
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-crystallisation as a modular approach to the discovery of spin-crossover materials†
Lee T.
Birchall
 ,
Giada
Truccolo
,
Lewis
Jackson
and
Helena J.
Shepherd
*
,
Giada
Truccolo
,
Lewis
Jackson
and
Helena J.
Shepherd
*
School of Physical Sciences, University of Kent, Canterbury, UK. E-mail: h.j.shepherd@kent.ac.uk
First published on 18th February 2022
Abstract
Herein we present co-crystallisation as a strategy for materials discovery in the field of switchable spin crossover (SCO) systems. Using [Fe(3-bpp)2]·2A (where 3-bpp = 2,6-bis(pyrazol-3-yl)pyridine, A = BF4−/PF6−) as a starting point, a total of 11 new cocrystals have been synthesised with five different dipyridyl coformers. Eight of these systems show spin crossover behaviour, and all show dramatically different switching properties from the parent complex. The cocrystals have been studied by variable temperature single-crystal X-ray diffraction and SQUID magnetometry to develop structure–property relationships. The supramolecular architecture of the cocrystals depends on the properties of the coformer. With linear, rigid coformer molecules leading to 1D supramolecular hydrogen-bonded chains, while flexible coformers form 2D sheets and bent coformers yield 3D network structures. The SCO behaviour of the cocrystals can be modified through changing the coformer and thus co-crystallisation presents a rapid, facile and highly modular tool for the discovery of new switchable materials. The wider applicability of this strategy to the design of hybrid multifunctional materials is also discussed.
Introduction
The spin-crossover (SCO) phenomenon is observed in first row transition metal complexes (d4–d7), where changes in temperature, pressure, light, and the presence of guest molecules can result in a reversible change between high spin (HS) and low spin (LS) states.1,2 This spin state switching leads to changes in magnetic properties and colour of the material as well as significant differences in the structure. Such switchable properties lead to potential applications for these materials in sensing,3 display4 and actuator technologies.5 SCO properties in the solid state are strongly dependent on the structure of the material, with abrupt spin transitions occurring in systems with a high degree of communication between SCO sites.1 Highly cooperative systems can result in hysteresis in the switching temperature, leading to a ‘molecular memory’ effect.6 Communication between SCO centres takes the form of elastic interactions in the solid state, usually interpreted as strong intermolecular interactions between switching centres.7 Where cooperativity is weaker, more gradual and incomplete transitions are observed, while more complex effects lead to multi-step SCO profiles.One of the most significant challenges in the field of SCO is predicting the temperature at which individual materials undergo switching as well as tailoring it towards the requirements of each application. In some cases it has been possible to relate the ligand field strength with the temperature at which SCO occurs (T½),8 but intra- and intermolecular interactions also play a substantial role in changing the relative stability of HS and LS states,9,10 making prediction of properties difficult. This is clearly illustrated by the fact that different polymorphs of the same molecular SCO material are known to have significantly different transition temperatures or degrees of cooperativity.11 Previously explored strategies for tuning the SCO behaviour of materials without changing the coordination environment around the metal centre include the incorporation of different solvent molecules into the crystal structure,12–14 changing non-coordinating anions15–19 and modifying remote ligand substituents.9,20 However, each of these approaches has limitations. For example, solvates of molecular materials tend to have low thermal stability compared to the parent compound and the choice of solvent incorporated is limited by solubility considerations. Changing the anion is limited by the comparatively low number of small, non-coordinating anions commercially available, and modifying ligands adds synthetic complexity, which is a significant disadvantage for large-scale applications of these systems. Here we propose using co-crystallisation of SCO materials with commercially available coformers to change the supramolecular organization of switching units as a simple and effective way to modify SCO properties and simultaneously improve our understanding of the factors affecting SCO in the solid-state.
Co-crystallisation involves supramolecular aggregation of two or more different molecules to achieve a range of outcomes including solid solutions, solvates, molecular salts and cocrystals.21 The term ‘cocrystal’ has been rather more difficult to define, due in part to the regulatory environment of the pharmaceutical industry in which much early work into cocrystals was conducted.22 Without wishing to reiterate the arguments involved in that discussion, for clarity we define a ‘SCO-cocrystal’ as a crystallographically ordered combination of a SCO-active material (either salt or neutral species) and a second material known as a coformer, which is a solid at ambient conditions. One significant advantage of co-crystallisation that has been exploited extensively in organic materials and pharmaceutical applications is that there is an element of rational design in their synthesis through the use of functional groups with known supramolecular complementarity (so-called supramolecular synthons) of the material and coformer.23 Such a degree of control over molecular aggregation may one day offer the ability to design molecular SCO materials with properties tailored to specific applications. Changing the coformer is significantly more facile than covalent modification of ligands and thus there is huge scope for rapid materials discovery, investigation of structure–property correlations in a systematic manner and even the development of hybrid materials through the use of active coformers.
Previously, an elegant series of halogen-bonded SCO-cocrystal networks have been presented in which the structure-directing halogen bonding interactions occur in the second coordination sphere between the coformers, anions, and solvent, without directly bridging SCO moieties.24–27 Studies of SCO-cocrystals where the coformer interacts directly with the SCO complex also exist, but focus on the effect of chemical modification of the material rather than modifying the supramolecular architecture,28 or were discovered serendipitously rather than as a result of systematic investigation.29 There are also examples of what may be considered ionic co-crystals, in which the organic coformer also acts as a counterion.18,19,30 In some cases, for example, when TCNQδ− was used, additional properties such as (semi)conducting behaviour have been achieved.31–38 However, examples of ionic SCO-cocrystals are often highly solvated, and difficulties with reproducibility and elucidation of structure–property correlations have been associated with desolvation effects. The use of neutral rather than charged molecules would allow the incorporation of a much wider range of coformers to be employed, increasing the modularity of the approach.
The SCO system we focus on here is [Fe(3-bpp)2]·2A (where 3-bpp = 2,6-di(pyrazol-3-yl)pyridine, A = BF4− (1a) or PF6− (1b)), which (along with many derivatives) is well-known in the field of SCO materials.39,40 These systems have been investigated for several decades and their SCO behaviour is highly dependent on the presence of water or other solvents of crystallisation, as well as the anion present.39 In recent work, this cation has been paired with carboxylate anions to form salts in which the anion bridges directly between cations.18,19,30 The unsolvated parent complexes used here, 1a and 1b have both been shown to be SCO active. 1a undergoes an abrupt spin transition with a 10 K hysteresis (T½ = 173 K on cooling and 183 on heating).411b shows a relatively abrupt, but incomplete SCO at 177 K without hysteresis.42
The system was chosen due to the presence of four hydrogen-bond donating pyrazole moieties with a well-defined relative disposition. Both the BF4− and PF6− counter-ions were used due to their relatively low charge density to minimize potential competition for hydrogen bonding to the pyrazole hydrogen bond donors. The ditopic pyridine-based molecules 4,4′-bipyridine (bipy), 1,2-di(4-pyridyl)ethylene (bpe), 1,2-bis(4-pyridyl)ethane (bpa), 4,4′-azopyridine (azp) and 2,2′-dipyridyl disulfide (dpds) (Fig. 1) were selected as coformers due to the expected formation of pyrazole–pyridine hydrogen bonds as supramolecular synthons. Three of these coformers (bipy, bpe and azp) are expected to be relatively linear and rigid in solution as well as in the solid state. The alkyl-containing bpa is expected to be more flexible, while the bent structure of the dpds allows for a greater diversity of possible supramolecular architectures. The aim of this specific study is to modify the crystal packing via co-crystallisation with a series of neutral coformers in a modular and systematic manner, and then to determine how the structural modifications affect switching behaviour. The long-term goal of this new approach is to use co-crystallisation (particularly its demonstrated elements of supramolecular control) to design SCO systems with specific properties tailored to individual applications.
 | ||
| Fig. 1 Structures of (a) 2,6-bis(pyrazol-3-yl)pyridine (3-bpp), (b) 1,2-di(4-pyridyl)ethylene (bpe), (c) 4,4′azopyridine (azp) (d) 4,4′-bipyridine (bipy), (e) 1,2-bis(4-pyridyl)ethane (bpa), and (f) 2,2′-dipyridyl disulfide (dpds). | ||
Results and discussion
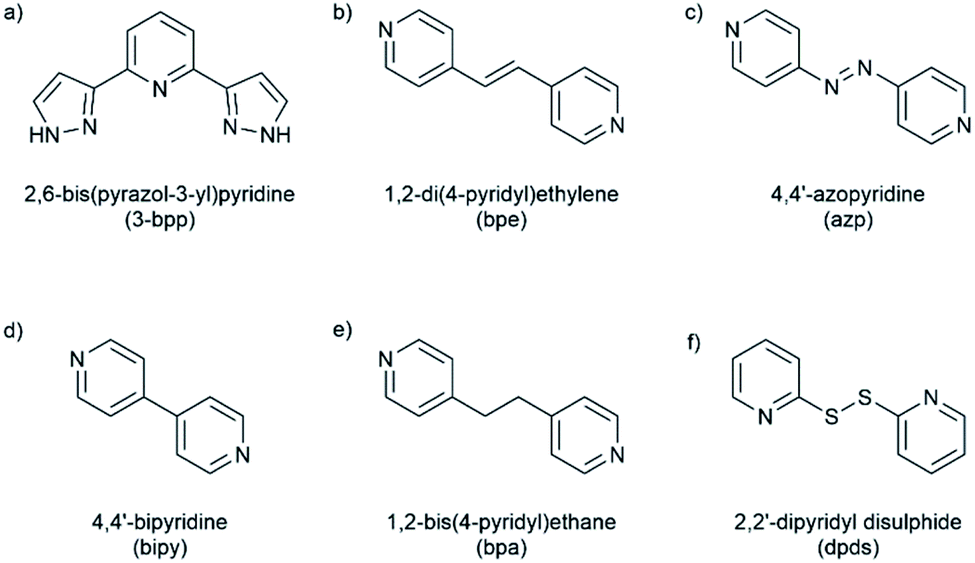
All cocrystals were grown through slow evaporation of a methanolic solution of 1a or 1b and the appropriate coformer in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometric ratio. Full experimental details, including synthetic protocols are provided in the experimental section of the ESI. Eleven novel cocrystals were successfully synthesized through co-crystallisation of [Fe(3-bpp)2][BF4]2 (1a) and [Fe(3-bpp)2][PF6]2 (1b) with the ditopic coformers shown in Fig. 1. In 1, the 3-bpp ligands are orthogonal and the H-bond donors on the two ligands point in opposite directions. In all cocrystals, the coformers bridge between adjacent cations of 1via hydrogen-bonding to form infinite one-dimensional (1D) chains, 2D sheets or 3D networks, examples of which are shown in Fig. 2.
:2 stoichiometric ratio. Full experimental details, including synthetic protocols are provided in the experimental section of the ESI. Eleven novel cocrystals were successfully synthesized through co-crystallisation of [Fe(3-bpp)2][BF4]2 (1a) and [Fe(3-bpp)2][PF6]2 (1b) with the ditopic coformers shown in Fig. 1. In 1, the 3-bpp ligands are orthogonal and the H-bond donors on the two ligands point in opposite directions. In all cocrystals, the coformers bridge between adjacent cations of 1via hydrogen-bonding to form infinite one-dimensional (1D) chains, 2D sheets or 3D networks, examples of which are shown in Fig. 2.
 | ||
| Fig. 2 Structures of (a) 1a·bipy at 250 K showing AB-type 1D chain structure (b) 1b·azp(β) showing CC-type 1D chain structure, (c); 1b·bpa showing 2D sheet structure and (d) 1b·dpds showing 3D network formation. Iron atoms are shown as red spheres, 3-bpp ligands are shown in blue, coformers are shown in green and hydrogen bonding interactions are shown as red dotted lines. Symmetry equivalent coformers are coloured the same shade of green. All counterions and hydrogen atoms not involved in hydrogen bonding are omitted for clarity. | ||
When 1a and 1b are co-crystallised with bipy, bpe and azp, they form infinite 1D chains through hydrogen bonding interactions. When bpa is used, the cocrystals consist of 2D hydrogen-bonded sheets, while the dpds coformer leads to a 3D hydrogen-bonded network with 1b. The supramolecular architecture of each of the cocrystals along with their crystallographic space groups and SCO properties are summarized in Table 1. Eight of the cocrystals are SCO-active, with three in the HS state at all investigated temperatures. In all cases the SCO properties are distinct from the parent complex, demonstrating that co-crystallisation can be used to modify SCO properties. We begin this section with a discussion of the different supramolecular architectures observed across the series before describing the details of each compound separately. We then address structure–property relationships across the series, looking at the effect of coformer choice on the SCO properties.
| Coformer | Linear, rigid | Flexible | Bent | ||||
|---|---|---|---|---|---|---|---|
| bpe | azp | bipy | bpa | dpds | |||
| [Fe(3-bpp)2][BF4]2 | Co-crystal abbreviation | 1a·bpe | 1a·azp(α) | 1a·azp(β) | 1a·bipy | 1a·bpa | — |
| Supramolecular structure | 1D chain CC | 1D chain AB | 1D chain CC | 1D chain AB (<340 K) CC (≥340 K, irreversible) | 2D layers | — | |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P (<340 K), P21/n (≥340 K, irreversible) |
I2/a | — | |
| SCO activity | HS at RT. Gradual incomplete SCO | HS at RT. Stepped SCO | HS at RT. Incomplete SCO | Gradual incomplete SCO. Profile changes after irreversible transition | HS at RT. No SCO observed | — | |
| [Fe(3-bpp)2][PF6]2 | Co-crystal abbreviation | 1b·bpe | 1b·azp(α) | 1a·azp(β) | 1b·bipy | 1b·bpa | 1b·dpds |
| Supramolecular structure | 1D chain AB | 1D chain AB | 1D chain CC | 1D chain AB | 2D layers | 3D network | |
| Space group |
P |
P |
P21/n | C2/m | P21 | P3221 | |
| SCO activity | HS at RT. Abrupt stepped SCO with hysteresis | Stepped SCO with small hysteresis | HS at RT. Not SCO active | LS at RT. SCO above 420 K | HS at RT. Abrupt SCO with hysteresis | HS at RT. Not SCO active | |
Supramolecular architecture
The eight 1D chain cocrystals can be divided into two sub-groups, denoted AB- and CC-type chains, depending on the crystallographic equivalence of different coformers. In the chains labelled AB, the pair of coformers that bridge between any two cations are crystallographically equivalent. These pairs are crystallographically distinct from the adjacent pair along the chain. As a consequence, AB-type chains have two distinct Fe–Fe distances along the chain. By contrast, the chains labelled CC are formed through pairs of inequivalent coformers bridging between two cations, but in this case coformer pairs are identical along the chain and thus all Fe–Fe distances along the chain are identical. AB-type chains have P symmetry, while CC-type chains have P21/c (or P21/n) space group symmetry. In both cases all cations of 1 are equivalent at room temperature, with one [Fe(3-bpp)2]2+ cation, two anions and two coformers in the asymmetric unit. The exception to this classification is 1b·bipy (AB-type), which crystallises in the space group C2/m, with half a cation, two anions and two coformers (the anions and cations are located on special positions with occupancy of 50%) in the asymmetric unit. In this case, although AB-type ordering is observed with alternating Fe–Fe distances along the chain, there is additional internal symmetry imposed on the system due to the different space group. In some of the 1D chain cocrystals, the coformers show a ‘pedal motion’ type of disorder, which is common in these molecules in the solid state.43 Examples of AB- and CC-type 1D chains are presented in Fig. 2. BF4− or PF6− anions are located between chains and do not form any hydrogen-bonding interactions with either the cation or the coformers.
Cocrystals of [Fe(3-bpp)2][BF4]2 (1a)
Before general structure–property correlations can be drawn it is important to understand the SCO behaviour of each specific compound. A fragment of each crystal structure and graphs showing the thermal SCO behaviour for each cocrystal of 1a are shown in Fig. 3. | ||
| Fig. 3 Fragments of the crystal structures of each coformer of 1a (left) and curves indicating their SCO behaviour (right). (a) CC-type 1D chain structure observed in 1a·bpe at 290 K. (b) AB-type 1D chain structure of 1a·azp(α) at 270 K. (c) CC-type 1D chain structure of 1a·azp(β) at 270 K (d) AB-type 1D chain structure observed in 1a·bipy at 250 K (e) 2D supramolecular sheet structure of 1a·bpa at 150 K. Atomic displacement parameters are drawn at 50% probability. | ||
:2 stoichiometric ratio gave rise to the formation of concomitant cocrystal polymorphs, denoted 1a·azp(α) and 1a·azp(β). Crystals of each polymorph can be easily distinguished visually; crystals of the α-polymorph are yellow, with a plate-like morphology, while crystals of the β-polymorph are darker and form large block-like parallelepipeds. During crystallization, the β-polymorph is the major component, greatly outweighing the proportion of the α-polymorph obtained. Consequently, it was not possible to isolate a pure sample of the α-polymorph in sufficient quantities for analysis via magnetometry. A sample of the β-polymorph was analysed using squid magnetometry, although the possibility that this sample was contaminated with a very small quantity of the α-polymorph cannot be excluded.
The first polymorph, 1a·azp(α) crystallizes in the P space group with AB-type ordering of the 1D supramolecular chains. In the absence of a phase pure sample of the α polymorph, magnetometry measurements were not performed. Instead, the volume of the Fe–N6 coordination octahedron was determined as a function of temperature using single-crystal X-ray diffraction measurements as a way to monitor the SCO behaviour, as shown in Fig. 3b. At 270 K the material is in the HS state (VFe–N6 = 12.51(1) Å3) and undergoes a partial SCO centered around 210 K to a mixed state with c.a. 55% HS and 45% LS centres (VFe–N6 = 11.17(1) Å3). On further cooling the remaining HS sites undergo SCO, located around 120 K to a fully LS state (VFe–N6 = 9.87(1) Å3 at 80 K). Despite careful inspection of the diffraction pattern, no supercell reflections that might indicate a symmetry-breaking transition with an ordered mixed spin state intermediate phase were observed. However, an isostructural phase transition was observed between 130 and 120 K, associated with the second step in the SCO. The transition does not affect the number of independent Fe centres, and they remain equivalent throughout. The AB-type chain topology is also retained, but these chains become more linear during the transition to the low temperature phase, as shown in Fig. 4. Unit cell parameters for this and all other reported complexes are provided in the ESI.†
 | ||
| Fig. 4 1D chain structure of 1a·azp(α) in the (a) high temperature and (b) low temperature phases showing increased linearity of chains in low temperature phase. | ||
The second polymorph, 1a·azp(β), also forms 1D supramolecular chains, but with CC-type ordering of coformers rather than AB-type as observed in the α-polymorph. The difference between AB and CC-type ordering is explained in detail above and shown schematically in Fig. 2. At 270 K, the structure is in the HS state, as identified from the volume of the Fe–N6 octahedron (VFe–N6 = 12.43(1) Å3). On cooling the cocrystal undergoes a partial SCO to a mixed spin state comprising approximately 50% HS and 50% LS at 80 K (VFe–N6 = 10.89(1) Å3). Magnetometry measurements confirm this incomplete SCO behaviour, with a gradual SCO from the HS state to a mixed spin state located around 210 K, which is the same temperature range as the first step in the stepped SCO observed for the α-polymorph. The measurement was performed on a sample that may have been contaminated with a very small fraction of the α-polymorph, reflected in a small decrease in χMT around 125 K, which corresponds to the second step in the SCO of the α-polymorph. The β-polymorph does not show a second step in the SCO, remaining in the mixed spin state, as confirmed by single crystal diffraction measurements. A powder diffraction pattern of the measured sample and that simulated from the α and β-polymorphs is shown in the ESI, Fig. S8.† Again, it is worth noting that there was no evidence of symmetry breaking or ordering of the mixed spin state in this material.
On a second thermal cycling, the evolution of χMT is significantly different from that initially observed. The abrupt region of the SCO disappears and is instead replaced by a more gradual transition between 400 and 200 K. There is also a higher residual HS fraction at low temperatures recorded during the second thermal cycle, observed in both the magnetic data and with an increased VFe–N6 10.73(1) Å3 at 300 K after thermal cycling. Inspection of structural parameters provides an explanation as to the differences between the first and second thermal cycles observed in the magnetic data. On warming from room temperature, single crystals of 1a·bipy undergo an irreversible phase transition from P to P21/n at 340 K. The effect is to transform the 1D chain structure from AB-type observed at temperatures below 340 K to CC-type at higher temperatures. On cooling back to 300 K, the CC-type structure is preserved, revealing the phase transition to be irreversible. The CC-type chain structure has a more gradual transition than the initially observed AB-type, and it may be assumed that it is the more thermodynamically stable phase in this specific system due to the irreversible nature of this transition.
1a·bpa (Fig. 3e) crystallises in the space group I2/a, with only half of the complex and one coformer present in the asymmetric unit. The structure consists of 2D supramolecular layers formed through the hydrogen bonding between 1a complexes and bpa coformers, a fragment of which is shown in Fig. 5e. Magnetic measurements reveal that the cocrystal does not exhibit SCO behaviour and remains in the HS state on cooling from 290 K, where it has a χMT value of 3.17 cm3 K mol−1.
 | ||
| Fig. 5 Fragments of the crystal structures of each coformer of 1b (left) and curves indicating their SCO behaviour (right). (a) AB-type 1D chain structure observed in 1b·bpe at 200 K (on cooling). (b) AB-type 1D chain structure of 1b·azp(α) at 270 K. (c) CC-type 1D chain structure of 1b·azp(β) at 250 K (d) AB-type 1D chain structure observed in 1b·bipy at 298 K (e) 2D supramolecular sheet structure of 1b·bpa at 190 K (f) 3D supramolecular network structure of 1b·dpds at 80 K. Atomic displacement parameters are drawn at 50% probability. | ||
Cocrystals of [Fe(3-bpp)2][PF6]2 (1b)
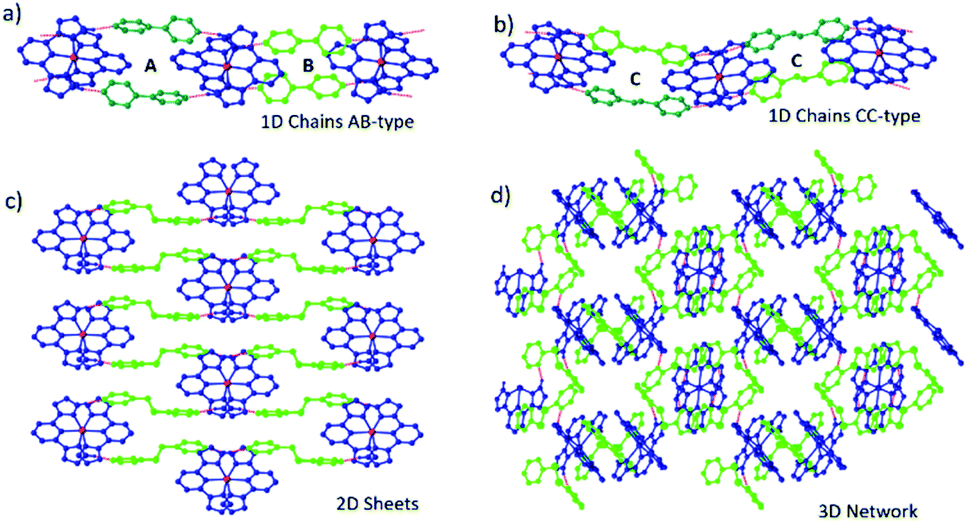
space group with AB ordering of the 1D supramolecular chains, unlike its tetrafluoroborate analogue. Squid magnetometry data show that at room temperature the cocrystal is in the HS state and undergoes an abrupt spin transition on cooling to a 50% HS, 50% LS intermediate phase (IP), which is centred at 178 K. On further cooling, the material undergoes a further abrupt spin transition to a fully LS phase at 155 K. Both transitions are reversible on heating, with hystereses of c.a. 5 K and 34 K for the low and high temperature steps respectively.
Single crystal diffraction data reveal that both steps in the spin transition of 1b·bpe are accompanied by structural phase transitions, with the fully HS and LS phases being isostructural, and in each case all iron centres are equivalent. By contrast, the diffraction pattern of the IP is characterized by supercell reflections, which result in a doubling of the unit cell volume, indicative of a symmetry breaking phase transition as shown in Fig. 6. There are two crystallographically distinct iron centres in the IP, one of which is HS and the other LS, leading to long-range ordering of –HS–LS– sites along the supramolecular chains. In the IP, the 1D hydrogen-bonded chain structure persists, but due to the symmetry breaking phase transition it is neither AB nor CC-type, despite apparent similarities with both. Each pair of coformers that bridge between the same two cations are crystallographically distinct as in the case of CC-type chains. However, adjacent pairs of coformers along the chain are also crystallographically distinct, as in the case of AB-type chains.
 | ||
| Fig. 6 (a) Schematic illustration of structural pathway between HS and LS phases, which both display AB supramolecular chain architectures and the IP, which contains four crystallographically distinct coformers. (b) Variation in unit cell lengths as a function of temperature. Unit cell angles are provided in the ESI† (c) view of the cocrystal in the IP showing long-range order of HS (yellow) and LS (red) sites along the supramolecular chains. | ||
Further detail regarding single crystal diffraction measurements is provided in the ESI.† Small differences in the absolute temperature of the transition measured using diffraction or magnetometry methods are attributed to ensemble averaging effects in the polycrystalline sample used for squid magnetometry and different regimes of cooling/heating between the two measurements.
1b·azp(α) crystallizes in P with AB ordering in the 1D supramolecular chains. In general the structural and SCO behaviour are very similar to those of the BF4− analogue, with a stepped SCO profile without observed structural phase transitions or symmetry breaking. The difference from the BF4− analogue is that the SCO in 1b·azp(α) is shifted to higher temperatures by approximately 100 K, with the high temperature step being centred around 298 K and the low temperature step around 233 K. There is no hysteresis in the low temperature step, but the high temperature step shows an asymmetry with a more gradual profile and shift to higher temperature (309 K) on heating.
The 1D supramolecular chains in 1b·azp(β) are of the CC type and the structure crystallizes in the P21/n space group. The cocrystal was analysed by SCXRD and found to be in the HS state at 250 K. The crystal was cooled down slowly to 100 K, but no evidence of SCO was observed. In the absence of a pure sample to measure magnetometry data it is assumed that if 1b·azp(β) does undergo partial SCO as in the case of the 1a analogue, it must occur below 100 K.
1b·bipy is the only material in the series to crystallise in the LS state at room temperature, and shows a high symmetry form of the AB-type 1D chain topology. Upon heating up to 400 K, no evidence of SCO was observed using X-ray diffraction measurements. On heating to approximately 430 K the crystal changes colour from red to yellow, which is the typical colour change observed on SCO in these materials. Unfortunately, this colour change was accompanied by a severe deterioration in the quality of the diffraction pattern and further analysis was not possible. The SCO behaviour of the cocrystal was assessed using SQUID magnetometry, which showed the crystal remains essentially in the LS state between 0 K and 400 K, with a small HS component. Regrettably it was not possible to measure higher temperatures using magnetometry, but we are confident that the material is SCO active and also undergoes an irreversible phase transition in an analogous manner to the 1a analogue. The colour of the crystal on cooling from 430 K to room temperature is very different from that observed prior to heating, as shown in ESI Fig. S21.† The argument for the presence of a high temperature SCO is supported in light of the high temperature of the SCO observed in the analogous 1a·bipy material and the fact that on moving from 1a to 1b, the SCO shifts 100 K higher in temperature for the azp(α) polymorphs, which also show AB-type ordering.
1b·bpa crystallises in the P21 space group with one [Fe(3-bpp)2]2+ cation and two crystallographically distinct bpa coformers present in the asymmetric unit. Each cation is bridged to four others through the hydrogen bonded coformers, resulting in 2D supramolecular sheets, with the same hydrogen-bonding topology as those seen in the BF4− analogue 1a·bpa, despite the difference in crystallographic space group and contents of the asymmetric unit. Differences in symmetry from the 1a analogue result from different conformations of the flexible C2H4 linking moiety of the bpa coformer. Explicit comparisons of the packing in 1b·bpa and 1a·bpa are shown in the ESI Fig. S23.†
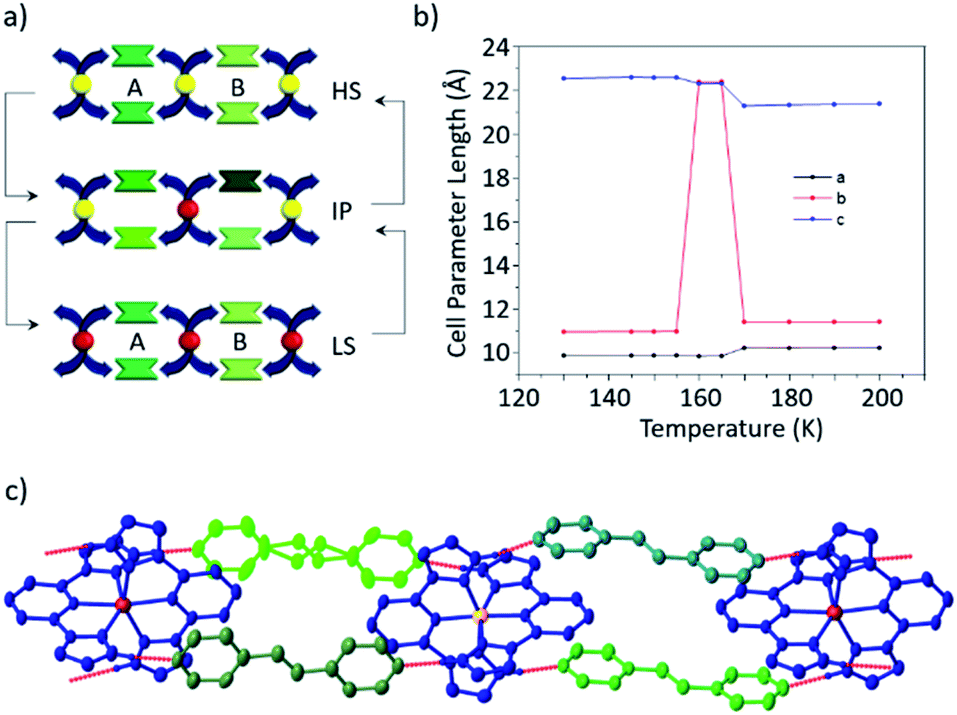
Magnetometry measurements indicate that this sample shows an abrupt and reversible spin transition centred at 128 K on cooling and 180 K on warming, resulting in a hysteresis of 50 K. Crystallographic investigations reveal a very abrupt transition in single crystal samples (Fig. 7), but with a comparatively smaller hysteresis of 30 K. We attribute these differences to kinetic effects observed in this sample (vide infra). The spin transition is accompanied by a crystallographic phase transition to a unit cell with double the volume and concomitant twinning. The result is that adjacent 2D supramolecular layers are no longer crystallographically equivalent, despite the fact that both Fe atoms are in the LS state.
 | ||
| Fig. 7 (a) Side-view of 2D layers of 1b·bpa in the HS state where all Fe centres are identical (b) side-view of 2D layers of 1b·bpa in the LS state where adjacent layers are non-equivalent. Different shades of blue represent crystallographic equivalence of cations. Other colours are as in Fig. 2. (c) Octahedral volume as a function of temperature derived from single crystal diffraction measurements. Points shown in blue correspond to measurements made on warming after flash freezing to 80 K. Where there are two independent cations in the LS state the volume has been determined by an average of the two independent values. | ||
During investigation of the structural properties, it became clear that flash freezing single crystals of 1b·bpa leads to isolation of a metastable HS state as a result of thermally-induced excited spin-state trapping (TIESST) effects, which have previously been observed in solid solutions of 1a.45 The metastable HS state recorded after flash freezing the crystal from room temperature to 80 K is isostructural with the thermal HS structure at higher temperatures. The metastable HS state decays to the thermal LS ground state by 95 K on heating. We attribute the asymmetry of the thermal transition curve observed via magnetometry and small residual HS component observed in those measurements to the close thermal proximity of T½ on cooling and TTIESST, as has been discussed previously.45,46
 | ||
| Fig. 8 Distortion of the [Fe(3-bpp)2]2+ cation in the HS state structures of 1b·dpds and 1b·bpa at 80 K. Both molecules are viewed down one Fe–N{pyridine} bond. | ||
Structure–property correlations
We attribute the difference in supramolecular architecture to the difference in geometry and flexibility of the coformer molecules, examples of which are shown in Fig. 9. When 1a and 1b are co-crystallised with the rigid, linear coformers (bipy, bpe and azp), they form infinite 1D chains through hydrogen bonding interactions. When the flexible bpa coformer is used, the cocrystals consist of 2D hydrogen-bonded sheets, while the bent dpds coformer leads to a 3D hydrogen-bonded network with 1b. Clearly changing the relative disposition of the two hydrogen-bond acceptors on the dipyridyl coformers leads to a change in the supramolecular assembly. Careful choice of coformers with specific geometries and properties (length, degree of conjugation, flexibility etc.) should lead to the ability to design the supramolecular architecture to yield specific SCO properties. It is clear however that many more examples of SCO-cocrystals, along with computational modelling will be required to achieve this goal. | ||
| Fig. 9 Examples of (a) linear, rigid coformer (bpe), (b) linear, flexible coformer (bpa) and (c) the bent dpds coformer. | ||
Two of the cocrystals (1b·bpa and 1b·bpe) show remarkably abrupt spin transitions, indicative of a high degree of cooperativity within the structures. This is perhaps surprising when considering the lower density of SCO centres in a SCO-cocrystal than in the pure SCO material. However, the careful choice of complimentary coformers ensures that SCO sites are linked together through structure-directing hydrogen-bonding interactions, providing a pathway for propagation of strain during the SCO. In such cases, cooperativity can clearly be maintained in SCO-cocrystals.
The 1D chain cocrystals can be categorized into two types, AB and CC, depending on the crystallographic equivalence of bridging coformers as described above. Those with AB-type ordering of coformers (1a·azp(α), 1b·azp(α) and 1b·bpe) all undergo stepped spin crossover. By contrast, those with CC-type crystallographic ordering (1a·azp(β) and 1b·bpe) undergo a variety of SCO behaviours, but are generally more gradual and incomplete, or in the case of 1b·azp(β), do not show SCO properties. This distinction is confirmed by 1a·bipy, which shows stepped SCO when it has AB-type ordering, but changes to a smooth, gradual and incomplete behaviour after undergoing a structural phase transition to CC-type ordering. We attribute the stepped SCO in AB-type chains to be the result of the alternating pattern of interactions between coformer pairs and SCO sites that occur along the chain, which give rise to two different Fe–Fe distances. As all interactions between pairs of coformers and SCO sites along the chain are equivalent for CC-type chains, SCO proceeds in a single step. Clearly the relative energies of the AB and CC-type ordering are very similar, as evidenced by the formation of cocrystal polymorphs in the case of the azp coformer, and the interconversion between ordering types shown by 1a·bipy.
Most of the AB 1D chain cocrystals show a stepped SCO behavior, but there are significant differences in cooperativity, which is evidenced by the varying degrees of abruptness seen among their SCO curves. In an effort to understand the different degrees of cooperativity amongst this subset of cocrystals, we have investigated the distortion of the [Fe(3-bpp)2]2+ complex by calculating the Θ distortion parameter (see ESI Table S16†).49 Previous work has suggested that the extent of the distortion seen in HS complexes is due to crystal packing effects and intermolecular interactions within the structure that cause strain.47,48,50 In this isostructural subset of cocrystals, the differences in distortion of the cation must be attributed to the choice of coformer.
In the cocrystal with the most abrupt transition, 1b.bpe, the Θ distortion parameter changes from 540.45° in the HS state to 313.53° in the LS state. This is a very significant change in the distortion and shape of the [Fe(3-bpp)2]2+ complex and is attributed to the rigidity of the bpe molecule. This large difference in distortion between HS and LS states may also explain why the SCO propagates so quickly throughout the structure and generates an ordered intermediate phase. For the 1a·azp(α) and 1b·azp(α) cocrystals, the azp coformers appear more conformationally flexible than bpe. This results in decreased distortion of the complex in the HS state compared to 1b·bpe, with Θ distortion values of 478.25° and 483.71° respectively in the HS state and values of 345.66° and 311.83° respectively in the LS state. This decreased HS state distortion means that the change in shape and distortion of these complexes upon SCO is significantly less than in 1b·bpe, resulting in more gradual transitions.
It should be noted that the rationale for the differences in cooperativity mentioned here are only applicable to the AB 1D chain structures, as the propagation of SCO in different structure types will occur through unique cooperativity pathways. As more examples of SCO-cocrystals are reported using this methodology, the easier it will be to draw conclusions regarding how the structural attributes of the coformer affect the switching properties of the cocrystal. Detailed work to computationally model the strain propagation in these different types of cocrystal would help to further rationalize the varying degrees of cooperativity in these compounds, but is beyond the scope of the present study.
In general, changing anion has a significant effect on the SCO behaviour of the material, but drawing generalized conclusions for the [Fe(3-bpp)2]2+ family of materials has been complicated by the presence of differing degrees of solvation, which result from the unsatisfied hydrogen-bond donors on the pyrazole moieties. While the goal of this study is to explore the effect of co-crystallisation, it is worth noting the effects of moving from BF4− to PF6− in these cocrystal systems. A more comprehensive overview of anion effects can be found elsewhere.39,40,51,52 In the investigated series of cocrystals presented here, the effect of moving from BF4− to PF6− is to stabilize the LS state, either shifting the SCO to higher temperatures as in the case of the α polymorph of the azo cocrystals, or inducing SCO in PF6− systems where the BF4− analogue does not show SCO effects (bpa cocrystals). Perhaps a useful step to simplify this issue would be to use neutral SCO materials to remove the BF4− or PF6− anions and thus reduce the number of components interacting in the solid state. The co-crystallisation approach reported here may also provide a method for overcoming difficulties associated with evaluating anion effects in materials with unsatisfied hydrogen bond donors or acceptors. Coformers have the ability to block the otherwise free hydrogen bond donors of the 3-bpp ligand, inhibiting incorporation of solvent molecules into the lattice and thus allowing a more direct comparison across a series of unsolvated isostructural analogues.
Conclusions and outlook
Co-crystallisation of [Fe(3-bpp)2]2+ cations with dipyridyl coformers causes the formation of a variety of supramolecular architectures through self-assembly via hydrogen-bonding interactions. The relative disposition of the coformers determines the hydrogen-bonding topology as well as the supramolecular dimensionality of the resultant hybrid materials. The SCO properties of the cation are modified dramatically by these changes in crystal packing. Linear, rigid coformers produce 1D supramolecular chains, flexible coformers can lead to 2D sheets and bent coformers can produce 3D networks of hydrogen bonding in this specific system.The number of commercially available potential coformers with appropriate chemical functionality is vast and thus the potential for discovery of new SCO materials, using only the [Fe(3-bpp)2]2+ cation is huge. Building a library of SCO-cocrystals may allow the development of explicit correlations between chemical properties of the coformer and physical properties of the spin state switching, which can be further applied in the design of new materials.
Thus, of even greater importance to the wider materials science community is the potential for this strategy in the discovery of new systems with specific switching properties. Co-crystallisation can be applied to virtually any of the many hundreds of known switchable molecular materials. Any SCO material in which hydrogen bond donors and/or acceptors (or indeed any suitable, supramolecular interaction) are present in the structure can be a candidate for this approach. Using principles that are already well-established in the co-crystals literature23 will allow identification of appropriate coformers to form structure-directing supramolecular synthons. This imbues an element of modularity in the design of new SCO materials that is so far unprecedented. It does not require chemical modification of the SCO component, making the materials discovery process quicker, easier and cheaper, while also enhancing the potential for rational design of systems with specific properties.
Probably the most exciting opportunity for this strategy, and the direction of our current work lies in the design of multifunctional materials, which has long been a goal in the field of SCO.53 The ability to rapidly screen through a series of functional coformers makes the likelihood of finding hybrid systems with exploitable emergent properties much greater. The ultimate goal is the rational design of such materials via co-crystallisation but achieving this will certainly require further experimental and computational exploration of what is possible.
Data availability
Diffraction data are available from the CCDC. Other data presented here are available from the authors upon request.Author contributions
LTB synthesised the materials, collected and interpreted most of the data. LTB and HJS designed the experiments and wrote the manuscript. GT collected and interpreted some structural data. LJ ensured experiments were reproducible. HJS collected some SQUID data, conceived of the project, and secured funding to undertake it. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
HJS and LTB gratefully acknowledge the Leverhulme Trust for funding (RPG-2019-067).Notes and references
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC
.
- C. Bartual-Murgui, A. Akou, C. Thibault, G. Molnár, C. Vieu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2015, 3, 1277–1285 RSC
- Z. P. Ni, J. L. Liu, M. N. Hoque, W. Liu, J. Y. Li, Y. C. Chen and M. L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2017, 17003862 Search PubMed
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef
- A. Bousseksou and G. Molnár, C. R. Chim., 2003, 6, 1175–1183 CrossRef CAS
- M. G. Reeves, E. Tailleur, P. A. Wood, M. Marchivie, G. Chastanet, P. Guionneau and S. Parsons, Chem. Sci., 2021, 12, 1007–1015 RSC
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem. Int. Ed., 2016, 55, 4327–4331 CrossRef CAS PubMed
- C. Bartual-Murgui, S. Vela, O. Roubeau and G. Aromí, Dalton Trans., 2016, 45, 14058–14062 RSC
- S. Vela, M. Fumanal, J. Ribas-Arino and V. Robert, Phys. Chem. Chem. Phys., 2015, 17, 16306–16314 RSC
- J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703–737 RSC
- M. Fumanal, F. Jiménez-Grávalos, J. Ribas-Arino and S. Vela, Inorg. Chem., 2017, 56, 4474–4483 CrossRef CAS PubMed
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H. B. Bürgi, Angew. Chem. Int. Ed., 2004, 43, 4589–4594 CrossRef CAS PubMed
- X.-P. Sun, R.-J. Wei, Z.-S. Yao and J. Tao, Cryst. Growth Des., 2018, 18, 6862 Search PubMed
- M. Yamada, H. Hagiwara, H. Torigoe, N. Matsumoto, M. Kojima, F. Dahan, J. P. Tuchagues, N. Re and S. Iijima, Chem.–Eur. J., 2006, 12, 4536–4549 CrossRef CAS PubMed
- F.-X. Shen, Q. Pi, L. Shi, D. Shao, H.-Q. Li, Y.-C. Sun and X.-Y. Wang, Dalton Trans., 2019, 48, 8815–8825 RSC
- J. H. Askew and H. J. Shepherd, Dalton Trans., 2020, 49, 2966–2971 RSC
- V. Jornet-Mollá, C. Giménez-Saiz, B. J. C. Vieira, J. C. Waerenborgh and F. M. Romero, Dalton Trans., 2021, 50, 2536–2544 RSC
- V. Jornet-Mollá, C. Giménez-Saiz, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem.–Eur. J., 2021, 27, 740–750 CrossRef PubMed
- C. Atmani, F. El Hajj, S. Benmansour, M. Marchivie, S. Triki, F. Conan, V. Patinec, H. Handel, G. Dupouy and C. J. Gómez-García, Coord. Chem. Rev., 2010, 254, 1559–1569 CrossRef CAS
- S. Cherukuvada, R. Kaur and T. N. Guru Row, CrystEngComm, 2016, 18, 8528–8555 RSC
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. Roy Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. Kumar Goswami, N. Rajesh Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. Malla Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. Guru Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS
- G. R. Desiraju, Angew Chem. Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS
- I. R. Jeon, O. Jeannin, R. Clérac, M. Rouzières and M. Fourmigué, Chem. Commun., 2017, 53, 4989–4992 RSC
- M. C. Pfrunder, J. J. Whittaker, S. Parsons, B. Moubaraki, K. S. Murray, S. A. Moggach, N. Sharma, A. S. Micallef, J. K. Clegg and J. C. McMurtrie, Chem. Mater., 2020, 32, 3229–3234 CrossRef CAS
- A. R. Zuluaga, A. J. Brock, M. C. Pfrunder, W. Phonsri, K. S. Murray, P. Harding, A. S. Micallef, K. M. Mullen, J. K. Clegg, D. J. Harding and J. C. McMurtrie, Chem. Mater., 2020, 32, 10076–10083 CrossRef CAS
- H. Zenno, R. Akiyoshi, M. Nakamura, Y. Sekine and S. Hayami, Chem. Lett., 2021, 50, 1259–1262 CrossRef CAS
- T. Nakanishi and O. Sato, Crystals, 2016, 6, 131 CrossRef
- I. A. Gass, S. R. Batten, C. M. Forsyth, B. Moubaraki, C. J. Schneider and K. S. Murray, Coord. Chem. Rev., 2011, 255, 2058–2067 CrossRef CAS
- V. Jornet-Mollá, C. Giménez-Saiz, L. Cañadillas-Delgado, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem. Sci., 2021, 12, 1038–1053 RSC
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Angew. Chem. Int. Ed., 2015, 54, 823–827 CrossRef CAS PubMed
- X. Zhang, H. Xie, M. Ballesteros-Rivas, Z. X. Wang and K. R. Dunbar, J. Mater. Chem. C, 2015, 3, 9292–9298 RSC
- X. Zhang, Z. X. Wang, H. Xie, M. X. Li, T. J. Woods and K. R. Dunbar, Chem. Sci., 2016, 7, 1569–1574 RSC
- Y. N. Shvachko, D. V. Starichenko, A. V. Korolyov, A. I. Kotov, L. I. Buravov, V. N. Zverev, S. V. Simonov, L. V. Zorina and E. B. Yagubskii, Magnetochemistry, 2017, 3, 9 CrossRef
- J. Y. Zhang, L. J. Su, Q. J. Guo and J. Tao, Inorg. Chem. Commun., 2017, 82, 39–43 CrossRef CAS
- A. V. Kazakova, A. V. Tiunova, D. V. Korchagin, G. V. Shilov, E. B. Yagubskii, V. N. Zverev, S. C. Yang, J. Y. Lin, J. F. Lee, O. V. Maximova and A. N. Vasiliev, Chem.–Eur. J., 2019, 25, 10204–10213 CrossRef CAS PubMed
- H. Xie, K. R. Vignesh, X. Zhang and K. R. Dunbar, J. Mater. Chem. C, 2020, 8, 8135 RSC
- Ö. Üngör, E. S. Choi and M. Shatruk, Chem. Sci., 2021, 12, 10765–10779 RSC
- M. A. Halcrow, New J. Chem., 2014, 38, 1868–1882 RSC
- G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13–31 CrossRef CAS
- K. Sugiyarto and H. Goodwin, Aust. J. Chem., 1988, 41, 1645–1663 CrossRef CAS
- K. Sugiyarto, D. Craig, A. Rae and H. Goodwin, Aust. J. Chem., 1994, 47, 869 CrossRef CAS
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244–2252 RSC
- K. J. Meers, T. N. Tran, Q. Zheng, D. K. Unruh and K. M. Hutchins, Cryst. Growth Des., 2020, 20, 5048–5060 CrossRef CAS
- N. Paradis, G. Chastanet and J. F. Létard, Eur. J. Inorg. Chem., 2012, 3618–3624 CrossRef CAS
- J. F. Létard, S. Asthana, H. J. Shepherd, P. Guionneau, A. E. Goeta, N. Suemura, R. Ishikawa and S. Kaizaki, Chem.–Eur. J., 2012, 18, 5924–5934 CrossRef PubMed
- G. A. Craig, J. S. Costa, O. Roubeau, S. J. Teat and G. Aromí, Chem.–Eur. J., 2012, 18, 11703–11715 CrossRef CAS PubMed
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC
- M. Marchivie, P. Guionneau, J. F. Létard and D. Chasseau, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 479–486 CrossRef PubMed
- M. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC
- S. A. Barrett and M. A. Halcrow, RSC Adv., 2014, 4, 11240–11243 RSC
- K. Senthil Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: This includes experimental details, additional structural data and crystallographic tables. CCDC 2102711–2102733 and 2107483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04956a |
| This journal is © The Royal Society of Chemistry 2022 |