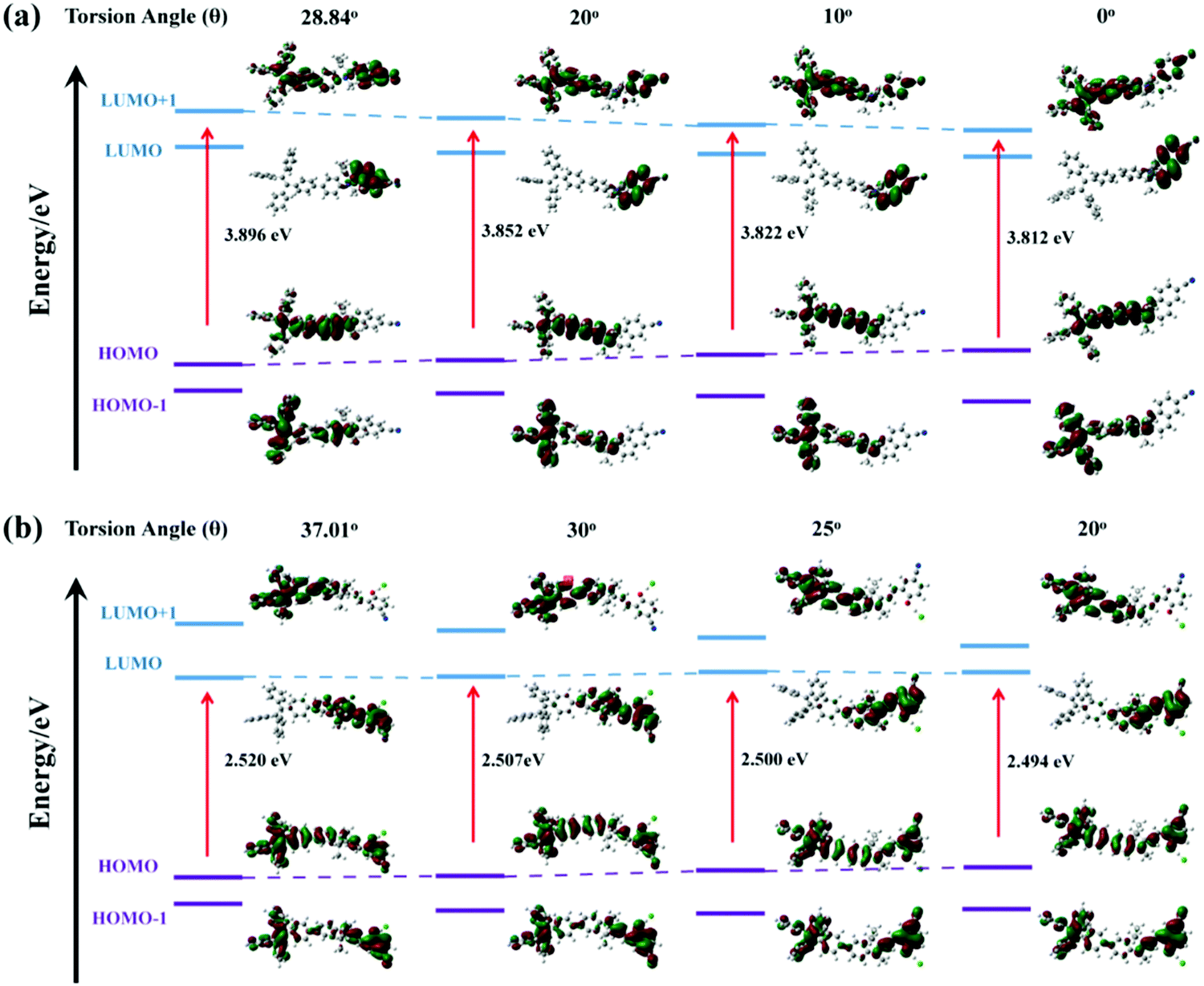
TPE-Sp-CN molecules because neither Sp nor TPE has a planar structure that is suitable for the formation of π–π interactions. The emission red shift can be attributed to the planarity of the molecules under pressure. For the TPE moiety, pressure-induced planarity has been reported to cause a red shift of the emission.
60 Meanwhile, a decrease of the torsion angle between the TPE moiety and indole would also result in a bathochromic shift of the emission. As depicted in
Fig. 7a, the pristine torsion angle of
TPE-Sp-CN in the single crystal was 28.84° at first. As this angle of the model structure decreased, the band gap tended to narrow gradually from 3.896 eV to 3.812 eV (torsion angle = 0°), which corresponds to the red-shifts of the
in situ optical spectra under increasing pressure. Thus, by analysing the emission wavelength changes of single crystal
TPE-Sp-CN, pressure can be sensed in the range from 0 to 1.94 GPa with a sensitivity of 3.025 nm GPa
−1.
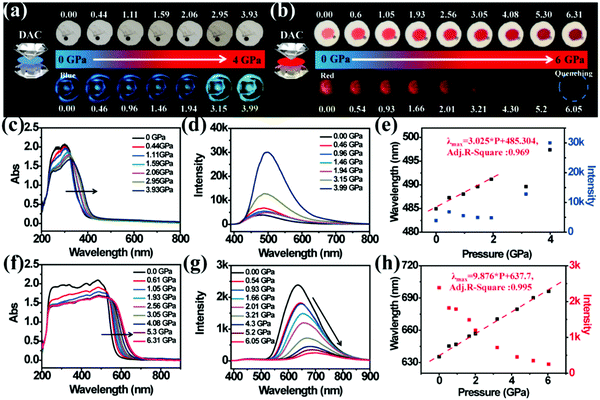 |
| | Fig. 6 (a) Visible light images (top) and fluorescence images (bottom) of single crystal TPE-Sp-CN under increasing hydrostatic pressure. (b) Visible light images (top) and fluorescence images (bottom) of cTPE-MCH-CN under increasing hydrostatic pressure. (c) The in situ absorption spectra and (d) emission spectra of single crystal TPE-Sp-CN under increasing hydrostatic pressure. (e) The correlation plots of emission wavelength and intensity of single crystal TPE-Sp-CN with hydrostatic pressure, respectively. (f) The in situ absorption spectra and (g) emission spectra of cTPE-MCH-CN under increasing pressure. (h) The correlation plots of emission wavelength and intensity of cTPE-MCH-CN against hydrostatic pressure, respectively. | |
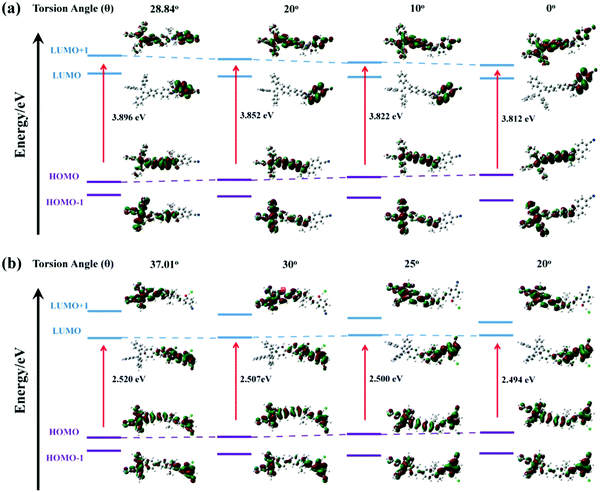 |
| | Fig. 7 The orbital amplitude plots and corresponding band gaps of (a) TPE-Sp-CN and (b) TPE-MCH-CN in fixed conformation with gradually decreasing torsion angle (θ) between TPE and the indole, depicted by B3LYP/6-31G (d) with Gaussian 09. | |
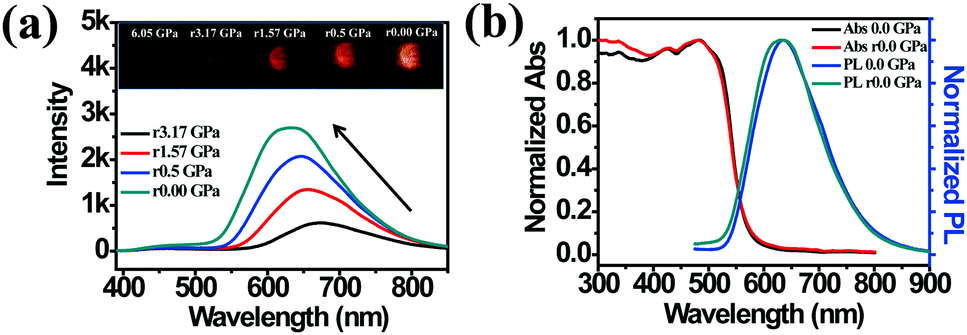
The high pressure sensing properties of TPE-MCH-CN were also investigated. Because a single crystal of TPE-MCH-CN was not obtained, cTPE-MCH-CN powder was used in the experiment. The results indicated that the powder exhibits good pressure sensing properties in the range from 0 GPa to 6 GPa. The colour of cTPE-MCH-CN changed from orange to red in ambient light (Fig. 6b), and the absorption band edge of cTPE-MCH-CN red shifted from 600 nm to nearly 700 nm when the pressure increased from 0 GPa to 6.31 GPa (Fig. 6f). This should result from the enhanced planarity of the molecules and π–π interactions formed under high pressure.61 Likewise, the emission of cTPE-MCH-CN exhibited a red shift under high pressure. The corresponding fluorescence red-shifted nearly 65 nm, from 635 nm at 0 GPa to nearly 700 nm at 6.05 GPa (Fig. 6g). Confirmed by theoretical calculations, the planarity also induced red-shifts of both the absorption and PL spectra of TPE-MCH-CN. As the torsion angle between TPE and MCH decreased from 37.01° to 20°, the band gap decreased from 2.520 eV to 2.494 eV (Fig. 7b). Meanwhile, the emissive intensity decreased 9.5 times and was finally nearly quenched. As shown in the correlation plot of emission wavelength and pressure (Fig. 6h), the pressure has a good linear relationship with the emission wavelength in the whole pressure range. The adj. R-square value is 0.995, and the slope and intercept for linear eqn (3) are 9.876 and 637.661, respectively. The larger slope and obvious emission color change indicate that TPE-MCH-CN has better sensitivity to pressure compared with single crystal TPE-Sp-CN, which can also be confirmed by the CIE coordinates (Fig. S19, ESI†). Moreover, after the pressure was removed, both the absorption and fluorescence gradually spectra blue shifted (Fig. 8a and Fig. S20, ESI†) and eventually recovered to the original state (Fig. 8b). High pressure IR spectra were recorded during the pressurization and depressurization process, and no obvious change was observed; this indicates the good stability and reversibility of cTPE-MCH-CN under pressure (Fig. S21, ESI†). Therefore, cTPE-MCH-CN (sensing range: 0 to 6.05 GPa, sensing sensitivity: 9.876 nm GPa−1) exhibits better pressure sensing properties compared with single crystal TPE-Sp-CN (sensing range: 0 to 1.94 GPa, sensing sensitivity: 3.025 nm GPa−1) because of its wider sensing range and higher sensitivity.
 |
| | Fig. 8 (a) The in situ PL spectra of cTPE-MCH-CN during the pressure releasing process. (b) Comparison of the absorption spectra and PL spectra between the pristine state and pressure-released state. | |
3. Conclusions
A new fluorescent molecular switch, TPE-Sp-CN, with multi-stimuli responsive properties was explored to quantitatively detect multi-parameters of pH value, sulfite anion, and hydrostatic pressure. The pH sensing range covers 0 to 4, with different aggregation-induced emission degrees of TPE-Sp-CN in acidic conditions. By optical spectra monitoring of the selective addition reaction between sulfite anion and the protonated ring-open form TPE-MCH-CN, sulfite anion can be distinguished from other anions, and the detection limit is 1.04 ppm. Meanwhile, solid state TPE-MCH-CN exhibited better pressure sensitivity than TPE-Sp-CN, and the hydrostatic pressure sensing range was 0 GPa to 6 GPa. All the external stimuli have good linear relationships with the optical signals of TPE-Sp-CN; therefore, this molecule exhibits great application potential in the field of multi-parameter analysis for complex environments, such as geological activity prediction. Furthermore, this work has pushed the limit of stable states that a small molecular switch can possess and has demonstrated how every fragment of the molecule can be used to enrich its sensing ability. In addition to its applications in sensing, this molecule has application potential in other fields, such as logic gates, data storage, and super-resolution imaging; these are also under investigation.
4. Experimental section
4.1. Instruments
1H NMR spectra were recorded on a 500 MHz Bruker Avance spectrometer, while the 13C NMR spectra were recorded on a 126 MHz Bruker Avance spectrometer. Deuterated solvents, including CDCl3 and DMSO-d6, were used with tetramethylsilane (TMS) as an internal standard (δ = 0.00 ppm). Fluorescence spectra of all samples (solid and solution) were measured with a RF-5301PC spectrofluorometer. UV-Vis spectra were measured on a Shimadzu UV-2550 spectrophotometer. Kubelka–Munk diffuse reflectance absorption spectra were obtained from a Maya 2000PRO fiber optical spectrometer with an Ocean DH-2000-BAL UV/Vis/NIR light source using BaSO4 as the background. Powder XRD patterns were obtained from a PANalytical B.V. Empyrean X-ray diffractomer with Cu-Kα radiation (λ = 1.5418 Å) at 25 °C (scan range: 5 to 45°). DSC data were obtained from a NETZSCH DSC 204 instrument at a scanning rate of 10 K min−1. The time of flight mass spectra were recorded using a Kratos MALDI-TOF mass system. The fluorescence quantum yields (Φf) and lifetime were measured on an FLS 920 lifetime and steady state spectrometer. Photographs were taken using a Canon camera. High pressure experiments were performed using diamond anvil cells at room temperature. The samples were placed in holes (diameter: 150 μm) with a small ruby chip used for in situ pressure calibration according to the R1 ruby fluorescence method. Silicon oil was used as the pressure transmitting medium for both single crystal TPE-Sp-CN and cTPE-MCH-CN in the high pressure experiments. Both the in situ photoluminescence spectra and UV/Vis absorption spectra under high pressure were obtained using a QE65000 Scientific-grade spectrometer. The images were taken using a camera (Canon Eos 5D mark II) equipped on a microscope (Ecilipse TI-U, Nikon) in the high pressure experiments. Absorption spectra were measured in the exciton absorption band region using a deuterium−halogen light source. The excitation source for the PL measurements was the 355 nm line of a UV DPSS laser. The optical fiber spectrometer was an Ocean Optics QE65000 spectrometer. IR microspectroscopy of cTPE-MCH-CN was carried out with a Nicolet iN10 microscope spectrometer (Thermo Fisher Scientific, USA) using a liquid-nitrogen-cooled detector.
4.2. Materials
THF, 1,4-dioxane, and toluene were treated with calcium hydride and sodium, then distilled before use. Trifluoroacetic acid, n-butyllithium, iodomethane, bis(pinacolato)diboron, PdCl2(dppf), Pd(Ph3P)4, diphenylmethane, 4-bomobenzophenone, tosilate, 4-bromophenylhydrazine hydrochloride, 3-methyl-2-butanone were purchased from Energy Chemical. Hexamethylenetetramine was purchased from Aladdin. 4-Cyanophenol was bought from Ourchem. Ethanol, hexane, dichloromethane, ethyl acetate, potassium acetate and potassium carbonate were purchased from Beijing Reagents. Acetonitrile and triethylamine were purchased from Xilong Reagents. All these chemicals were used without any further purification.
4.3. General synthesis information
The detailed synthetic route is depicted in Scheme 2. Compounds M1–M3 were synthesized using the prevalent methods according to literature procedures.53,62,63
 |
| | Scheme 2 Synthetic route of TPE-Sp-CN. | |
Synthesis of 2,3,3-trimethyl-5-(4-(1,2,2-triphenylvinyl) phenyl)-3H-indole (M4).
To a 250 mL round-bottom flask full of nitrogen gas were added 4.11 g (10 mmol) of M2, 2.94 g (10 mmol) of M3 and 3.33 g (24.6 mmol) of potassium carbonate (K2CO3). 40 mL deaerated tetrahydrofuran and 8 mL ultrapure water were added to the flask. Then, the uniformly stirred solution was frozen with liquid nitrogen, and 130 mg tetrakis (triphenylphosphine) palladium(0) were added. The flask was evaporated for 1 min and nitrogen was inlet to remove residual oxygen. After the solution recovered to room temperature, the reaction liquid was heated to reflux overnight. Water was added to the flask, and the solution was extracted with ethyl ether. The organic layer was dried with anhydrous sodium sulfate and evaporated to remove the solvent. The crude product was purified by flash column chromatography (ethyl acetate:hexane = 1:4). Finally, a white solid powder was obtained with a yield of 67.7% (3.31 g). 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 8 Hz, 1H), 7.50 (dd, J = 8, 1.5 Hz, 1H), 7.46 (d, J = 1 Hz, 1H), 7.37 (d, J = 8 Hz, 2H), 7.10 (m, 17H), 2.30 (s, 3H), 1.33 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 188.26, 152.90, 146.15, 143.73, 143.74, 143.69, 142.53, 141.05, 140.48, 138.99, 137.88, 131.76, 131.38, 131.31, 127.73, 127.64, 127.60, 126.47, 126.43, 126.38, 126.20, 119.87, 53.68, 23.14, 15.44. LC-HRMS (ESI): m/z: 490.2594 [M + H]+, calcd: 490.2490.
Synthesis of 1,2,3,3-tetramethyl-5-(4-(1,2,2-triphenylvinyl) phenyl)-3H-indol-1-ium iodide (M5).
M4 (0.98 g, 2 mmol), CH3I (0.71 g, 5 mmol) and 15 mL ethyl acetate were added to a flask and ventilated with nitrogen. Then, the reaction liquid was heated to reflux for 12 h. After the solution recovered to ambient temperature, a grey solid precipitated. Pure product was obtained by further washing the precipitate with ethyl acetate in a yield of 89.5% (1.13 g). 1H NMR (500 MHz, CDCl3) δ 7.70 (dd, J = 20.5, 8.5 Hz, 2H), 7.61 (s, 1H), 7.31 (d, J = 8 Hz, 2H), 7.17–7.00 (m, 17H), 4.30 (s, 3H), 3.11 (s, 3H), 1.67 (s, 6H).13C NMR (126 MHz, CDCl3) δ 195.39, 144.25, 143.53, 143.50, 143.45, 143.27, 141.92, 141.77, 140.93, 140.05, 136.81, 132.14, 131.36, 131.33, 131.28, 128.21, 127.88, 127.80, 127.69, 126.70, 126.64, 126.63, 126.60, 121.38, 115.50, 54.61, 37.32, 23.25, 17.05. LC-HRMS (ESI): m/z: 504.2682 [M + H]+, calcd: 504.2686.
Synthesis of 1′,3′,3′-trimethyl-5′-(4-(1,2,2-triphenylvinyl) phenyl)spiro[chromene-2,2′-indoline]-6-carbonitrile (TPE-Sp-CN).
M1 (1.2 mmol, 0.18 g), M5 (1 mmol, 0.63 g), and anhydrous EtOH (10 mL) were added to a flask. The reaction solution was heated at reflux for 36 h, and the solvent was evaporated to obtain a red powder as the crude product. The red powder was further purified by re-crystallization from CH2Cl2/MeOH to obtain TPE-MCH-CN iodide. Then, flash column chromatography (DCM:TEA:hexane = 1:2:50) was used to obtain 0.41 g of TPE-Sp-CN as a white solid powder. Yield: 64.9%. 1H NMR (500 MHz, DMSO-d6) δ 7.73 (s, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.41 (m, 4H), 7.18–6.98 (m, 18H), 6.84 (d, J = 8 Hz, 1H), 6.64 (d, J = 9 Hz, 1H), 5.94 (d, J = 10.5 Hz, 1H), 2.69 (s, 3H), 1.24 (s, 3H), 1.13 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.50, 147.23, 143.40, 143.33, 143.28, 143.07, 141.07, 140.46, 138.61, 136.82, 134.08, 131.23, 131.20, 130.97, 130.81, 130.73, 128.13, 127.97, 127.89, 127.85, 126.64, 126.61, 126.55, 126.53, 126.03, 125.21, 121.04, 120.03, 119.63, 119.02, 115.88, 115.86, 107.30, 105.62, 102.59, 54.97, 51.93, 28.59, 25.59, 19.67. LC-HRMS (ESI): m/z: 633.2865 [M + H]+, calcd: 633.2861.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (20160414002GH, 51303063, 41722405, 21574058, 41374075, 20180201017GX and 21725304), the key project of the National Instrumentation Project under Grant 2011YQ030133, and the Jilin Outstanding Professor Plan under Grant 20150519008JH and Grant 20140204022GX.
Notes and references
- J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC.
- T. Chen, Z.-Q. Chen, W.-L. Gong, C. Li and M.-Q. Zhu, Mater. Chem. Front., 2017, 1, 1841–1846 RSC.
- D. Hu, T. Zhang, S. Li, T. Yu, X. Zhang, R. Hu, J. Feng, S. Wang, T. Liang, J. Chen, L. N. Sobenina, B. A. Trofimov, Y. Li, J. Ma and G. Yang, Nat. Commun., 2018, 9, 362 CrossRef PubMed.
- L. Guo, M. Wang, X. Zeng and D. Cao, Mater. Chem. Front., 2017, 1, 2643–2650 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951–13953 CrossRef CAS PubMed.
- M. Hu, W. Kang, Y. Zhao, J. Shi and B. Cheng, RSC Adv., 2017, 7, 26849–26856 RSC.
- X. Cheng, H. Jia, T. Long, J. Feng, J. Qin and Z. Li, Chem. Commun., 2011, 47, 11978–11980 RSC.
- A. Panja and K. Ghosh, Mater. Chem. Front., 2018, 2, 1866–1875 RSC.
- A. Fermi, G. Bergamini, M. Roy, M. Gingras and P. Ceroni, J. Am. Chem. Soc., 2014, 136, 6395–6400 CrossRef CAS PubMed.
- J. Dong, K. Zhang, X. Li, Y. Qian, H. Zhu, D. Yuan, Q. H. Xu, J. Jiang and D. Zhao, Nat. Commun., 2017, 8, 1142 CrossRef PubMed.
- X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276–15286 CrossRef CAS PubMed.
- X.-G. Liu, C.-L. Tao, H.-Q. Yu, B. Chen, Z. Liu, G.-P. Zhu, Z. Zhao, L. Shen and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 2983–2988 RSC.
- W. Lee, D. Lee, J.-Y. Kim, S. Lee and J. Yoon, Mater. Chem. Front., 2018, 2, 291–295 RSC.
- Q. Feng, Y. Li, L. Wang, C. Li, J. Wang, Y. Liu, K. Li and H. Hou, Chem. Commun., 2016, 52, 3123–3126 RSC.
- J. M. Han, M. Xu, B. Wang, N. Wu, X. Yang, H. Yang, B. J. Salter and L. Zang, J. Am. Chem. Soc., 2014, 136, 5090–5096 CrossRef CAS PubMed.
- J. Xie, Y. Wang, W. Liu, X. Yin, L. Chen, Y. Zou, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt, G. Liu and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 7500–7504 CrossRef CAS PubMed.
- M. Gao, X. Jia, G. Kuang, Y. Li, D. Liang and Y. Wei, Macromolecules, 2009, 42, 4273–4281 CrossRef CAS.
- Z. Guo, W. Zhu, Y. Xiong and H. Tian, Macromolecules, 2009, 42, 1448–1453 CrossRef CAS.
- H. Mori, K. Nishino, K. Wada, Y. Morisaki, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2018, 2, 573–579 RSC.
- J. Feng, K. Tian, D. Hu, S. Wang, S. Li, Y. Zeng, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2011, 50, 8072–8076 CrossRef CAS PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed.
- X. Meng, C. Chen, G. Qi, X. Li, K. Wang, B. Zou and Y. Ma, ChemNanoMat, 2017, 3, 569–574 CrossRef CAS.
- Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed.
- S. Zhu, M. Li, L. Sheng, P. Chen, Y. Zhang and S. X.-A. Zhang, Analyst, 2012, 137, 5581–5585 RSC.
- I. Zhang, Y. Wang, C. Wan, Z. Xing, W. Li, M. Li and S. X.-A. Zhang, RSC Adv., 2015, 5, 66416–66419 RSC.
- Z. Tian, W. Wu, W. Wan and A. D. Li, J. Am. Chem. Soc., 2011, 133, 16092–16100 CrossRef CAS PubMed.
- S. M. Borisov, R. Seifner and I. Klimant, Appl. Microbiol. Biotechnol., 2011, 400, 2463–2474 CAS.
- Q. Chen, D. Zhang, G. Zhang, X. Yang, Y. Feng, Q. Fan and D. Zhu, Adv. Funct. Mater., 2010, 20, 3244–3251 CrossRef CAS.
- H. J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed.
- I. Gallardo, G. Guirado, J. Hernando, S. Morais and G. Prats, Chem. Sci., 2016, 7, 1819–1825 RSC.
- G. Szaloki, G. Sevez, J. Berthet, J. L. Pozzo and S. Delbaere, J. Am. Chem. Soc., 2014, 136, 13510–13513 CrossRef CAS PubMed.
- H.-R. Zheng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, RSC Adv., 2016, 6, 41002–41006 RSC.
- S. Z. Pu, Q. Sun, C. B. Fan, R. J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4, 3075–3093 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- R. Zhang, Z. Wang, Y. Wu, H. Fu and J. Yao, Org. Lett., 2008, 10, 3065–3068 CrossRef CAS PubMed.
- S. Mo, Q. Meng, S. Wan, Z. Su, H. Yan, B. Z. Tang and M. Yin, Adv. Funct. Mater., 2017, 27, 1701210 CrossRef.
- J. Hu, L. Dai and S. Liu, Macromolecules, 2011, 44, 4699–4710 CrossRef CAS.
- P. Wei, J.-X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 1966–1975 CrossRef CAS PubMed.
- J. Su, T. Fukaminato, J.-P. Placial, T. Onodera, R. Suzuki, H. Oikawa, A. Brosseau, F. Brisset, R. Pansu, K. Nakatani and R. Métivier, Angew. Chem., Int. Ed., 2016, 55, 3662–3666 CrossRef CAS PubMed.
- X. Su, Y. Ji, W. Pan, S. Chen, Y.-M. Zhang, T. Lin, L. Liu, M. Li, Y. Liu and S. X.-A. Zhang, J. Mater. Chem. C, 2018, 6, 6940–6948 RSC.
- J.-X. Liu, B. Xin, C. Li, W.-L. Gong, Z.-L. Huang, B.-Z. Tang and M.-Q. Zhu, J. Mater. Chem. C, 2017, 5, 9339–9344 RSC.
- S. Tang, Y. Zhang, P. Dhakal, L. Ravelo, C. L. Anderson, K. M. Collins and F. M. Raymo, J. Am. Chem. Soc., 2018, 140, 4485–4488 CrossRef CAS PubMed.
- X. Su, Y. Wang, X. Fang, Y. M. Zhang, T. Zhang, M. Li, Y. Liu, T. Lin and S. X.-A. Zhang, Chem. – Asian J., 2016, 11, 3205–3212 CrossRef CAS PubMed.
- M. Rosas-Carbajal, J. C. Komorowski, F. Nicollin and D. Gibert, Sci. Rep., 2016, 6, 29899 CrossRef CAS PubMed.
- D. A. Butterfield, K. i. Nakamura, B. Takano, M. D. Lilley, J. E. Lupton, J. A. Resing and K. K. Roe, Geology, 2011, 39, 803–806 CrossRef CAS.
- T. Feseker, A. Boetius, F. Wenzhofer, J. Blandin, K. Olu, D. R. Yoerger, R. Camilli, C. R. German and D. de Beer, Nat. Commun., 2014, 5, 5385 CrossRef CAS PubMed.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed.
- S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906–6910 CrossRef CAS PubMed.
- X. Su, Q. Yu, T. Zhang, Y.-M. Zhang, L. Yu, I. Zhang, M. Li, Y. Liu and S. X.-A. Zhang, RSC Adv., 2016, 6, 90305–90309 RSC.
- Y. Zhang, L. Guan, H. Yu, Y. Yan, L. Du, Y. Liu, M. Sun, D. Huang and S. Wang, Anal. Chem., 2016, 88, 4426–4431 CrossRef CAS PubMed.
- Y. Sun, D. Zhao, S. Fan, L. Duan and R. Li, J. Agric. Food Chem., 2014, 62, 3405–3409 CrossRef CAS PubMed.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- F. Wang, C. A. DeRosa, M. L. Daly, D. Song, M. Sabat and C. L. Fraser, Mater. Chem. Front., 2017, 1, 1866–1874 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS PubMed.
- S. Zhang, Y. Dai, S. Luo, Y. Gao, N. Gao, K. Wang, B. Zou, B. Yang and Y. Ma, Adv. Funct. Mater., 2017, 27, 1602276 CrossRef.
- E. I. Balmond, B. K. Tautges, A. L. Faulkner, V. W. Or, B. M. Hodur, J. T. Shaw and A. Y. Louie, J. Org. Chem., 2016, 81, 8744–8758 CrossRef CAS PubMed.
- J. Oriou, F. Ng, G. Hadziioannou, G. Garbay, M. Bousquet, L. Vignau, E. Cloutet and C. Brochon, Polym. Chem., 2014, 5, 7100–7108 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1821887. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00544c |
| ‡ Tingting Lin and Xing Su contributed equally to this work. |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Minjie
Li
c,
Minjie
Li