DOI:
10.1039/D4DT00749B
(Paper)
Dalton Trans., 2024,
53, 8642-8656
Dibenzoazepine hydrazine is a building block for N-alkene hybrid ligands: exploratory syntheses of complexes of Cu, Fe, and Li†
Received
12th March 2024
, Accepted 17th April 2024
First published on 2nd May 2024
Abstract
The new hydrazine 5H-dibenzo[b,f]azepin-5-amine (2) reacts with P- and Si-electrophiles via deprotonation to afford P(III)-, P(V)-, and TMS-hydrazides 3–8 and with carbonyl electrophiles via acid-free condensation to the N-substituted hydrazones 9–12 that are potential N-alkene ligands. While β-ketohydrazone 9 and α-dihydrazone 10 react with [Mes(Cu)]4, [Cu(NCCCH3)4]2PF6, and FeCl2(THF)1.5 to afford complexes devoid of alkene interaction, [Cu(OTf)]2·C6H6 reacts with the α-keto hydrazone 11 or with N,N dimethyl-hydrazone 12 to form the neutral dimeric Cu(I) complex 18 with bridging Cu(I)–alkene interactions or the tetrahedral cationic complex 19 in which 12 binds as a bidentate hydrazone-alkene ligand, respectively. The surprising stability of the alkene coordination in complexes 18 and 19 prevents substitutions with, e.g., PPh3.
Introduction
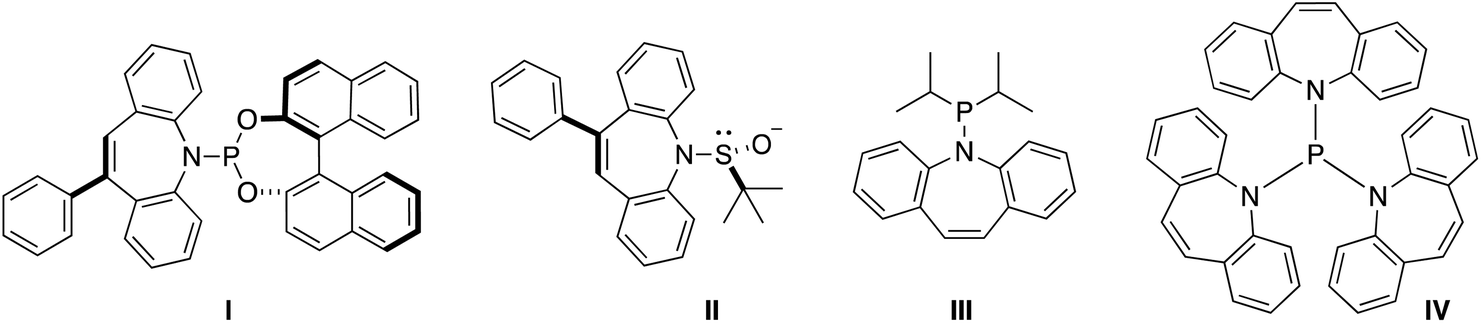
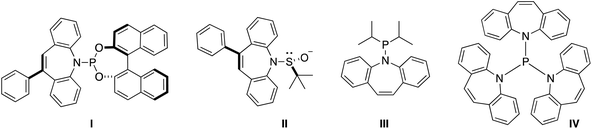
Hydrazones are a versatile synthetic tool in organic chemistry.1 In particular, metallated hydrazones are key intermediates in C–C bond forming reactions2 and reductions such as the Shapiro reaction.3 However, their application as supporting ligands for metal complexes in synthesis and catalysis is less developed.4 For example, hydrazones corresponding to α- and β-diimines (e.g. nacnac type ligands) have different electronic properties through the second hydrazine nitrogen atom.5 Some time ago, we and others have introduced 5H-dibenzo[b,f]azepine as a building block for the synthesis of chiral6 and achiral7 hemilabile P- and S-alkene hybrid ligands (Chart 1): Ligands I and II feature both point and planar chirality, while the superbulky ligand IV displays a crystallographic Tolman cone angle of 229°. In order to extend the versatility of this building block we envisaged the corresponding hydrazine 5H-dibenzo[b,f]azepin-5-amine (2) inspired by Grützmacher's 5-amino-5H-dibenzo[a,d]cycloheptene (tropNH2).8 In this paper, we disclose a multi-gram scale preparation of the new hydrazine 2 and its use for the synthesis of hydrazone-based N-alkene hybrid ligands. The coordination chemistry with Li, Fe(II), Cu(II), and Cu(I) is presented. In particular, the Cu(I) species are shown to be additionally stabilized by alkene coordination of the alkene function of the benzoazepine moiety.
 |
| | Chart 1 | |
Results and discussion
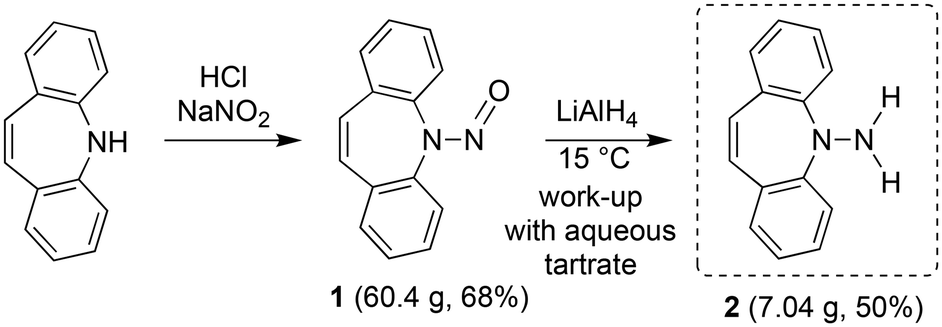
For the realization of new N-alkene hybrid ligands, the accessibility of the primary hydrazine 2 on a gram scale had first to be demonstrated: dibenzo[b,f]azepine reacts with NaNO2 under acidic conditions following literature procedures for similar systems9 to afford nitrosoamine 1 as yellowish crystals in good yields (Scheme 1). The reduction of 1 with LiAlH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 can be performed on a multi gram scale and yields 2 as a bright yellow crystalline solid. 2 decomposes by N–N bond cleavage under acidic conditions, which must be avoided.
10 can be performed on a multi gram scale and yields 2 as a bright yellow crystalline solid. 2 decomposes by N–N bond cleavage under acidic conditions, which must be avoided.
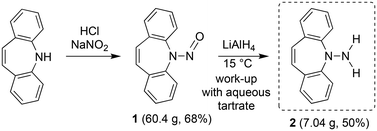 |
| | Scheme 1 Synthesis of the new dibenzo[b,f]azepine derived hydrazine 2. | |
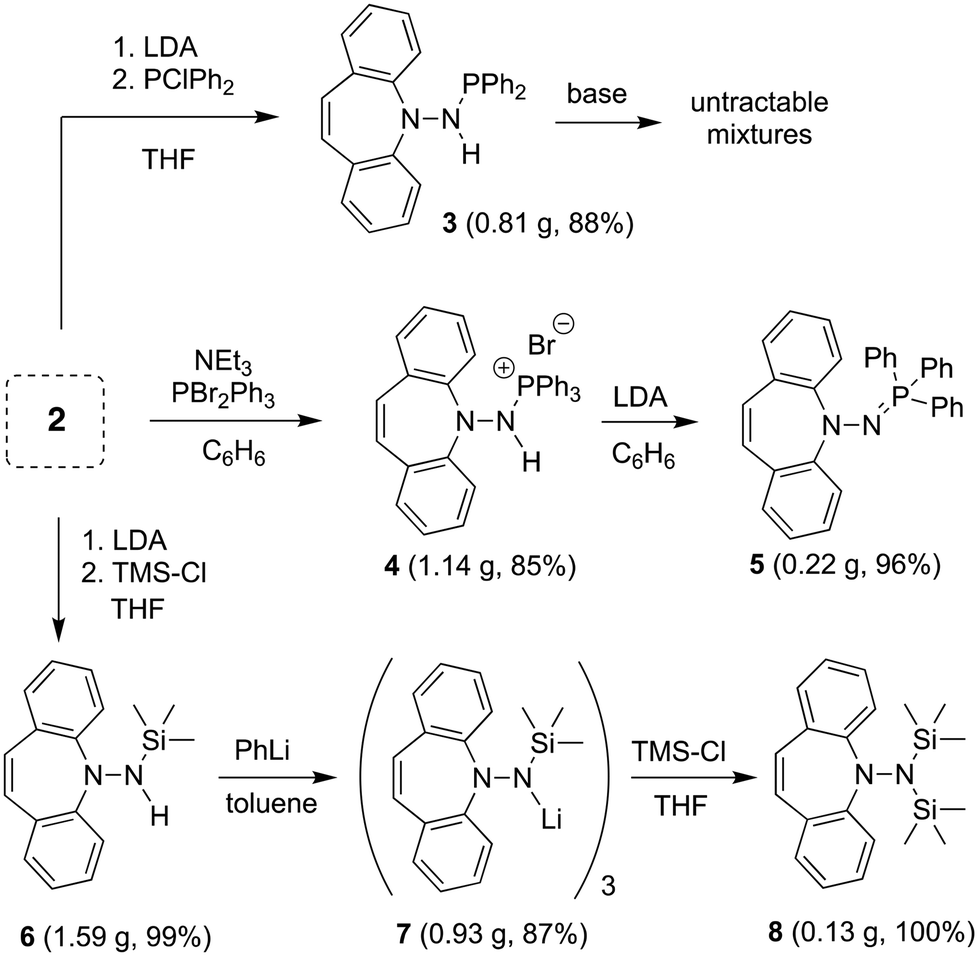
The reaction of 2 with Si- and P-electrophiles is outlined in Scheme 2. Yellow 2 is deprotonated with LDA in THF at RT to afford a red suspension of an orange solid, which is quenched in situ with a slight excess of Ph2PCl or TMSCl to afford 311 and 6, respectively, in very good isolated yields. Similarly, 2 reacts with PBr2Ph312 in the presence of 1.5 equiv. of NEt3 to afford the phosphonium bromide 4. The 1H NMR spectra for 3 and 4 show characteristic doublets at 4.95 and 11.38 ppm, respectively, for the amine protons due to 2J-coupling to phosphorus. In 6 the NH proton resonates at 3.75 ppm as a board singlet. The 31P NMR spectra of 3 and 4 show a singlet resonance at 49.1 and 38.9 ppm, respectively (2J-couplings are not resolved).
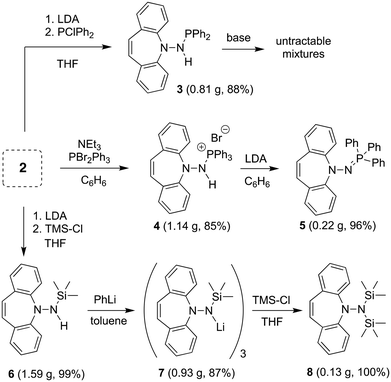 |
| | Scheme 2 Reactions of 2 with P(III), P(V), and the TMS-Cl electrophiles. | |
While the deprotonation of 3 to the corresponding amidophosphine does not afford clean, isolable products with a variety of bases, such as PhLi,13 from 4, the second equivalent of HBr is eliminated with LDA liberating the iminophosphine 5 as a yellow powder in practically quantitative yields. Contrary to N-alkyliminophosphines,145 is air-stable and resistant to hydrolysis. 5 was needed in this project as potential imidation reagent15 for carbonyl compounds (vide infra). 6 is smoothly deprotonated by PhLi in toluene to form the trimeric lithium complex 7 as an orange powder in good yield,16 whose 1H NMR reveals broad signals due to fluxional behavior. Attempts to prepare the analogous potassium salt by reacting 6 with benzyl potassium or KH afforded untractable products. Reacting 7 with an additional equivalent of TMSCl quantitatively yields pure 8 that does not need further purification.171H NMR shows one singlet at 0.25 ppm for the TMS groups and a characteristic singlet at 5.99 ppm for the alkene protons of the azepine. As with 5, 8 is a potential hydrazide transfer reagent (vide infra).18
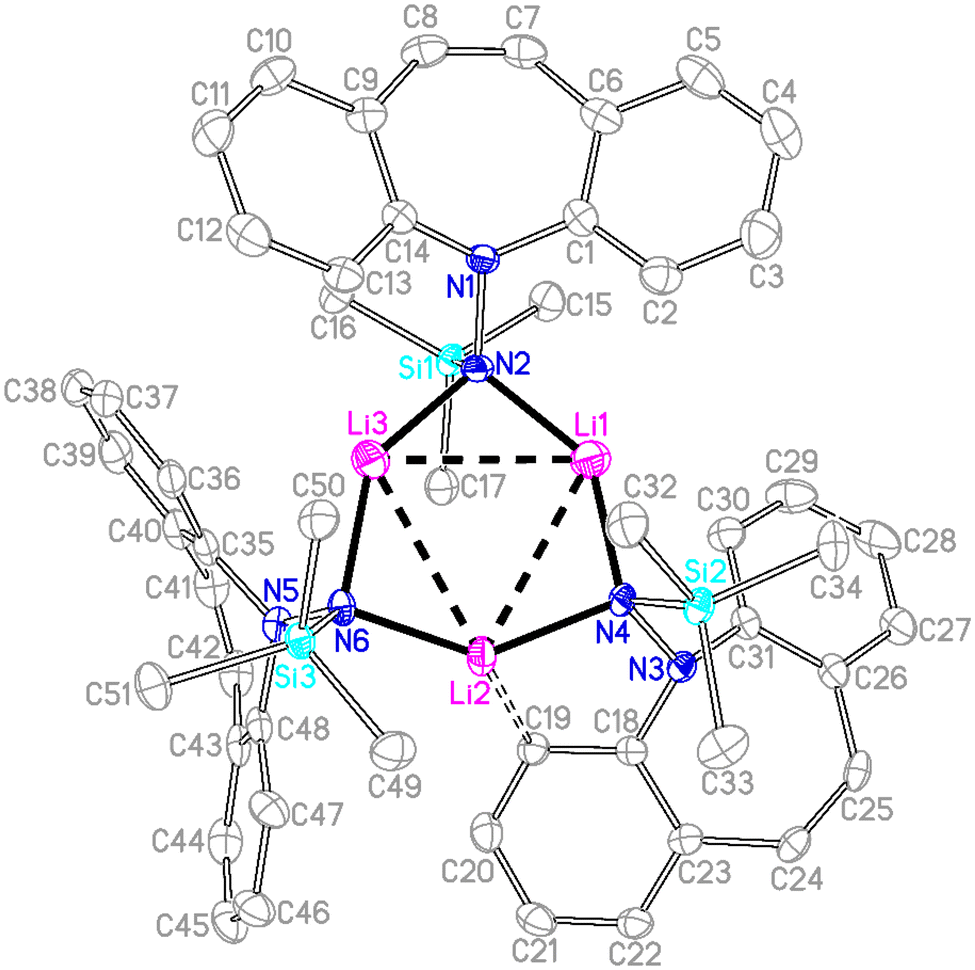
Single crystal X-ray diffraction analysis of 7 revealed a C3-symmetric trimeric structure containing three lithium atoms forming an equilateral triangle (Fig. 1). The average Li–N bond length of 1.988 Å is in the range of reported values19,16b and similar to the corresponding bond in complex 13 (vide infra). The lithium-bridging hydrazido N-donors are tetrahedral, while the azepine ring N-atoms N1, N3 and N5 are trigonal planar (sum of bond angles ∑∢(N) ≈ 360°). One of the two independent molecules in the asymmetric unit exhibits an anagostic interaction (Li2⋯C19) at a distance of 2.431 Å.20 Similar Agostic interactions in this position of the dibenzo[b,f]azepine backbone have also been observed for Ru(II) complexes with P-alkene ligands.21
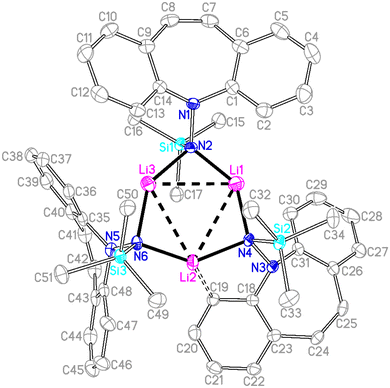 |
| | Fig. 1 Crystal structure of one of two independent molecules of 7 (50% probability ORTEP, H atoms are omitted). Selected bond lengths (Å) and angles (deg): Li1–N2 1.933(7), Li3–N2 1.989(7), Li1–Li3 2.960(9), N1–N2 1.479(4), Si1–N2 1.736(3), Si1–C16 1.866(4), Li2–C19 2.429(7) Å; C1–N1–C14 114.8(3), C1–N1–N2 117.2(3), Li1–N2–Li3 98.0(3), N2–Li1–N4 139.8(4), N6–Li2–N4 138.1(3). | |
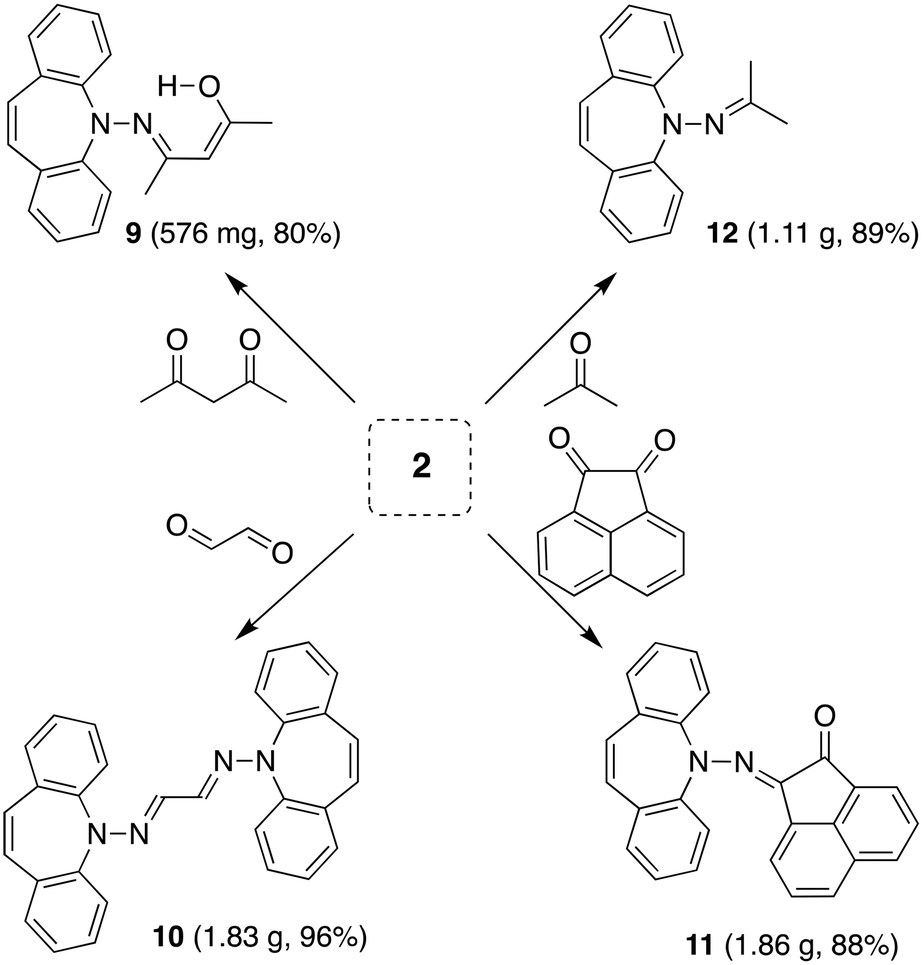
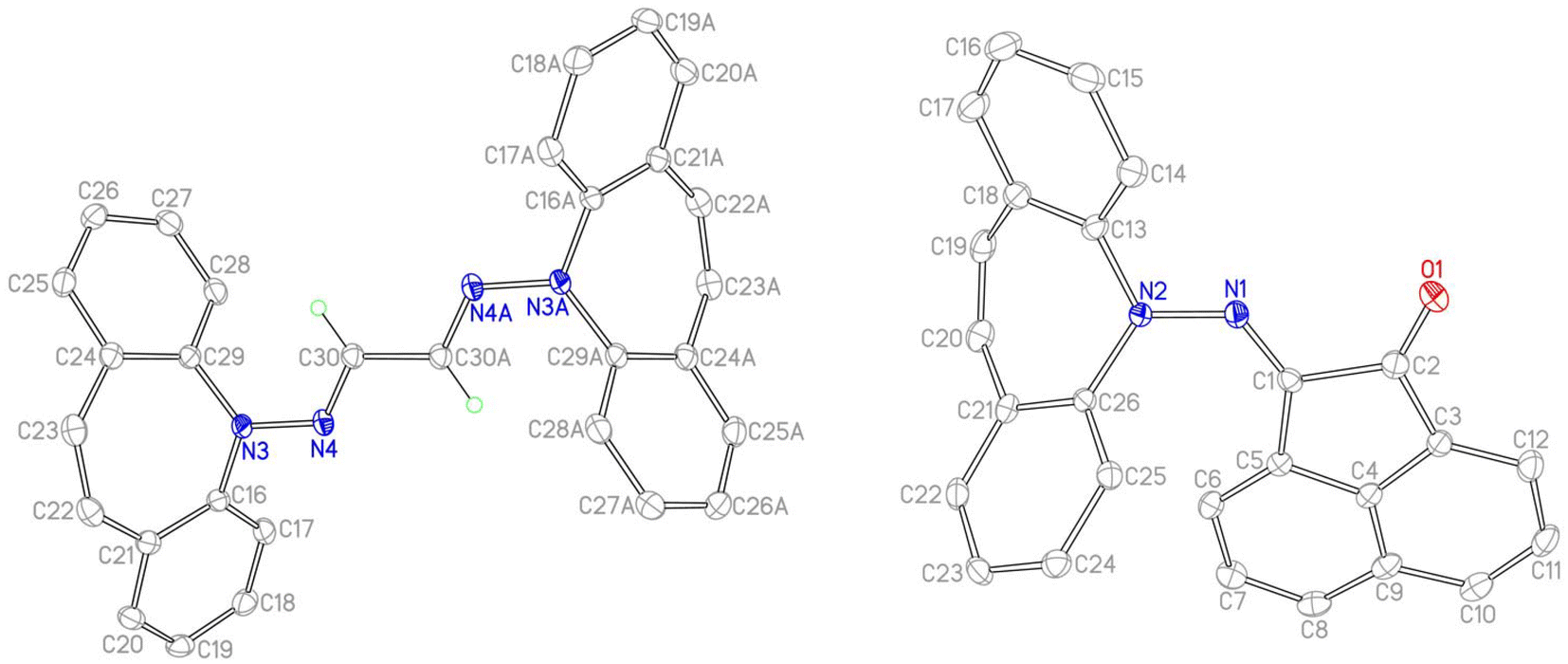
The reaction of 2 with carbonyl electrophiles is summarized in Scheme 3. With acetylacetone the mono substituted β-ketohydrazone 9 is isolated in high yield after stirring for 10 d in EtOH at room temperature and crystallization. Long reaction times are necessary because the usual catalysts cannot be used due to the instability of 2 towards Brønsted acids.22 The 1H NMR spectrum shows characteristic resonances for the OH and alkene protons at 13.1 and 6.75 ppm, respectively. Condensation of a second equivalent of 2 was attempted in three ways: (i) in situ alkylation of 9 with [OMe3][BF4] followed by addition of 2 under basic conditions,23 (ii) reaction of 9 with bis-silyl hydrazide 8 at 60 °C (with the intention to drive the reaction by forming (TMS)2O), and (iii) by an acid-free aza-Wittig reaction2 employing imino-phosphine 5 (Scheme 2). While the first two methods gave no reaction, a mixture of acetylacetone with 5 led to decomposition at 90 °C (at lower T no reaction takes place), even though P(O)Ph3 was identified in the crude product mixture. With hexafluoroacetylacetone, on the other hand, 5 reacts as a base in THF at 50 °C to afford the iminium salt [5-H][F3C(O)CCC(O)CF3] (an analogue of 4).24 Condensation of 2 with glyoxal quantitatively affords 10, which turned out to be hydrolysis resistant and which precipitates as a mixture of isomers as evinced by its 1H NMR spectrum. Such yellow CDCl3 NMR solutions of 10 turn orange within hours and show the expected peak pattern of one sole symmetrical isomer.25 XRD analysis revealed the exact configuration of 10 in the crystal and shows the trans-isomer with a perfectly planar tetraazahexadiene chain (Fig. 3). The terminal azepine N-atoms are very slightly pyramidalized (∑∢(N) = 356°), and the C15–N2 bond length of 1.290 Å is in the expected range.26 With acenaphthenequinone, 2 reacts only in a one-to-one ratio to the α-ketohydrazone 11,27 albeit in excellent yields. The 1H NMR spectrum shows a characteristic singlet at 6.9 ppm suggesting CS symmetry in solution. In the crystal, the symmetry is lower with the dibenzoazepine moiety tending into coplanarity with the acenaphthenequinone substitutent contrasting aniline-based BIANs (Fig. 2, right).28 The nitrogen atoms lie in the quinone plane within ca. 5°, while the dibenzo[b,f]azepine moiety is twisted out of plane by 23° (average torsion angle along C5–C1–C19–C20 in four independent molecules). The N1–N2 bond distance of 1.37 Å indicates double bond character. Finally, hydrazone 12 forms upon condensation of 2 with acetone. The 1H NMR spectrum shows two singlets at 1.98 and 2.28 ppm for the methyl groups and a singlet at 7.09 ppm for the alkene protons.
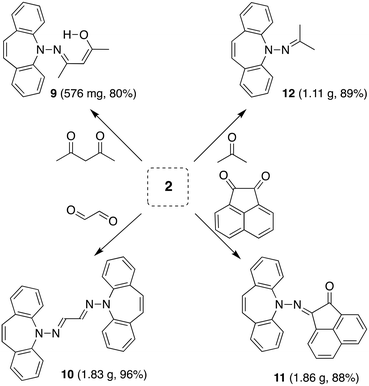 |
| | Scheme 3 Condensation of 2 with carbonyl compounds to hydrazones. | |
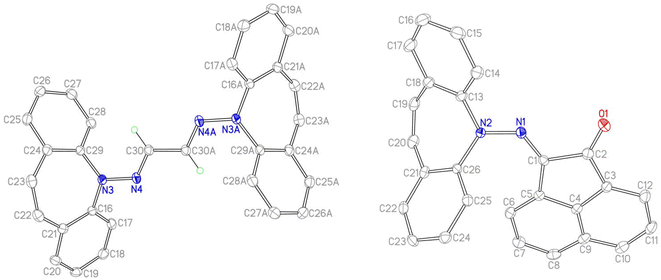 |
| | Fig. 2 Crystal structures of 10 (left, one of two independent molecules) and 11 (right, one of four independent molecules). ORTEPs are drawn at 50% probability, H atoms are omitted. Selected bond lengths (Å) and angles (deg) for 10: C15–C15A 1.444(2), N1–N2 1.3741(12), N2–C15 1.2904(13), N1–C1 1.4278(13), C7–C8 1.3426(17), N2–N1–C1 113.37(8), N2–N1–C14 123.22(8), C15–N2–N1 119.95(9); for 11: C2–O1 1.214(3), N1–N2 1.354(2), C1–N1 1.296(3), N2–C13 1.437(3), C1–C2 1.517(3), C19–C20 1.340(3), N2–N1–C1 124.17(17), N1–C1–C2 114.39(17). | |
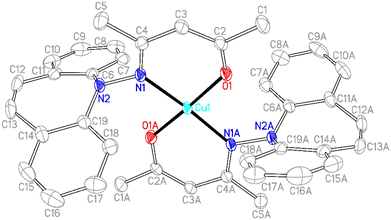 |
| | Fig. 3 Crystal structure of 15 (50% probability ORTEP, H atoms are omitted). Selected bond lengths (Å) and angles (deg): Cu1–O1 1.8903(14), N1–N2 1.432(2) Cu1–N1, 2.0041(16), O1–C2 1.277(3), N1–C4 1.323(3), C2–C3 1.376(3), C3–C4 1.418(3), O1–Cu1–O1A 173.98(13), N1–Cu1–N1A 172.26(12), O1–Cu1–N1 90.80(6), O1–Cu1–N1A 89.60(6), C4–N1–N2 112.89(17), C2–C3–C4 125.5(2). | |
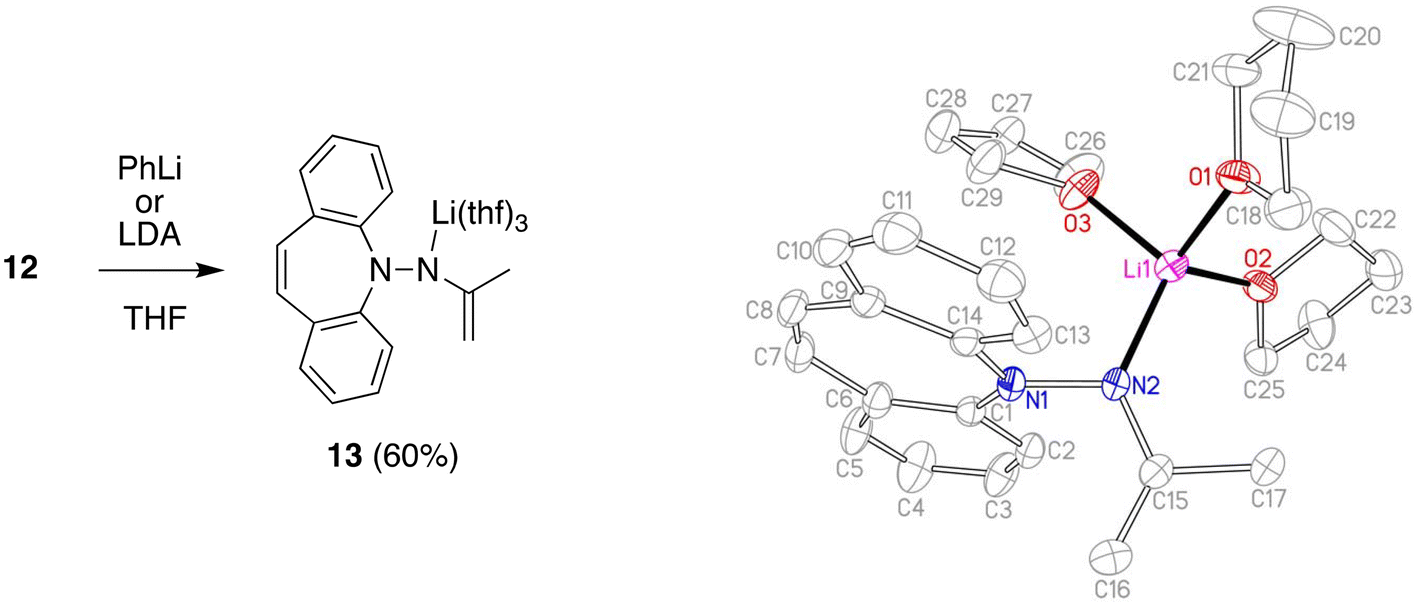
Deprotonation of 12 with lithium di-isoprpopyl-amide (LDA) or PhLi in THF solution affords the deep red aza-enolate 13 (eqn (1)). The enamine protons resonate as singlets at 2.75 and 2.96 ppm in the 1H NMR spectrum. There is no indication for conjugation between the π-systems of the enamide and the dibenzo[b,f]azepine. The crystal structure of 13 has approximate CS symmetry consistent with its solution structure (Scheme 4). Solvation by three THF molecules creates together with the hydrazido N-donor a tetrahedral coordination geometry around lithium and prevents higher aggregation (cf. complex 7 above). The N atoms are trigonal planar with ∑∢(N1) = 358.5° and ∑∢(N2) = 360.0°, and the N–Li distance of 1.956 Å parallels the values observed in 7.29 The delocalized nature of the enamide function is reflected in its planar geometry and the short N2–C15 bond distance of 1.370 Å indicating double bond character.30 The enamide moiety is oriented perpendicular to the unusually planar dibenzo[b,f]azepine backbone with the C15–C16 double bond (1.346 Å) pointing towards it. Normally, the azepine ring displays a boat configuration due to its antiaromatic character,31 which has been used to great effect to install stable planar chirality in similar P- and S-alkene ligand systems.32 In addition, the C7–C8 bond (1.327 Å) clearly has double bond character and is not in conjugation with the benzene rings. Conjugative effects between the π-systems are also excluded because the N1–N2 distance (1.443 Å) identifies it as a single bond and because of the aforementioned perpendicular orientation of the N-substituents. The planarity of the system is therefore explained in terms of steric, rather than electronic, preference. Nevertheless, this is a rare example, in which the planar structure is stable enough to be caught in the crystal.
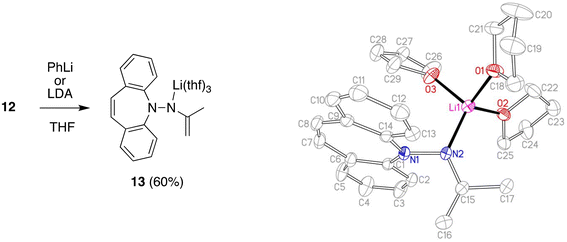 |
| | Scheme 4 Synthesis and crystal structure of 13 (50% probability ORTEP, H atoms are omitted). Selected bond lengths (Å) and angles (deg): Li1–N2 1.956(4), N2–C15 1.370(2), C15–C16 1.346(3), C15–C17 1.514(3), N1–N2 1.443(2), N1–C1 1.413(2), C7–C8 1.327(3); N1–N2–Li1 114.44(14), C15–N2–Li1 132.58(16), C16–C15–C17 119.77(18). | |
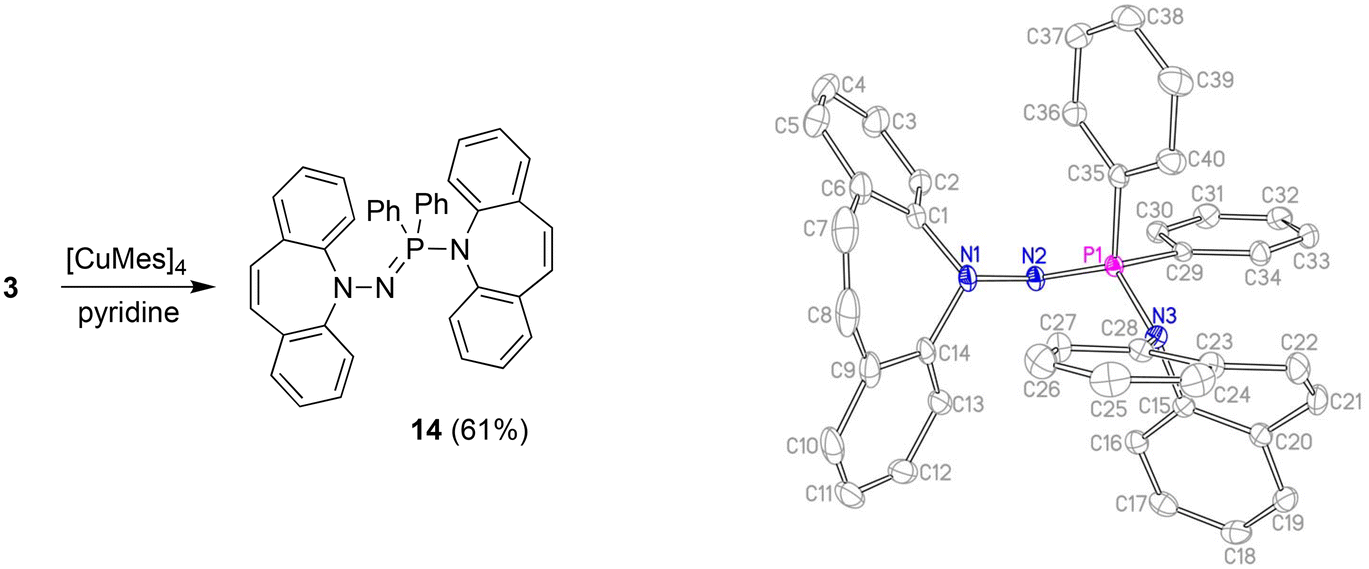
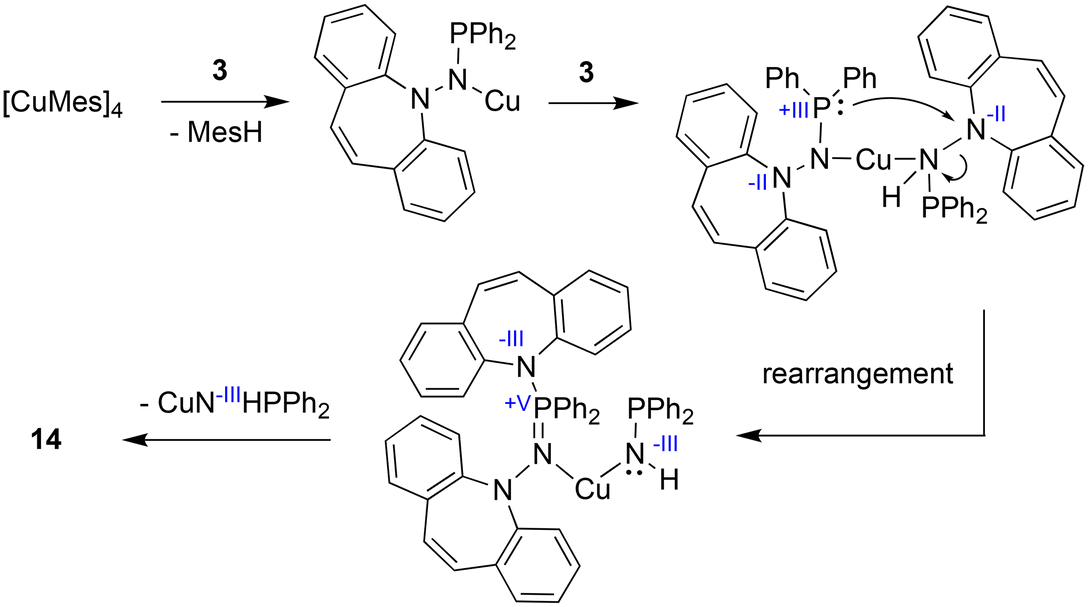
With the well characterized potential ligands 3, 5, 7–13 at hand, their coordination chemistry with iron and copper was explored, because, ultimately, we are interested in the stabilization of reactive hydrides of first-row transition metal-hydrides for the reduction of CO2.33 The synthesis of Cu(I) amido complexes was attempted with ligands 3, 6–8 (Scheme 2). 3 reacts with [Cu(Mes)]4 in pyridine or THF solution and a beige solid is isolated (Scheme 5). Against expectations, crystallographic analysis revealed the metal-free imino-phosphine 14, in which the tetrahedral phosphorus atom is bound to two phenyl groups, one dibenzo[b,f]azepine, and one hydrazine dibenzo[b,f]azepine moiety. The P1–N2 bond distance of 1.598 Å corresponds to a double bond,34 whereas the P1–N3 single bond measures 1.692 Å.35 The 31P nucleus resonates as a singlet at 16.9 ppm. The mechanism outlined in Scheme 6 rationalizes the outcome of the reaction of [Cu(Mes)]4 with two equivalents of 3. NMR control experiments of the reaction revealed rapid formation of mesitylene on deprotonation of 3 by [CuMes]4 in the proton spectrum and a broad 31P resonance at 58 ppm of a major side product, which we attribute to CuNHPPh2. We assume that the initially formed unsaturated copper amido complex is stabilized by coordination of a second equivalent of neutral ligand, triggering the intramolecular redox rearrangement to 14 upon elimination of CuNHPPh2. The N–N cleavage reduces two nitrogen atoms from -II to -III, while the P(III) is oxidized to P(V), so overall this reaction could be termed an oxidative addition of an N–N bond to P(III). Oxidation of phosphorous by Cu(I) is excluded because no precipitation of metallic Cu was observed.
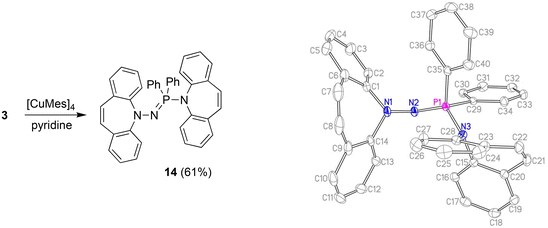 |
| | Scheme 5 Synthesis and crystal structure of 14 (50% probability ORTEP, H atoms are omitted). Selected bond lengths (Å) and angles (deg): P1–N2 1.5977(12), P1–N3 1.6919(12), N1–N2 1.4611(16), N1–C1 1.4381(19), N3–C15 1.4453(18), N1–N2–P1 111.31(9), N2–P1–N3 122.47(6), N3–P1–C29 103.55(6), N3–P1–C35 104.05(6), C29–P1–C35 105.14(7), C15–N3–P1 118.54(9), C1–N1–N2–P1 117.32(11). | |
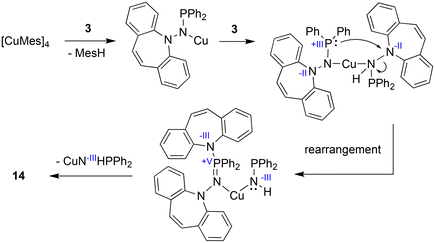 |
| | Scheme 6 Proposed mechanism for the formation of 14. | |
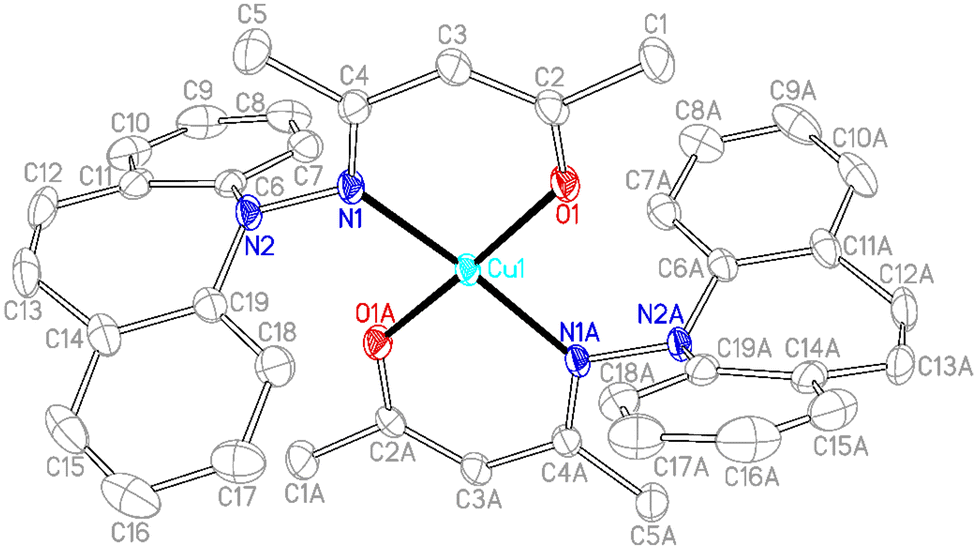
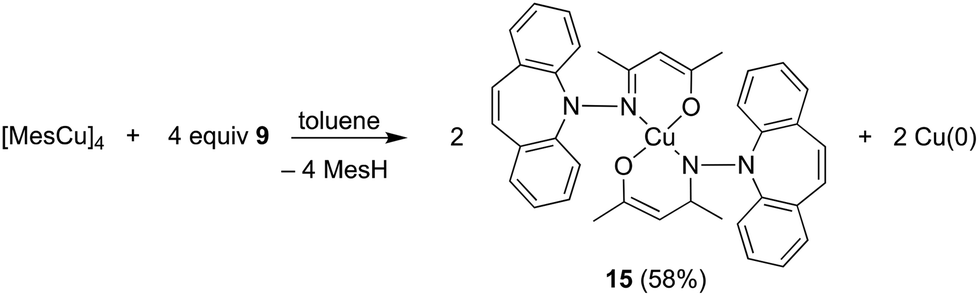
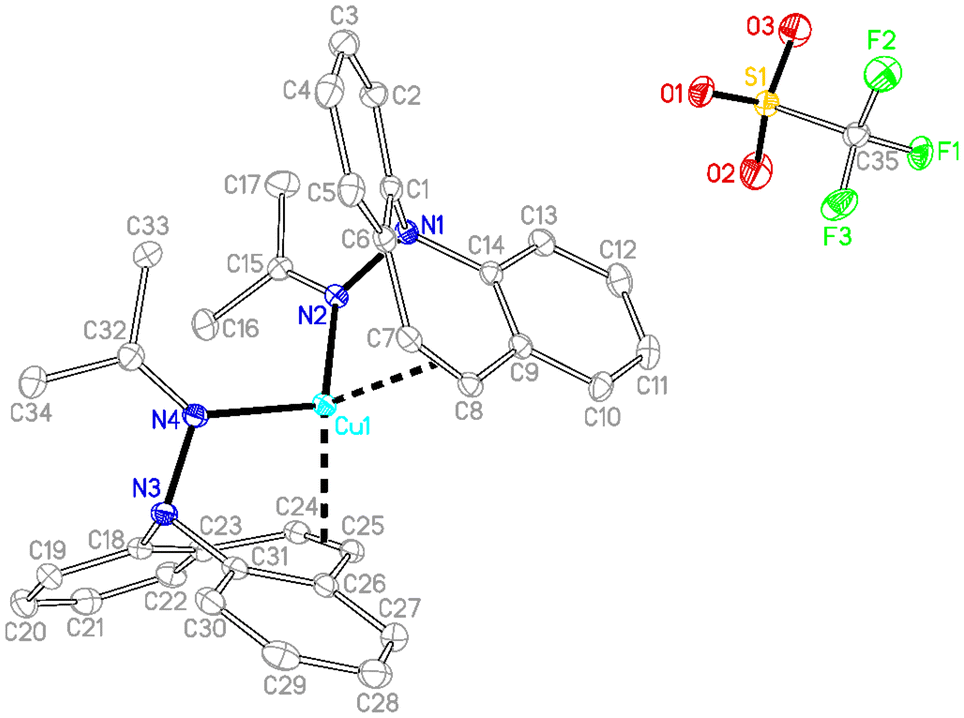
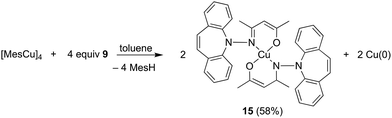
In contrast to phosphine hydrazide 3, the silyl hydrazide 6 does not react with the Cu(I) precursors [MesCu]4 (Mes = mesityl anion) and [Cu(OtBu)]4 to the Cu(I) silyl amido complex,36 presumably because the copper precursors are not basic enough. Also 7 and 8 were tried in reactions with CuCl in a bid to obtain the copper amide via salt metathesis or under the elimination of TMSCl, respectively.37 The reactions with 7 in varying metal to ligand ratios (cuprates were also envisioned) resulted in untractable mixtures,38 while 8 did not react with CuCl even at 70 °C. The reaction of 9 with [CuMes]4 results in dark brown reaction mixtures that show no 1H NMR signals, indicative of a disproportionation reaction to metallic copper and a Cu(II) complex 15 (eqn (1)),39 which was confirmed by single crystal X-ray diffraction analysis (Fig. 3).40 The roughly square planar complex (dihedral angle between coordination planes: 9.7° (ref. 41)) is supported by two molecules of deprotonated N,O-coordinated 9 and has crystallographic centrosymmetry. The trans positioned O and N donors span angles of 172–174° and their distances to the metal center are in the range of reported values.42 The Cu–N bond is slightly longer due to the steric pressure exerted by the dibenzo[b,f]azepine residue, which is oriented perpendicularly to the Cu–nacac plane.
| |  | (1) |
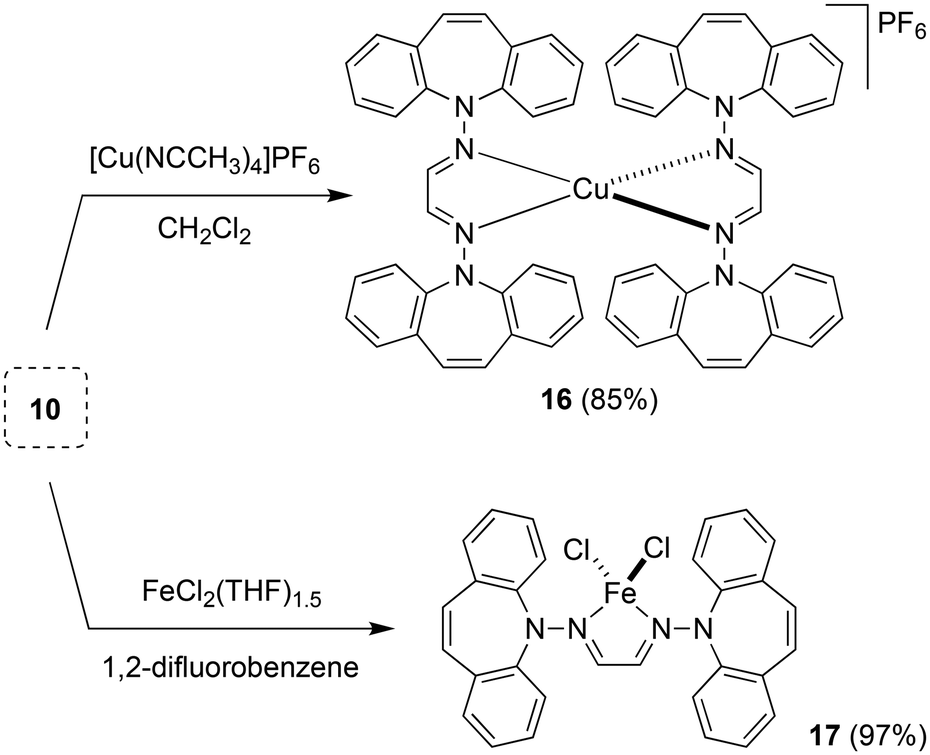
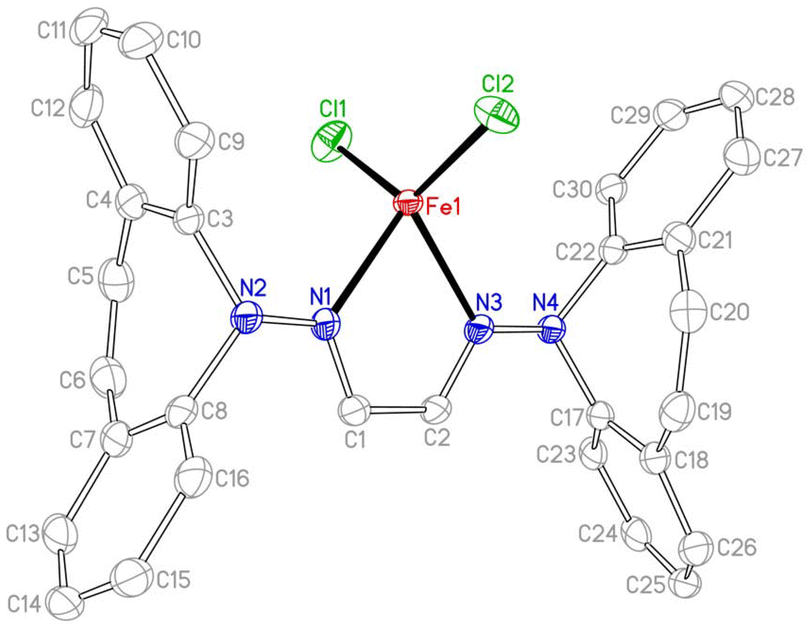
Two equivalents of 10 react with [Cu(NCCH3)4]PF6 to yield complex 16 as an orange powder (Scheme 7).43 Long reaction times (ca. 3 d) are required to ensure the coordination of the second ligand equivalent. NMR spectra do not indicate ligand alkene coordination and support a complex of high symmetry in solution. 10 also reacts with FeCl2(THF)1.5 to afford red-brown crystals of 17 in excellent yield.44 The crystal structure suffered from substantial disorder, caused by the presence of different conformers. The major conformer, which occupied 78% of the lattice sites, showed a transoid arrangement of the dibenzoazepine moieties, and a distorted tetrahedral coordination geometry around iron (Fig. 4). The conformers are defined by the relative orientations of the dibenzo[b,f]azepine moieties to each other and suggest low energy barriers between them.45 The nitrogen, iron, and imine carbon atoms are coplanar, and azepine N atoms are pyramidallized (∑∢(N2) = 345.41°, ∑∢(N4) = 343.05°). The C1–N1 and C1–C1 distances do not significantly change compared to the free ligand, indicating redox-neutral coordination.
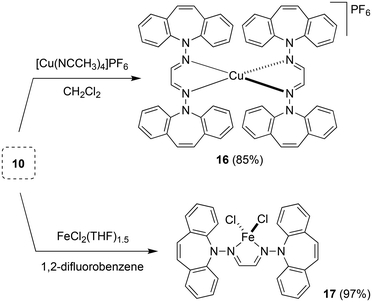 |
| | Scheme 7 Synthesis of the tetrahedral Cu(I) and Fe(II) complexes 16 and 17. | |
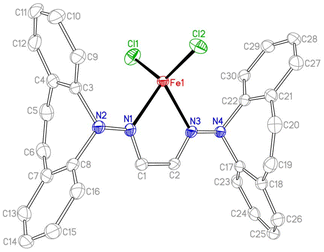 |
| | Fig. 4 Crystal structure of one of three conformers of 17 (50% probability ORTEP, H atoms and solvent molecules are omitted). Selected bond lengths (Å) and angles (deg): Fe1–Cl1 2.220(2), Fe1–N1 2.137(2), N1–C1 1.292(3), C1–C2 1.458(3), Cl1–Fe1–Cl2 126.68(6), N1–Fe1–N3 77.14(7), N1–N2–C3 113.22(16), N1–N2–C8 117.00(17), C3–N2–C8 115.19(16), C1–N1–N2 120.75(18), N2–N1–Fe1 125.85(13), C1–N1–Fe1 113.33(14). | |
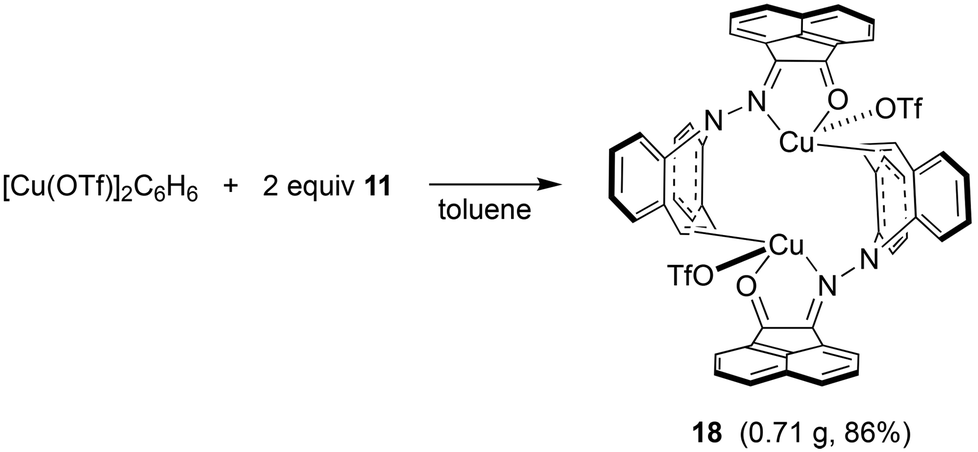
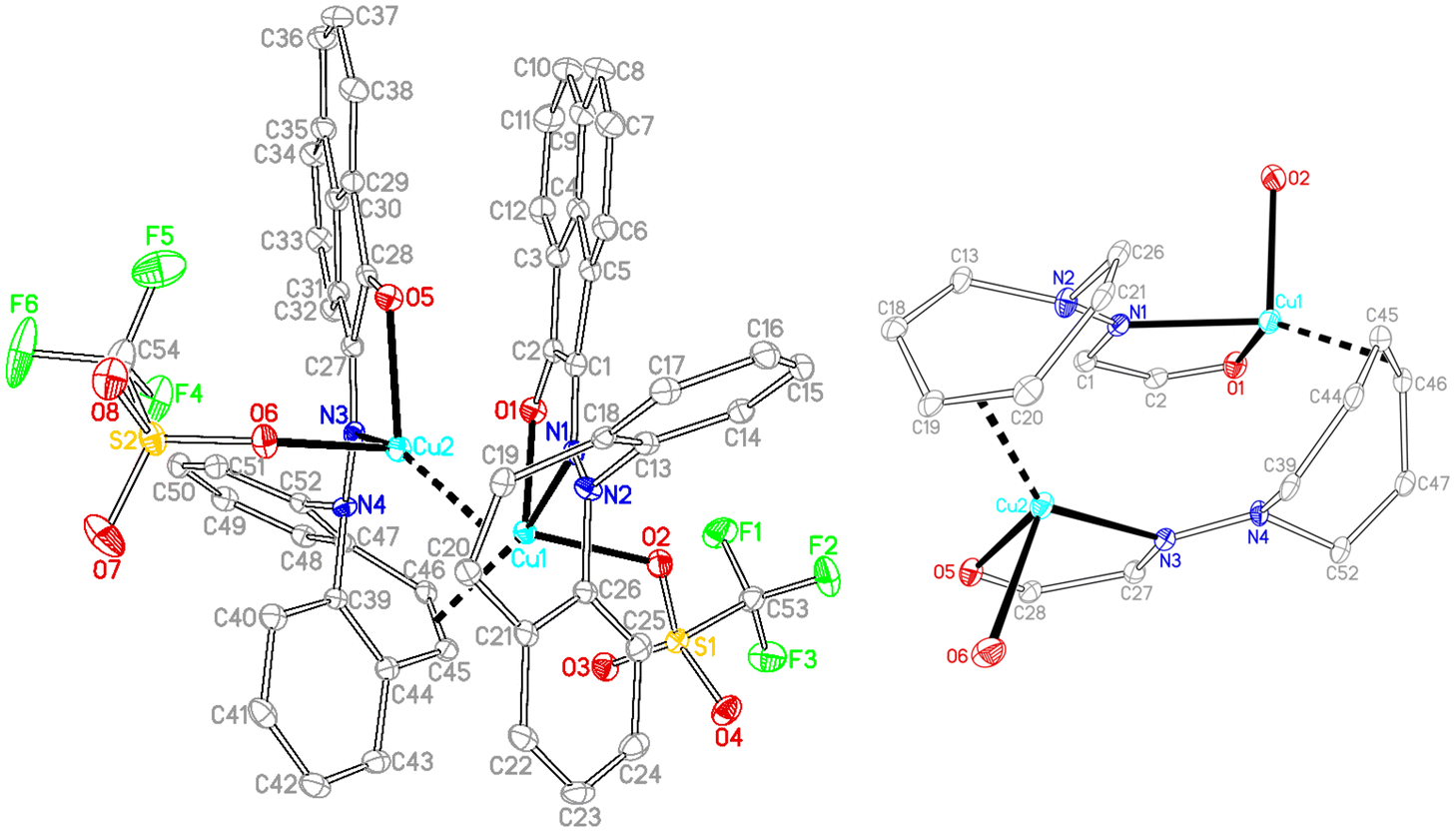
In contrast to the synthesis of complex 15, disproportionation reactivity is suppressed by the use of the α-ketohydrazone 11. It reacts with [Cu(OTf)]2·C6H6 to form the dinuclear complex 18 within minutes (eqn (2), Cu(I) halides are unreactive).46 The 1H NMR spectrum shows 16 individual proton resonances indicating a low-symmetry, rigid structure. The diastereotopic dibenzoazepine alkene protons resonate as doublets at 6.27 and 4.74 ppm. Indeed, the crystal structure reveals a C2-symmetric dinuclear complex with pseudo-tetrahedral copper centers reminiscent of triene–Cu(I)–triflates (Fig. 5).47 Each α-keto-hydrazone coordinates one copper atom in a bidentate manner, while the alkene function bridges onto the neighboring copper atom. The coordinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds are elongated from 1.341 Å in the free ligand to 1.381 Å, which is on the long side of comparable values.48 The copper atoms are separated by 4.58 Å, and the naphthalene moieties are π-stacked at roughly 3.5 Å (C4⋯C30) and offset.49 Due to the fact that the ligands bridge the metals, there is no need for pyramidalization of the dibenzoazepine N atoms (∑∢(N2) = 355°, ∑∢(N4) = 353°), contrasting their usual sp3-hybridization encountered in bidentate coordination modes in mononuclear complexes of such hybrid ligands (cf.19 below). The remaining coordination sites are occupied by the triflate anion with a Cu–O contact of 2.232 Å,50 thereby securing an 18 valence electron count.51
C double bonds are elongated from 1.341 Å in the free ligand to 1.381 Å, which is on the long side of comparable values.48 The copper atoms are separated by 4.58 Å, and the naphthalene moieties are π-stacked at roughly 3.5 Å (C4⋯C30) and offset.49 Due to the fact that the ligands bridge the metals, there is no need for pyramidalization of the dibenzoazepine N atoms (∑∢(N2) = 355°, ∑∢(N4) = 353°), contrasting their usual sp3-hybridization encountered in bidentate coordination modes in mononuclear complexes of such hybrid ligands (cf.19 below). The remaining coordination sites are occupied by the triflate anion with a Cu–O contact of 2.232 Å,50 thereby securing an 18 valence electron count.51
| |  | (2) |
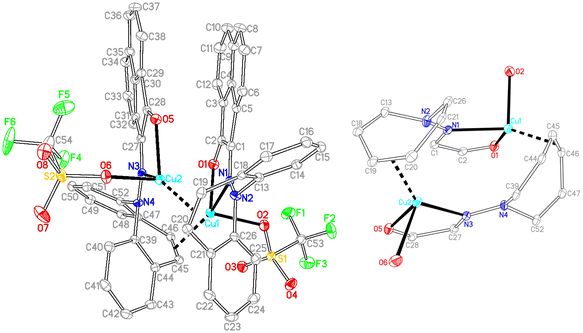 |
| | Fig. 5 Crystal structure of 18 with its core structure to the right (50% probability ORTEP, H atoms and solvent molecules are omitted). Selected bond lengths (Å) and angles (deg): Cu1–O1 2.0590(13), Cu1–O2 2.2263(13), Cu1–N1 2.1358(15), Cu1–C45 2.1026(18), Cu1–C46 2.0518(18), C45–C46 1.381(3), Cu2–O5 2.0537(13), Cu2–O6 2.2391(13), Cu2–N3 2.1121(15), Cu2–C19 2.0559(18), Cu2–C20 2.0872(18), C19–C20 1.381(2), N1–N2 1.344(2), N3–N4 1.344(2), O1–Cu1–N1 80.23(5). | |
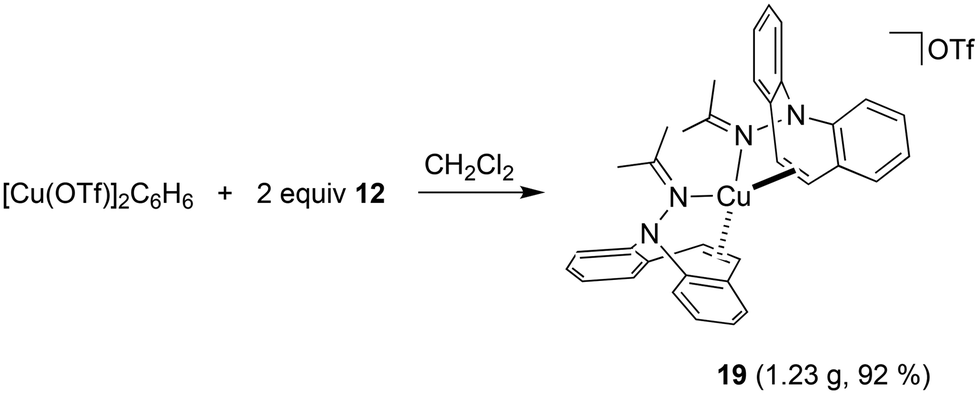
Hydrazone 12 cleanly reacts52 with [Cu(OTf)]2·C6H6 in CH2Cl2 solution to complex 19, which is isolated as gold-yellow powder in excellent yields (eqn (3)). The NMR spectra reflect a highly symmetric complex, and the low frequency shift of the singlet resonance of the alkene protons at 6.54 ppm by 0.5 ppm compared to the free ligand indicates alkene coordination. In fact, X-ray analysis authenticated the bidentate coordination of the ligands through the imine nitrogen and the azepine CC double bond (Fig. 6). The copper atom has tetrahedral coordination geometry with an outer-sphere OTf− counterion. While the Cu–N distances are in the expected range for this type of structures,53 the Cu–C bonds are slightly longer than in most reported structures at 2.194–2.288 Å.54 The dibenzo[b,f]azepine nitrogen atoms are pyramidalized (∑∢(N1) = 333° and ∑∢(N2) = 335°) due to the bidentate coordination mode of the ligand, which is also a recurring observation in complexes of bidentate dibenzoazepine-based chiral P- and S-alkene ligands.55 The average C–C double bond is similar to the values found in complex 18 and is slightly elongated to 1.368 Å compared with the uncoordinated alkene in the aza-enolate 13 (1.327 Å). This is consistent with Cu(I) alkene coordination involving mostly σ-donation and very little π-back donation.56 However, the coordinated alkene functions in complexes 18 and 19 resist substitution by PPh3, CO, CO2, phenyl isocyanate, or dicyclohexylcarbodiimide.
| | 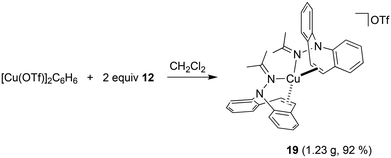 | (3) |
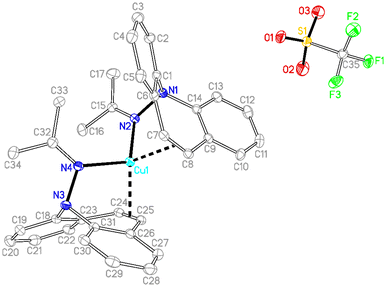 |
| | Fig. 6 Crystal structure of 19 (50% probability ORTEP, H atoms are omitted). Selected bond lengths (Å) and angles (deg): Cu1–N2 2.0460(18), Cu1–N4 2.0823(19), Cu1–C7 2.223(2), C7–C8 1.370(3), C24–C25 1.366(3), Cu1–C8 2.280(2), Cu1–C24 2.268(2), Cu1–C25 2.194(2), N1–N2 1.470(3), N3–N4 1.471(3), N4–C32 1.286(3), N2–C15 1.285(3), N2–Cu1–N4 113.89(7), N2–Cu1–C7 92.26(8), N4–Cu1–C24 90.33(8), C32–N4–Cu1 135.03(16), C15–N2–Cu1 127.37(16). | |
Conclusion
The new hydrazine 2 is accessible in multi-gram scales and is a useful building block for the synthesis of N- and P-alkene hybrid ligands and iminophosphines 5 and 14, the latter being accessible by a Cu(I) mediated reaction. The neutral and anionic hydrazone derived N-alkene ligands are readily prepared on gram-scales and show a versatile coordination behaviour. While 8 was not suitable for Cu(I) complexes, a biscoordinated Cu(II) complex was isolated. α-keto hydrazone 11 and the N,N-dimethyl hydrazone 12 form the alkene coordinated Cu(I) complexes 18 and 19, which sport unexpectedly strong alkene interactions that resist PPh3 and CO substitution. Moreover, we think that the tricyclic hydrazine 2 is of interest to pharmaceutical/medicinal chemistry and developments along these lines are currently being pursued in our laboratories.
Experimental details
All reactions were carried out under anaerobic and anhydrous conditions, using standard Schlenk and glovebox techniques unless otherwise stated. Et2O, THF, benzene, n-hexane and n-pentane were distilled from purple Na/benzophenone solutions, toluene from Na, C6D6 from Na/K alloy, CH2Cl2 and 1,2-difluorobenzene from CaH2, and NEt3 from K. CDCl3 and CD2Cl2 were degassed with three freeze–pump–thaw cycles and then kept over activated molecular sieves (4 Å) in the glovebox. LiAlH4 was purified by filtration of a concentrated Et2O solution over glass fiber filter (Whatman GF/B) and subsequent evaporation to a snow-white solid. [CuMes]4,57 [Cu(NCCH3)4]PF658 and [Cu(OTf)]2·C6H659 were prepared according to published procedures. Technical solvents were pre-purified by distillation on a rotary evaporator. NMR spectra were recorded on a JOEL EX 400 (400.5 MHz), JOEL EX 270 (270 MHz), Bruker Avance DPX300 NB (300 MHz) and Bruker Avance III HD (600 MHz). Chemical shifts δ are indicated in ppm and 1H- and 13C NMR shifts refer to the solvent signal as an internal standard (CDCl3, δ = 7.25; C6D6, δ = 7.16).60 Signal multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), oct (octet) or m (multiplet). The raw data were processed with Mestrec. The elemental analyses (EAs) of new products were determined with a Euro EA 3000 (Euro Vector).
5-Nitroso-5H-dibenzo[b,f]azepine (1)
Dibenzo[b,f]azepine (77.7 g, 402 mmol) was suspended in EtOH (800 ml) and cooled to 0 °C before conc. HCl (280 ml) was added. After 15 min a solution of NaNO2 (30.5 g, 442 mmol) in H2O (250 ml) was added dropwise via addition funnel over 40 min. The reaction mixture turned brown before a brown precipitate formed. The suspension was stirred for 1 h at 0 °C and filtered with a frit. The solid was slurried in EtOH (300 ml), filtered and washed with petrol ether (200 ml). The crude was extracted with DCM (400 ml) and filtered over activated charcoal to remove remaining inorganic salts. The brownish solution was concentrated to 150 ml and n-pentane was added to promote crystallization. After cooling overnight at −30 °C, the mother liquor was removed, and the yellowish crystals were dried in HV. The product was recrystallized from boiling toluene (400 ml) to obtain slightly yellowish crystals. The solvent was decanted, and the crystals were washed with n-pentane (2 × 50 ml) before drying in HV (60.4 g, 68%). 1H NMR: (600 MHz, CDCl3) δ (ppm) = 7.62 (d, 1H, ArH, 3JH,H = 6 Hz), 7.56–7.53 (m, 1H, ArH), 7.49–7.38 (m, 5H, ArH), 7.15 (d, 1H, ArH, 3JH,H = 6 Hz), 6.89; 6.84 (dd, 2H, ArH, 4JH,H = 12 Hz, 3JH,H = 30 Hz). 13C NMR (101 MHz, CDCl3) δ (ppm) = 140.83 (s), 136.25 (s), 132.81 (s), 132.16 (s), 130.49 (s), 130.43 (s), 130.17 (s), 129.85 (s), 129.61 (s), 129.23 (s), 129.16 (s), 128.94 (s), 127.61 (s), 126.09 (s). EA found: C 75.39%, H 4.46%, N 12.47%; calculated for C14H10N2O: C 75.66%, H 4.54%, N 12.60%.
5H-Dibenzo[b,f]azepin-5-amine (2)
1 (15.4 g, 68.4 mmol) was dissolved in dry THF (250 ml) and cooled to 15 °C. A solution of LiAlH4 (2.72 g, 71.8 mmol) in THF (30 ml) was added dropwise, so that the gas formed at a controlled rate. The resulting dark greenish solution was stirred for 1 h at 15 °C. The cooling bath was removed, and stirring was continued for 15 h at RT. The reaction mixture was quenched with aqueous sodium tartrate solution (20 g of C4H4Na2O6·2H2O in 180 ml of H2O) and Et2O (300 ml) was added. The phases were separated, and the aqueous phase was extracted with Et2O (3 × 200 ml). The combined organic phases were dried over MgSO4, and the solvent was removed. The product was purified by column chromatography with deactivated silica (Hex/EtOAc/NEt3, 8:1:1) and obtained as a yellow crystalline solid (7.04 g, 50%).1H NMR: (600.13 MHz, CDCl3) δ 7.33–7.23 (m, 4H, ArH), 7.09–7.01 (m, 4H, ArH), 6.77 (s, 2H, ArH), 4.29 (s (br), 2H, NH2). 13C NMR: (150.9 MHz, CDCl3) δ 152.7 (s), 132.4 (s), 130.9 (s), 129.2 (s), 129.1 (s), 123.7 (s). EA found: C 81.15%, H 5.81%, N 13.07%; calculated for C14H12N2: C 80.74%, H 5.81%, N 13.45%.
N-(Diphenylphosphanyl)-5H-dibenzo[b,f]azepin-5-amine (3)
A solution of LDA (249 mg, 2.33 mmol) in THF (3 ml) was added dropwise to a solution of 2 (530 mg, 2.33 mmol) in THF (5 ml). While stirring for 1 h, the reaction mixture turned red and an orange precipitate formed. The reaction mixture was cooled to −30 °C and was added dropwise to a solution of PPh2Cl (525 mg, 2.38 mmol) in THF (5 ml), which was cooled to −30 °C. The formed yellow solution was stirred for additional 2 h. The solvent was removed, the residue was extracted with benzene (15 ml) and filtered over cotton and Celite in a pipette. The yellow solution was cooled to −30 °C and subsequently the frozen benzene was sublimed in HV. Afterwards the product was slurried in n-pentane (15 ml) for 1 h, cooled to −30 °C and filtered cold. The precipitate was washed with n-pentane (6 ml), filtered again and dried in HV to afford a yellow powder (811 mg, 88%). 1H NMR: (400 MHz, C6D6) δ 7.45–7.41 (m, 4H, ArH), 6.93–6.87 (m, 8H, ArH), 6.73–6.65 (m, 6H, ArH), 6.33 (s, 2H, ArH), 4.95 (d, 1H, NH, 2JH,P = 32 Hz) 13C NMR: (101 MHz, C6D6) δ 152.8 (s), 139.6 (s), 139.5 (s), 132.8 (s), 132.6 (s), 132.3 (s), 131.0 (s), 128.74 (s), 128.4 (s), 128.5 (s), 127.7 (s), 123.4 (s), 119.2 (s). 31P NMR: (162 MHz, C6D6) δ 49.1 (s). EA found: C 79.88%, H 5.38%, N 6.84%; calculated for C26H21N2P: C 79.58%, H 5.39%, N 7.14%.
Dibromotriphenyl-λ5-phosphine (PBr2Ph3)61
Triphenylphosphine (3.00 g, 11.4 mmol) was suspended in acetonitrile (15 ml) and the mixture was cooled down to 0 °C. Bromine (1.82 g; 11.4 mmol; 0.58 ml) was added dropwise via syringe and stirring was continued for 1 h. To remove the excess of bromine, a solution of triphenyl phosphine (0.60 g diluted in 10 ml acetonitrile) was added. The white precipitate was separated by filtration, washed with acetonitrile (15 ml) and dried under reduced pressure to yield an off-white powder (4.67 g, 97%). 1H NMR (600 MHz, DMSO-d6) δ 7.64–7.57 (m, 9H, ArH), 7.57–7.51 (m, 6H, ArH). 31P NMR: (243 MHz; DMSO-d6) δ 25.5 (s).
((5H-Dibenzo[b,f]azepin-5-yl)amino)triphenyl-phosphonium bromide (4)62
PBr2Ph3 (1.03 g, 2.43 mmol) was dissolved in benzene (7 ml) and a solution of 2 (505 mg, 2.43 mmol) and NEt3 (369 mg, 3.65 mmol) in benzene (7 ml) was added slowly. The yellow reaction mixture was stirred for 20 hours, then the solvent was removed by sublimation. The air stable light-yellow solid was washed with water (50 ml), Et2O (40 ml) and dried under reduced pressure to obtain an off-white powder (1.14 g, 85%) 1H NMR (400 MHz, CDCl3) δ 11.38 (d, J = 41.6 Hz, 1H, NH), 8.47 (d, J = 8.2 Hz, 2H), 7.69 (ddd, J = 13.1, 8.2, 1.4 Hz, 6H), 7.61 (td, J = 7.5, 1.6 Hz, 3H), 7.42 (td, J = 7.8, 3.5 Hz, 6H), 7.31 (ddd, J = 8.4, 7.3, 1.7 Hz, 2H), 6.93 (td, J = 7.5, 1.0 Hz, 2H), 6.84 (dd, J = 7.6, 1.6 Hz, 2H), 6.46 (s, 2H). 31P NMR (162 MHz, CDCl3) δ 39.06 (d, J = 38.0 Hz). 13C NMR (101 MHz, CDCl3) δ 149.54 (d, J = 2.5 Hz), 134.77 (d, J = 2.9 Hz), 134.33 (s), 134.2 (s), 131.9 (s), 130.2 (s), 130.0 (s), 129.4 (s), 129.2 (s), 128.1 (s), 124.7 (s), 122.5 (s), 120.2 (s), 119.14 (s). EA found: C 70.05%, H 4.67%, N 5.02%; calculated for C32H26N2PBr: C 69.95%, H 4.77%, N 5.10%.
N-(5H-Dibenzo[b,f]azepin-5-yl)-1,1,1-triphenyl-λ5-phosphanimine (5)
4 (263 mg, 0.476 mmol) and LDA (51.0 mg, 0.476 mmol) were placed in a vial and suspended in benzene (10 ml). The initial colorless suspension turned immediately yellow and stirring was continued for 2.5 h. Afterwards the reaction mixture was centrifuged at 6000 rpm for 6 minutes to separate the LiBr from the mother liquor. The benzene was removed by sublimation and the remaining solid was dried under reduced pressure. The product is obtained as a yellow solid (215 mg, 96%). 1H NMR (600 MHz, C6D6) δ 8.81 (dd, J = 8.2, 1.2 Hz, 2H), 8.04–7.95 (m, 6H), 7.43–7.32 (m, 3H), 7.23–7.18 (m, 3H), 7.15 (d, J = 1.4 Hz, 6H), 7.00 (dd, J = 7.5, 1.7 Hz, 2H), 6.96 (td, J = 7.3, 1.2 Hz, 2H), 6.73 (s, 2H). 31P-NMR (243 MHz, C6D6) δ 17.30 (s). 13C NMR (151 MHz, C6D6) δ 157.13 (s), 133.17 (d, J = 8.6 Hz), 132.73 (s), 131.11 (s), 130.82 (s), 130.62 (s), 130.22 (s), 128.43 (s), 128.00 (s), 127.82 (s), 127.60 (s), 121.94 (s), 121.72(s). EA found: C 81.68%, H 5.63%, N 5.82%; calculated for C32H25N2P: C 82.03%, H 5.38%, N 5.98%.
N-(Trimethylsilyl)-5H-dibenzo[b,f]azepin-5-amine (6)
A solution of LDA (0.625 g, 5.84 mmol) in THF (6 ml) was added dropwise to a solution of 2 (1.20 g, 5.74 mmol) in THF (10 ml). While stirring for 2 h, the reaction mixture turned red and an orange precipitate formed. A solution of TMSCl (0.750 g, 6.87 mmol, 0.88 ml, 1.2 eq.) in THF (4 ml) was added dropwise and the yellow solution was stirred for an additional 1 h. The solvent was removed, and the residue was extracted with benzene (10 ml) and centrifuged for 5 min at 5300 rpm. The decanted solution was cooled to −30 °C and subsequently the frozen benzene was sublimed in HV to afford a yellow powder (1.59 g, 99%). 1H NMR: (270 MHz, C6D6) δ 7.07–7.00 (m, 2H, ArH), 6.88–6.85 (m, 2H, ArH), 6.76–6.74 (m, 4H, ArH), 6.49 (s, 2H, ArH), 3.75 (s, 1H, NH), 0.05 (s, 9H, Si(CH3)3). 13C NMR: (68 MHz, CDCl3) δ 154.4 (s), 132.5 (s), 130.9 (s), 128.9 (s), 128.6 (s), 123.1(s), 118.8 (s), −0.99 (s). EA found: C 72.65%, H 7.08%, N 9.50%; calculated for C17H20N2Si: C 72.81%, H 7.19%, N 9.99%.
Tris-(lithium{N-(trimethylsilyl)-5H-dibenzo[b,f]azepin-5-amide}) (7)
PhLi (309 mg, 3.63 mmol) was suspended in toluene (13 ml) and added dropwise to a solution of 6 (1.01 g, 3.60 mmol) in toluene (3 ml). The red solution was stirred for 4 h at RT. Subsequently the solvent was removed to dryness to give a red foam and the crude was slurried in n-pentane (12 ml) for 6 h. The orange precipitate (933 mg, 87%) was filtered and dried in HV. 1H NMR: (400 MHz, C6D6) δ 7.46 (m, 2H, ArH), 7.12 (m, 2H, ArH), 6.85 (m, 4H, ArH), 6.51 (m, 2H, ArH), 0.12 (br. s, 9H, Si(CH3)3). 13C NMR: (101 MHz, C6D6) δ 155.9 (s), 133.0 (s), 131.9 (s), 129.79, (s), 124.8 (s), 123.7(s), 2.43. EA found: C 70.89%, H 6.73%, N 9.33%; calculated for C17H19N2SiLi: C 71.30%, H 6.69%, N 9.78%.
N,N-Bis(trimethylsilyl)-5H-dibenzo[b,f]azepin-5-amine (8)
A solution of TMSCl (38.2 mg, 0.352 mmol) in THF (2 ml) was added dropwise to a solution of 7 (103 mg, 0.352 mmol) in THF (1.5 ml). The resulting red solution was stirred for 45 min at RT. Afterwards the solvent was removed to dryness and the crude was extracted with benzene (5 ml) and filtered over cotton and Celite in a pipette. The solution was cooled to −30 °C and the frozen benzene was sublimed in HV to afford the product as a yellow solid (125 mg, 100%). 1H NMR: (400 MHz, C6D6) δ 7.09 (dd, 2H, ArH, 4JH,H = 1 Hz, 3JH,H = 8 Hz), 6.95–6.91 (m, 2H, ArH), 6.65 (dt, 2H, ArH, 4JH,H = 1 Hz, 3JH,H = 8 Hz), 6.58 (dd, 2H, ArH, 4JH,H = 1 Hz, 3JH,H = 8 Hz), 6.05 (s, 2H, ArH), 0.31 (s, 18H, Si(CH3)3). 13C NMR: (68 MHz, C6D6) δ 154.7, 133.8, 130.9, 130.8, 127.8, 122.6, 121.6, 2.25. EA found: C 67.97%, H 7.89%, N 7.51%; calculated for C20H28N2Si2: C 68.12%, H 8.00%, N 7.94%.
(2Z,4E)-4-((5H-Dibenzo[b,f]azepin-5-yl)imino)pent-2-en-2-ol (9)
2 (502 mg, 2.41 mmol) was dissolved in EtOH (6 ml) and acetyl acetonate (0.250 ml, 2.41 mmol) was added. The yellow reaction solution was stirred for 10 d at RT and reaction progress was monitored by TLC. The solvent was removed and the remaining solid was dissolved in n-pentane. The solution was cooled to −30 °C in the fridge overnight. The formed crystals were filtered and dried to obtain the product as yellow crystals (576 mg, 80%). 1H NMR: (400.13 MHz, CDCl3) δ 13.1 (br. s, 1H, OH), 7.32–7.02 (m, 8H, ArH), 6.75 (s, 2H, ArH), 5.06 (s, 1H, CH), 2.11 (s, 3H, CH3) 1.95 (s, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 196.88(s), 163.90 (s), 149.84 (s), 132.19 (s), 130.57 (s), 129.49 (s), 129.41 (s), 124.61 (s), 118.91 (s), 96.16 (s), 29.11 (s), 17.96 (s). EA found: C 78.78%, H 6.32%, N 9.54%; calculated for C17H18N2O: C 78.59%, H 6.25%, N 9.65%.
(1E,2E)-N1,N2-Bis(5H-dibenzo[b,f]azepin-5-yl)ethane-1,2-diimine (10)
2 (2.00 g, 9.60 mmol) was dissolved in EtOH (50 ml) and a glyoxal solution (0.55 ml, 4.80 mmol, 40 wt% in H2O) was added via syringe. After 10 min a yellow precipitate formed, and the reaction mixture was stirred for additional 15 h. The solvent was removed and the solid was slurried in hexane (20 ml). The product was filtered, washed with cold hexane (3 × 5 ml) and dried in HV to obtain a yellow powder (1.83 g, 87%). 1H NMR: (600.13 MHz, CDCl3) δ 7.27–7.23 (m, 9H, ArH), 7.17–7.15 (m, 5H, ArH), 7.11–7.08 (m, 4H, ArH), 6.69 (s, 4H, ArH). 13C NMR: (150.9 MHz, CDCl3) δ 144.6 (s), 135.7 (s), 134.3 (s), 130.9 (s), 130.0 (s), 129.6 (s), 127.0 (s), 126.8 (s). EA found: C 82.51%, H 5.09%, N 12.84%; calculated for C30H22N4: C 82.17%, H 5.06%, N 12.78%.
(E)-2-((5H-Dibenzo[b,f]azepin-5-yl)imino)acenaphthylen-1(2H)-one (11)
Crystalline acenaphthylene-1,2-dione (1.03 g, 5.73 mmol) and 2 (1.22 g, 5.84 mmol) were placed in a flask with MeOH (40 ml). The suspension turned orange, before an orange precipitate formed. Stirring was continued for 15 h, the solvent was removed, and the crude was washed with n-hexane (2 × 20 ml). After drying in HV the product was obtained as an orange powder (1.86 g, 87%) 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.06 (d, J = 7.0 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.77–7.54 (m, 4H), 7.38–7.21 (m, 5H), 7.17 (td, J = 7.4, 1.1 Hz, 3H), 7.04 (s, 2H), 6.10 (d, J = 7.3 Hz, 1H) 13C NMR (101 MHz, CDCl3) δ 189.96 (s), 149.00 (s), 141.22 (s), 140.09 (s), 131.86 (s), 131.59 (s), 131.52 (s), 131.38 (s), 130.41 (s), 129.80 (s), 129.38 (s), 127.93 (s), 127.89 (s), 127.64 (s), 126.70 (s), 126.02 (s), 123.72 (s), 123.07 (s), 121.71 (s). EA found: C 83.65%, H 4.17%, N 7.32%; calculated for C26H16N2O C 83.85%, H 4.33%, N 7.52%.
N-(5H-Dibenzo[b,f]azepin-5-yl)propan-2-imine (12)
2 (1.04 g, 5.00 mmol) was dissolved in MeOH (20 ml) and acetone (0.400 ml, 5.50 mmol,) was added via syringe. The reaction mixture was stirred for 3 d, during which time a yellow precipitate formed. Reaction progress was monitored by TLC and after complete conversion the solvent was removed and the crude washed with n-hexane (5 ml) and dried in HV to a bright yellow powder (1.11 g, 89%) 1H NMR (600 MHz, CDCl3) δ (ppm) = 7.75 (dd, J = 8.3, 1.2 Hz, 2H), 7.51 (ddd, J = 8.4, 7.2, 1.6 Hz, 2H), 7.36 (dd, J = 7.7, 1.6 Hz, 2H), 7.27–7.21 (m, 1H), 7.09 (s, 2H), 2.39 (s, 3H), 2.00 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 166.16 (s), 152.46 (s), 132.26 (s), 130.71 (s), 129.08 (s), 128.88 (s), 123.27 (s), 119.85 (s), 25.22 (s), 19.91 (s). EA found: C 82.40%, H 6.43%, N 11.35%; calculated for C17H16N2: C 82.22%, H 6.49%, N 11.28%.
Lithium N-(5H-dibenzo[b,f]azepin-5-yl)propene-2-amide tris-tetrahydrofuranate (13)
12 (151 mg, 0.606 mmol) and LDA (68.2 mg, 0.636 mmol) were placed in a vial and THF (5 ml) was added. The deep red reaction mixture was stirred for 15 h, layered with n-hexane (6 ml), and cooled to −30 °C for crystallization. The mother liquor was decanted off and the remaining deep red crystals washed with n-hexane (4 ml) and dried in HV (153 mg, 60%). Despite measuring crystalline 13, NMR spectra always showed traces of 12 as impurity, which we attribute to partial hydrolysis of the product by adventitious water. 1H NMR (400 MHz, THF-d8) δ 6.93 (d, J = 8.6 Hz, 2H), 6.45 (t, J = 8.8 Hz, 2H), 6.6 (d, J = 7.2 H, 2H), 5.97 (d, J = 7.2, 2H), 5.20 (s, 2H), 3.64–3.51 (m, 4H, THF), 2.96 (s, 1H), 2.75 (s, 1H), 1.83–1.70 (m, 4H, THF), 1.52 (s, 3H). EA found: C 74.50%, H 7.75%, N 7.00%, calculated for C17H15N2Li·2.35 C4H8O: C 74.84%, H 8.04%, N 6.61%.
N,1-Bis(5H-dibenzo[b,f]azepin-5-yl)-1,1-diphenyl-λ5-phosphine-imine (14)
To a suspension of [MesCu]4 (101 mg, 0.423 mmol) in pyridine (5 ml), which was cooled to −30 °C, a cold solution (−30 °C) of 3 (168 mg, 0.422 mmol) in pyridine (2 ml) was added dropwise. The yellow solution turned red brown within 30 min. After stirring overnight at RT, the solvent was removed to dryness and the crude was slurried in n-pentane (8 ml) for 2.5 h. The precipitate was filtered, washed with n-pentane (5 ml) and dried in HV to afford the product as a light brown solid (172 mg, 61% with respect to 3). 1H NMR: (600 MHz, C6D6) δ 8.64 (d, 2H, ArH, 3JH,H = 12 Hz), 8.16–8.11 (m, 6H, ArH), 7.28–7.25 (m, 3H, ArH), 6.97–6.68 (m, 19H, ArH), 31P NMR: (243 MHz, C6D6) δ 16.9 (s).13C NMR: (68 MHz, C6D6) δ 156.9 (d, 3JC,P = 9 Hz), 144.0 (d, 2JC,P = 5 Hz), 135.1 (s), 135.1 (s), 133.7 (s), 133.5 (s), 132.7 (s), 131.6 (s), 130.9 (s), 130.8 (s), 130.6 (t, 1JC,P = 1 Hz), 129.0 (s), 128.9 (s), 128.8 (s), 128.0 (s), 127.6 (s), 127.6 (s), 125.7 (s), 122.3 (s), 121.8 (s).
[Cu((2Z,4E)-4-((5H-Dibenzo[b,f]azepin-5-yl)imino)pentenoate)2] (15)
9 (111 mg, 0.381 mmol) and mesityl copper (91 mg, 0.38 mmol) were placed in a vial and toluene (5 ml) was added. The initial yellow reaction solution turned slowly from orange to deep brown. After stirring for 15 h the reaction solution was filtered over cotton and Celite to remove metallic Cu. The filtrate concentrated to half of the initial volume and layered with n-hexane (6 ml) at −30 °C. The complex was obtained as red brownish crystals (71 mg, 0.11 mmol, 58%) The paramagnetic complex is NMR silent. EA: found C 71.14%, H 5.37%, N 8.54%; calculated for C38H34N4O2Cu: C 71.06%, H 5.34%, N 8.72%.
[Cu(10)2]PF6 (16)
A solution of 10 (150 mg, 0.343 mmol) in DCM (4 ml) was added dropwise to a suspension of [Cu(CNCH3)4]PF6 (128 mg, 0.343 mmol) in DCM (2 ml). An orange precipitate formed, and stirring was continued for 3 d. The solvent volume was reduced to a third and n-hexane (6 ml) was added to complete precipitation. The reaction mixture was cooled to −30 °C, filtered and washed with cold n-hexane (2 × 2 ml). After drying in HV the product was obtained as an orange powder (213 mg, 85%). 1H NMR: (400 MHz, CD2Cl2) δ 7.48–7.32 (m, 25H, ArH), 6.97 (s, 8H, ArH), 6.97–6.88 (m, 7H, ArH), 6.32 (s, 4H, ArH). 13C NMR: (101 MHz, CD2Cl2) δ 134.6 (s), 130.7 (s), 130.4 (s), 129.9 (s), 128.4 (s), 128.1 (s), 127.8 (s). EA found: C 64.74%, H 3.89%, N 10.22%; calculated for C60H44CuN8PF6: C 64.80%, H 4.03%, N 10.01%.
[FeCl2(10)] (17)
10 (300 mg, 0.684 mmol) and FeCl2·(THF)1.5 (161 mg, 0.684 mmol) were placed in a vial, suspended in 1,2-difluorobenzene (5 ml) and stirred overnight, while the suspension changed color from yellow to dark brown. For crystallization, the reaction vial was warmed with a heat gun until a clear solution was obtained and crystallization was completed by cooling down to −30 °C overnight. The liquid was decanted, and the crystals were washed with n-hexane (2 ml). The product was dried under vacuum and dark reddish crystals were obtained (374 mg, 97%). 1H NMR (400 MHz, C6D6) δ 4.95 (br. s), 3.71 (br. s) EA found: C 64.02%; H, 3.89%; N, 9.76%; calculated for C36H28Cl2FeN4: C 63.74%; H 3.92%; N 9.91%.
[Cu(OTf)(11)]2 (18)
A suspension of 11 (500 mg, 1.34 mmol) in toluene (10 ml) was added dropwise to [Cu(OTf)]2·C6H6 (338 mg, 0.671 mmol) suspended in toluene (4 ml). Immediately an orange-brown precipitate formed, and the reaction mixture was stirred for 15 h. The solvent was removed, and the precipitate was washed with toluene (6 ml) and n-pentane (2 × 5 ml). After drying in HV, the complex was obtained as an orange powder (705 mg, 86%). 1H NMR (400 MHz, CD2Cl2) δ 8.29 (d, J = 8.0 Hz, 2H), 7.85–7.72 (m, 4H), 7.58 (d, J = 8.0 Hz, 2H), 7.43–7.24 (m, 8H), 7.18 (dd, J = 11.1, 9.0 Hz, 4H), 7.07 (t, J = 7.8 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 6.80 (d, J = 7.8 Hz, 2H), 6.72 (t, J = 7.9 Hz, 2H), 6.25 (d, J = 9.8 Hz, 2H), 4.74 (d, J = 7.5 Hz, 2H). 19F NMR (400 MHz, CD2Cl2) δ −78.37 (s). 13C NMR (101 MHz, CD2Cl2) δ (ppm) = 194.25 (s), 146.25 (s), 141.28 (s), 139.86 (s), 138.46 (s), 133.29 (s), 133.24 (s), 131.38 (s), 130.24 (s), 130.07 (s), 129.86 (s), 128.91 (s), 128.87(s), 128.53 (s), 128.41 (s), 128.18 (s), 127.57 (s), 127.12 (s), 126.59 (s), 125.49 (s), 125.10 (s), 124.04 (s), 123.32 (s), 104.25 (s), 102.06 (s). EA found: C 54.00%, H 2.95%, N 4.57% S 5.39%; calculated for C27H16N2O4SF3Cu·0.25 CH2Cl2: C 53.99%, H 2.74%, N 4.62%, S 5.29%.
[Cu(12)2]OTf (19)
A solution of 12 (945 mg, 3.81 mmol) in CH2Cl2 (15 ml) was added to a stirred slurry of [Cu(OTf)]2·C6H6 (485 mg, 0.950 mmol) in CH2Cl2 (5 ml). After stirring the orange solution for 2 h at room temperature, the reaction solution was filtered over cotton and Celite. The dark yellow filtrate was concentrated to half of its volume and the product was precipitated by adding n-hexane (15 ml). The solid was filtered, washed with n-hexane (2 × 6 ml), and dried in HV to afford a yellow powder (1.23 g, 91%). 1H NMR (400 MHz, CDCl3) δ 7.55 (dd, J = 8.3, 1.4 Hz, 2H), 7.47 (m, 4H), 7.36 (td, J = 7.3, 1.4 Hz, 2H), 6.58 (s, 2H), 2.47 (s, 3H), 1.62 (s, 3H).13C NMR (151 MHz, CDCl3) δ 145.34 (s), 133.49 (s), 131.09 (s), 129.6 (s), 129.00 (s), 128.32 (s), 119.04 (s), 26.95 (s), 21.45 (s). EA found: C 59.14%, H 4.56%, N 7.93%, S 4.31%; calculated for C35H32N4CuSO3F3: C 59.27%, H 4.55%, N 7.90%, S 4.52%. Diffraction quality single crystals were grown by layering a CH2Cl2 solution with n-pentane.
Single crystal X-ray structure determinations
Single crystal X-ray structure determinations were performed on a Bruker Kappa IμS Duo diffractometer using MoKα radiation (λ = 0.71073 Å) and QUAZAR focusing Montel optics (for 7, 13, 14, and 19), a Bruker Smart diffractometer using MoKα radiation (λ = 0.71073 Å, curved graphite monochromator) for 10, an Agilent SuperNova dual radiation diffractometer with microfocus X-ray sources and mirror optics using MoKα radiation (λ = 0.71073 Å for 11 and 18) or CuKα radiation (λ = 1.54184 Å for 15 and 17). All intensity data sets were collected at a temperature of 100 K. The structures were solved with SHELXT63 and were refined on F2 with SHELXL 2018/3.64 Further details are summarized in the ESI.†
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgements
We thank Mses Christina Wronna and Antigone Roth for carrying out the elemental analyses. Generous financial support for AG by Siemens Energy within the framework of the Energy Campus Erlangen-Nuremberg are gratefully acknowledged.
References
- R. Lazny and A. Nodzewska,
N,N-Dialkylhydrazones in Organic Synthesis. From Simple N,N-Dimethylhydrazones to Supported Chiral Auxiliaries, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed
 .
.
-
(a) D. Enders and E. J. Corey, Herstellung Und Synthetische Verwendung von Metallierten Dimethylhydrazonen Regio- Und Stereoselektive Alkylierung von Carbonylverbindungen, Chem. Ber., 1978, 111, 1337–1361 CrossRef ;
(b) J. S. M. Lehn, S. Javed and D. M. Hoffman, Synthesis of Zirconium, Hafnium, and Tantalum Complexes with Sterically Demanding Hydrazide Ligands, Inorg. Chem., 2007, 46, 993–1000 CrossRef CAS PubMed .
-
(a) R. H. Shapiro and M. J. Heath, Tosylhydrazones. V. Reaction of Tosylhydrazones with Alkyllithium Reagents. A New Olefin Synthesis, J. Am. Chem. Soc., 1967, 89, 5735 Search PubMed . For a catalytic example, see:
(b) K. Maruoka, M. Oishi and H. Yamamoto, The Catalytic Shapiro Reaction, J. Am. Chem. Soc., 1996, 118, 2289–2290 CrossRef CAS .
-
(a) A. Bermejo, A. Ros, R. Fernández and J. M. Lassaletta,
C
2-Symmetric Bis-Hydrazones as Ligands in the Asymmetric Suzuki-Miyaura Cross-Coupling, J. Am. Chem. Soc., 2008, 130, 15798–15799 CrossRef CAS PubMed ;
(b) B. Sedai, M. J. Heeg and C. H. Winter, Volatility Enhancement in Calcium, Strontium, and Barium Complexes Containing β-Diketiminate Ligands with Dimethylamino Groups on the Ligand Core Nitrogen Atoms, Organometallics, 2009, 28, 1032–1038 CrossRef CAS and ref. 2b;
(c) D. F.-J. Piesik, R. Stadler, S. Range and S. Harder, Binuclear Magnesium, Calcium, and Zinc Complexes Based on Nitrogen–Nitrogen-Coupled Salicylaldiminate and β-Diketiminate Ligands, Eur. J. Inorg. Chem., 2009, 3569–3576 CrossRef CAS ;
(d) J. Intemann, J. Spielmann, P. Sirsch and S. Harder, Well-Defined Molecular Magnesium Hydride Clusters: Relationship between Size and Hydrogen-Elimination Temperature, Chem. – Eur. J., 2013, 19, 8478–8489 CrossRef CAS PubMed ;
(e) J. Intemann, P. Sirsch and S. Harder, Comparison of Hydrogen Elimination from Molecular Zinc and Magnesium Hydride Clusters, Chem. – Eur. J., 2014, 20, 11204–11213 CrossRef CAS PubMed . For the use of monodentate hydrazide ligands, see for example:
(f) J. S. M. Lehn, S. Javed and D. M. Hoffman, Synthesis of Zirconium, Hafnium, and Tantalum Complexes with Sterically Demanding Hydrazide Ligands, Inorg. Chem., 2007, 46, 993–1000 CrossRef CAS PubMed . For applications in catalysis, see for example:
(g) T. Mino, Y. Shirae, Y. Sasai, M. Sakamoto and T. Fujita, Phosphine-Free Palladium Catalyzed Mizoroki-Heck Reaction Using Hydrazone as a Ligand, J. Org. Chem., 2006, 71, 6834–6839 CrossRef CAS PubMed ;
(h) T. Mino, M. Shibuya, S. Suzuki, K. Hirai, M. Sakamoto and T. Fujita, Palladium-Catalyzed Mizoroki-Heck Type Reaction with Aryl Trialkoxysilanes Using Hydrazone Ligands, Tetrahedron, 2012, 68, 429–432 CrossRef CAS .
- For historical examples of α-dihydrazone ligands, see:
(a) H. D. Hausen and K. Krogmann, Die Kristallstruktur von Dicarbonyl-diacetylbis(dimethylhydrazon)-Ni0), Z. Anorg. Allg. Chem., 1972, 389, 247–253 CrossRef CAS ;
(b) P. Ammendaola, M. R. Ciajolo, A. Panunzi and A. Tuzi, Trigonal bipyramidal platinum complexes as models for Ziegler-Natta catalytic sites, J. Organomet. Chem., 1983, 254, 389–397 CrossRef ;
(c) J. Ehlers and H. Tom Dieck, Modellkomplexe Mit Hydrazonchelaten von (S)- und (R)-1-Amino-2-Methoxymethylpyrrolidin (“SAMP” and “RAMP”) Und Dimethylhydrazine - Struktur Des (Pyridinaldehyd-RAMP-Hydrazon-Tetracarbonyl-Molybdäns, Z. Anorg. Allg. Chem., 1988, 560, 80–92 CrossRef CAS . For β-dihydrazone ligands, see:
(d) G. Bocelli, A. Cantoni and G. Tosi, Structure of Butanedione Bis(Methylphenylhydrazone), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1041–1043 CrossRef .
-
(a) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139 CrossRef CAS PubMed ;
(b) A. Briceño and R. Dorta, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1718–m1719 CrossRef ;
(c) R. Mariz, A. Briceno, R. Dorta and R. Dorta, Organometallics, 2008, 27, 6605 CrossRef CAS ;
(d) E. Drinkel, A. Briceno, R. Dorta and R. Dorta, Organometallics, 2010, 29, 2503 CrossRef CAS ;
(e) T. J. Hoffman and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 10670 CrossRef CAS PubMed ;
(f) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568 CrossRef CAS PubMed ;
(g) M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470 CrossRef CAS PubMed ;
(h) A. Herrera, A. Grasruck, F. W. Heinemann, A. Scheurer, A. Chelouan, S. Frieß, F. Seidel and R. Dorta, Organometallics, 2017, 36, 714 CrossRef CAS ;
(i) A. Chelouan, S. Bao, S. Frieß, A. Herrera, F. W. Heinemann, A. Escalona, A. Grasruck and R. Dorta, Organometallics, 2018, 37, 3983 CrossRef CAS ;
(j) L. Leinauer, G. Parla, J. Messelberger, A. Herrera, F. W. Heinemann, J. Langer, I. Chuchelkin, A. Grasruck, S. Frieß, A. Chelouan, K. Gavrilov and R. Dorta, Evolution of a ‘privileged’ P-alkene ligand: added planar chirality beats BINOL axial chirality in catalytic asymmetric C–C bond formation, Chem. Commun., 2023, 59, 13879–13882 RSC .
-
(a) A. Grasruck, G. Parla, F. W. Heineman, J. Langer, A. Herrera, S. Frieß, G. Schmid and R. Dorta, New J. Chem., 2023, 47, 2307 RSC ;
(b) A. Grasruck, G. Parla, L. Lou, F. W. Heineman, J. Langer, C. Christian Neiß, A. Herrera, S. Frieß, A. Görling, G. Schmid and R. Dorta, Trapping of soluble, KCl-stabilized Cu(I) hydrides with CO2 gives crystalline formates, Chem. Commun., 2023, 59, 13879–13882 RSC ;
(c) A. Grasruck, G. Parla, F. W. Heineman, J. Langer, A. Herrera, S. Frieß, G. Schmid and R. Dorta, Developing bulky P-alkene ligands: stabilization of copper complexes with 14 valence electrons, New J. Chem., 2023, 47, 2307–2323 RSC .
-
(a) F. Breher, C. Böhler, G. Frison, J. Harmer, L. Liesum, A. Schweiger and H. Grützmacher, TROPDAD: A New Ligand for the Synthesis of Water-Stable Paramagnetic [16+1]-Electron Rhodium and Iridium Complexes, Chem. – Eur. J., 2003, 9, 3859–3866 CrossRef CAS PubMed ;
(b) T. Büttner, F. Breher and H. Grützmacher, Amine olefin rhodium(I) complexes: pKa and NH bond strength, Chem. Commun., 2004, 2820–2821 RSC .
-
(a) D. Bogdal, Carbazolyl Nitrenium Cations: Generation and Reaction, Heterocycles, 2000, 53, 1–29 CrossRef ;
(b) A. H. Winter, H. H. Gibson and D. E. Falvey, Carbazolyl Nitrenium Ion: Electron Configuration and Antiaromaticity Assessed by Laser Flash Photolysis, Trapping Rate Constants, Product Analysis, and Computational Studies, J. Org. Chem., 2007, 72, 8186–8195 CrossRef CAS PubMed .
- I. A. Tonks, A. C. Durrell, H. B. Gray and J. E. Bercaw, Groups 5 and 6 Terminal Hydrazido(2-) Complexes: Nβ Substituent Effects on Ligand-to-Metal Charge-Transfer Energies and Oxidation States, J. Am. Chem. Soc., 2012, 134, 7301–7304 CrossRef CAS PubMed .
- R. P. Nielsen and H. H. Sisler, Hydrazinophosphorus Compounds. I. Preparation, of Some Substituted Hydrazinophosphines, Hydrazinophosphine Oxides, and Hydrazinophosphine Sulfides, Inorg. Chem., 1963, 2, 753–760 CrossRef CAS .
- PBr2Ph3 was not used in situ as usual for such reactions and was isolated in 97% yield (see experimental section). See also: J. P. Schaefer, J. G. Higgins and P. K. Shenoy, Cinnamyl Bromide, Org. Synth., 1968, 48, 51 CrossRef CAS .
- T. Bauer, S. Schulz, M. Nieger and U. Kessler, Bis(Alkylamino)Phosphanes: Deprotonation Reactions and Reactivity of t-BuP(NH-t-Bu)2 toward Group 13 Metalloorganics, Organometallics, 2003, 22, 3134–3142 CrossRef CAS .
-
(a) H. Zimmer and G. Singh, Reaktion von Triphenyl-Phosphiniminen Mit Nitrosylchlorid, Angew. Chem., Int. Ed. Engl., 1963, 75(12), 574–574 CrossRef CAS ;
(b) H. Zimmer, M. Jayawant and P. Gutsch, Synthesis of Secondary Amines via Triphenylphosphinimines, J. Org. Chem., 1970, 35, 2826–2828 CrossRef CAS .
-
(a) H. Zimmer and G. Singh, Synthesis and Some Reactions of Triphenylphosphinamino- and (β-N-Disubstituted Amino)Imines, J. Org. Chem., 1964, 29(6), 1579–1581 CrossRef CAS ;
(b) M. T. Reetz and J. Rheinheimer, Synthesis and Properties of Cyclopropanone Hydrazones, J. Org. Chem., 1986, 51, 5465–5467 CrossRef CAS .
-
(a) O. J. Scherer and W. Gläßel, 1-Tert.-Butyl-3-Trimethylsilyl-4,4-Dimethyl-2-Phospha-1-Tetrazen, Angew. Chem., Int. Ed. Engl., 1975, 87, 667–668 CrossRef CAS ;
(b) M. F. Lappert, M. J. Slade, A. Singh, J. L. Atwood, R. D. Rogers and R. Shakir, Structure and Reactivity of Sterically Hindered Lithium Amides and Their Diethyl Etherates: Crystal and Molecular Structures of [Li(N(SiMe3)2}(OEt2)]2 and Li(NCMe2CH2CH2CH2CMe2)]4, J. Am. Chem. Soc., 1983, 105, 302–304 CrossRef CAS .
-
(a) B. Gemünd, H. Nöth, H. Sachdev and M. Schmidt, Structural Chemistry of Alkali Metal Phenylhydrazides, Chem. Ber., 1996, 129, 1335–1344 CrossRef ;
(b) M. G. Voronkov, B. A. Gostevskij, B. A. Shajnyan, V. I. Rakhlin, R. G. Mirskov and O. S. Makarova, Reaction of Organosilicon Derivatives of 1,1-Dimethylhydrazine with Methyliodide, Dokl. Chem., 2005, 400, 17–20 CrossRef CAS ;
(c) C. Keck, C. Maichle-Mossmer and H. F. Bettinger, Photo Electron Transfer Induced Desilylation of: N,N-Bis(Trimethylsilyl)Aminodibenzoborole to Aminodibenzoborole, Chem. Commun., 2019, 55, 7470–7473 RSC .
- J. Ru Hwu and N. Wang, Counterattack Reagents:Hexamethyldisilane and 1,2-Dimethyl-1,1,2,2-Tetraphenyldisilane in the Synthesis of Polysilylated Hydrazines, Tetrahedron, 1988, 44, 4181–4196 CrossRef .
- D. R. Armstrong, K. W. Henderson, A. R. Kennedy, W. J. Kerr, F. S. Mair, J. H. Moir, P. H. Moran and R. Snaith, Structural Studies of the Chiral Lithium Amides [{PhC(H)Me}2NLi] and [PhCH2{PhC(H)Me}NLi·THF] Derived from α-Methylbenzylamine, J. Chem. Soc., Dalton Trans., 1999, 4063–4068 RSC .
- For a good discussion and definitions of agostic vs. anagostic interactions, see:
(a) M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed ;
(b) D. Braga, F. Grepioni, K. Biradha and G. R. Desiraju, Agostic Interactions in Organometallic Compounds. A Cambridge Structural Database Study, J. Chem. Soc., Dalton Trans., 1996, 3925–3930 RSC .
- A. Herrera, A. Grasruck, F. W. Heinemann, A. Scheurer, A. Chelouan, S. Frieß, F. Seidel and R. Dorta, Developing P-Stereogenic, Planar-Chiral P-Alkene Ligands: Monodentate, Bidentate, and Double Agostic Coordination Modes on Ru(II), Organometallics, 2017, 36, 714–720 CrossRef CAS .
-
(a) M. Stender, R. J. Wright, B. E. Eichler, J. Prust, M. M. Olmstead, H. W. Roesky and P. P. Power, The Synthesis and Structure of Lithium Derivatives of the Sterically Encumbered β-Diketiminate Ligand [{(2,6-Pri2H3C6)N(CH3)C}2CH]-, and a Modified Synthesis of the Aminoimine Precursor, J. Chem. Soc., Dalton Trans., 2001,(23), 3465–3469 RSC ;
(b) J. Feldman, S. J. McLain, A. Parthasarathy, W. J. Marshall, J. C. Calabrese and S. D. Arthur, Electrophilic Metal Precursors and a β-Diimine Ligand for Nickel(II)- and Palladium(II)-Catalyzed Ethylene Polymerization, Organometallics, 1997, 16, 1514–1516 CrossRef CAS .
-
(a) N. Kuhn and H. Lanfermann, Synthese von N-Tert-Butyl-4-(Tert-Butylimino)-2-Penten-2-Amin, Liebigs Ann. Chem., 1987, 6, 727–728 CrossRef ;
(b) D. F. J. Piesik, R. Stadler, S. Range and S. Harder, Binuclear Magnesium, Calcium, and Zinc Complexes Based on Nitrogen-Nitrogen-Coupled Salicylaldiminate and β-Diketiminate Ligands, Eur. J. Inorg. Chem., 2009,(24), 3569–3576 CrossRef CAS .
- J. Vicente, M. T. Chicote, A. J. Martínez-Martínez, D. Bautista and P. G. Jones, Synthesis of a Family of 3-Alkyl- or 3-Aryl-Substituted 1,2-Dihydroquinazolinium Salts and Their Isomerization to 4-Iminium-1,2,3,4- Tetrahydroquinolines, Org. Biomol. Chem., 2011, 9(7), 2279–2285 RSC .
- Assignment of the cis/trans conformation is prevented by overlapping signals of the aromatic and imine protons.
-
(a) N. Muresan, C. C. Lu, M. Ghosh, J. C. Peters, M. Abe, L. M. Henling, T. Weyhermöller, E. Bill and K. Wieghardt, Bis(α-Diimine)Iron Complexes: Electronic Structure Determination by Spectroscopy and Broken Symmetry Density Functional Theoretical Calculations, Inorg. Chem., 2008, 47, 4579–4590 CrossRef CAS PubMed ;
(b) V. C. Gibson, R. K. O'Reilly, W. Reed, D. F. Wass, A. J. P. White and D. J. Williams, Four-Coordinate Iron Complexes Bearing α-Diimine Ligands: Efficient Catalysts for Atom Transfer Radical Polymerisation (ATRP), Chem. Commun., 2002, 8, 1850–1851 RSC .
- Acenaphthoquinone usually reacts with 2 equiv. of amine to afford very stable BIAN (bis(aryl)acenaphthene quinonediimine) α-diimines.
(a) M. Gasperini, F. Ragaini and S. Cenini, Synthesis of Ar-BIAN Ligands (Ar-BIAN = Bis(Aryl)Acenaphthenequinonediimine) Having Strong Electron-Withdrawing Substituents on the Aryl Rings and Their Relative Coordination Strength toward Palladium(0) and-(II) Complexes, Organometallics, 2002, 21, 2950–2957 CrossRef CAS . For examples of catalytic applications of BIAN ligands, see:
(b) M. Laren and C. J. van Elsevier, Selective Homogeneous Palladium (0)-Catalyzed Hydrogenation of Alkynes to (Z)-Alkenes, Angew. Chem., Int. Ed., 1999, 38, 3715–3717 CrossRef ;
(c) F. Ragaini, S. Cenini, S. Tollari, G. Tummolillo and R. Beltrami, Allylic Amination of Unactivated Olefins by Nitroarenes, Catalyzed by Ruthenium Complexes. A Reaction Involving an Intermolecular C-H Functionalization, Organometallics, 1999, 18, 928–942 CrossRef CAS ;
(d) F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Sumner, G. P. McGovern and M. Findlater, Iron-Catalyzed Hydrosilylation of Aldehydes and Ketones under Solvent-Free Conditions, Organometallics, 2015, 34, 5051–5056 CrossRef CAS .
- The C2–O1, C1–N1, and C1–C2 distances are in the range of reported values of comparable structures. Cf. e.g.: J. Kovach, M. Peralta, W. W. Brennessel and W. D. Jones, Synthesis and X-Ray Crystallographic Characterization of Substituted Aryl Imines, J. Mol. Struct., 2011, 992, 33–38 CrossRef CAS . ref. [5b].
-
(a) R. Von Bülow, H. Gornitzka, T. Kottke and D. Stalke, Dimers of Thf-Solvated Lithium Anilide and Lithium Pentafluoroanilide: Basic Building Blocks of Lithium Amide Ladder Structures, Chem. Commun., 1996, 982(14), 1639–1640 RSC ;
(b) D. B. Grotjahn, P. M. Sheridan, I. Al Jihad and L. M. Ziurys, First Synthesis and Structural Determination of a Monomeric, Unsolvated Lithium Amide, LiNH2, J. Am. Chem. Soc., 2001, 123, 5489–5494 CrossRef CAS PubMed .
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of Bond Lengths Determined by X-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds, J. Chem. Soc., Perkin Trans., 1987, 12, 1–19 Search PubMed .
- M. P. Sadashiva, B. H. Doreswamy, K. S. Basappa, K. S. Rangappa, M. A. Sridhar and J. Shashidhara Prasad, Synthesis
and Crystal Structure of 5-Allyl-5H-dibenzo[b,f]azepine, J. Chem. Crystallogr., 2005, 35(3), 171–175 CrossRef CAS .
- A. Herrera, A. Grasruck, F. W. Heinemann, A. Scheurer, A. Chelouan, S. Frieß, F. Seidel and R. Dorta, Developing P-Stereogenic, Planar-Chiral P-Alkene Ligands: Monodentate, Bidentate, and Double Agostic Coordination Modes on Ru(II), Organometallics, 2017, 36, 714–720 CrossRef CAS .
- For a recent example of highly reactive, P-alkene-stabilized Cu(I) hydrides for CO2 reduction, see ref. 7.
-
(a) S. D. J. Brown, W. Henderson, K. J. Kilpin and B. K. Nicholson, Orthomercurated and Cycloaurated Derivatives of the Iminophosphorane Ph3P=NPh, Inorg. Chim. Acta, 2007, 360, 1310–1315 CrossRef CAS ;
(b) K. T. K. Chan, L. P. Spencer, J. D. Masuda, J. S. J. McCahill, P. Wei and D. W. Stephan, Anionic Phosphinimine-Chelate Complexes of Rhodium and Iridium: Steric and Electronic Influences on Oxidative Addition of CH2Cl2, Organometallics, 2004, 23, 381–390 CrossRef CAS .
- C. Hettstedt, P. Köstler, E. Ceylan and K. Karaghiosoff, Synthesis of the First Representatives of Amino Bis(Picolyl) and Amino Bis(Quinaldinyl) Phosphines, Tetrahedron, 2016, 72, 3162–3170 CrossRef CAS .
-
(a) M. Stollenz and F. Meyer, Mesitylcopper - A Powerful Tool in Synthetic Chemistry, Organometallics, 2012, 31(22), 7708–7727 CrossRef CAS ;
(b) T. Tsuda, T. Hashimoto and T. Saegusa, Cuprous Tert-Butoxide., a New and Useful Metalation Reagent, J. Am. Chem. Soc., 1972, 94(2), 658–659 CrossRef .
-
(a) A. R. Cowley, J. R. Dilworth, A. K. Nairn and A. J. Robbie, Preparation and Characterization of Diarylphosphazene and Diarylphosphinohydrazide Complexes of Titanium, Tungsten and Ruthenium and Phosphorylketimido Complexes of Rhenium, Dalton Trans., 2005,(4), 680–693 RSC ;
(b) S. K. Vasisht, G. Singh and P. K. Verma, Reactions of Bis (Trimethylsilyl) Amine and -Amide with MoOCl4, Monatsh. Chem., 1986, 117, 177–183 CrossRef CAS .
-
(a) A. J. Peel, J. Slaughter and A. E. H. Wheatley, ,New Options in Directed Cupration: Studies in Heteroleptic Bis(Amido)Cuprate Formation Dedicated to the Memory of Jack Lewis, J. Organomet. Chem., 2016, 812, 259–267 CrossRef CAS ;
(b) J. Haywood, J. V. Morey, E. H. A. Wheatley, C. Y. Liu, S. Yasuike, J. Kurita, M. Uchiyama and P. R. Raithby, Gilman-Type versus Lipshutz-Type Reagents: Competition in Lithiocuprate Chemistry, Organometallics, 2009, 28, 38–41 CrossRef CAS ;
(c) P. J. Harford, A. J. Peel, F. Chevallier, R. Takita, F. Mongin, M. Uchiyama and A. E. H. Wheatley, New Avenues in the Directed Deprotometallation of Aromatics: Recent Advances in Directed Cupration, Dalton Trans., 2014, 43, 14181–14203 RSC .
- Disproportionation reactions are often observed with related β-diketonate ligands systems and Cu(I) bases:
(a) A. T. Larson, A. S. Crossman, S. M. Krajewski and M. P. Marshak, Copper(II) as a Platform for Probing the Steric Demand of Bulky β-Diketonates, Inorg. Chem., 2020, 59, 423–432 CrossRef CAS PubMed ;
(b) B. Cheng, H. Yi, C. He, C. Liu and A. Lei, Revealing the Ligand Effect on Copper(I) Disproportionation via Operando IR Spectra, Organometallics, 2015, 34, 206–211 CrossRef CAS .
- For examples of monomeric nacnac-copper complexes, see:
(a) D. S. Laitar, C. J. N. Mathison, W. M. Davis and J. P. Sadighi, Copper(I) Complexes of a Heavily Fluorinated β-Diketiminate Ligand: Synthesis, Electronic Properties, and Intramolecular Aerobic Hydroxylation, Inorg. Chem., 2003, 42, 7354–7356 CrossRef CAS PubMed ;
(b) A. K. Gupta and W. B. Tolman, Cu(I)/O2 Chemistry Using a β-Diketiminate Supporting Ligand Derived from N,N-Dimethylhydrazine: A [Cu3O2] 3+ Complex with Novel Reactivity, Inorg. Chem., 2012, 51, 1881–1888 CrossRef CAS PubMed ;
(c) J. Takaichi, K. Ohkubo, H. Sugimoto, M. Nakano, D. Usa, H. Maekawa, N. Fujieda, N. Nishiwaki, S. Seki and S. Fukuzumi,
et al., Copper Complexes of the Non-Innocent β-Diketiminate Ligand Containing Phenol Groups, Dalton Trans., 2013, 42, 2438–2444 RSC .
- D. Gao, M. Parvez and T. G. Back, Synthesis of Indoles by Conjugate Addition and Ligand-Free Copper-Catalyzed Intramolecular Arylation of Activated Acetylenes with o-Haloanilines, Chem. – Eur. J., 2010, 16(48), 14281–14284 CrossRef CAS PubMed .
-
(a) P. A. Stabnikov, I. A. Baidina, S. V. Sysoev, N. S. Vanina, N. B. Morozova and I. K. Igumenov, Saturated Vapor Pressure and Crystal Structure of Bis-(2-Imino-4- Pentanonato)Copper(II), J. Struct. Chem., 2003, 44(6), 1054–1061 CrossRef CAS ;
(b) G. R. Clark, D. Hall and T. N. Waters, The Colour Isomerism and Structure of Some Copper Co-Ordination Compounds. Part XV. The Crystal Structure of N,N-Ethylene- Bis(Acetylacetoneiminato)Copper(II) Hemihydrate, J. Chem. Soc., 1968, 1959, 203–206 Search PubMed .
- For similar cationic Cu(I)-α-diimine complexes, see for example: B. Zelenay, R. Frutos-Pedreño, J. Markalain-Barta, E. Vega-Isa, A. J. P. White and S. Díez-González, Homo- and Heteroleptic Copper(I) Complexes with Diazabutadiene Ligands: Synthesis, Solution- and Solid-State Structural Studies, Eur. J. Inorg. Chem., 2016, 4649–4658 CrossRef CAS .
- For selected examples of iron-α-diimine complexes, see ref. 22 and:
(a) S. C. Bart, E. J. Hawrelak, A. K. Schmisseur, E. Lobkovsky and P. J. Chirik, Synthesis, Reactivity, and Solid State Structures of Four-Coordinate Iron(II) and Manganese(II) Alkyl Complexes, Organometallics, 2004, 23(2), 237–246 CrossRef CAS ;
(b) H. Lee, M. G. Campbell, R. Hernández Sánchez, J. Börgel, J. Raynaud, S. E. Parker and T. Ritter, Mechanistic Insight into High-Spin Iron(I)-Catalyzed Butadiene Dimerization, Organometallics, 2016, 35, 2923–2929 CrossRef CAS .
- For a discussion regarding the ring flip of the dibenzo[b,f]azepine rings, see: J. C. Calderón, A. Herrera, F. W. Heinemann, J. Langer, A. Linden, A. Chelouan, A. Grasruck, R. Añez, T. Clark and R. Dorta, Stereochemical Stability of Planar-Chiral Benzazepine Tricyclics: Inversion Energies of P- and S–Alkene Ligands, J. Org. Chem., 2023, 88, 16144–16154 CrossRef PubMed .
- For a ligand that reacts with Cu(I) halide, see ref. 5b.
- E. S. Chernyshova, R. Goddard and K.-R. Pörschke,
cis,cis,cis-1,5,9-Cyclododecatriene-Metal Complexes, Organometallics, 2007, 26, 4872–4880 CrossRef CAS .
- For a stilbenyl–Cu(I) triflate, see:
(a) B. J. Barrett and V. Iluc, Coordination of a Hemilabile Pincer Ligand with an Olefinic Backbone to Mid-to-Late Transition Metals, Inorg. Chem., 2014, 53, 7248–7259 CrossRef CAS PubMed . For Cu(I)–ethylene structures, see for example:
(b) P. Ebrahimpour, M. F. Haddow and D. F. Wass, New Cu(I)-Ethylene Complexes Based on Tridentate Imine Ligands: Synthesis and Structure, Inorg. Chem., 2013, 52, 3765–3771 CrossRef CAS PubMed ;
(c) H. Masuda, N. Yamamoto, T. Taga and S. Kitagawa, Structural Studies of Copper(I) [Cu(2,2′-Biydridine)(Ethylene)]ClO4, and [Cu(1,10-Phenantroline)(Ethylene)]ClO4, J. Organomet. Chem., 1987, 322, 121–129 CrossRef CAS .
- C. R. Martinez and B. L. Iverson, Rethinking the Term “Pi-Stacking”, Chem. Sci., 2012, 3(7), 2191–2201 RSC .
- This is slightly longer than in comparable [Cu(L)CO(OTf)] complexes:
(a) G. Doyle, K. A. Eriksen and D. V. Engen, Preparation of Copper(I) Carbonyl Complexes Derived from Sulfonic Acids. Crystal Structure of Cu(CO)C2H5SO3, Inorg. Chem., 1983,(22), 2892–2895 CrossRef CAS ;
(b) P. J. J. A. Timmermans, A. Mackor, A. L. Spek and B. Kojic-Prodic, The Crystal and Molecular Structure of Copper(I) Trifluoromethanesulphonate Cyclohexene Complex, J. Organomet. Chem., 1984, 276, 287–295 CrossRef CAS . But shorter compared to poly-alkene or -alkyne supported structures: J. D. Ferrara, C. Tessler-Youngs and W. J. Youngs, Synthesis and Characterization of a Copper(I) Trifiate Complex of 1,2:5,6:9,10-Trlbenzo-Cyclododeca-1,5,9-Trlene-3,7,11-Triyne, Organometallics, 1987, 6, 676–678 Search PubMed .
- There is only a limited number of published Cu(I) BIAN complexes.
(a) L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. Amélia, N. D. A. Lemos, M. T. Duarte, T. Avilés and P. T. Gomes, Synthesis and Structural Characterisation of (Aryl-BIAN)Copper(I) Complexes and Their Application as Catalysts for the Cycloaddition of Azides and Alkynes, Dalton Trans., 2012, 41, 5144–5154 RSC ;
(b) V. Rosa, C. I. M. Santos, R. Welter, G. Aullón, C. Lodeiro and T. Avilés, Comparison of the Structure and Stability of New α-Diimine Complexes of Copper(I) and Silver(I): Density Functional Theory versus Experimental, Inorg. Chem., 2010, 49, 8699–8708 CrossRef CAS PubMed .
- Complexation of mesityl copper with ligand 7 only afforded untractable reaction products.
-
(a) S. Fraysse, V. von Zelewsky and H. Stoeckli-Evans, A Stable Chiral Diolefin Complex of CuI with Predetermined Configuration at the Metal Centre, New J. Chem., 2001, 25, 1374–1375 RSC ;
(b) J. Zhang, R. G. Xiong, J. L. Zuo, C. M. Che and X. Z. You, Highly Stable Copper(I)-Olefin Coordination Polymers Capable of Co-Existing with Water and Acid, J. Chem. Soc., Dalton Trans., 2000,(17), 2898–2900 RSC .
-
(a) B. Gustafsson, M. Håkansson and S. Jagner, Copper(II) Is Harder than Copper(I): A Novel Mixed-Valence Example from Alkoxide Chemistry, New J. Chem., 2003, 27, 459–461 RSC ;
(b) G. Santiso-Quiñones, A. Reisinger, J. Slattery and I. Krossing, Homoleptic Cu-Phosphorus and Cu-Ethene Complexes, Chem. Commun., 2007, 47, 5046–5048 RSC .
-
(a) A. Linden, L. Llovera, J. Herrera, R. Dorta, G. Agrifoglio and R. Dorta, Organometallics, 2012, 31, 6162 CrossRef CAS ;
(b) R. Mariz, A. Briceño, R. Dorta and R. Dorta, Organometallics, 2008, 27, 6605–6613 CrossRef CAS .
- J. J. Allen and A. R. Barron, Olefin, Coordination in Copper(I) Complexes of Bis(2-Pyridyl)Amine, Dalton Trans., 2009, 878–890 RSC .
- C. Floriani, E. M. Meyer, S. Gambarotta, A. Chiesi-Villa and C. Guastini, Polynuclear Aryl Derivatives of Group 11 Metals: Synthesis, Solid State-Solution Structural Relationship, and Reactivity with Phosphines, Organometallics, 1989, 8, 1067–1079 CrossRef .
-
J. Kubas, Tetrakis(Acetonitrile)Copper(l) Hexafluorophosphate(1-), in Inorganic synthesis, ed. R. J. Angelici, 1990, vol. 28, pp. 68–70 Search PubMed .
- T. Cohen, R. J. Ruffner, D. W. Shull, E. R. Fogel and J. Falck, Vinyl Sulfides From Thioacetals With Copper(I) Trifluoromethanesulfonate: (Z)-2-Methoxy-1-Phenylthio-1,3-Butadiene, Org. Synth., 1979, 59, 202 CrossRef .
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, Organometallics, 2010, 29(9), 2176–2179 CrossRef CAS .
- J. P. Schaefer, J. G. Higgins and P. K. Shenoy, Cinnamyl Bromide, Org. Synth., 1968, 48, 51 CrossRef CAS .
- H. Zimmer and G. Singh, Synthesis and Some Reactions of Triphenylphosphinamino- and (β-N-Disubstituted Amino)Imines, J. Org. Chem., 1964, 29(6), 1579–1581 CrossRef CAS .
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic information, NMR spectra. CCDC 2338469–2338477. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00749b |
| ‡ Current address: IBU-tec advanced materials AG, Hainweg 9-11, 99425 Weimar, Germany. |
| § Present adresss: 3M Dach, 40721 Hilden, Germany. |
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jens
Langer
a,
Jens
Langer