
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the complex configurational landscape of the intralayer cavities in a crystalline carbon nitride†
Magnus
Pauly
a,
Julia
Kröger
 bd,
Viola
Duppel
b,
Corban
Murphey
c,
James
Cahoon
c,
Bettina V.
Lotsch
*bd and
Paul A.
Maggard
*a
bd,
Viola
Duppel
b,
Corban
Murphey
c,
James
Cahoon
c,
Bettina V.
Lotsch
*bd and
Paul A.
Maggard
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695, USA. E-mail: Paul_Maggard@ncsu.edu
bMax Planck Institute for Solid State Research, Stuttgart, 70569, Germany
cDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA
dUniversity of Munich (LMU), Butenandtstraße 5-13 (Haus D), Munich, 81377, Germany
First published on 15th February 2022
Abstract
The in-depth understanding of the reported photoelectrochemical properties of the layered carbon nitride, poly(triazine imide)/LiCl (PTI/LiCl), has been limited by the apparent disorder of the Li/H atoms within its framework. To understand and resolve the current structural ambiguities, an optimized one-step flux synthesis (470 °C, 36 h, LiCl/KCl flux) was used to prepare PTI/LiCl and deuterated-PTI/LiCl in high purity. Its structure was characterized by a combination of neutron/X-ray diffraction and transmission electron microscopy. The range of possible Li/H atomic configurations was enumerated for the first time and, combined with total energy calculations, reveals a more complex energetic landscape than previously considered. Experimental data were fitted against all possible structural models, exhibiting the most consistency with a new orthorhombic model (Sp. Grp. Ama2) that also has the lowest total energy. In addition, a new Cu(I)-containing PTI (PTI/CuCl) was prepared with the more strongly scattering Cu(I) cations in place of Li, and most closely matching with the partially-disorder structure in Cmc21. Thus, a complex configurational landscape of PTI is revealed to consist of a number of ordered crystalline structures that are new potential synthetic targets, such as with the use of metal-exchange reactions.
Introduction
Carbon nitrides have been the subject of intense recent investigations as semiconducting catalysts, such as for solar energy conversion.1–5 Generally, this class of materials includes covalent polymers based upon triazine and heptazine monomers forming a backbone consisting of strictly alternating carbon and nitrogen atoms (carrying NHx moieties, but no C–H bonds). Advantages of carbon nitrides include their lower cost, ease of synthesis and functionalization, low toxicity, as well as their greater kinetic stability as compared to most inorganic semiconductors. Their structures are typically layered, with the occurrence of intralayer cavities that can host solvent molecules, functional groups, and/or coordination sites for metal cations. The arrays of micropores typically embedded within covalent CNx frameworks thus represent a complex configurational landscape that can impact their physical properties. This feature has typically not been well characterized nor explored in carbon nitrides.While structural characterization of most carbon nitrides is hindered by their partially amorphous character, a crystalline carbon nitride has been discovered in recent years, poly(triazine imide)/LiCl (i.e., PTI/LiCl). PTI/LiCl has garnered intense interest for its photocatalytic activity and utility in solar energy conversion. This includes as a photocatalyst for the reduction of water to dihydrogen,6–11 and/or for the oxidation of water to dioxygen when its surface is loaded with a Pt and/or CoOx co-catalyst.7 It has also been investigated as a photocatalyst for the degradation of organic dyes in wastewater,12 and for use as a counter electrode in dye-sensitized solar cells.13 Notably, these reports include conflicting values for its band gap (2.95 to 3.15 eV), band energies and other photocatalytic properties.
Despite the well-defined carbon nitride substructure of PTI/LiCl, its intralayer cavities accommodate Li/H atoms (Fig. 1a) that have yielded conflicting reports regarding their ordered or disordered nature. The ‘C6N9’ subcomponent of PTI was first characterized as exhibiting a hexagonal-based structure in space group P63cm.14 While this model is consistent with a complete disordering of the Li/H atoms within its cavities, subsequent investigations have revealed evidence for the occurrence of partial ordering in the orthorhombic space group Cmc21 or a complete ordering in P212121.15,16 A clear resolution of the Li/H atoms is obscured by the weak X-ray scattering of these ions, as well as complicated by the high sensitivity of the crystallinity with reaction conditions.17 The latter is suggestive of a number of possible structural configurations, including both ordered and disordered, that is the possible origin of the seemingly differing structure reports and variances in its physical properties.18 The full extent of alternate possible structural configurations has yet to be considered.
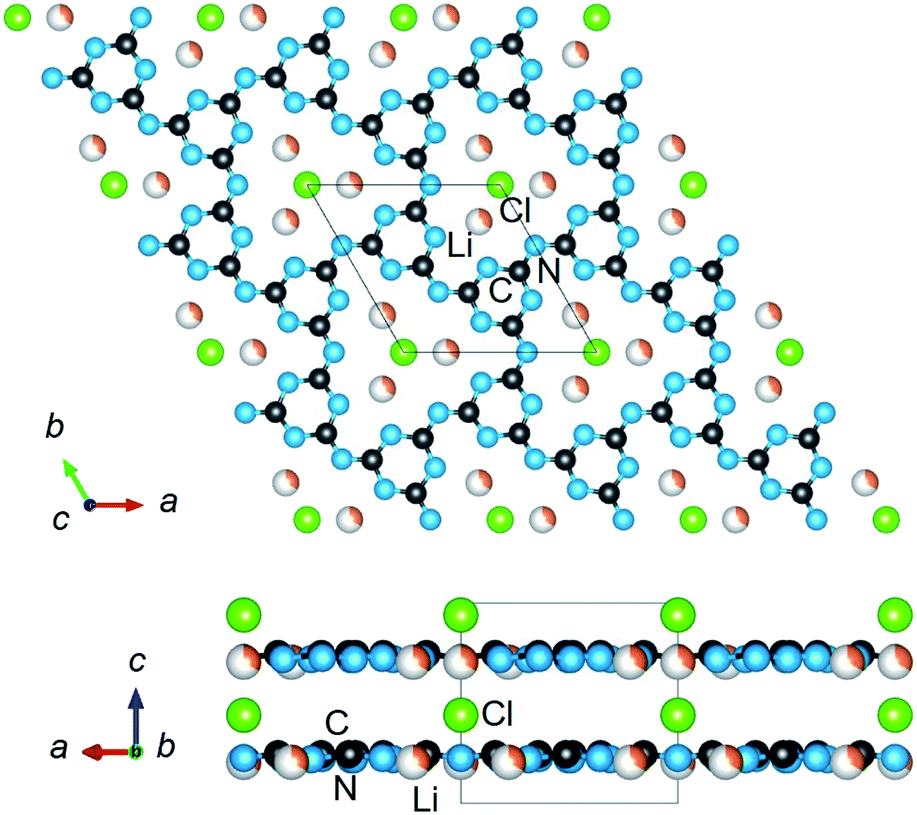 | ||
| Fig. 1 The crystalline structure of PTI (C6N9H2Li2Cl) in space group P63cm, showing the disordered intralayer cavities (upper) containing Li/H atoms, and a structural view along the layers (lower). | ||
Presented herein is an enumeration of potential structural models in the configurational landscape of Li/H atoms within the PTI/LiCl intralayer cavities and their relative total energies calculated by density-functional theory methods. These structural models have each been experimentally fitted against electron microscopy and X-ray and neutron diffraction data for PTI/LiCl, d-PTI/LiCl (i.e., deuterated) and PTI/CuCl. The latter contains the more strongly scattering Cu(I) in partial replacement of Li(I) cations. A single-step synthesis of PTI/LiCl in high-crystallinity was targeted in which melamine was reacted in a eutectic LiCl/KCl flux (∼300 mg) in a molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (melamine:flux) at a temperature of 470 °C for 36 h and cooled at a rate of 2 °C h−1 within a sealed, evacuated fused-silica tube (∼80 ml). To prepare d-PTI/LiCl, the protons in melamine were exchanged by D2O soaking prior to the flux synthesis. The use of a eutectic LiCl/KCl flux in the synthesis enables the crystallization of PTI/LiCl, which has been posited to result from the stabilization of small molecule intermediates by the Li cation.17 Reaction conditions are thus critical, with both single step and multi-step reaction pathways reported to typically result in a wide possible range of crystallinity. The PTI/CuCl was prepared by reacting PTI with a (60/40 molar) CuCl/KCl eutectic in a molar ratio of 1.2:1 (CuCl:PTI; ∼300 mg) at a temperature of 450 °C for 24 h within a sealed, evacuated fused-silica tube. All products were washed multiple times in deionized water and then collected by centrifugation.
:1 (melamine:flux) at a temperature of 470 °C for 36 h and cooled at a rate of 2 °C h−1 within a sealed, evacuated fused-silica tube (∼80 ml). To prepare d-PTI/LiCl, the protons in melamine were exchanged by D2O soaking prior to the flux synthesis. The use of a eutectic LiCl/KCl flux in the synthesis enables the crystallization of PTI/LiCl, which has been posited to result from the stabilization of small molecule intermediates by the Li cation.17 Reaction conditions are thus critical, with both single step and multi-step reaction pathways reported to typically result in a wide possible range of crystallinity. The PTI/CuCl was prepared by reacting PTI with a (60/40 molar) CuCl/KCl eutectic in a molar ratio of 1.2:1 (CuCl:PTI; ∼300 mg) at a temperature of 450 °C for 24 h within a sealed, evacuated fused-silica tube. All products were washed multiple times in deionized water and then collected by centrifugation.
Results and discussion
The crystalline structure of PTI/LiCl in the highest-symmetry hexagonal system is plotted in Fig. 1, as found in its first reported crystalline structure.14 Generally, its crystal structure contains triazine rings fused together by imide functional groups. These layers are stacked and held together by the chloride anions located above and below the intralayer cavities, Fig. 1 (lower). It has the composition [(C3N3)2(NHxLi1−x)3·LiCl], in which C3N3 represents the triazine species and NHxLi1−x the imide bridging groups. Elemental analysis, Table S1,† of the PTI/LiCl product gives x = 2/3, and a nominal formula of C6N9H2Li2Cl that is in line with prior studies that have shown partial deprotonation of the NH groups under similar reaction conditions.14–16For this hexagonal symmetry, two protons and two lithium cations are each disordered over three symmetry-equivalent coordination sites in the intralayer cavities, shown in Fig. 1 (upper). Alternative ordered structural models were fully enumerated according to three criteria, i.e., the coordination patterns of the (i) lithium cations, (ii) hydrogen atoms, and (iii) layer-to-layer orientation of the Li/H atoms. These ordered configurations were constructed from a metrically orthorhombic unit cell that derives from the hexagonal unit cell, as described previously,15,16,19 and can be indexed to the powder X-ray diffraction data (described below). This unit cell provides for two symmetry-unique intralayer cavities within the unit cell that leads to three possible Li-cation configurations, Fig. 2. Each of these configurations contain two Li cations per intralayer cavity that are coordinated to 2/3 of the chelating triazine groups. Additionally, two H atoms are contained within each cavity, and which can coordinate to either the N-triazine or N-imide groups that are opposite to the Li cations in the cavities. From one layer to the next, the orientation of the coordinated Li/H atoms can be rotated by either 180° or 60°. Representative examples of the different types of ordered structural configurations are provided in the ESI, Fig. S4–S8.† Total electronic energies for all structural models were calculated for the geometry-relaxed structures using density functional theory in VASP,20,21 using the generalized gradient approximation of Perdew, Burke and Ernzerhof.22 A k-point mesh of 4 × 4 × 2 was used and dispersion force corrections were included within the DFT-D3(BJ) scheme and Becke–Johnson damping.23,24
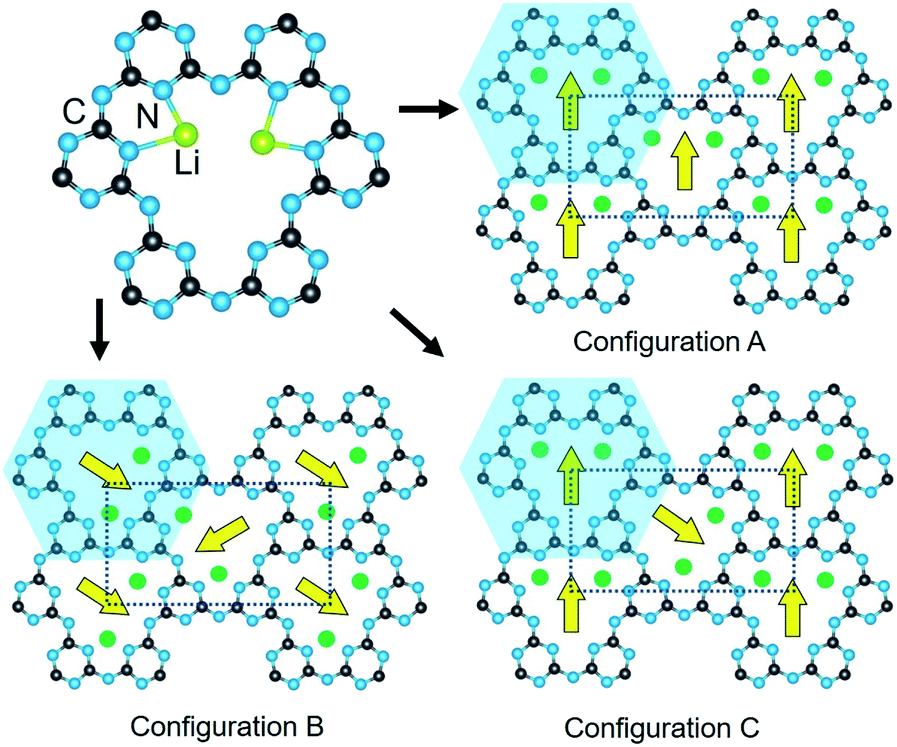 | ||
| Fig. 2 The Li-cation coordination within a cavity (upper), and different orientations between the cavities in the plane of the layer labelled as the configurations A, B, and C. Yellow arrows indicate the intracavity orientation of the coordinated Li cations, and atoms are labelled for C (black), N (blue), and Li (green). | ||
Listed in Table 1, the enumeration of all ordered structural configurations using the above procedures resulted in 18 different crystalline structures preserving a minimum symmetry of monoclinic. The space group of each structural model, inclusive of the hydrogen atoms, was determined using PLATON and transformed to the standard unit cell settings and atomic coordinates within VESTA.25,26 This resulted in six different structural models for each of the configurations in Fig. 2(A–C).27 Configuration A exhibits the highest space group symmetries (e.g., Cmcm, Ama2) with a single symmetry-unique cavity with all Li cations aligned identically within the pores, while configurations B and C yielded lower symmetries (e.g., Pbcm, P21/m) and have two symmetry-inequivalent cavities. The crystallographic information files of all structure models are included in the ESI.†
| Classa (Li) | Space group | Rotate (°) | H on N-imide | H on N-triazine | Relative energyb (eV f.u.−1) | Li⋯Hc (%) |
|---|---|---|---|---|---|---|
| a Class of Li orientation with reference to Fig. 1, with A and B groups indicating the pattern of H-atoms bonded to N-imide groups. b All energies are relative to −603.123 eV calculated for the A1a model. c Percent of Li cations across from a protonated imide bridge. | ||||||
| A1 | Ama2 | 60 | 2 | 0 | 0.932 | 100 |
| A1a | Ama2 | 60 | 1 | 1 | 0 | 50 |
| A1b | Pma2 | 60 | 1 | 1 | 0.157 | 50 |
| A2 | Cmcm | 180 | 2 | 0 | 1.097 | 100 |
| A2a | Pnma | 180 | 1 | 1 | 0.076 | 50 |
| A2b | P21/m | 180 | 1 | 1 | 0.134 | 50 |
| B1 | Pbcm | 60 | 2 | 0 | 0.605 | 50 |
| B1a | Pbcm | 60 | 1 | 1 | 1.941 | 0 |
| B1b | Pbcm | 60 | 1 | 1 | 0.322 | 50 |
| B2 | Pnma | 180 | 2 | 0 | 0.883 | 50 |
| B2a | Pnma | 180 | 1 | 1 | 0.290 | 50 |
| B2b | Pnma | 180 | 1 | 1 | 1.383 | 0 |
| C1 | P21/m | 60 | 2 | 0 | 0.816 | 75 |
| C1a | P2 1 /m | 60 | 1 | 1 | 0.126 | 50 |
| C1b | P21/m | 60 | 1 | 1 | 0.807 | 25 |
| C2 | P21/m | 180 | 2 | 0 | 1.331 | 75 |
| C2a | P21/m | 180 | 1 | 1 | 0.284 | 50 |
| C2b | P21/m | 180 | 1 | 1 | 0.668 | 25 |
A few important trends between the structural features and the total electronic energies are evident. For example, there is a significant decrease in electronic energy as one H atom per cavity is shifted from bonding an N-imide to an N-triazine group, i.e., in Table 1 compare A1 with A1a and A1b, or compare A2 with A2a and A2b. The lowered energy with deprotonation of half of the imide bridges occurs for most of the related models and is partly related to the stabilization of the resonance structures which place the formal charge closer to the Li cations in the cavity, as shown in Scheme 1. In Table 1 this corresponds to the Li⋯H column, which gives the percentage of Li atoms across from protonated imide bridges, yielding lower electronic energies for lower percentages. This effect also couples to the H-tautomerism (not shown), wherein a shift of a H atoms to the N-triazine groups also leads to a shift of the electron density to nearby Li cations. Shown in Fig. 3 are the geometry relaxed structural models for A1 (H on N-imides only) and the lowest energy A1a (H on N-imide and N-triazine). Displacement of the H atoms from the N-triazine to N-imide groups can only result in a minimum of 50% Li⋯H in category A. However, in category C the Li⋯H decreases to 25%, but also has a larger number of close contacts between layers.
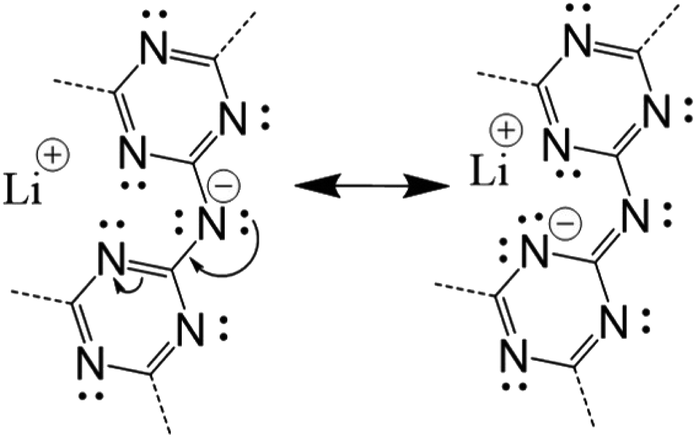 | ||
| Scheme 1 Two resonance structures for a deprotonated imide bridge, including formal charges. Alternative resonance structures locate the formal charge on other N-triazine groups near to Li cations in this and neighbouring cavities. | ||
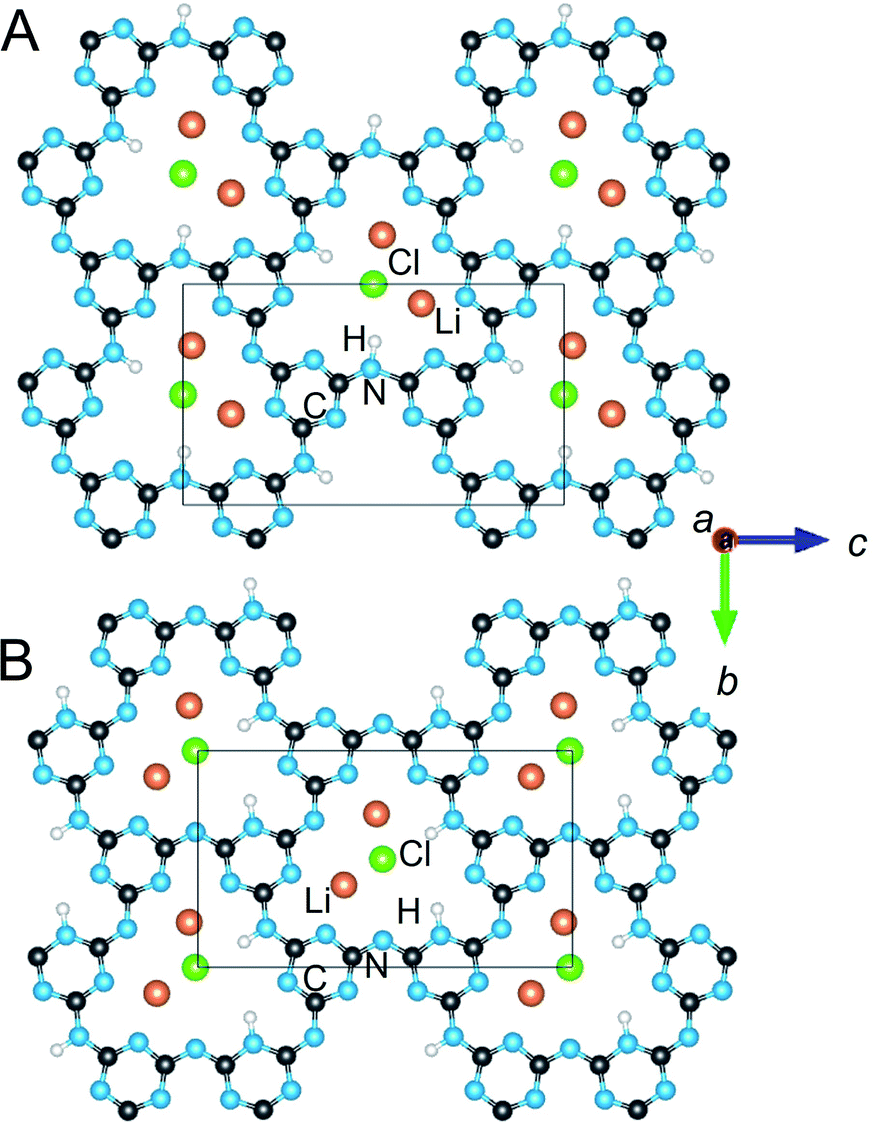 | ||
| Fig. 3 Crystalline structures of ordered PTI/LiCl models for A1 (A) and the lowest energy A1a (B) in the Ama2 space group for each. Atoms are labelled for C (black), N (blue), Li (orange), Cl (green) and H (white). | ||
The A2a and C1a models in Pnma and P21/m constitute the second and third lowest relative electronic energies as compared to A1a, with all three in bold font in Table 1. The model in Pnma is consistent with the previously reported Li/H ordering model within the lower-symmetry (non-isomorphic) subgroup of P212121. However, the energetic differences between the lowest-energy structural models must be considered as relatively small as compared to the experimental range of synthesizability for carbon nitrides (∼0.2 eV per atom above the ground state).28 Thus, the experimentally-observed crystallization of different ordered or disordered PTI/LiCl structures, with only small changes in reaction conditions, is very likely owing to the fact that the nucleation of carbon nitrides is strongly kinetically-driven. The conflicting structures reported in the literature may only reflect the dependence of the Li/H ordering on the synthetic conditions. For example, higher temperatures, shorter reaction times with subsequent quenching lead to less well-defined Bragg diffraction peaks.
Rietveld refinements of the PTI/LiCl powder XRD based on the structural models all yield similarly good fits owing to the weak scattering of the Li/H atoms. To better probe the Li/H ordered structures, neutron diffraction data were taken on deuterated-PTI/LiCl (d-PTI/LiCl) that was prepared using reaction conditions that yielded the highest crystallinity. The deuterium exchange was confirmed by peak shifting present in the FTIR data in Fig. S2 (ESI†). Rietveld refinements for d-PTI/LiCl were performed for each of the ordered structure models in Table 1. The best fit was observed for the lowest-energy A1a model in Ama2, plotted in Fig. 4, as judged by the weighted residual and the absence of additional diffraction peaks predicted by the other models in Table 1. For example, as a result of different ordering of the Li/H positions in alternative models, additional (non-observed) diffraction peaks were predicted at d-spacings of ∼4.5 Å for A1b, A2, and A2a, or at ∼5.5 Å for C1a, C2 and C2a. The A1a model only exhibited peaks which could be indexed by the structure model in the neutron diffraction pattern. However, similarly low refinement residuals of ∼7 to 8% were found for all models, except for the disordered phases Cmc21 and P63cm which yielded larger residuals of >10%.
 | ||
| Fig. 4 Results of Rietveld refinement of neutron diffraction data for d-PTI/LiCl in space group Ama2 (A1a model; wR% = 7.5; a = 6.7321(4) Å, b = 8.4597(21) Å, and c = 14.614(4) Å). | ||
To introduce the more strongly scattering Cu(I) cation in the cavities of PTI/LiCl, i.e., to prepare PTI/CuCl, flux exchange reactions were performed according to reaction (1):
| CuCl + KCl + C6N9H2Li2Cl → xLiCl + (1 − x)CuCl + KCl + C6N9H2Li2−xCuxCl | (1) |
The addition of KCl to the mixture lowers its melting point, and the salt products are subsequently removed by washing in aqueous solution. The reaction is thermodynamically driven by the more negative heat of formation of LiCl versus CuCl (i.e., −408 kJ mol−1versus −138 kJ mol−1, respectively). It is also driven by the greater enthalpy of N-ligand binding of Li versus Cu (e.g., M(I)-pyridine of −180 kJ mol−1 and −260 kJ mol−1 for M = Li and Cu).29,30 Elemental analysis of the products, Table S1,† shows that the product contains ∼36.9 mol% Cu(I) cations and maintains the PTI structure by powder XRD. A higher concentration of the Cu(I) cation can be introduced, but this results in a new crystalline phase of PTI/CuCl that is currently under investigation.
Powder XRD refinements were performed on the PTI/CuCl product using the structural models in Table 1, with the partial replacement of Cu for Li cations in the cavities. The best fits were obtained for the A1a (Ama2), B1 (Pbcm) and B2 (Pnma) structural models, with weighted refinement residuals of 6.6%, 7.2% and 7.3%, respectively. The Cu(I)-content of C6N9H2Li2−xCuxCl refined consistently across all models, giving x = 0.65(1), 0.65(2) and 0.65(2), respectively. Alternative structural models resulted in additional (non-observed) peaks in the simulated diffraction patterns and relatively larger weighted residuals. The partially disordered model Cmc21 resulted in low weighted residual of ∼6.0%. The Cu cation occupancy is statistically disordered for x = 0.68(2), yielding a formula of C6N9H2Li1.32Cu0.68Cl with ∼34 mol% Cu and ∼66 mol% Li cations, consistent with the elemental analysis. Additional details are provided in the ESI.†
HRTEM images and precession electron diffraction data were also taken on PTI/CuCl in order to better distinguish between the possible structure models. Images of some of the selected crystallites that were relatively small and thin are shown in Fig. 5 and given in Fig. S12 and S13.† The in-plane layer is clearly shown in the hexagonally-shaped crystallite and is homogeneous. The view down the [100] direction of the stacking of layers is shown in Fig. 5B and exhibits few detectable stacking faults. The carbon nitride substructure is thus preserved after the Cu(I) exchange reactions with a homogeneous metal cation distribution, as is also corroborated by FTIR and PXRD data given in the ESI.† From the precession electron diffraction, the structural models B1 (Pbcm) and B2 (Pnma) could be excluded from consideration based upon the observed intensity distribution down the [110] and [011] zone axes, Fig. S11.† The two remaining models found to be most consistent with the precession electron diffraction were the ordered A1a (Ama2) and the partially H/Li-disordered structure previously reported in Cmc21.15
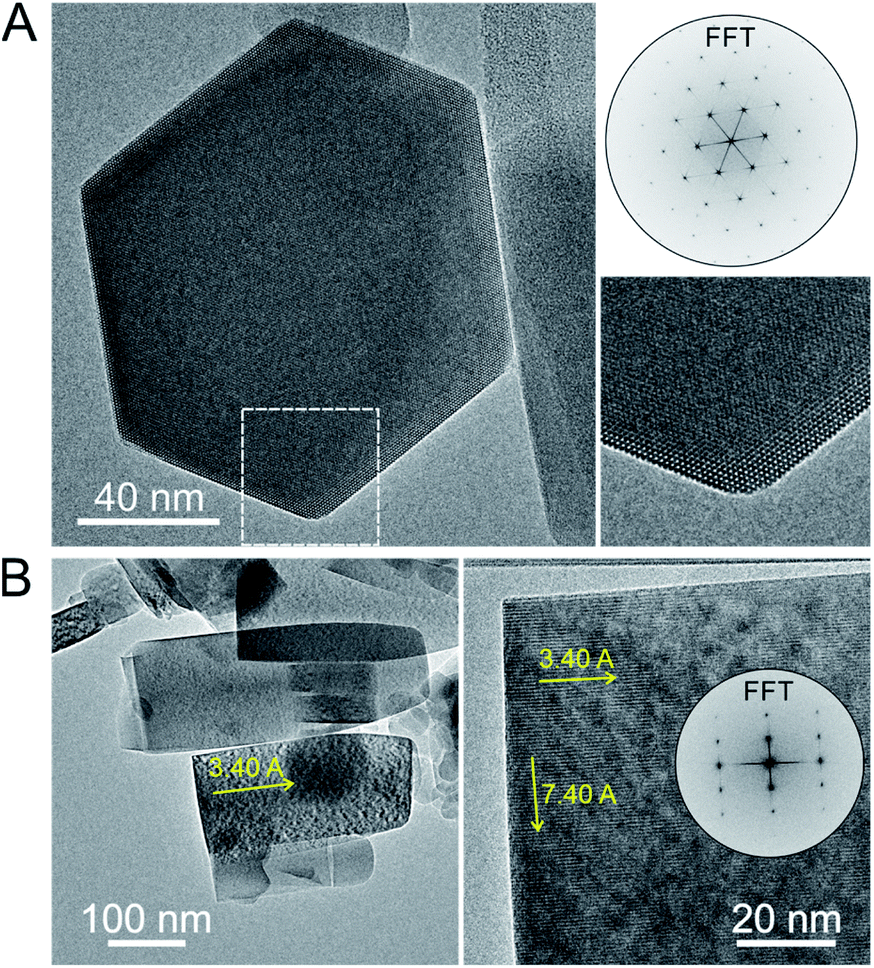 | ||
| Fig. 5 TEM images of PTI/CuCl viewed down the [001] (A) and the [100] zone axis (B) with the interlayer (7.40 Å) and intercavity spacings (3.40 Å) labeled with yellow arrows. | ||
Rietveld refinements of the XRD data according to the two above models showed consistently better fits and peak matching to the latter, partially H/Li-disordered, Cmc21 structural model. This structure shows the closest consistency with the fully ordered A2 model (Cmcm) in terms of the interlayer and intralayer Li arrangements (i.e., category A and 180° layer orientation in Table 1). However, this model is also >1.0 eV f.u.−1 higher than the lowest energy structure, Table 1, owing to the fact that the H atoms are bonded only to the N-imide groups. Using synchrotron X-ray data,15 it was noted that the structure refinements in Cmc21 were insensitive to the H-atom positions. The refined structure of PTI/CuCl in Cmc21 thus likely corresponds instead to a partially-disordered structural model derived from shifting 50% of the H atoms in A2 (Cmcm) to the N-triazine groups, e.g., to A2a (Pnma). Shown in Fig. 6 is a comparison of the structural models in Cmc21 and Pnma. The models are basically the same within and between the intralayer cavities, except for a disordering of the H-atom positions and a slight shifting of the metal-cation positions. It should also be noted that the P212121 model is also consistent with these structures after some subtle Cl-atom shifting that causes further symmetry breaking.
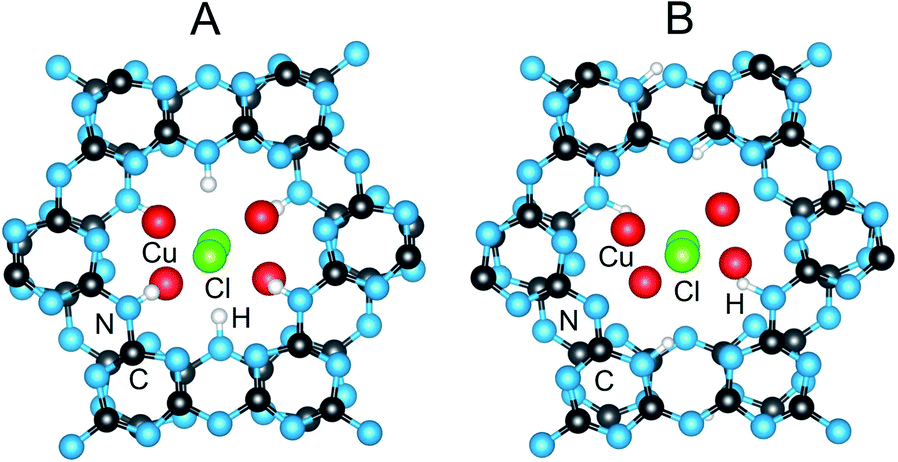 | ||
| Fig. 6 Crystalline structures of PTI/CuCl in the partially-disordered model in Cmc21 ((A); H sites are only partially occupied) and the fully ordered A2a model in Pnma ((B); H sites are fully occupied). This illustrates the only significant difference between the models is the disordered H-atom positions in the former. Atoms are labelled for C (black), N (blue), Li (green), Cl (green), Cu (red), and H (white). | ||
Thus, while evidence for an ordered structure is found for d-PTI/LiCl in A1a (Ama2) that corresponds to the lowest energy model, the flux exchange reaction has caused a partial disordering of the intralayer cavities in PTI/CuCl. A complete disorder of the metal/H atoms within the cavities, including partial occupancies of each of the crystallographic sites, would eventually yield the highest-symmetry P63cm structure as described in the original structural report.14 Given the number of competing low energy structures in Table 1, a disordered structure could easily arise with the use of slightly more harsh reaction conditions (e.g., higher temperatures and reaction quenching) that tend to favor a higher degree of disorder. The configurational entropy will also favor disordered structural arrangements at higher temperatures. For example, preliminary evidence for the complete disorder of intralayer cavities has recently been found with the exchange of divalent metal cations into the structure, e.g., in PTI/ZnCl, and is currently under investigation.
Solid-state NMR data were also collected to further probe the PTI/LiCl and PTI/CuCl structures, shown in the ESI.† Briefly, the 1H, 13C and 15N spectra indicate that the local structures of PTI/LiCl and PTI/CuCl are closely related, both in terms of proton distribution patterns and relative signal intensities. More specifically, the proton NMR of PTI/CuCl shows two low-field signals at essentially the same chemical shifts as PTI/LiCl which are consistent with protonated N-triazine and protonated N-imide groups in both materials.14–16 Similar to PTI/LiCl, the PTI/CuCl also exhibits three signals in the 13C NMR, also consistent with the presence of different carbon signals in the vicinity to protonated (163 and 158 ppm) and non-protonated (168 ppm) nitrogen atoms. The relative signal intensities differ for PTI/LiCl and PTI/CuCl, and likely caused by the slightly difference chemical environments in both samples. The 15N NMR spectra of both compounds are again very similar, with only very slight differences in the chemical shifts and relative intensities, indicative of slightly different protonation patterns. Finally, the 7Li spectrum shows a reduced amount of Li remaining in the sample after Cu exchange, consistent with the elemental analysis, Table S1,† and the Rietveld refinement.
Conclusions
The intralayer cavities within the crystalline PTI/LiCl have been found to provide a complex configuration landscape for metal cations and H atoms. Starting from a metrically orthorhombic unit cell, the arrangements of Li/H within its cavities results in 18 distinct structural models that maintained a minimum of monoclinic symmetry. Total energy calculations revealed that the lowest energy models possessed structural features which included, (i) displacement of H atoms from the N-imide to N-triazine groups, (ii) Li coordination to triazine groups that have a deprotonated N-imide bridge, and (iii) the fewest number of interlayer Li⋯H interactions. Refinements of the structural models against neutron diffraction data of d-PTI/LiCl gave a best fit to the new, lowest-energy structural model A1a in space group Ama2. While both this structure and its synthesis are different from prior literature, the presence of other similarly low-energy structures also likely leads to a strong dependence of its crystalline structure on the reaction conditions. The partial replacement of monovalent Li for Cu cations using flux-based reactions, giving PTI/CuCl, was best matched to the partially disordered structure in Cmc21. Finally, a more complete elucidation of the range of possible structural Li/H arrangements in PTI/LiCl can be used to analyse and more fully understand the reported range of photoelectrochemical and photocatalytic properties for this class of crystalline carbon nitrides in the future.Data availability
Raw data are available from the corresponding authors upon request.Author contributions
The conceptualization was performed by M. P., B. V. L. and P. M., the funding acquisition, resources, and supervision by J. C., B. V. L. and P. M., the investigation and data discussion by all authors, the data acquisition by M. P., J. K., V. D. and C. M., the formal analysis and writing (original draft, reviewing and editing) by M. P., J. K., V. D., J. C., B. V. L., and P. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the Center for Hybrid Approaches in Solar Energy to Liquid Fuels (CHASE), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021173, supporting M. P., J. C. and P. M for the synthetic work and the structural modeling and calculations. A portion of this research also used resources at the POWGEN Powder Diffractometer at Beamline 11A for structural characterization of the deuterated PTI/LICl that was sent to the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. HRTEM data and images were collected at the Max Planck Institute für Festkörperforschung (B. V. L., J. K., V. D.) with acknowledgment of infrastructure support for these studies by the Cluster of Excellence e-conversion (EXC 2089) and the Center for Nanoscience (CeNS). The authors thank Igor Moudrakovski for collection of the solid-state NMR data and Marie-Luise Schreiber for the elemental analysis data.Notes and references
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- V. Briega-Martos, A. Ferre-Vilaplana, A. de la Peña, J. L. Segura, F. Zamora, J. M. Feliu and E. Herrero, ACS Catal., 2017, 7, 1015–1024 CrossRef CAS.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- Y. Ham, K. Maeda, D. Cha and K. Takanabe, Chem.–Asian J., 2013, 8, 218–224 CrossRef CAS PubMed.
- L. Lin, C. Wang, W. Ren, H. Ou, Y. Zhang and X. Wang, Chem. Sci., 2017, 8, 5506–5511 RSC.
- L. Heymann, S. C. Bittinger and C. Klinke, ACS Omega, 2018, 3, 17042–17048 CrossRef CAS PubMed.
- K. Schwinghammer, B. Tuffy, M. B. Mesch, E. Wirnhier, C. Martineau, F. Taulelle, W. Schnick, J. Senker and B. V. Lotsch, Angew. Chem., Int. Ed., 2013, 52, 2435–2439 CrossRef CAS PubMed.
- Y. Huang, M. Su, Y. Zhou, D. Chen, Z. Xu, H. Zhang and C. Liao, Ceram. Int., 2020, 46, 26492–26501 CrossRef CAS.
- L. Tian, J. Li, F. Liang, J. Wang, S. Li, H. Zhang and S. Zhang, Appl. Catal., B, 2018, 225, 307–313 CrossRef CAS.
- W. Lee, Y.-S. Jun, J. Park and G. D. Stucky, J. Mater. Chem. A, 2015, 3, 24232–24236 RSC.
- E. Wirnhier, M. Döblinger, D. Gunzelmann, J. Senker, B. Lotsch and W. Schnick, Chem.–Eur. J., 2011, 17, 3213–3221 CrossRef CAS PubMed.
- C.-Z. Liao, V. W-hei Lau, M. Su, S. Ma, C. Liu, C.-K. Chang, H.-S. Sheu, J. Zhang and K. Shih, Inorg. Chem., 2019, 58, 15880–15888 CrossRef CAS PubMed.
- M. B. Mesch, K. Bärwinkel, Y. Krysiak, C. Martineau, F. Taulelle, R. B. Neder, U. Kolb and J. Senker, Chem.–Eur. J., 2016, 22, 16878–16890 CrossRef CAS PubMed.
- F. K. Kessler and W. Schnick, Z. Anorg. Allg. Chem., 2019, 645, 857–862 CrossRef CAS.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- J. Wang, D. Hao, J. Ye and N. Umezawa, Chem. Mater., 2017, 29, 2694–2707 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- A. J. Spek, PLATON. A Multipurpose Crystallographic Tool, Utrecht University, The Netherlands, 2002, http://www.platonsoft.nl/platon/ Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS , https://jp-minerals.org/vesta/en/.
- It should be noted that helical arrangements of the Li/H positions were also modelled and resulted in similar electronic energies as compared to the nonhelical arrangements. This required a tripling of the cell dimension perpendicular to the layers, as illustrated for one example in the ESI in Fig. S10.†.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
- N. S. Rannulu and M. T. Rodgers, J. Phys. Chem. A, 2007, 111, 3465–3479 CrossRef CAS PubMed.
- M. T. Rodgers and P. B. Armentrout, Mass Spectrom. Rev., 2000, 19, 215–247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04648a |
| This journal is © The Royal Society of Chemistry 2022 |