
A nuclei-rich strategy for highly reversible dendrite-free zinc metal anodes†
Qingli
Zou
,
Zhuojian
Liang
 ,
Wanwan
Wang
,
Dejian
Dong
and
Yi-Chun
Lu
*
,
Wanwan
Wang
,
Dejian
Dong
and
Yi-Chun
Lu
*
Electrochemical Energy and Interfaces Laboratory, Department of Mechanical and Automation Engineering, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong SAR, China. E-mail: yichunlu@mae.cuhk.edu.hk
First published on 24th October 2023
Abstract
Aqueous zinc (Zn) metal batteries are promising candidates for large-scale energy storage owing to their low cost and high safety. However, dendrite formation on Zn metal electrodes leads to short circuits and low coulombic efficiency. Zinc deposition tends to grow into large hexagonal plates with sharp corners and edges. To address this, we propose nuclei-rich Zn plating in which nucleation is promoted over growth to achieve dendrite-free cycling. By fabricating a hydroxyapatite-based nuclei-incubating interface with high ion conductivity and nucleation catalytic activity on the Zn surface, the plating reaction of Zn is fundamentally modified to persistently generate a large number of nuclei that grow into densely packed Zn crystals with small radii and high uniformity. Owing to this high-rate progressive nucleation strategy, the nuclei-rich Zn electrode enables stable cycling for over 1200 h with a coulombic efficiency of 99.7% under the harsh conditions of 10 mA h cm−2 areal capacity at 15 mA cm−2, based on which a Zn–MnO2 full cell with a low N/P ratio of 1.9 and a long cycle life of 3000 cycles is demonstrated. This strategy highlights the profound impact of the growth mechanism on metal morphology and can be applied to address dendrite issues in other metal electrodes such as lithium.
Broader contextDue to the increasing energy demand and the utilization of renewable energy, the development of advanced rechargeable battery systems has become an urgent requirement. However, state-of-the-art lithium-ion batteries are insufficient to meet the demands of large-scale applications due to the known safety issues and limited availability of lithium resources. Aqueous zinc metal batteries, known for their high safety and low cost, have received significant interest as promising alternatives to lithium-ion batteries. Both aqueous electrolyte and earth-abundant zinc metal are much lower in cost. In addition, the application of aqueous electrolytes effectively mitigates the risk of fire and explosion. However, the cycling performance of zinc metal batteries has been limited by the formation of dendrites on the zinc metal anode during cycling. Zinc deposition tends to grow into large hexagonal plates with sharp corners and edges. These sharp dendrites penetrate the separator, leading to internal shorting failures and limited cycle life. Here, we demonstrate the nuclei-rich plating strategy to achieve dendrite-free cycling of Zn anodes. With zinc nucleation promoted by a nuclei-incubating interface, the growth of large plates and sharp corners is inhibited. The electrode shows a large amount of densely packed zinc crystals with significantly reduced radii. The nuclei-rich Zn electrode enables stable cycling for over 1200 h, which is 30 times longer than that of conventional electrodes, with a coulombic efficiency of 99.7% under the harsh conditions of 10 mA h cm−2 areal capacity at 15 mA cm−2. Using the nuclei-rich Zn electrode, a Zn–MnO2 full cell with a low N/P ratio (the ratio between the capacity of the negative electrode to the positive electrode, ideally being 1) exhibits a cycle life of 3000 cycles. Our work highlights the profound impact of the growth mechanism on metal morphology and we anticipate that the nuclei-rich plating strategy can inspire future exploration of mechanism modifications in the plating–stripping process of Zn and other important metal anodes, such as lithium and sodium. |
Introduction
Advanced electrochemical energy storage technologies are in high demand due to the increasing use of renewable energy electrification of vehicles. The lthium-ion battery (LIB) is one of the most developed electrochemical energy storage technologies but it is insufficient for meeting the demands of large-scale applications due to the known safety issues and limited resource availability of lithium and transition metals (e.g., Co).1,2Aqueous Zn metal batteries (ZMBs) have received significant interest due to an appealing combination of advantages over LIBs.3,4 Both aqueous electrolytes and the earth-abundant Zn metal5 are much lower in cost compared to the materials used in LIBs. Benefiting from the high safety of aqueous electrolytes, the risk of fire or explosion is effectively mitigated in ZMBs.6 Moreover, aqueous electrolytes deliver superior power outputs due to their fast ion-transport nature.7,8 However, ZMBs have long been suffering from limited cycling life and low coulombic efficiency (CE) due to the formation of dendrites on the Zn metal anode during cycling.4,8–10 Sharp dendrites penetrate the separator, leading to internal shorting failures. The loss of their connection to the anode during the stripping of Zn converts them into ‘dead Zn’, resulting in a decrease of CE.
The majority of the research efforts aimed at addressing the Zn dendrite issue focus on regulating the electric field.11,12 Similar to studies on other metal anodes, Zn dendrite formation has long been considered to originate from an uneven distribution of the electric field, which leads to geometric inhomogeneity during the Zn plating process.9,13 Many methods have been proposed following this understanding, including the introduction of electrolyte additives,14–16 construction of artificial interlayers,17–19 and modification of Zn electrodes.20–24 The application of these methods successfully regulated the charge distribution on the Zn surface, guiding homogenous ion diffusion and local current which further promoted uniformity in Zn plating.
However, the intrinsic crystal properties of Zn pose a high risk of dendrite formation during its growth even though the electric field is regulated (Fig. 1a). As a hexagonal close-packed (hcp) metal, Zn tends to grow into hexagonal tablets as the (002) plane on the six edges is the closest-packed plane exposed and exhibits the lowest thermodynamic free energy.25,26 As the small tablets grow, their aspect ratio grows, resulting in the formation of larger hexagonal plates with sharper edges and corners. The random orientation of Zn plates inevitably leads to local inhomogeneity and uneven current distribution, which is exacerbated by the “tip effect”27 on the corners where preferential growth occurs, further increasing inhomogeneity, before eventually penetrating the separator and leading to battery failure. This growth mode has been widely observed during Zn plating.26,28,29 Therefore, the intrinsic growth of Zn crystals poses a great challenge to the realization of high cycling stability of ZMBs, especially when cycling at high areal capacity. A fundamental modification of the Zn crystal plating mechanism is necessary to achieve dendrite-free Zn anodes.
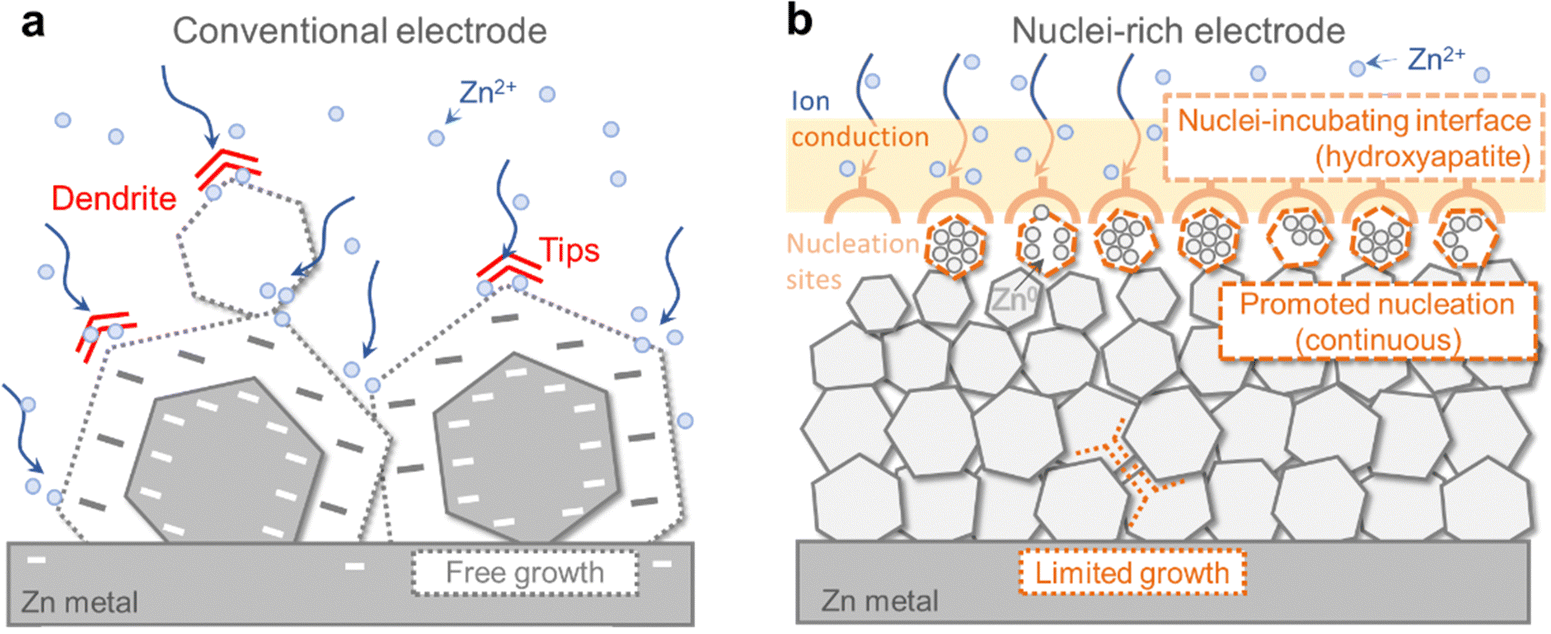 | ||
| Fig. 1 Comparison between the conventional and nuclei-rich Zn plating strategy. Schematic of Zn plating on (a) conventional and (b) nuclei-rich Zn electrodes. | ||
How could the formation of large hexagonal Zn plates be inhibited? We propose nuclei-rich plating of Zn metal as a solution. Plating of metals involves initial nucleation followed by subsequent growth, during which nuclei grow into larger crystals. Preferential promotion of nucleation over growth could result in a larger number of grains and thereby smaller crystals (Fig. 1b). Consider a special scenario where nuclei are constantly generated and piled up at an interface at a fast rate, with the concentration of nuclei becoming so high that there is little room for their subsequent growth. Their grain boundaries meet each other as soon as their growth starts, resulting in a collection of densely packed small crystals whose growth is limited by the depletion of space. As a result, no large hexagonal plates can be generated. Such nuclei-rich plating, if realized, could possibly be a strategy to inhibit Zn dendrite formation.
Here, we demonstrate the nuclei-rich plating strategy to achieve dendrite-free cycling of Zn anodes. We construct a nuclei-incubating interface by fabricating a hydroxyapatite layer on the Zn surface, which changes the plating mode of Zn from conventional instantaneous nucleation to progressive nucleation, with Zn nuclei forming throughout the whole plating process at high rates. As described above, the promotion of nucleation results in uniform Zn plating, consisting of a large number of densely packed crystals with significantly reduced radii. The inhibition of crystal growth eliminates sharp tips or large plates even at a high current density of 15 mA cm−2 and a high areal capacity of 10 mA h cm−2, enabling stable cycling for over 1200 h in a symmetric cell with the nuclei-rich Zn electrode, which is 30 times longer than that of a conventional one. Using the nuclei-rich Zn electrode, a Zn–MnO2 full cell with a low N/P ratio (the ratio between the capacity of the negative electrode to the positive electrode, ideally being 1 but a value below 2 is seldom demonstrated) of 1.9 exhibits a cycle life of 3000 cycles.
Results and discussion
A nuclei-rich Zn electrode
To reveal how Zn crystal growth is correlated with dendrite formation, we first probe the morphology of a conventional Zn-electrolyte interface during cycling. Here, current densities of 1, 5, and 15 mA cm−2 were applied for 40 min (reaching areal capacities of 0.67, 3.33, and 10 mA h cm−2) to plate and strip Zn in symmetric cells. The SEM images of plated and stripped Zn are shown in Fig. 2 and Fig. S1, S2 (ESI†). | ||
| Fig. 2 Morphology comparison between conventional and nuclei-rich Zn electrodes. The (a), (c), (e) and (g) top-view and (b), (d), (f) and (h) sectional-view SEM image of (a) and (d) conventional and (e) and (h) nuclei-rich Zn electrodes after (a), (b), (e) and (f) plating and (c), (d), (g) and (h) stripping. The current density and areal capacity for plating/stripping are 15 mA cm−2 and 10 mA h cm−2, respectively. To preserve the morphologies most effectively, section-view samples were prepared by cracking the electrodes after freezing them in liquid N2. | ||
On conventional Zn electrodes, uneven distribution of Zn starts during the plating process. On the electrode surface (Fig. 2a), the plated Zn forms islands with different heights instead of a uniform film that covers the Zn substrate, even at areal capacities as high as 10 mA h cm−2. The section view (Fig. 2b) shows that the islands are clusters of large Zn plates of up to tens of microns, which are similar in size and shape to the Zn plates in the Zn substrate. Large protrusions with tips (yellow arrow in Fig. 2b) form in the plated layer, which may have already penetrated the quartz fiber separator as fibers are found interspersed in the plated Zn (Fig. 2b and Fig. S1b, ESI†). The uneven distribution of plated Zn on the plane could be related to the inhomogeneous initial nucleation. The tips, more importantly, are attributed to the hexagonal shape of Zn plates which is inevitable during the growth of Zn crystals. Although they are not as sharp as the needle-like dendrites on Li metal electrodes, they also present a risk of short circuits.
The inhomogeneity of Zn metal further increases after stripping. As shown in Fig. 2c, instead of having the islands removed, dendrites and holes emerge in some zones on the electrode surface, which are attributed to the incomplete stripping of the plated Zn and stripping of the original Zn substrate, respectively. With similar crystal size and crystallinity, the plated Zn and original Zn substrate are stripped in parallel due to similar stripping kinetics. The sectional SEM image (Fig. 2d) gives a clearer view of the dendrites and holes divided by a clear line which is the base plane of the Zn substrate. With an increase in plating-stripping capacity (comparing Fig. 2d and Fig. S2, ESI†), the stripped Zn electrode shows larger holes under the line and sharper residual plated Zn consisting of thin plates or even needles (Fig. 2d inset), which manifest the high-aspect-ratio characteristic of the grown Zn crystals. These sharp tips could cause the further growth of dendrites in the next plating and stripping cycles due to the tip effect, that is metal deposits faster on tips due to their stronger local electric field.
To regulate the plating of Zn and thereby eliminate the formation of dendrites, we designed and constructed a nuclei-incubating interface as discussed above, by interfacing an immediately attached layer of hydroxyapatite particles with the Zn electrode surface. Hydroxyapatite is chosen due to its high Zn2+ adsorb capability.30 The XRD (Fig. S3a, ESI†) and XPS (Fig. S4, ESI†) profiles of this layer show obvious signals of hydroxyapatite. And according to the SEM images (Fig. S3b, ESI†), these nanoparticles are distributed uniformly on the surface of Zn and remain stable upon contact. The thickness of the layer was optimized to around 15 μm (Fig.S3c, ESI†).
The Zn electrode with an incubating interface demonstrates a significantly different morphology. Unlike the roughened surface of a conventional electrode (Fig. 2a), no changes are observed here from the top view (Fig. 2e) because the plated Zn forms a uniform and flat layer consisting of a large number of small crystals measuring 0.5–3 μm in a densely packed structure (Fig. 2f and Fig. S5d, ESI†) without forming any sharp tips or large plates. The formation of densely packed small nuclei under high current density (15 mA cm−2) and high areal capacity (10 mA h cm−2) has not been reported before since such harsh cycling conditions usually lead to Zn dendrite formation. In addition, the plated layer is about 30% thinner compared with that on a conventional Zn electrode, indicating higher density and lower porosity. EDS mapping (Fig. S6, ESI†) of the plated electrode revealed a distinct boundary between the Zn and Ca distribution regions, confirming that the deposited Zn was confined within the plated layer and did not traverse the incubating interface. The absence of Zn signals beyond the interface verifies the incubating layer's effectiveness in blocking dendritic Zn growth. We note that in some cases, the electrode undergoes initial stripping, which increases surface roughness. However, the incubating interface enables the electrode to recover after re-plating, reconstructing a flat interface (Fig. S7, ESI†). We further compared the morphology obtained at different plating capacities (Fig. 2f and Fig. S5, ESI†). As the capacity increases from 0.67 to 10 mA h cm−2, the thickness of the plated layer also increases. Intriguingly, it is the number of grains that significantly increases with the increasing capacity (Fig. 2fvs. Fig. S5b, S14 times higher capacity, ESI†), while the crystal size remains similar. This contrasts strongly with the conventional interface (Fig. 2bvs. Fig. S1d, ESI†), where increasing capacity leads to an increase in grain size. This difference in plating morphology with and without the incubating interface becomes even more pronounced when the plating capacity was increased under same current density (Fig. S8, ESI†). The increased number of grains and significantly reduced crystal size compared with that of the conventional Zn electrode demonstrate the unique effect of the nuclei-incubating interface: it constantly promotes nucleation and suppresses crystal growth. This nuclei-rich plating mode, as discussed in the Introduction section, eliminates the risky formation of large and high-aspect-ratio hexagonal Zn crystals.
In addition to uniform plating, the nuclei-rich electrode displays uniform stripping as well. After striping (Fig. 2h and Fig. S9, ESI†), the electrode and nuclei-incubating interface are completely restored to their state before plating, indicating a fully reversible cycle. Due to the substantially smaller size (up to 99% smaller) of the plated Zn in compared to the Zn substrate, the stripping of the former is overwhelmingly favored in reaction kinetics. This fully restored morphology could be observed even after multiple cycles (Fig. S10b–d, ESI†) with the XRD patterns remaining unchanged and no by-products are observed (Fig. S10a, ESI†). The XPS spectra of the Zn 2p2/3 core-level (Fig. S11d, ESI†) could be deconvoluted into two components located at 1021.6 and 1022.6 eV, which are ascribed to the adsorption of Zn2+ on PO33− and OH−, respectively. In addition, the Ca 2p, P 2p, and O 1s core-level spectra (Fig S11a–c, ESI†) show no significant change after cycling. These findings indicate the high stability of the nuclei-rich electrode and the corresponding Zn plating and stripping reactions, which successfully enables a dendrite-free reversible cycle of the Zn electrode.
High-rate progressive nucleation
To reveal the formation mechanism of the nuclei-rich morphologies, we analyzed the nucleation-growth behavior of Zn at the conventional and nuclei–incubating interfaces with chronoamperometry (details described in Methods). A gradient series of overpotentials from 60 mV to 160 mV was applied and the resulting current responses (i–t curves) were measured (Fig. S12, ESI†). At the conventional interface, a minimum overpotential of 140 mV is required to initiate nucleation. However, at the nuclei-incubating interface, a typical diffusion-controlled nucleation feature was observed, starting from a much lower overpotential of 80 mV, indicating promoted nucleation.These data were further normalized to (I/Im)2–(t/tm) curves and analyzed using the Scharifker–Hills model,31 which describes two nucleation modes:
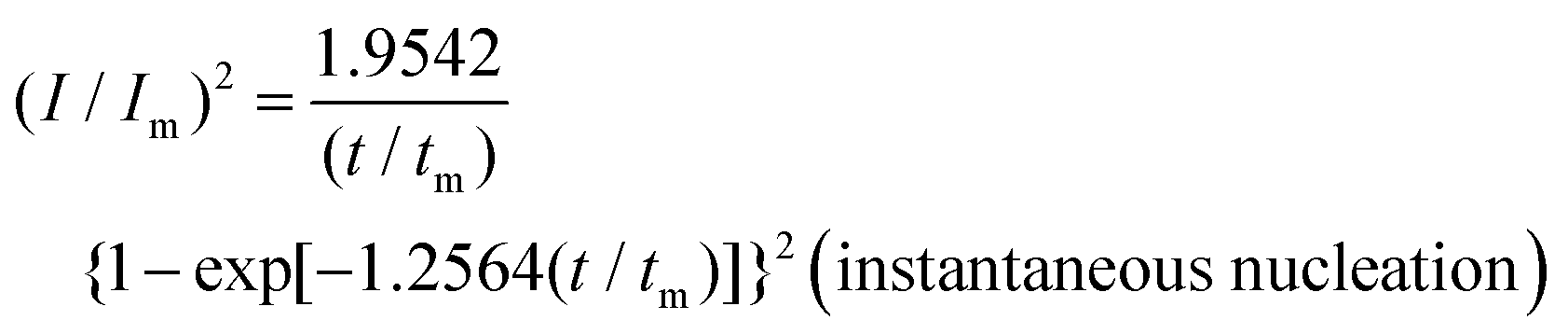 | (1) |
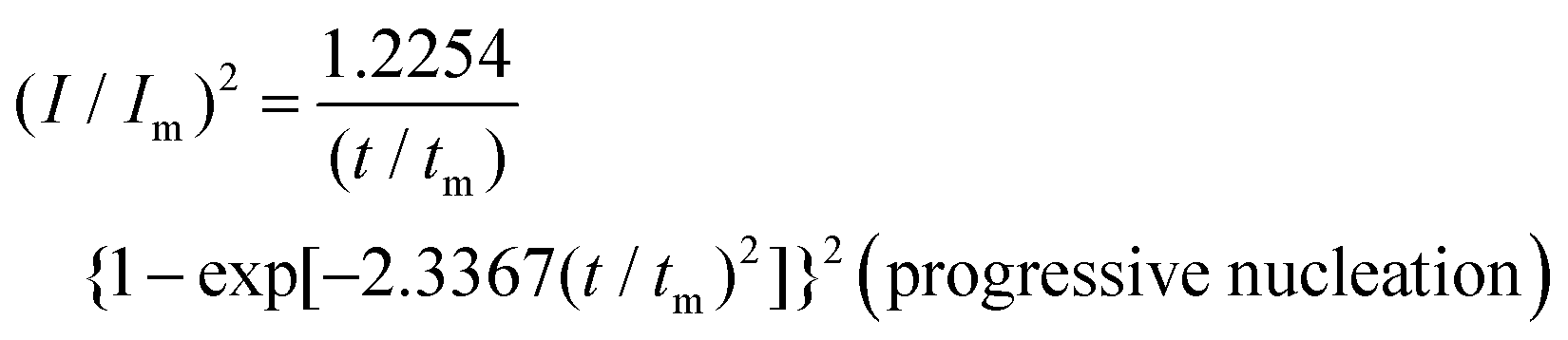 | (2) |
In general, instantaneous nucleation corresponds to fast nucleation that exhausts all nucleation sites at the very beginning of plating, while progressive nucleation is relatively sluggish and takes a longer time to occupy the nucleation sites.31–33 As shown in Fig. 3, the dimensionless curves of Zn plating at a conventional interface fit perfectly with the theoretical model of instantaneous nucleation (eqn (1)). Calculations based on the model reveal a small nucleation density of 6 × 10−3 μm−2 at an overpotential of 160 mV (Fig. 3c) with an initial nuclei radius of 7 μm (Fig. 3d; the nuclei then grow larger), which is in agreement with the nuclei-sparse feature of the observed morphology (Fig. 3b).
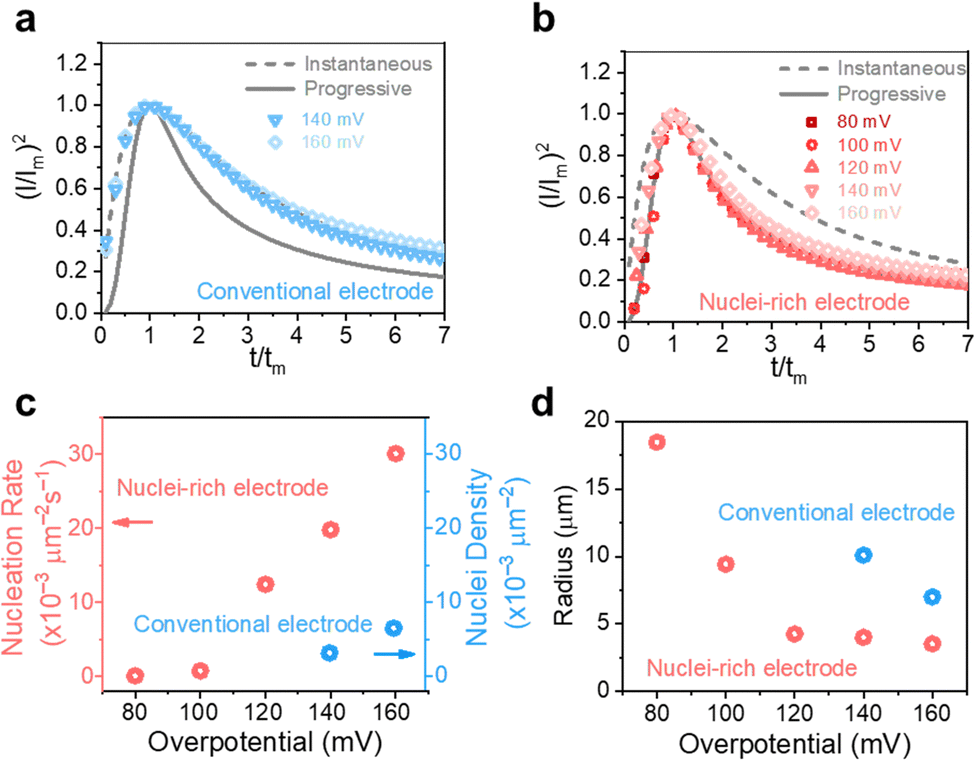 | ||
| Fig. 3 High-rate progressive nucleation enabled by the nuclei-incubating interface. (I/Im)2–(t/tm) curves derived from the current response in chronoamperometry plating of Zn at (a) conventional and (b) nuclei-incubating interfaces are compared with theoretical instantaneous (grey solid line) and progressive nucleation (grey dashed line) curves described in the Scharifker–Hills model. (c) Nucleation rate/density and (d) radius of plated Zn at the conventional and nuclei-incubating interfaces. | ||
In contrast, Zn plating at the nuclei-incubating interface follows the progressive nucleation mode in the whole investigated range of overpotentials (Fig. 3b), indicating a longer-lasting generation of nuclei. Interestingly, unlike the common progressive nucleation system that displays sluggish nucleation, a high nucleation rate of 30 × 10−3 μm−2 s−1 was found (Fig. 3c), which means that within the first second of plating the number of nuclei generated at the nuclei-incubating interface is already 5 times of that at the conventional one, and keeps increasing with time. The drastically larger number of grains, and the resulting reduction in nuclei radius to ∼3 μm (Fig. 3d), align well with the observed morphology (Fig. 2f), which validates the analysis of the interfaces using this model. Accordingly, high-rate progressive nucleation is the key to achieving the nuclei-rich morphology.
However, high-rate progressive nucleation is rare in the electro-deposition of metals. How is it realized here? We believe that the fast nucleation kinetics of the nucleation sites and their persistent activity are the critical enabling factors for this special and effective nuclei-incubating interface.
We first studied the plating kinetics at the conventional and nuclei-incubating interfaces comprehensively. Galvanostatic voltage profiles were collected in two electrode symmetric cells (Fig. 4a). The potential dips at the beginning of the plating process indicate nucleation energy barriers. With the nuclei-incubating interface, the nucleation barrier is reduced to half of the conventional one, suggesting promoted nucleation kinetics. This improvement is also demonstrated in cyclic voltammograms in three-electrode symmetric cells (Fig. 4b), where the starting point of Zn plating is shifted by +10 mV with the introduction of the nuclei-incubating interface. Nucleation is the rate-limiting step in phase transformation,34 like plating. Consequently, the overall plating kinetics is substantially improved, leading to an enhancement of ∼50% in plating current and even more in the following stripping current, as well as an increase in the exchange current density from 10.19 mA cm−2 to 17.04 mA cm−2 according to the Tafel plot (Fig. 4c). Benefiting from the enhanced Zn plating kinetics, the competing H2 evolution reaction is suppressed. We monitored gas evolution from the linear sweep voltammetry test using online electrochemical mass spectrometry (OEMS). The H2 evolution from the nuclei-incubating interface during the entire process, including the idling time before and after plating, is significantly reduced (Fig. S13, ESI†). The promoted nucleation and overall plating kinetics both could be attributed to the decrease in activation energy. According to the Arrhenius curves (Fig. 4d) based on a temperature-dependent variation of the electrochemical impedance spectra (Fig. S14, ESI†), the introduction of the nuclei-incubating interface reduces the activation energy of plating from 66.0 kJ mol−1 to 48.6 kJ mol−1. This reduction in activation energy is a result of the significantly enhanced adsorption of Zn0 on the hydroxyapatite nanoparticles. Density functional theory (DFT) calculations show that the adsorption energy of Zn0 on the Zn surface is −0.78 eV, while hydroxyapatite displays an extraordinary −5.05 eV adsorption energy (Fig. S15 (ESI†), representing a strong tendency of nucleation on the hydroxyapatite surface.) Additionally, hydroxyapatite's strong affinity35 for Zn2+ could promote Zn2+ desolvation and thus enhance nucleation kinetics.36 The Raman spectra collected at the electrolyte–hydroxyapatite interface (Fig. S16c, ESI†) exhibited a pronounced peak shift for the phosphate vibration from 959.5 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37(Fig. S16a, ESI†) to a higher Raman shift regime, along with the appearance of a new peak at 963 cm−1. This shift implies an interaction between hydroxyapatite and Zn2+ that could lead to dehydration of the solvation sheath, evidenced by the decreased fraction of separated ion pairs (SSIPs).38 These compelling enhancement in nucleation kinetics drives the high nucleation rate at the nuclei-incubating interface.
37(Fig. S16a, ESI†) to a higher Raman shift regime, along with the appearance of a new peak at 963 cm−1. This shift implies an interaction between hydroxyapatite and Zn2+ that could lead to dehydration of the solvation sheath, evidenced by the decreased fraction of separated ion pairs (SSIPs).38 These compelling enhancement in nucleation kinetics drives the high nucleation rate at the nuclei-incubating interface.
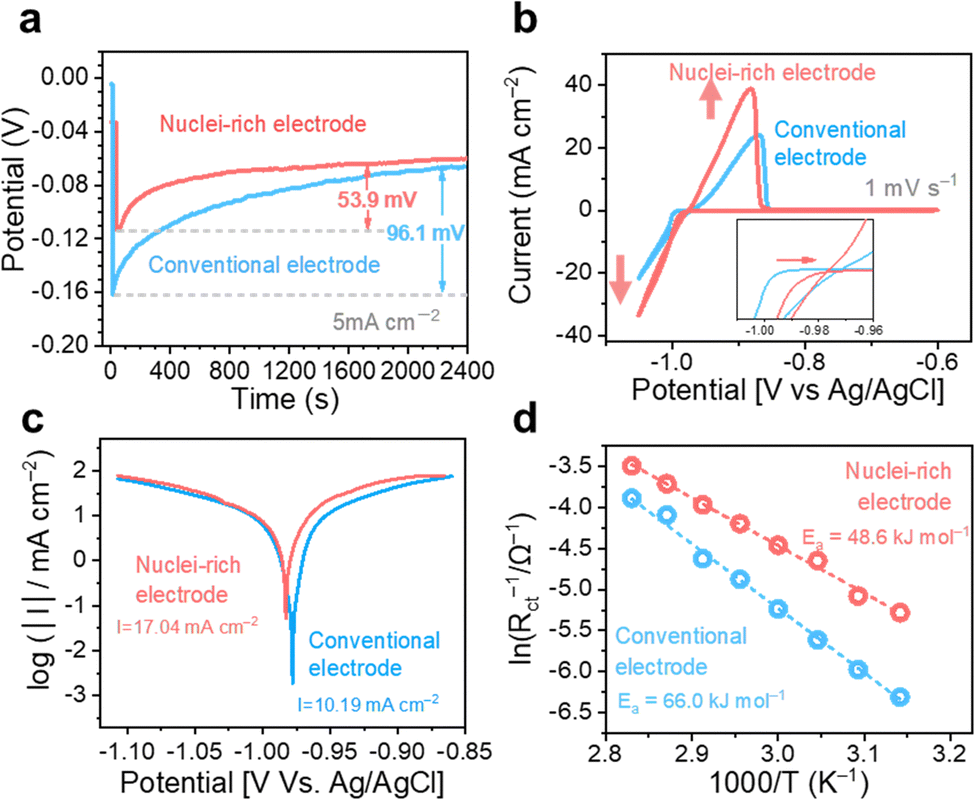 | ||
| Fig. 4 Enhanced Zn plating and stripping kinetics. (a) Galvanostatic voltage profiles, (b) cyclic voltammograms curves, (c) Tafel plots, and (d) Arrhenius plots of Zn plating and stripping on conventional and nuclei-rich electrodes. | ||
Maintaining the activity of nucleation sites is another prerequisite for effective nuclei incubation. Zn nucleation sites require access to electrons and Zn2+ ions to function. Therefore, potential nucleation sites must make immediate contact with the Zn surface, where electrons are available; however, access to Zn2+ from the liquid electrolyte could be blocked by the attachment of Zn nuclei to the nucleation sites, which is termed “self-poisoning”39 of the active sites, leading to an instantaneous nucleation event. To keep the original nucleation sites active, Zn2+ access must be provided via other pathways, for example, diffusion through the site-containing solid itself. A high Zn2+ diffusion coefficient was found in the hydroxyapatite layer, which is consistent with the literature.40 The high Zn2+ conductivity enables constant access to Zn2+ through a solid pathway even though the nucleation sites are already occupied by Zn crystals. As a result, these sites remain active to progressively incubate nuclei. To confirm the importance of high Zn2+ conductivity, we fabricated a layer of zinc carbonate basic (ZCB) on the Zn electrode, which is a common SEI component41,42 showing one-fourth the Zn2+ conductivity of hydroxyapatite (Fig. S19, ESI†). By regulating the surface charge with the ZCB layer, the plated Zn (Fig. S17a, ESI†) appeared to be more uniform than that on a conventional Zn electrode (Fig. 2). However, the morphologies resemble those found at a conventional interface. The plated Zn displays a large grain size (Fig. S17c, ESI†), which is almost indistinguishable from the Zn substrate; large protrusions of plated Zn deform the ZCB layer (Fig. S17d, ESI†); the plated Zn penetrates the ZCB layer and even the separator, as suggested by the emergence of Zn tablets inside (Fig. S17b, ESI†) and above the layer (Fig. S17e and f, ESI†), and the attachment of quartz fibers on the sample (Fig. S17e and f, ESI†). Such morphologies originate from the occupation of the nucleation sites by the plated Zn. After the initial instantaneous nucleation (Fig. S18, ESI†), the nuclei remain on the ZCB particles and grow; further nucleation must occur on the separator-facing side (where Zn2+ comes from) of these initial Zn crystals, and then the subsequent growth starts the penetration through the ZCB layer.
We note that, in addition to the high-rate progressive nucleation, the uniformized charge distribution on the nuclei–incubating interface, which led to the homogeneous distribution of small nuclei, is also necessary for the formation of dense-packed nuclei-rich structure.
Stable cycling of nuclei-rich Zn electrodes and batteries
With high-rate progressive nucleation at the designed interface, the nuclei-rich Zn electrode demonstrates long-term cycling even under high current and high capacity. Zn symmetric cells of the two types of electrodes were cycled from 1 mA cm−2 to 1 mA h cm−2 and 15 mA cm−2 to 10 mA h cm−2, respectively, to investigate the cycling stability (Fig. 5). At 1 mA cm−2, the conventional Zn electrodes show a sudden voltage drop after 252 h of cycling, indicating an internal short circuit. With the increase in current density and areal capacity to 15 mA cm−2 and 10 mA h cm−2, respectively, the cycling life of conventional Zn electrodes decreases to less than 40 h.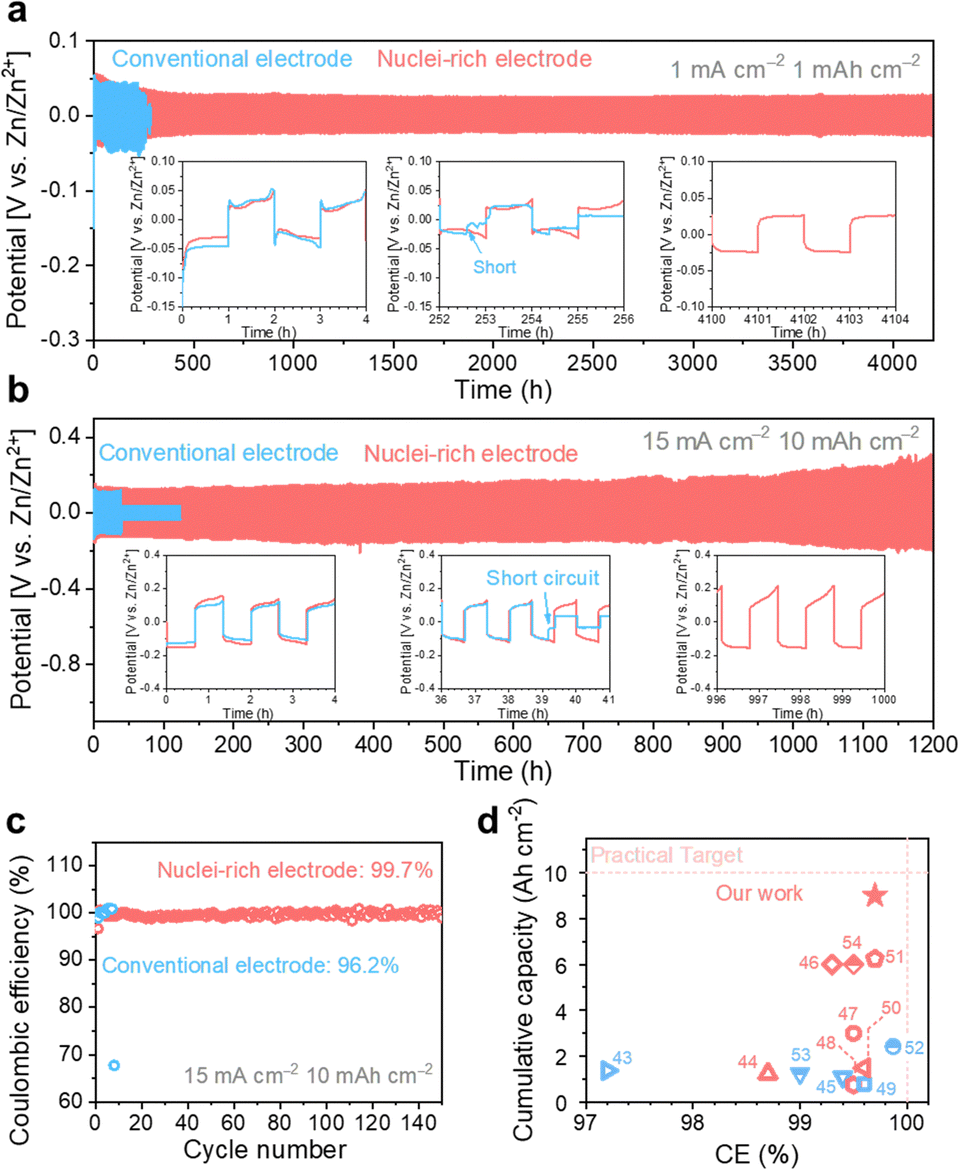 | ||
| Fig. 5 High plating and striping stability of the nuclei-rich Zn electrode during long-term cycling. Long-term cycling profiles of Zn symmetric cells with current density and areal capacity of (a) 1 mA cm−2 and 1 mA h cm−2, and (b) 15 mA cm−2 and 10 mA h cm−2. (c) Coulombic efficiency of Zn–Cu cells. (d) Summary of published Zn plating/stripping performance with electrode modifications.43–54 Red dash lines represent a practically competitive performance target for ZMBs (a cumulative capacity of 10 Ah cm−2, an ideal CE of 100%, a current density of 10 mA cm−2, and an areal capacity of 5 mA h cm−2). Accordingly, the color red or blue here (d) indicates current densities in the references meeting or below the target. | ||
With the introduction of the nuclei-incubating interface, the cycle life of the Zn symmetric cell was significantly increased to more than 4000 h at 1 mA cm−2. The increase in cycle life is even more pronounced at high current density and high areal capacity due to the high-rate progressive nucleation strategy. The nuclei-rich electrode demonstrates over 1200 h of stable cycling at 15 mA cm−2 without any sign of short circuit, which is more than 30 times longer than the conventional Zn electrodes. To quantify the CE of Zn plating and stripping (Fig. 5b), Zn–Cu cells were discharged under the same condition and charged to 0.5 V. The nuclei-incubating interface displays an excellent long-term CE of 99.7%, which can be attributed to the inhibition of H2 evolution and the prevention of ‘dead Zn’ formation on the nuclei-rich electrodes. Compared with recently reported Zn electrodes with surface modifications (Fig. 5c), the nuclei-rich strategy demonstrates outstanding comprehensive performance in terms of cumulative capacity, Coulombic efficiency, and current density. We note that the same strategy can also be applied to other metal electrodes that suffer from the dendrite issues. As shown in Fig. S20 (ESI†), the enhanced cycling stability of Li symmetric cells with the nuclei-incubating layer indicates the feasibility of this strategy for inhibiting the formation of lithium dendrites.
We coupled the nuclei-rich Zn anode with an α-MnO2 cathode (2 mg cm−2, XRD and SEM of α-MnO2 shown in Fig. S21, ESI†) and iodide catholyte respectively to evaluate its performance in full cells. The voltage profiles of Zn–MnO2 batteries operated at 1C and 10C rates (1C = 300 mA g−1) show two discharge plateaus (Fig. 6a), consistent with the reported profiles.55 With the nuclei-rich Zn anode, the cell displays excellent performance (Table S1, ESI†) of over 300 mA h g−1 capacity at 1C and 220 mA h g−1 at 10C, which is more than 50 mA h g−1 higher than that with a conventional Zn anode. This could be related to the suppressed H2 evolution reaction on the nuclei-rich anode, which facilitates the H+ intercalation and/or conversion reactions on the cathode. The nuclei-rich Zn–MnO2 full cell display stable cycling for over 3000 cycles at 10C with an average capacity of 215 mA h g−1 and little capacity fading, which is a substantial improvement over the conventional Zn cell showing significant capacity fading starting from ∼1200 cycles (Fig. 6b). The voltage profiles of these cells are shown in Fig. S22 (ESI†). Similar improvements could be observed in Zn–I2 batteries (Fig. S23, ESI†). The nuclei-rich Zn anode dramatically mitigated capacity fading, which is the primary challenge plaguing Zn–I2 systems. This enhanced stability is attributed not only to suppressed Zn dendrite growth but also to the prevention of direct reactions between the Zn anode and soluble reactants or intermediates.
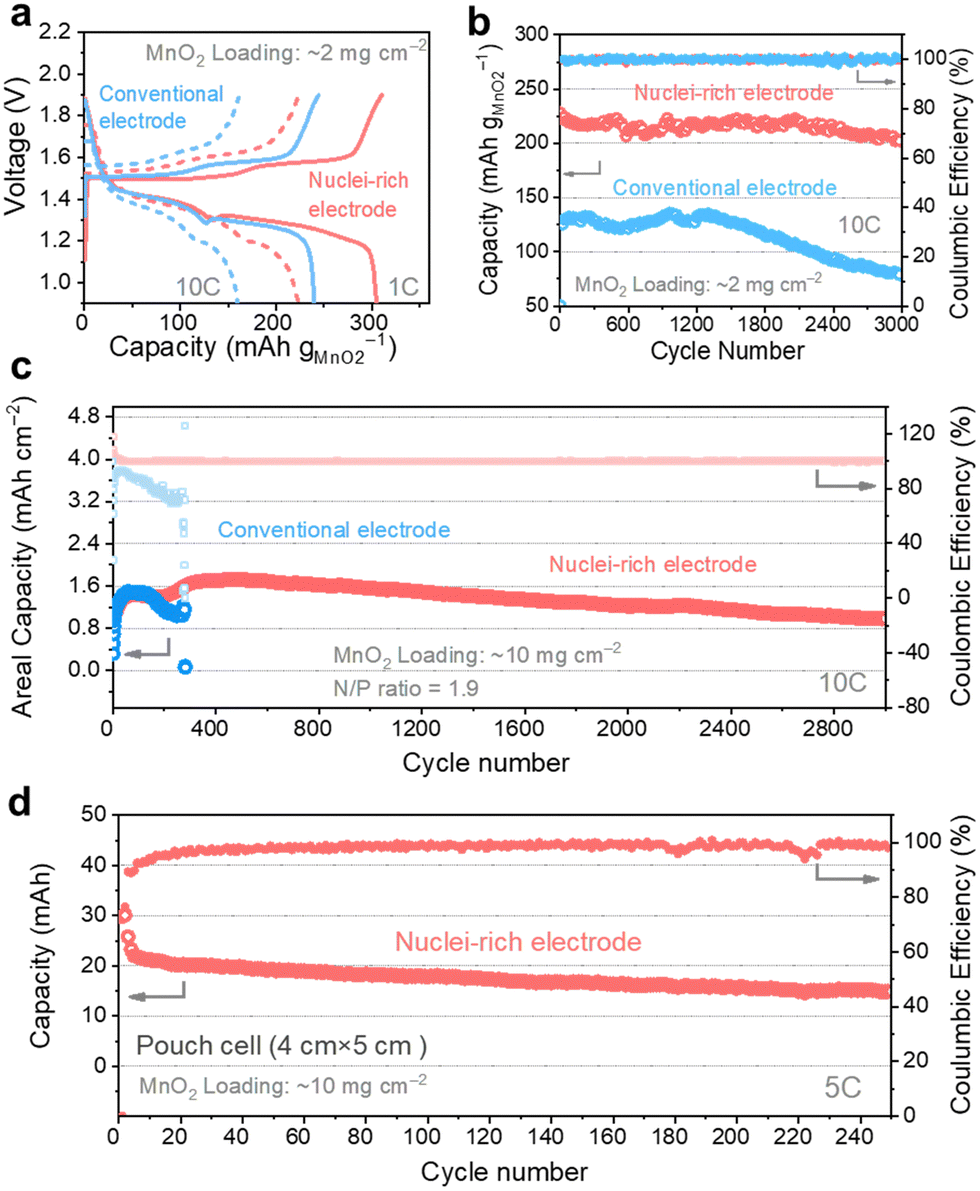 | ||
| Fig. 6 High capacity and high stability Zn–MnO2 full cells enabled by the nuclei-rich anode. (a) Voltage profile of Zn–MnO2 full cells with conventional and nuclei-rich Zn electrodes. The long-term-cycling profile of Zn–MnO2 full cells under different conditions: (b) MnO2 loading at 2 mg cm−2, (c) MnO2 loading at 10 mg cm−2, and Zn foil thickness at 0.01 mm (N/P ratio = 1.9), and (d) MnO2 loading at 10 mg cm−2 in pouch cells (single layer). | ||
To demonstrate the practical feasibility of the nuclei-rich Zn electrode, we decreased the N/P ratio to as low as 1.9 by using a high MnO2 loading of 10 mg cm−2 (3.08 mA h cm−2 theoretical capacity) and a small Zn foil thickness of 0.01 mm (5.86 mA h cm−2 theoretical capacity). At a high operation rate of 10C (Fig. 6c), the nuclei-rich low N/P ratio cell still delivers an average capacity of 1.4 mA h cm−2 for 3000 cycles with a CE of 99.97%. The capacity increase during the first tens of cycles could be attributed to the activation of the cathode material. In comparison, the low N/P ratio cell with a conventional Zn anode can only survive less than 400 cycles under the same condition, which is attributed to the depletion of Zn metal or internal short circuits. To further demonstrate the scalability, we assembled a 20 cm2 (4 cm × 5 cm) single-layer pouch cell. With a 10 mg cm−2 MnO2 cathode (Fig. 6d), the cell delivers a capacity of around 20 mA h at a 5C rate (around 0.3 A). The dendrite-free and cathode-friendly nuclei-rich Zn anode enable stable cycling for more than 250 cycles.
Conclusions
In this work, we propose nuclei-rich Zn plating as a new strategy to inhibit dendrite formation in Zn anodes. By establishing immediate contact between the Zn surface and hydroxyapatite, which possesses high Zn0 adsorption energy and Zn2+ conductivity, a nuclei-incubating interface is constructed to progressively promote nucleation at high rates. The grain size naturally decreases due to the increase in the number of grains, and its further growth is prevented by the depletion of space as fast nucleation results in a high volumetric concentration of nuclei. These small Zn grains are preferentially removed during the subsequent stripping process due to favored oxidation kinetics, forming a reversible cycle. The promoted Zn reduction kinetics also suppresses the competing H2 evolution, and when combined with inhibited dendrite growth, results in a high coulombic efficiency of 99.7%. The high-rate and high-stability nuclei-rich Zn anode is coupled with a MnO2 cathode to demonstrate a low N/P ratio full cell that delivers a capacity of 1.4 mA h cm−1 capacity for 3000 cycles at a high operation rate of 10C. We anticipate that the nuclei-rich plating strategy can inspire future exploration of mechanism modifications in the plating–stripping process of Zn and other important metal anodes, such as lithium and sodium.Author contributions
Q. Z., Z. L., and Y.-C. Lu conceived the project. Q. Z. and Y.-C. Lu designed the experiments. Q. Z. and W. W. performed the SEM characterization. Z. L. performed the OEMS experiments. Q. Z. and D. D. synthesized MnO2. Q. Z. performed other experiments and analysis. Q. Z. and Y.-C. Lu wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work described in this paper was supported by two grants from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region (HK SAR), China, under no. CUHK14308321, and C1002-21G.Notes and references
- C. Grey and J. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- A. Yaroshevsky, Geochem. Int., 2006, 44, 48–55 CrossRef.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Angew. Chem., Int. Ed., 2021, 60, 598–616 CrossRef CAS PubMed.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- W. Lu, C. Xie, H. Zhang and X. Li, ChemSusChem, 2018, 11, 3996–4006 CrossRef CAS PubMed.
- Z. Cao, P. Zhuang, X. Zhang, M. Ye, J. Shen and P. M. Ajayan, Adv. Energy Mater., 2020, 10, 2001599 CrossRef CAS.
- Y. Zuo, K. Wang, P. Pei, M. Wei, X. Liu, Y. Xiao and P. Zhang, Mater. Today Energy, 2021, 20, 100692 CrossRef CAS.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- A. Bayaguud, X. Luo, Y. Fu and C. Zhu, ACS Energy Lett., 2020, 5, 3012–3020 CrossRef CAS.
- Q. Zhang, J. Luan, L. Fu, S. Wu, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15841–15847 CrossRef CAS PubMed.
- G. Chang, S. Liu, Y. Fu, X. Hao, W. Jin, X. Ji and J. Hu, Adv. Mater. Interfaces, 2019, 6, 1901358 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- Y. Fang, X. Xie, B. Zhang, Y. Chai, B. Lu, M. Liu, J. Zhou and S. Liang, Adv. Funct. Mater., 2022, 32, 2109671 CrossRef CAS.
- J. Cao, D. Zhang, C. Gu, X. Wang, S. Wang, X. Zhang, J. Qin and Z. S. Wu, Adv. Energy Mater., 2021, 11, 2101299 CrossRef CAS.
- L. Zeng, H. He, H. Chen, D. Luo, J. He and C. Zhang, Adv. Energy Mater., 2022, 12, 2103708 CrossRef CAS.
- G. Liang, J. Zhu, B. Yan, Q. Li, A. Chen, Z. Chen, X. Wang, B. Xiong, J. Fan, J. Xu and C. Zhi, Energy Environ. Sci., 2022, 15, 1086–1096 RSC.
- C. Deng, X. Xie, J. Han, Y. Tang, J. Gao, C. Liu, X. Shi, J. Zhou and S. Liang, Adv. Funct. Mater., 2020, 30, 2000599 CrossRef CAS.
- Z. Li, W. Deng, C. Li, W. Wang, Z. Zhou, Y. Li, X. Yuan, J. Hu, M. Zhang, J. Zhu, W. Tang, X. Wang and R. Li, J. Mater. Chem. A, 2020, 8, 17725–17731 RSC.
- Y. Yin, S. Wang, Q. Zhang, Y. Song, N. Chang, Y. Pan, H. Zhang and X. Li, Adv. Mater., 2020, 32, 1906803 CrossRef CAS PubMed.
- R. Sato, J. Electrochem. Soc., 1959, 106, 206 CrossRef CAS.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang and D. C. Bock, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- L. Enze, J. Phy. D. Appl. Phys., 1986, 19, 1 CrossRef CAS.
- Y. Zhang, X. Han, R. Liu, Z. Yang, S. Zhang, Y. Zhang, H. Wang, Y. Cao, A. Chen and J. Sun, Small, 2022, 18, 2105978 CrossRef CAS PubMed.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- C. Stötzel, F. Müller, F. Reinert, F. Niederdraenk, J. Barralet and U. Gbureck, Colloids Surf. B., 2009, 74, 91–95 CrossRef PubMed.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- M. Tylka, J. Willit and M. Williamson, J. Electrochem. Soc., 2017, 164, H5327 CrossRef CAS.
- Z. Li, Y. Zhou, Y. Wang and Y. C. Lu, Adv. Energy Mater., 2019, 9, 1802207 CrossRef.
- C. K. Kwok and S. B. Desu, J. Mater. Res., 1993, 8, 339–344 CrossRef.
- A. Nayak and B. Bhushan, Mater. Today: Proc., 2021, 46, 11029–11034 CAS.
- L. Geng, J. Meng, X. Wang, C. Han, K. Han, Z. Xiao, M. Huang, P. Xu, L. Zhang, L. Zhou and L. Mai, Angew. Chem. Int. Ed., 2022, 61, e202206717 CrossRef CAS PubMed.
- M. D. Morris and G. S. Mandair, Clin. Orthop. Relat. Res., 2011, 469, 2160–2169 CrossRef PubMed.
- H. Yang, Z. Chang, Y. Qiao, H. Deng, X. Mu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2020, 132, 9463–9467 CrossRef.
- J. D. Schmit and K. Dill, J. Am. Chem. Soc., 2012, 134, 3934–3937 CrossRef CAS PubMed.
- K. Qi, W. Zhu, X. Zhang, M. Liu, H. Ao, X. Wu and Y. Zhu, ACS Nano, 2022, 16, 9461–9471 CrossRef CAS PubMed.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- H. He and J. Liu, J. Mater. Chem. A, 2020, 8, 22100–22110 RSC.
- Y. Tian, Y. An, C. Liu, S. Xiong, J. Feng and Y. Qian, Energy Storage Mater., 2021, 41, 343–353 CrossRef.
- S. Zhou, Y. Wang, H. Lu, Y. Zhang, C. Fu, I. Usman, Z. Liu, M. Feng, G. Fang and X. Cao, Adv. Funct. Mater., 2021, 31, 2104361 CrossRef CAS.
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2021, 31, 2001867 CrossRef CAS.
- S. H. Park, S. Y. Byeon, J.-H. Park and C. Kim, ACS Energy Lett., 2021, 6, 3078–3085 CrossRef CAS.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, 2007406 CrossRef CAS PubMed.
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, R. Huang, G. Wei, A. Liu, L. Li and R. Chen, Adv. Mater., 2021, 33, 2101649 CrossRef CAS PubMed.
- Z. Cai, Y. Ou, B. Zhang, J. Wang, L. Fu, M. Wan, G. Li, W. Wang, L. Wang and J. Jiang, J. Am. Chem. Soc., 2021, 143, 3143–3152 CrossRef CAS PubMed.
- Z. Guo, L. Fan, C. Zhao, A. Chen, N. Liu, Y. Zhang and N. Zhang, Adv. Mater., 2022, 34, 2105133 CrossRef CAS PubMed.
- D. Wang, H. Liu, D. Lv, C. Wang, J. Yang and Y. Qian, Adv. Mater., 2023, 35, 2207908 CrossRef CAS PubMed.
- G. Li, X. Wang, S. Lv, J. Wang, W. Yu, X. Dong and D. Liu, Adv. Funct. Mater., 2023, 33, 2208288 CrossRef CAS.
- Q. Cao, Y. Gao, J. Pu, X. Zhao, Y. Wang, J. Chen and C. Guan, Nat. Commun., 2023, 14, 641 CrossRef CAS PubMed.
- W. Xu, X. Liao, W. Xu, K. Zhao, G. Yao and Q. Wu, Adv. Energy Mater., 2023, 13, 2300283 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li and P. Bhattacharya, Nat. Energy, 2016, 1, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03246a |
| This journal is © The Royal Society of Chemistry 2023 |