
Dapansutrile OLT1177 suppresses foreign body response inflammation while preserving vascularisation of implanted materials†
Alex H. P.
Chan
ab,
Xueying S.
Xu
ab,
Ian L.
Chin
ab,
Angus J.
Grant
ab,
Kieran
Lau
ab,
Yunfei
Hu
ab,
Praveesuda L.
Michael
ab,
Yuen Ting
Lam
ab,
Steven G.
Wise
 ab and
Richard P.
Tan
*ab
ab and
Richard P.
Tan
*ab
aSchool of Medical Sciences, Faculty of Health and Medicine, University of Sydney, NSW 2006, Australia
bCharles Perkins Centre, University of Sydney, NSW 2006, Australia. E-mail: richard.tan@sydney.edu.au; Tel: +61-2-86279425
First published on 28th June 2024
Abstract
Mitigating inflammation associated with the foreign body response (FBR) remains a significant challenge in enhancing the performance of implantable medical devices. Current anti-inflammatory approaches aim to suppress implant fibrosis, the major outcome of the FBR, but also inadvertently inhibit beneficial immune signalling necessary for tissue healing and vascularization. In a previous study, we demonstrated the feasibility of ‘selective’ immunosuppression targeting the NLRP3 inflammasome using the small molecule inhibitor MCC950, leading to reduced implant fibrosis without compromising healing and leading to enhanced vascularization. However, the clinical potential of MCC950 is severely limited due to its failure to pass Phase I clinical safety trials. This has triggered substantial efforts to develop safer analogues of NLRP3 inhibitors. Dapansutrile (OLT1177) is emerging as a leading candidate amongst current NLRP3 inhibitors, demonstrating both safety and effectiveness in a growing number of clinical indications and Phase 2 trials. While the anti-inflammatory effects of OLT1177 have been shown, validation of these effects in the context of implanted materials and the FBR have not yet been demonstrated. In this study, we show OLT1177 possesses beneficial effects on key cell types which drive FBR outcomes, including macrophages, fibroblasts, and smooth muscle cells. Evaluation of OLT1177 in a 28 day subcutaneous implantation model showed OLT1177 reduced fibrotic capsule formation while promoting implant vascularization. Mechanistic studies revealed that this occurred through activation of early pro-angiogenic markers while suppressing late-stage anti-angiogenic markers. These findings establish OLT1177 as a promising therapeutic approach for mitigating implant fibrosis while supporting vascularisation, suggesting a highly promising selective immunosuppressive strategy for the FBR warranting further research to explore its optimal integration into medical materials and devices.
 Richard P. Tan | Dr Richard Tan is a Lecturer and Group Leader at the University of Sydney in Australia. He completed his undergraduate degree at the Georgia Institute of Technology (USA) and went on to complete his PhD in 2019 at the University of Sydney with a focus on Immunomodulatory Biomaterials. His group focuses on understanding the immunology surrounding implanted cardiovascular biomaterials and devices and developing novel engineering solutions that favorably modulate these responses for improved patient outcomes. He is the lead inventor of several international patents and is the co-founder of Nanomedx hoping to commercialise a new nanotechnology therapy for vascular disease. He is the recipient of a National Heart Foundation of Australia Postdoctoral Fellow and Vanguard Awards, as well as funded by New South Wales Health (NSW Health). |
1. Introduction
The challenge posed by the foreign body response (FBR) to implanted biomaterials is widely acknowledged and their impacts on the performance of medical devices are well-characterised.1 Given that the FBR is primarily driven by inflammation, halting its progression is largely accomplished using anti-inflammatory approaches which can involve optimizing both the physical and chemical properties of the material and/or utilizing drugs to reduce immune cell interaction at the tissue-implant interface and minimize the degree of fibrotic encapsulation.2,3 While these methods effectively suppress fibrosis by antagonising the major pathways of inflammation, this also inactivates other beneficial aspects of immune signalling, such as those that drive tissue healing and associated processes like vascularization.4 This poses a particular challenge for implants whose functions specifically rely not only on reduced fibrosis but also simultaneously require enhanced vascularisation, such as biosensors, insulin pumps, or cell encapsulation devices.5 The broad limitation of current anti-inflammatory approaches lies in the assumption that all inflammation during the FBR is detrimental, and that inflammation related to fibrosis cannot be selectively targeted without affecting immune-driven healing processes.In a recent previous study, we demonstrated that ‘selective’ immunosuppression in the context of implanted materials is feasible and that it can effectively mitigate the traditional outcomes of the FBR while enhancing vascularization.6 We achieved this by targeting the NLRP3 inflammasome using the first generation inhibitor MCC950.7 Our findings revealed that MCC950 inhibited immune cell infiltration leading to reduced implant fibrosis and more notably, this was achieved at levels comparable to the conventional broad-acting anti-inflammatory, dexamethasone.6 This highlighted that even though MCC950 only targeted a specific pathway of inflammation, it was just as effective as a broader anti-inflammatory approach, suggesting that inflammation linked to the NLRP3 inflammasome pathway is a significant driver of implant fibrosis. More importantly, MCC950 uniquely induced angiogenesis and stable vascularization while maintaining this suppression of fibrosis, showcasing the potential for selective immunosuppressive approaches in dealing with the FBR.6
Continued research efforts supporting the integration of NLRP3 inhibitors into emerging FBR therapies may pave the way for the enhanced performance of implanted materials and devices.8,9 As new inhibitors are developed, selection of the appropriate candidate will become critical to suitability of these therapies for human use. Previous clinical trials with MCC950 showed that escalating doses of the drug were associated with increasing liver toxicity, halting its commercial development during Phase 1 safety testing.10 This setback has since prompted a concerted effort both by industry and academia to develop alternative analogues of MCC950 with reduced toxicity. Notable pharma companies are actively pursuing small-molecule NLRP3 inflammasome inhibitors within their drug discovery pipelines. Due largely to public disclosure of its chemical structure, Dapansutrile (OLT1177) is arguably the most independently researched of these second generation NLRP3 inhibitors.11 OLT1177 is an orally available drug, currently in Phase 2 trials for gout,12 with demonstrated Phase 1 safety in patients with cardiovascular disease.13 Preclinical data also shows efficacy in treating multiple sclerosis,14 Alzheimer's disease,15 melanoma,16 reperfusion injury,17 intracerebral hemorrhage18 and osteoarthritis.19 As the evidence of its safety continues to grow, OLT1177 is emerging as an exciting new candidate. However, whether OLT1177 can replicate the beneficial effects of NLRP3 inhibition in the context of materials requires investigation and is crucial for the prospect of a clinically-feasible selective immunosuppressive approach for the FBR.
In this study, we conduct an analysis of OLT1177 as a localised therapy for mitigating the FBR to subcutaneous implants. We first show in vitro that OLT1177 drives direct beneficial effects on the three key cell types that drive FBR outcomes: macrophages, fibroblasts, and endothelial cells. Further evaluation of OLT1177 using a 28 day subcutaneous implantation model showed that treatment of implanted scaffolds resulted in thinner fibrotic capsules alongside increased capillary and arteriole formation around the implants. We further revealed that OLT1177 promoted this vascularisation by activating a unique temporal angiogenic signalling pattern consisting of early activation of pro-angiogenic markers followed by a suppress of late-stage anti-angiogenic markers. These findings provide the first evidence of the effectiveness of OLT1177 as a therapeutic approach to mitigate implant fibrosis while maintaining implant vascularisation, offering promise for a selective and clinically-feasible immunosuppressive approach for the FBR.
2. Methods
2.1 Cell culture
J774a.1 mouse macrophages (from Sigma Aldrich, MA, USA), human dermal fibroblasts (hFB, obtained from ThermoFisher Scientific, Waltham, MA, USA), and human coronary artery endothelial cells (hCAECs, sourced from Cell Applications, San Diego, CA, USA) were maintained in a CO2 (5%) humidified incubator at 37 °C. Macrophages and hFB were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin). hCAECs were cultured in MesoEndo medium (Merck, 212-500, Kenilworth, NJ, USA) without additional supplements. The culture media was changed every 2–3 days, and cells were passaged when they reached a confluency of ≥ 85%. Evaluation for each cell type was conducted in 2D using tissue culture plates or 3D using electrospun PCL scaffolds placed into tissue culture plates, prior to cell seeding.2.2 Electrospinning PCL scaffolds
Polycaprolactone (PCL) scaffolds were fabricated using electrospinning (IME Medical Electrospinning, Waalre, Netherlands) as previously described.20 A PCL solution was prepared by dissolving PCL in 1,1,1,3,3,3-hexafluoroisopropanol (HFP) at a concentration of 10% w/v, with overnight mixing at room temperature. The solution was then loaded into a syringe (Terumo Corp., Tokyo, Japan) and pumped at a rate of 4 mL per hour through a 20G needle under an applied voltage of 20 kV. The needle was positioned at a distance of 16 cm from a 10 cm diameter stainless steel drum rotating at 500 rpm. After electrospinning, the scaffolds were removed from the drum, washed, and air-dried to eliminate any residual solvent. They were subsequently cut into circular discs using a 6 mm diameter biopsy punch. Disc samples were used for evaluation in both cell culture and in vivo animal experiments.2.3 Alamar blue
The cytotoxic effects of OLT1177 were evaluated using Alamar blue cell viability assays (ThermoFisher Scientific, Waltham, MA, USA). Following specific culture conditions, cells were incubated in a mixture of alamarBlue reagent and culture media (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio) for 2 hours, and fluorescence was measured using a Tecan M-1000 plate reader (Ex. 560/Em. 590 nm). Cell viability was quantified relative to the untreated control group. Based on the concentration-response curve, a concentration of 500 μm was selected for subsequent assays.
:9 ratio) for 2 hours, and fluorescence was measured using a Tecan M-1000 plate reader (Ex. 560/Em. 590 nm). Cell viability was quantified relative to the untreated control group. Based on the concentration-response curve, a concentration of 500 μm was selected for subsequent assays.
2.4 Macrophage stimulation
J774a.1 cells were plated in 96-well plates (3 × 104 cells per well/scaffold), and lipopolysaccharide (LPS) (1 μg mL−1) simultaneously added to the culture media. After 90 minutes of LPS stimulation, cells were treated with adenosine triphosphate (ATP) (1.25 mM) along with OLT1177 (500 μM), followed by a 24-hour incubation period. Supernatants were then collected and analysed using ELISA (Abcam, Cambridge, UK) for IL-1β (ab197742) according to the manufacturer's instructions. Once supernatants were removed, cells were then stained for F-actin using phalloidin and pyroptosis assessed by examining the degradation of F-actin staining.2.5 Myofibroblast transdifferentiation
Human dermal fibroblasts (hFBs) underwent myofibroblast transdifferentiation induction by treatment with transforming growth factor β (TGF-β).21 First, hFBs were seeded into 96-well plates (3000 cells per well/scaffold) and allowed to adhere overnight. TGF-β (2 ng mL−1) was then added to each well, and cells were incubated for 24 hours. hFBs were then exposed to OLT1177 (500 μM) and further incubated for 24 hours while TGF-β remained in the cell media. hFBs were then fixed and subjected to immunostaining for SMα actin (1:500, Sigma, F3777).
2.6 Western blot
NLRP3 and Pro-IL1β expression was examined for changes with stimulation in J7 murine macrophages. Collagen type 1 expression in transdifferentiated myofibroblasts was quantified using western blot. The same myofibroblast differentiation protocol described previously was performed in a 12-well plate. Protein lysates were extracted using Mammalian Cell Lysis Buffer (Sigma). Protein concentration was determined using a standard bicinchoninic acid (BCA) assay (Thermo Fisher Scientific) and 5 μg of protein was loaded onto NuPAGE Novex 4–12% Bis-Tris gels (Life Technologies) and separated via SDS-PAGE electrophoresis. The proteins were then transferred to polyvinylidene fluoride membranes using a semidry iBlot gel transfer system (Life Technologies). The membrane was blocked with 5% BSA in PBS for 1 hour at room temperature. Primary antibodies against NLRP3 (1:500, Novus Bio., NBP2-12446), Pro-IL1β (1:500, Abcam, ab9722), Col I (1:500, Abcam, ab2413) and α-tubulin (1:2000, Abcam, ab7291) as a loading control were incubated overnight at 4 °C in 1% BSA at appropriate dilutions. The membrane was washed with PBST (0.1% Tween20) and then incubated with a secondary antibody conjugated with horseradish peroxidase (HRP) for 2 hours at room temperature in 1% skim milk in PBS. Protein bands were detected using Luminata Crescendo Western HRP substrate (Millipore), and western blot densitometry was analyzed using Image Lab Software 6.1 (Bio-Rad).
2.7 Endothelial dysfunction
Endothelial dysfunction was modelled using human coronary artery endothelial cells (hCAECs) treated with TNF-α.22 HCAECs were seeded into 96-well plates (3000 cells per well). Cells were allowed to grow to grow to confluence before stimulation with TNF-α (100 ng mL−1). OLT1177 (500 μM) was also added at the same time and allowed to incubate for 24 hours. HCAECs were then fixed and immunostained for vascular endothelial-cadherin (VE-Cad; 1:250, Abcam, ab76198) and intercellular adhesion molecule-1 (ICAM-1; 1:250, Abcam, ab282575) to visualise and quantify endothelial junction formation and activation, respectively.
2.8 Tubulogenesis assay
Matrigel-coated 96-well plates (Corning, 354 248) were used for culturing HCAECs (3 × 103 cells per well). OLT1177 (500 μM) was combined with fresh HCAEC growth media to achieve the specified final concentrations before being added to the Matrigel cultures. Cell behaviour was observed over a 16-hour period, with time-lapse images captured using an IncuCyte S5X live cell imager (Essen Bioscience). To assess the capillary network, four representative images were selected at the 6-hour mark post-seeding from each condition, with five replicate wells per condition. The number of meshes, segments, and total branch length were quantified using the angiogenesis analyzer plugin in ImageJ.2.9 Mouse subcutaneous implant
The study received approval from the University of Sydney Animal Ethics Committee (protocol number 2023/2283) and was conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Male C57BL/6 mice, aged 9–10 weeks and weighing 25 ± 2 g, were purchased from Animal BioResources (Moss Vale, NSW, Australia). OLT1177 was assessed in vivo using a subcutaneous mouse implantation model as previously outlined.23 Briefly, each mouse received two scaffolds spaced 2 cm apart subcutaneously across their back: a control (untreated) PCL scaffold, and passively absorbed overnight at 4 °C with OLT1177 (0.5 mM in PBS). At the endpoints of day 14 and 28 post-implantation, mice were euthanized, and samples were collected for histological processing.2.10 Histology and immunostaining
Explanted scaffolds underwent fixation overnight at room temperature with 4% paraformaldehyde (PFA). Samples were then dehydrated using an ascending ethanol gradient, embedded in paraffin, and sectioned at 5 μm thickness. For histological staining, the sections were deparaffinized, rehydrated, and subjected to staining using hematoxylin and eosin as well as Masson's trichrome. For immunohistochemistry staining, primary antibodies against CD68 (1:500, Abcam, ab125212) for macrophages CD206 (1:200, Abcam, ab64693) for M2 macrophage, MHCII (1:200, Abcam, ab180779) for M1 macrophage, vimentin (1:200, Abcam, ab45939) for fibroblasts, CD31 (1:250, Abcam, ab182981) for endothelial cells, and smooth muscle α-actin (1:500, Sigma, F3777) for smooth muscle cells were utilized. Following staining, the sections were counterstained for nuclei using mounting media containing DAPI (Sigma, F6057) and imaged on a Zeiss AxioScan microscope at 20× magnification.
2.11 Image analysis
Image analysis was conducted using ImageJ. For pyroptosis, total cell number per image was quantified by particle counts for DAPI staining, while cells with positive actin staining were manually counted. Experiments in vitro involving immunohistochemistry were measured by positive staining quantified as a percentage of the total area based on a common threshold. For in vivo histological analysis, analysis for macrophages, fibroblasts, SMA-α, and M1/M2 markers were quantified as a percentage of the total scaffold area using a common threshold. Fibrotic capsule thickness was measured manually at three different points on the scaffold using H&E staining. For collagen, positive staining using the Milligan's trichrome was identified with skin tissue then quantified as a percentage of total area based on common threshold. Capillary (CD31+ only) and arteriole (CD31+ and SMα+) density were manually counted for vessels with defined lumens in the area of interest at the scaffold site. For native arteriole formation, the size of arterioles was manually measured. Arteriole localisation was measured by the perpendicular distance from the scaffold.2.12 Angiogenesis antibody array
Samples from in vivo study were explanted at day 7 and 14 and snap frozen in liquid nitrogen. Tissue samples were homogenized in lysis buffer on the Precellys Evolution with Cyrolys cooling system (Bertin Technology) and protein concentration quantify via BCA assay. 250 μg (total protein) of protein lysates were incubated with array membranes. Angiogenic antibody array (Abcam, ab139697) was conducted following the manufacturer instructions, labels in ESI† figure (Fig. S1). Densitometry was analyzed using Image Lab Software 6.1 (Bio-Rad).2.13 Statistical analysis
Analyses was conducted using GraphPad Prism 9 (Graphpad Software, San Diego, California). Data are presented as mean ± standard error of the mean (SEM). Statistical significance was determined using Student's t-test for comparisons between two groups, or one-way analysis of variance (ANOVA) followed by post hoc Dunnett's multiple comparisons test for datasets with more than two groups, or two-way ANOVA with Tukey's multiple comparison test for datasets with more than two groups and multiple timepoints. A significance level of P < 0.05 was considered statistically significant. Symbols *, **, ***, and **** indicate P-values less than 0.05, 0.01, 0.001, and 0.0001, respectively.3. Results
3.1 OLT1177 inhibits NLRP3 inflammasome activation in macrophages
The effects of OLT1177 on NLRP3 inflammasome activation in vitro was studied using murine macrophages. Firstly, escalating doses of OLT1177 (0–500 μM) did not show any significant decreases in viability compared to control (Fig. 1A), enabling the use of the highest dose 500 μM for all subsequent in vitro experiments.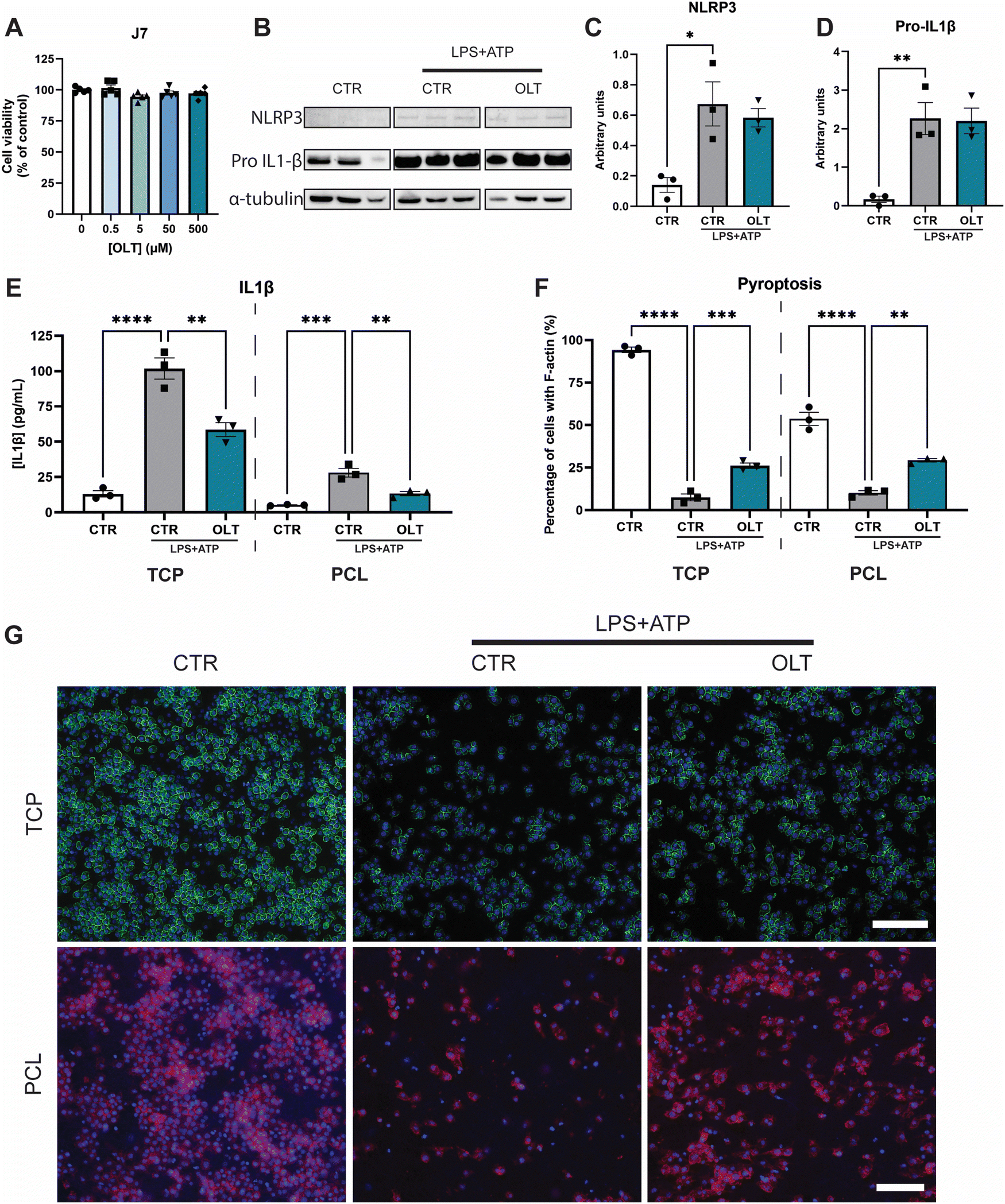 | ||
| Fig. 1 OLT1177 inhibits NLRP3 pathway in vitro. (A) alamarBlue Cytotoxicity of OLT1177 on J774.1 murine macrophages following 3 days exposure to increasing concentration of OLT1177. (B) Western blot of J774.1 murine macrophage after stimulation of NLRP3 pathway using LPS and ATP. (C) Quantification of NLRP3 expression. (D) Quantification of Pro-IL1β expression. (E) ELISA quantification of IL1β release in vitro on TCP or PCL scaffolds following stimulation with LPS + ATP, then treated with OLT1177. (F) Quantification of pyroptosis of J774.1 murine macrophage on TCP or PCL scaffolds following stimulation with LPS + ATP. Data represented as percentage of cells with complete F-actin membrane staining. (G) Representative images of pyroptosis, F-actin stained in green or red and nuclei counter stained in blue. Scale bar = 100 μm. Statistical significance was determined using Dunnett's multiple comparison one-way ANOVA test relative to stimulated group (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
To validate the inhibitory mechanisms of OLT1177 underlying these effects, western blots were performed to quantify the expression levels of NLRP3 and pro-IL1β (Fig. 1B). Both NLRP3 and pro-IL1β expression were found to be increased as a result of combined LPS and ATP immune activation. However, when treated with OLT1177 no significant decreases were observed compared to stimulated levels of expression (Fig. 1C and D). This demonstrated that the mechanism of OLT1177 action was not to suppress expression of components of the NLRP3 inflammasome but rather its downstream formation/oligomerisation.
Further examination of the anti-inflammatory effects of OLT1177 was conducted by analysing canonical downstream products of NLRP3 inflammasome activation. Quantification of IL-1β in macrophage supernatants (Fig. 1E) showed a 33% reduction in IL-1β secretion in OLT1177 treated 2D cultures compared to stimulated controls (58.63 ± 4.89 pg mL−1vs. 87.83 ± 7.48 pg mL−1). Similar reductions were observed when macrophages were cultured on 3D PCL substrates. OLT1177 treated cultures on PCL showed 53% lower IL-1β secretion compared to stimulated controls (13.31 ± 1.46 pg mL−1vs. 28.13 ± 3.05 pg mL−1).
Pyroptosis is an inflammatory triggered programmed cell death that is initiated by NLRP3 activation followed by caspase activity. Morphologically, this is characterised by the loss of F-actin in the cell membrane. Similarly, OLT1177 decreased pyroptosis in both 2D and 3D cultures of NLRP3 inflammasome-activated macrophages (Fig. 1F). This was indicated by an increase in F-actin staining of 247% and 188% in 2D and 3D cultures, respectively, compared to stimulated controls (26.13 ± 1.60% vs. 7.53 ± 2.01% and 29.28 ± 0.97% vs. 10.17 ± 1.28%). F-actin staining showed that OLT1177 treated cultures significantly preserved cytoskeleton integrity (Fig. 1G).
3.2 OLT1177 prevents myofibroblast differentiation
The direct effects of OLT1177 on fibroblast behaviour was evaluated using a myofibroblast differentiation protocol. Escalating doses of OLT1177 up to 500 μM had no impact on the viability of fibroblast cultures (Fig. 2A). TGF-β stimulated fibroblasts express high levels of SMA-α when fully transdifferentiated to myofibroblasts. However, when treated with OLT1177, levels of SMA-α were reduced by 65% and 96% in 2D and 3D cultures respectively, when compared to their stimulated controls (26.57 ± 5.30% vs. 75.11 ± 9.30% and 0.92 ± 0.45% vs. 22.91 ± 6.48%) (Fig. 2B). Staining of SMα actin showed that this reduction occurred without any effect on overall cell viability (Fig. 2C).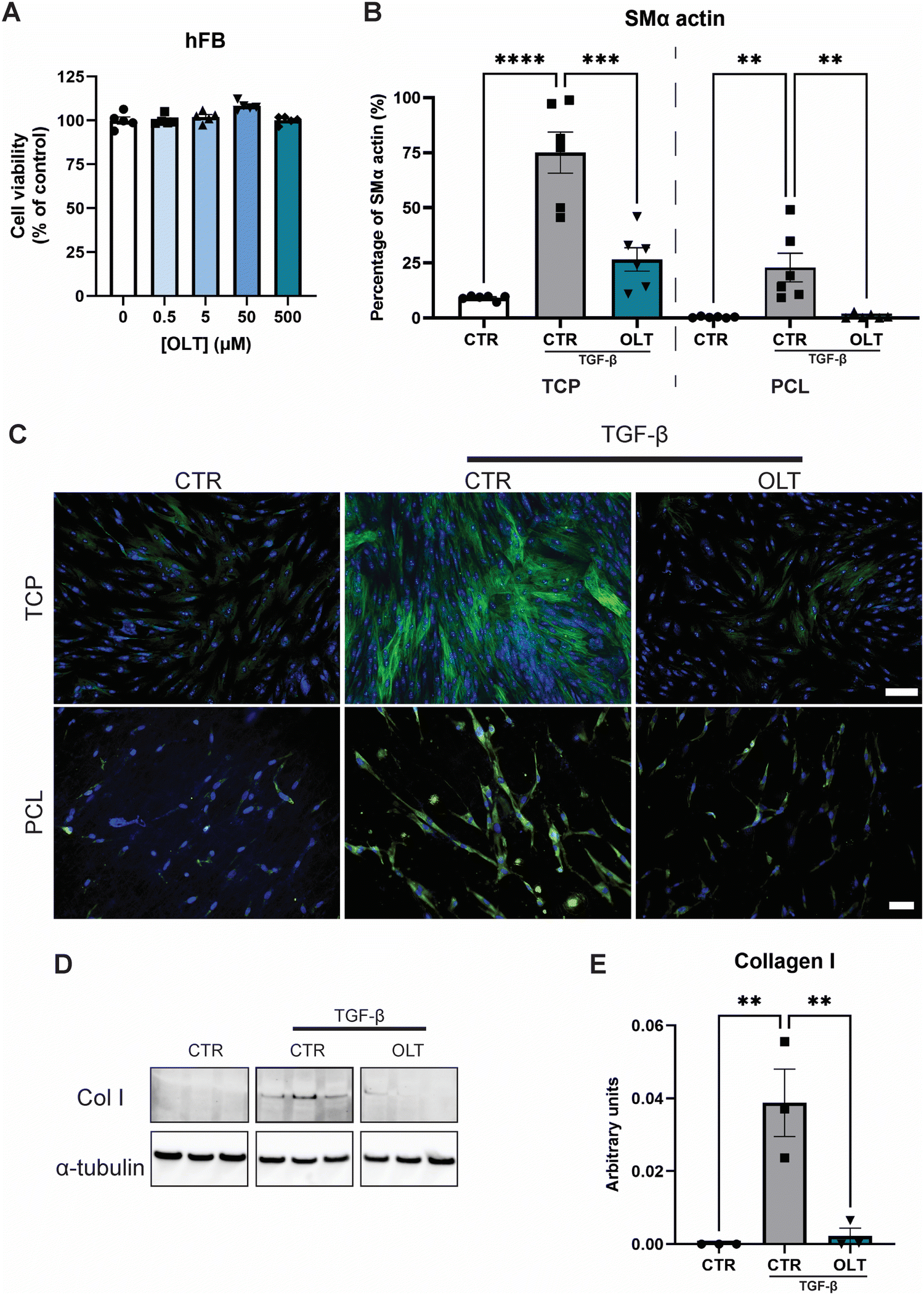 | ||
| Fig. 2 OLT1177 supresses myofibroblast transdifferentation. (A) alamarBlue Cytotoxicity of OLT1177 on human dermal fibroblast following 3 days exposure to increasing concentration of OLT1177. (B) Quantification of myofibroblast transdifferentiation, SMα staining of fibroblast stimulated with TGFβ then treated with OLT1177. Data represented as percentage area of SMα positive staining. (C) Representative images of myofibroblast transdifferentiation, SMα stained in green and nuclei counter stained in blue. Scale bar = 100 μm (TCP), 50 μm (PCL). (D) Western blot of fibroblast stimulated with TGFβ then treated with OLT1177. (E) Quantification of collagen I expression. Statistical significance was determined using Dunnett's multiple comparison one-way ANOVA test relative to stimulated group (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
Given that myofibroblast function is to produce neomatrix components of fibrotic capsules, Collagen I (Col I) expression was evaluated using western blot (Fig. 2D). In OLT1177 treated cultures, Col I expression was reduced by 88% (Fig. 2E) compared to stimulated controls (38.8 ± 9.2 × 10−3vs. 2.23 ± 2.1 × 10−3). These results confirmed that OLT1177 had direct effects on both myofibroblast differentiation and downstream function.
3.3 OLT1177 protects against endothelial activation and dysfunction
OLT1177 was evaluated for protective effects on endothelial cells against TNF-α induced dysfunction. Increasing doses of OLT1177 up to 500 μM also had no impact on endothelial cell viability (Fig. 3A). TNF-α stimulation of endothelial cells induced a significant reduction in vascular endothelial-cadherin (VE-Cad) alongside an increase in endothelial activation, marked by intracellular adhesion molecule (ICAM) expression However, when treated with OLT1177, endothelial cultures showed both protection against VE-Cad breakdown and suppressed ICAM expression. This was indicated by an 82% increase in VE-Cad (22.30 ± 1.84% vs. 12.25 ± 1.94%), and a 53% (6.61 ± 0.67 vs. 14.12 ± 2.09% and) decrease in ICAM compared to stimulated controls (Fig. 3B and C).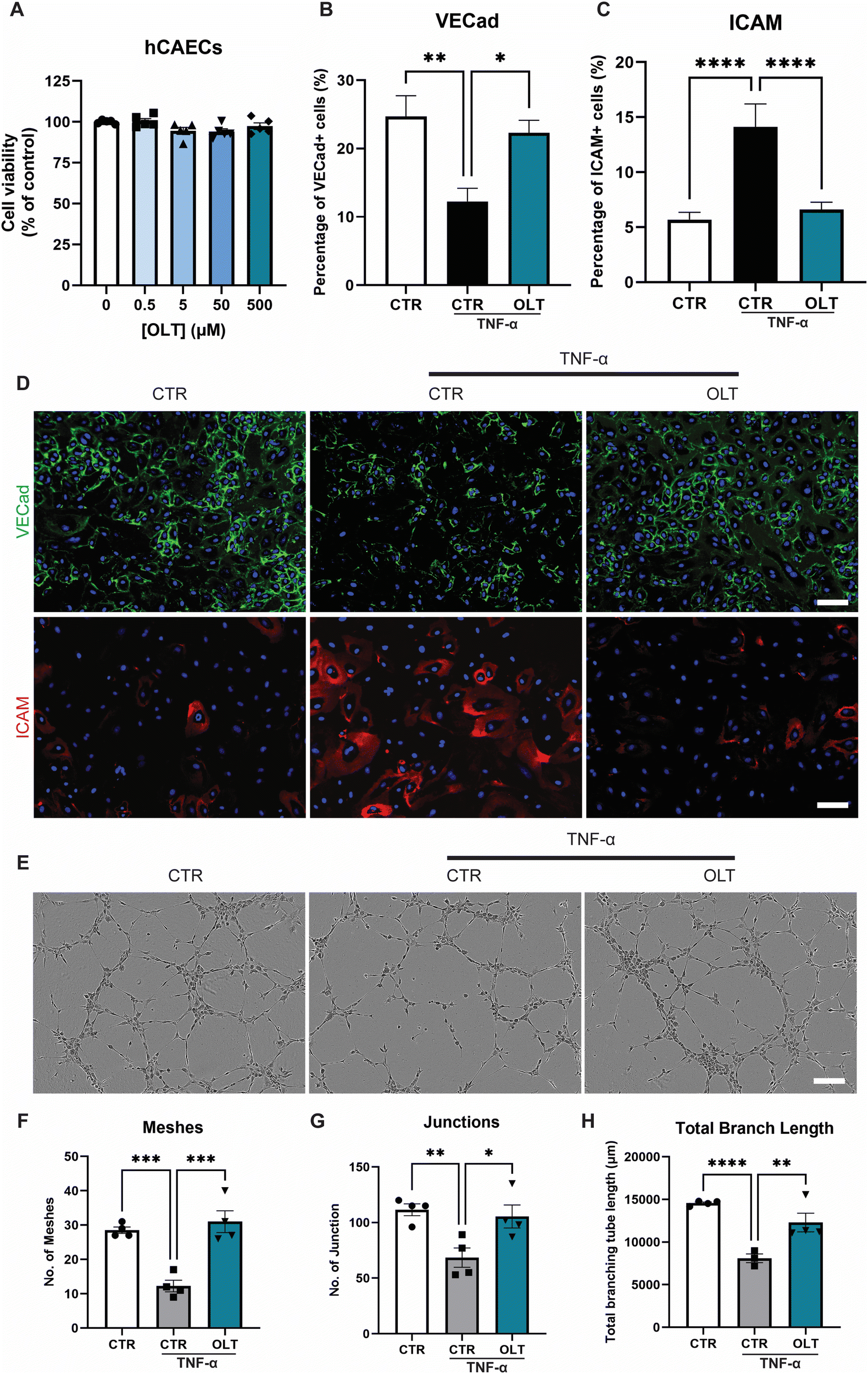 | ||
| Fig. 3 OLT1177 rescues inflammatory driven endothelial dysfunction in vitro. (A) alamarBlue Cytotoxicity of OLT1177 on human coronary artery endothelial cells following 3 days exposure to increasing concentration. (B) Quantification of VE-cadherin, VE-Cad staining of hCAECs stimulated with TNFα then treated with OLT1177. (C) Quantification of ICAM+ cells, ICAM staining of hCAECs stimulated with TNFα then treated with OLT1177. (D) Representative images of endothelial cells, VE-Cad stained in green, ICAM in red and nuclei counter stained in blue. Scale bar = 100 μm. (E) Representative of tubulogenesis at 6 hours post seeding. Scale bar = 200 μm. (F) Quantification of number of meshes per image. (G). Quantification of number of junctions per image. (H). Quantification of total branch length per image. Statistical significance was determined using Dunnett's multiple comparison one-way ANOVA test relative to stimulated group (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
Tubulogenesis assays were next used to determine whether the benefits of OLT1177 translated into favourable impacts on endothelial function in 3D matrigels. Endothelial cultures stimulated with TNF-α showed significant impairments in tubule formation which appeared rescued when treated with OLT1177 (Fig. 3E). This was confirmed by tubule quantification readouts which revealed a 153% increase in meshes (31.00 ± 3.19 vs. 12.25 ± 1.70), 54% increase in junctions (105.5 ± 10.3 vs. 68.50 ± 8.73), and 52% increase in total branch length (12301 ± 1088 vs. 8096 ± 513) when treated with OLT1177 compared to stimulated controls (Fig. 3F–H). These results showed that OLT1177 had direct protective effects on endothelial cells against TNF-α induced activation and dysfunction.
3.4 Cellular remodelling by OLT1177 in subcutaneous implants
Evaluation of OLT1177 as an approach for mitigating the FBR was conducted using a 28 day model of subcutaneous implantation of PCL scaffolds with or without passively adsorbed OLT1177. Scaffold cross-sections were then immunostained to determine changes in cellular remodelling as a result of OLT1177 treatment.Macrophages were stained using the CD68 marker and appeared localised mainly within the body of implanted scaffolds (Fig. 4A). Control scaffolds showed a resolving macrophage recruitment profile over time. However, this occurred more rapidly in scaffolds treated with OLT1177, indicated by a 48% decrease in CD68 (Fig. 4B) staining at day 14 (3.92 ± 0.75% vs. 7.53 ± 1.16%). Further analysis of macrophage phenotype revealed using the CD206 and MHC II markers for the M2 and M1 phenotype, respectively, was conducted and calculated as a M2/M1 ratio (Fig. 4C). Across days 14 and 28, no differences were observed in the M2/M1 ratio.
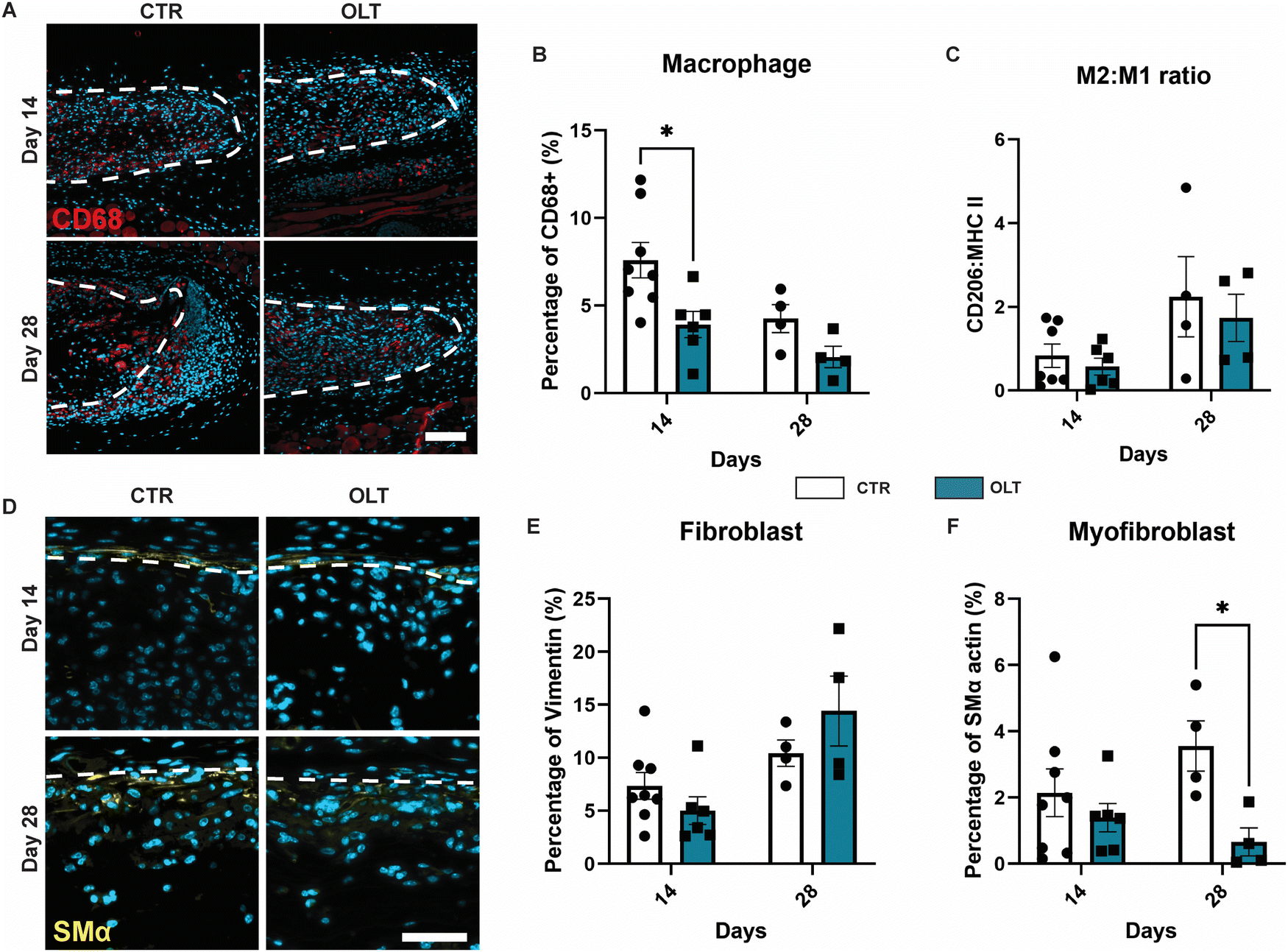 | ||
| Fig. 4 OLT1177 decreases macrophage accumulation and myofibroblast in subcutaneous PCL implants on the backs of C57BL/6 mice. (A) Representative of cross section electrospun PCL scaffold (white dotted line) stained for macrophage. CD68 in red, nuclei in blue. Scale bar = 100 μm. (B) Quantification of area percentage of CD68 staining. (C) Quantification of macrophage polarization, data represented as a ratio of area percentage of CD206 staining and area percentage of MHCII staining. (D) Representative of cross section electrospun PCL scaffold (white dotted line) stained for myofibroblast, SMα in yellow, nuclei in blue. Scale bar = 50 μm. (E) Quantification of area percentage of vimentin staining. (F) Quantification of area percentage of non-arteriole SMα staining. Statistical significance was determined using t-test at each time point between control and OLT117 group. (*p < 0.05). | ||
Fibroblasts and myofibroblast were assessed using vimentin and SMα actin staining, respectively, and also appeared mainly infiltrated into the scaffold body (Fig. 4D). Although the profile of fibroblast recruitment was found to increase over time, no significant differences were observed between control and OLT1177 treated scaffolds (Fig. 4E). However, further analysis revealed a significant decrease in the myofibroblast phenotype at day 28, indicated by a 81% reduction in SMA-α expression (Fig. 4F) at day 28 (0.66 ± 0.42% vs. 3.56 ± 0.76%). Together this showed that effects of OLT1177 on local cell remodelling was primarily at reducing early macrophage recruitment and suppressing late stage myofibroblast differentiation.
3.5 OLT1177 suppresses fibrotic encapsulation while enhancing angiogenesis
The end-stage outcomes of OLT1177 treatment on the FBR was evaluated using histopathology and immunostaining to assess fibrotic capsule formation and angiogenesis, respectively. H&E staining revealed that while fibrotic capsule thickness increased over time in control scaffolds, this was suppressed in OLT1177 treated scaffolds throughout the duration of implantation (Fig. 5A). This was indicated by a 54% and 51% decrease in capsule thickness at days 14 and 28 (Fig. 5B), respectively, in OLT1177 scaffolds when compared to control (19.46 ± 0.62 μm vs. 41.87 ± 1.26 μm and 36.14 ± 6.62 μm vs. 73.11 ± 6.64 μm). Despite reduction in capsule thickness, OLT1177 treatment was not consistent with changes in collagen density within the formed capsules (Fig. 5C).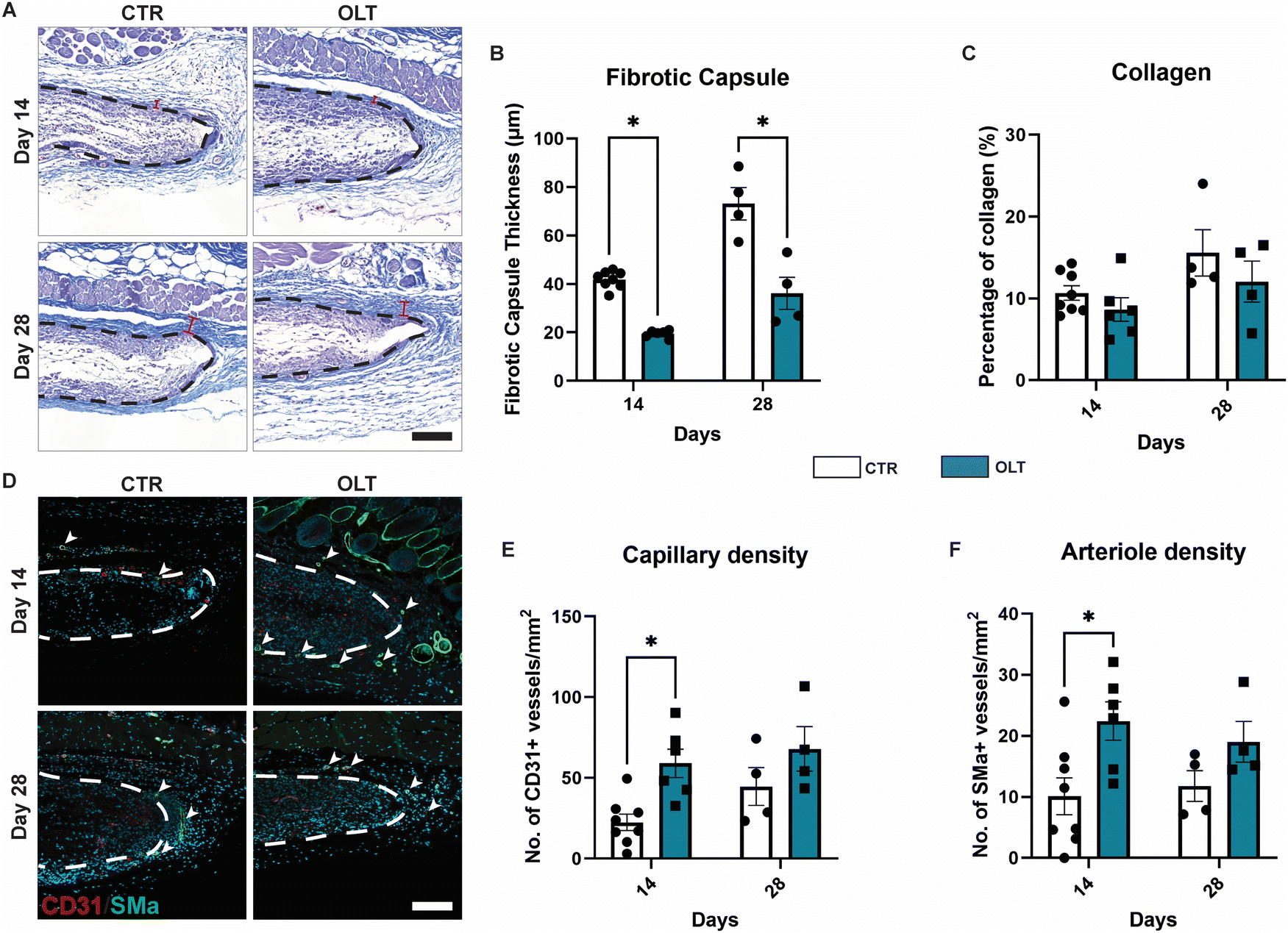 | ||
| Fig. 5 OLT1177 functionalised scaffolds decreases fibrotic response and maintains angiogenesis in vivo. (A) Representative images of Masson's trichrome staining of electrospun PCL scaffold (black dotted line). Collagen in blue, muscle in purple, nuclei in black. Scale bar = 100 μm. (B) Quantification of fibrotic capsule thickness. (C) Quantification of collagen in the fibrotic capsule and implant. Data represented as area percentage of blue staining. (D) Representative of cross section electrospun PCL scaffold (white dotted line) co-stained for capillaries and arterioles. CD31 stained in red, SMα stained in green, nuclei countered stained in blue. Scale bar = 100 μm. (E) Quantification of capillary density, data represented as counts CD31+ vessels per mm2. (F) Quantification of arteriole density, data represented as counts CD31+ and SMα+ vessels per mm2. Statistical significance was determined using t-test at each time point between control and OLT117 group. (*p < 0.05). | ||
Suppression of fibrotic encapsulation was evaluated alongside changes in vascularisation by staining for capillary formation through CD31+ staining alone and arteriole formation through CD31+/SMA-α+ co-staining (Fig. 5D). Increasing capillary formation was observed in control scaffolds, however this occurred more rapidly in OLT1177 treated scaffolds (Fig. 5E), indicated by a 63% increase in CD31+ vessels at day 14 (57.41 ± 10.59 counts mm−2vs. 21.26 ± 5.68 counts mm−2). Similar trends were observed for arteriole formation, with increasing formation over time in control scaffolds that occurred more rapidly when treated with OLT1177. This was similarly indicated by a 55% increase in the density of CD31+/SMA-α+ co-positive vessels (22.43 ± 3.15 counts mm−2vs. 10.10 ± 3.03 counts mm−2, Fig. 5F). This showed that OLT1177 treatment of in vivo scaffolds provided sustained suppression of fibrotic encapsulation meanwhile enhancing early vascularisation.
3.6 Mechanism of OLT1177 mediated enhanced vascularisation
Closer examination into the mechanisms of enhanced vascularisation by OLT1177 revealed distinct differences between the local vascular beds of control and OLT1177 scaffolds. Quantification of arteriole size revealed that at both days 14 and 28, OLT1177 scaffolds had an increase of 43% and 60% in average arteriole cross-sectional area compared to control (219.3 ± 20.25 μm2 and 153.5 ± 15.75 μm2vs. 303.4 ± 40.96 μm2 and 189.7 ± 20.46 μm2, respectively, Fig. 6A). Violin plots of the measured arterial sizes showed that OLT1177 treated scaffolds had a wider distribution of arterioles sized 100–500 μm2 when compared to control at days 14 and 28 (Fig. 6B and C). This analysis supported that OLT1177 was stimulating significantly improved vascular remodelling around implanted scaffolds.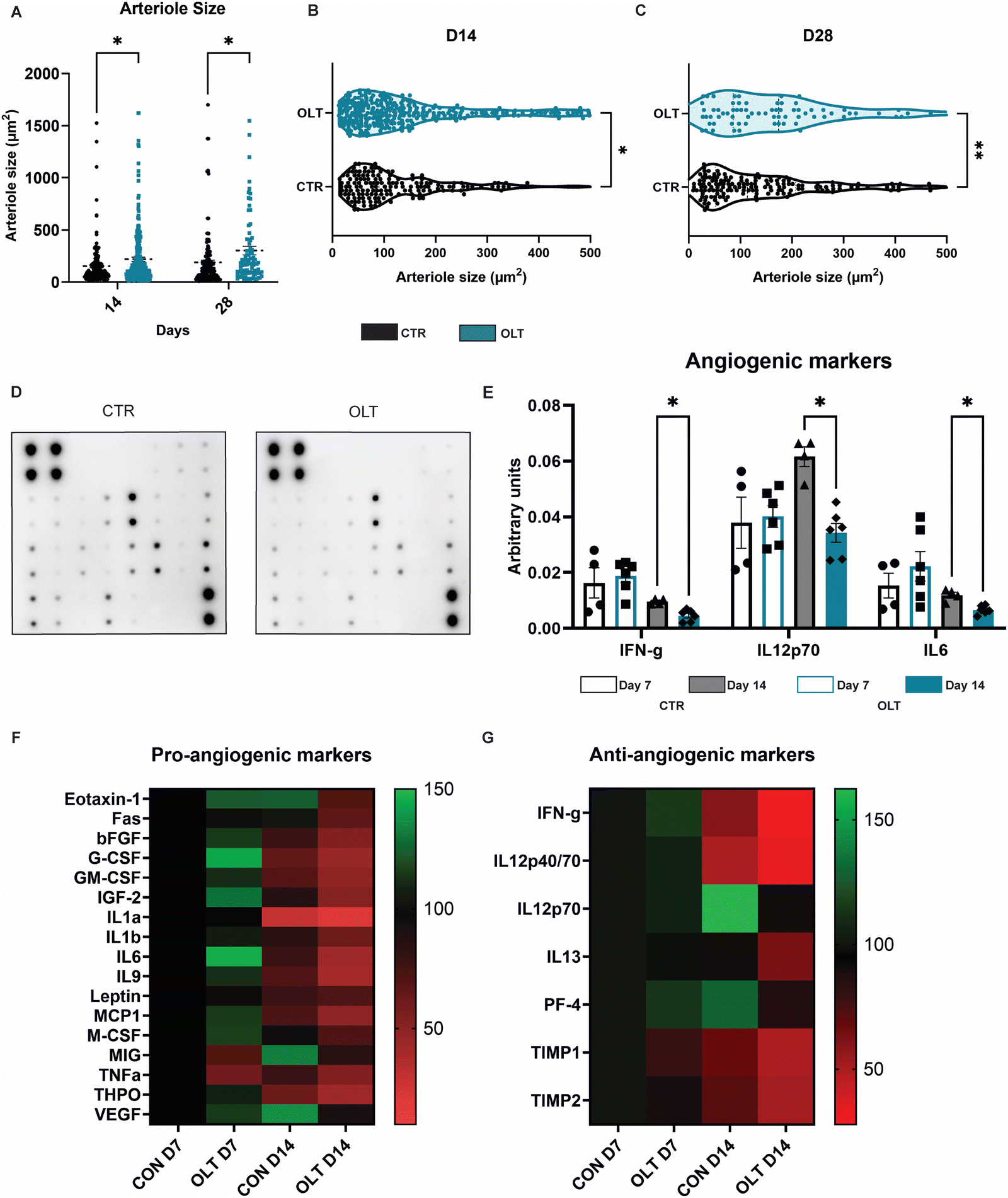 | ||
| Fig. 6 OLT1177 enhance angiogenesis. (A) Quantification of arteriole size. (B) Violin plot of arteriole size at day 14. (C) Violin plot of arteriole size at day 28. (D) Representative images of angiogenic dot blot. (E) Quantification of angiogenic dot blot. (F) Heat map of pro-angiogenic markers. (G) Heat map of anti-angiogenic markers. Statistical significance was determined using t-test at each time point between control and OLT117 group. (*p < 0.05, **p < 0.01). | ||
Further analysis across a spectrum of angiogenic signalling was conducted using angiogenesis antibody arrays (Fig. 6D). Across 24 combined pro-angiogenic and anti-angiogenic markers, significant differences were only observed in IFN-γ, IL12p70 and IL6. These differences were only observed at day 14 as reductions of 50%, 45%, and 42%, respectively, in OLT1177 treated scaffolds compared to control scaffolds (Fig. 6E). IFN and IL12p70 are considered anti-angiogenic markers, whereas IL-6 is classified as a pro-angiogenic marker. Examining across the entire array of markers showed that when compared to control at day 7, OLT1177 had greater relative increases in pro-angiogenic markers than anti-angiogenic markers (Fig. 6F). Interestingly at day 14 however, it appeared OLT1177 induced greater relative decreases in both pro- and anti-angiogenic markers. These results were more supportive that pro-angiogenic and anti-angiogenic signalling pathways were operating independently rather than being interconnected. Our findings suggest that during early stages of vascular remodelling induced by OLT1177, there is a propensity towards supporting pro-angiogenic signalling, whereas later stages exhibit a shift towards greater support of anti-angiogenic signalling. Together this reveals a potential mechanism for enhanced vascularisation of OLT1177 treated scaffolds.
4. Discussion
OLT1177 has been extensively demonstrated to inhibit NLRP3 inflammasome immune activation in both in vitro and in vivo settings. Some of the earliest validations of OLT1177 were conducted in human and murine models of macrophage activation.11 Similarly, the earliest clinical trials involving OLT1177 were targeted against immune-related conditions such as gout flare and osteoarthritis.12 Our study distinguishes itself having examined OLT1177 within the context of inflammation associated with implanted biomaterials, a process we have previously shown to involve NLRP3 inflammasome activation throughout its progression. We provide findings on the direct impacts of OLT1177 within key cell types driving outcomes in the foreign body response to determine its effectiveness as a potential therapeutic approach for mitigating fibrosis to implanted biomaterials.Our initial findings in macrophages validated several previous studies showing that OLT1177, rather than suppressing expression of the NLRP3 inflammasome, instead acts to limit NLRP3 inflammasome formation and thereby reduce its downstream outputs that include both IL-1β secretion and induction of pyroptosis. However, the most novel aspect of our findings is the amplified inhibitory effect of OLT1177 on both IL-1β secretion and pyroptosis, observed in macrophages cultured on 3D PCL scaffolds. However, the most novel aspect of our findings is the amplified inhibitory effect of OLT1177 on both IL-1β secretion and pyroptosis, observed in macrophages cultured on 3D PCL scaffolds. Further investigation of this effect may require studying NLRP3 activation in different physical environment contexts. Such considerations are crucial when addressing foreign body inflammation, as immune cells and their activation have been shown to be impacted by and to respond to physical attributes that include material architecture, roughness, porosity, and stiffness.24,25 If OLT1177 is to advance as a selective immunosuppressive strategy for the FBR, these discoveries bear significant implications for further research aimed at elucidating the role of NLRP3 in immune responses to physical aspects of 3D biomaterials.
Despite its primary association with canonical inflammation pathways, our previous work implicated the direct involvement of the NLRP3 inflammasome in fibroblast-to-myofibroblast differentiation.6 Here, we demonstrated that OLT1177 exhibits direct anti-fibrotic effects, inhibiting SMA-α expression-mediated differentiation and suppressing collagen I production. This suggests that OLT1177 treatment may lead to reduced thickness and/or less collagen in fibrotic capsules surrounding implanted materials. Similar to our findings in macrophages, this inhibitory effect was notably enhanced in fibroblasts cultured on PCL scaffolds. Numerous studies have established that fibroblasts respond to substrate stiffness,26,27 which influences cell differentiation and gene expression. Separately, reductions in NLRP3 inflammasome-mediated caspase-1 activity in fibroblasts have shown to decrease SMA-α expression.28 This suggests that fibroblast responses to physical attributes of materials heavily involve NLRP3, and its inhibition by OLT1177 would have even more pronounced effects. These findings carry implications for using OLT1177 as an anti-fibrotic therapy in various tissues and more importantly underscores the potential impact of OLT1177 on capsule development, independent of its effects on inflammation.
The inflammation-independent benefits of OLT1177 were also observed in endothelial cells. Distinct from our previous research findings demonstrating that NLRP3 inhibition does not compromise endothelial function,8 this study further revealed that NLRP3 inhibition can in fact protect against TNF-α-induced dysfunction. Endothelial cultures treated with OLT1177 after TNF-α stimulation were protected against VE-cadherin degradation and were prevented from expressing ICAM. Both markers are classically indicative of endothelial dysfunction, as VE-cadherin breakdown leads to leaky junctions allowing immune cell infiltration, while increased ICAM expression facilitates immune cell attachment to these areas.29 Growing evidence suggests direct NLRP3 activity in endothelial cells, driving endothelial activation and dysfunction. Our findings support this notion as OLT1177 appears to offer protection across all aspects of endothelial dysfunction. Similarly, these protective effects appear more pronounced in 3D Matrigel cultures, implying that the local environment influences NLRP3-induced endothelial cell dysfunction. This has significant implications for the potential of OLT1177 to support enhanced vascularization in the context of implanted materials, further underlining its therapeutic relevance beyond its anti-inflammatory properties.
The performance of OLT1177 was then assessed as a FBR therapy in vivo using a 28-day subcutaneous implantation model. It was observed that prior in vitro effects extended to the in vivo environment, evidenced by changes in local cell remodelling following OLT1177 treatment. There was a more rapid resolution of macrophages recruited to implanted scaffolds, demonstrating the direct anti-inflammatory effects of OLT1177. Despite a decrease in overall macrophage numbers, there was no apparent impact on macrophage polarization. While there are studies linking NLRP3 signalling with macrophage polarisation whereby production of IL1-β promotes M1 polarisation,30,31 this was not apparent in the context of implanted biomaterials. The faster macrophage resolution did not result in a changes in fibroblast recruitment, we did observe a significant decrease in the myofibroblast phenotype in late stages of OLT1177 treated scaffolds. These collective finding aligns with our in vitro results and suggests two mechanistic possibilities. Either reduced early macrophage recruitment altered the local immune microenvironment, resulting in lower levels of cytokines inducing myofibroblast differentiation, or the sustained activity of OLT1177 over the 28-day period directly suppressed myofibroblast differentiation in the mid-term. Future studies investigating optimal drug delivery and presentation of OLT1177 within the context of implantable materials could provide further insights into these mechanisms. Regardless of mechanism, our study is the only other finding to report anti-fibrotic effects of OLT1177 in vivo. The other study, focusing on ischemic reperfusion injury in mice, showed OLT1177 to reduce infarct size and myocardial fibrosis.17 This highlights the broader therapeutic potential of OLT1177 to mitigate the key cellular remodelling events that drive the FBR towards its critical end stage outcomes.
Implant fibrosis represents the culmination of these responses and its severity often measured through analysis of the surrounding fibrotic capsule. Throughout the implantation period, OLT1177 treatment consistently suppressed the development of the fibrotic capsule, maintaining a baseline thickness compared to controls. This suppression however did not involve a reduction in collagen content within the capsule. Despite this, our findings of fibrotic capsule reduction by OLT1177 contributes to a limited body of evidence showing that OLT1177 can reduce fibrosis in vivo, and is the first to demonstrate this within the context of implanted materials, underscoring its potential as an FBR therapy. More importantly, in line with our previous findings with MCC950,6 OLT1177 preserved and enhanced scaffold vascularization. This was evidenced by significant early increases in local capillary and arteriole formation, indicating its ability to promote and maintain blood vessel growth around the implant site.
Extending from these outcomes, we explored the mechanisms underlying enhanced vascularization as a result of NLPR3 inhibition. The average overall size of formed arterioles was significantly larger as a result of OLT1177 treatment. Larger vessels have been shown to possess a higher capacity for essential nutrient and oxygen transport, crucial for supporting cellular metabolism, tissue growth, and repair.32 This would further help to limit tissue damage and inflammation around the implant. OLT1177 promoted a broader distribution of blood vessels within the 100–500 μm2 range at both time points. This range aligns with the average size of mature cutaneous blood vessels in mice,33 suggesting a level of vascularisation critical for native tissue repair and function.
Antibody arrays were used to assess angiogenic signalling across 24 combined pro- and anti-angiogenic signalling markers. The main significant differences were observed in IFN-γ, IL12p70 and IL6 all at day 28. Both IFN-γ and IL12p70 are classically characterised as anti-angiogenic supporting the notion that OLT1177 was functioning to remove suppressors of angiogenesis. IL6 has been previously reported to have both pro- and anti-inflammatory functions which are heavily context dependent.34 Its pro-angiogenic functions acts directly on endothelial cells to promote proliferation and expression of angiogenic growth factors. However within more complex contexts in vivo, IL-6 has been shown to promote pro-inflammatory environments that limit angiogenic potential.35 A broader look at the array perhaps gives a general hypothesis of OLT1177 action. OLT1177 increases pro-angiogenic signalling at day 7 then at day 14 enhances anti-angiogenic signalling. This suggests that OLT1177 functions in early stages by promoting pro-angiogenic signalling and sustains this vascularisation in late stages by removing suppressors of angiogenesis, rather than by continuing to sustain pro-angiogenic signalling. It is important to note however that these mechanistic results could potentially change depending upon the manner in which OLT1177 is presented through varying delivery strategies and/or slow-release platforms. In any case, this supported the notion the OLT1177 provided a local environment which is conducive for angiogenesis around implants. This enhanced vascularisation has positive implications for implantable materials and devices that require close proximity to vascular beds for effective therapeutic function.
More research is needed to fully understand and optimize OLT1177's clinical application. Its promising safety profile and now demonstrated efficacy suggest a valuable addition to the therapeutic options for managing the FBR. Future integration into clinical practice will require longer-term evaluation (greater than 3 months)36 and employ pre-clinical large animal models (pig subcutaneous implant)37 to ensure sustained safety and efficacy profiles. This would undoubtedly rely on redefining delivery strategies with more sophisticated local controlled-release platforms. Such platforms would enable detailed pharmacokinetic data essential for sustaining OLT1177 presentation during the FBR, optimizing dosing schedules, and maintaining therapeutic levels over time in clinical settings. Addressing these aspects would help progress OLT1177 through key translational studies to validate its clinical feasibility for improving the effectiveness, longevity, and safety of implantable medical devices.
5. Conclusion
Selective immunosuppression targeting the NLRP3 inflammasome using the clinically-safe analogue OLT1177 represents a significant advance in managing the FBR. This approach not only reduces fibrosis but also enhances vascularisation, offering an improvement in performance for a wide range of implantable materials and devices. Growing interest and investment from both industry and research underscore the urgency and potential of further research into selective immunosuppressive strategies. Continued understanding of the most optimal forms of drug presentation and release will help pave the way for these approaches into clinical practice, potentially revolutionizing the field of biomaterials and offering a promise of improved health outcomes for patients who rely on implantable medical devices.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
We have no financial disclosures or industry relationships to disclose.Acknowledgements
This work was supported by NSW Health in the form of a NSW Cardiovascular Early-Mid Career Researcher Grant (S. G. W. H20/28248 & R. P. T. H21/174585). R. P. T. receives funding as a National Heart Foundation Postdoctoral Fellow (106625) and Vanguard Project Grant (107274). S. G. W. receives funding as a National Heart Foundation Future Leader Fellow (105622). The authors acknowledge the facilities as well as scientific and technical assistance at the Australian Center for Microscopy and Microanalysis, The University of Sydney.References
- D. Zhang, Q. Chen, C. Shi, M. Chen, K. Ma, J. Wan and R. Liu, Adv. Funct. Mater., 2021, 31, 2007226 CrossRef CAS.
- L. Davenport Huyer, S. Pascual-Gil, Y. Wang, S. Mandla, B. Yee and M. Radisic, Adv. Funct. Mater., 2020, 30, 1909331 CrossRef CAS.
- X. Zhou, Y. Wang, J. Ji and P. Zhang, Adv. Healthcare Mater., 2024, e2304478, DOI:10.1002/adhm.202304478.
- M. Cenciarini, M. Valentino, S. Belia, L. Sforna, P. Rosa, S. Ronchetti, M. C. D'Adamo and M. Pessia, Front. Mol. Neurosci., 2019, 12, 65 CrossRef CAS.
- Y. Wang, S. Vaddiraju, B. Gu, F. Papadimitrakopoulos and D. J. Burgess, J. Diabetes Sci. Technol., 2015, 9, 966–977 CrossRef CAS PubMed.
- A. H. P. Chan, M. J. Moore, A. J. Grant, Y. T. M. Lam, M. V. Darnell, P. L. Michael, S. G. Wise and R. P. Tan, Adv. Healthcare Mater., 2023, 12, e2301571 CrossRef PubMed.
- R. C. Coll, A. A. Robertson, J. J. Chae, S. C. Higgins, R. Muñoz-Planillo, M. C. Inserra, I. Vetter, L. S. Dungan, B. G. Monks, A. Stutz, D. E. Croker, M. S. Butler, M. Haneklaus, C. E. Sutton, G. Núñez, E. Latz, D. L. Kastner, K. H. Mills, S. L. Masters, K. Schroder, M. A. Cooper and L. A. O'Neill, Nat. Med., 2015, 21, 248–255 CrossRef CAS PubMed.
- A. J. Grant, N. Yang, M. J. Moore, Y. T. Lam, P. L. Michael, A. H. P. Chan, M. Santos, J. Rnjak-Kovacina, R. P. Tan and S. G. Wise, Adv. Sci., 2023, 10, e2300521 CrossRef.
- D. G. Barone, A. Carnicer-Lombarte, P. Tourlomousis, R. S. Hamilton, M. Prater, A. L. Rutz, I. B. Dimov, G. G. Malliaras, S. P. Lacour, A. A. B. Robertson, K. Franze, J. W. Fawcett and C. E. Bryant, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2115857119 CrossRef CAS.
- M. S. J. Mangan, E. J. Olhava, W. R. Roush, H. M. Seidel, G. D. Glick and E. Latz, Nat. Rev. Drug Discovery, 2018, 17, 588–606 CrossRef CAS PubMed.
- C. Marchetti, B. Swartzwelter, F. Gamboni, C. P. Neff, K. Richter, T. Azam, S. Carta, I. Tengesdal, T. Nemkov, A. D'Alessandro, C. Henry, G. S. Jones, S. A. Goodrich, J. P. Laurent, T. M. Jones, C. L. Scribner, R. B. Barrow, R. D. Altman, D. B. Skouras, M. Gattorno, V. Grau, S. Janciauskiene, A. Rubartelli, L. A. B. Joosten and C. A. Dinarello, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1530–E1539 CrossRef CAS.
- V. Klück, T. Jansen, M. Janssen, A. Comarniceanu, M. Efdé, I. W. Tengesdal, K. Schraa, M. C. P. Cleophas, C. L. Scribner, D. B. Skouras, C. Marchetti, C. A. Dinarello and L. A. B. Joosten, Lancet Rheumatol., 2020, 2, e270–e280 CrossRef PubMed.
- G. F. Wohlford, B. W. Van Tassell, H. E. Billingsley, D. Kadariya, J. M. Canada, S. Carbone, V. L. Mihalick, A. Bonaventura, A. Vecchié, J. G. Chiabrando, E. Bressi, G. Thomas, A. C. Ho, A. A. Marawan, M. Dell, C. R. Trankle, J. Turlington, R. Markley and A. Abbate, J. Cardiovasc. Pharmacol., 2020, 77, 49–60 CrossRef.
- A. Sánchez-Fernández, D. B. Skouras, C. A. Dinarello and R. López-Vales, Front Immunol., 2019, 10, 2578 CrossRef PubMed.
- N. Lonnemann, S. Hosseini, C. Marchetti, D. B. Skouras, D. Stefanoni, A. D'Alessandro, C. A. Dinarello and M. Korte, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32145–32154 CrossRef CAS PubMed.
- A. Dinarello, T. S. Mills, I. W. Tengesdal, N. E. Powers, T. Azam and C. A. Dinarello, Cells, 2023, 12(2), 294 CrossRef CAS PubMed.
- S. Toldo, A. G. Mauro, Z. Cutter, B. W. Van Tassell, E. Mezzaroma, M. G. Del Buono, A. Prestamburgo, N. Potere and A. Abbate, J. Cardiovasc. Pharmacol., 2019, 73, 215–222 CrossRef CAS.
- M. Fang, F. Xia, J. Wang, C. Wang, B. Teng, S. You, M. Li, X. Chen and X. Hu, Int. Immunopharmacol., 2024, 131, 111869 CrossRef CAS PubMed.
- C. Marchetti, B. Swartzwelter, M. I. Koenders, T. Azam, I. W. Tengesdal, N. Powers, D. M. de Graaf, C. A. Dinarello and L. A. B. Joosten, Arthritis Res. Ther., 2018, 20, 169 CrossRef PubMed.
- M. J. Moore, Y. T. Lam, M. Santos, R. P. Tan, N. Yang, J. Hung, Z. Li, K. A. Kilian, J. Rnjak-Kovacina, J. B. Pitts, H. Menzel and S. G. Wise, ACS Biomater. Sci. Eng., 2023, 9, 3320–3334 CrossRef CAS PubMed.
- F. S. Younesi, D. O. Son, J. Firmino and B. Hinz, Methods Mol. Biol., 2021, 2299, 17–47 CrossRef CAS PubMed.
- Y. Liu, W. Wei, Y. Wang, C. Wan, Y. Bai, X. Sun, J. Ma and F. Zheng, Inflammation Res., 2019, 68, 597–611 CrossRef CAS.
- R. P. Tan, B. S. L. Lee, A. H. P. Chan, S. C. G. Yuen, J. Hung, S. G. Wise and M. K. C. Ng, Acta Biomater., 2017, 53, 378–388 CrossRef CAS PubMed.
- D. Wang, Y. Zhang, X. Xu, J. Wu, Y. Peng, J. Li, R. Luo, L. Huang, L. Liu, S. Yu, N. Zhang, B. Lu and K. Zhao, Nat. Commun., 2021, 12, 2674 CrossRef CAS PubMed.
- J. C. Escolano, A. V. Taubenberger, S. Abuhattum, C. Schweitzer, A. Farrukh, A. Del Campo, C. E. Bryant and J. Guck, Front. Cell. Dev. Biol., 2021, 9, 639815 CrossRef PubMed.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802–812 CrossRef CAS.
- B. K. Verma, A. Chatterjee, P. Kondaiah and N. Gundiah, Bioengineering, 2023, 10, 998 CrossRef CAS.
- C. M. Artlett, S. Sassi-Gaha, J. L. Rieger, A. C. Boesteanu, C. A. Feghali-Bostwick and P. D. Katsikis, Arthritis Rheum., 2011, 63, 3563–3574 CrossRef CAS.
- T. M. Bui, H. L. Wiesolek and R. Sumagin, J. Leukocyte Biol., 2020, 108, 787–799 CrossRef CAS PubMed.
- P. Wisitpongpun, P. Potup and K. Usuwanthim, Front Immunol., 2022, 13, 856296 CrossRef CAS.
- J. Zhang, X. Liu, C. Wan, Y. Liu, Y. Wang, C. Meng, Y. Zhang and C. Jiang, J. Clin. Periodontol., 2020, 47, 451–460 CrossRef CAS.
- S. A. Eming, P. Martin and M. Tomic-Canic, Sci. Transl. Med., 2014, 6, 265sr266 Search PubMed.
- A. J. Deegan and R. K. Wang, Phys. Med. Biol., 2019, 64, 07tr01 CrossRef PubMed.
- M. Lindkvist, M. M. Zegeye, M. Grenegård and L. U. Ljungberg, Int. J. Mol. Sci., 2022, 23, 1448 CrossRef CAS PubMed.
- M. M. Zegeye, B. Andersson, A. Sirsjö and L. U. Ljungberg, Cells, 2020, 9(6), 1414 CrossRef CAS PubMed.
- L. Zhang, Z. Cao, T. Bai, L. Carr, J. R. Ella-Menye, C. Irvin, B. D. Ratner and S. Jiang, Nat. Biotechnol., 2013, 31, 553–556 CrossRef CAS.
- I. Ribitsch, P. M. Baptista, A. Lange-Consiglio, L. Melotti, M. Patruno, F. Jenner, E. Schnabl-Feichter, L. C. Dutton, D. J. Connolly, F. G. van Steenbeek, J. Dudhia and L. C. Penning, Front. Bioeng. Biotechnol., 2020, 8, 972 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00705k |
| This journal is © The Royal Society of Chemistry 2024 |