
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile plasmonic-effects at the interface of inverted perovskite solar cells†
Ahmed Esmail
Shalan
a,
Tomoya
Oshikiri
a,
Hiroki
Sawayanagi
a,
Keisuke
Nakamura
a,
Kosei
Ueno
a,
Quan
Sun
a,
Hui-Ping
Wu
b,
Eric Wei-Guang
Diau
*b and
Hiroaki
Misawa
*ab
aResearch Institute for Electronic Science, Hokkaido University, N21, W10, Kita-ku, 001-0021, Sapporo, Japan
bDepartment of Applied Chemistry & Institute of Molecular Science, National Chiao Tung University, 1001 Ta Hsueh R., Hsinchu 30010, Taiwan, Republic of China. E-mail: misawa@es.hokudai.ac.jp; diau@mail.nctu.edu.tw
First published on 13th December 2016
Abstract
Plasmonics is a highly promising approach to enhancing the light-harvesting properties of hybrid organic/inorganic perovskite solar cells. In the present work, our cells have a p–i–n inverted planar structure. An ultrathin NiO film with two different thicknesses of 5 and 10 nm prepared by a pulsed laser deposition process on an ITO substrate with a faceted and furrowed surface enabled the formation of a continuous and compact layer of well-crystallized CH3NH3PbI3via an anti-solvent chlorobenzene process. The coverage mechanism of the NiO film on the ITO was clearly demonstrated through the J–V and external quantum efficiency (EQE) curves. Moreover, the results demonstrated that the gold nanoislands (Au NIs) increased the power conversion efficiency to 5.1%, almost double that of the samples without Au NIs. This result is due to the excitation of surface plasmons, which is characterized by strong scattering and enhancement of the electric field in the vicinity of the Au NIs loaded at the interface between the NiO and perovskite films. Additionally, we observed an enhancement of the EQE at wavelengths shorter than the plasmon resonance peak. In the current state, we speculate that the plasmoelectric potential effect is considered to be a good explanation of the photocurrent enhancement at the off-resonance region. Our work provides good guidance for the design and fabrication of solar-energy-related devices employing NiO electrodes and plasmonic Au NIs.
Introduction
The beneficial characteristics of perovskite (PSK) materials, namely, an appropriate direct band gap, a high absorption coefficient, and propensity to form thin films with excellent carrier transport properties and an apparent tolerance of defects,1,2 have facilitated the rapid increase in power conversion efficiencies (PCEs) of PSK solar cells (PSCs) from 3.8% to 20% within 6 years.3–6 The reported PSCs can be mainly divided into two classes based on their geometric structures. The first class evolves from solid-state dye-sensitized solar cells that contain a mesoporous film as a support layer of the PSK, such as TiO2 and Al2O3 mesoporous films.7–9 By contrast, the other class comprises planar heterojunction (PHJ) solar cells, which have a similar structure as n–i–p or p–i–n silicon solar cells.10,11 In typical PHJ PSCs, a PSK absorption layer is sandwiched between a hole transport layer (HTL) and an electron transport layer (ETL). Poly-(3,4-ethylenedioxythiophene):poly (styrenesulfonic acid) (PEDOT:PSS) and phenyl-C71-butyric acid methylester (PCBM) are frequently employed as the HTL and ETL, respectively, in p–i–n-type devices.12–14 Such types of devices are known as inverted PHJ solar cells, and a PCE as high as 18.1% has recently been reported for such cells.15 Unfortunately, research on organic photovoltaic and light-emitting diode devices has already demonstrated that PEDOT:PSS is not good as HTL in terms of the long-term stability of the device due to its high acidity and hygroscopicity.16 Hence, many inorganic semiconductors, including MoOx, NiOx, V2O3, and WO3,16–18 have been employed to replace PEDOT:PSS. In particular, NiO is an inexpensive material with superior thermal and chemical stability and has also been demonstrated to be a good hole-selective contact for PSCs due to its suitable work function and high conduction band edge position, which can transport holes and block electrons efficiently.19 Several approaches have been used to prepare a NiO (NiOx) film, including sol–gel processes, electro-deposition, and sputtering, which could be adopted for the fabrication of organic photovoltaic devices and dye-sensitized solar cells.20,21 Indeed, in this study, we prepared a well-ordered nanostructured NiO film with different ultrathin thicknesses of 5 and 10 nm on the ITO substrate, which achieves a good optical transparency through a pulsed laser deposition (PLD) method. Furthermore, in this study, we focus on the p–i–n heterojunction because it could feasibly obtain a charge generation profile with more effective hole extraction, and the planar structure simplifies the device fabrication process at reduced material cost.Regarding the p–i–n planar structure, the incident light is irradiated from the NiO side to the PSK layer. The light with shorter wavelengths is absorbed around the interface between the NiO and the PSK. Subsequently, the holes generated at the interface easily transfer to the NiO layer. Simultaneously, the electrons move inside the PSK and then reach the ETL. However, longer wavelengths of light penetrate deeper into the PSK layer and are absorbed at deeper positions because the absorption cross section of the PSK is small at longer wavelengths. In such cases, excited holes have to move longer distances to inject NiO. Therefore, recombination will occur easily because the mobility of a hole in the PSK is substantially smaller than that of an electron.22,23 As a result, photocurrent and external quantum efficiency (EQE) will be small at longer wavelengths compared with the shorter wavelength region. Very recently, Ito and co-workers reported the effects of the crystal grain size of the PSK on the EQE.24 They noted that the EQE signals were almost saturated at more than 80% over the wavelength range from 350 to 750 nm for the device with the largest grain of approximately 500 nm. Their results also showed that the EQE decreased at wavelengths longer than 500 nm for devices with smaller grain sizes of approximately 100 nm.
To address this problem, it is necessary to find a new approach to increasing the EQE at longer wavelengths for small-grain-size PSK cells. Gold nanoislands (Au NIs) are considered to be a good candidate for operating as optical antennas, in which Au NIs strongly interact with the incident light and induce electric field enhancement and/or light scattering through localized surface plasmon resonance (LSPR).25–29 We attempt to develop Au NIs in the interface between the NiO and the PSK. The longer wavelength light can be localized near the NiO/PSK interface, and light is absorbed by the PSK near the interface via LSPR. As a characteristic point of this study, we deposited a NiO layer as the HTL with two different thicknesses on the ITO substrate by PLD. It is emphasizing, furthermore, that Au NIs are used for the enhancement of photocurrent generation. We speculate on the mechanism how the free carriers (electron and hole) move through the p–i–n planar PSCs in the absence and presence of Au NIs for the shorter and longer wavelengths, as shown in the schematic diagrams of Fig. 1, and we discuss about the plasmoelectric potential effect30 in addition to the near-field enhancement effect based on LSPR induced by Au NIs in the NiO/PSK interface.
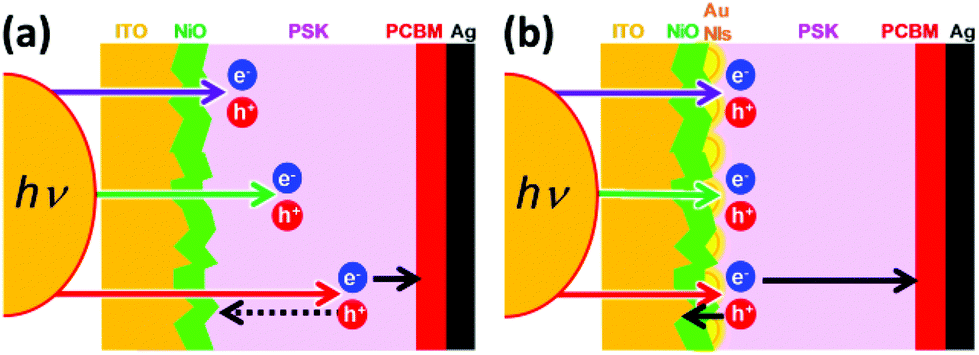 | ||
| Fig. 1 Schematics of the speculated mechanism of the free carrier (electrons and holes) pathway in the shorter and longer wavelengths in the absence (a) and presence (b) of Au NIs. | ||
Results and discussion
To investigate the efficacy of Au NIs on inverted PHJ PSCs with NiO as the HTL, the solar cell devices were fabricated in the presence of Au NIs. The detailed fabrication procedures for the devices in the presence and absence of Au NIs are described in the Experimental section. Fig. 2 presents a schematic of the preparation processes for PSCs with Au NIs layer by layer, illustrating the configuration of glass/ITO/NiO/Au NIs/PSK/PCBM/Ag. As shown in Fig. 2, it can be considered that the locations of the Au NIs loaded at the interface between the NiO and PSK layer may play an important role for the PCE by facilitating scattering of the light through LSPR.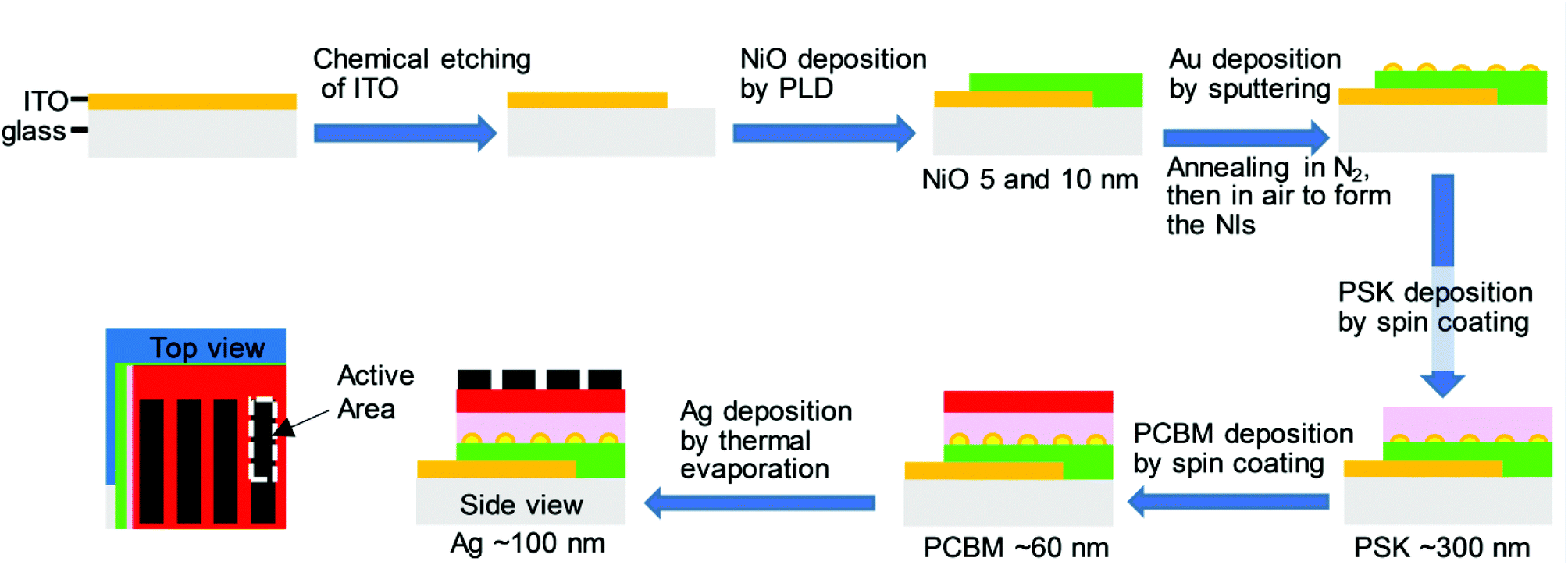 | ||
| Fig. 2 A schematic of the layer-by-layer fabrication process for the full-structure PSC with Au NIs with the configuration of glass/ITO/NiO/Au NIs/PSK/PCBM/Ag. | ||
To optimize the conditions of the NiO ultrathin film, we first evaluate the PCEs using two different thicknesses. We choose NiO with a thickness of 5 and 10 nm for comparison because it is expected that the thinner NiO as the HTL shortens the path of the holes and prevents recombination between holes and electrons. However, the results show that when the NiO film is thicker, i.e., 10 nm, the cell obtained better photovoltaic performances, as shown in Fig. 3.
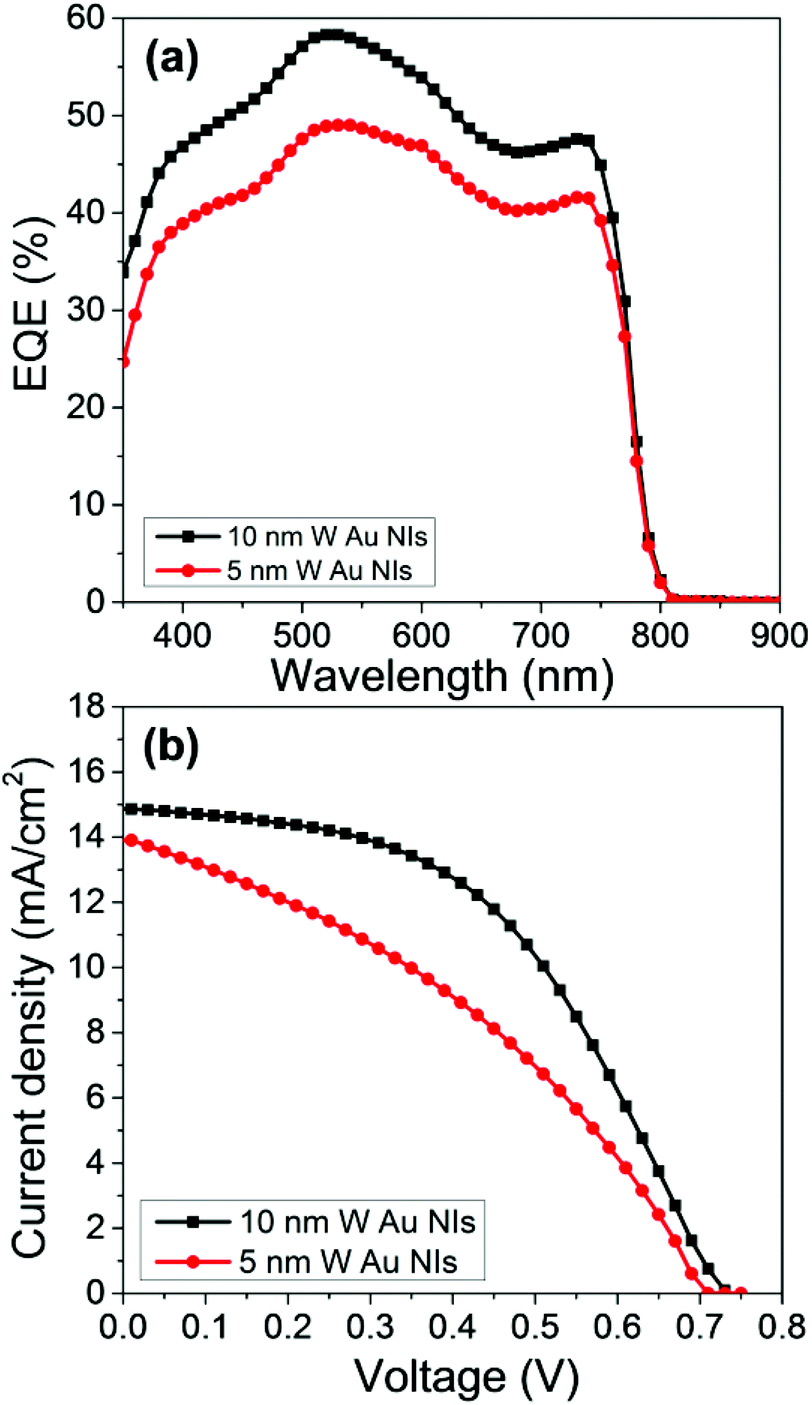 | ||
| Fig. 3 (a) EQE action spectra and (b) device performance for NiO ultrathin films with thicknesses of 5 and 10 nm with Au NIs based on the device structure of ITO/NiO/PSK/PCBM/Ag. | ||
To confirm the J–V characteristics and EQE results and to illustrate the effect of the thickness of the NiO film on the PCE, the roughness of the films was studied by atomic force microscopy (AFM). The root-mean-square roughness factor Rrms of the bare ITO and that of the 5- and 10 nm-thick NiO films on the ITOs are 3.2, 2.2, and 3.0 nm, respectively, as shown in Fig. 4(a–c). It is speculated that the photovoltaic properties of the samples with 5- and 10 nm-thick NiO can be affected by the roughness and morphological structures of the substrates.31–34 We can estimate the relationship between the roughness of the NiO surface and the coverage of the NiO film on the ITO. Previous studies confirmed the relationship between the Rrms and the coverage; that is, as the roughness of the film increases, the coverage of that film on the surface increases.35–37
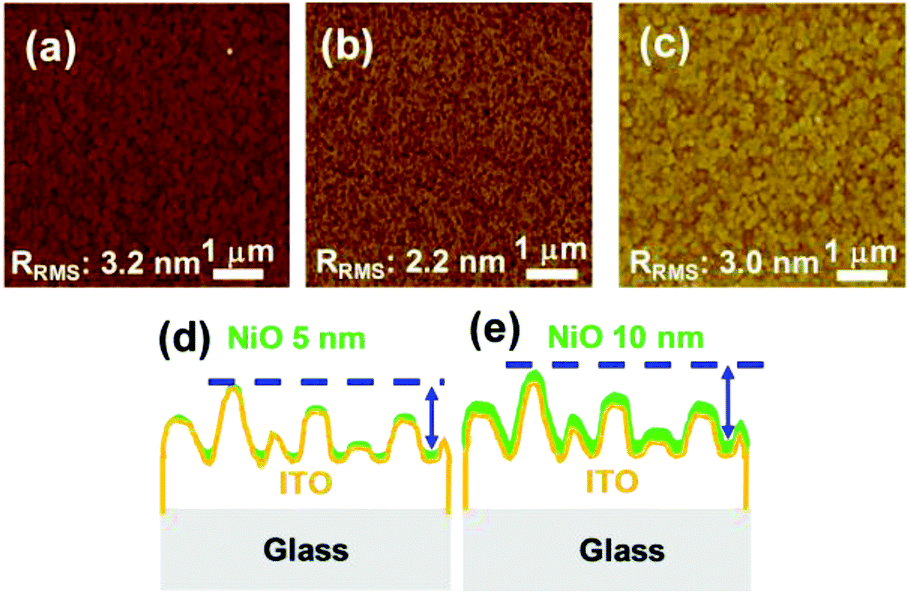 | ||
| Fig. 4 AFM images of (a) bare ITO substrate, (b) 5 nm-thick NiO/ITO, and (c) 10 nm-thick NiO/ITO. The magnification is ×12500, and the scale bar is 1 μm for all AFM figures. Schematic of the two different thicknesses of NiO PLD thin films, (d) 5 nm and (e) 10 nm, showing their coverage and roughness on the ITO surface. | ||
The AFM results demonstrate that the roughness of the 10 nm-thick NiO film is closer to that of bare ITO, whereas it is decreased in the case of the 5 nm-thick NiO film. From these results, we can suppose that the decrease in the roughness in the case of the 5 nm-thick NiO film is derived from the heterogeneous coverage, as represented in Fig. 4(d and e). For planar thin-film solar cells with the 5 nm-thick NiO film, the incomplete coverage of the NiO film in some areas of the substrates may result in low-resistance shunting paths, which affect the fill factor (FF).
Fig. 5a shows the EQE action spectra of the devices. The EQE of the device with the 10 nm-thick NiO with Au NIs steadily increases compared with that without Au NIs over the entire visible wavelength range. Moreover, it is clearly demonstrated that FF and the short-circuit current density (Jsc) increased after the Au NIs were incorporated into the devices, thereby leading to an increase in PCE, as shown in Fig. 5c. Compared with the maximum PCE of 2.6% for the devices without Au NIs, the maximum PCE for the devices with NIs reaches 5.1%, which represents an almost doubled enhancement measured under AM 1.5G (100 mW cm−2). The enhancement of Jsc and FF by loading Au NIs into the cells could be due to the improved utilization of visible light by electric field enhancement (near-field effect) or by scattering effects (far-field effects) via Au NIs embedded in the NiO/PSK interface. The Jsc shows an obvious increase, which is a result of the LSPR enhanced optical absorption of the PSK. Moreover, the increased absorption will contribute to the increased number of free carriers, which results in the corresponding FF and Jsc increases. Also, there is a possibility that an electron is injected from the photoexcited Au NIs to the conduction band of the PSK layer like Au-deposited TiO2 films.38,39 In this case, the photocurrent generation is induced according that the generated hole at the Au NIs is transferred to the valence band of NiO.40
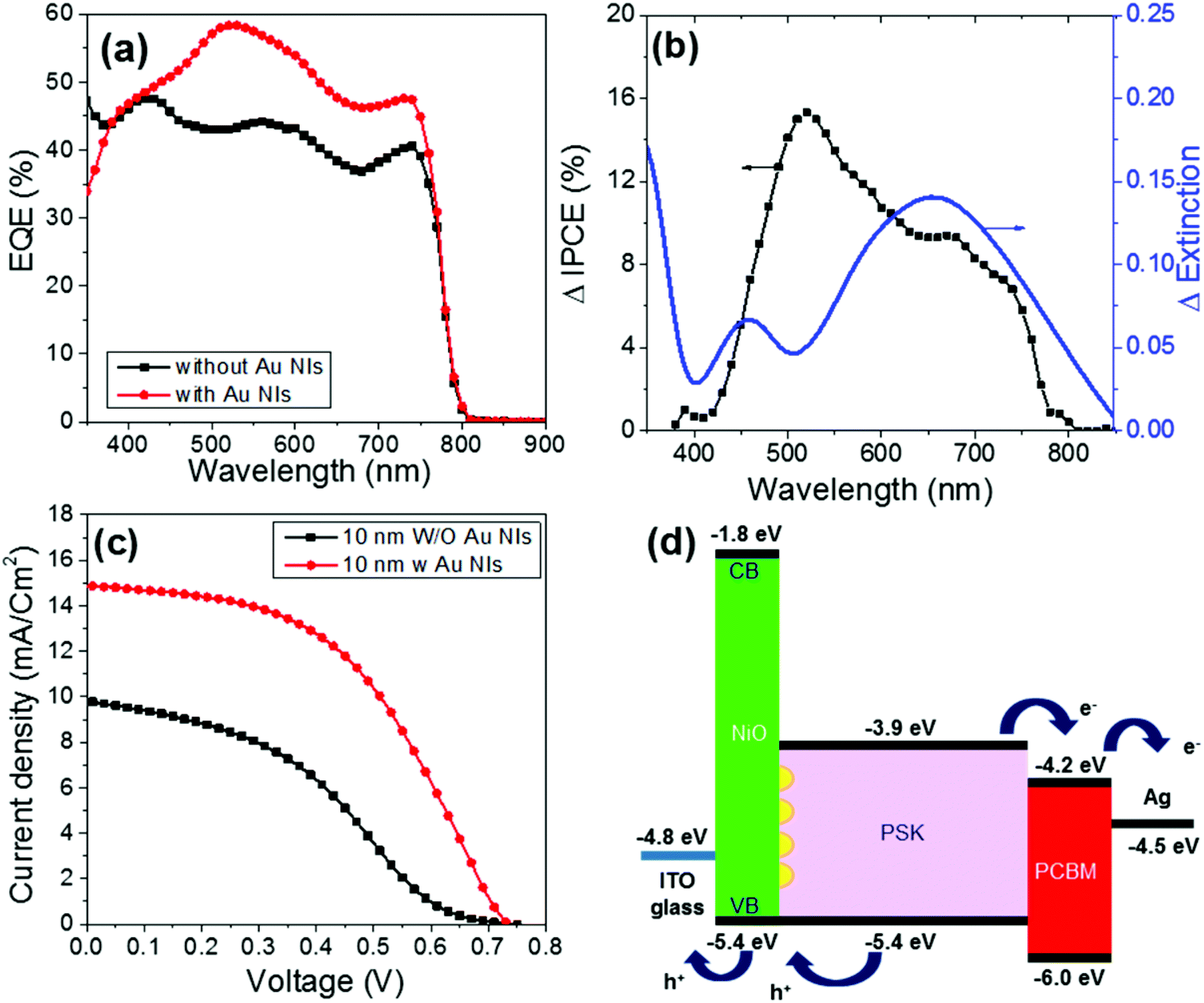 | ||
| Fig. 5 (a) EQE spectra of ITO/NiO/PSK/PCBM/Ag in the absence and presence of Au NIs. (b) Comparison of the ΔEQE action spectrum of the PSCs and the Δextinction between samples with and without Au NIs. (c) Device performances of ITO/NiO/PSK/PCBM/Ag in the absence and presence of Au NIs. (d) Energy level diagram of each layer in the presence of Au NIs based on the device structure of ITO/NiO/PSK/PCBM/Ag. The plots in black in (a) and (c) are the same data shown in Fig. 3. The thickness of the NiO was 10 nm in all cases. | ||
To determine the reproducibility and efficacy of the performance of the simple p–i–n planner PSCs, we fabricated and measured 24 separate devices with and without Au NIs using NiO ultrathin films with thicknesses of 5 nm and 10 nm. The devices’ average performances are shown in Table 1. The histograms of the photovoltaic parameter results are presented in Fig. S1.† These data indicate that the reproducibility of the device is very good and the performance enhancement by LSPR is significant. Furthermore, we measured the XRD of the PSK and estimated series resistances (Rs) of the devices with and without Au NIs to check the effect of the Au not involving LSPR. The XRD pattern of the PSK on the NiO film with Au NIs did not show remarkable difference compared with that without Au NIs as shown in Fig. S2.† Also, we estimated Rs of devices by the numerical fitting of J–V curves using the reported equations.41 The Rs of the devices with and without Au NIs were estimated as 11.5 and 9.2 Ω cm2, respectively. Although this result indicates that the Au NIs slightly increase (ca. 25%) the conductivity from the cathode to the anode, the increment cannot explain the significant improvement of the device performances.
| Sample | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| a The data which are classified in the table are averaged from 24 devices. | ||||
| NiO 10 nm with Au NIs | 15.1 ± 0.8 | 0.73 ± 0.02 | 48.5 ± 0.1 | 4.8 ± 0.4 |
| NiO 10 nm without Au NIs | 9.5 ± 0.5 | 0.79 ± 0.08 | 35.8 ± 0.8 | 2.2 ± 0.4 |
| NiO 5 nm with NIs | 15.3 ± 0.7 | 0.70 ± 0.01 | 37.2 ± 0.1 | 3.9 ± 0.3 |
| NiO 5 nm without Au NIs | 6.1 ± 0.9 | 0.75 ± 0.03 | 33.8 ± 0.3 | 1.6 ± 0.4 |
Then, the enhancement ratio of the EQE (ΔEQE) of the 10 nm-thick NiO devices was calculated and compared with their Δextinction spectrum, as shown in Fig. 5b. The Δextinction spectrum was extracted from the extinction spectra of samples of 10 nm-thick NiO with and without Au NIs, as shown in Fig. S3.† It is clear that the EQE of the plasmonic device increases sharply around the band edge of the PSK. The extinction spectrum wavelength of the NiO/PSK cells with a thickness of 10 nm correlate to overlapping with the ΔEQE band in the region of wavelengths greater than 650 nm, which is due to electric field enhancement or the scattering effects of the Au NIs.
Furthermore, from Fig. 5b, we note two peaks for ΔEQE. One peak is at longer wavelengths, which may be due to LSPR, localizing the light near the NiO/PSK interface, where light is absorbed by the PSK near the interface. This explanation was confirmed by the correlation between the Δextinction spectrum and ΔEQE in the region of longer wavelengths. The other peak for ΔEQE appears at shorter wavelengths, which is not in agreement with LSPR and forces us to consider another mechanism to explain this phenomenon.
Recently, the tuning of the plasmon resonance frequency of a nanostructure has been demonstrated by modulating the electron density.42–45 When a static electric potential was applied to inject or remove electrons from resonant structures, the plasmonic resonance was shifted to higher or lower frequencies, respectively. The electrical state of the conductor is coupled with the frequency of the plasmonic resonance. Atwater et al. demonstrated the reverse effect—the optical generation of an electrostatic potential due to an optically driven change in the carrier density in a plasmonic nanostructure.30 They observed that irradiation at wavelengths shorter and longer than the plasmon resonance peak created negative and positive surface potentials as a result of electron injection and ejection, respectively.
By applying this discovery to our results in the current work, we can speculate on the possible mechanism of EQE enhancement via plasmoelectric potential effects in the shorter wavelength region of LSPR. There may be two scenarios to explain the mechanism: the electron is transferred from the NiO or from the PSK to the Au NIs under irradiation at wavelengths shorter than the plasmon resonance peak. In the former scenario, the valence band (VB) of the NiO shifts to positive values, and the open-circuit voltage (Voc) values decrease. Simultaneously, the photocurrent increases as a result of efficient hole transport. In the latter scenario, the local electric field generated at the interface between the Au NIs and the PSK separates the electron and hole spatially.
To confirm the speculated mechanism, we measured J–V characteristics for the 10 nm-thick NiO sample at different wavelengths in the range of 400–800 nm in the presence of band-pass filters to check Voc at each wavelength and observe the difference in both shorter and longer wavelength regions (Fig. S4†). Additionally, we plotted the relationship between the Voc values and LSPR represented by the ΔEQE spectra, as shown in Fig. S5.† In the presence of Au NIs, the Voc decreases from the longer wavelengths to the shorter wavelengths and crosses the peak of LSPR. Voc decreases from the peak of LSPR to the shorter wavelengths, corresponding to the increase in Jsc, which is shown in Fig. 5b. It is considered that the small Voc in the range from 700 to 800 nm may be due to the small photocurrent. When the Au NIs are absent, there is no notable change in the Voc values, as shown in Fig. S5a.† These results indicate that the plasmoelectric potential could efficiently enhance the EQE in the shorter wavelength region compared to the LSPR peak.
To explain the experimental observations, we performed a numerical simulation using a finite-difference time-domain (FDTD) method. Fig. 6a and b show the SEM image of the Au NIs on the 10 nm-thick NiO film and their corresponding model for FDTD numerical simulations. Fig. 6c shows the 1-T-R spectrum and the action spectrum of near-field intensity monitored at the interface between the Au-NIs and the NiO film calculated by the FDTD numerical simulations based on Fig. 6b, where T and R indicate transmittance and reflectance of the Au NI loaded substrate, respectively. The simulated 1-T-R spectrum is strongly correlated with the Δextinction spectrum shown in Fig. 5b. The action spectrum of near-field intensity shows two main peaks. One peak is at 830 nm, which is off-resonance from the absorption of PSK. The other peak is at around 675 nm, which may contribute to the enhancement of EQE. However, the enhancement of EQE at shorter wavelengths in the range from 400 to 600 nm is not in agreement with the near-field spectrum. These results support that the enhancement of ΔEQE at the shorter wavelength region is derived from the plasmoelectric potential effect rather than the simple near-field enhancement effect.
 | ||
| Fig. 6 The SEM image of Au NIs on the 10 nm-thick NiO film (a) and its corresponding model for FDTD numerical simulations (b). The scale bar in the SEM image is 100 nm. The inset color region of (b) is a simulation area on the x–y plane. (c) The 1-T-R spectrum and the action spectrum of near-field intensity monitored at the interface between Au NIs and the NiO film calculated by the FDTD numerical simulations. | ||
Fig. 5d presents a diagram of the energy level of each layer in the case of cells with Au NIs. As shown in Fig. 5d, both the edge level of the conduction band (CB, −3.9 eV) and the VB (−5.4 eV) in the PSK exhibit a suitable distance from the LUMO level of the PCBM (−4.2 eV) and are in good agreement with the work function of NiO (−5.4 eV). Accordingly, it is anticipated that the minimum energy loss for the transfer or mobility of free carriers (electrons and holes) toward the ETL and HTL in the PSK/PCBM and NiO/PSK heterojunction interfaces enhances the photo-voltage output of the hybrid solar cells. On the other hand, NiO is a wide band-gap material with a CB edge level at approximately −1.8 eV, which is much higher than that of the PSK of approximately −3.9 eV; thus, injection of an electron into the NiO is not energetically favorable. In other words, NiO works as an electron-blocking layer.
Conclusion
In summary, we prepared highly transparent, nanostructured NiO films with two different thicknesses through PLD. The photovoltaic performance was explored using PLD-prepared NiO ultrathin films from a p–i–n-type device of a NiO/PSK/PCBM/Ag configuration. The ultrathin nature of the NiO film helps in reducing the material consumption and manufacture cost in the large-scale production of PSCs. Furthermore, the PCE enhancement in PSCs was achieved by incorporating plasmonic Au NIs at the interface of the NiO and PSK. This enhancement could be attributed to the LSPR excitation in the range of 650–800 nm via strong scattering and enhancement of the electric field in the vicinity of the metal nanoparticle. Furthermore, a plasmoelectric potential effect is proposed as a possible mechanism of EQE enhancement in the shorter wavelength region of the LSPR peak. These results demonstrate that the location of NIs plays an important role in improving the charge extraction properties, and the PCEs of the samples with Au NIs were 5.1%, which is almost double those values obtained with samples without Au NIs.Experimental
NiO film preparation (with or without Au NIs)
The glass substrates, with a coated ITO layer of 15 Ohm per square, were purchased from Pilkington Inc. ITO was partially removed from the substrate via etching with zinc powder and 2 mol dm−3 HCl to produce the desired pattern. The patterned ITO substrates were cleaned using ultra-sonication for 30 min in a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) mixture of acetone, soap water and iso-propyl alcohol (IPA) and then washed with de-ionized water. Then, the glass was dried with a N2 blow gun. The NiO films were deposited onto patterned glass/ITO substrates using a Pulsed Laser Deposition (PAC-LMBE, PASCAL CO.) method. This process was conducted at 200 °C at an oxygen pressure of 1.0 × 10−2 mTorr using a KrF excimer laser (λ = 248 nm) with a pulse energy density of 40 mJ cm−2 and a repetition rate of 10 Hz. The NiO (non-dope) ceramic target was prepared using high-purity NiO powder (99.99%, High Purity Chemicals, Japan). The film thickness was controlled by deposition time. In this study, films with thicknesses of 5 and 10 nm were used. Then, a 3 nm-thick gold film was deposited on the surface of NiO (5 or 10 nm) by helicon sputtering (ULVAC, MPS-4000) with a deposition rate of 1 Å s−1 and annealed at 300 °C for 1 h in a nitrogen atmosphere. Then, the sample was annealed at 450 °C for 1 h under an air atmosphere. After annealing, the Au-NIs formed on the surface of the NiO. The averages and standard deviations of the size of Au NIs on 5 nm-thick and 10 nm-thick NiO were estimated as 53.0 ± 35.8 and 40.1 ± 28.8 nm, respectively (Fig. S6†). All the substrates, both with and without Au, were annealed under the same conditions to facilitate their comparison.
:1:1) mixture of acetone, soap water and iso-propyl alcohol (IPA) and then washed with de-ionized water. Then, the glass was dried with a N2 blow gun. The NiO films were deposited onto patterned glass/ITO substrates using a Pulsed Laser Deposition (PAC-LMBE, PASCAL CO.) method. This process was conducted at 200 °C at an oxygen pressure of 1.0 × 10−2 mTorr using a KrF excimer laser (λ = 248 nm) with a pulse energy density of 40 mJ cm−2 and a repetition rate of 10 Hz. The NiO (non-dope) ceramic target was prepared using high-purity NiO powder (99.99%, High Purity Chemicals, Japan). The film thickness was controlled by deposition time. In this study, films with thicknesses of 5 and 10 nm were used. Then, a 3 nm-thick gold film was deposited on the surface of NiO (5 or 10 nm) by helicon sputtering (ULVAC, MPS-4000) with a deposition rate of 1 Å s−1 and annealed at 300 °C for 1 h in a nitrogen atmosphere. Then, the sample was annealed at 450 °C for 1 h under an air atmosphere. After annealing, the Au-NIs formed on the surface of the NiO. The averages and standard deviations of the size of Au NIs on 5 nm-thick and 10 nm-thick NiO were estimated as 53.0 ± 35.8 and 40.1 ± 28.8 nm, respectively (Fig. S6†). All the substrates, both with and without Au, were annealed under the same conditions to facilitate their comparison.
Fabrication of inverted thin-film (p–i–n) perovskite solar cells
The NiO (with and without Au NIs)/ITO substrates were subsequently exposed to ozone for 18 min via excimer lamp (PC-01-H, N-Cobo Co.) irradiation under an O2 atmosphere to remove the organic residues. Methylammonium iodide (CH3NH3I) was synthesized from 30 mL hydroiodic acid (57% in water, Aldrich) by reacting 27.86 mL methylamine (40% in methanol, Aldrich) in a 250 mL round-bottomed flask at 0 °C for 5 h by stirring. The precipitates were recovered by evaporation at 60 °C for 1 h. The product, CH3NH3I (MAI), was dissolved in ethanol, recrystallized from diethyl ether, and dried at 60 °C in a dryer oven for 24 h. MAI was synthesized according to a previous study in the literature.46–49 In brief, the 1 mol dm−3 perovskite (PSK)–CH3NH3PbI3 solution was prepared by reacting the synthesized MAI (198 mg mL−1) powder and PbI2 (574.3 mg mL−1) (99.99%, Aldrich) [1:1 molecular ratio] in dimethylformamide (1000 μl) (DMF, 99.9%, Aldrich) at 70 °C for 3 h. The solution was filtered with 0.45 μm PTFE filters before spin-coating. After the substrates were transferred into a N2-filled glovebox, the perovskite precursor solution was deposited onto an ultrathin NiO (with and without Au NIs)/ITO substrate via a spin-coating process at 5000 rpm for 15 s, and a few drops of chlorobenzene (CBZ) as an anti-solvent were dripped onto the substrate during spin coating (at a certain time, i.e., the 5th second for 45 wt% perovskite in DMF solution) in the second spin stage. Then, the perovskite–precursor-coated substrates were dried on a hot plate at 100 °C for 10 min. Subsequently, the phenyl-C71-butyric acid methyl ester (PCBM) layer (UniRegion Bio-Tech, Taiwan) was deposited by spin-coating (18 mg ml−1 in CBZ as solvent) at 1000 rpm for 30 s. Finally, the device was completed via thermal evaporation of 100 nm Ag contact electrodes in a high vacuum through a shadow mask. The active area of the Ag electrodes in the fabricated device was 0.05 cm2.
Structure characterization and other measurement
The crystallite phases were identified by X-ray diffraction (XRD) (RINT-2000/PC RIGAKU) using Cu Kα radiation and a scanning speed of 2° (2θ) min−1 within the 2θ range of 5 to 60°. The particle morphologies were obtained using a field emission scanning electron microscope (FE-SEM, JSM-6700FT, JEOL), whose maximum resolution at an electron acceleration voltage of 15 kV was 1 nm. Atomic force microscopy (AFM) was performed using a VN-8010 nanoscale hybrid microscope (Keyence microscope, Japan) instrument. The UV-vis absorption spectra and the extinction spectrum of the Au-NIs were monitored using a UV/vis spectrometer (UV-3100, Shimadzu Co.). An X-ray photoemission spectrometer (JPS-9200, JEOL) was used to obtain the X-ray photoemission spectrum (XPS) to analyze the chemical composition of the surfaces. The Al Kα line was used as the X-ray source. The C 1s signal was used as an internal reference (284.6 eV). The detailed characterization of the cells, XRD patterns, XPS spectra and the cross sectional SEM image are shown in the ESI and Fig. S7.†Photovoltaic performance measurement
Fabricated photovoltaic cells were characterized by their J–V characteristics and EQE. The J–V characteristic curve measurements were performed using the solar simulation (SAN-EL ELECTRIC, XES-40S1, 450 W Xe lamp, AM 1.5 global filter) equipped with an electrochemical workstation and a Keithley 2601 multimeter. The action spectra of the EQE were recorded with a system consisting of a Xe lamp (A-1010, PTi, 150 W), monochromator (PTi, 1200 grooves per mm blazed at 500 nm) and source meter (Keithley 2400, computer-controlled).Numerical simulations
The FDTD Solutions software package (Lumerical, Inc.) was used to numerically simulate the absorption spectrum and the near-field spectrum monitored at the interface between the Au NIs and the NiO film. The ITO covered glass substrate and the NiO film were assumed to behave as a dielectric with an average refractive index of n = 1.6 and 2.3, respectively. The thickness of Au NIs was assumed to be 20 nm. The FDTD simulations were performed on a discrete mesh with a maximum resolution of 1 nm.Acknowledgements
We thank Mr Hau-Shiang Shiu and Mr Kai-En Hung for their assistance in this work at the initial stage. This study was supported by funding from JSPS KAKENHI Grant Numbers JP23225006 and JP15H00856, the Nanotechnology Platform (Hokkaido University), and Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials (Five-Star Alliance) of MEXT. A. E. Shalan thanks the Elite Internship Program of National Chiao Tung University (Hsinchu, Taiwan) and the Research Institute for Electronic Science, Hokkaido University (Sapporo, Japan) for support of his visit to NCTU.Notes and references
- Q. Chen, H. P. Zhou, Z. R. Hong, S. Luo, H. S. Duan, H. H. Wang, Y. S. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622 CrossRef CAS PubMed.
- H.-S. Kim, S. H. Im and N.-G. Park, J. Phys. Chem. C, 2014, 118, 5615 CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef CAS PubMed.
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. M. Espallargas, M. Graetzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2014, 8, 128 CrossRef CAS.
- J. H. Kim, P.-W. Liang, S. T. Williams, N. Cho, C.-C. Chueh, M. S. Glaz, D. S. Ginger and A. K.-Y. Jen, Adv. Mater., 2015, 27, 695 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Graetzel, Nature, 2013, 499, 316 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739 CAS.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542 CrossRef CAS PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- P.-W. Liang, C.-Y. Liao, C.-C. Chueh, F. Zuo, S. T. Williams, X.-K. Xin, J. Lin and A. K. Y. Jen, Adv. Mater., 2014, 26, 3748 CrossRef CAS PubMed.
- X. Yin, M. Que, Y. Xing and W. Que, J. Mater. Chem. A, 2015, 3, 24495 CAS.
- J. H. Heo, H. J. Han, D. Kim, T. K. Ahn and S. H. Im, Energy Environ. Sci., 2015, 8, 1602 CAS.
- J. J. Jasieniak, J. Seifter, J. Jo, T. Mates and A. J. Heeger, Adv. Funct. Mater., 2012, 22, 2594 CrossRef CAS.
- D.-D. Zhang, R. Wang, Y.-Y. Ma, H.-X. Wei, Q.-D. Ou, Q.-K. Wang, L. Zhou, S.-T. Lee, Y.-Q. Li and J.-X. Tang, Org. Electron., 2014, 15, 961 CrossRef CAS.
- E. L. Ratcliff, J. Meyer, K. X. Steirer, A. Garcia, J. J. Berry, D. S. Ginley, D. C. Olson, A. Kahn and N. R. Armstrong, Chem. Mater., 2011, 23, 4988 CrossRef CAS.
- Z. Zhu, Y. Bai, T. Zhang, Z. Liu, X. Long, Z. Wei, Z. Wang, L. Zhang, J. Wang, F. Yan and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 12571 CAS.
- J. R. Manders, S. W. Tsang, M. J. Hartel, T. H. Lai, S. Chen, C. M. Am, J. R. Reynolds and F. So, Adv. Funct. Mater., 2013, 23, 2993 CrossRef CAS.
- J. H. Park, J. Seo, S. Park, S. S. Shin, Y. C. Kim, N. J. Jeon, H.-W. Shin, T. K. Ahn, J. H. Noh, S. C. Yoon, C. S. Hwang and S. Il Seok, Adv. Mater., 2015, 27, 4013 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344 CrossRef CAS PubMed.
- H. D. Kim, H. Ohkita, H. Benten and S. Ito, Adv. Mater., 2016, 28, 917 CrossRef CAS PubMed.
- X. Shi, K. Ueno, N. Takabayashi and H. Misawa, J. Phys. Chem. C, 2013, 117, 2494 CAS.
- Y. Zhong, K. Ueno, Y. Mori, X. Shi, T. Oshikiri, K. Murakoshi, H. Inoue and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 10350 CrossRef CAS PubMed.
- Y. Nishijima, K. Ueno, Y. Yokota, K. Murakoshi and H. Misawa, J. Phys. Chem. Lett., 2010, 1, 2031 CrossRef CAS.
- H. Aouani, O. Mahboub, N. Bonod, E. Devaux, E. Popov, H. Rigneault, T. W. Ebbesen and J. R. M. Wenger, Nano Lett., 2011, 11, 637 CrossRef CAS PubMed.
- Z. Lu, X. Pan, Y. Ma, Y. Li, L. Zheng, D. Zhang, Q. Xu, Z. Chen, S. Wang, B. Qu, F. Liu, Y. Huang, L. Xiao and Q. Gong, RSC Adv., 2015, 5, 11175 RSC.
- M. T. Sheldon, J. van de Groep, A. M. Brown, A. Polman and H. A. Atwater, Science, 2014, 346, 828 CrossRef CAS PubMed.
- H. P. Kim, A. R. Yusoff, H. M. Kim, H. J. Lee, G. J. Seo and J. Jang, Nanoscale Res. Lett., 2014, 9, 150 CrossRef PubMed.
- S. W. Cho, Y. T. Kim, W. H. Shim, S. Y. Park, K. D. Kim, H. O. Seo, N. K. Dey, J. H. Lim, Y. Jeong, K. H. Lee, Y. D. Kim and D. C. Lim, Appl. Phys. Lett., 2011, 98, 023102 CrossRef.
- K. H. Kim, C. Takahashi, T. Okubo, Y. Abe and M. Kawamura, Appl. Surf. Sci., 2012, 258, 7809 CrossRef CAS.
- Z. Ma, Z. Tang, E. Wang, M. R. Andersson, O. Inganas and F. Zhang, J. Phys. Chem. C, 2012, 116, 24462 CAS.
- Z. Yilei, Retrospective Theses and Dissertations, Paper 2007, 15934.
- L. Pons, M.-L. Délia and A. Bergel, Bioresour. Technol., 2011, 102, 2678 CrossRef CAS PubMed.
- O. Malinkiewicz, C. Roldan-Carmona, A. Soriano, E. Bandiello, L. Camacho, M. K. Nazeeruddin and H. J. Bolink, Adv. Energy Mater., 2014, 4, 1400345 CrossRef.
- G. Zhao, H. Kozuka and T. Yoko, Thin Solid Films, 1996, 277, 147 CrossRef CAS.
- Y. Nishijima, K. Ueno, Y. Yokota, K. Murakoshi and H. Misawa, J. Phys. Chem. Lett., 2010, 1, 2031 CrossRef CAS.
- J. Halme, P. Vahermaa, K. Miettunen and P. Lund, Adv. Mater., 2010, 22, E210 CrossRef CAS PubMed.
- K. Nakamura, T. Oshikiri, K. Ueno, Y. Wang, Y. Kamata, Y. Kotake and H. Misawa, J. Phys. Chem. Lett., 2016, 7, 1004 CrossRef CAS PubMed.
- A. Polman, Science, 2008, 322, 868 CrossRef PubMed.
- C. Novo, A. M. Funston, A. K. Gooding and P. Mulvaney, J. Am. Chem. Soc., 2009, 131, 14664 CrossRef CAS PubMed.
- S. K. Dondapati, M. Ludemann, R. Müller, S. Schwieger, A. Schwemer, B. Händel, D. Kwiatkowski, M. Djiango, E. Runge and T. A. Klar, Nano Lett., 2012, 12, 1247 CrossRef CAS PubMed.
- G. Garcia, R. Buonsanti, E. L. Runnerstrom, R. J. Mendelsberg, A. Llordes, A. Anders, T. J. Richardson and D. J. Milliron, Nano Lett., 2011, 11, 4415 CrossRef CAS PubMed.
- Z. Yuan, Z. Wu, S. Bai, Z. Xia, W. Xu, T. Song, H. Wu, L. Xu, J. Si, Y. Jin and B. Sun, Adv. Energy Mater., 2015, 5, 1500038 CrossRef.
- J.-H. Im, C.-R. Lee, J.-W. Lee, S.-W. Park and N.-G. Park, Nanoscale, 2011, 3, 4088 RSC.
- L. Etgar, P. Gao, Z. Xue, Q. Peng, A. K. Chandiran, B. Liu, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2012, 134, 17396 CrossRef CAS PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr06741g |
| This journal is © The Royal Society of Chemistry 2017 |