
Emission mechanism understanding and tunable persistent room temperature phosphorescence of amorphous nonaromatic polymers†
Qing
Zhou
 ,
Ziyi
Wang
,
Xueyu
Dou
,
Yunzhong
Wang
,
Saier
Liu
,
Yongming
Zhang
and
Wang Zhang
Yuan
*
,
Ziyi
Wang
,
Xueyu
Dou
,
Yunzhong
Wang
,
Saier
Liu
,
Yongming
Zhang
and
Wang Zhang
Yuan
*
School of Chemistry and Chemical Engineering, Shanghai Key Lab of Electrical Insulation and Thermal Aging, Shanghai Electrochemical Energy Devices Research Center, Shanghai Jiao Tong University, No. 800 Dongchuan Rd, Minhang District, Shanghai 200240, China. E-mail: wzhyuan@sjtu.edu.cn
First published on 4th December 2018
Abstract
Deciphering the emission mechanism of nonconventional luminogens and achieving persistent room temperature phosphorescence (p-RTP) from pure organic compounds have drawn increasing attention due to their significant fundamental importance and promising applications. Previous reports on nonconventional luminogens, however, mainly focus on fluorescence, while advances in pure organic p-RTP are generally restricted to aromatic crystals or host–guest systems. Herein, we report the unique intrinsic emission and moreover p-RTP in amorphous nonaromatic polymers of poly(acrylic acid) (PAA), polyacrylamide (PAM) and poly(N-isopropylacrylamide) (PNIPAM). These polymers are nonluminescent in dilute solutions, while being highly emissive in concentrated solutions, nanosuspensions and solid powders/films. This can be rationalized by the clustering-triggered emission (CTE) mechanism, as supported by further thermoresponsive emission, cryogenic and aggregation-induced emission (AIE) experiments, alongside single crystal analysis. Furthermore, PAA and PAM solids under ambient conditions, and PNIPAM solids under vacuum or under nitrogen, demonstrate distinct p-RTP, which can be enhanced through further ionization or pressurization. These results not only refresh our understanding of the emission mechanism of nonaromatic polymers, but also enable the facile fabrication and application of pure organic p-RTP luminogens from readily available compounds, thus providing an important step forward in both nonconventional luminogens and p-RTP.
Introduction
Nonconventional luminogens without classic conjugates are currently attracting increasing attention owing to their fundamental significance and diverse technical applications. These luminogens normally enjoy the merits of good hydrophilicity, facile preparation, environmental-friendliness and outstanding biocompatibility, which render them highly suitable for biological and medical applications.1–10 Despite different systems being reported, such as poly(amidoamine)s (PAMAM),6,10 poly(amino ester)s (PAE),11 poly(ether amide)s (PEA),12 polyethylenimines (PEI)13 and peptides,14 thus far, their emission mechanism remains a controversial issue, with various assumptions being suggested.5a,14 For example, some researchers argue the essential roles of oxidation,6a,10b crosslinking and molecular architecture,13 while others emphasize the external stimuli4b and hydrogen bonding,14 making it difficult to reach a consensus. Previously, based on the observation of intrinsic emissions in rice, starch and cellulose, we proposed the clustering-triggered emission (CTE) mechanism,3a which can also explain other nonconventional luminogens such as natural products,3 synthetic compounds1,5a and biomolecules.15 In this scenario, the clustering of nonconventional chromophores with lone pairs (n) and/or π electrons results in effective through-space electronic communications, which give rise to extended electron delocalization and simultaneously rigidified conformations;1,3,5a,b,15 consequently, the clusters can be easily excited to generate remarkable emissions. Further insights into nonconventional luminogens will be beneficial to the understanding of their emission mechanism and future exploration of novel emitters.Meanwhile, as excellent alternatives to organometallic complexes, pure organic luminogens with room temperature phosphoresce (RTP) have also received considerable attention owing to their unprecedented potential for optoelectronic and biomedical applications.16–25 Generally, to overcome the spin-forbidden nature of singlet–triplet transitions and to suppress the nonradiative processes, on one hand, researchers have tried to enhance spin–orbit coupling (SOC), and thus promote intersystem crossing (ISC) by incorporation of aromatic carbonyl, heavy atoms or hetero atoms.22 On the other hand, they have endeavored to construct a rigid environment through crystallization,17a,18 embedding in a matrix16a,19,20 or supramolecular interactions.21 Thus far, reported RTP systems, however, are predominantly aromatics, with emphasis on crystalline compounds.17a,22 Less attention has been paid to amorphous nonconventional luminogens, particularly those with persistent RTP (p-RTP). Such a situation might stem from the active vibrational stretching in amorphous states, and moreover, the previously preconceived notion of the nonluminescence for nonconjugated compounds. It would be interesting to obtain p-RTP from amorphous nonconventional luminogens, which could inspire new applications in view of their unique photophysical properties, and offer further insights into the emission mechanism and the origin of p-RTP.6,22
p-RTP from amorphous nonaromatic luminogens, nevertheless, remains in its infancy.3b,15,24 Recently, we observed such emissions from an example nonaromatic poly(amino acid) of ε-poly-L-lysine (ε-PLL)15 and sodium alginates (SA).3b Considering the prevalence of RTP and persistent phosphorescence at 77 K in nonconventional luminogens,5,15 it is rational to speculate the possibility of realizing p-RTP in nonaromatic compounds with effective intra- and intermolecular interactions. In view of this, herein, three amorphous polymers of poly(acrylic acid) (PAA), polyacrylamide (PAM) and poly(N-isopropylacrylamide) (PNIPAM) were synthesized and studied (Fig. 1A). They were carefully selected based on the following considerations: first, clustering of the pendants may generate intense emission;1 second, intra- and intermolecular interactions could be modulated by the changing structure; finally, p-RTP might be achieved and finely tuned due to the involvement of n electrons and the variation of pendants. Indeed, they emit intense blue lights in concentrated solutions and solid states. Amazingly, bright p-RTP are clearly visualized in PAA and PAM solids, even at ambient conditions, which can be enhanced through ionization. Furthermore, despite the fact that no RTP is detected for PNIPAM in air, isolation of oxygen endows it with noticeable p-RTP. These results strongly suggest that no aromatics are a prerequisite for the light emission of such nonconjugated polymers. Moreover, the unique p-RTP from these amorphous and polymeric nonaromatics not only provides more opportunities toward emerging advanced applications, but also offers new aspects for deciphering the emission mechanism and the origin of triplet excitons.
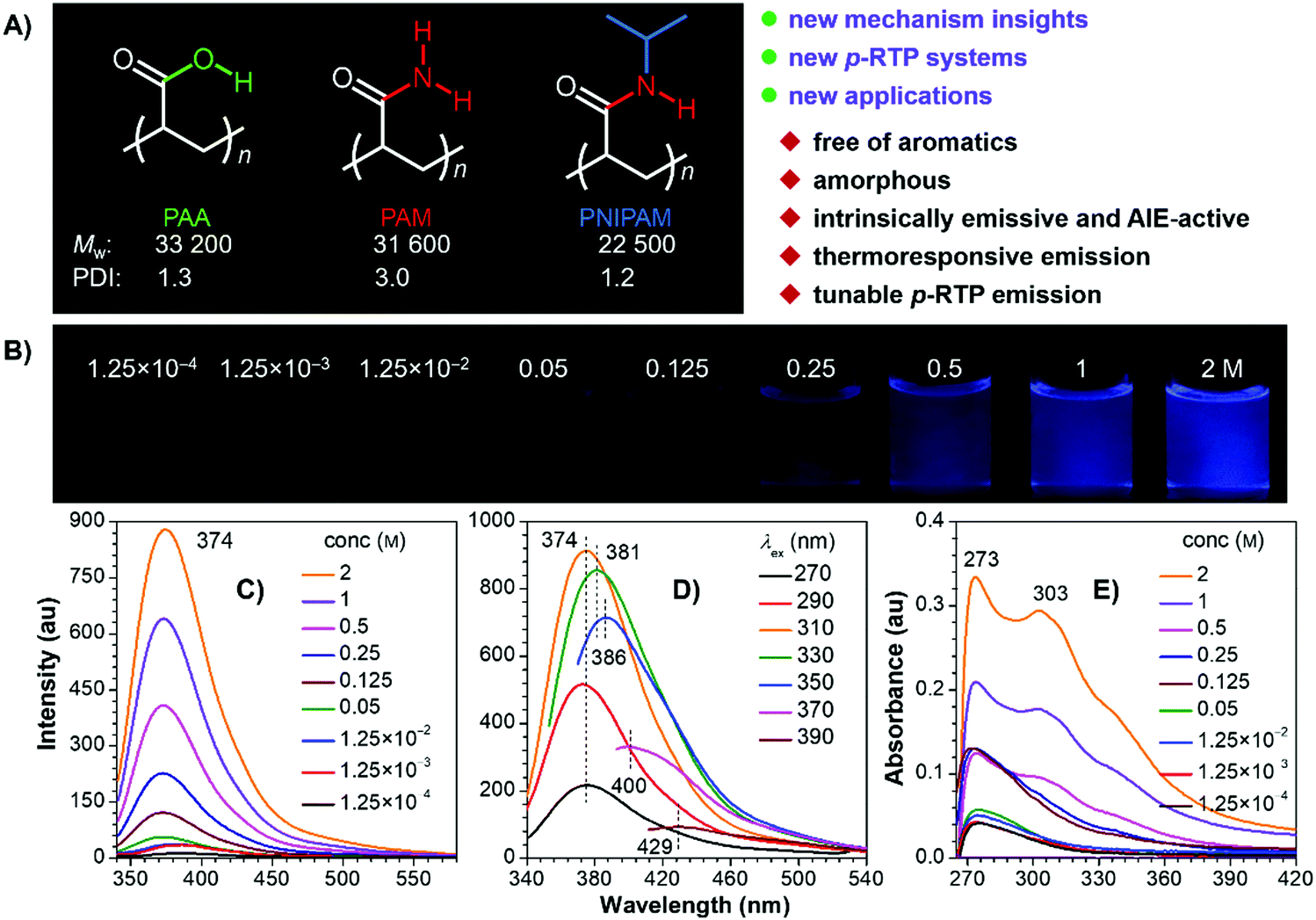 | ||
| Fig. 1 (A) Structures, Mw, PDI and features of PAA, PAM and PNIPAM. (B) Different PNIPAM/DMF solutions taken under 365 nm UV light. PL spectra of (C) different PNIPAM/DMF solutions (λex = 310 nm) and (D) 2 M solution with different λexs. (E) Absorption spectra of different PNIPAM/DMF solutions. | ||
Results and discussion
The polymers were facilely prepared by radical polymerization using 2,2′-azobisisobutyronitrile (AIBN) as the initiator (Scheme S1, ESI†), aiming to avoid the introduction of any aromatics. 1H and 13C NMR spectra clearly suggest the successful preparation of the target polymers (Fig. S1, ESI†), whose weight-averaged molecular weight (Mw) and polydispersity index (PDI) are 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200/31600/22500 and 1.3/3.0/1.2 for PAA/PAM/PNIPAM (Fig. 1A), respectively. They are nonluminescent in dilute solutions, but become emissive when concentrated (Fig. 1B and C and Fig. S2–S5, ESI†), exhibiting concentration-enhanced emission characteristics, which are similar to those observed in other nonconventional luminogens.1,3b,5a Taking PNIPAM as an example, its dilute dimethylformamide (DMF) solutions (i.e. 1.25 × 10−3 M) display no visible emission, with rather low photoluminescence (PL) signals being recorded (Fig. 1B and C). Weak but visible PL around 374 nm is detected when the concentration grows to 0.125 M, and even brighter emission is observed for the 2 M solution, accompanying the greatly boosted quantum efficiency (Φ) from nearly zero (1.25 × 10−4 M) to 8.9%. Meanwhile, when irradiated with different excitation wavelength (λexs), the 2 M solution emits with changing maxima at 374, 381, 386, 400 and 429 nm (Fig. 1D). Such excitation-dependent emission strongly indicates the presence of multiple emissive species. In addition, progressively enhanced absorption with extended edge is observed as the concentration increases (Fig. 1E), which implies the formation of new species with enlarged conjugations. Similar photophysical behaviors are also found in other DMF and/or aqueous solutions of the polymers (Fig. S2–S5, ESI†), thus suggesting their general underlying mechanism.
200/31600/22500 and 1.3/3.0/1.2 for PAA/PAM/PNIPAM (Fig. 1A), respectively. They are nonluminescent in dilute solutions, but become emissive when concentrated (Fig. 1B and C and Fig. S2–S5, ESI†), exhibiting concentration-enhanced emission characteristics, which are similar to those observed in other nonconventional luminogens.1,3b,5a Taking PNIPAM as an example, its dilute dimethylformamide (DMF) solutions (i.e. 1.25 × 10−3 M) display no visible emission, with rather low photoluminescence (PL) signals being recorded (Fig. 1B and C). Weak but visible PL around 374 nm is detected when the concentration grows to 0.125 M, and even brighter emission is observed for the 2 M solution, accompanying the greatly boosted quantum efficiency (Φ) from nearly zero (1.25 × 10−4 M) to 8.9%. Meanwhile, when irradiated with different excitation wavelength (λexs), the 2 M solution emits with changing maxima at 374, 381, 386, 400 and 429 nm (Fig. 1D). Such excitation-dependent emission strongly indicates the presence of multiple emissive species. In addition, progressively enhanced absorption with extended edge is observed as the concentration increases (Fig. 1E), which implies the formation of new species with enlarged conjugations. Similar photophysical behaviors are also found in other DMF and/or aqueous solutions of the polymers (Fig. S2–S5, ESI†), thus suggesting their general underlying mechanism.
The preceding results clearly indicate that the emission of these nonconventional luminogens does not rely on any aromatics, and is also irrelevant with oxidation or specific structures, all of which were previously regarded as essential roles for the emission of different systems.6a,9b,10b,13 These behaviors, however, can well be rationalized by the CTE mechanism. When dissolved in dilute solutions, COOH, CONH2 and CONH(i-Pr) groups are predominantly dispersed along the polymer chains as individuals, which are not easily excited. On the contrary, in concentrated solutions, polymer chains are collapsed, allowing the pendant moieties to approach one another in close proximity. The clustering of pendant groups facilitates effective intra- and intermolecular interactions among π and n electrons, resulting in chromophores with effective through-space electronic communications and consequently extended delocalization and simultaneously rigidified conformations; therefore readily being excited to generate remarkable emissions. Notably, DMF solutions have much higher efficiencies compared with those of their aqueous solutions,26 presumably because the hydrogen bonds between water and pendants are unfavorable to the emission.
To further probe the emission mechanism, additional experiments were conducted. As an archetypal thermoresponsive polymer, PNIPAM has a lower critical solution temperature (LCST) of around 32 °C.27 Its aqueous solutions are highly transparent at 25 °C, whose emission is hardly visualized until the concentration reaches 0.25 M (Fig. 2A and B). When heated to 40 °C, apparent turbidity stars at 1.25 × 10−3 M, with greatly decreased transmittance and remarkably enhanced emission (Fig. 2B–D). This phenomenon is associated with a coil-to-globule transition of the polymer chains, which results in the formation of aggregates (Fig. 2E). Notably, despite the 0.125 M solution remaining nonemissive at 25 °C, its even more dilute counterparts (1.25 × 10−2 and 0.05 M) display feeble yet visible PL at 40 °C, which verifies the crucial role of pendant aggregation for the emission. Meanwhile, upon cooling to 77 K, no obvious emission is noticed for the dilute solutions (i.e. 1.25 × 10−2 M) (Fig. 2F and Fig. S6, ESI†); however, for the 1.25 × 10−3 M solution, upon aggregation in 5/95 tetrahydrofuran (THF)/n-hexane, bright PL is observed at room temperature, exhibiting typical aggregation-induced emission (AIE) characteristics (Fig. 2G).28 Similar results are also found in other polymers (Fig. S7–S10, ESI†), which indicates that without clustering of the pendants, sole conformation rigidification is impossible to generate effective emission. It is also noted that remarkably boosted PL along with persistent phosphorescence is observed for the concentrated solutions, owing to further conformation rigidification of existing clusters. Additionally, after ceasing the 365 and 312 nm ultraviolet (UV) irradiation, green and blue afterglows are observed (Fig. 2F and Fig. S6–S8, ESI†), respectively, thus testifying the presence of heterogeneous populations of emission species.
 | ||
| Fig. 2 Photographs of PNIPAM/water solutions taken under (A) room light and (B) 365 nm UV light at 25 and 40 °C. (C) Transmittance, (D) emission spectra and (E) dynamic light scattering result of 1.25 × 10−3 M PNIPAM/water solution at 25 and 40 °C. (F) Photographs of PNIPAM/DMF solutions taken under 365 and 312 nm UV lights or after ceasing the irradiation at 77 K. (G) PL spectra (λex = 340 nm) and photographs taken under 365 nm UV light of PNIPAM in THF and 5/95 THF/n-hexane mixture (1.25 × 10−3 M). | ||
The unique intrinsic emission and AIE behavior of the polymers prompt us to further investigate their solid emissions. As can be seen from Fig. 3 and Fig. S11 (ESI†), PAA/PAM/PNIPAM powders and cast films depict excitation-dependent luminescence and emit bright blue lights under UV irradiation, with Φ (%) values of 4.5/8.3/9.6 and 5.7/13.7/12.4 (Table S1, ESI†), respectively. These values are much higher than those of their solutions, presumably owing to the stronger chain entanglement with more effective intra- and intermolecular interactions. Meanwhile, the higher efficiencies of the films compared with the powders strongly suggest more compact molecular packing and consequently depressed molecular motions in the former. Moreover, PAA/PAM powders and films demonstrate cyan p-RTP after ceasing the UV irradiation (Fig. 3B and Video S1, ESI†), with emission maxima (λem) and lifetimes (〈τ〉p) of 488/482, 504/489 nm and 41.8/97.6, 54.4/117.0 ms (Fig. 4 and Table S2, ESI†),29 respectively. X-ray diffraction (XRD) patterns of these solids show weak and broad diffusion diffractions, which clearly suggest their amorphous nature (Fig. S12, ESI†). Such p-RTP emissions from nonaromatic luminogens with well-defined chemical structures remain rare cases,3b,15 particularly at the amorphous state. Closer insights into these systems are not only beneficial to the in-depth mechanism understanding, but also offer opportunities for the development of new p-RTP luminogens with emerging applications. Based on the CTE mechanism, the origin of p-RTP can be understood as described below. First, the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups as well as O and N heteroatoms can promote the SOC; second, heterogeneous clusters provide enriched energy levels and thus narrowed energy gaps between excited singlet and triplet states. The above factors are favorable for the ISC processes to generate considerable triplet excitons, which can be stabilized through conformation rigidification and isolation from quenchers, thus yielding remarkable p-RTP. Meanwhile, a much longer 〈τ〉ps of PAM solids compared with those of PAA solids indicate their more rigidified conformations, which are consistent with the much higher glass transition temperatures (Tgs) of PAM powders/films (185.5/188.7 °C, 129.5/131.6 °C for PAA, Fig. S13, ESI†). No p-RTP, however, is observed for PNIPAM solids in air (Video S1, ESI†), which might be associated with its bulky i-Pr pendants (vide infra).
O groups as well as O and N heteroatoms can promote the SOC; second, heterogeneous clusters provide enriched energy levels and thus narrowed energy gaps between excited singlet and triplet states. The above factors are favorable for the ISC processes to generate considerable triplet excitons, which can be stabilized through conformation rigidification and isolation from quenchers, thus yielding remarkable p-RTP. Meanwhile, a much longer 〈τ〉ps of PAM solids compared with those of PAA solids indicate their more rigidified conformations, which are consistent with the much higher glass transition temperatures (Tgs) of PAM powders/films (185.5/188.7 °C, 129.5/131.6 °C for PAA, Fig. S13, ESI†). No p-RTP, however, is observed for PNIPAM solids in air (Video S1, ESI†), which might be associated with its bulky i-Pr pendants (vide infra).
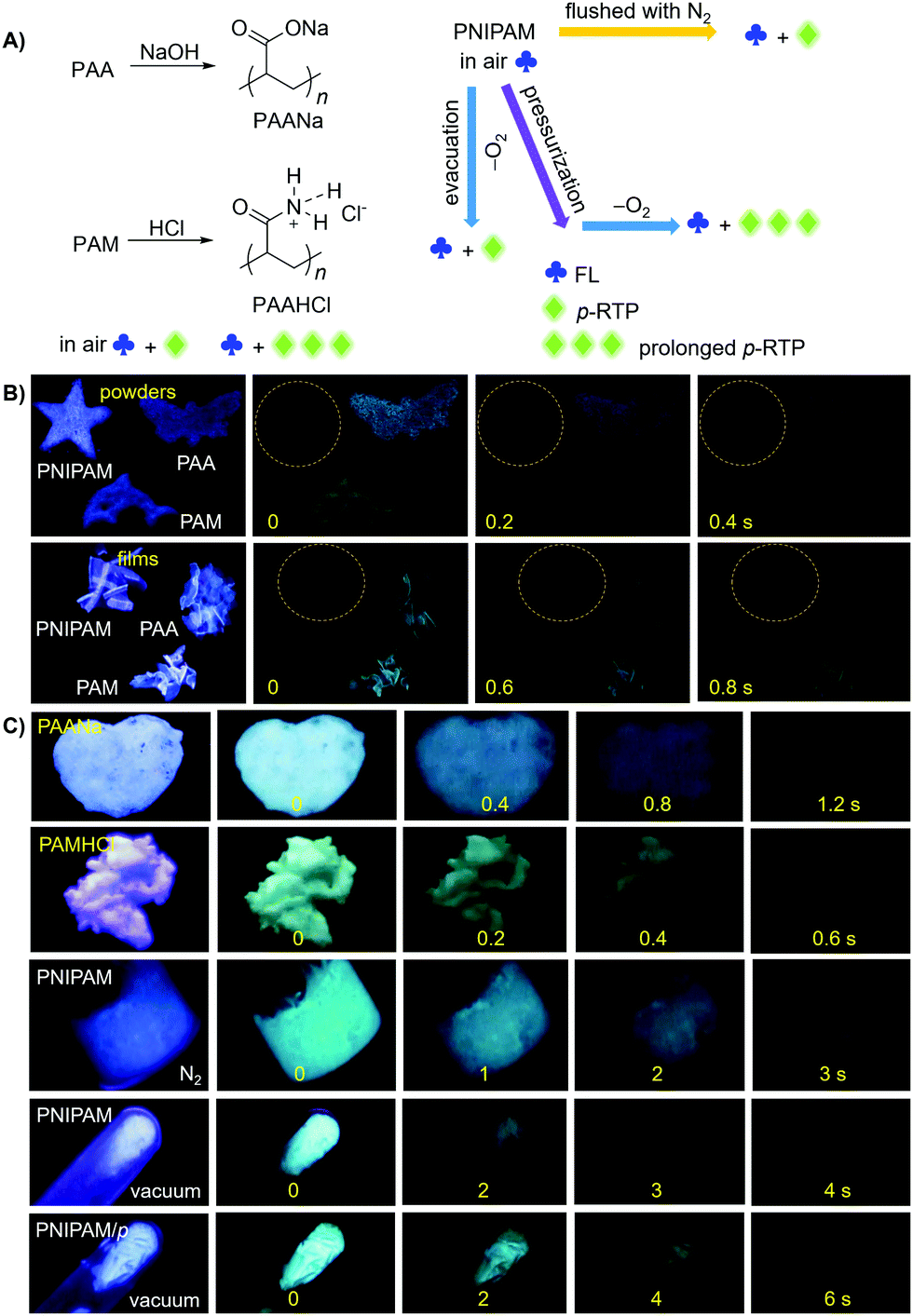 | ||
| Fig. 3 (A) Schematic illustration of the modulation of p-RTP properties of different polymers. (B and C) Photographs of different powders and films taken under 312 nm UV light or after ceasing the UV irradiation at ambient conditions, under nitrogen or in vacuum. PNIPAM/p: pressed PNIPAM powders under a pressure of 2000 kg cm−2 for 1 min. | ||
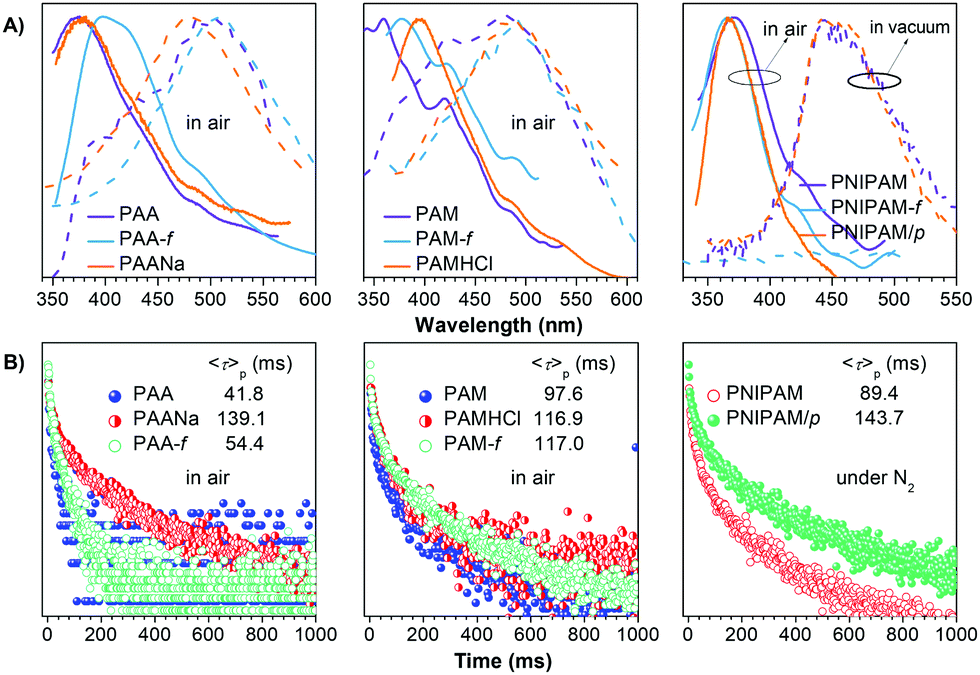 | ||
| Fig. 4 (A) Emission spectra with td of 0 (solid line) and 0.1 ms (dash line) and (B) RTP decay curves of different films (f) and powders in air, under nitrogen, or in vacuum. Detailed λexs for the emission measurement and monitored wavelengths for the lifetime test are listed in Tables S1 and S2 (ESI†). | ||
Notably, upon neutralization with NaOH or fuming with HCl (Fig. 3A), the resulting PAANa and PAMHCl powders show greatly enhanced efficiency and prolonged p-RTP (Fig. 3C), with Φ/〈τ〉p values of 7.6%/139.1 ms and 16.7%/116.9 ms (Fig. 4B), respectively, which should be ascribed to the stiffening of conformations through electrostatic interaction. Such ionization-enhanced p-RTP is further confirmed by the results of PAACa solids (Fig. S14, ESI†). Compared with PAANa and PAMHCl salts, even longer p-RTP is observed, which might be ascribed to the much stronger coordination between Ca2+ ions and carboxyls, thus resulting in clusters with more rigidified conformations. Surprisingly, when placed under nitrogen or in vacuum, p-RTP persisting for more than 3 s (〈τ〉p = 89.4 ms) is observed for PNIPAM powders (Fig. 3C, 4B and Video S2, ESI†),30 strongly indicating the predominant role of oxygen quenching rather than vibrational dissipations to the disappearance of p-RTP at ambient conditions. Such results should be attributed to the relatively bulky structure of the i-Pr group, which would decrease the packing density and subsequently increase the oxygen penetration in solids. Pressurization is expected to ensure much denser molecular packing, thus shortening the distance of pendants within clusters and making it more resistant to oxygen. Indeed, p-RTP persisting for ∼6 s (〈τ〉p = 143.7 ms) is recorded when pressurized PNIPAM powders are placed in vacuum (Fig. 3C, 4B and Video S2, ESI†). A dingle crystal structure of the monomers provides exact molecular packings, which afford significant implications on how polymer pendants interact with one another in aggregates, therefore beneficial to deciphering the emission mechanism. Both acrylamide (AM) and N-isopropyl acrylamide (NIPAM) single crystals were thus cultured, which generate blue lights accompanying multiple peaks under UV illumination (Fig. 5A and Fig. S15, ESI†). For AM, a nearly planar structure is found in crystals (Fig. 5B).31 There are large numbers of N–H⋯OC (1.977, 2.074 Å) and N–H⋯CO (2.895 Å) hydrogen bonds, which not only stiffen the conformations, but also facilitate the H–N⋯OC (2.854, 2.941 Å) electronic communications among N atoms and CO units (Fig. 5B). Consequently, an effective 3D through-space electronic communication channel is formed (Fig. 5C), thereby offering optically excitable conjugates with extended delocalization and rigidified conformations. Similarly, in NIPAM crystals, there are also abundant N–H⋯OC (2.004, 2.054 Å), C–H⋯OC (2.485, 2.543, 2.673 Å), and moreover H–N⋯OC (2.866, 2.918 Å) short contacts (Fig. S16, ESI†).
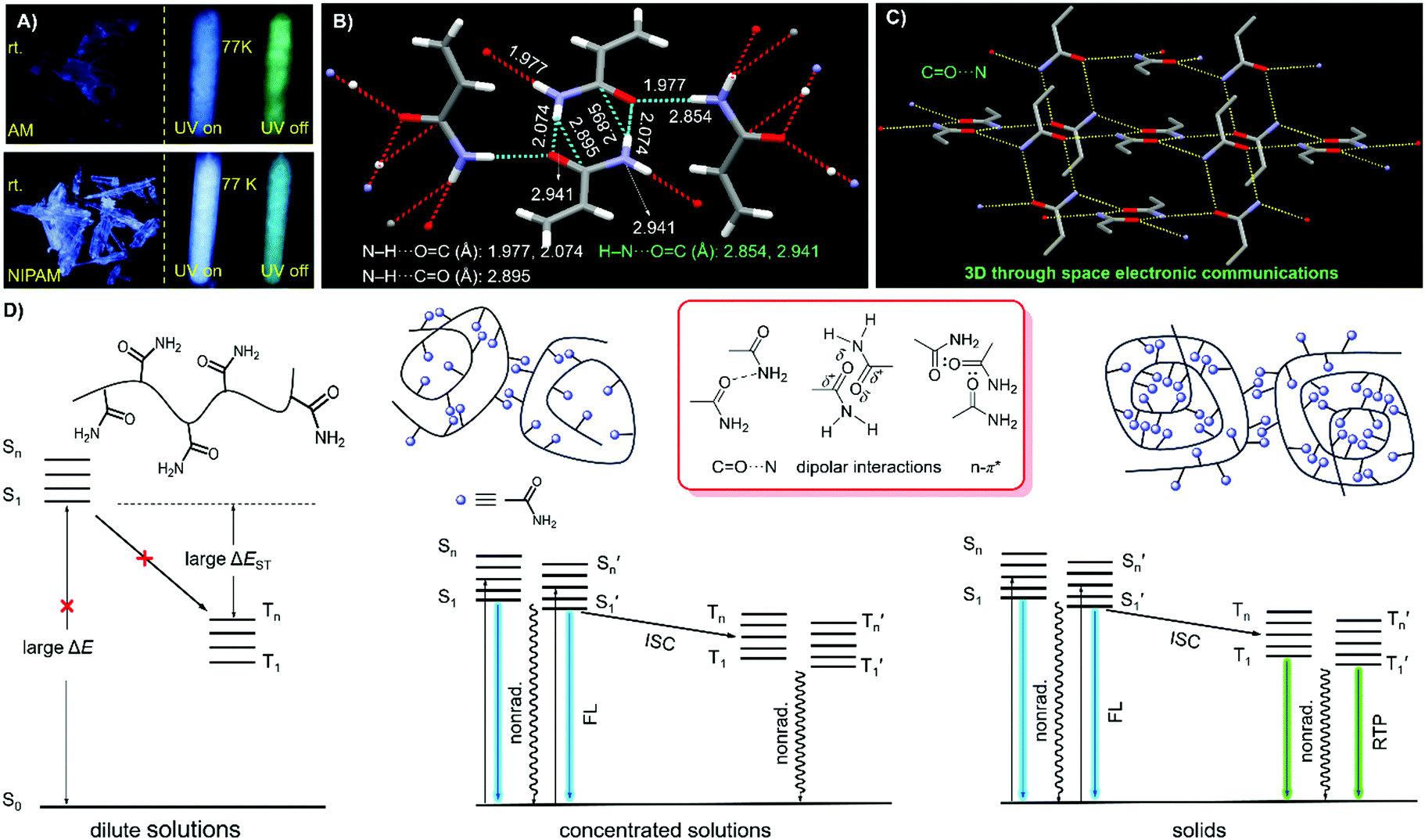 | ||
| Fig. 5 (A) Photographs of AM and NIPAM single crystals taken under 312 nm UV light or after ceasing the irradiation at room temperature (rt) and 77 K. Fragmental molecular packing of AM crystals with denoted (B) intermolecular interactions and (C) specific CO⋯N through-space electronic communications. (D) Schematic illustration and Jablonski diagram of the polymers (i.e. PAM) from isolated to aggregated states. | ||
Meanwhile, despite being placed in vacuum, no p-RTP can be detected for the monomer crystals. However, upon cooling to 77 K, bright and persistent green phosphorescence enduring for ∼3 s (AM) and ∼15 s (NIPAM) can be observed after ceasing the UV irradiation (Fig. 5A and Video S3, ESI†). It is also noted that while no emission of distilled AA can be observed under 312 nm UV light, bright blue light is observed at 77 K, along with green p-RTP after ceasing the irradiation (Fig. S17, ESI†). These results clearly suggest a powerful polymer effect, which should be ascribed to the conformation rigidification and decreased oxygen quenching induced by the chain entanglement and effective intra-/intermolecular interactions.
Considering all of the above results, it is rational to explain the photophysical properties of these nonaromatic polymers by the schematic illustration and corresponding Jablonski diagram demonstrated in Fig. 5D. In dilute solutions, individual chromophores with large energy gaps (ΔE) are not easily excited, therefore, no emission can be observed even at cryogenic temperatures. In the concentrated solutions, clustered chromophores with effective through-space electronic communications, such as CO⋯N, n–π* and dipolar interactions, are formed. The heterogeneity of the clusters provides diverse energy levels of both singlets and triplets, which may approach one another or even overlap, thus making the ISC processes highly possible. RTP emission, however, is difficult to achieve in solution due to active molecular motions and external quenching. In the solid state, bright emissions with both fluorescence and remarkable p-RTP can be realized in case of sufficient conformation rigidity and proper isolation from quenchers such as oxygen.
The combined properties of intrinsic emission with p-RTP features, biocompatibility and film-forming ability make these polymers applicable in anticounterfeiting, encryption, bioimaging, etc. Using the polymers and commercial highlighters, anticounterfeiting and information encryption have been demonstrated with respect to applications in security protection. As shown in Fig. 6A, a colorful bird becomes luminescent in another color model under UV light. After ceasing the irradiation, only the skeleton of the bird is visible, exhibiting multimodal anticounterfeiting properties. Meanwhile, a blue “CENTER” pattern composed of PAA, PAANa, PAM and PNIPAM powders is visible under 312 nm UV light; only green “CTE”, however, is seen because of their different RTP emission features. Additionally, these polymers are also promising for bioapplications. After incubation with 1 M PAM in McCoy's5A for 2 h, HCT116 cells exhibit bright blue emission under confocal microscopy, whereas no obvious PL signal is observed for the control (Fig. 6B and Fig. S18, ESI†), which suggest PAM is ready to stain cells. Furthermore, closer scrutinization reveals that PAM demonstrates somewhat specific imaging of endosomes, which is important to biomedical studies.
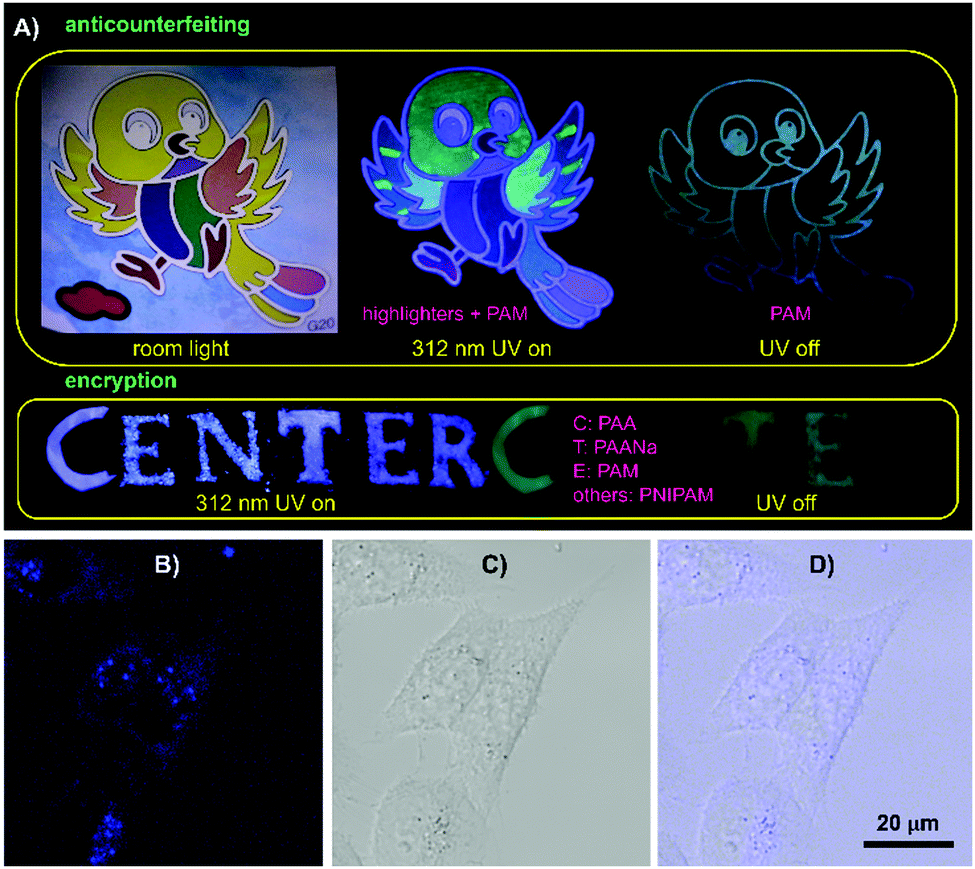 | ||
| Fig. 6 (A) Photographs of graphic security and information encryption made from varying polymers and commercial highlighters. Confocal luminescence images of HCT116 cells after incubation with 1 M PAM/McCoy's5A solution for 1.5 h. (B) Confocal image recorded under excitation at 405 nm, (C) bright field image and (D) corresponding overlay image. | ||
Conclusions
In summary, despite lacking any aromatics, intrinsic emission and moreover p-RTP are observed from a group of amorphous polymers. Concentration-enhanced emission features, and thermoresponsive emission of PNIPAM, as well as AIE and cryogenic experiments strongly suggest the CTE mechanism, which is further supported by the implications from emission and single crystal analysis of the monomers. Interestingly, while PAA and PAM solids show bright p-RTP in air, PNIPAM does not demonstrate noticeable RTP signals, owing to oxygen quenching. Furthermore, the p-RTP can be finely modulated by ionization, isolation of oxygen and pressurization (Fig. 3A), which indicates the tunability of p-RTP in such nonconjugated systems through the control of intra- and intermolecular interactions and isolation from or exposure to triplet quenchers. Their nonaromatic structure, biocompatibility, film-forming ability and p-RTP emission offer high potential for diverse applications, such as anticounterfeiting, encryption and bioimaging. These results, on one hand, shed new light on the origin of the unique emission from nonconventional luminogens, which is instructive for future discovery and fabrication of novel luminogens. On the other hand, the results also provide opportunities to achieve metal-free p-RTP from amorphous compounds, thus paving the way for the fundamental study and emerging applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51473092 and 51822303) and the Shanghai Rising-Star Program (15QA1402500).Notes and references
- (a) W. Z. Yuan and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 560 CrossRef; (b) Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef PubMed.
- (a) E. Zhao, J. W. Y. Lam, L. Meng, Y. Hong, H. Deng, G. Bai, X. Huang, J. Hao and B. Z. Tang, Macromolecules, 2015, 48, 64 CrossRef CAS; (b) A. Pucci, R. Rausa and F. Ciardell, Macromol. Chem. Phys., 2008, 209, 900 CrossRef CAS; (c) M. Fang, J. Yang, X. Xiang, Y. Xie, Y. Q. Dong, Q. Peng, Q. Li and Z. Li, Mater. Chem. Front., 2018, 2, 2124 RSC.
- (a) Y. Gong, Y. Tan, J. Mei, Y. Zhang, W. Z. Yuan, Y. Zhang, J. Sun and B. Z. Tang, Sci. China: Chem., 2013, 56, 1178 CrossRef CAS; (b) X. Dou, Q. Zhou, X. Chen, Y. Tan, X. He, P. Lu, K. Sui, B. Z. Tang, Y. Zhang and W. Z. Yuan, Biomacromolecules, 2018, 19, 2014 CrossRef CAS PubMed.
- (a) M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581 CrossRef CAS PubMed; (b) R. B. Restani, P. I. Morgado, M. P. Ribeiro, I. J. Correia, A. Aguiar-Ricardo and V. D. B. Bonifacio, Angew. Chem., Int. Ed., 2012, 51, 5162 CrossRef CAS PubMed.
- (a) Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586 CrossRef CAS PubMed; (b) Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Macromol. Rapid Commun., 2018, 39, 1800528 CrossRef PubMed; (c) N. Jiang, G. F. Li, B. H. Zhang, D. X. Zhu, Z. M. Su and M. R. Bryce, Macromolecules, 2018, 51, 4178 CrossRef CAS.
- (a) W. I. Lee, Y. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358 CrossRef CAS PubMed; (b) D. Wang and T. Imae, J. Am. Chem. Soc., 2004, 126, 13204 CrossRef CAS PubMed; (c) H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476 CrossRef CAS.
- (a) Q. Zhang, Q. Mao, C. Shang, Y. N. Chen, X. Peng, H. Tan and H. Wang, J. Mater. Chem. C, 2017, 5, 3699 RSC; (b) X. Guan, D. Zhang, T. Jia, Y. Zhang, L. Meng, Q. Jin, H. Ma, D. Lu, S. Lai and Z. Lei, Ind. Eng. Chem. Res., 2017, 56, 3913 CrossRef CAS; (c) Z. Guo, Y. Ru, W. Song, Z. Liu, X. Zhang and J. Qiao, Macromol. Rapid Commun., 2017, 38, 1700099 CrossRef PubMed.
- (a) Y. Du, H. Yan, W. Huang, F. Chai and S. Niu, ACS Sustainable Chem. Eng., 2017, 5, 6139 CrossRef CAS; (b) Z. Zhang, S. Feng and J. Zhang, Macromol. Rapid Commun., 2016, 37, 318 CrossRef CAS PubMed; (c) W. Yu, Y. Wu, J. Chen, X. Duan, X. F. Jiang, X. Qiu and Y. Li, RSC Adv., 2016, 6, 51257 RSC.
- (a) R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard III and B. Z. Tang, Polym. Chem., 2017, 8, 1722 RSC; (b) J.-J. Yan, Z.-K. Wang, X.-S. Lin, C.-Y. Hong, H.-J. Liang, C.-Y. Pan and Y.-Z. You, Adv. Mater., 2012, 24, 5617 CrossRef CAS PubMed.
- (a) Y. J. Tsai, C. C. Hu, C. C. Chu and T. Imae, Biomacromolecules, 2011, 12, 4283 CrossRef CAS PubMed; (b) L. Cao, S. Y. Lin, T. H. Wu, Y. C. Jao, C. P. Liu, H. Y. Lin, L. W. Lo and C. S. Yang, Chem. – Eur. J., 2011, 17, 7158 CrossRef PubMed.
- D. Wu, Y. Liu, C. He and S. H. Goh, Macromolecules, 2005, 38, 9906 CrossRef CAS.
- Y. Lin, J. W. Gao, H. W. Liu and Y. S. Li, Macromolecules, 2009, 42, 3237 CrossRef CAS.
- L. Pastor-Perez, Y. Chen, Z. Shen, A. Lahoz and S. E. Stiriba, Macromol. Rapid Commun., 2007, 28, 1404 CrossRef CAS.
- (a) A. Shukla, S. Mukherjee, S. Sharma, V. Agrawal, K. V. R. Kishan and P. Guptasarma, Arch. Biochem. Biophys., 2004, 428, 144 CrossRef CAS PubMed; (b) D. Pinotsi, L. Grisanti, P. Mahou, R. Gebauer, C. F. Kaminski, A. Hassanali and G. S. K. Schierle, J. Am. Chem. Soc., 2016, 138, 3046 CrossRef CAS PubMed.
- X. Chen, W. Luo, H. Ma, Q. Peng, W. Z. Yuan and Y. Zhang, Sci. China: Chem., 2018, 61, 351 CrossRef CAS.
- (a) W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef CAS; (b) O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed; (c) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed.
- (a) Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195 CrossRef CAS PubMed; (b) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- (a) R. Kabe and C. Adachi, Nature, 2017, 550, 384 CrossRef CAS PubMed; (b) Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed; (c) J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- (a) S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386 CrossRef CAS; (b) D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916 CrossRef CAS PubMed; (c) D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325 CrossRef CAS PubMed; (d) Y. Katsurada, S. Hirata, K. Totani, T. Watanabe and M. Vacha, Adv. Opt. Mater., 2015, 3, 1726 CrossRef CAS.
- (a) H. Chen, X. Yao, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1397 CrossRef CAS; (b) C. Zhou, T. Xie, R. Zhou, C. O. Trindle, Y. Tikman, X. Zhang and G. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 17209 CrossRef CAS PubMed; (c) Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed; (d) M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- Y. Gong, H. Chen, X. Ma and H. Tian, ChemPhysChem, 2016, 17, 1934 CrossRef CAS PubMed.
- For recent reviews, see: (a) M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081 RSC; (b) S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef; (c) S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920 CrossRef CAS PubMed.
- (a) G. He, W. T. Delgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587 CrossRef CAS PubMed; (b) T. Ogoshi, H. Tsuchida, T. Kakuta, T. A. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- (a) S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Angew. Chem., 2018, 130, 2417 CrossRef; (b) Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181 CrossRef CAS PubMed; (c) G. Bergamini, A. Fermi, C. Botta, U. Giovanella, S. DiMotta, F. Negri, R. Peresutti, M. Gingras and P. Ceroni, J. Mater. Chem. C, 2013, 1, 2717 RSC.
- (a) W. Zhao, Z. He, J. W. Lam, Q. Peng, H. Ma, Z. G. Shuai, X. Bai, J. H. Hao. and B. Z. Tang, Chem, 2016, 1, 592 CrossRef CAS; (b) G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed; (c) G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942 CrossRef CAS PubMed.
- While the Φ (%) values of 2 M DMF solutions of PAA/PAM/PNIPAM are 4.2/—/8.9, those for their aqueous counterparts are 2.2/3.1/6.2, respectively. The Φ value of PAM in DMF cannot be obtained since it is insoluble in DMF.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627 CrossRef CAS.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (b) J. Yang, L. Li, Y. Yu, Z. Ren, Q. Peng, S. Ye, Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91 RSC.
- With a td no less than 0.1 ms, all short-lived nanosecond signals can be excluded. Two 〈τ〉ps are obtained for each decay curve (Table S2, ESI†), herein, we give the much longer one since it determines the duration of the afterglow.
- Due to the limitation of the instrument to achieve vacuum, 〈τ〉p values of PNIPAM solids were only measured under nitrogen.
- CCDC 1842339 (AM) and 1842340 (NIPAM) contain the supplementary crystallographic data for this paper†.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterisation data including 1H and 13C NMR, emission spectra, lifetimes, photographs and chemical structure of samples. See DOI: 10.1039/c8qm00528a |
| This journal is © the Partner Organisations 2019 |