
Vasculogenesis and angiogenesis in modular collagen–fibrin microtissues†
A. W.
Peterson
,
D. J.
Caldwell
,
A. Y.
Rioja
,
R. R.
Rao
,
A. J.
Putnam
and
J. P.
Stegemann
*
Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI, USA. E-mail: jpsteg@umich.edu
First published on 22nd July 2014
Abstract
The process of new blood vessel formation is critical in tissue development, remodeling and regeneration. Modular tissue engineering approaches have been developed to enable the bottom-up assembly of more complex tissues, including vascular networks. In this study, collagen–fibrin composite microbeads (100–300 μm in diameter) were fabricated using a water-in-oil emulsion technique. Human endothelial cells and human fibroblasts were embedded directly in the microbead matrix at the time of fabrication. Microbead populations were characterized and cultured for 14 days either as free-floating populations or embedded in a surrounding fibrin gel. The collagen–fibrin matrix efficiently entrapped cells and supported their viability and spreading. By 7 days in culture, endothelial cell networks were evident within microbeads, and these structures became more prominent by day 14. Fibroblasts co-localized with endothelial cells, suggesting a pericyte-like function, and laminin deposition indicated maturation of the vessel networks over time. Microbeads embedded in a fibrin gel immediately after fabrication showed the emergence of cells and the coalescence of vessel structures in the surrounding matrix by day 7. By day 14, inosculation of neighboring cords and prominent vessel structures were observed. Microbeads pre-cultured for 7 days prior to embedding in fibrin gave rise to vessel networks that emanated radially from the microbead by day 7, and developed into connected networks by day 14. Lumen formation in endothelial cell networks was confirmed using confocal sectioning. These data show that collagen–fibrin composite microbeads support vascular network formation. Microbeads embedded directly after fabrication emulated the process of vasculogenesis, while the branching and joining of vessels from pre-cultured microbeads resembled angiogenesis. This modular microtissue system has utility in studying the processes involved in new vessel formation, and may be developed into a therapy for the treatment of ischemic conditions.
Introduction
Vasculogenesis and angiogenesis are the processes by which new blood vessels form during tissue development, remodeling, and regeneration. Vasculogenesis is generally defined as the de novo formation of vessels from the coalescence of cells of mesodermal lineage into tubular structures, and is a hallmark of tissue development.1 Angiogenesis is the term used to describe the formation of new vessels from an existing vasculature via a process of sprouting and elongation, or via intussusception of the lumen.2 These processes are associated with tissue remodeling and repair, and unregulated angiogenesis is a hallmark of cancer and other pathological conditions.3,4 Clearly, understanding the processes of vasculogenesis and angiogenesis has broad implications for biology, physiology, and medicine. More recently, as the field of tissue engineering has progressed to developing implantable therapies, there has been an increased interest in creating appropriately vascularized tissues that can quickly integrate with the host.5,6Cell-based approaches have been used to recapitulate some of the key features of both the vasculogenic and angiogenic processes, and have been applied to generating new vasculature for therapeutic applications. A common model for studying vasculogenesis is the combination of endothelial and stromal cell populations, which has been shown to generate functional capillary networks.7,8 The initiation and progression of angiogenesis is often simulated using a “bead assay” in which endothelial cells are coated onto the outside of polystyrene microspheres, which are then embedded in a surrounding protein matrix (often the clotting protein fibrin).9 The endothelial cells can extend and migrate from the surface of the microsphere, and in some cases these studies are performed in the presence of stromal cell populations either directly in or on top of the surrounding gel construct.10 It has been shown that endothelial networks and vascular structures are stimulated by a variety of stromal cell types, including human lung fibroblasts,11 marrow-derived mesenchymal stem cells,12,13 adipose-derived mesenchymal stem cells,14 and smooth muscle cells.15 The role of stromal cell populations as paracrine signal sources and as pericyte-like cells is still being investigated, and there is evidence that both the endothelial cell type16 and the stromal cell type17,18 can influence the type and rate of vessel formation.
The concept of modular tissue engineering has emerged over the last decade as a way to create large and complex tissue structures via the assembly of more basic building blocks.19–21 A key motivation for the modular approach is to avoid the mass transfer limitations associated with larger tissues, since it has been estimated that the effective diffusion limit in tissues is 150–200 μm.5,6 Another advantage of the modular methods is that microtissue building blocks can be delivered minimally invasively to be assembled in situ. Such approaches have now been applied to a variety of tissue systems including bone,22,23 cartilage,24 cardiac tissue,25,26 as well as more complex organs.27,28 Controlled combination of multiple microtissue types has more recently been used to create multiphase tissues,29,30 and the challenge of tissue vascularization has also been addressed using modular microtissues. Purely cellular “spheroids” produced by directed aggregation of cells,20,31 as well as various forms of micromolding to create defined sizes and shapes of microtissues32,33 have been investigated for this purpose. These approaches also have been used to deliver EC as modules for vascularization, either in spheroid form34 or as coatings on molded protein modules.35,36 In these cases, the modules have been used as a vehicle for endothelial cell delivery, and more recently for co-delivery of endothelial cells and mesenchymal stem cells.37–39
A variety of materials have been used to create modular microtissues. Synthetic hydrogels40 and ionically- or thermally-gelling polysaccharides41,42 are attractive for this purpose due to their dimensional and temporal stability. Natural polymers and extracellular matrix proteins have the advantage that cells can recognize and bind to them, and these proteins can be used to support and direct cell function.43 In particular, the proteins collagen Type I and fibrin have been used widely in regenerative medicine because of their abundance, versatility, and biological relevance (recently reviewed in ref. 44 and 45). The modular approach has been adapted to create collagen-based modules,46,47 including for promoting vascularization.48,49 However, fibrin has been shown to generally be a more permissive matrix for vessel formation, and is commonly used in studies of both vasculogenesis and angiogenesis.50 Our previous work has shown that collagen–fibrin composite matrices are well suited to cell encapsulation and have superior mechanical properties as compared to the pure protein materials.51 In addition, we recently have shown that collagen–fibrin matrices support vasculogenesis.52,53
The study presented here combines several elements of the modular microtissue approach to create cell-seeded biomaterial building blocks that promote new vessel formation. Human endothelial cells and fibroblasts were embedded directly in microbeads (100–300 μm in diameter) composed of a collagen–fibrin matrix. Fig. 1 schematically depicts the microbead fabrication process and the experimental plan. Collagen–fibrin microbeads and corresponding bulk hydrogel matrices were compared in terms of the efficiency of cell encapsulation and their ability to sustain cell viability. Microbeads were then cultured over time either as free-floating populations, or were embedded in a surrounding fibrin hydrogel, and vessel formation within the microbead matrix and in the surrounding hydrogel was monitored over 14 days. In the spirit of this celebration issue of Biomaterials Science, this study was motivated by the pioneering work of Dr Michael V. Sefton in the field of modular tissue engineering. The approach we describe may have utility in understanding the processes of vasculogenesis and angiogenesis, and may be developed into a therapeutic approach for the reversal of ischemia in tissues.
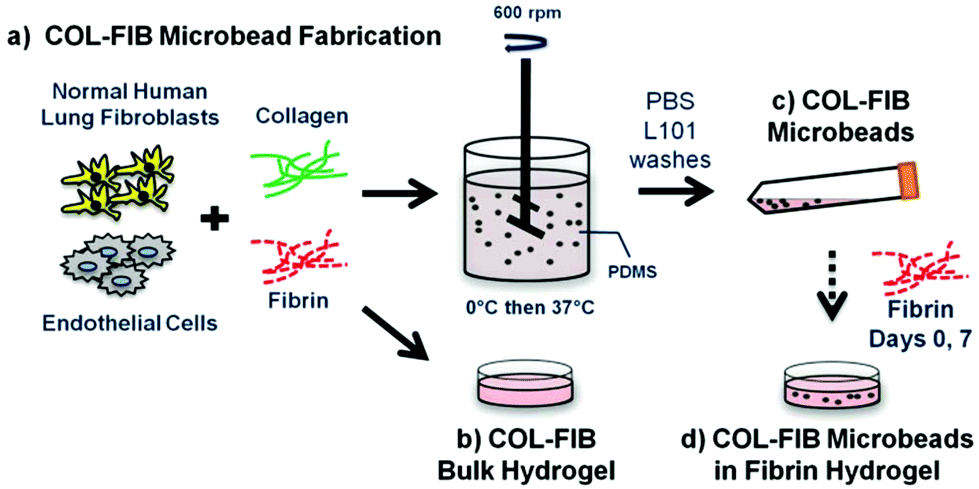 | ||
| Fig. 1 Schematic of (a) COL–FIB microbead fabrication process, and experimental design. In separate experiments, COL–FIB microbeads were (b) compared to bulk hydrogels, (c) cultured as populations, and (d) embedded in a surrounding fibrin hydrogel. | ||
Materials and methods
Cell sourcing and culture
Normal human lung fibroblasts (NHLF; Lonza Inc., Walkersville, MD) were cultured in Medium 199 medium (M199; Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Life Technologies) and 1% penicillin and streptomycin (PS; Life Technologies). NHLF were used at P9. Media was changed every other day.Human umbilical vein endothelial cells (HUVEC) were isolated as previously described.8 Briefly, donated umbilical veins were irrigated with sterile phosphate buffered saline (PBS) and then incubated with 0.1% collagenase (Type I, Worthington Biochemical, Lakewood, NJ) at 37 °C for 20 min. The digestion product was collected, the vein was washed with PBS, and the resulting suspension was centrifuged. The cell pellet was re-suspended in Endothelial Growth Medium-2 (EGM-2, Lonza) and plated into flasks. After 24 hours, the cells were washed with PBS to remove residual erythrocytes. HUVEC were cultured in EGM-2 with medium changed every other day, and were used at P4.
Fabrication of microbeads and bulk hydrogels
Collagen–fibrin (COL–FIB) matrices were generated using a variation of a previously described procedure,54 shown schematically in Fig. 1. Briefly, this method involves suspension of cells in a solution of collagen and fibrin, followed by emulsification of the cell suspension in a bath of silicone fluid by controlled stirring. The temperature is then raised to allow gelation of the collagen–fibrin matrix, resulting in the formation of spheroidal microbeads with cells entrapped directly within the matrix. The microbeads can be collected from the emulsification bath to yield a population of modular microtissues. Details of the procedure used in this study are provided in the following paragraphs.The stock matrix mixture was prepared by combining solubilized collagen and fibrin with additives required for matrix gelation and support of cell viability. Bovine collagen type I (MP Biomedicals, Solon, OH) was dissolved in 0.02 N acetic acid to yield a 4.0 mg mL−1 stock solution. Bovine fibrinogen (Sigma Aldrich, St. Louis, MO) was dissolved at a concentration of 4.0 mg mL−1 clottable protein in serum-free EGM-2. The matrix mixture used to create microbeads and constructs consisted of collagen (1.0 mg mL−1) and fibrinogen (1.5 mg mL−1) solutions combined with FBS (10%), 5X-concentrated serum-free Dulbecco's modified Eagle's medium, NaOH (0.02 N), bovine thrombin (1.0 U mL−1; Sigma), serum-free EGM-2 (9.1%), and glyoxal (1.0 mM; Sigma) at 4 °C. Glyoxal was used to partially crosslink the matrix, as described previously.55 Cells were trypsinized using 0.05% trypsin-EDTA (Life Technologies) and added directly to the solubilized protein matrix mixture at a concentration of 3.0 × 105 HUVEC mL−1 and 3.0 × 105 NHLF mL−1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HUVEC to NHLF, 6.0 × 105 cells mL−1 total). To facilitate visualization of the microbeads embedded in fibrin gels, FITC-labeled fibrinogen (Life Technologies) was added at 9 μg mL−1 to the filtered fibrinogen mixture. When using FITC-fibrinogen, samples were kept in the dark to preserve fluorescent signal intensity.
:1 HUVEC to NHLF, 6.0 × 105 cells mL−1 total). To facilitate visualization of the microbeads embedded in fibrin gels, FITC-labeled fibrinogen (Life Technologies) was added at 9 μg mL−1 to the filtered fibrinogen mixture. When using FITC-fibrinogen, samples were kept in the dark to preserve fluorescent signal intensity.
To create microbeads, the cell–matrix mixture was pipetted into a pre-cooled bath containing 100 cSt polydimethylsiloxane (PDMS; Xiameter, Dow Corning, Midland, MI) stirred at 600 RPM. After 5 minutes of mixing at 0 °C, the temperature was increased to 37 °C for 25 minutes to initiate and allow for full gelation of the microbead matrix. Microbeads were collected using PBS containing Pluronic L101 (PBS-L101; BASF, Florham Park, NJ) and centrifugation at 200g for 5 min. The collected microbeads were then washed three times in PBS-L101, and were then resuspended in EGM-2. Microbeads were cultured in 15 mL centrifuge tubes in EGM-2 with a loose cap at a slight angle to allow gas exchange in a standard cell culture incubator at 37 °C and 5% CO2. Culture medium was changed every other day.
Bulk COL–FIB gels were created using the same matrix mixture as for microbeads. A small volume (250 μL for DNA content, and 500 μL for all other gels) of matrix mixture with suspended cells was pipetted into a 24 well plate, and the solution was allowed to gel at 37 °C for 30 min. Bulk gels were washed three times in PBS (5 min per wash). Gels were either stored in PBS (acellular samples) or used for DNA analysis.
Embedding of microbeads in 3D fibrin hydrogels
COL–FIB microbeads were embedded in fibrin hydrogels at a 1:1 ratio of microbeads to surrounding fibrin matrix, with the volume of microbeads determined by the starting volume of the matrix mixture. Microbeads were collected by suspension in medium and then centrifuged to create a concentrated microbead preparation of defined volume. Following removal of the supernatant medium, an appropriate volume of a solution of cold 2.5 mg mL−1 fibrinogen, 10% FBS, 1.0 U mL−1 thrombin and serum-free EGM-2 was added to match the volume of microbeads. This solution was then pipetted into the wells of a 24-well plate (500 μL per well). Following gelation at 37 °C for 30 min, constructs were cultured in EGM-2 with medium changes every other day.
Characterization and imaging of acellular microbeads
To determine the size of acellular microbeads, a population of COL–FIB microbeads was stained overnight in Coomassie Blue reagent. Representative digital images were captured on a conventional light microscope (Olympus America, Center Valley, PA). The diameter of 255 microbeads in these images was then measured using ImageJ software (National Institutes of Health, Bethesda, MD). The morphology and architecture of the microbead and bulk gel matrix were examined using confocal reflectance microscopy on a laser scanning confocal microscope (Olympus).Characterization of NHLF-HUVEC in microbeads
Cell incorporation into microbeads and bulk gels was assessed by measuring DNA content using an established assay (PicoGreen®; Life Technologies). Briefly, samples were collected and digested overnight in 10 mM Tris-HCl (Sigma) containing 0.6 mg mL−1 collagenase type I (MP Biomedicals), 0.2% IGEPAL (Sigma), and 2.0 mM phenylmethanesulfonylfluoride (Sigma). Samples were measured fluorometrically according to the manufacturer's instructions, and were compared to a DNA standard curve to determine absolute DNA quantity. Comparison of the DNA content of bulk gel and microbead samples provided an estimate of the efficiency of cell embedding in microbeads.Cell viability was assessed at days 1 and 7 using a vital stain (Live/Dead®, Life Technologies). Briefly, NHLF-HUVEC-containing microbeads were collected and washed three times in PBS (5 min per wash) at 37 °C. Samples were then incubated for 45 min at 37 °C in 4.0 μM calcein-AM and 4.0 μM ethidium homodimer-1 dissolved in PBS. Microbeads were then washed twice in PBS and imaged using a laser scanning confocal microscope (Nikon Instruments, Melville, USA). For each sample, five images were captured and viability was quantified by comparing the total number of green-stained cells (live) to the number of red-stained nuclei (dead).
Immunohistochemical staining of microbeads and bulk gels
Microbeads and bulk gels were washed twice (5 min per wash) at 37 °C in phosphate buffered saline (PBS) to remove excess medium. All samples were then fixed with zinc-buffered formalin (Z-Fix; Anatech, Battle Creek, MI) for 10 min at 4 °C, washed twice in PBS, and cell membranes were subsequently permeabilized using 0.5% Triton X-100 (Sigma) in PBS for 20 min at room temperature, followed by two washes in PBS (5 min per wash).Samples used to examine cellular components were stained with 165 nM Alexa Fluor® 488 phalloidin (Life Technologies), 20 μg ml−1 rhodamine-labeled Ulex Europaeus Agglutinin I (UEA-1; Vector Laboratories, Burlingame, CA), and 10 nM DAPI (Life Technologies) for 1.5–4.5 h at room temperature in PBS containing 1% bovine serum albumin (BSA; Sigma). Excess staining solution was removed by two washes in PBS (5 min per wash).
Samples used to identify laminin or alpha-smooth muscle actin (α-SMA) were blocked with 2% BSA in PBS for 1 h at room temperature following permeabilization. After three PBS washes (5 min per wash), samples were stained either with 1:100 mouse anti-human α-SMA (Abcam, Cambridge, MA) for 1 h at room temperature, or with 1:100 rabbit anti-human laminin (Abcam) overnight at 4 °C, in PBS with 2% BSA. Following three PBS washes (5 min each), anti-mouse Alexa Fluor® 488 IgG (Life Technologies) or anti-rabbit Alexa Fluor® 488 (Life Technologies) were added at 1:400 in 2% BSA in PBS for 1 h at room temperature. Samples were washed twice with PBS, counterstained with 20 μg ml−1 UEA-1 and 10 nM DAPI for 1.5 h at room temperature in PBS with 1% BSA, and rinsed two more times with PBS before imaging.
Confocal imaging of NHLF-HUVEC microbeads
Microbead samples were aliquoted into microscope imaging dishes (0 rating, In Vitro Scientific, Sunnyvale, CA) prior to imaging using a laser scanning confocal microscope (Nikon A-1). Images were acquired using multiple objectives (10–60× magnification), depending on the feature size being examined. Maximum intensity projection images were created by obtaining z-stacks of areas of interest and flattening them to produce a single projection. As appropriate, image brightness was balanced between image stacks to better represent the cellular content of microbeads. Z-stacks were also used to create three-dimensional renderings of microbead and construct volumes using Nikon Elements software.Statistical analysis
Statistical significance was determined using Student's t-test at a level of α = 0.05. The n value refers to the number of separate experiments performed. Numerical data are reported as the mean ± standard deviation, and error bars on graphs represent the standard deviation.Results and discussion
Characterization of COL–FIB microbeads
The size, morphology and matrix architecture of acellular 40–60 COL–FIB microbeads is displayed in Fig. 2. The large majority of microbeads were in the range of 100–300 μm in diameter, with an average of 205 ± 71 μm (Fig. 2a). The COL–FIB microbeads were generally spheroidal in shape, and could be well dispersed in aqueous medium immediately following fabrication (Fig. 2a, inset). The microbead matrix (Fig. 2b) was characterized by more densely packed and closely associated protein fibers, compared to the matrix in bulk hydrogels of the same starting protein composition (Fig. 2c), as assessed by confocal reflection microscopy. Since these matrices were acellular, the difference cannot be attributed to cell-mediated compaction, and is therefore likely to be a result of the fabrication and processing. The denser matrix architecture in microbeads resembled that of bulk gel matrices fabricated at a higher initial protein content,52 and this difference may correspondingly affect cell behavior.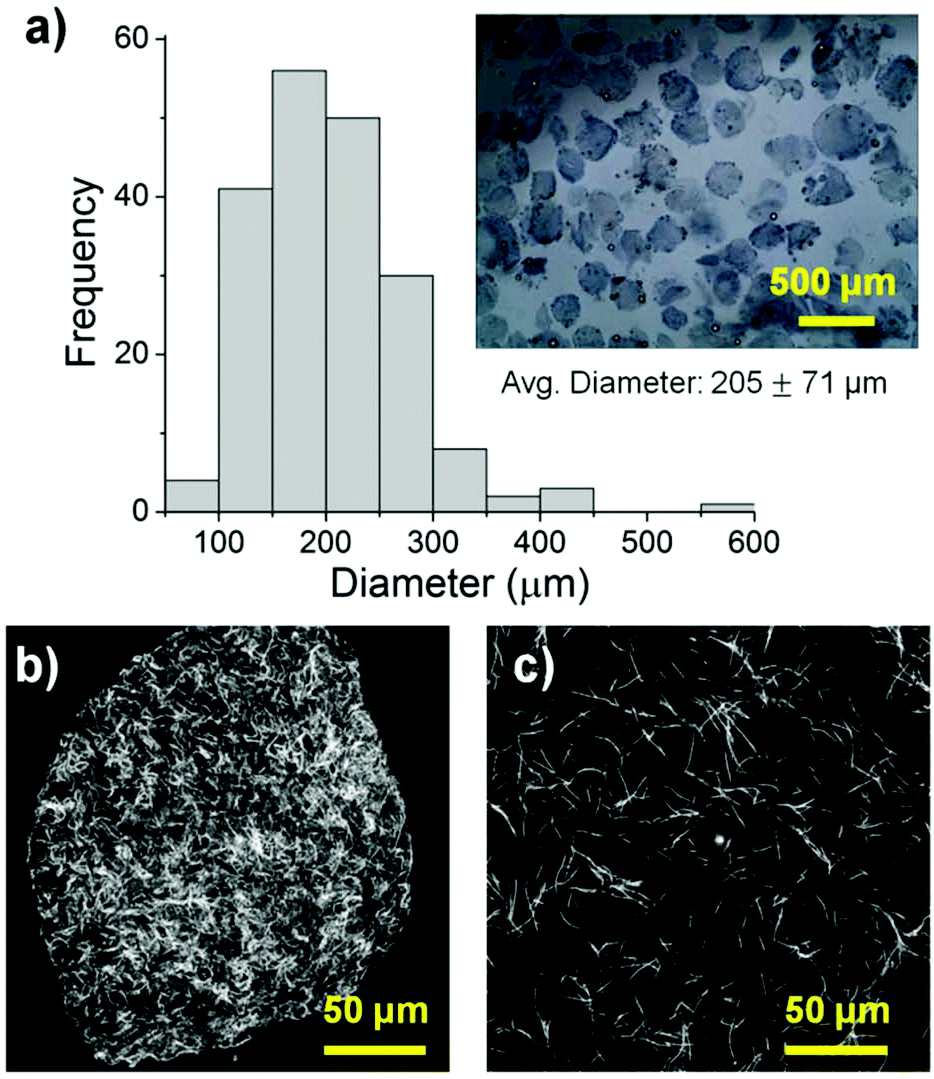 | ||
| Fig. 2 (a) Size, size distribution, and morphology of acellular 40–60 COL–FIB microbead populations. Confocal reflectance images of protein matrix architecture in (b) COL–FIB microbeads and (c) a COL–FIB bulk gel made at equal initial protein concentrations. | ||
Cells were incorporated into cellular COL–FIB composite materials at the time of fabrication, such that they were fully embedded in the matrix. Fig. 3 shows the efficiency of cell incorporation and cell viability for both bulk gel and microbead matrices. DNA content was used to track cell content, and showed that cell incorporation into COL–FIB microbead populations was approximately 72% of that in corresponding COL–FIB bulk gels (Fig. 3a). The lower number of cells in microbead preparations was probably caused by loss of both cells and matrix material during the microbead fabrication process, which was negligible when producing bulk gels. Reducing this loss is a process optimization challenge, and it is expected that the relative loss would be smaller as the process is scaled up.
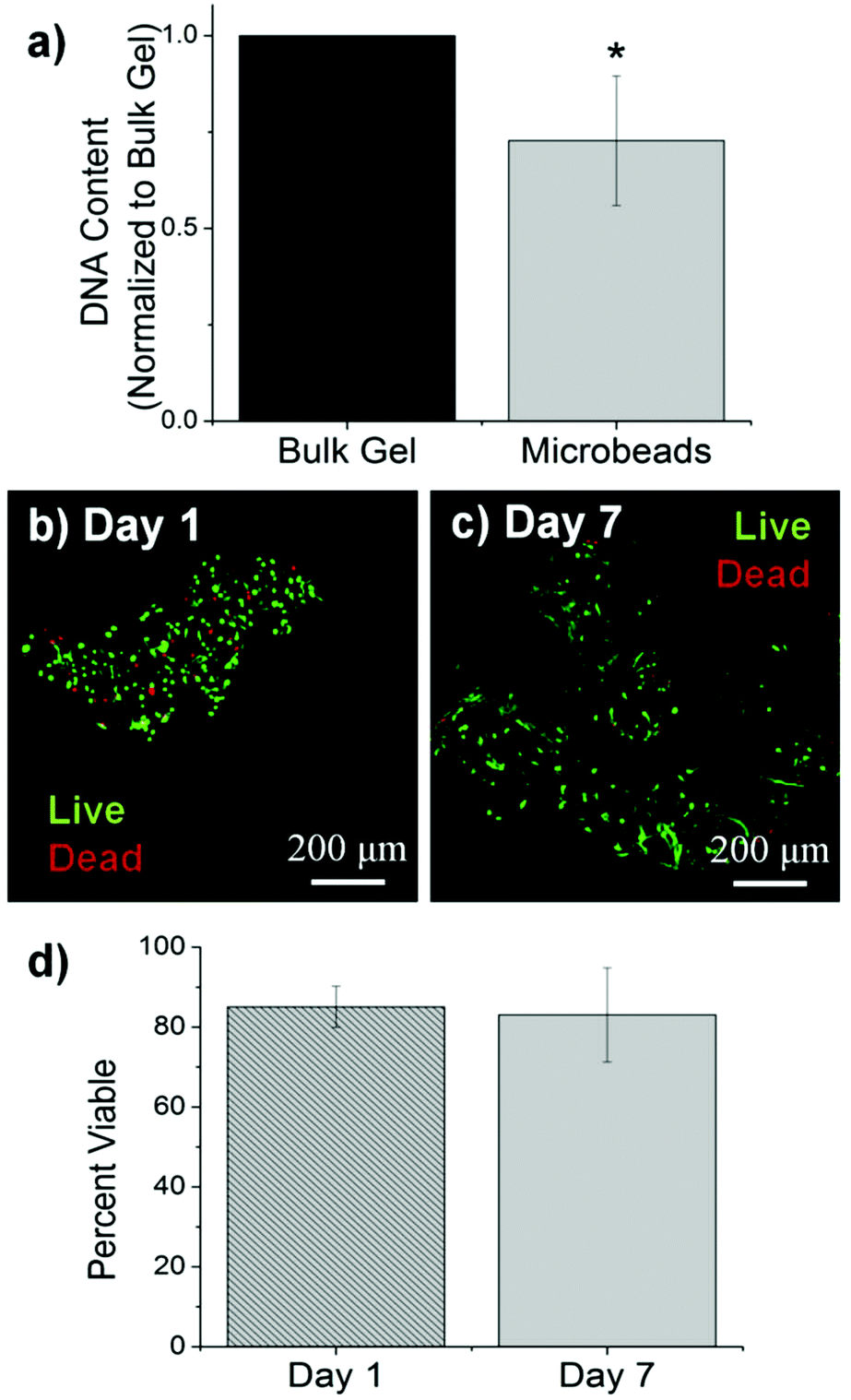 | ||
| Fig. 3 Cell incorporation and viability in COL–FIB microbeads. (a) DNA content showed incorporation of cells into microbeads as compared to bulk gels. (b) Images of viability of HUVEC and NHLF within loose clumps of microbeads at day 1 and (c) day 7. Live cells are stained green, dead cells are stained green. (d) Quantification of cell viability by image analysis. *Statistical significance vs. bulk gel (p < 0.05). Best viewed in color. | ||
The viability of HUVEC and NHLF in COL–FIB microbeads was high after fabrication and remained high over the week in culture during which these samples were followed. Vital staining and confocal imaging showed that cells were >80% viable at day 1 (Fig. 3b) and that viability was uniform throughout the microbeads. Similarly, cell viability at day 7 (Fig. 3c) was >80% and spatially uniform. These results are similar to those typically obtained in 3D collagen–fibrin bulk gels.56 Quantification of these observations using image analysis confirmed that there was no statistically significant change in cell viability in microbeads over time (Fig. 3d). Taken together, these microbead characterization data indicate that multiple cell types can be efficiently incorporated into microbeads, and that cell viability is maintained in culture. This allows microbead populations to be cultured as dispersed suspensions, and to be stimulated or manipulated in culture, prior to being used for cell delivery or to assemble larger tissue constructs.
Endothelial network formation and maturation within COL–FIB microbeads
Microbeads containing mixed populations of HUVEC and NHLF were cultured as dispersed populations for two weeks to examine the process and extent of endothelial network formation within individual microtissue units. Fig. 4 shows the analysis of cell morphology and distribution, with HUVEC stained specifically with UEA-1 (red), and the actin cytoskeleton and nucleus of all cells stained with phalloidin (green) and DAPI (blue), respectively. By day 7 in culture (Fig. 4a), cells were clearly spread with the COL–FIB matrix, and three-dimensional endothelial networks had begun to form. These networks were more developed and extended throughout the 3D matrix by Day 14 (Fig. 4b). Microbead and cell co-localization can be seen in the merged images, as well as in the 3D reconstruction of image stacks at day 7 (Fig. 4c) and day 14 (Fig. 4d). NHLF were dispersed throughout the microbeads and association between NHLF and HUVEC was observed. It should be noted that in this system the microbeads tended to aggregate over time to form larger structures when cultured under static conditions. In addition, fibroblasts on the surface of microbeads exhibited a more spread morphology and began to cover the surface. This effect was more evident in the day 14 samples, which exhibited endothelial networks that spanned across adjacent microbeads, as well as highly spread and numerous NHLF on the surface of microbead assemblies. This type of aggregation can potentially be inhibited by using stirred suspension culture; however in the present study aggregation was not actively prevented.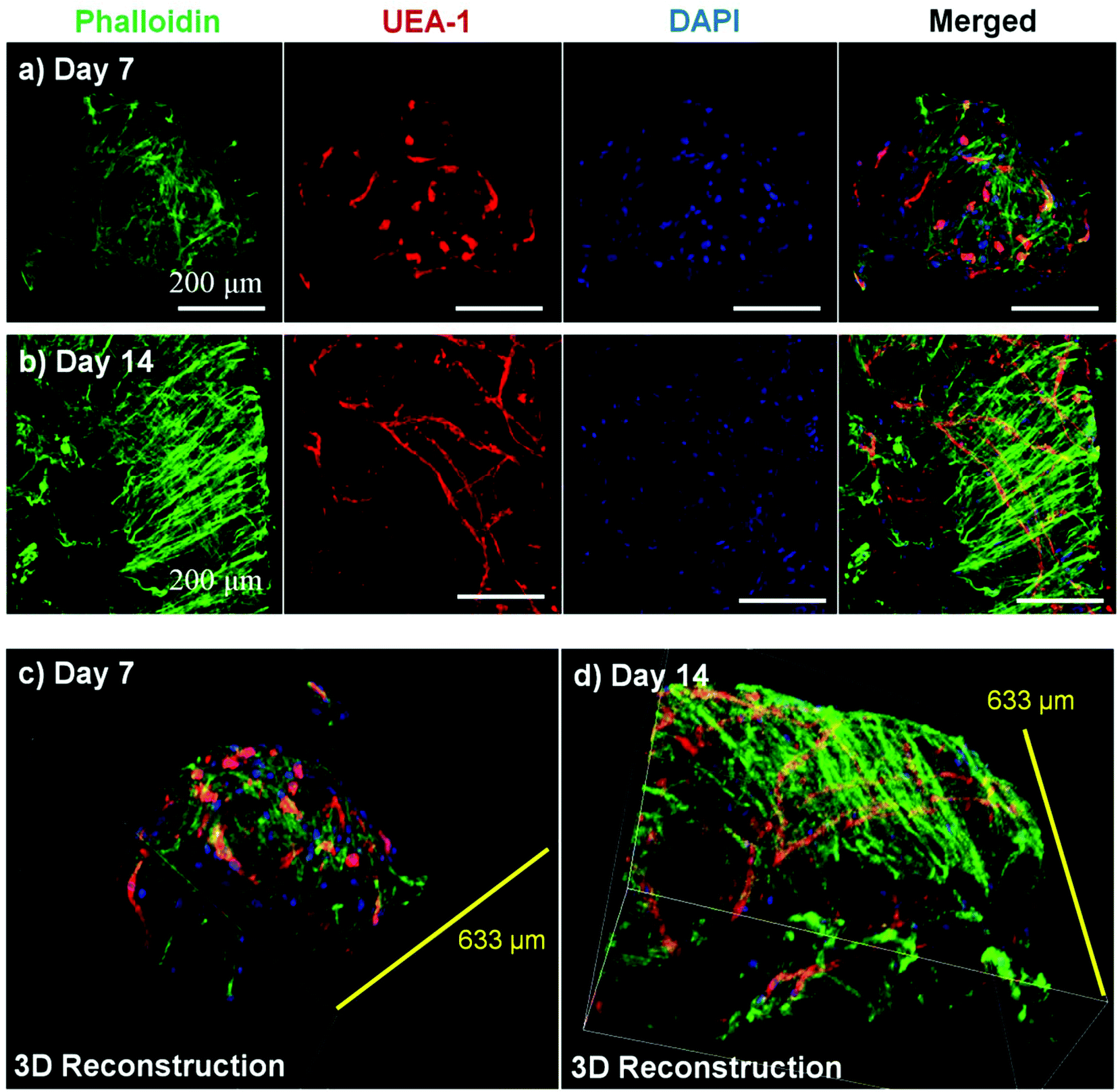 | ||
| Fig. 4 Network formation by endothelial cells within COL–FIB microbeads cultured as free-floating populations. Maximum projections of cell staining for the actin cytoskeleton (phalloidin, green), endothelial cells (UEA-1, red), and cell nuclei (DAPI, blue) at (a) day 7 and (b) day 14. Three-dimensional reconstructions of microbeads at (c) day 7 and (d) day 14. Best viewed in color. | ||
Fig. 5 shows cell-seeded COL–FIB microbeads that were cultured under static conditions for 7 days, and were then stained for α-SMA to identify NHLF and with UEA-1 to identify HUVEC. In addition, laminin deposition was visualized by immunostaining in these samples. It can be seen that NHLF co-localized with HUVEC and exhibited α-SMA staining (Fig. 5a), suggesting they perform a pericyte function in these microtissues. Laminin deposition was also robust in these samples (Fig. 5b and 5c) and was similarly co-located with endothelial networks. Some laminin staining was evident in areas where EC were not clearly present, and this observation may be due to the contribution of out-of-plane EC, or by EC that had retracted. Alternately, it is possible that the fibroblasts, which also co-located with the EC, produced or stimulated the assembly of the laminin, as has been reported.57,58 In both cases the presence of laminin can be interpreted as the development of a basement membrane, which in turn is indicative of maturing microvasculature.59 Overall, the 3D reconstruction images of these samples (Fig. 5d–5f), show robust endothelial networks characterized by both EC-associated pericyte-like cells as well as deposition of a basement membrane-associated extracellular matrix, indicative of a maturing vascular network.
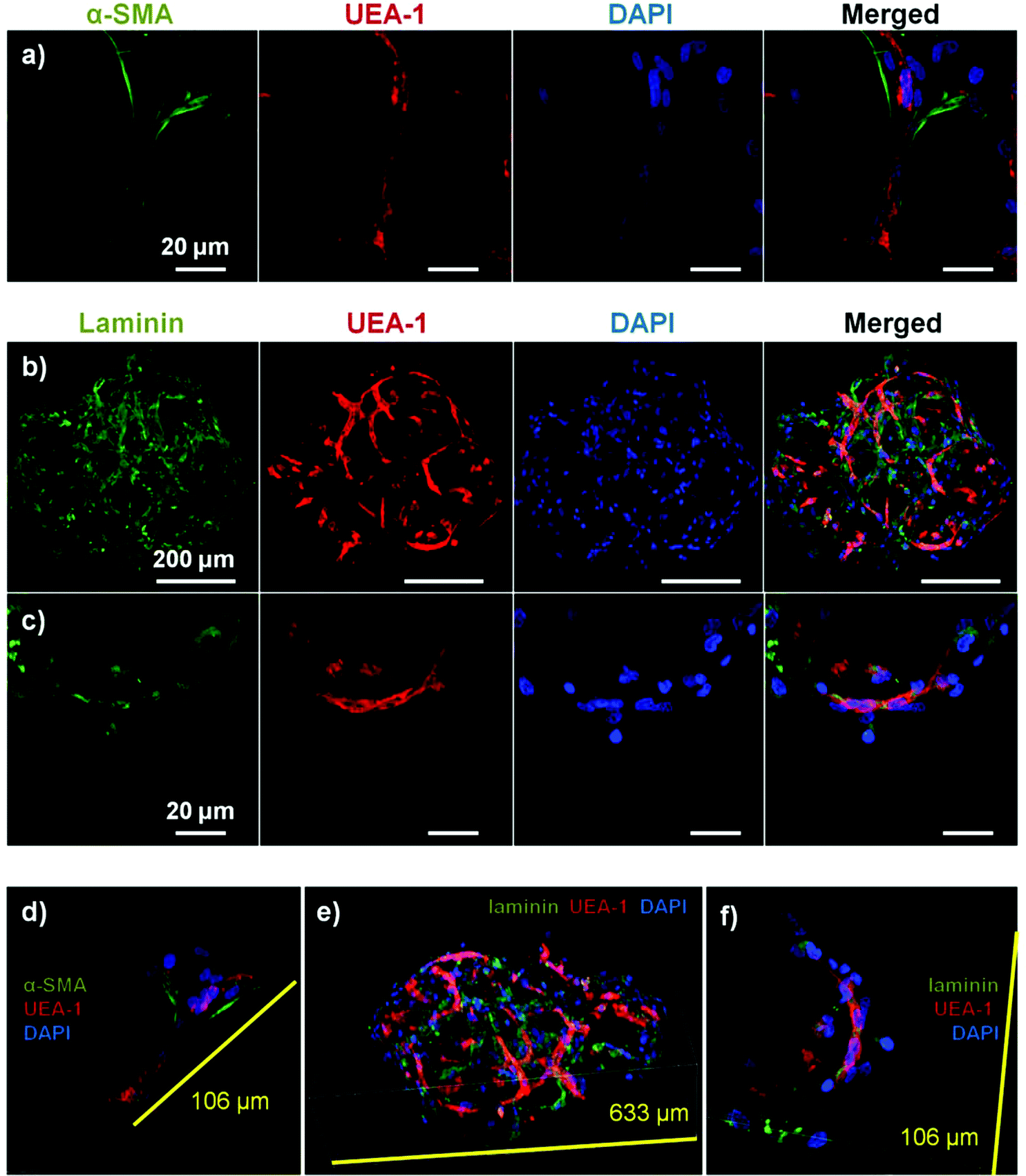 | ||
| Fig. 5 Maturity of endothelial networks formed in COL–FTB microbeads cultured as free-floating populations. (a) NHLF stained with α-SMA (green) co-localized with endothelial networks by day 7. Laminin deposition (green) also co-localized with endothelial networks, shown at (b) low and (c) high magnification. Three-dimensional reconstructions of stained micro beads are shown in panels d–f. Best viewed in color. | ||
Endothelial network formation by COL–FIB microbeads embedded in fibrin gel
To assess the ability of endothelial networks to emerge from microbeads and into surrounding matrices and tissues, COL–FIB microbeads seeded with HUVEC and NHLF were embedded in bulk fibrin (FIB) hydrogels. Microbeads that were embedded immediately after fabrication are shown in Fig. 6. Distribution of the microbeads in the surrounding hydrogel was visualized by using FITC-fibrinogen (green) as the microbead matrix material, to distinguish them from the external, unstained fibrin matrix (Fig. 6a and 6b). HUVEC were stained with UEA-1 (red), and the nuclei of all cells were stained with DAPI (blue). By day 7 (Fig. 6a), HUVEC networks were evident in the surrounding fibrin gel. Endothelial cords were generally localized near microbeads, but in some cases were entirely outside of the microbead matrix and separate from microbeads. By day 14 (Fig. 6b), inosculation of neighboring cords and more prominent vessel structures were observed. Staining of the actin cytoskeleton with phalloidin (green, Fig. 6c and 6d) showed that NHLF migrated out of the microbeads and populated the surrounding gel. This effect was observed at the earlier day 7 time point, and was especially prevalent at the later day 14 time point. The merged images in Fig. 6 (right column) show the microbead and cell distribution, as well as network formation from these embedded COL–FIB microbeads.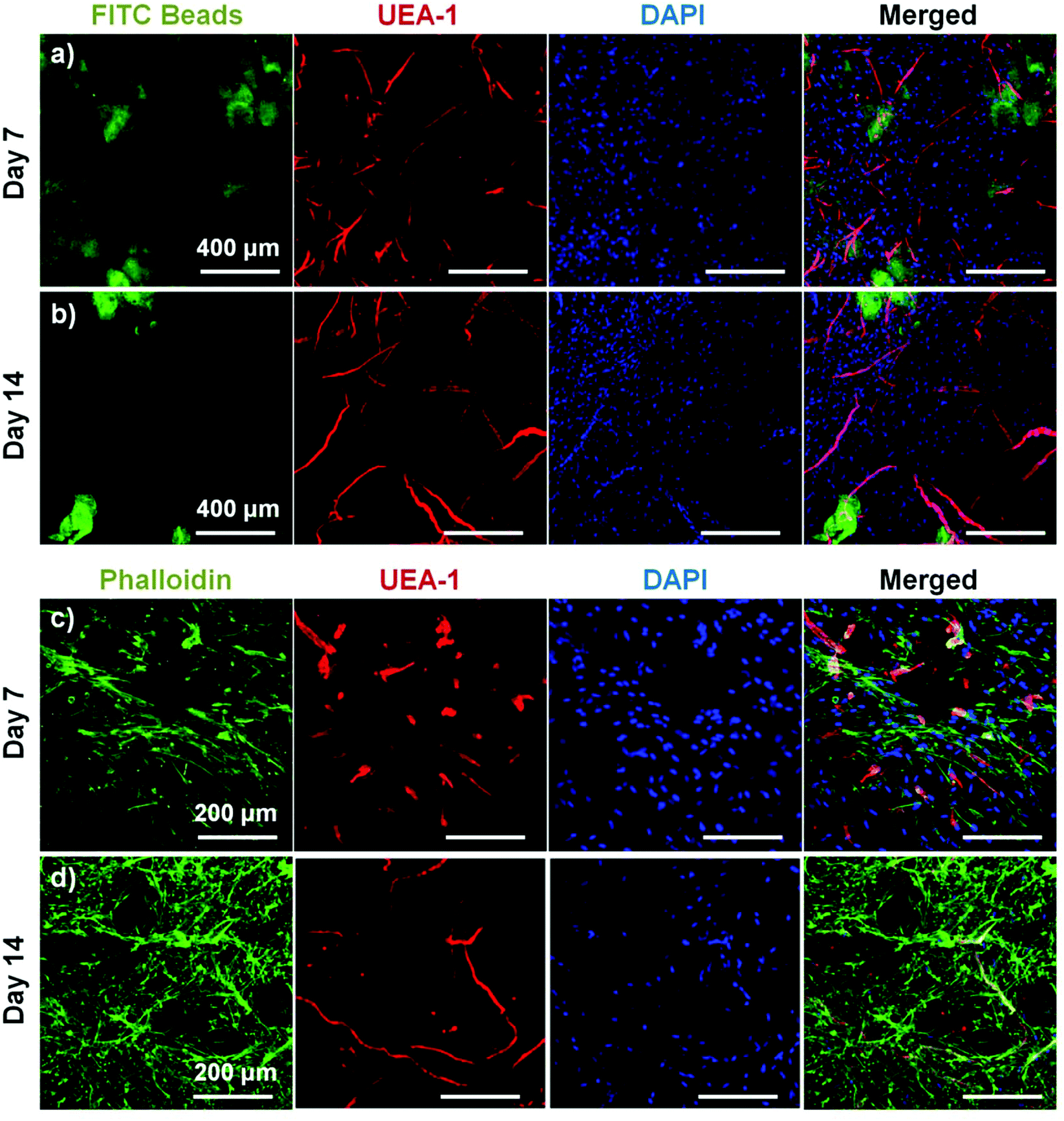 | ||
| Fig. 6 COL–FIB microbeads embedded in a surrounding FIB hydrogel immediately after fabrication. Microbeads produced endothelial network formation in the matrix at (a, c) day 7 and (b, d) day 14. HUVECs were stained with UEA-1 (red) and nuclei with DAPI (blue). In (a, b) microbeads were created using FITC-fibrinogen (green). In (c, d) actin was stained with phalloidin (green). Best viewed in color. | ||
The effect of pre-culture of COL–FIB microbeads before embedding in a fibrin hydrogel was also assessed, as shown in Fig. 7. In this case, microbeads were cultured for 7 days to allow for endothelial network formation in the microbead matrix prior to embedding in bulk fibrin gels. Such pre-vascularized microbeads also gave rise to endothelial networks, though a less homogenous distribution of cells within the bulk hydrogel environment was observed, compared to microbeads embedded immediately after fabrication. At day 7 post-embedding, HUVEC networks (red, Fig. 7a) were clearly more localized to microbeads (outlined in white), with some cords protruding radially outward into the surrounding matrix. Invasion of the external fibrin gel by NHLF was also evident in these studies. By day 14, the radial endothelial networks were more developed (Fig. 7b) and in regions with multiple microbeads (Fig. 7c) had begun to inosculate with networks from neighboring microbeads.
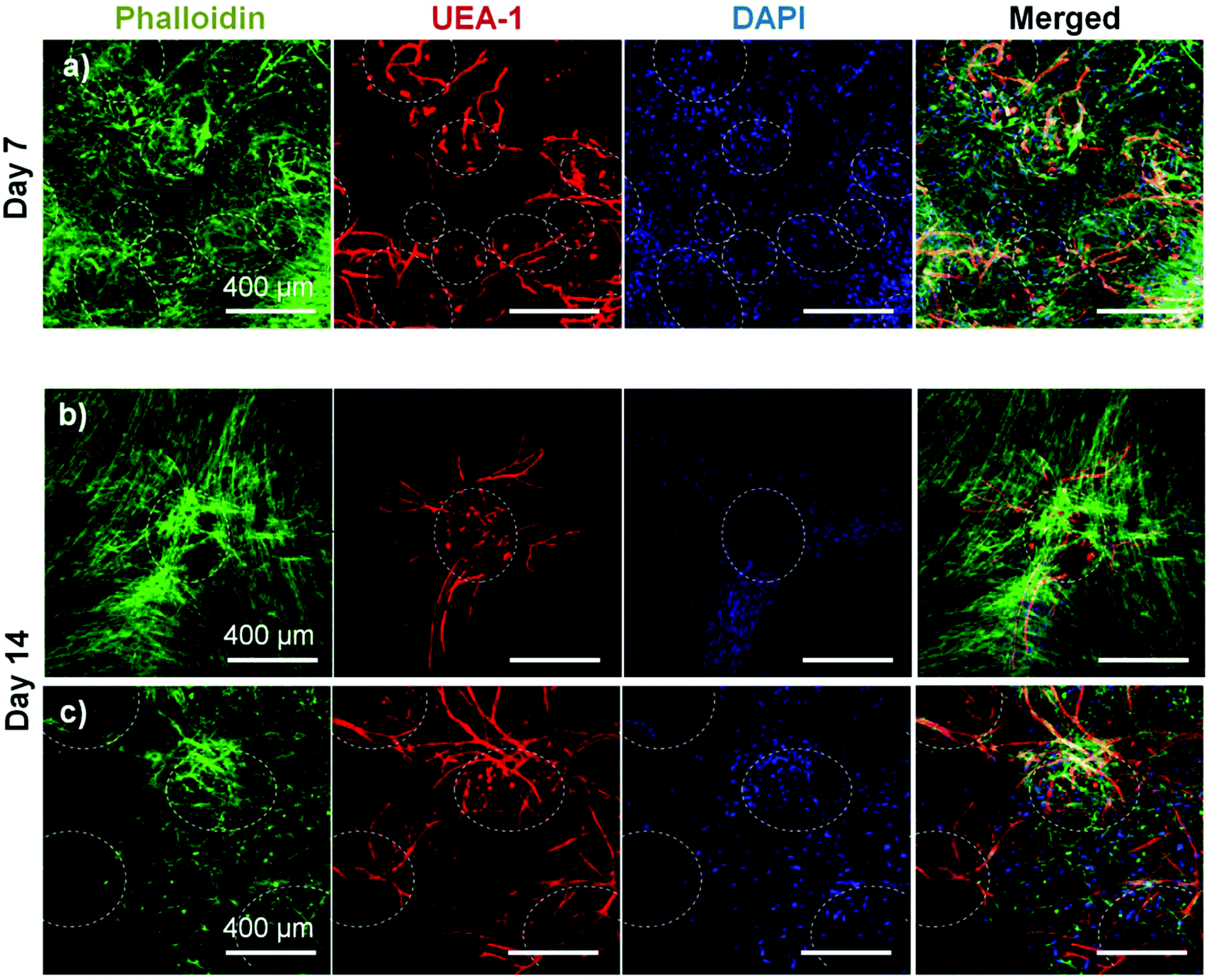 | ||
| Fig. 7 COL–FIB microbeads embedded in a 3D bulk fibrin gel after 7 days of pre-culture. Radial endothelial networks emanating from the pre-cultured microbeads were evident at (a) day 7 and were more prominent at (b, c) day 14. A region of relatively low microbead density is shown in (b) and a region of higher microbead density is shown in (c). Approximate microbead locations determined from transmitted light images (not shown) are outlined in white. Actin was stained with phalloidin (green), HUVEC with UEA-1 (red) and nuclei with DAPI (blue). Best viewed in color. | ||
Lumen formation in endothelial networks
COL–FIB microbeads generated robust endothelial networks when embedded in a surrounding fibrin hydrogel. The development of lumens is an indication of maturation of vessel networks, and was investigated using confocal microscopy. Three-dimensional confocal z-stacks were acquired, allowing visualization of specified x–y, x–z, and y–z planes that intersected endothelial cord structures, as shown in Fig. 8. At day 7, there was little evidence of lumen formation from microbeads embedded immediately after fabrication (Fig. 8a), and pre-cultured samples showed only the very early stages of cell hollowing (Fig. 8b). By day 14 after embedding in fibrin, clear lumens were observed in both samples embedded immediately after fabrication (Fig. 8c) and those that were pre-cultured prior to embedding (Fig. 8d). These hollow structures were multicellular and exhibited branching. The formation of lumens was also associated with more mature and highly developed vascular structures that were observed over time (Fig. 8e–8h, which show the maximum intensity projections of the z-stacks used in Fig. 8a–8d). | ||
| Fig. 8 Examination of lumen formation by analysis of orthogonal sections of a confocal z-stacks. At day 7, lumens were not evident in samples from (a) microbeads embedded immediately after fabrication, though (b) pre-cultured samples exhibited evidence of cell hollowing. By day 14, lumens were distinct in both samples from (c) microbeads embedded immediately after fabrication, and (d) microbeads pre-cultured before embedding. Maximum projects of the samples shown in panels a–d are shown in panels e–h. Best viewed in color. | ||
Vasculogenesis and angiogenesis using the COL–FIB microbead system
The morphology of the endothelial networks that were formed in the 3D microbead–hydrogel system depended on whether the microbeads were embedded immediately after fabrication, or were pre-cultured prior to embedding. Immediately-embedded microbeads produced networks that coalesced from within the microbead matrix and also created vascular structures that were not associated with the originating microbeads. In this case, the microbeads appear to have served as cell carriers that allowed the de novo formation of endothelial networks in the surrounding fibrin hydrogel. In contrast, vessel networks derived from pre-cultured microbeads were more closely localized to microbead locations, and radiated out from within the microbeads. In this case, network formation appeared to occur via extension and branching of existing cords within the microbead matrix.The system of using either newly fabricated or pre-cultured composite microbeads embedded in a surrounding hydrogel may have utility in studying aspects of vasculogenesis and angiogenesis. By varying the period of pre-culture, the maturity of the endothelial networks within microbeads can be controlled, and upon embedding in a surrounding hydrogel the process by which they extend and form interconnected networks can be studied. Embedding microbeads immediately after fabrication represents the limiting case in which endothelial network formation has not been initiated, and therefore mimics new vessel formation via vasculogenesis. Embedding of pre-cultured microbeads containing pre-formed endothelial cords more closely represents network formation by sprouting, elongation, and joining of existing vessels, thereby mimicking aspects of angiogenesis. The microbead system therefore complements other approaches that have been developed to study blood vessel formation, including the use of cells embedded in bulk gels,60,61 “bead assays” using cell-coated microspheres,9 and spheroid culture.62,63
The microbead approach is also flexible in that the composition, content, and properties of both the embedded microbeads and the surrounding hydrogel can be varied to investigate how these elements affect new vessel formation. The composite matrix used in the current study has been shown to support vasculogenesis in bulk gels,52 but the type and ratio of cells could be varied to examine their effects on endothelial network formation. Similarly, other cell types can be used to create microbeads, either as co-cultures within the same microbead, or as separate microbead populations. Fibrin was used as the surrounding matrix in these studies because of its proven and widely leveraged ability to support endothelial networks. However, the composition and properties of the surrounding matrix also can be altered, for example to examine the effects of matrix density or stiffness, or to investigate the role of growth factors in the development and maturation of vessel networks. This flexibility provides a rich platform with which to study the effects of cell type, neovessel maturity, and matrix properties on the process of neovascularization.
Modular tissue engineering for therapeutic vascularization
A primary motivation behind creating small modular tissue units is to avoid the diffusive mass transfer limitations that can cause cell damage or death in larger tissue constructs with dimensions greater than 100–200 μm.6 Each microbead has dimensions on a scale at which diffusional transport will not limit the function or viability of embedded cells, and populations of microbeads contain inter-bead channels that can supply nutrients. The bottom-up approach of modular tissue engineering aims to direct the assembly of such small tissue building blocks to create larger structures.64 Vasculogenesis is inherently such a process, since new blood vessels are formed when groups of cells and matrix coalesce to form primitive networks, which subsequently link up to form more mature and stable vessels. Angiogenesis can also be thought of in these terms, since it relies on the linking of small vessels to create larger networks that infiltrate tissues.The microbead approach presented here can be applied in vivo to deliver cells and pre-formed tissues to sites requiring tissue repair. Delivery of microtissue preparations has been demonstrated and used to support the differentiation and function of cells,23,54 including in vascular applications.65 In some applications, the use of microbeads as a delivery vehicle may be sufficient to keep cells viable and promote their retention and engraftment. However, a major advantage of the modular approach described here is that microbeads can be pre-cultured to promote differentiated function or tissue formation prior to implant. In the case of revascularization therapies, there may be a significant advantage to delivering pre-formed vascular units to ischemic regions. Such pre-formed structures may anastomose with host vasculature more rapidly than dispersed cells delivered alone. Rapid reperfusion of the implant site would help the transplanted microtissues survive, which in turn may improve the chances of rescuing ischemic tissue.
Fabrication, delivery and assembly of modular microtissues to create larger and more complex tissue structures are promising developments in the fields of biomaterials science and tissue engineering. A number of approaches have been developed for a variety of indications, with materials, cells, and modules tailored for specific tissue types. The present study has shown that biomaterials-based microbeads can support the viability and function of endothelial and pericyte-like cells, and can be used to create vascularized microtissues. Such modules may find utility in studying the processes of angiogenesis and vasculogenesis, and may also be used therapeutically to vascularize damaged and ischemic tissues, or larger transplanted engineered constructs. The flexibility and simplicity of the modular format offers a wide range of possible therapeutic targets.
Conclusions
This study shows that a facile water-in-oil emulsion process can be used to fabricate uniform populations of spheroidal collagen–fibrin microbeads containing viable co-cultures of endothelial cells and fibroblasts. Such microtissues support the formation of endothelial cell networks over time in culture, and the fibroblasts act to potentiate and stabilize the nascent vessels. Embedding of microbeads in a surrounding fibrin hydrogel showed that vessel networks could populate the matrix outside of the embedded microtissues. Microbeads embedded directly after fabrication mimicked the process of vasculogenesis, in which vessels are formed de novo from dispersed cells. Pre-culture of microbeads prior to embedding resulted in vessel sprouting, branching, and inosculation in a process that resembled angiogenesis. The protein composite microbead concept presented here may have utility in studying vascular network formation in vitro. It may also provide a method of vascularizing tissues in vivo, and in particular may offer the ability to more rapidly recapitulate the vasculature that is needed to rescue and regenerate ischemic tissues.Acknowledgements
This work was supported in part by National Heart, Lung and Blood Institute grant R01-HL118259 (to AJP and JPS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- L. C. Goldie, M. K. Nix and K. K. Hirschi, Organogenesis, 2008, 4, 257–263 CrossRef PubMed.
- R. K. Jain, Nat. Med., 2003, 9, 685–693 CrossRef CAS PubMed.
- O. C. Velazquez, J. Vasc. Surg., 2007, 45, A39–A47 CrossRef PubMed.
- S. M. Bauer, R. J. Bauer and O. C. Velazquez, Vasc. Endovascular Surg., 2005, 39, 293–306 CrossRef PubMed.
- E. C. Novosel, C. Kleinhans and P. J. Kluger, Adv. Drug Delivery Rev., 2011, 63, 300–311 CrossRef CAS PubMed.
- R. K. Jain, P. Au, J. Tam, D. G. Duda and D. Fukumura, Nat. Biotechnol., 2005, 23, 821–823 CrossRef CAS PubMed.
- C. J. Kirkpatrick, S. Fuchs and R. E. Unger, Adv. Drug Delivery Rev., 2011, 63, 291–299 CrossRef CAS PubMed.
- C. M. Ghajar, K. S. Blevins, C. C. Hughes, S. C. George and A. J. Putnam, Tissue Eng., 2006, 12, 2875–2888 CrossRef CAS PubMed.
- M. N. Nakatsu, J. Davis and C. C. Hughes, J. Vis. Exp., 2007, 3, 186 Search PubMed.
- B. Carrion, Y. P. Kong, D. Kaigler and A. J. Putnam, Exp. Cell Res., 2013, 319, 2964–2976 CrossRef CAS PubMed.
- X. Chen, A. Aledia, C. Ghajar, C. Griffith, A. J. Putnam, C. C. Hughes and S. C. George, Tissue Eng., Part A, 2009, 15, 1363–1371 CrossRef CAS PubMed.
- P. Au, J. Tam, D. Fukumura and R. Jain, Blood, 2008, 111, 4551–4558 CrossRef CAS PubMed.
- E. Kniazeva, S. Kachgal and A. J. Putnam, Tissue Eng., Part A, 2011, 17, 905–914 CrossRef CAS PubMed.
- S. Merfeld-Clauss, N. Gollahalli, K. March and D. Traktuev, Tissue Eng., Part A, 2010, 16, 2953–2966 CrossRef CAS PubMed.
- B. Shepherd, S. Jay, W. Saltzman, G. Tellides and J. Pober, Tissue Eng., Part A, 2009, 15, 165–173 CrossRef CAS PubMed.
- P. Au, L. M. Daheron, D. G. Duda, K. S. Cohen, J. A. Tyrrell, R. M. Lanning, D. Fukumura, D. T. Scadden and R. K. Jain, Blood, 2008, 111, 1302–1305 CrossRef CAS PubMed.
- S. J. Grainger and A. J. Putnam, PLoS One, 2011, 6, e22086 CAS.
- S. Kachgal and A. J. Putnam, Angiogenesis, 2011, 14, 47–59 CrossRef CAS PubMed.
- A. P. McGuigan and M. V. Sefton, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11461–11466 CrossRef CAS PubMed.
- J. M. Kelm, V. Djonov, L. M. Ittner, D. Fluri, W. Born, S. P. Hoerstrup and M. Fussenegger, Tissue Eng., 2006, 12, 2151–2160 CrossRef PubMed.
- D. M. Dean, A. P. Napolitano, J. Youssef and J. R. Morgan, FASEB J., 2007, 21, 4005–4012 CrossRef CAS PubMed.
- J. K. Wise, A. I. Alford, S. A. Goldstein and J. P. Stegemann, Tissue Eng., Part A, 2014, 20, 210–224 CrossRef CAS PubMed.
- L. Wang, R. R. Rao and J. P. Stegemann, Cells Tissues Organs, 2013, 197, 333–343 CrossRef CAS PubMed.
- H. W. Cheng, K. D. Luk, K. M. Cheun and B. P. Chan, Biomaterials, 2011, 32, 1526–1535 CrossRef CAS PubMed.
- B. M. Leung, Y. Miyagi, R. K. Li and M. V. Sefton, J. Tissue Eng. Regen. Med., 2014 DOI:10.1002/term.1724.
- B. R. Desroches, P. Zhang, B. R. Choi, M. E. King, A. E. Maldonado, W. Li, A. Rago, G. Liu, N. Nath, K. M. Hartmann, B. Yang, G. Koren, J. R. Morgan and U. Mende, Am. J. Physiol. Heart Circ. Physiol., 2012, 302, H2031–H2042 CrossRef CAS PubMed.
- R. Tiruvannamalai-Annamalai, D. R. Armant and H. W. Matthew, PLoS One, 2014, 9, e84287 Search PubMed.
- R. Gupta and M. V. Sefton, Tissue Eng., Part A, 2011, 17, 2005–2015 CrossRef CAS PubMed.
- D. J. Caldwell, R. R. Rao and J. P. Stegemann, Adv. Healthcare Mater., 2013, 2, 673–677 CrossRef CAS PubMed.
- R. Gauvin and A. Khademhosseini, ACS Nano, 2011, 5, 4258–4264 CrossRef CAS PubMed.
- V. Mironov, R. P. Visconti, V. Kasyanov, G. Forgacs, C. J. Drake and R. R. Markwald, Biomaterials, 2009, 30, 2164–2174 CrossRef CAS PubMed.
- R. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522–9527 CrossRef PubMed.
- A. P. McGuigan, D. A. Bruzewicz, A. Glavan, M. J. Butte and G. M. Whitesides, PLoS One, 2008, 3, e2258 Search PubMed.
- A. Alajati, A. M. Laib, H. Weber, A. M. Boos, A. Bartol, K. Ikenberg, T. Korff, H. Zentgraf, C. Obodozie, R. Graeser, S. Christian, G. Finkenzeller, G. B. Stark, M. Heroult and H. G. Augustin, Nat. Methods, 2008, 5, 439–445 CrossRef CAS PubMed.
- R. Gupta, N. Van Rooijen and M. V. Sefton, Tissue Eng., 2009, 15, 2875–2887 CrossRef CAS PubMed.
- O. F. Khan and M. V. Sefton, Biomaterials, 2010, 31, 8254–8261 CrossRef CAS PubMed.
- M. J. Butler and M. V. Sefton, Tissue Eng., Part A, 2012, 18, 1628–1641 CrossRef CAS PubMed.
- M. D. Chamberlain, R. Gupta and M. V. Sefton, Tissue Eng., 2012, 18, 285–294 CrossRef CAS PubMed.
- E. C. Ciucurel and M. V. Sefton, Tissue Eng., Part A, 2014, 20, 1235–1252 CrossRef CAS PubMed.
- Y. Du, M. Ghodousi, H. Qi, N. Haas, W. Xiao and A. Khademhosseini, Biotechnol. Bioeng., 2011, 108, 1693–1703 CrossRef CAS PubMed.
- H. F. Chan, Y. Zhang, Y. P. Ho, Y. L. Chiu, Y. Jung and K. W. Leong, Sci Rep., 2013, 3, 3462 Search PubMed.
- Z. Chen, L. Wang and J. P. Stegemann, J. Microencapsulation, 2011, 28, 344–352 CrossRef CAS PubMed.
- A. W. Lund, B. Yener, J. P. Stegemann and G. E. Plopper, Tissue Eng., Part B, 2009, 15, 371–380 CrossRef PubMed.
- B. D. Walters and J. P. Stegemann, Acta Biomater., 2014, 10, 1488–1501 CrossRef CAS PubMed.
- J. Ceccarelli and A. J. Putnam, Acta Biomater., 2014, 10, 1515–1523 CrossRef CAS PubMed.
- B. P. Chan, T. Y. Hui, C. W. Yeun, J. Li, I. Mo and G. C. Chan, Biomaterials, 2007, 28, 4652–4666 CrossRef CAS PubMed.
- A. Batorsky, J. Liao, A. W. Lund, G. E. Plopper and J. P. Stegemann, Biotechnol. Bioeng., 2005, 92, 492–500 CrossRef CAS PubMed.
- T. P. Cooper and M. V. Sefton, Acta Biomater., 2011, 7, 1072–1083 CrossRef CAS PubMed.
- E. C. Ciucurel, A. E. Vlahos and M. V. Sefton, Tissue Eng., Part A, 2014, 20, 1222–1234 CrossRef CAS PubMed.
- K. T. Morin and R. T. Tranquillo, Exp. Cell Res., 2013, 319, 2409–2417 CrossRef CAS PubMed.
- C. L. Cummings, D. Gawlitta, R. M. Nerem and J. P. Stegemann, Biomaterials, 2004, 25, 3699–3706 CrossRef CAS PubMed.
- R. R. Rao, A. W. Peterson, J. Ceccarelli, A. J. Putnam and J. P. Stegemann, Angiogenesis, 2012, 15, 253–264 CrossRef CAS PubMed.
- R. R. Rao, J. Ceccarelli, M. L. Vigen, M. Gudur, R. Singh, C. X. Deng, A. J. Putnam and J. P. Stegemann, Acta Biomater., 2014, 10, 3091–3097 CrossRef CAS PubMed.
- R. R. Rao, A. W. Peterson and J. P. Stegemann, J. Biomed. Mater. Res., Part A, 2013, 101, 1531–1538 CrossRef PubMed.
- L. Wang and J. P. Stegemann, Acta Biomater., 2011, 7, 2410–2417 CrossRef CAS PubMed.
- C. M. Voge, M. Kariolis, R. A. MacDonald and J. P. Stegemann, J. Biomed. Mater. Res., Part A, 2008, 86, 269–277 CrossRef PubMed.
- S. Paquet-Fifield, H. Schlüter, A. Li, T. Aitken, P. Gangatirkar, D. Blashki, R. Koelmeyer, N. Pouliot, M. Palatsides, S. Ellis, N. Brouard, A. Zannettino, N. Saunders, N. Thompson, J. Li and P. Kaur, J. Clin. Invest., 2009, 119, 2795–2806 CAS.
- A. N. Stratman, K. M. Malotte, R. D. Mahan, M. J. Davis and G. E. Davis, Blood, 2009, 114, 5091–5101 CrossRef CAS PubMed.
- A. N. Stratman and G. E. Davis, Microsc. Microanal., 2012, 18, 68–80 CrossRef CAS PubMed.
- A. O. Smith, S. L. Bowers, A. N. Stratman and G. E. Davis, PLoS One, 2013, 8, e85147 Search PubMed.
- G. E. Davis, D. J. Kim, C. X. Meng, P. R. Norden, K. R. Speichinger, M. T. Davis, A. O. Smith, S. L. Bowers and A. N. Stratman, Methods Mol. Biol., 2013, 1066, 17–28 CAS.
- C. W. Eckermann, K. Lehle, S. A. Schmid, D. N. Wheatley and L. A. Kunz-Schughart, Cell Biol. Int., 2011, 35, 1097–1110 CrossRef PubMed.
- A. P. Napolitano, P. Chai, D. M. Dean and J. R. Morgan, Tissue Eng., 2007, 13, 2087–2094 CrossRef CAS PubMed.
- O. F. Khan, D. N. Voice, B. M. Leung and M. V. Sefton, Adv. Healthcare Mater., 2014 DOI:10.1002/adhm.201400150.
- K. J. Portalska, M. D. Chamberlain, C. Lo, C. van Blitterswijk, M. V. Sefton and J. de Boer, J. Tissue Eng. Regen. Med., 2013 DOI:10.1002/term.1738.
Footnote |
| † This paper is part of a collection of scientific reports celebrating Professor Michael Sefton's 65th birthday, as well as his many contributions to biomaterials science and tissue engineering. Dr Sefton's career to date has included important contributions to enzyme immobilization, controlled release, hemocompatibility, wound healing, polymer synthesis, cell encapsulation, and modular tissue engineering. A common thread in his approach to all of these areas is the innovative but rigorous application of the principles of biomaterials science. The authors sincerely thank Professor Sefton for his leadership in the field, and in particular his valuable insights into many of the important issues in the development of cell-based therapies. |
| This journal is © The Royal Society of Chemistry 2014 |