
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of μ-conotoxin lt5d†
A. M. B. Naraga ,
O. J. V. Belleza and
A. J. L. Villaraza*
,
O. J. V. Belleza and
A. J. L. Villaraza*
Institute of Chemistry, College of Science, National Science Complex, University of the Philippines, Diliman, Quezon City, 1100, Philippines. E-mail: alvillaraza@up.edu.ph
First published on 30th October 2018
Abstract
The total synthesis of μ-conotoxin lt5d is presented for the first time employing two different strategies. One involves glutathione-assisted oxidation where all disulphide linkages are formed simultaneously. Another involves orthogonal protection of cysteine residues, allowing the controlled formation of disulphide linkages sequentially. Both methods achieve the same peptide.
Liu et al. previously described the isolation and characterization of a neuroactive μ-conotoxin lt5d from Conus litteratus.1 Such peptides are of interest since conopeptides isolated from Conus snail venoms have high specificity and activity towards neuronal ligand- and voltage-gated channels, receptors and transporters, making them potential peptidic drug candidates for neurodegenerative and chronic pain conditions.2 For instance, one of which is the FDA approved ω-conotoxin MVIIA, commercially available as Prialt®, used for the management of severe chronic pain experienced by patients suffering from cancer, diabetes, multiple sclerosis, and HIV/AIDS.3–6 However, there are over 800 marine snails of genus Conus distributed throughout tropical and subtropical waters, each having approximately 50 to 200 conopeptides with only a few overlaps among species. Such biodiversity presents a rich source of novel and bioactive compounds.2,7
Lt5d, a 12-residue conotoxin belonging to the T-superfamily with cysteine framework V, was previously reported to have dose-dependent inhibitory activity against tetrodotoxin (TTX)-sensitive Na channel receptors in adult dorsal root ganglion (DRG) neurons.1,8,9 However, the total chemical synthesis of lt5d has yet to be described. In this study, we report the total chemical synthesis of lt5d using two different peptide folding strategies.
Scheme 1 summarizes the first method employed (i.e., Method 1). To achieve synthetic lt5d, the linear peptide was first synthesized on solid phase using N-Fmoc protected amino acids, in which the Cys residues were protected with the trityl (Trt) group. Afterwards, 1 mM oxidized glutathione (GSSG), together with equimolar amounts of reduced glutathione (GSH) and EDTA, were combined with a 20 μM solution of linear peptide to yield the folded product.10 GSSG acts as the oxidizing agent while GSH acts as the reducing agent, since GSH is known for correcting misbridged cysteine residues of proteins in the endoplasmic reticulum where proteins are folded in vivo.11,12 These conditions are widely adopted to mimic physiological conditions favouring the formation of thermodynamically stable folded proteins and cysteine-rich peptides in vitro.13–15
 | ||
| Scheme 1 Lt5d synthesis using glutathione-assisted oxidative folding (Method 1). | ||
Fig. 1 presents the HPLC chromatograms (C18) of pure linear and folded lt5d. A shift in retention time from tR = 24.3 min to 23.1 min is indicative of folding as the shape and the number of exposed residues change. This leads to less interaction with the hydrophobic C18 stationary phase and hence earlier elution of the folded peptide.
 | ||
| Fig. 1 RP-HPLC chromatograms of pure (a) linear and (b) folded lt5d, tR = 24.3 min and tR = 23.1 min, respectively. Analytical C18 column was used to profile the sample with a linear gradient from 15% to 45% solution B (90% MeCN in H2O with 0.1% TFA) for 35 min at 1 mL min−1 flow rate. Absorbance was monitored at 220 nm. | ||
The identities of the products were confirmed by high resolution electron spray ionization mass spectrometry in positive mode (HR-ESI(+)-MS). Fig. 2 shows the [M + H]+ molecular ion peaks at m/z = 1279.6920 and 1275.6353 which correspond to the calculated molecular weights of linear (1278.52 Da) and folded peptide (1274.49 Da), respectively. The 4 Da mass difference is due to the loss of four hydrogen atoms upon the formation of the two disulphide bridges.
 | ||
| Fig. 2 HR-ESI(+) mass spectra of pure (a) linear lt5d with the molecular ion peak at 1279.6920, and (b) folded lt5d at 1275.6353. Calculated monoisotopic mass of target linear and folded peptide is 1278.5229 and 1274.4916 Da, respectively. | ||
However, Method 1 assumes that disulphide bonds Cys2–Cys9 and Cys3–Cys10 were formed in solution by glutathione oxidation. In order to verify this, an alternative method in which the disulphide bonds are sequentially formed in a controlled manner by orthogonal cysteine protection was devised (i.e., Method 2), as described in Scheme 2.16,17 In brief, two different protecting groups, trityl (Trt) and acetoxymethyl (Acm), were used in each pair of Cys residues. Upon acidolytic cleavage of the peptide from the resin, the Trt protecting groups were removed from Cys2 and Cys9. As the Acm protecting groups are resistant to TFA cleavage, Cys3 and Cys10 remained protected under these conditions. Since the HPLC chromatogram (C18) of “crude” linear peptide 1 (Fig. 3a) showed a single peak at 21.39 min, the peptide was folded without further purification: linear peptide 1 was dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 DMSO/water and the oxidative folding reaction stirred for 24 h. The reaction was quenched by addition of an equal volume of acetic acid to the reaction mixture, forming partially folded peptide 2. The HPLC chromatogram (C18) of 2 (Fig. 3b) showed a slight decrease in the retention time of the major product (21.14 min), indicating the formation of the first disulphide bond, Cys2–Cys9. In order to form the second disulphide bond Cys3–Cys10, the Acm protecting groups were cleaved using iodine. This reaction was carried out by adding a 5-fold excess of iodine to the solution, and quenched by the addition of ascorbic acid, yielding fully folded lt5d peptide, as evidenced by mass spectrometry and HPLC (Fig. 3c). In contrast with the previous observation that a shorter retention time is observed for the folded peptide, in this case the retention time is slightly increased (23.20 min) due to the removal of the two hydrophilic Acm groups. The major peaks in the HPLC chromatograms of crude partially folded peptide 2 and fully folded lt5d were collected and analyzed using HR-ESI(+)-MS. Fig. 4 shows the molecular ion peaks observed at m/z = 1419.5576 for 2 (one disulphide bond, i.e., Cys2–Cys9) and m/z = 1275.4716 for lt5d (two disulphide bonds, i.e., Cys2–Cys9 and Cys3–Cys10). The mass difference of 144 Da between these compounds corresponds to the removal of the two Acm groups.
:2 DMSO/water and the oxidative folding reaction stirred for 24 h. The reaction was quenched by addition of an equal volume of acetic acid to the reaction mixture, forming partially folded peptide 2. The HPLC chromatogram (C18) of 2 (Fig. 3b) showed a slight decrease in the retention time of the major product (21.14 min), indicating the formation of the first disulphide bond, Cys2–Cys9. In order to form the second disulphide bond Cys3–Cys10, the Acm protecting groups were cleaved using iodine. This reaction was carried out by adding a 5-fold excess of iodine to the solution, and quenched by the addition of ascorbic acid, yielding fully folded lt5d peptide, as evidenced by mass spectrometry and HPLC (Fig. 3c). In contrast with the previous observation that a shorter retention time is observed for the folded peptide, in this case the retention time is slightly increased (23.20 min) due to the removal of the two hydrophilic Acm groups. The major peaks in the HPLC chromatograms of crude partially folded peptide 2 and fully folded lt5d were collected and analyzed using HR-ESI(+)-MS. Fig. 4 shows the molecular ion peaks observed at m/z = 1419.5576 for 2 (one disulphide bond, i.e., Cys2–Cys9) and m/z = 1275.4716 for lt5d (two disulphide bonds, i.e., Cys2–Cys9 and Cys3–Cys10). The mass difference of 144 Da between these compounds corresponds to the removal of the two Acm groups.
 | ||
| Scheme 2 Lt5d synthesis using orthogonal cysteine protection (Method 2). | ||
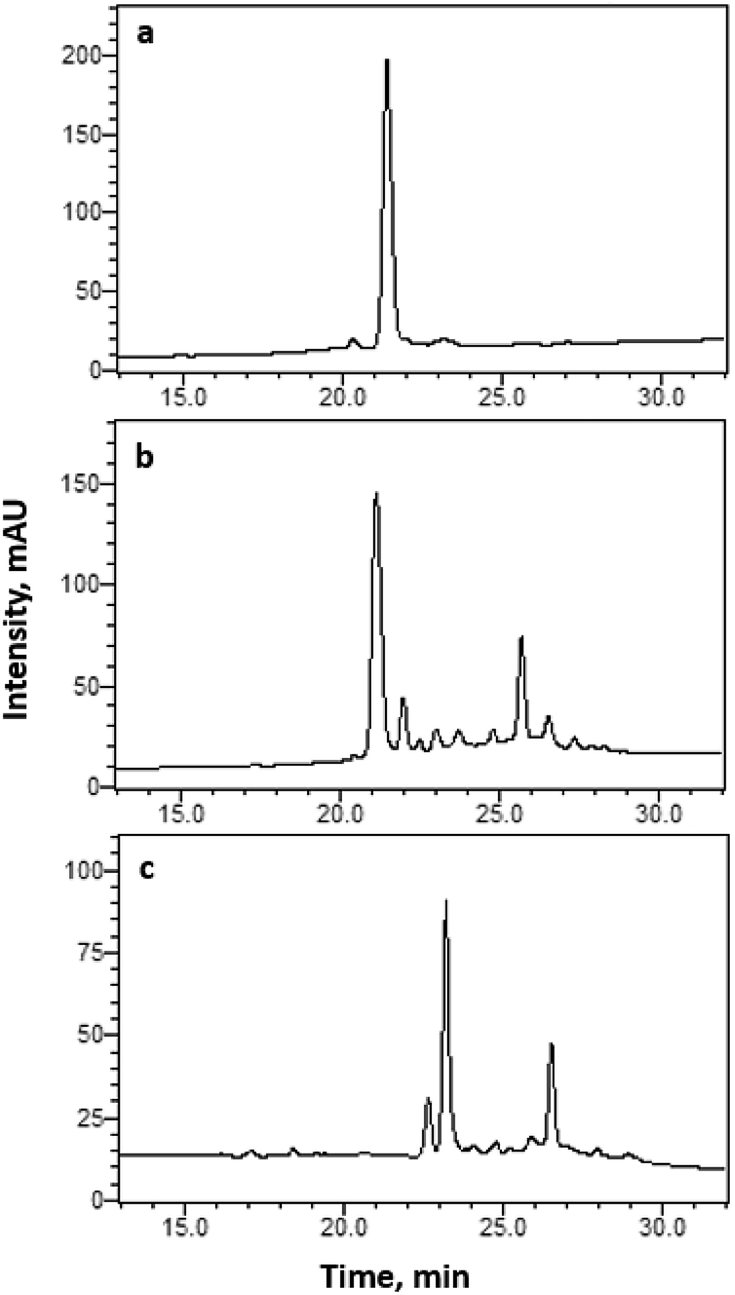 | ||
| Fig. 3 RP-HPLC chromatograms of crude (a) 1, (b) 2, and (c) lt5d with tR = 21.39, 21.14 and 23.20 min, respectively. Analytical C18 column was used to profile the sample with a linear gradient from 15% to 45% solution B (90% MeCN in H2O with 0.1% TFA) for 35 min at 1 mL min−1 flow rate. Absorbance was monitored at 220 nm. | ||
 | ||
| Fig. 4 HR-ESI(+) mass spectra of pure (a) 2 at 1419.5576, and (b) folded lt5d, at 1275.4716. Calculated monoisotopic mass of 2 and lt5d is 1418.6278 and 1274.4916 Da, respectively. | ||
In order to confirm the structural similarity of the peptides obtained from Method 1 and Method 2, the purified peptide products from the two methods were co-injected and analyzed by HPLC. The chromatograms in Fig. 5 show that the two compounds exhibit the same retention time, confirming that the synthesized peptides have the same folding configuration. Since Method 2 guarantees the controlled, sequential formation of disulphide bonds Cys2–Cys9 and Cys3–Cys10, its co-elution with the glutathione-folded product from Method 1 confirms that the same disulphide linkages were formed. Furthermore, co-injection of the two peptide products in HR-ESI(+)-MS also yielded the same molecular ion peaks (Fig. S1†).
 | ||
| Fig. 5 RP-HPLC chromatograms of lt5d synthesized using (a) Method 1, glutathione folding (tR = 22.43 min), (b) Method 2, orthogonal cysteine protection (tR = 22.57 min) and their (c) co-injection (tR = 22.53 min). Analytical C18 column was used to profile the samples with a linear gradient from 15% to 45% solution B (90% MeCN in H2O with 0.1% TFA) for 35 min at 1 mL min−1 flow rate. Absorbance was monitored at 220 nm. | ||
Finally, both peptides were also characterized by 1H NMR spectroscopy (500 MHz) in DMSO-d6, and the similarities in chemical shifts, peak shape, and integration further confirm that the peptides obtained from both methods are identical (Fig. S2†). The 1H NMR spectrum of the peptide obtained from Method 2 exhibited peaks of better resolution, therefore was characterized further using 2D NMR techniques. In particular, H–H COSY spectroscopy permitted the identification of ten (10) amino acids through the cross-peaks observed in the fingerprint region of the NMR spectrum (Fig. 6).18 These correspond to the ten (10) amino acids in the sequence of lt5d, with the exception of the two (2) Pro residues. A more detailed assignment of individual spin systems to these cross-peaks can be derived from the TOCSY spectrum (Fig. S3†). For instance, the NMR signals arising from Leu were readily identifiable based on the cross-peaks of amide protons at 8.19 and 7.36 ppm with the resonance peaks of aliphatic protons at 0.84, 1.45, and 1.54 ppm. Pro was identified based on the cross-peaks between 1.80 and 3.50 ppm, and the absence of cross-peaks between Hα protons in the ∼4.10 to 4.80 ppm region with amide protons in ∼6.80 to 8.77 region. The four (4) Cys residues are identifiable based on the signature cross-peaks of NH, Hα and Hβ signals, although the sequence-specific resonance of each individual Cys was indistinguishable. The results of these NMR analyses provide further structural confirmation of the target peptide lt5d.
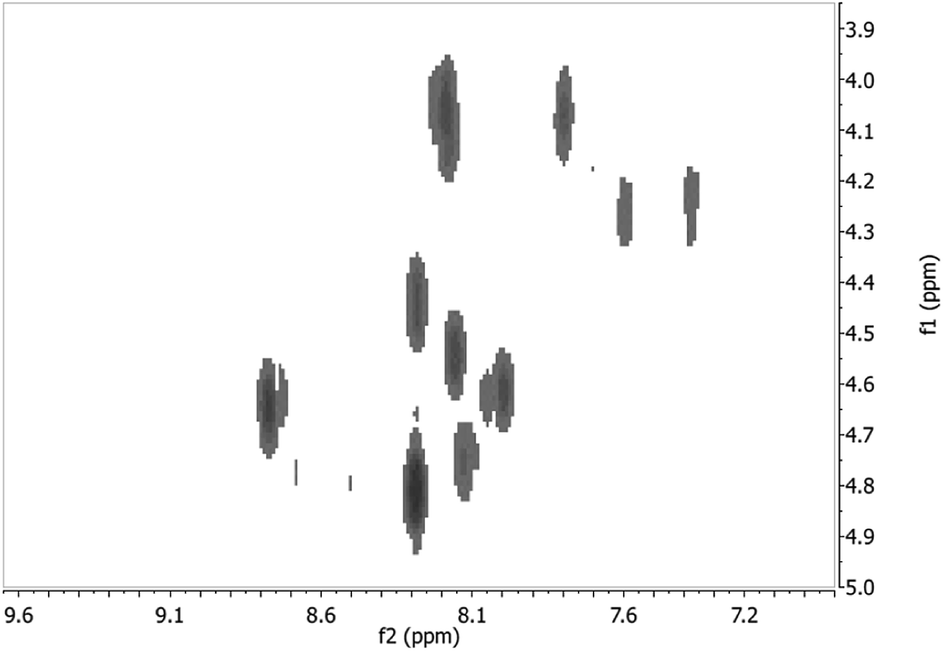 | ||
| Fig. 6 Fingerprint region of the H–H COSY NMR spectrum of lt5d obtained from Method 2 (500 MHz, DMSO-d6). The ten (10) cross-peaks observed correspond to the correlations of NH protons with ten (10) of the twelve (12) Hα protons of the lt5d amino acid sequence. As Pro has no amide proton, correlations for the two (2) Pro residues in the sequence are absent. | ||
Conclusions
The successful synthesis of lt5d is reported for the first time. It has been shown that the two strategies described above, i.e., glutathione-assisted oxidative folding (Method 1) and orthogonal cysteine protection (Method 2), yield the same product as confirmed by HPLC, MS, and NMR data. The capability of producing large amounts of purified peptide is particularly necessary in order to conduct a comprehensive in vitro and in vivo pharmacological assessment of the peptide. For instance, in order to determine its specific molecular target, lt5d can be assessed by different bioactivity assays, such as electrophysiology using cells expressing a specific type of TTX-sensitive Na channel isoform (e.g. Nav1.1–4, Nav1.6 and Nav1.7).2,19,20 Voltage-gated sodium channel isoforms Nav1.3 and Nav1.7 are among those identified to be involved in neuropathic pain.21 As lt5d has been previously reported to have high inhibitory activity against TTX-sensitive Na channels (IC50 = 156.16 nM),1 it is a peptidic drug candidate worth pursuing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Department of Science and Technology – Philippine Council for Health Research and Development (DOST PCHRD, FP140015 “Project 2: Anti-Pain and Anti-Neurodegeneration Drug Candidates: Discovery and Development” under the “Discovery and Development of Health Products (DDHP) Marine Component”). Special thanks to Dessa Camille Batoctoy, Vicenzo Paolo Torreno and Ramoncito Luis de Boda for generating the ESI-MS data.Notes and references
- J. Liu, Q. Wu, C. Pi, Y. Zhou, M. Zhou, L. Wang, S. Chen and A. Xu, Peptides, 2007, 28, 2313–2319 CrossRef CAS PubMed.
- K. B. Akondi, M. Muttenthaler, S. Dutertre, Q. Kaas, D. J. Craik, R. J. Lewis and P. F. Alewood, Chem. Rev., 2014, 114, 5815–5847 CrossRef CAS PubMed.
- P. Anand, A. O'Neil, E. Lin, T. Douglas and M. Holford, Sci. Rep., 2015, 5, 12497 CrossRef CAS PubMed.
- M. Sanford, CNS Drugs, 2013, 27, 989–1002 CrossRef CAS PubMed.
- D. P. Wermeling, Pharmacotherapy, 2005, 25, 1084–1094 CrossRef CAS PubMed.
- S. V. Phan and J. M. Waldfogel, General Hospital Psychiatry, 2015, 37, 97.e11–97.e12 CrossRef PubMed.
- WoRMS Editorial Board, World Register of Marine Species, available from http://www.marinespecies.org at VLIZ, 2017, DOI:10.14284/170.
- Q. Kaas, R. Yu, A. H. Jin, S. Dutertre and D. J. Craik, Nucleic Acids Res., 2012, 40, D325–D330 CrossRef CAS PubMed.
- Q. Kaas, J. C. Westermann, R. Halai, C. K. Wang and D. J. Craik, Bioinformatics, 2008, 24, 445–446 CrossRef CAS PubMed.
- K. K. Khoo, K. Gupta, B. R. Green, M.-M. Zhang, M. Watkins, B. M. Olivera, P. Balaram, D. Yoshikami, G. Bulaj and R. S. Norton, Biochemistry, 2012, 51, 9826–9835 CrossRef CAS PubMed.
- M. Okumura, M. Saiki, H. Yamaguchi and Y. Hidaka, FEBS J., 2011, 278, 1137–1144 CrossRef CAS PubMed.
- S. Chakravarthi, C. E. Jessop and N. J. Bulleid, EMBO Rep., 2006, 7, 271–275 CrossRef CAS PubMed.
- S. A. Esperante, G. Covaleda, S. A. Trejo, S. Bronsoms, F. X. Aviles and S. Ventura, Sci. Rep., 2017, 7, 5457 CrossRef PubMed.
- G. Bulaj, O. Buczek, I. Goodsell, E. C. Jimenez, J. Kranski, J. S. Nielsen, J. E. Garrett and B. M. Olivera, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14562–14568 CrossRef CAS PubMed.
- E. Fuller, B. R. Green, P. Catlin, O. Buczek, J. S. Nielsen, B. M. Olivera and G. Bulaj, FEBS J., 2005, 272, 1727–1738 CrossRef CAS PubMed.
- A. Cuthbertson and B. Indrevoll, Org. Lett., 2003, 5, 2955–2957 CrossRef CAS PubMed.
- E. Klüver, S. Schulz-Maronde, S. Scheid, B. Meyer, W.-G. Forssmannm and K. Adermann, Biochemistry, 2005, 44, 9804–9816 CrossRef PubMed.
- C. Redfield and C. M. Dobson, Biochemistry, 1988, 27, 122–136 CrossRef CAS PubMed.
- J. R. Prashanth, N. Hasaballah and I. Vetter, Neuropharmacology, 2017, 127, 4–19 CrossRef CAS PubMed.
- C. H. Lee and P. C. Ruben, Channels, 2008, 2(6), 407–412 CrossRef PubMed.
- S. D. Dib-Hajj, J. A. Black and S. G. Waxman, Pain Med., 2009, 10, 1260–1269 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03706j |
| This journal is © The Royal Society of Chemistry 2018 |