
Development of an hydrophobic fluoro-silica surface for studying homotypic cancer cell aggregation–disaggregation as a single dynamic process in vitro†
Matthew
Nicklin
ab,
Robert C.
Rees
b,
A. Graham
Pockley
b and
Carole C.
Perry
*a
aInterdisciplinary Biomedical Research Centre, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: Carole.Perry@NTU.AC.UK
bJohn van Geest Cancer Research Centre, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK
First published on 22nd July 2014
Abstract
Under normal conditions the detachment of anchorage-dependant cells from their extracellular matrix typically induces programmed cell death which is mediated through a pathway referred to as anoikis. However, a resistance to anoikis in cancer enables the migration of cells from the primary tumour and the establishment of aggressive metastatic disease. Although cancer cell aggregation is known to be an important mechanism within anoikis resistance, research into the underlying mechanisms that govern this process remain problematic as commercially available tissue culture material can only sustain 2D monolayer or 3D aggregate/spheroid cultures. This necessitates the development of a system that can accommodate for cancer cell aggregation–disaggregation as a single dynamic process, without the disruption of passaging cells between alternate substrates. This study describes a procedure for modifying tissue culture polystyrene (TCP) to produce a fluoro-silica (FS) surface which preferentially promotes the deposition of a distinct profile of proteins/factors from serum which mediate the transient aggregation of human breast cancer cell lines. This modified surface therefore provides an experimental platform for better understanding cancer cell aggregation–disaggregation events in vitro, and their influence on the establishment of metastatic disease in patients with cancer.
1. Introduction
Changes in cell adhesive properties are believed to play a key role in tumour invasiveness, progression and metastasis.1 The two key hypotheses describing cancer metastasis – termed the “seed and soil” and the “mechanical trapping” theories – emphasise the adherence of heterotypic tumour cells to the blood vessel endothelia.1–3 Furthermore, both include cancer cell aggregation as a key component of the metastatic cascade.1,3,4 Previous work has demonstrated that highly metastatic cells exhibit a superior ability to form homotypic aggregates than their low metastatic counterparts.1,4 A phenomenon which may provide a means of cellular separation/isolation yet to be exploited.In normal epithelial tissue, key survival signals are modulated by cell adhesion to extracellular matrix components, with cells undergoing intrinsically programmed cell death via apoptosis (a process known as anoikis) in their absence.5 This pathway of anoikis represents a homeostatic process which regulates and maintains a steady state within defined tissues and organs, whilst also acting as a barrier for preventing the metastatic spread of anchorage-dependant cells.5,6 In contrast, cells within tumours gain resistance to anoikis and are able to survive the harsh non-adherent conditions of the blood and lymph vessels.7 Indeed, the dysregulation of anoikis is an emerging hallmark of cancer, and has been reported to contribute to its mechanism of metastatic spread.6,8 For instance, the escape of multicellular aggregates from primary tumours facilitates the formation of emboli within the lymph and blood vessels, thereby contributing to haematogenous spread in several cancers.1,4,5 Despite the fundamental importance of these events, the underlying mechanisms governing cancer cell aggregation remain largely unknown.5 Furthermore, the physiological cues, which modulate the adherent capacity of the tumour cell within its microenvironment remain poorly understood.9
Cells in the body are typically surrounded by an extracellular matrix and are in direct physical contact with surrounding cells, conditions that are poorly replicated within 2D culture. Growing evidence suggests 3D spheroid culture to provide a more accurate representative model of in vivo tissues than traditional 2D monolayers.10 For example, chondrocytes and hepatocytes lose their regular phenotype when cultured in a 2D system, a process that can be attenuated or even reversed using 3D culture methodology.11 Currently, methods such as the liquid overlay, hanging drop, static liquid overlay and centrifugation techniques, amongst others, can be used for producing 3D spheroids.11 However, most of these techniques are impractical, too costly or require specialist equipment to conduct on a large scale and/or are often poorly reflective of in vivo conditions. Moreover, studies in the area of cell aggregation events remain problematic, as no current system can facilitate cellular aggregation–disaggregation as a single dynamic process without the disruption of passaging cells between substrates.
As cell–surface interactions are governed by surface properties, the study of cell responses towards alternate surface chemistry profiles is a plausible means to investigating important behavioural phenomena, such as adhesive and aggregation events.12 The adhesion of cells is mediated by the pre-absorbed proteins at the surface, with protein adsorption being influenced by surface properties, such as charge, topography and wettability.12–14 Extracellular matrix (ECM) proteins present structures on the nano-meter-scale which are believed to contribute towards cell-matrix signalling.15,16 Cells interact directly with the surface nanotopography, with the cell response towards such structures being mediated via a process referred to as contact guidance. This phenomenon of contact guidance is mediated by the organisation of ECM proteins and has been shown to be an essential component in regulating cell fate, migration and organelle formation.15–17 By manipulating surface properties such as wettability and topology, biointerfaces have been produced for directing cell adhesion, spread, motility and survival.17 Through enhancing topological interactions between cell surface structures and the substrate; researchers have produced interfaces for enhanced cell adhesion, as well as highly efficient target cell adhesion substrates for use in cancer diagnosis and treatment.17,18
We have previously reported on a protocol for functionalising TCP with a silica film.19 In the present study, the procedure has been modified and implemented alongside a successive step to functionalise the silica film with a fluorosilane in order to produce a hydrophobic, low energy FS surface. Using normal human mammary epithelial (MCF-10a) and breast cancer (MCF-7) cells, serum-dependent, transient aggregate formation on the FS surface was observed, with no evidence of altered cell proliferation. Protein adsorption studies suggest that the observed aggregation/disaggregation of cells is driven by phased competitive adsorption of proteins to the surface in a time dependant manner, a process often referred to as the Vroman effect.20
2. Materials and methods
2.1 Silica film preparation
The silica surface was prepared using an adaptation of a previously described protocol.19,21 Briefly, untreated tissue culture polystyrene (Sarstedt) was coated with a polyaniline film by reacting 0.25 M aniline hydrochloride (Sigma A8524) in 1 M HCl with 0.08 M ammonium persulfate (Sigma 248614) in double distilled (dd)H2O. This was conducted at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio with total coating volumes being 5 mL, 1 mL and 200 μL for petri dishes (83.1801.003), 24-well (83.1836) and 96-well (83.1835) plates respectively. Plates were incubated at room temperature (RT) for 15 min followed by three washes with an excess of (dd)H2O. Maintaining the same coating volumes, plates were treated with 2% v/v glutaric dialdehyde (Sigma 340855) for 2 h at 57 °C followed by washing.
:1 volumetric ratio with total coating volumes being 5 mL, 1 mL and 200 μL for petri dishes (83.1801.003), 24-well (83.1836) and 96-well (83.1835) plates respectively. Plates were incubated at room temperature (RT) for 15 min followed by three washes with an excess of (dd)H2O. Maintaining the same coating volumes, plates were treated with 2% v/v glutaric dialdehyde (Sigma 340855) for 2 h at 57 °C followed by washing.
Plates were then treated with 1 mg mL−1 lysozyme (Sigma 62971) in Dulbecco's phosphate buffered saline (DPBS) (Lonza 17-512F) for 2 h at RT and then washed. To prepare the silica surface, 0.5 M tetramethyl orthosilicate (Sigma 218472) in (dd)H2O was pre-hydrolysed in 1 mM HCl for 15 min. The pH of this solution was raised to 4 by adding 0.1 M NaOH in a drop-wise fashion prior to surface application. Plates were incubated for 2 h at RT to allow for silica film formation and were then washed once more.
2.2 Hydrophobic FS surface preparation
Silica surfaces were then used for FS functionalisation via a silane reaction using 1H,1H,2H,2H-perfluorodecyltriethoxysilane (Sigma 658758) (FDTES). This was pre-hydrolysed at a 10 mM concentration in 75% v/v ethanol and 5 mM HCl for 30 min. The pH of the coating solution was then raised to 8 by adding 0.25 M NH4OH (Sigma 318612) in a drop wise fashion prior to application. Plates were incubated for 2 h at 57 °C, washed three times with an excess of ethanol and three times with (dd)H2O, followed by curing for 24 h at 57 °C. All surfaces were UV sterilised for 15 min immediately prior to characterisation or use in cell culture. During the initial acid hydrolysis of the fluorinated alkoxysilane, FDTES, ethoxy groups (CH3CH2O) are cleaved to produce free silanol groups (Si–OH), with the silane adopting chemical structures schematically represented in Fig. 1. However, condensation to the surface occurs at an inherently low rate under these acidic conditions (∼pH 2.6) due to the close proximity of the reaction medium to the isoelectric point of Si at ∼pH 2.22,23 As the pH is raised to 8, silanol groups become deprotonated and the condensation rate is increased due to an increased tendency for nucleophilic attack.23 As a consequence, covalent Si–O–Si (siloxane) bonds are formed between the aqueous silane and the free silanol groups localised at the surface. | ||
| Fig. 1 Functionalisation of the silica film with FDTES via silanol group condensation. For simplicity the silica film is shown as a monolayer. | ||
2.3 Contact angle analysis
To assess surface wettability, 5 μL drops of (dd)H2O were applied to each surface using an Attension Theta Lite tensiometer. Measurements were taken after 5 seconds to allow for droplet stabilisation and the data was analysed using OneAttension software. Triplicate measurements were taken for each surface, with each surface being tested in triplicate (9 replicates in total). Repeat measurements using formamide (Sigma 47670) and diodomethane (Sigma 158429) liquid phases allowed the surface energy to be calculated using the FOWKES equation.242.4 Attenuated total reflectance infrared spectroscopy (ATR-IR)
FDTES functionalisation was confirmed by identifying specific chemical bonds such as C–F and Si–O–Si by ATR-IR using a PerkinElmer spectrum 100 spectrometer with samples analysed in petri-dish format. Spectra were plotted and labelled using Origin 7.5 following acquisition.2.5 X-ray photoelectron spectroscopy (XPS)
The specific chemical composition of TCP and the FS surface was investigated by XPS using a VG Scientific ESCALab MkII X-ray photoelectron spectrometer with Al Kα X-ray source (hν = 1483.6 eV). Surfaces (in petri-dish format) were dissected into 4 × 4 cm2 samples using a heated scalpel and mounted onto standard sample holders. Survey spectra covering the full binding energy (BE) range from 0–1200 eV with a step size of 0.2 eV and pass energy (PE) of 50 eV were collected. All binding energies were corrected to the saturated hydrocarbon C 1s peak at 285.0 eV in order to eliminate surface charging effects.2.6 Scanning electron microscopy (SEM)
SEM was used to analyse surface morphology (petri-dish format) using a JEOL JSM-840A scanning electron microscope in secondary electron mode with an accelerating voltage of 20–25 kV and working distances between 15 and 35 mm. Samples were gold coated using an Edwards Sputter Coater S150B. Energy dispersive X-ray analysis (EDXa) using an Oxford Instruments INCA X-sight system with a count rate of 3 kcounts s−1 was used to detect Si and F upon the FS surface.2.7 Atomic force microscopy (AFM)
Surface topology and roughness were detected by AFM using a Pacific Nano-R2 atomic force microscope in close contact mode with Pacific Nanotechnology Close Contact Mounted Cantilevers (petri-dish format). Root mean square (RMS) roughness measurements were calculated using Nano rule software following a triplicate of levelled scans per surface.2.8 Fluorescamine assay
Fluorescamine rapidly reacts with primary amines of proteins to produce a highly fluorescent product, with an emission intensity which directly correlates with protein concentration.25 For FS and TCP surfaces, 4 replicate wells (in 96-well format) were treated with Dulbecco's modified eagles medium (DMEM, LONZA 12-604Q) + 10% v/v foetal bovine serum (FBS, Thermo Scientific SV30160.03) and 1% non-essential amino acids (NEAA) (Lonza BE13-114E) for 2 or 24 h under cell culture conditions. Wells were washed three times with (dd)H2O and dried using compressed helium. For each test, a standard dose response curve of bovine serum albumin (BSA, 5 to 0.05 mg mL−1 + blank) (Thermo Scientific 23209) in DPBS was prepared. Each standard was applied at a volume of 50 μL to 4 separate (untreated) wells of the plate, resulting in a standard curve range from 250–2.5 μg per well, plus a blank. DPBS (50 μL) was added to each test well. A 4 mg mL−1 stock solution of fluorescamine (Sigma F9015) was prepared in acetone (Sigma 320110), this was diluted to 1.33 mg mL−1 in DPBS immediately prior to use in order to prevent degradation of the TCP plate. To each standard/test well 150 μL of this solution was added, resulting in 200 μL of 1 mg mL−1 fluorescamine in each well. Plates were mixed for 1 min prior to an immediate fluorescence intensity measurement using a TECAN ULTRA spectrophotometer at excitation and emission detection wavelengths of 390 and 465 nm respectively. The average signal produced from control wells was subtracted from each corresponding test/standard well. The amount of protein present within each test well was then calculated by reference to the standards and the amount of protein per cm2 calculated using the surface area occupied by 200 μL in a standard well of a 96-well plate.2.9 Indirect ELISA assays
Fibronectin and albumin adsorption to the TCP and the FS surfaces was compared using an indirect enzyme immunoassay (ELISA, 96-well plate format) using the rabbit polyclonal antibodies F3648, and B1520 (Sigma) for the primary detection of the respective proteins. All samples/ELISA reagents were applied at a 200 μL volume unless otherwise stated, plates were washed 3 times using 300 μL of DPBS + 1% v/v Tween 20 (Sigma P9416) after each stage. For each test, a total of 4 replicate wells of the FS surface and TCP were treated with DMEM media + 10% v/v FBS and 1% NEAA, or DPBS for 2 or 24 h periods under cell culture conditions. Wells were blocked with 1% w/v casein solution (Novagen 70955) for 2 h before the application of the primary detection antibody at 50 ng mL−1. The goat anti-rabbit biotinylated secondary antibody (DAKO E0432) was then applied at a concentration of 50 ng mL−1 for 2 h. Horseradish peroxidase (HRP) conjugated streptavidin (R & D systems 4800-30-60) was diluted 1 in 200 in DPBS and applied to each well for 20 min at RT. Tetramethylbenzidine substrate solution (R & D systems DY999) was added at a volume of 250 μL per well and plates were incubated at RT for 20 min, after which a 200 μL aliquot from each well was transferred to a clean 96-well plate. To each well, 100 μL of 0.5 M H2SO4 (Acros 7664-93-9) was added and absorbance was immediately measured at 450 nm with correction at 570 nm using a TECAN ULTRA spectrophotometer. The average signal from the control wells was then subtracted from each corresponding test well.2.10 Cell lines
MCF-7 (ATCC HTB-22) and MCF-10a (ATCC CRL-10317) cells were purchased from the American Type Culture Collection (ATCC) and used within 5 passages of the master stock. Cell culture medium was obtained from LONZA: DMEM media supplemented with 10% v/v FBS and 1% NEAA was used for MCF-7 cells. Mammary epithelium growth factor medium (MEGM) plus growth factors (CC-3150, with exception of the gentamycin–amphotericin B mix) was used for MCF-10a cells. This medium was supplemented with 50 ng mL−1 cholera toxin (Sigma C8052) prior to use, following the recommended culture guidelines of the ATCC. It should be noted that MCF-10a cells are engineered to grow in both serum-free (MCF-10a S−) and serum supplemented (MCF-10a S+) conditions, with 10% v/v FBS supplemented medium being used in the latter case. In contrast, serum supplementation of DMEM medium is compulsory for the culture of MCF-7 cells in order to provide the necessary growth factors required for their growth and survival. As with most commercial sera, fibrinogen and other clotting factors are deliberately removed prior to sale, which was the case here.2.11 MTT proliferation assay
In order to assess cellular proliferation (96-well format); 1 × 104 cells were seeded in 200 μL of medium per well, with 4 replicates for each surface/time point. Similarly, 200 μL of cell free medium was added to 4 wells (control). Plates were incubated at 37 °C in 5% v/v CO2 for 24–72 h, with cell viability being assessed every 24 h through the MTT assay. To each well, 20 μL of 5 mg mL−1 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (Sigma M5655) in DPBS was added, after which plates were incubated for 1 h under cell culture conditions. In order to minimise the disturbance of weakly adhered aggregate cultures, the culture medium was carefully removed from each well using a multichannel pipette. The insoluble product was dissolved by incubating each well with 250 μL of acidified isopropanol in sealed plates for 15 min at RT. A 200 μL aliquot was taken from each well and seeded in a clear flat bottom 96-well plate prior to an immediate absorbance measurement at 570 nm using a TECAN ULTRA spectrophotometer. The average absorbance values from control wells was then subtracted from corresponding test wells to remove background signal.2.12 Cell adhesion assays
For each test, 20000 cells were seeded in 150 μL of medium to 4 wells of each surface (96-well format). In order to determine background signal, 150 μL of medium was added to 4 wells of each surface. Plates were incubated for 2 or 24 h under cell culture conditions. Following incubation, the culture medium was carefully removed from each well followed by three delicate wash cycles with 200 μL of DPBS using a multichannel pipette. To each well, 200 μL of fresh (pre-warmed) medium was then added and plates were incubated for 2 h under cell culture conditions to allow cell recovery from washing. To each well 20 μL of 5 mg mL−1 MTT in DPBS was added and plates were incubated for 1 h under cell culture conditions prior to removal of the culture medium. Isopropanol (250 μL, Fisher Scientific 10366430) was added to each well, and the plates were sealed and incubated for 15 min at RT. A 200 μL aliquot from each well was transferred into wells of a clear flat bottom 96-well plate and the absorbance at 570 nm was immediately determined using a TECAN ULTRA spectrophotometer. The average absorbance values from control wells was subtracted from corresponding test wells to remove background signal.
2.13 Live imaging/cell tracking
Live imaging was conducted for 48 h under standard cell culture conditions, using a Leica TCS SP5 inverted confocal microscope programmed to take bright field images every 300 s (×100). Each experiment was repeated 3 times for each surface/cell line, with 1 × 104 cells being seeded per well in a 96-well format. Cells were incubated for 1 h before initiation of the experiment in order to allow cell sedimentation. Videos were compiled from each focal plane using LAS-AF software. Individual cells were tracked using ImageJ software (1.47v) and the manual tracking plugin, set at 300 s intervals with an x/y calibration of 3.02. A total of 4 cells (selected at random) were tracked for each surface experiment (12 in total). The centre of the cell was used as a focal point, and the position of the cell within the aggregate was tracked during cellular aggregation until disaggregation had occurred. The total distance travelled and maximum observed velocity was then plotted for each cell.2.14 Media pre-treatment of surfaces
FS surfaces (96-well format) were pre-treated for 24 h with 200 μL of standard DMEM media, or 10% v/v FBS supplemented DMEM media under standard cell culture conditions, after which they were washed three times with 300 μL DPBS under sterile conditions and used immediately as a substrate for cell culture. For each cell line, 1 × 104 cells were seeded per well in 200 μL of medium. Morphology was assessed 24 h post seeding using light microscopy, and is presented within Fig. 8.2.15 Statistical analysis
Error bars display standard deviation and statistical differences between relevant experimental groups were examined using the unpaired t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). All figures were prepared using GraphPad Prism 6, with the exception of the EDXa and ATR-IR spectra.3. Results and discussion
3.1 Surface characterisation
The FS surface was shown to hold a contact angle of ∼115° in comparison to ∼65° for parental TCP (Fig. 2). The total surface energy was significantly reduced following FS functionalisation, and was shown to be almost exclusively composed of non-polar, dispersive forces. The increase in observed hydrophobicity results from a reduction of hydrogen bonding sites due to the addition of fluorinated moieties to the surface. This finding is also suggestive of a high level of functionalisation, as nearly all of the polar component of the TCP is absent from the modified FS surface. | ||
| Fig. 2 Surface wettability is shown in A, with images of droplet spreading upon TCP and the FS surface shown in B and C respectively. Surface energy is shown in D (n = 9). | ||
The ATR-IR spectra of TCP and the FS surface are shown in Fig. 3. The signals arising from C–H and C–C vibrations produced by TCP in the regions of 3115–2815 cm−1 and 1650 and 1445 cm−1 (Fig. 3 peaks labelled A + B) were reduced on the FS surface spectra, suggesting a high level of surface coverage following modification.26 The strong doublet peak between 1120–1350 cm−1 (Fig. 3 peak labelled C) confirms C–F stretching vibrations, with the shape of the signal being typical of the presence of multi-fluorinated hydrocarbons.27 The FS surface shows a strong peak arising from the asymmetric stretching vibration of Si at ∼1075 cm−1 alongside an asymmetric broad shoulder at 960 cm−1, which arises from the presence of Si–OH groups (Fig. 3 peaks labelled D + E).28,29 The peak at ∼800 cm−1 (Fig. 3 peak labelled F) is associated with the symmetric stretch of Si–O–Si bonds.28
 | ||
| Fig. 3 ATR-IR spectra of the FS surface (top) and TCP (bottom) confirms the presence of both Si and F upon the FS surface. Stretching vibrations of the C–H bond are shown at positions A and B respectively, C–F vibrations (doublet) are shown at position C, stretching vibration involving Si are shown in positions D and E and the peak shown at position F is associated with the stretching of the Si–O–Si bond. | ||
Quantitative XPS analysis was performed to determine the chemical composition of the top 10 nm of TCP and FS samples. The survey spectra produced (ESI† Fig. 1A) shows TCP to be composed exclusively of C and O with 89.3% and 10.7% atomic concentrations respectively, when using C 1s and O 1s peaks. It should be noted that H holds an atomic number below the detection limit of this technique.30 The FS surface showed additional peaks for Si and F (ESI† Fig. 1B) with atomic concentrations of 2.9 and 58.8% using Si2p and F 1s peaks respectively. The low atomic percentage of Si within the FDTES silane (2.7% excluding H) suggests that the outermost 10 nm of the sample primarily consists of surface bound silane. Similarly, the absence of an N peak shows that the lysozyme protein used to generate the underlying silica layer has been effectively covered.19 These data, alongside those shown in Fig. 2 and 3, confirm the successful functionalisation of the silica film with FDTES silane.
Atomic force microscopy (AFM) studies (ESI Fig. 2†) of the surfaces showed TCP to have a relatively smooth surface profile with a RMS roughness of ∼20 nm, whereas the RMS roughness of the silica and FS surfaces was higher at ∼205 and ∼119 nm respectively. An increase in roughness is known to exacerbate surface hydrophilicity or hydrophobicity, depending upon the specific chemical functionality present.22,27 Thus, the greater surface roughness produced via FS functionalisation of TCP will contribute to the increase in contact angle illustrated in Fig. 2. Although a greater surface roughness has also been reported to enhance cellular attachment, this is largely mediated by the specific surface chemistry of the substrate and the conformational presentation of adsorbed serum proteins.31–33 AFM scans and SEM images (ESI Fig. 2†) show that the FS surface has a topological profile composed of a non-uniform array of clefts and ridges with interconnected nodules of functionalised silica. EDXa analysis (ESI Fig. 2†) supports previous characterisation data showing the presence of Si and F upon the FS surface. In addition, elemental mapping shows a strong F signal localised to the FS sample surface.
Total protein adsorption was quantified using the fluorescamine assay. As shown in Fig. 4, the uptake following 2 h of exposure to serum supplemented media was greater for the FS surface (∼2.6 μg cm−2vs. ∼2 μg cm−2 for TCP). Although protein levels for the FS surface remained relatively constant between the 2 and 24 h time points, they increased to ∼2.5 μg cm−2 following 24 h of incubation for TCP. These data show that, despite the inert and low surface energy profile of the FS film, protein uptake occurs at an increased rate in comparison with TCP. Other systems which promote cancer cell aggregation (such as poly HEMA coated plates) are based on an abrogation to protein adsorption, resulting in a disruption to cellular adherence.11 The data shown in Fig. 4 indicate this to not be the case for the FS surface. An increased uptake of serum proteins has previously been observed upon hydrophobic surfaces.34–36 Furthermore, protein adsorption has also been reported to be dominated by hydrophobic interactions at ∼pH 7.4 (cell culture conditions), with an absorbed mass of protein steadily increasing as surface wettability decreases.36 Previous work has also shown protein adsorption to increase with increasing surface roughness, resulting from a change in the geometrical arrangement of proteins at the surface.37 All of these findings correlate with the results presented here.
 | ||
| Fig. 4 Total protein adsorption upon TCP and the FS surface following 2 and 24 h of serum supplemented media exposure is shown in A (n = 4). A comparison of fibronectin and albumin adsorption after 2 and 24 h of serum supplemented media exposure is shown in B and C respectively (n = 4). | ||
Indirect ELISA assays revealed that the FS surface promoted a different profile of fibronectin and albumin adsorption to that observed for the TCP surface, Fig. 4B and C. There was a significantly higher adsorption of serum albumin to the FS surface at the 2 h time point, with no significant differences between the TCP and the FS surfaces being detected after 24 h of incubation, the latter result coinciding with an increase in the total protein adsorbed by TCP. The increase in albumin adsorption shown upon the FS surface after 2 h of incubation can be expected due to an overall increase in the total protein adsorbed, coupled with the relative abundance of albumin within FBS. However, in addition, albumin adsorption has been specifically shown to occur at an increased rate upon surfaces of reduced wettability, a finding which is governed by an increased level of hydrophobic interaction.36,38 Increased hydrophobic interactions have also been shown to produce a stronger surface-albumin association, resulting in an enhanced level of resistance to albumin displacement by cell adhesion proteins, including fibronectin.39 In this study we show the levels of albumin adsorption upon the FS surface to only approximate to those on TCP after 24 h of incubation. The levels of fibronectin adsorption were indeed shown to be significantly lower upon the FS surface in comparison to TCP at both 2 and 24 h time points tested, a finding which may be attributed to a decreased affinity for albumin displacement as discussed above. Previous research has also shown an increased tendency for albumin to adsorb over fibronectin upon hydrophobic surfaces in a competitive system, thereby resulting in disruption to cellular attachment.40
Factors affecting competitive protein adsorption include rates of transport, unfolding and protein binding affinity.41 The initial stages of competitive adsorption are mainly influenced by the rate of transport with abundant proteins of low binding affinity adsorbing first, being later displaced by proteins of a higher binding affinity.20 Upon hydrophobic surfaces (such as the FS surface), the main driving force for adsorption is the hydrophobic interaction, which is known to induce large conformational change to the protein.12,41 The globular and flexible nature of serum albumin facilitates a comparatively rapid denaturisation during adsorption, in comparison to the higher molecular weight fibronectin.41 Once bound to the FS surface, albumin displacement is likely delayed by a strong surface association which is mediated via hydrophobic interactions. In contrast, the main driving forces for interaction on more hydrophilic surfaces (such as TCP) are hydrogen bonding and electrostatic interactions, forces demanding less conformational change to the proteins adsorbed.41
3.2 Cell culture application
Both MCF-7 and MCF-10a S+ were shown to have a significantly reduced level of adherence after both 2 and 24 h of culture on the FS surface in comparison to cells cultured upon TCP (Fig. 5). However, this was not observed for MCF-10a S− cells, which showed a near equal level of adherence to their TCP cultured counterparts at both the 2 and 24 h time points tested. This finding is indicative of an obligatory role for serum proteins in the mechanism of disrupted cellular adherence. The levels of cell adhesion upon the FS surface increased between the 2 and 24 h periods of culture, suggesting the FS surface (and the serum proteins that it interacts with) to only delay cellular adhesion rather than prevent it completely. | ||
| Fig. 5 Relative absorbance values representing surface adhesion of MCF-7 (A), MCF-10a S+ (B) and MCF-10a S− (C) when cultured upon TCP and the FS surface for periods of 2 and 24 h, as outlined in section 2.12. | ||
The morphology of MCF-7, MCF-10a S+ and MCF-10a S− was assessed during FS surface culture, with both MCF-7 and MCF-10a S+ being shown to undergo aggregate formation (Fig. 6). As this was not observed for MCF-10a S− cells, an obligatory role for serum proteins in the transient disruption to cellular adherence is shown once more. It can also be noted that MCF-7 cells produced well defined spheroids after 24 h of culture upon the FS surface, with a complete return to a 2D monolayer after 72 h of culture. In comparison, MCF-10a S+ produced less demarcated, more irregularly shaped aggregates after 24 h of culture on the FS surface. Furthermore, a proportion of the cells remained as spheroids at the 72 h time point. Varying aggregation rates between different cell lines has been reported previously, with recent studies showing that metastatic potential, E-cadherin and HER2 expression enhance spheroid formation rates.42,43 The results of this study correlate with this finding, with the HER2+ MCF-7 cancer cells undergoing complete aggregation–disaggregation at a faster rate than the “normal transformed”, HER2− MCF-10a cells. The precursor to the FS surface (the silica surface), is rougher than both TCP and the FS surface, but did not induce cellular aggregation (ESI† Fig. 2 and 3). This suggests the FS surface induced cellular aggregation to be at least partly due to FDTES chemical functionalisation, as opposed to increasing surface roughness.
 | ||
| Fig. 6 Morphology of MCF-7, MCF-10a S+ and MCF-10a S− cells when cultured upon TCP and the FS surface (2–72 h). Using MCF-10a S±; spheroid formation was shown to be serum dependant, occurring in a transient manner for only MCF-7 and MCF-10a S+ cells. Images taken at ×100 magnification with scale bars representing 100 μm. | ||
Disruption to cellular adherence is likely to decrease cellular proliferation rates, with multi-cellular spheroids being reported to display reduced proliferation in comparison to 2D cultures.41,44 However, in this study, the transient generation of aggregates had no significant effect on cellular proliferation up to the 72 h time point (ESI Fig. 4†). This also shows the FS surface to exert no significant toxicity towards both MCF-7 and MCF-10a cells, as would be expected given the inert nature of the carbon fluorine bonds in the FDTES silane.
Live imaging was used to capture dynamic changes to cell morphology during FS surface culture, again only showing MCF-7 and MCF-10a S+ cells to form aggregates upon the FS surface (ESI video set 1†). The videos produced gave visual evidence of a delayed adhesion to the FS surface but only in the case of MCF-7 and MCF-10a S+ cells, reinforcing the data shown in Fig. 5 and 6. Quantitative tracking data (Fig. 7) indicated that MCF-7 and MCF-10a S+ cells migrate for significantly greater distances and travel at a greater maximum observed velocity over the 48 h period on FS surfaces, a likely consequence of poor adherence conditions/aggregate formation. The differential response level between each cell line may be attributable to the different aggregation–disaggregation rates, as has been discussed above.
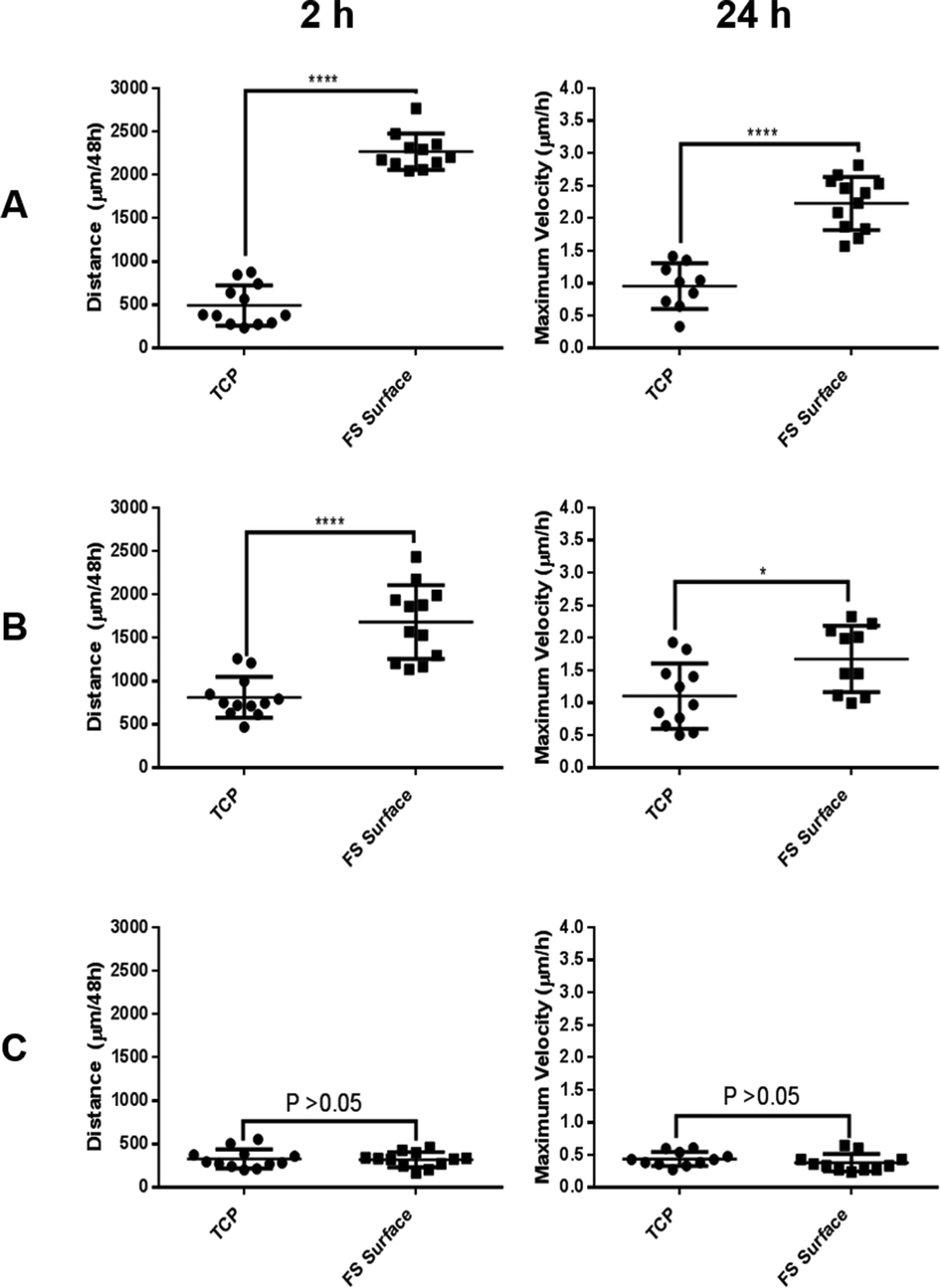 | ||
| Fig. 7 Total distance travelled and maximum velocity observed for MCF-7 (A), MCF-10a S+ (B) and MCF-10a S− (C) during 48 h of culture upon TCP and the FS surfaces. | ||
In order to further investigate the role of serum proteins within cellular aggregate formation; FS surfaces were pre-treated with either serum supplemented DMEM or serum free DMEM medium prior to use as substrates for cell culture, as outlined in section 2.14. Although aggregation occurred upon the FS surface pre-treated with standard DMEM medium, MCF-7 and MCF-10a S+ cells formed a 2D monolayer on FS surfaces that had been pre-treated with serum supplemented medium, and exhibited a morphology which was similar to that of cells cultured upon standard TCP (Fig. 8). This observation suggests that surface induced aggregation can be inhibited by pre-treating the surface with serum containing medium. This observation also suggests that the transient nature of aggregate formation may reflect a competitive exchange of proteins over time. For FS surfaces pre-treated with FBS supplemented medium, any exchange in surface bound proteins has progressed to near completion prior to the application of cells. As a consequence, cells are introduced to a substrate which can accommodate and support their adherence in a shorter time frame, in comparison to standard DMEM pre-treated FS surfaces. It can also be noted that MCF-10a S− cells formed aggregates upon the FS surface which had been pre-treated with FBS supplemented medium only. As FBS supplemented media was removed prior to the application of these cells any dynamic exchange of proteins at the surface would be inhibited, resulting in a prolonged exposure to a surface-protein profile which is poorly accommodating for cellular adhesion.
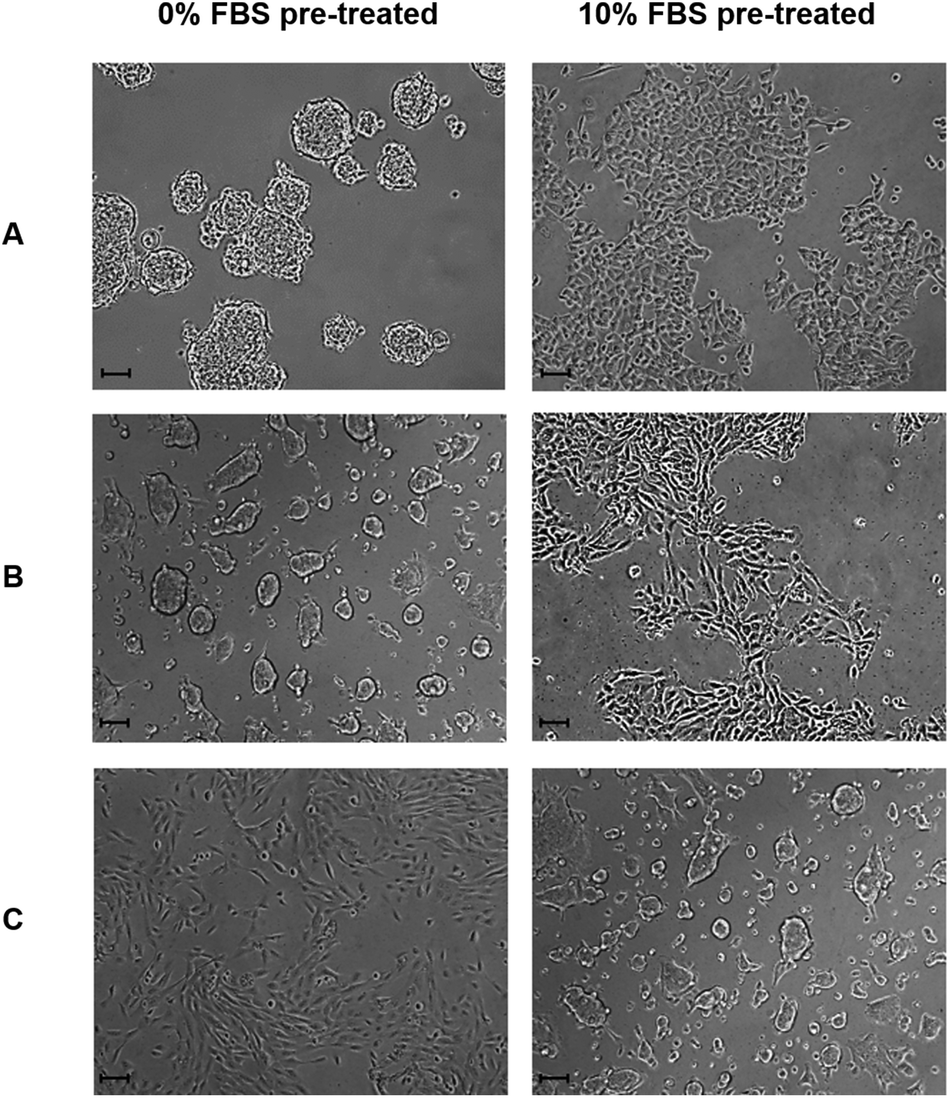 | ||
| Fig. 8 MCF-7 (A), MCF-10a S+ (B) and MCF-10a S− (C) morphology 24 h post seeding upon the FS surface which has been pre-treated with serum free or 10% v/v FBS supplemented DMEM media for 24 h, as outlined in section 2.14. Images taken at ×100 magnification and scale bars represent 100 μm. | ||
The competitive exchange of proteins upon surfaces was first noted by Vroman and Adams in the early 1960s, who observed the displacement of absorbed proteins over time by proteins that subsequently arrive at the material surface.20,45 The Vroman effect explains how abundant, motile, low molecular weight proteins saturate a surface during the initial stages of protein exposure.20 These are subsequently displaced by less abundant, less motile, higher molecular weight proteins which exhibit a greater affinity for surface binding.20,45,46 The Vroman effect is known to be influenced by several parameters such as pH, temperature, protein concentration and the specific surface chemistry of the substrate.47
The data produced in this study suggest that the inert properties of the FS surface provide the necessary conditions to selectively adsorb serum proteins that can disrupt cellular adhesion during the early stages of culture from the medium. As only MCF-10a S+ (and not S−) form aggregates upon the FS surface, the observed aggregation phenomenon appears to be reliant on both the FS surface and the serum proteins with which it interacts. The composition/conformation of the adsorbed proteins may then change during the later stages of culture as a consequence of the Vroman effect, facilitating cellular attachment as a monolayer. The data presented in Fig. 4 demonstrate that the FS surface promotes a protein adsorption profile which is rich in albumin, a serum protein which has previously been reported to disrupt cellular adherence during the early stages of culture.47,48 The negative impact which this has upon cellular adherence is likely to be exacerbated by the decreased uptake of fibronectin, a serum protein that plays a major role in the adherence of many cell lines.48,49 Over time, the levels of other unknown cell adhesion proteins may then begin to rise, leading to an increase in the cell–surface binding affinity and a facilitation of cellular adherence as a monolayer. A simplified illustration showing this process is shown in Fig. 9. As Fig. 8 shows that the pre-treatment of the FS surface with serum supplemented media encourages monolayer formation, we postulate that the proteins/factors facilitating cell adhesion derive from the serum itself, as opposed to cell secretion. However, this does not rule out the possibility that a combination of secretory and serum derived proteins may act to facilitate cellular attachment during culture.
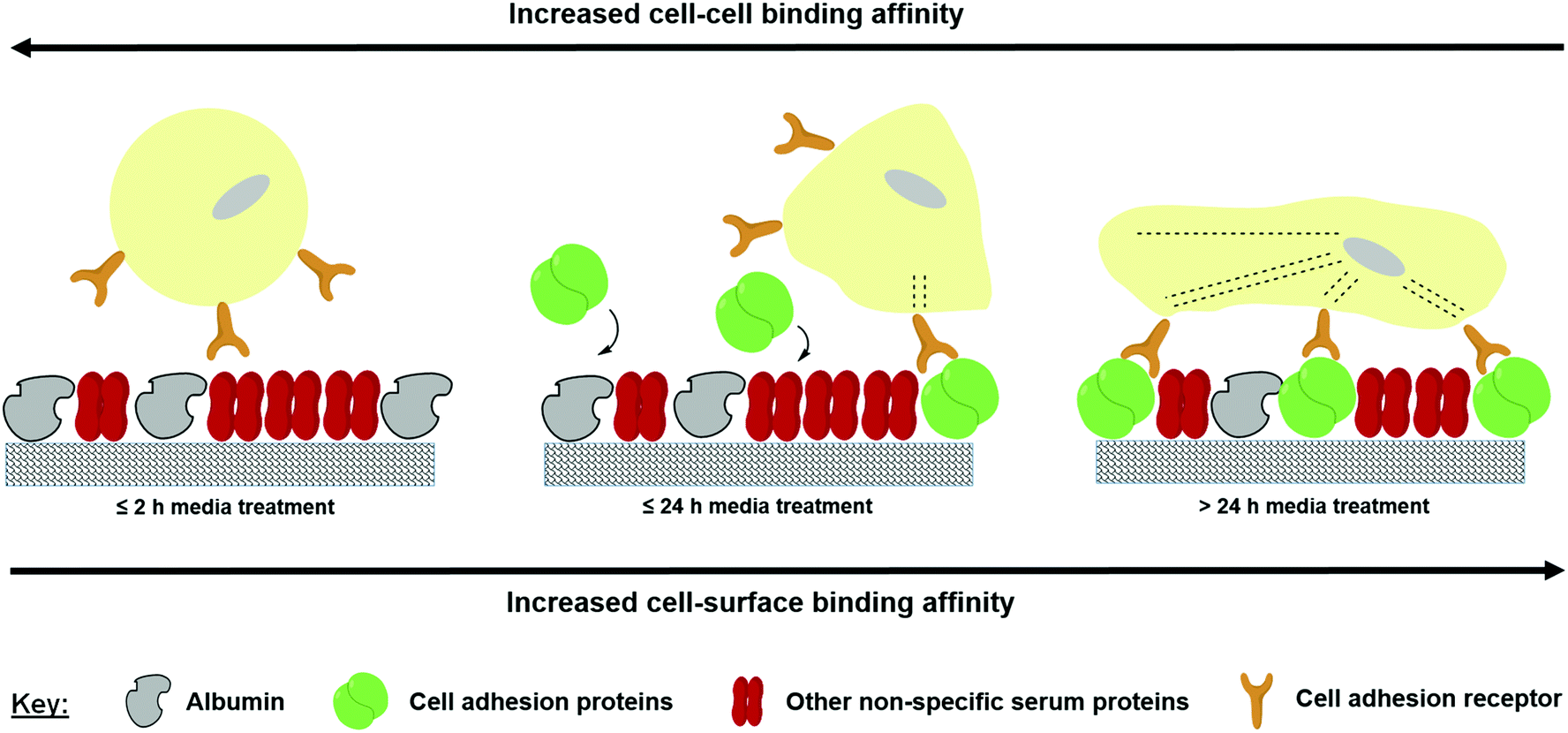 | ||
| Fig. 9 Competitive protein exchange occurring upon the FS surface during cell culture in serum supplemented medium. | ||
4. Conclusions
We show serum-dependant, transient, homotypic cellular aggregation to take place upon the hydrophobic, low energy FS cell culture surface developed. Both cell lines used for FS surface culture exhibited a reduced adherence capacity and greater motility when cultured in serum supplemented medium, in comparison to cells cultured on the TCP surface. Cells undergoing aggregation–disaggregation show no significant difference in proliferation rate in comparison to standard 2D cultures. The distinct differences in the surface chemistry between the FS surface and TCP result in a differential adsorption rate of serum albumin and fibronectin and the transient nature of aggregate formation reflects the changes in the adsorbed serum proteins over time. The material developed supports the study of cancer cell aggregation–disaggregation as a single dynamic process and to our knowledge represents the first report of such a system. The ability to control and study cancer cell aggregation–disaggregation events has potential applications for the study of cell signalling, cytoskeletal changes, cell surface marker expression and cancer cell–cell recognition during this process. Fabrication of the surface directly onto commonly available TCP makes the approach broadly accessible to the cell culture community. Herein, we present an example of how surface chemistry can be fine-tuned in order to manipulate serum protein adsorption, and as a consequence directly manipulate cellular behaviour in vitro.Acknowledgements
MN acknowledges support of a Vice-Chancellors Bursary from Nottingham Trent University. Additional funding was provided by the John van Geest Cancer Research Centre and the Biomolecular Materials and Interfaces Research Group, both at Nottingham Trent University. Martin Roe at Nottingham University is thanked for performing the XPS analysis.Notes and references
- V. V. Glinsky, G. V. Glinsky, O. V. Glinskii, J. R. Turk, W. Mossine and S. L. Deutscher, et al. , Cancer Res., 2003, 63, 3805–3811 CAS.
- C. H. Qian and B. T. Teh, Encycl. Cancer, 2012, 3354–3355 Search PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed.
- I. Saiki, S. Naito, J. Yoneda, I. Azuma, J. E. Price and I. J. Fidler, Clin. Exp. Metastasis, 1991, 9, 551–566 CrossRef CAS.
- X. Zhong and F. J. Rescorla, Cell. Signalling, 2012, 24, 393–401 CrossRef CAS PubMed.
- C. D. Simpson, K. Anyiwe and A. D. Schimmer, Cancer Lett., 2008, 272, 177–185 CrossRef CAS PubMed.
- M. C. Guadamillas, A. Cerezo and M. A. Del Pozo, J. Cell Sci., 2011, 124, 3189–3197 CrossRef CAS PubMed.
- P. Paoli, E. Giannoni and P. Chiarugi, Biochim. Biophys. Acta, Mol. Cell. Res., 2013, 1833, 3481–3498 CrossRef CAS PubMed.
- R. Morimoto-Katama, S. I. Mizoguchi, T. Ichisugi and S. Yui, Mediators Inflammation, 2012, 456462 Search PubMed.
- L. L. Yi Lu, J. Cancer, 2011, 2, 458–466 Search PubMed.
- E. Fennema, N. Rivron, J. Rouwkema, C. Blitterswijk and J. Boer, Trends Biotechnol., 2013, 31, 108–115 CrossRef CAS PubMed.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS PubMed.
- M. Wahlgren and T. Arnebrant, Trends Biotechnol., 1991, 9, 201–208 CrossRef CAS.
- C. A. Hayens and W. Norde, J. Colloid Sci., 1994, 169, 313–328 CrossRef.
- C. J. Bettinger, R. Langer and J. T. Borenstein, Angew. Chem., Int. Ed., 2009, 48, 5406–5415 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- X. Liu and S. Wang, Chem. Soc. Rev., 2014, 43, 2385–2401 RSC.
- N. A. Zhang, Y. L. Deng, Q. D. Tai, B. R. Cheng, L. B. Zhao, Q. L. Shen, R. X. He, L. Y. Hong, W. Liu, S. S. Guo, K. Liu, H. R. Tseng, B. Xiong and X. Z. Zhao, Adv. Mater., 2012, 24, 2756–2760 CrossRef CAS PubMed.
- G. J. Hickman, A. Rai, D. J. Boocock, R. Rees and C. C. Perry, J. Mater. Chem., 2012, 22, 12142–12148 RSC.
- L. Vroman, A. L. Adams, G. C. Fischer and P. C. Munoz, Blood, 1980, 55, 156–159 CAS.
- A. Rai and C. C. Perry, J. Mater. Chem., 2012, 22, 4790–4796 RSC.
- C. J. Brinker, J. Non-Cryst. Solids, 1988, 100, 31–50 CAS.
- S. H. Wu, C. Y. Mou and H. P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- F. M. Fowkes, Ind. Eng. Chem. Res., 1964, 56, 40–52 CrossRef CAS.
- P. Held, Nat. Methods, 2006 DOI:10.1038/an1794.
- E. Jabbari and N. A. Peppas, Macromolecules, 1993, 16, 2175–2186 CrossRef.
- A. Hozumi and O. Takai, Appl. Surf. Sci., 1996, 103, 431–441 CrossRef CAS.
- V. Purcar, D. Donescu, I. C. Spataru, M. Ghiurea, S. Caprarescu and C. Petcu, et al. , Rev. Roum. Chim., 2013, 58, 37–42 CAS.
- S. S. Latthe, H. Imai, V. Ganesan and A. V. Rao, Appl. Surf. Sci., 2009, 256, 217–222 CrossRef CAS PubMed.
- M. S. Wagner, S. L. McArthur, M. Shen, T. A. Horbett and D. G. Castner, J. Biomater. Sci., Polym. Ed., 2002, 13, 407–428 CrossRef CAS PubMed.
- E. Biazar, M. Heidari, A. Asefnezhad and N. Montazeri, Int. J. Nanomed., 2011, 6, 631–639 CrossRef CAS PubMed.
- B. G. Keselowsky, D. M. Collard and A. J. Garcia, J. Biomed. Mater. Res., Part A, 2003, 66, 47–59 Search PubMed.
- R. N. Wenzel, Ind. Eng. Chem. Res., 1936, 28, 988–994 CrossRef CAS.
- Y. T. Phung, D. Barbone, C. V. Broaddus and M. Ho, J. Cancer, 2011, 2, 507–514 CrossRef CAS.
- G. B. Sigal, M. Mrksich and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 3464–3473 CrossRef CAS.
- X. Wang, G. Liu and G. Zhang, Langmuir, 2012, 28, 14642–14653 CrossRef CAS PubMed.
- K. Rechendorff, M. B. Hovgaard, M. Foss, V. P. Zhdanov and F. Besenbacher, Langmuir, 2006, 22, 10885–10888 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- J. Wei, T. Igarashi, N. Okumori, T. Igarashi, T. Maetani, B. Lui and M. Yoshinari, Biomed. Mater., 2009, 4, 045002 CrossRef PubMed.
- P. Ying, G. Jin and Z. Tao, Colloids Surf., B, 2004, 33, 259–263 CrossRef CAS PubMed.
- A. K. Takagi, M. Watanabe, Y. Ishii, J. Morita, Y. Hirowkawa and T. Matsuzaki, et al. , Anticancer Res., 2007, 27, 45–54 CAS.
- S. Zhao, S. Ohara, Y. Kanno, Y. Midorikawa, M. Nakayama and M. Makimura, et al. , Cancer Lett., 2013, 330, 41–48 CrossRef CAS PubMed.
- J. M. Iglesias, I. Beloqui, F. Garcia-Garcia, O. Leis, A. Vazquez-Martin and A. Eguiara, et al. , PLoS One, 2013, 8, e77281 CAS.
- A. Ivascu and M. Kubbies, J. Biomol. Screening, 2006, 11, 922–932 CrossRef CAS PubMed.
- S. L. Hirsh, D. R. McKenzie, N. J. Nosworthy, J. A. Denman, O. U. Sezerman and M. M. Bilek, Colloids Surf., B, 2013, 103, 395–404 CrossRef CAS PubMed.
- P. Vilaseca, K. A. Dawson and G. Franzese, Soft Matter, 2013, 9, 6978–6985 RSC.
- P. A. Netland and B. R. Zetter, Biochem. Biophys. Res. Commun., 1986, 139, 515–522 CrossRef CAS.
- E. Ruoslahti, Cancer Metastasis Rev., 1984, 3, 43–51 CrossRef CAS.
- J. Sottile, D. C. Hocking and P. J. Swiatek, J. Cell Sci., 1998, 111, 2933–2943 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS, AFM, SEM, EDXA, MTT proliferation data, cell morphology upon the silica surface, video stills and live cell imaging are all available. See DOI: 10.1039/c4bm00194j |
| This journal is © The Royal Society of Chemistry 2014 |