
Synthetic approaches for copolymers containing nucleic acids and analogues: challenges and opportunities
Hao
Lu
,
Jiansong
Cai
and
Ke
Zhang
 *
*
Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. E-mail: k.zhang@northeastern.edu
First published on 29th March 2021
Abstract
Deep integration of nucleic acids with other classes of materials has become the basis of many useful technologies. Among these biohybrids, nucleic acid-containing copolymers have seen rapid development in both chemistry and applications. This review focuses on the various synthetic approaches for accessing nucleic acid–polymer biohybrids spanning post-polymerization conjugation, nucleic acids in polymerization, solid-phase synthesis, and nucleoside/nucleobase-functionalized polymers. We highlight the challenges associated with working with nucleic acids with each approach and the ingenuity of the solutions, with the hope of lowering the entry barrier and inspiring further investigations in this exciting area.
Introduction
Nucleic acids are a brilliant gift from Nature. Since the elucidation of the B-form DNA structure in the 1953 landmark paper by Watson and Crick,1 synthetic nucleic acids and their analogues have populated many fields of explorations including materials science,2 nanotechnology,3 and medicine.4 The diversity of their application reflects the powerful traits exhibited by these materials: highly predictable and programmable base pairing, ability to work with a plethora of natural or engineered enzymes, and unique photochemical and energy transfer properties. The key to many of these applications is the ability to interface nucleic acids with other types of materials, either covalently or non-covalently. For example, oligonucleotides are used to decorate gold nanoparticles via the thiol–gold bond to form “spherical nucleic acids” (SNAs), which are being investigated as “atom equivalents” in crystal engineering and as therapeutic/prophylactic agents for disease treatment/prevention.5,6One fruitful area of nucleic acid–material integration involves the formation of different classes of copolymers—block copolymers, stars, bottlebrushes, cross-linked networks, etc. Pegaptanib,7,8 a Y-shaped poly(ethylene glycol) (PEG)–oligonucleotide conjugate, was approved in 2004 by the U.S. Food and Drug Administration for the treatment of age-related macular degeneration, where the PEG component serves to improve ocular retention after intravitreal injection. Similar linear PEG conjugates are also being used as supports in liquid-phase oligonucleotide synthesis.9 While these simple conjugates, characterized as having a hydrophilic polymer component and a single oligonucleotide strand, are relatively straightforward to synthesize, the synthetic methodology for more architecturally complex copolymers and amphiphilic copolymers, which are oftentimes at the forefront of scientific and technological explorations, is far from trivial. Workarounds are often needed to circumvent the difficulties associated with nucleic acids. For example, nucleic acids are inherently highly anionic due to their charged phosphate backbone and therefore generally lack solubility in organic solvents,10,11 which puts a severe limitation on the chemistries that can be used to assemble copolymers. Additionally, nucleobases contain nucleophilic exocyclic amines and basic aromatic nitrogen atoms, which may disrupt transition metal catalysts. Nucleotides are also subject to damage due to various reactivities such as acid depurination, oxidation (e.g. by Cu(I)/O2), reaction with strong nucleophiles/electrophiles, reaction with radicals, photodimerization, etc.12,13 Consequently, a variety of strategies have been developed to access useful copolymers containing nucleic acids. This review will summarize these strategies, focusing on the chemistries used, the structural features of the resulting copolymer, and challenges faced/overcome.
Post-polymerization modification
The earliest development of nucleic acid-containing polymers primarily involves the “grafting-onto” strategy, i.e. direct conjugation of the nucleic acid to the synthetic polymer (Table 1). This strategy may allow the polymer and the nucleic acid to be separately prepared, purified, and fully characterized prior to the coupling reaction. However, removal of the unreacted nucleic acid and/or the polymer after the conjugation can be difficult, requiring various chromatography protocols (size exclusion, reversed-phase, anionic exchange, etc.) to be developed. Additionally, a severe limitation involves the requirement for the polymer component to be soluble in water or a mixed solvent with water content for homogeneous coupling, as unmodified DNA or RNA has limited solubility in almost all organic solvents.10,14,15 Therefore, highly hydrophobic, non-polar polymers are generally thought to be incompatible for conjugation with DNA, although extensive optimizations have achieved reasonable yields.16| Reaction type | Polymer structure | Polymer Mn (kDa) | Nucleic acid | Conversion (%) | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|
| Amidation |

|
10 | 15-mer DNA | 75 | Sodium borate buffer (pH 8.0) | 17 |
| Terminal amine | DMSO, R.T., 12 h | |||||
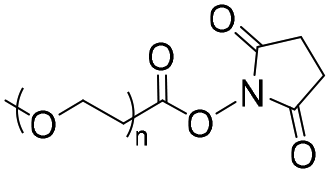
|
2 | 18-mer DNA | 65 | Phosphate buffer (pH 7.0) | 18 and 19 | |
| Terminal amine | R.T., 1.5 h | |||||
| Michael addition |

|
4.8 | 19-mer DNA | 63 | 10 mM Tris-HCl buffer (pH 8.0) | 20 |
| 3′-Thiol | ||||||
|
5.1 | 17-mer DNA/RNA | 79 | 10 mM Tris-HCl buffer (pH 8.0) | 21 and 22 | |
| 3′-Thiol | R.T. | |||||
| CuAAC |

|
167 | 18–21-mer DNA | — | Tris buffer (pH 8.0) | 28 |
| Terminal alkyne | Sodium ascorbate acid, ACN | |||||
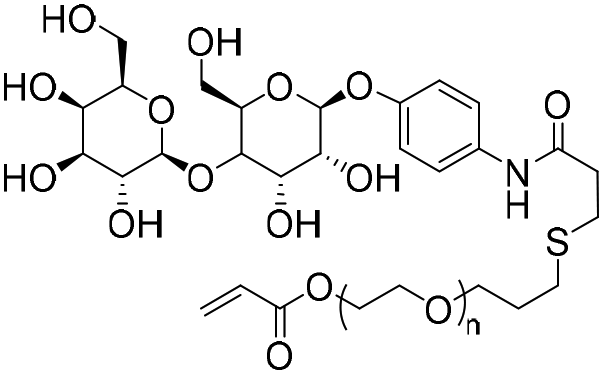
|
122 | 63-mer DNA | 79–90 | Iodo(triethyl phosphite)copper(I) | 31 | |
| Terminal alkyne | DMF or DMSO or NMP, R.T., overnight | |||||
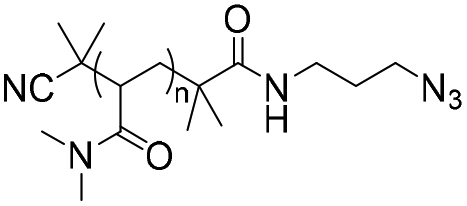
|
3.9–14 | 6–26-mer DNA on CPG | 56–99 | CuI/DIPEA/acetic acid | 44 | |
| 5′ alkyne | DCM, R.T., 8 h | |||||
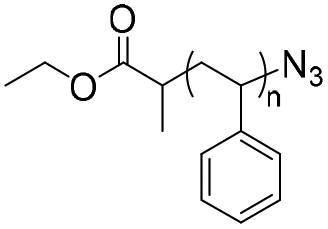
| SPAAC |
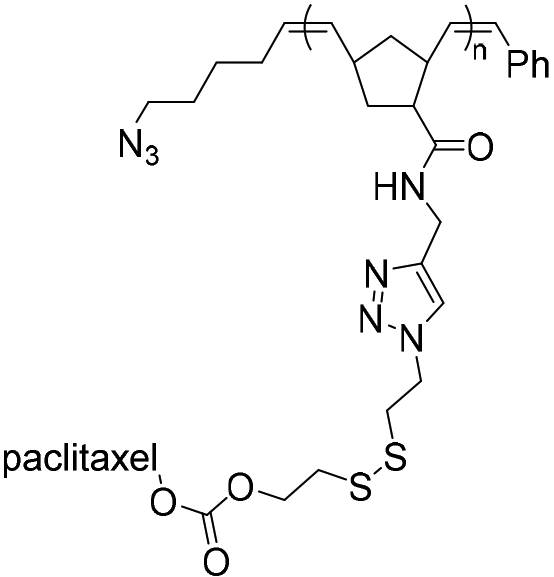
|
10.8 | 21-mer DNA | 40 | Water/DMSO, 40 °C, 48 h | 34 |
| 5′ DBCO | ||||||
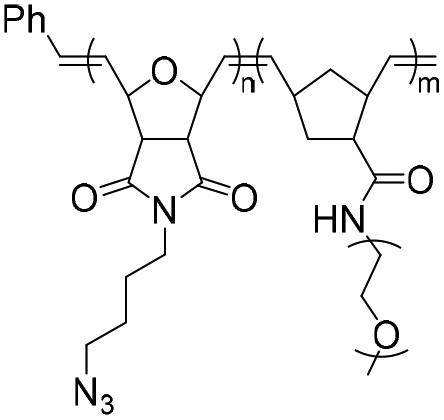
|
178.8 | 18–21-mer DNA/RNA | >99 | 2 M NaCl/water, 40 °C, 48 h | 35 and 36 | |
| 285.5 | 5′ DBCO | 38 and 39 | ||||
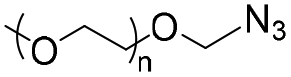
|
10.0 | 41–58-mer hairpin DNA | — | 3 M NaCl/water, 40 °C, 48 h | 37 | |
| Terminal DBCO | ||||||
| Phosphoramidite |
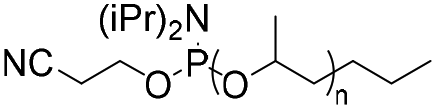
|
1–6.8 | 22-mer DNA on CPG | 41–32 | Standard DNA synthesis with extra (1 min) coupling time | 42 and 43 |
With regard to the bioconjugation reaction, amidation is among the first which is studied. Park and co-workers coupled poly(D,L-lactic-co-glycolic acid) (PLGA) with an antisense oligonucleotide to form an amphiphilic diblock copolymer, which self-assembled into micelles in aqueous buffer.17 The reaction was carried out between a 5′ primary amine-terminated oligonucleotide and the ω-carboxylic acid of PLGA under dicyclohexylcarbodiimide/N-hydroxysuccinimide (DCC/NHS) conditions in a predominantly dimethyl sulfoxide (DMSO) solution with 70% conjugation efficiency. The same group also synthesized poly(ethylene glycol) (PEG)–oligonucleotide conjugates using NHS-activated PEG and a 5′ amine-derivatized oligonucleotide in sodium phosphate buffer with 65% yield after HPLC.18,19 For nucleic acids lacking a 5′ amine, one can be added by reacting 5′ phosphate with ethylene diamine in the presence of a carbodiimide and imidazole.18
Aside from amidation, thiol–maleimide and thiol–acrylate(mide) Michael addition reactions have also been used to synthesize DNA-containing copolymers. Kataoka developed a series of oligonucleotide–PEG conjugates with varying substituents at the α-terminus of the PEG.20–22 The Michael reaction between a thiol-terminated oligonucleotide and acrylate-modified PEG results in an acid-labile linkage (β-propionate), while a reaction with a maleimide gives a non-releasable thioether, both in 60–80% yields. Of note is that the thioether linkages are subject to β-elimination (retro-Michael) reactions under basic conditions, and thus should be avoided if subsequent treatment with certain bases (NaOH, ammonia, etc.) is needed.23 Additionally, strong Michael acceptors (e.g. acrylonitrile) may also alkylate sites of the nucleobases (e.g. N3 of thymine); the reaction is often a source of impurity that takes place during the deprotection step of oligonucleotide chemical synthesis.24,25 Other than thiols, amine-nucleophiles have also been used to prepare nucleic acid–polymer conjugates via amination. For example, Nguyen and co-workers conjugated amine-terminated oligonucleotides to amphiphilic block copolymer micelles with surface-exposed tosylates, producing SNA-like structures.26 Because SNAs are the desired final products, yields for the conjugation reaction were not reported.
The discovery of copper(I)-catalyzed and strain-promoted azide–alkyne cycloaddition (CuAAC and SPAAC) click chemistry paved a new way to bioconjugates.27,28 The reaction shows very high selectivity and reaction yields. Matyjaszewski and Das have used CuAAC to synthesize a multiarm star conjugate consisting of polyacrylate-g-oligo(ethylene oxide) arms and multiple DNA strands on the periphery of the star.29 The conjugation was performed in aqueous buffer using an optimized “ligandless” condition, with near-quantitative yield as suggested by the loss of azide vibrations in infrared (IR) spectroscopy.30 O'Reilly et al. conducted a comprehensive optimization study for the CuAAC conjugation between hydrophobic polymers and a 22-mer oligonucleotide, and achieved exceptional yields (>50%) for permanently hydrophobic polymers such as polystyrene, poly(dimethylacrylamide), and poly(4-acryloylmorpholine).31 It is suggested that the record-breaking yields are in part due to the use of copper iodide triethylphosphite as the catalyst (CuI·P(OEt)3), which has a pre-complexed copper(I) that increases the reaction efficiency relative to traditional catalysts which require in situ formation of the complex. Of note is that prolonged treatment of DNA or RNA with Cu(I) can lead to extensive oxidative lesions mediated by reactive oxygen species such as hydroxyl radicals. However, the damage can be dramatically suppressed by introducing DMSO, a known radical scavenger, to the reaction system.32 Alternatively, the oxidation issue can be completely bypassed by switching to SPAAC, where ring strain is used to promote cycloaddition instead of Cu(I). Our group has routinely used this reaction to conjugate a variety of dibenzocyclooctyne (DBCO)-modified nucleic acids and analogues to azide-derivatized polymers, even hydrophobic ones,33,34 often with near-quantitative yields.35–39
One challenge associated with bioconjugation is the removal of unreacted polymers and nucleic acids. Mirkin and co-workers circumvented this difficulty by performing the conjugation reaction on a solid support. The group converted hydroxy-terminated polystyrene to a phosphoramidite by reacting it with chlorophosphoramidite.40 The resulting polymer was incorporated into DNA during solid-phase synthesis (SPS), and unreacted polymers were simply removed during the washing step. When polymers bearing multiple phosphoramidite functionalities are used during SPS, polyvalent DNA–polymer conjugates are expected. Indeed, Mirkin and Nguyen found that such a strategy can achieve up to 30% occupation of the potential polymer attachment sites by DNA strands.41 The Herrmann group also adopted the solid-state reaction for the preparation of a series of DNA-b-poly(propylene oxide) (PPO) diblock copolymers, which can form well-ordered structures in aqueous buffer due to their amphiphilicity.42,43 Along the same line, our group has performed solid-phase conjugation using the CuAAC click reaction to obtain a library of polystyrene-b-DNA and poly(tBu acrylate)-b-DNA conjugates with varying DNA and polymer lengths.44 While quantitative yields were achieved with very short DNA (6-mer), longer DNA and polymer lengths decrease the yield, likely due to the limited pore size of the controlled pore glass support (CPG), with the lowest yield (56%) observed for a 26-mer DNA and a 14-kDa polystyrene. One advantage of this strategy is that failed DNA strands during SPS will not be coupled to the polymer, as they lack the 5′ functionality used for coupling (assuming 3′–5′ DNA SPS). This feature saves the traditional purification step needed for DNA.
Nucleic acids as initiators or macromonomers for polymerization
While bioconjugation techniques have improved the accessibility of nucleic acid-containing copolymers considerably, certain constructs, such as high-density (e.g. 100% grafting density) multivalent DNA conjugates and amphiphilic conjugates, are still difficult to achieve using a post-polymerization conjugation methodology due to the strong repulsive interaction of the negatively charged phosphates in the nucleic acid backbone. Thus, methods are being developed to directly involve nucleic acids in the polymerization reaction and thereby access new types of nucleic acid-containing biomaterials.The He group studied living polymerization using DNA as a macroinitiator (Fig. 1A).45,46 The initiator DNA strand is immobilized on a surface by hybridization with a capture strand. The anchored dsDNA macroinitiator then initiates the polymerization of hydroxyethyl methacrylate (HEMA) in water in the presence of Cu(I). The growth of the surface brush can be visualized as a change in substrate opacity, which serves as a means for signal detection. Interestingly, polymer growth was accelerated with DNA, possibly due to the charged backbone promoting Cu(I) association. Matyjaszewski and Das expanded this methodology by developing an ATRP initiator phosphoramidite, allowing for the initiating site(s) to be incorporated anywhere in the oligonucleotide sequence (Fig. 1B).47,48 The polymerization reaction can then be performed from the DNA macroinitiator, either in solution or off the solid support.
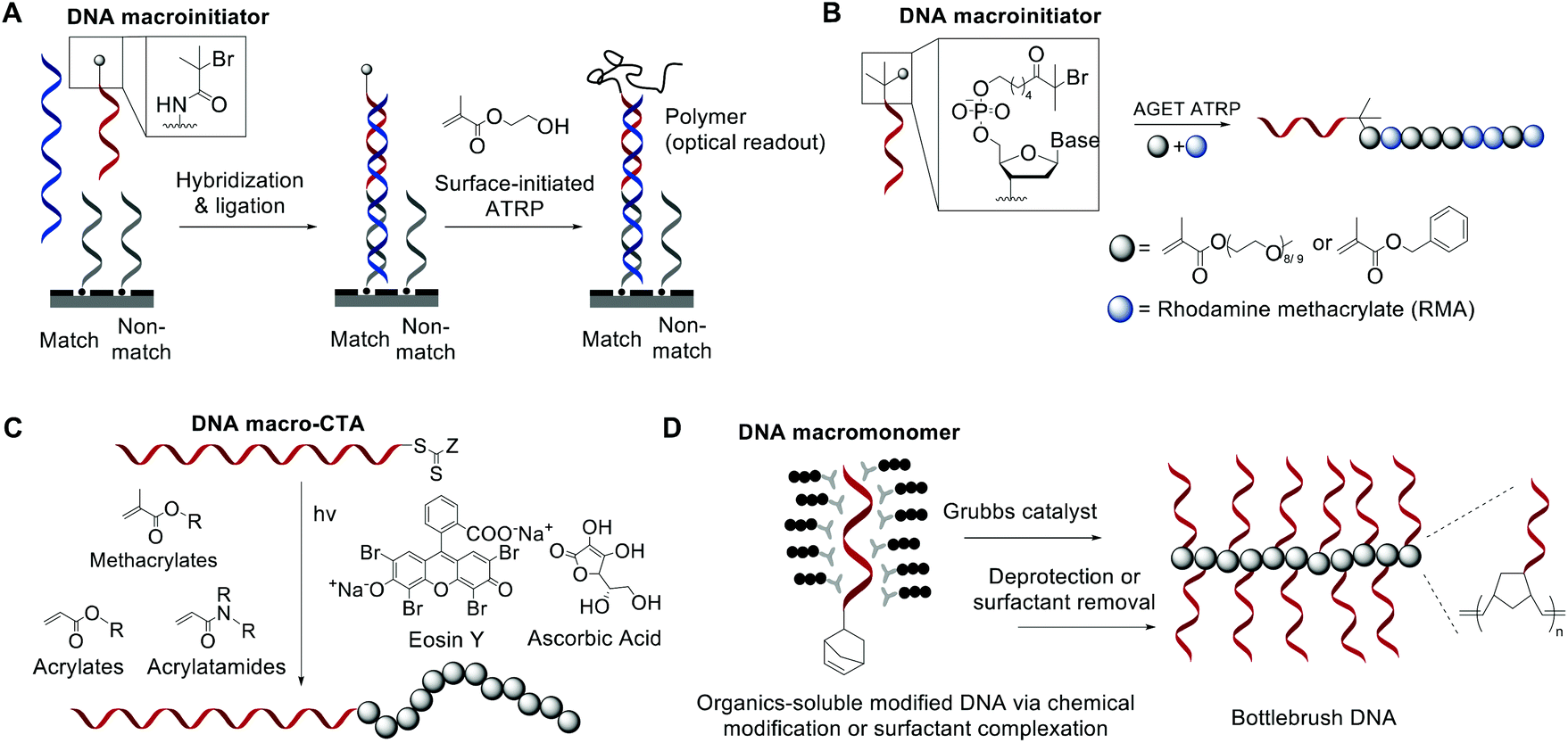 | ||
| Fig. 1 Living polymerization approaches to nucleic acid–polymer conjugates. The nucleic acid can be a macroinitiator for (A) surface-initiated ATRP45 or (B) solution-phase ATRP,48 a macro-CTA for (C) RAFT polymerization,53 and a macromonomer for (D) ROMP.23,54,55 | ||
In order to minimize DNA damage from Cu(I), increase oxygen tolerance, and enable better control of the polymerization reaction, improved ATRP methods such as activator generated by electron transfer (AGET) ATRP and photochemically mediated ATRP (photoATRP) have been applied in the synthesis of DNA-containing copolymers. For example, He and co-workers enhanced their DNA detection system with AGET ATRP using ascorbic acid as the reducing agent for Cu(II), allowing for a lower concentration of Cu(II) (0.3 mM) to be used.49 Maynard et al. reported a series of siRNA–polymer conjugates that were synthesized by AGET ATRP of two monomers, poly(ethylene glycol) methyl ether methacrylate (PEGMA) and di(ethylene glycol) methyl ether methacrylate (DEGMA) via the grafting-from method.50 However, only ∼30–50% yield was observed, which is lower than that of the grafting-onto method (∼50–80%, compared side-by-side). Matyjaszewski and co-workers developed an automated system for photoATRP using a DNA synthesizer.47 After the irradiation by UV light (365 nm), copper(II) was photoreduced in the presence of electron-donor ligands to initiate the reaction. The reaction only requires ppm levels of a copper catalyst at ambient temperature, and can be carried out by non-experts in synthetic polymer chemistry to obtain DNA–polymer conjugates without degassing procedures.
The general grafting-from method has been expanded to include reversible addition–fragmentation chain transfer (RAFT) polymerization, where the oligonucleotide is attached to a RAFT chain transfer agent (CTA). For instance, He and coworkers polymerized oligo(ethylene glycol) methacrylate (OEGMA) monomers via surface-anchored, trithiocarbonate-derivatized DNA and demonstrated successful growth by ellipsometric and IR measurements.51,52 Similarly, Weil et al. reported a solution-phase photoinduced RAFT polymerization to synthesize DNA–polymer hybrids (Fig. 1C).53 DNA macro-CTAs were synthesized by coupling tri- and dithiol carbonate-based CTAs to amine-terminated, single-stranded DNA. Upon irradiation with blue light (470 nm) in the presence of the photocatalyst, Eosin Y, and ascorbic acid, three monomer families (i.e., methacrylates, acrylates, and acrylamides) were successfully polymerized onto DNA.
Apart from using a nucleic acid as an initiator in polymerization, a limited number of successes were achieved in directly polymerizing oligonucleotides as a macromonomer. Nucleic acid-based macromonomers present a significant difficulty for polymerization. Not only must the oligonucleotide be soluble in the solvent of choice, the polymerized product must also be soluble. Additionally, oligonucleotides are bulky and highly anionic, resulting in steric issues during propagation. Gianneschi and co-workers solved these problems using a non-charged DNA analogue, peptide nucleic acid (PNA), as the macromonomer, which has great solubility in dimethylformamide (DMF). The team was able to polymerize a norbornene-functionalized decamer PNA by ring-opening metathesis polymerization (ROMP) using a 3rd generation Grubbs catalyst, achieving quantitative yields in some instances.54 Herrmann et al. was the first to achieve ROMP of natural, phosphodiester-based DNA macromonomers. To overcome the problems associated with solubility and charge-repulsion, Herrmann used a cationic surfactant, didodecyldimethylammonium bromide (DDAB), to form an electrostatic complex with a 7- or 14-mer oligonucleotide in aqueous solution. The isolated complex is dried and then redissolved in an organic solvent such as DMF, DMSO, tetrahydrofuran (THF), or chloroform, where polymerization was carried out (Fig. 1D).55 The strategy works well not only for polymerization but also for coupling reactions with hydrophobic ligands. However, complete removal of the surfactant from the product may be difficult for applications that require high purity. Our group also approached this problem, albeit from a different angle. Instead of using surfactants to neutralize the negative charge associated with DNA, we recognized that typical oligonucleotides are synthesized with various protecting groups attached to the exocyclic amine of nucleobases and the phosphates in the triester form, which make the oligonucleotide highly hydrophobic and charge-neutral. The protected form of DNA (protDNA) can be removed from the solid CPG support without affecting the protecting groups using the triphenylphosphine cleavage of a disulfide linker, and the isolated protDNA is soluble in dichloromethane. The ROMP of a 15-mer protDNA modified with a terminal norbornene yielded a bottlebrush-type conjugate with 70–90% yields and a high molecular weight (highest Mn: ∼300 kDa, Fig. 1D).23 After the polymerization, treatment with methanolic ammonia for 4 h cleanly removes the protecting groups. Collectively, substantial progress has been made to integrate nucleic acids with polymerization. With the vast knowledge base of both polymer chemistry and nucleic acid chemistry, we anticipate that a deeper merge of the two fields will greatly expand current materials possibilities.
Solid-phase synthesis
Instead of relying on polymerization to generate the polymer component of the nucleic acid–polymer biohybrid, it is also possible to use solid-phase reactions, typically used for oligonucleotide synthesis, to assemble the polymer. By designing appropriate phosphoramidite monomers, non-nucleotide units can be easily incorporated. In contrast to polymerization, SPS is carried out in a stepwise, iterative fashion, where the addition of each monomer involves a set of deprotection, coupling, capping, and oxidation steps. However, the complex synthesis can be automated, and the result is unmatched control in the monomer sequence, degree of polymerization, polydispersity, and relative position of DNA on the polymer chain. Nonetheless, while SPS may be ideal for linear or slightly branched DNA–polymer conjugates, multivalent architectures (i.e. multiple oligonucleotide strands) such as stars and brushes are currently not accessible via SPS. In addition, if phosphoramidite chemistry is to be used for the SPS, the monomers must be free of incompatible functionalities such as unprotected amines and hydroxyl groups, and the resulting polymer segment will have a phosphorus-containing backbone. Finally, the size of the DNA–polymer conjugate is limited due to the exponentially decreasing yields as the coupling number increases. For example, for a combined length of the nucleotides and polymer repeat units of 50, even when each coupling step enjoys 98% yield, the overall yield is a mere 36%.Early work in this field revolves around introducing functional residues to oligonucleotides to improve their properties. For instance, Behr and coworkers reported a series of oligonucleotide-b-oligospermine diblock copolymers, termed zip nucleic acids (ZNAs). These structures are produced by introducing varying numbers (1–6) of trifluoroacetyl-protected spermine phosphoramidite monomers during DNA synthesis, providing a conjugate with a cationic tail after deprotection. Spermines are outside edge binders, adding an ionic contribution to the overall stability of DNA duplexes (in addition to Watson–Crick base pairing). This contribution massively increases the duplex melting temperature (ΔTm as high as 15 °C), which is the function of the number of spermine residues (Fig. 2).56 Later, Corey et al. synthesized ZNAs using either DNA or locked nucleic acids (LNAs) as antisense agents to target human huntingtin (HTT) and human progesterone receptors (PRs). The spermine segment serves to enhance intracellular delivery and target binding, leading to carrier-free, high-efficacy transfection.57 Similarly, Remy and Kotera et al. synthesized di- and triblock siRNA–oligospermine conjugates to target the cell-constitutive natural lamin A/C gene. Interestingly, a more potent activity was observed when 30 spermine residues are attached singly to the 5′ of the sense strand (diblock) compared to having 15 residues at each of the termini (triblock).58
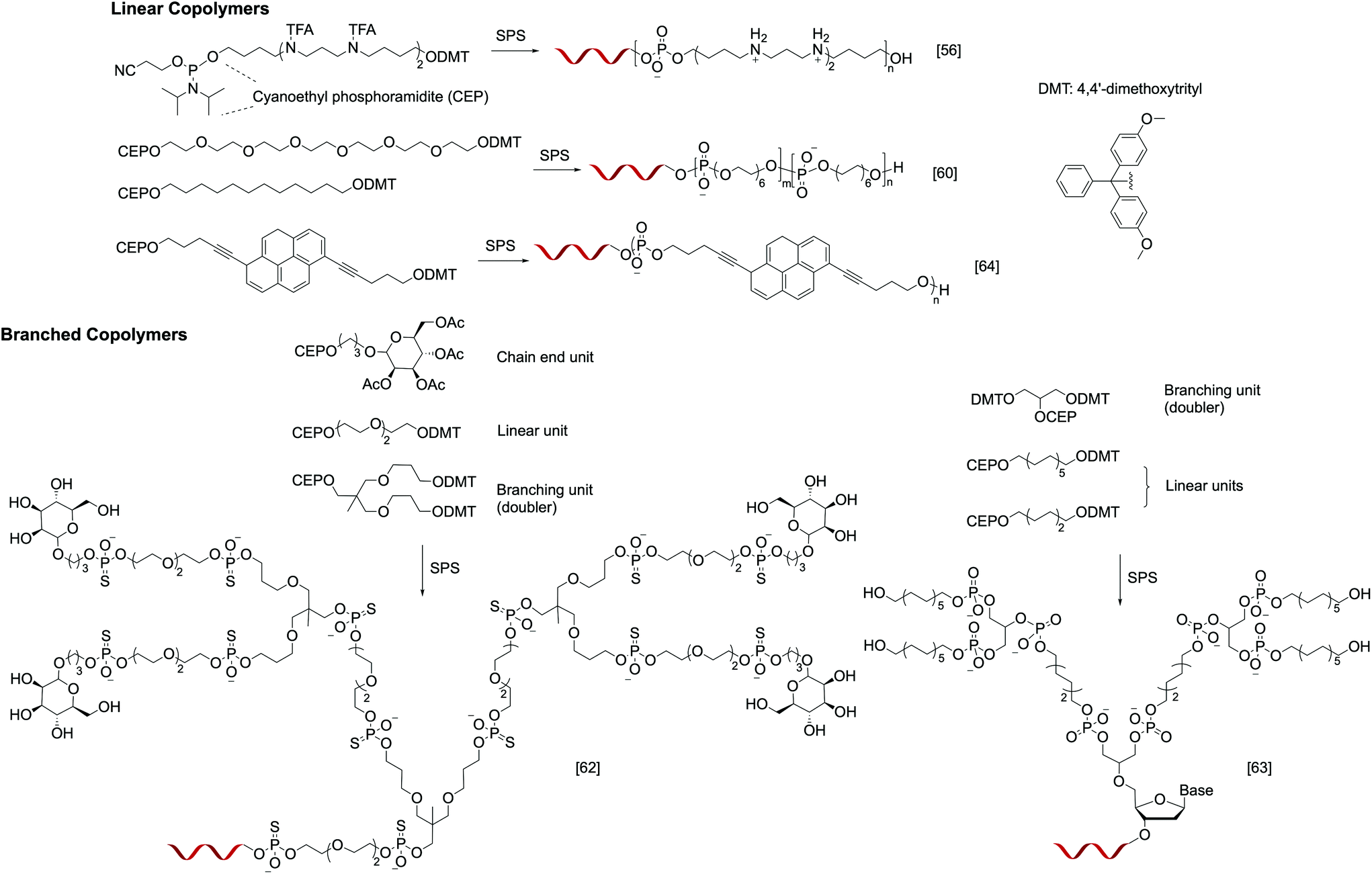 | ||
| Fig. 2 Examples of linear and branched nucleic acid–polymer conjugates prepared via solid-phase synthesis. | ||
More recently, with emerging interest in studying the organization of oligonucleotides via hybridization, hydrophobic self-assembly, or both,59 SPS is being utilized to access well-defined, amphiphilic oligonucleotide–polymer conjugates. Selman et al. reported several monodisperse, sequence/length-controlled DNA-b-polymer amphiphiles, where hexaethylene (HE) and hexaethylene glycol (HEG) units were sequentially introduced to the oligonucleotide during DNA synthesis (Fig. 2). When 12 HE/HEG monomer units were added onto a 19-mer oligonucleotide, the combined 31 coupling steps resulted in isolated yields ranging from 19 to 29%. These novel materials add a new dimension to the field of DNA nanotechnology by providing a secondary interaction that can be used to organize the nanostructures, encapsulate small molecules, etc. (Fig. 2).60 Subsequently, the same group expanded the monomer scope using a variety of phosphoramidites bearing a tertiary amine core. Two substituents of the tertiary amine are used for SPS coupling, while the third substituent carries a desired functionality. Using this method, monomers containing β-D-glucose, alkyne, carboxylate, and phenylalanine derivatives have been successfully added onto oligonucleotides.61
In addition to linear structures, conjugates involving dendritic polymers have also been successfully synthesized via SPS. Fréchet et al. reported G1 and G2 dendrimers bearing mannosylated chain ends and an oligonucleotide focal point (Fig. 2). In order to create the branching units, “doubler” and “tripler” phosphoramidites were used, which bear two and three dimethoxytrityl-capped hydroxyl groups, respectively. Upon deprotection, each of the free nucleophilic sites can lead to chain extension. The glycodendron with four mannoses conjugated to a thiolated 21-mer oligonucleotide exhibits a precisely measured mass of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 575.7 Da.62 Sleiman et al. recently reported an analogous G2 dendrimer (Fig. 2), but with a hydrophobic, alkylated dendron. Remarkably, after the conjugate is hybridized to the eight corners of a cube-shaped DNA nanoscaffold, the dendrons fold into a cube to interact with each other via hydrophobic interactions, forming essentially a cube-shaped micelle.63
575.7 Da.62 Sleiman et al. recently reported an analogous G2 dendrimer (Fig. 2), but with a hydrophobic, alkylated dendron. Remarkably, after the conjugate is hybridized to the eight corners of a cube-shaped DNA nanoscaffold, the dendrons fold into a cube to interact with each other via hydrophobic interactions, forming essentially a cube-shaped micelle.63
Other than fully covalent oligonucleotide–polymer conjugates, SPS-based methods also provide access to a class of supramolecular polymers, where a DNA–ligand system is used as a monomer for crystallization into higher-order structures. For instance, Häner et al. reported an amphiphilic chimeric pyrene–DNA oligomer (Fig. 2), which, when immersed in an aqueous medium, can assemble into a helical ribbon supramolecular polymer reaching up to several hundred nanometers in length. The number of pyrene units in the conjugate is critical for the formation of a elongated supramolecular polymer; while seven pyrenes per conjugate yielded the assembly, having four or less did not.64 In these supramolecular polymers, the DNA component remains able to hybridize with the complementary strand.65 When two supramolecular polymers bearing complementary DNA sequences are mixed, network formation was observed. Upon thermal disassembly and reannealing, the initial suprapolymer blend is converted to a supramolecular random copolymer, losing the ability to aggregate.66
Nucleoside/nucleobase-functionalized polymers
While SPS provides unparalleled control over the structure of the nucleic acid-containing copolymer, it is limited by the accessible architectures as well as the scale of synthesis. A strategy that in principle can solve these problems involves nucleobase-functionalized synthetic polymers. Unlike DNA and RNA, the backbone of nucleobase-functionalized polymers usually consists of enzymatically and hydrolytically stable bonds such as carbon–carbon bonds, amides, thioethers, etc. The backbone is often sufficiently flexible to accommodate the geometry requirements for base stacking. Using living polymerization techniques, sequential click chemistry, and post-polymerization modification, nucleoside/nucleobase-functionalized polymers can be prepared easily and in large quantities, and a variety of architectures can be achieved. In addition, the nucleobase can be expanded significantly beyond the four-letter library associated with natural nucleic acids. However, a severe downside is the difficulty in controlling the base sequence precisely and oftentimes limited solubility in aqueous solutions. Here, we focus on the various polymerization chemistries used; for more detailed reading and potential applications, there is a recent review on this topic by Tang and Zan.67As early as the 1960s, T'so and Takemoto independently reported the first synthetic nucleic acid analogs, which were achieved via free radical polymerization using nucleobases containing N-vinyl derivatives.68,69 However, only homopolymers or random copolymers with poor polydispersity can be synthesized due to the uncontrolled nature of conventional free radical polymerization. This general approach received renewed interest with the emergence of living polymerization. Haddleton et al. successfully synthesized uridine- and adenosine-functionalized polymers through ATRP in solution and on a solid support using nucleoside-substituted methacrylate monomers (Fig. 3).70,71 Remarkably, poly(5′-acryloyluridine) can act as a template in the radical polymerization of the complementary 5′-acryloyladenosine in the presence of the noncomplementary 5′-acryloyluridine.72 The work was later expanded to include the synthesis of water-soluble triblock and pentablock poly(methacryloyl nucleosides) by using bi-functional PEG macroinitiators (Fig. 3).73 Van Hest et al. adopted a similar strategy in the polymerization of methacryloyl-derivatized monomers bearing all four nucleobases (thymine, adenine, cytosine, and guanine).74 Interestingly, in the case of cytosine, a stronger copper-binding ligand (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) was used to gain control over the polymerization, which implies that the basic nature of the nucleobases may be problematic in reactions with transition metal complexes.
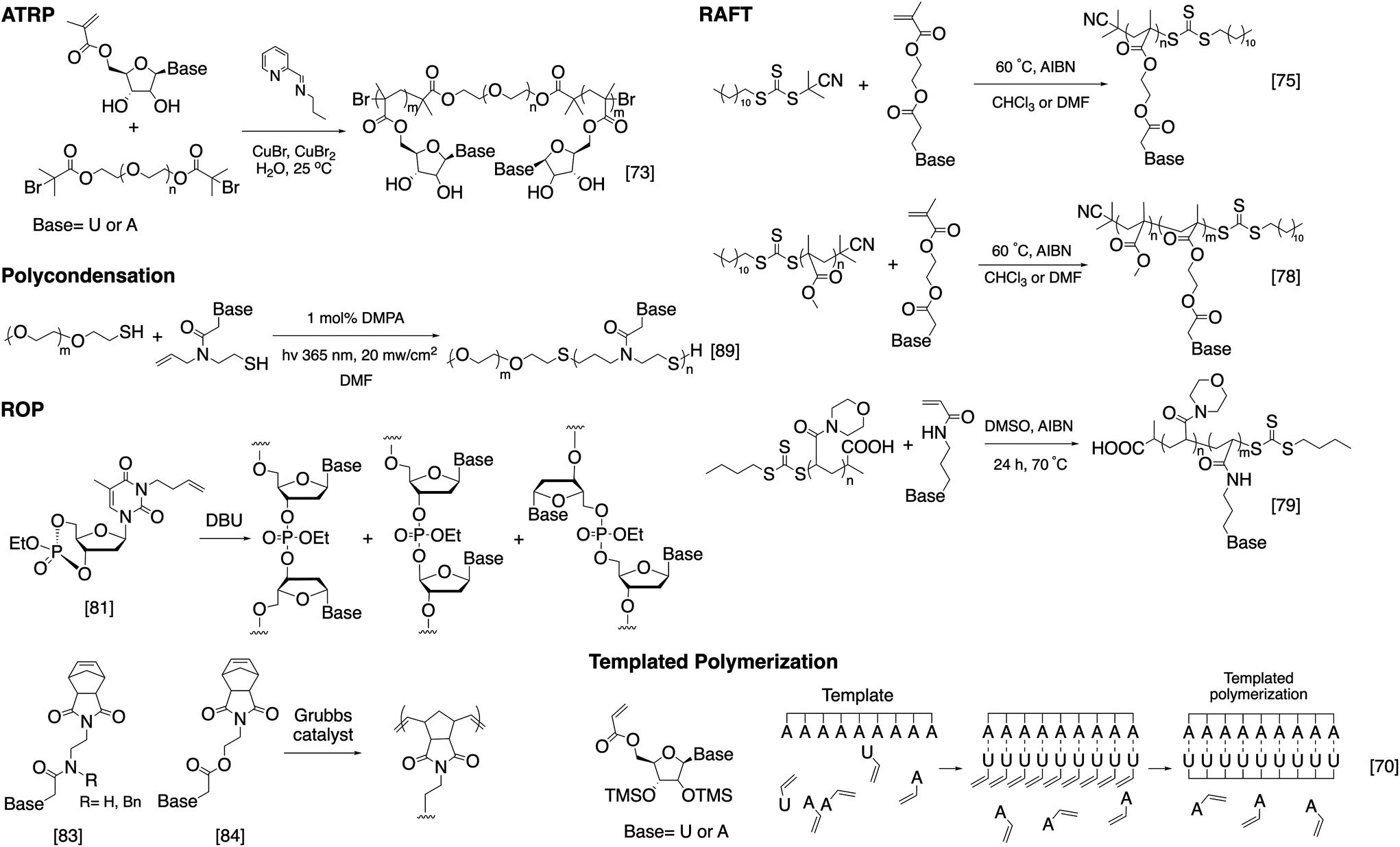 | ||
| Fig. 3 Examples of the polymerization strategies to access nucleoside/nucleobase-functionalized polymers. | ||
Unlike ATRP, RAFT polymerization does not involve a transition metal complex and may be more compatible with nucleobase-functionalized monomers. O'Reilly et al. have extensively investigated nucleobase-conjugated methacrylates for use in RAFT polymerization (Fig. 3).75–79 Several amphiphilic block copolymers consisting of poly(acryloylmorpholine) or PEG as the hydrophilic block and a nucleobase-containing polymer as the hydrophobic block were synthesized. Upon self-assembly in aqueous buffer, the nucleobase functionality forms the core of the micellar nanoparticles. Unexpectedly, upon mixing with a micelle containing the complementary nucleobase copolymer, the nucleobases are still able to interact despite being sequestered in the core, causing particle morphological and/or size changes. The group also explored the assembly of a fully hydrophobic diblock copolymer, poly(methylmethacrylate)-b-poly(methacryloylthymine), in organic solvents. Again, in the presence of an adenine-containing mediator, N9-hexyladenine, the morphology and/or size of the assembled particles are different from the assemblies without the mediator, suggesting that the hydrogen bonding interaction is sufficient to offset the reduced base stacking in organic solvents.78 This principle was used by Long et al. in the work on “supramolecular adhesives”, where adenine- and thymine-containing polymers form supramolecular crosslinks in chloroform.80 The increased importance of hydrogen bonding relative to base stacking is important even at the monomer stage: it was observed that adenine- and thymine-containing monomers pre-associate in chloroform, causing the polymerization to yield alternating copolymers.75
Another polymerization technique adapted for building nucleobase-functionalized polymers involves ring-opening polymerization. Wooley et al. reported a cyclic nucleotide monomer (3-butenylthymidine 3′,5′-cyclic monophosphate triester), which can be polymerized anionically to give a homopolymer with 70% 3′–5′ linkages (the rest are other isomeric forms).81,82 These polymers bear high structural similarity to natural DNA, with the exception of the modified thymine and a phosphotriester backbone. Gibson and co-workers first reported the synthesis of norbornene conjugated thymine, adenine, cytosine, and guanine monomers and their polymerization into homopolymers through ROMP (Fig. 3).83,84 However, a high degree of polymerization was not achieved (DPn = 5–8) due to solubility limitations. Subsequently, Sleiman et al. expanded upon Gibson's earlier work to synthesize well-defined, adenine-containing block copolymers and investigated their self-assembly. Similar block copolymers containing either thymine or diaminopyrimidine, a nucleobase analogue, were later used to template the polymerization of monomers with complementary hydrogen bonding character.85,86
The idea of DNA-templated polymerization was carefully examined by Liu et al. In order to create a higher degree of sequence control, the Liu group designed a four-base PNA monomer, aldehyde–TCAG–amine, which recognizes “codons” (5′-AGTC-3′) on a template DNA strand. Reductive amination yields repeats of the four-base PNA sequence with the length determined precisely by the DNA template.87 The same technique was later used to template the synthesis of synthetic polymers without structural analogy to nucleic acids, which was achieved by linking the “anticodon” region of the monomer (a pentamer PNA) to the polymerizable region via a disulfide linker. Cleavage of the linker post-polymerization yields the final polymer lacking any nucleobases.88 Of note is that these polymers require a step-growth mechanism to assemble, which can be carried out in solution as well, such as Bowman's series of “clickable nucleic acids” synthesized via thiol–ene click reactions (Fig. 3).89,90
Conclusions and outlook
To summarize, we have presented various synthetic strategies for nucleic acid-containing copolymers. Post-polymerization conjugation remains the most flexible approach regarding the types of nucleic acid/polymers that can be used and the accessible architectures of the conjugate. However, when it comes to amphiphilic conjugates and conjugates with a high-density arrangement of nucleic acids, direct involvement of a nucleic acid as a macromonomer or initiator for polymerization can achieve better results, although the solubility of nucleic acids in organic solvents is generally poor and requires extra steps to improve. The highest-quality copolymers are synthesized via solid-phase reactions, which incorporate monomers into the polymer in the same fashion as the incorporation of nucleotides, resulting in absolute control over the degree of polymerization, polydispersity, and polymer architecture. However, solid-phase approaches suffer from poor overall yields for structures requiring high coupling numbers, difficulty in synthesizing copolymers with multiple nucleic acid strands, and limited reaction scales. Conversely, it is possible to synthesize large scales of nucleoside- or nucleotide-functionalized polymers using conventional polymerization reactions, where the backbone of the nucleic acid is replaced with a synthetic polymer. With this approach, it is possible to synthesize novel polymers with unusual features such as non-natural nucleobases, an enzymatically stable backbone, solubility in organic solvents, and the ability to form hydrogen bonding in bulk. A serious downside, however, is the lost ability to control the base sequence.It is clear that each method has its own limitations, which require delicate balancing according to specific scenarios. However, every challenge is an opportunity. This area of study is advancing rapidly, and limitations may become irrelevant with new techniques. One way to further advance the field would be to integrate different approaches and thereby maximizing their respective advantages. It is also promising to combine efficient and compatible polymerization reactions with organic-soluble, protected forms of nucleic acids to expand the material space of the latter.
Author contributions
All authors contributed to the writing of the manuscript. H. L. and J. C. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. Z. acknowledges the support from the National Institute of General Medical Sciences (Award Number 1R01GM121612), the National Cancer Institute (Award Number 1R01CA251730), and the National Science Foundation (DMR Award Number 2004947).References
- J. D. Watson and F. H. Crick, Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid, Nature, 1953, 171(4356), 737–738 CrossRef CAS PubMed
.
- N. C. Seeman, DNA in a material world, Nature, 2003, 421(6921), 427–431 CrossRef PubMed
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, DNA nanotechnology from the test tube to the cell, Nat. Nanotechnol., 2015, 10(9), 748–760 CrossRef CAS PubMed
- S. D. Patil, D. G. Rhodes and D. J. Burgess, DNA-based therapeutics and DNA delivery systems: a comprehensive review, AAPS J., 2005, 7(1), E61–E77 CrossRef CAS PubMed
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, A DNA-based method for rationally assembling nanoparticles into macroscopic materials, Nature, 1996, 382(6592), 607–609 CrossRef CAS PubMed
- J. I. Cutler, E. Auyeung and C. A. Mirkin, Spherical Nucleic Acids, J. Am. Chem. Soc., 2012, 134(3), 1376–1391 CrossRef CAS PubMed
- M. A. A. Siddiqui and G. M. Keating, Pegaptanib, Drugs, 2005, 65(11), 1571–1577 CrossRef CAS PubMed
- E. W. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease, Nat. Rev. Drug Discovery, 2006, 5(2), 123–132 CrossRef CAS PubMed
- G. M. Bonora, E. Ivanova, V. Zarytova, B. Burcovich and F. M. Veronese, Synthesis and Characterization of High-Molecular Mass Polyethylene Glycol-Conjugated Oligonucleotides, Bioconjugate Chem., 1997, 8(6), 793–797 CrossRef CAS PubMed
- S.-I. Nakano and N. Sugimoto, The structural stability and catalytic activity of DNA and RNA oligonucleotides in the presence of organic solvents, Biophys. Rev., 2016, 8(1), 11–23 CrossRef CAS PubMed
- S. M. Mel'nikov, M. O. Khan, B. Lindman and B. Jönsson, Phase Behavior of Single DNA in Mixed Solvents, J. Am. Chem. Soc., 1999, 121(6), 1130–1136 CrossRef
- M. Dizdaroglu, Oxidatively induced DNA damage: Mechanisms, repair and disease, Cancer Lett., 2012, 327(1), 26–47 CrossRef CAS PubMed
- C. Angelé-Martínez, C. Goodman and J. Brumaghim, Metal-mediated DNA damage and cell death: mechanisms, detection methods, and cellular consequences, Metallomics, 2014, 6(8), 1358–1381 CrossRef PubMed
- M. Kwak and A. Herrmann, Nucleic acid amphiphiles: synthesis and self-assembled nanostructures, Chem. Soc. Rev., 2011, 40(12), 5745–5755 RSC
- J. Winkler, Oligonucleotide conjugates for therapeutic applications, Ther. Delivery, 2013, 4(7), 791–809 CrossRef CAS PubMed
- T. R. Wilks and R. K. O'Reilly, Efficient DNA-Polymer Coupling in Organic Solvents: A Survey of Amide Coupling, Thiol-Ene and Tetrazine-Norbornene Chemistries Applied to Conjugation of Poly(N-Isopropylacrylamide), Sci. Rep., 2016, 6, 39192 CrossRef CAS PubMed
- J. H. Jeong and T. G. Park, Novel Polymer−DNA Hybrid Polymeric Micelles Composed of Hydrophobic Poly(d,l-lactic-co-glycolic Acid) and Hydrophilic Oligonucleotides, Bioconjugate Chem., 2001, 12(6), 917–923 CrossRef CAS PubMed
- J. H. Jeong, S. W. Kim and T. G. Park, Novel Intracellular Delivery System of Antisense Oligonucleotide by Self-Assembled Hybrid Micelles Composed of DNA/PEG Conjugate and Cationic Fusogenic Peptide, Bioconjugate Chem., 2003, 14(2), 473–479 CrossRef CAS PubMed
- J. H. Jeong, S. H. Kim, S. W. Kim and T. G. Park, Polyelectrolyte Complex Micelles Composed of c-raf Antisense Oligodeoxynucleotide−Poly(ethylene glycol) Conjugate and Poly(ethylenimine): Effect of Systemic Administration on Tumor Growth, Bioconjugate Chem., 2005, 16(4), 1034–1037 CrossRef CAS PubMed
- M. Oishi, S. Sasaki, Y. Nagasaki and K. Kataoka, pH-Responsive Oligodeoxynucleotide (ODN)−Poly(Ethylene Glycol) Conjugate through Acid-Labile β-Thiopropionate Linkage: Preparation and Polyion Complex Micelle Formation, Biomacromolecules, 2003, 4(5), 1426–1432 CrossRef CAS PubMed
- M. Oishi, F. Nagatsugi, S. Sasaki, Y. Nagasaki and K. Kataoka, Smart Polyion Complex Micelles for Targeted Intracellular Delivery of PEGylated Antisense Oligonucleotides Containing Acid-Labile Linkages, ChemBioChem, 2005, 6(4), 718–725 CrossRef CAS PubMed
- M. Oishi, Y. Nagasaki, K. Itaka, N. Nishiyama and K. Kataoka, Lactosylated Poly(ethylene glycol)-siRNA Conjugate through Acid-Labile β-Thiopropionate Linkage to Construct pH-Sensitive Polyion Complex Micelles Achieving Enhanced Gene Silencing in Hepatoma Cells, J. Am. Chem. Soc., 2005, 127(6), 1624–1625 CrossRef CAS PubMed
- X. Tan, H. Lu, Y. Sun, X. Chen, D. Wang, F. Jia and K. Zhang, Expanding the Materials Space of DNA via Organic-Phase Ring-Opening Metathesis Polymerization, Chem, 2019, 5(6), 1584–1596 CAS
- A. Wilk, A. Grajkowski, L. R. Phillips and S. L. Beaucage, The 4-[N-Methyl-N-(2,2,2-trifluoroacetyl)amino]butyl Group as an Alternative to the 2-Cyanoethyl Group for Phosphate Protection in the Synthesis of Oligodeoxyribonucleotides, J. Org. Chem., 1999, 64(20), 7515–7522 CrossRef CAS
- D. C. Capaldi, H. Gaus, A. H. Krotz, J. Arnold, R. L. Carty, M. N. Moore, A. N. Scozzari, K. Lowery, D. L. Cole and V. T. Ravikumar, Synthesis of High-Quality Antisense Drugs. Addition of Acrylonitrile to Phosphorothioate Oligonucleotides: Adduct Characterization and Avoidance, Org. Process Res. Dev., 2003, 7(6), 832–838 CrossRef CAS
- P. A. Bertin, J. M. Gibbs, C. K. F. Shen, C. S. Thaxton, W. A. Russin, C. A. Mirkin and S. T. Nguyen, Multifunctional polymeric nanoparticles from diverse bioactive agents, J. Am. Chem. Soc., 2006, 128(13), 4168–4169 CrossRef CAS PubMed
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40(11), 2004–2021 CrossRef CAS PubMed
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Efficiency and Fidelity in a Click-Chemistry Route to Triazole Dendrimers by the Copper(I)-Catalyzed Ligation of Azides and Alkynes, Angew. Chem., Int. Ed., 2004, 43(30), 3928–3932 CrossRef CAS PubMed
- S. Averick, E. Paredes, W. Li, K. Matyjaszewski and S. R. Das, Direct DNA Conjugation to Star Polymers for Controlled Reversible Assemblies, Bioconjugate Chem., 2011, 22(10), 2030–2037 CrossRef CAS PubMed
- E. Paredes and S. R. Das, Click Chemistry for Rapid Labeling and Ligation of RNA, ChemBioChem, 2011, 12(1), 125–131 CrossRef CAS PubMed
- T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, “Giant Surfactants” Created by the Fast and Efficient Functionalization of a DNA Tetrahedron with a Temperature-Responsive Polymer, ACS Nano, 2013, 7(10), 8561–8572 CrossRef CAS PubMed
- G. R. Abel, Z. A. Calabrese, J. Ayco, J. E. Hein and T. Ye, Measuring and Suppressing the Oxidative Damage to DNA During Cu(I)-Catalyzed Azide–Alkyne Cycloaddition, Bioconjugate Chem., 2016, 27(3), 698–704 CrossRef CAS PubMed
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, Light-Triggered, Self-Immolative Nucleic Acid-Drug Nanostructures, J. Am. Chem. Soc., 2015, 137(19), 6112–6115 CrossRef CAS PubMed
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, Blurring the Role of Oligonucleotides: Spherical Nucleic Acids as a Drug Delivery Vehicle, J. Am. Chem. Soc., 2016, 138(34), 10834–10837 CrossRef CAS PubMed
- X. Lu, F. Jia, X. Tan, D. Wang, X. Cao, J. Zheng and K. Zhang, Effective Antisense Gene Regulation via Noncationic, Polyethylene Glycol Brushes, J. Am. Chem. Soc., 2016, 138(29), 9097–9100 CrossRef CAS PubMed
- F. Jia, X. Lu, D. Wang, X. Cao, X. Tan, H. Lu and K. Zhang, Depth-Profiling the Nuclease Stability and the Gene Silencing Efficacy of Brush-Architectured Poly(ethylene glycol)-DNA Conjugates, J. Am. Chem. Soc., 2017, 139(31), 10605–10608 CrossRef CAS PubMed
- Y. Wang, D. Wang, F. Jia, A. Miller, X. Tan, P. Chen, L. Zhang, H. Lu, Y. Fang, X. Kang, J. Cai, M. Ren and K. Zhang, Self-Assembled DNA–PEG Bottlebrushes Enhance Antisense Activity and Pharmacokinetics of Oligonucleotides, ACS Appl. Mater. Interfaces, 2020, 12(41), 45830–45837 CrossRef CAS PubMed
- X. Lu, H. Fu, K. C. Shih, F. Jia, Y. Sun, D. Wang, Y. Wang, S. Ekatan, M. P. Nieh, Y. Lin and K. Zhang, DNA-Mediated Step-Growth Polymerization of Bottlebrush Macromonomers, J. Am. Chem. Soc., 2020, 142(23), 10297–10301 CrossRef CAS PubMed
- D. Wang, J. Lin, F. Jia, X. Tan, Y. Wang, X. Sun, X. Cao, F. Che, H. Lu, X. Gao, J. C. Shimkonis, Z. Nyoni, X. Lu and K. Zhang, Bottlebrush-architectured poly(ethylene glycol) as an efficient vector for RNA interference in vivo, Sci. Adv., 2019, 5(2), eaav9322 CrossRef CAS PubMed
- Z. Li, Y. Zhang, P. Fullhart and C. A. Mirkin, Reversible and chemically programmable micelle assembly with DNA block-copolymer amphiphiles, Nano Lett., 2004, 4(6), 1055–1058 CrossRef CAS
- K. J. Watson, S. J. Park, J. H. Im, S. T. Nguyen and C. A. Mirkin, DNA-block copolymer conjugates, J. Am. Chem. Soc., 2001, 123(23), 5592–5593 CrossRef CAS PubMed
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, DNA-Templated Synthesis in Three Dimensions: Introducing a Micellar Scaffold for Organic Reactions, Angew. Chem., Int. Ed., 2006, 45(25), 4206–4210 CrossRef CAS PubMed
- F. E. Alemdaroglu, N. C. Alemdaroglu, P. Langguth and A. Herrmann, DNA Block Copolymer Micelles – A Combinatorial Tool for Cancer Nanotechnology, Adv. Mater., 2008, 20(5), 899–902 CrossRef CAS
- F. Jia, X. Lu, X. Tan and K. Zhang, Facile synthesis of nucleic acid-polymer amphiphiles and their self-assembly, Chem. Commun., 2015, 51(37), 7843–7846 RSC
- X. Lou, M. S. Lewis, C. B. Gorman and L. He, Detection of DNA Point Mutation by Atom Transfer Radical Polymerization, Anal. Chem., 2005, 77(15), 4698–4705 CrossRef CAS PubMed
- X. Lou and L. He, DNA-Accelerated Atom Transfer Radical Polymerization on a Gold Surface, Langmuir, 2006, 22(6), 2640–2646 CrossRef CAS PubMed
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Automated Synthesis of Well-Defined Polymers and Biohybrids by Atom Transfer Radical Polymerization Using a DNA Synthesizer, Angew. Chem., Int. Ed., 2017, 56(10), 2740–2743 CrossRef CAS PubMed
- S. E. Averick, S. K. Dey, D. Grahacharya, K. Matyjaszewski and S. R. Das, Solid-Phase Incorporation of an ATRP Initiator for Polymer–DNA Biohybrids, Angew. Chem., Int. Ed., 2014, 53(10), 2739–2744 CrossRef CAS PubMed
- Y. Wu, S. Liu and L. He, Electrochemical Biosensing Using Amplification-by-Polymerization, Anal. Chem., 2009, 81(16), 7015–7021 CrossRef CAS PubMed
- E.-W. Lin and H. D. Maynard, Grafting from Small Interfering Ribonucleic Acid (siRNA) as an Alternative Synthesis Route to siRNA–Polymer Conjugates, Macromolecules, 2015, 48(16), 5640–5647 CrossRef CAS
- P. He and L. He, Synthesis of Surface-Anchored DNA−Polymer Bioconjugates Using Reversible Addition−Fragmentation Chain Transfer Polymerization, Biomacromolecules, 2009, 10(7), 1804–1809 CrossRef CAS PubMed
- P. He, W. Zheng, E. Z. Tucker, C. B. Gorman and L. He, Reversible Addition−Fragmentation Chain Transfer Polymerization in DNA Biosensing, Anal. Chem., 2008, 80(10), 3633–3639 CrossRef CAS PubMed
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, DNA–Polymer Conjugates by Photoinduced RAFT Polymerization, Biomacromolecules, 2019, 20(1), 212–221 CrossRef CAS PubMed
- C. R. James, A. M. Rush, T. Insley, L. Vukovic, L. Adamiak, P. Kral and N. C. Gianneschi, Poly(oligonucleotide), J. Am. Chem. Soc., 2014, 136(32), 11216–11219 CrossRef CAS PubMed
- K. Liu, L. Zheng, Q. Liu, J. W. de Vries, J. Y. Gerasimov and A. Herrmann, Nucleic acid chemistry in the organic phase: from functionalized oligonucleotides to DNA side chain polymers, J. Am. Chem. Soc., 2014, 136(40), 14255–14262 CrossRef CAS PubMed
- R. Noir, M. Kotera, B. Pons, J.-S. Remy and J.-P. Behr, Oligonucleotide−Oligospermine Conjugates (Zip Nucleic Acids): A Convenient Means of Finely Tuning Hybridization Temperatures, J. Am. Chem. Soc., 2008, 130(40), 13500–13505 CrossRef CAS PubMed
- K. T. Gagnon, J. K. Watts, H. M. Pendergraff, C. Montaillier, D. Thai, P. Potier and D. R. Corey, Antisense and antigene inhibition of gene expression by cell-permeable oligonucleotide-oligospermine conjugates, J. Am. Chem. Soc., 2011, 133(22), 8404–8407 CrossRef CAS PubMed
- M. Nothisen, J. Bagilet, J. P. Behr, J. S. Remy and M. Kotera, Structure Tuning of Cationic Oligospermine-siRNA Conjugates for Carrier-Free Gene Silencing, Mol. Pharm., 2016, 13(8), 2718–2728 CrossRef CAS PubMed
- F. Jia, H. Li, R. Chen and K. Zhang, Self-Assembly of DNA-Containing Copolymers, Bioconjugate Chem., 2019, 30(7), 1880–1888 CrossRef CAS PubMed
- T. G. W. Edwardson, K. M. M. Carneiro, C. J. Serpell and H. F. Sleiman, An Efficient and Modular Route to Sequence-Defined Polymers Appended to DNA, Angew. Chem., Int. Ed., 2014, 53(18), 4567–4571 CrossRef CAS PubMed
- D. de Rochambeau, Y. Sun, M. Barlog, H. S. Bazzi and H. F. Sleiman, Modular Strategy To Expand the Chemical Diversity of DNA and Sequence-Controlled Polymers, J. Org. Chem., 2018, 83(17), 9774–9786 CrossRef CAS PubMed
- M. Dubber and J. M. J. Fréchet, Solid-Phase Synthesis of Multivalent Glycoconjugates on a DNA Synthesizer, Bioconjugate Chem., 2003, 14(1), 239–246 CrossRef CAS
- T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Site-specific positioning of dendritic alkyl chains on DNA cages enables their geometry-dependent self-assembly, Nat. Chem., 2013, 5(10), 868–875 CrossRef CAS PubMed
- Y. Vyborna, M. Vybornyi, A. V. Rudnev and R. Häner, DNA-Grafted Supramolecular Polymers: Helical Ribbon Structures Formed by Self-Assembly of Pyrene–DNA Chimeric Oligomers, Angew. Chem., Int. Ed., 2015, 54(27), 7934–7938 CrossRef CAS PubMed
- Y. Vyborna, M. Vybornyi and R. Haner, Functional DNA-grafted supramolecular polymers - chirality, cargo binding and hierarchical organization, Chem. Commun., 2017, 53(37), 5179–5181 RSC
- Y. Vyborna, M. Vybornyi and R. Haner, Pathway Diversity in the Self-Assembly of DNA-Derived Bioconjugates, Bioconjugate Chem., 2016, 27(11), 2755–2761 CrossRef CAS
- J. Li, Z. Wang, Z. Hua and C. Tang, Supramolecular nucleobase-functionalized polymers: synthesis and potential biological applications, J. Mater. Chem. B, 2020, 8(8), 1576–1588 RSC
- J. Pitha and P. O. P. Ts'o, N-Vinyl derivatives of substituted pyrimidines and purines, J. Org. Chem., 1968, 33(4), 1341–1344 CrossRef CAS PubMed
- N. Ueda, K. Konda, M. Kono, K. Takemoto and M. Imoto, Vinyl polymerization. 217. Vinyl compounds of nucleic acid basis. I. Synthesis of N-vinylthymine, and N-vinyladenine, Makromol. Chem., 1968, 120(1), 13–20 CrossRef CAS
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Hydrogen Bond Template-Directed Polymerization of Protected 5′-Acryloylnucleosides, Macromolecules, 1999, 32(20), 6560–6564 CrossRef CAS
- A. Marsh, A. Khan, M. Garcia and D. M. Haddleton, Copper(I) mediated radical polymerisation of uridine and adenosine monomers on a silica support, Chem. Commun., 2000,(21), 2083–2084 RSC
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Templated polymerizations on solid supports mediated by complementary nucleoside interactions, Polym. Chem., 2015, 6(11), 1944–1951 RSC
- M. Garcia, M. P. Beecham, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Water soluble triblock and pentablock poly(methacryloyl nucleosides) from copper-mediated living radical polymerisation using PEG macroinitiators, Eur. Polym. J., 2015, 66, 444–451 CrossRef CAS
- H. J. Spijker, F. L. van Delft and J. C. M. van Hest, Atom Transfer Radical Polymerization of Adenine, Thymine, Cytosine, and Guanine Nucleobase Monomers, Macromolecules, 2007, 40(1), 12–18 CrossRef CAS
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, Effect of Complementary Nucleobase Interactions on the Copolymer Composition of RAFT Copolymerizations, ACS Macro Lett., 2013, 2(7), 581–586 CrossRef CAS
- Y. Kang, A. Pitto-Barry, S. Rolph, M. Hua, Z. Hands-Portman, I. Kirby, N. O'Reilly and R. K, Use of complementary nucleobase-containing synthetic polymers to prepare complex self-assembled morphologies in water, Polym. Chem., 2016, 7(16), 2836–2846 RSC
- Y. Kang, A. Pitto-Barry, H. Willcock, W.-D. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Exploiting nucleobase-containing materials – from monomers to complex morphologies using RAFT dispersion polymerization, Polym. Chem., 2015, 6(1), 106–117 RSC
- Y. Kang, A. Pitto-Barry, A. Maitland and R. K. O'Reilly, RAFT dispersion polymerization: a method to tune the morphology of thymine-containing self-assemblies, Polym. Chem., 2015, 6(27), 4984–4992 RSC
- Z. Hua, A. Pitto-Barry, Y. Kang, N. Kirby, T. R. Wilks and R. K. O'Reilly, Micellar nanoparticles with tuneable morphologies through interactions between nucleobase-containing synthetic polymers in aqueous solution, Polym. Chem., 2016, 7(25), 4254–4262 RSC
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Nucleobase Self-Assembly in Supramolecular Adhesives, Macromolecules, 2012, 45(2), 805–812 CrossRef CAS
- Y.-Y. T. Tsao, T. H. Smith and K. L. Wooley, Regioisomeric Preference in Ring-Opening Polymerization of 3′,5′-Cyclic Phosphoesters of Functional Thymidine DNA Analogues, ACS Macro Lett., 2018, 7(2), 153–158 CrossRef CAS
- Y. T. Tsao and K. L. Wooley, Synthetic, Functional Thymidine-Derived Polydeoxyribonucleotide Analogues from a Six-Membered Cyclic Phosphoester, J. Am. Chem. Soc., 2017, 139(15), 5467–5473 CrossRef CAS PubMed
- C. Gibson, V. L, E. Marshall, M. North, A. Robson, D. J and P. Williams, Thymine functionalised polymers via living ring-opening metathesis polymerisation, Chem. Commun., 1997,(12), 1095–1096 RSC
- G. Davies, R. Gibson, V. C. Hursthouse, M. B. Light, M. E. Marshall, E. L. North, M. Robson, D. A. Thompson, I. White, A. J. P. Williams, D. J. Williams and P. J, Synthesis of nucleic-acid base containing norbornene derivatives as monomers for ring-opening-metathesis–polymerization, J. Chem. Soc., Perkin Trans. 1, 2001,(24), 3365–3381 Search PubMed
- H. S. Bazzi and H. F. Sleiman, Adenine-Containing Block Copolymers via Ring-Opening Metathesis Polymerization: Synthesis and Self-Assembly into Rod Morphologies, Macromolecules, 2002, 35(26), 9617–9620 CrossRef CAS
- P. K. Lo and H. F. Sleiman, Nucleobase-Templated Polymerization: Copying the Chain Length and Polydispersity of Living Polymers into Conjugated Polymers, J. Am. Chem. Soc., 2009, 131(12), 4182–4183 CrossRef CAS PubMed
- D. M. Rosenbaum and D. R. Liu, Efficient and Sequence-Specific DNA-Templated Polymerization of Peptide Nucleic Acid Aldehydes, J. Am. Chem. Soc., 2003, 125(46), 13924–13925 CrossRef CAS PubMed
- J. Niu, R. Hili and D. R. Liu, Enzyme-free translation of DNA into sequence-defined synthetic polymers structurally unrelated to nucleic acids, Nat. Chem., 2013, 5(4), 282–292 CrossRef CAS PubMed
- W. Xi, S. Pattanayak, C. Wang, B. Fairbanks, T. Gong, J. Wagner, C. J. Kloxin and C. N. Bowman, Clickable Nucleic Acids: Sequence-Controlled Periodic Copolymer/Oligomer Synthesis by Orthogonal Thiol-X Reactions, Angew. Chem., Int. Ed., 2015, 54(48), 14462–14467 CrossRef CAS PubMed
- Z. Liu, B. Fairbanks, L. He, T. Liu, P. Shah, J. N. Cha, J. W. Stansbury and C. N. Bowman, Water-soluble clickable nucleic acid (CNA) polymer synthesis by functionalizing the pendant hydroxyl, Chem. Commun., 2017, 53(73), 10156–10159 RSC
| This journal is © The Royal Society of Chemistry 2021 |