
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElaboration and mechanical properties of elastomeric fibrous scaffolds based on crosslinked poly(glycerol sebacate) and cyclodextrin for soft tissue engineering†
Florence
Flaig
,
Caroline
Faria Bellani
,
Özgün
Uyumaz
,
Guy
Schlatter
 * and
Anne
Hébraud
*
* and
Anne
Hébraud
*
ICPEES, Institut de Chimie et Procédés pour l’Energie, l’Environnement et la Santé, CNRS UMR 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: guy.schlatter@unistra.fr; anne.hebraud@unistra.fr
First published on 4th January 2021
Abstract
Poly(glycerol sebacate) (PGS) is a promising material designed for soft tissue engineering. Several examples of its use to prepare electrospun scaffolds can be found in the literature. However, the fabrication of nanofibrous scaffolds based on crosslinked PGS remains challenging because of its thermoset nature. Here, we propose a new method to electrospin PGS in blend with polyvinylpyrrolidone (PVP) and hydroxypropyl-β-cyclodextrin (HPβCD) as carrier molecules. After electrospinning, the PGS prepolymers were crosslinked by thermal treatment under vacuum, resulting in elastomeric fibers. Finally, unreacted molecules were removed by immersion of the fibers in water. Our PGS/HPβCD-based nanofibrous scaffolds are good candidates for soft tissue engineering with a Young's modulus of 2 MPa and strain at break of 390% in the dry state, reaching 120 to 170 kPa and 70% in the wet state.
Introduction
Tissue engineering is a promising field for the regeneration of soft tissues, in particular for cardiac regeneration.1 In this approach, cells are seeded on scaffolds designed to promote cell differentiation, growth and spreading, before in vivo implantation. Scaffolds should mimic the extracellular matrix to influence the cell behavior and the use of biomaterials having mechanical properties matching the host tissue's properties is highly desirable.2 The design of elastomeric scaffolds is thus of great interest for soft tissue engineering. Poly(glycerol sebacate) (PGS), a biodegradable elastomer prepared from two natural metabolites of the human body, has been developed for this purpose.3 Its mechanical properties and degradation kinetics can be tuned by varying the synthesis conditions.4 Scaffolds with Young's moduli matching heart's stiffness have thus been prepared and used as patches for the delivery of embryonic stems cells to the heart.5 They showed good compatibility in vitro as well as in vivo. Besides, the therapeutic properties of the scaffolds were improved by the structuration of the PGS constructs: for example, accordion-like honeycomb structures gave interesting results6 as they mimic myocardium's anisotropy. The structuration of the scaffold in a nanofibrous mat, mimicking the extracellular matrix, can also be achieved by electrospinning and has shown to enhance tissue regeneration.7 Electrospun scaffolds have thus become promising candidates for tissue engineering.7–10However, as PGS is an insoluble thermoset elastomer, it is not possible to electrospin it directly. Thus, PGS scaffolds have to be prepared in at least 3 steps. In the first step, the polycondensation of glycerol and sebacic acid leads to a soluble low-molar mass prepolymer (pPGS). In the second step, pPGS should be melted or dissolved in order to process it by electrospinning. Finally, a curing step at 120–180 °C under vacuum allows the crosslinking of pPGS into a 3D macromolecular elastomeric network.
However, because of its low molar mass, pPGS does not provide entanglements in solution, which is necessary to prevent the breaking of the jet into droplets during electrospinning.11 Different carrier polymers able to provide entanglements have thus been added into the pPGS solution to produce continuous nanofibers by electrospinning. As examples, proteins such as zein, gelatin or collagen were used for their good biocompatibility. Zein12 or gelatin13,14 was blended with pPGS to prepare fibers with up to two thirds of pPGS in the case of gelatin, while collagen was used as the shell material in the coaxial electrospinning of core–shell fibers containing pPGS in the core.15 In both cases, pPGS was not crosslinked into the PGS elastomer because proteins are not heat resistant and the mats cannot therefore withstand thermal treatment. To reach relatively high strains, proteins were crosslinked with EDC/NHS solutions for gelatin13 or under glutaraldehyde vapors for collagen.15 However, an additional toxicity is brought by these reactants, whereas the final material does not benefit from PGS elastomeric properties. Several works also focused on electrospinning of pPGS blended with the thermoplastic poly(ε-caprolactone) (PCL) to prepare cardiac patches,16,17 but in this case too, no curing step is possible due to the low melting temperature of the carrier polymer (60 °C). Electrospinning of pPGS blended with poly(lactic acid) (PLA) was also carried out for heart tissue engineering applications.18,19 However, although these scaffolds achieved excellent biological properties, PLA was not removed and the overall mechanical properties were not elastomeric. As a matter of fact, the chosen carrier polymer should have a glass transition temperature (Tg) or melting temperature (Tm) higher than the curing temperature in order to maintain the shape of the fibers and prevent the low-viscosity melted pPGS from leaking out of the fibers during crosslinking. Moreover, after crosslinking, it should be possible to dissolve the carrier polymer to obtain pure crosslinked PGS fibers. In this context, You et al.20,21 used PLA as the carrier polymer in coaxial electrospinning. They electrospun a core solution of pPGS and a shell solution of PLA (Tm = 160 °C). Polyethylene oxide (PEO, Tm = 60 °C) was added to the shell in order to decrease the modulus of the final material. The PEO had to be removed by dissolution in water before curing at 120 °C, and PLA was removed by dissolution in chloroform after crosslinking. The resultant fibrous mat had a Young's modulus of 0.47 MPa in the dry state which is suitable for soft tissues. However, in this method, a toxic solvent is employed to purify the PGS fibers: a water-soluble carrier polymer would be a better choice. Poly(vinyl alcohol) (PVA) was chosen by Xu et al.22 to electrospin coaxial fibers with PVA as the shell and pPGS as the core. After crosslinking, the PVA was partially removed by dissolution in water, leading to a material having a Young's modulus close to 35 MPa and a strain at break of 130% in the wet state. The blend strategy was preferred by Jeffries et al.23 whose work led, after curing and dissolution of PVA in water, to fibrous mats having, in the wet state, a Young's modulus close to 1 MPa and strain at break potentially higher than 750% under the weakest crosslinking conditions. Nevertheless, in these last examples, it was not possible to entirely remove the carrier polymer. This could be explained by a chemical reaction occurring during curing between the alcohols on PVA and the carboxylic acids on PGS.
In the present study, polyvinylpyrrolidone (PVP) was chosen as the carrier polymer for blend electrospinning as it has a Tg of 180 °C allowing thermal post-treatments, is water soluble and presents no reactive sites for PGS. Moreover, it is approved by the FDA for biomedical applications. PVP was also used with pPGS in blend electrospinning by Keirouz et al.24 to prepare fibers with up to 40% pPGS. But in this work, PGS was not totally crosslinked in the thermoset elastomer. Yet, uncrosslinked pPGS is known to possibly induce toxicity due to the acid groups.25
In our work, in order to increase the content of PGS in the fibers while preventing the fibers to merge together during electrospinning or crosslinking, addition of hydroxypropyl-β-cyclodextrin (HPβCD) in the processed mixture was proposed. Indeed, HPβCD has a high degradation temperature (278 °C), is biocompatible and is known to have the ability to stabilize the electrospinning process.26–28 Besides, its hydrophobic cavity can be exploited to encapsulate drugs by host–guest inclusion complexes.29 Different blends of pPGS, PVP and HPβCD were electrospun to form materials with 50% of pPGS and various proportions of PVP and HPβCD. The minimal HPβCD content necessary to electrospin regular fibers in a stable way was identified. These fibers were cured following two different heating settings in order to tune their mechanical properties. Finally, they were washed in water to remove the PVP and unreacted HPβCD and obtain elastomeric mats. Their mechanical properties were compared to those of PGS-based scaffolds prepared with PVA as the carrier polymer in order to evaluate their suitability for soft tissue engineering.
Experimental
Materials
Glycerol (>99.96%, Fisher Chemicals), sebacic acid (98%, Acros Organics), d-DMSO (99.8%D, Euriso-top), polyvinylpyrrolidone (PVP, 1300 kg mol−1, Aldrich), hydroxypropyl-β-cyclodextrin (HPβCD, 97%, degree of substitution: 2 to 6, Acros Organics), dimethylformamide (DMF, >99.8%, Sigma-Aldrich), absolute anhydrous ethanol (>99.9%, Carlo Erba) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, >99.0%, TCI Chemicals) were used as received. Poly(vinyl alcohol) (PVA, Gohsenol® EG-05PW, saponification value 86.5–89.0 mol%) was generously donated by Nippon Gohsei™ and used as received.Synthesis and characterization of the prepolymer (pPGS)
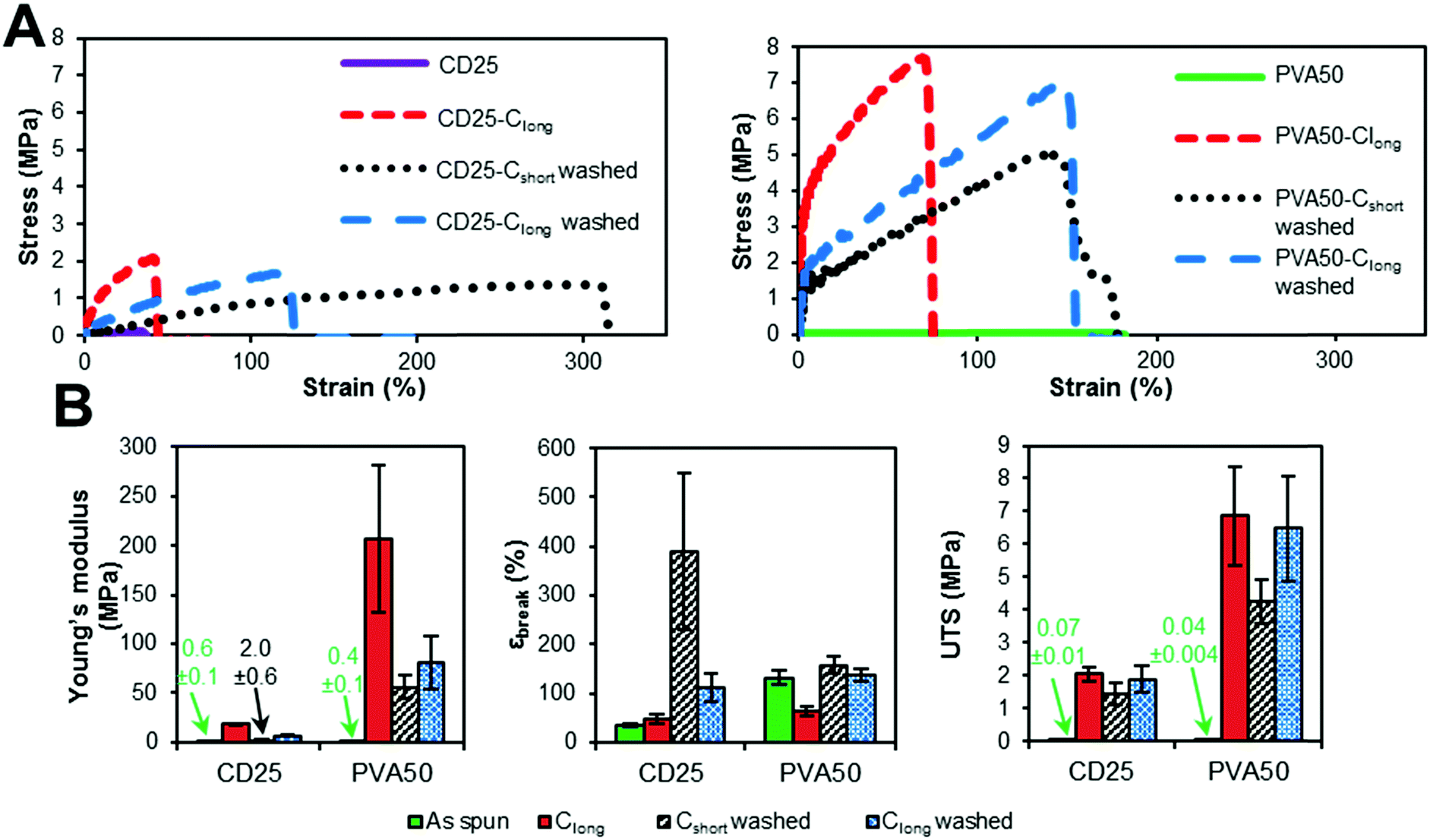
PGS synthesis was adapted from previous procedures.30,31 Briefly, 6.987 g (75.9 mmol, 2 equivalents) of glycerol, 23.013 g (113.9 mmol, 3 equivalents) of sebacic acid and a stirrer bar were introduced into a 100 mL round-bottomed flask. An open glass extender tube was connected to the flask to let the water vapor leave the reactor. The flask was placed in a microwave oven (Discover SP, CEM) and heated at 150 °C with a maximal power of 200 W. The reaction was stopped before complete crosslinking of PGS, otherwise neither the dissolution nor the subsequent process could be envisaged. After 400 minutes at 150 °C, a pale-yellow liquid is obtained. After cooling to room temperature, pPGS was pasty. The flask was placed under vacuum for 1 h at room temperature to evaporate excess of water without important loss of potential remaining glycerol. The sample was taken for 1H NMR (400 MHz, Bruker) analysis in d-DMSO. The degree of esterification was calculated from NMR analysis (Fig. S1, ESI†) according to the following equation:with Iacids and Iesters are the integration of the peaks on the spectra obtained by 1H NMR, respectively at 2.18 ppm for the carboxylic acids (–C
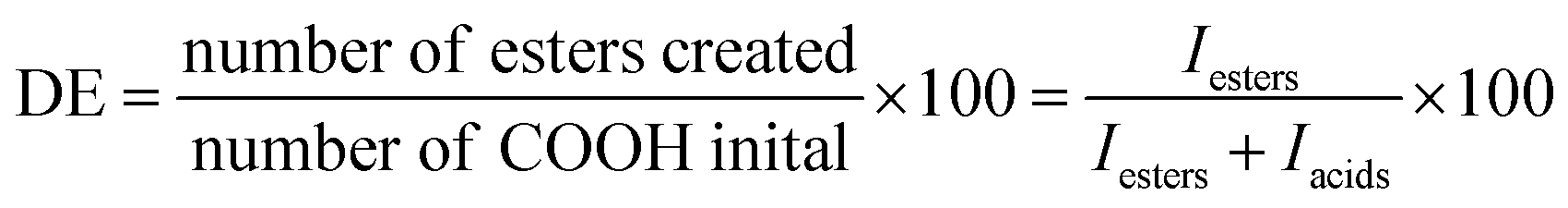
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) 2–COOH) and 2.28 ppm for the esters (–C2–COOR). The prepolymer used for the electrospinning process had a DE of 72%.
2–COOH) and 2.28 ppm for the esters (–C2–COOR). The prepolymer used for the electrospinning process had a DE of 72%.
Electrospinning of pPGS/HPβCD/PVP blends and optimization
pPGS, PVP and HPβCD are all soluble in DMF and ethanol. Electrospinning solutions were thus prepared 24 h prior to electrospinning, by dissolving the three components in DMF/ethanol 3/7 to reach 13 wt% of total solid in the solution.This total solid content was composed of 50% of pPGS, X% of HPβCD (X = 0, 5, 10, 15, 20 or 25) and (50–X)% of PVP. The compositions of the solutions are summarized in Table 1. Samples are named CDX with X being the percentage of HPβCD in the solid expressed in percentage of weight by weight of solid. A home-made vertical electrospinning set-up including a cylindrical rotating collector, covered by backing paper, and translating needles was used. Two metallic 18 gauge needles were fed with a solution at 2.4 mL h−1 by using a syringe pump (PHD 2000, Harvard Apparatus). The collector was placed 15 cm above the tip of the needles and rotated at 150 rpm. High voltage was applied to the collector (−5 kV) and to the needles (14 kV) by two power supplies (SL10, Spellman). Large membranes (15 × 30 cm2) suitable for post-treatments were electrospun for 5 h.
| Sample name | pPGS (%) | HPβCD (%) | PVP (%) |
|---|---|---|---|
| CD0 | 6.5 | 0 | 6.5 |
| CD5 | 6.5 | 0.65 | 5.85 |
| CD10 | 6.5 | 1.3 | 5.2 |
| CD15 | 6.5 | 1.95 | 4.55 |
| CD20 | 6.5 | 2.6 | 3.9 |
| CD25 | 6.5 | 3.25 | 3.25 |
Electrospinning of pPGS/PVA blends
Fibrous mats with 50 wt% of pPGS and 50 wt% of PVA were prepared by blend electrospinning using HFIP as the solvent. The solution, containing 3.2 wt% of pPGS and 3.2 wt% of PVA in HFIP, was prepared 24 h prior to electrospinning. The same electrospinning set-up was used as for pPGS/HPβCD/PVP samples. The distance between the needles and the collector was fixed at 18 cm and the applied voltage was 25 kV at the needles and −1 kV at the collector. The solution was infused at 2.0 mL h−1 in each syringe. Because of its low stability, the process was stopped after 1 h but it was enough to obtain detachable and easy to handle samples. All other parameters were kept the same as for pPGS/HPβCD/PVP. These samples containing 50% of PVA were named PVA50.Crosslinking of PGS and purification of the fibers
After peeling from the paper, the samples were cut into appropriate shapes and placed between two frames tightened together by clamps to limit membrane shrinkage during curing. They were put in a pre-heated vacuum oven (Vacutherm VT6025, Fisher Scientific) at 120 °C. The curing step was then conducted under a pressure below 10−2 mbar following different curing schemes: a short one with a maximal reached temperature of 140 °C named Cshort (24 h at 120 °C followed by 48 h at 140 °C) and a long one with a maximal temperature of 170 °C named Clong (24 h at 120 °C followed by 48 h at 140 °C and 24 h at 170 °C).After curing, the CD25 and PVA50 samples were immersed in gently stirred osmosed water. After 1 h, 2 h 30 minutes and 19 h of immersion, the water was changed and the membranes were rinsed. After 24 h of washing, they were dried overnight under vacuum at ambient temperature.
Characterization of PGS-based mats
The surface of the samples was observed by scanning electron microscopy (SEM) after coating with a thin layer of gold. The SEM (Vega 3, Tescan) was used in high vacuum mode with a voltage of 5 kV. The mean diameter of fibers was calculated over 100 diameter measurements using ImageJ software. The mean values are given with the standard deviation.The efficiency of the mat purification was first evaluated by measuring the mass loss after washing. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy analyses were also performed (Nicolet 380, Thermo Fisher) before and after washing to characterize the PVP and PVA removal. Results were compared with raw materials’ spectra.
The mechanical properties in the dry state of the as-spun, cured, and washed mats were determined by uniaxial tensile tests using the linear tension geometry of a rheometer (Discovery HR3, TA Instruments). Rectangular samples were cut with 5 mm width and about 20 mm long. The purified samples were also characterized in the wet state after two days of immersion in osmosed water. The width and thickness were then measured after hydration. The samples were fixed in the clamps with an initial gap of 10 mm and then pre-loaded with an axial force of about 0.01 N. The gap after preloading was set as the initial length. The tensile tests were conducted at 150 μm s−1 until breaking. 5 measurements were performed for each type of sample. The Young's modulus was calculated as the slope of the curve in the initial (between 0 and 1% of deformation) linear part.
Cyclic tests were conducted on cured and washed samples at dry and wet states to evaluate elastic recovery. Specimens were cut and their dimensions were measured in the same way as that for tensile tests. After pre-loading, they were stretched at 150 μm s−1 until a strain of 15%, 15% elongation being a typical value for cardiac muscles.32,33 After stretching, the samples were released at the same rate until 0% elongation to conclude one cycle, and a new cycle was started. The loading–release cycle was repeated 10 times.
Results and discussion
In the strategy presented here, fibrous scaffolds are prepared by blend electrospinning of pPGS with either PVP and HPβCD or PVA followed by a crosslinking and a washing step (Fig. 1A). A pPGS prepolymer was first synthetized, with a degree of esterification (DE) equal to 72% prepared with a molar ratio glycerol/sebacic acid fixed at 2/3, corresponding to the stoichiometric ratio (Fig. 1B). This ratio, favoring the reaction of the glycerol secondary alcohol during crosslinking, combined with a high DE, leads to a pPGS very close to its gel point. It results in a fast crosslinking of PGS, leaving no time for the pPGS to flow outside of the fibers during thermal treatment.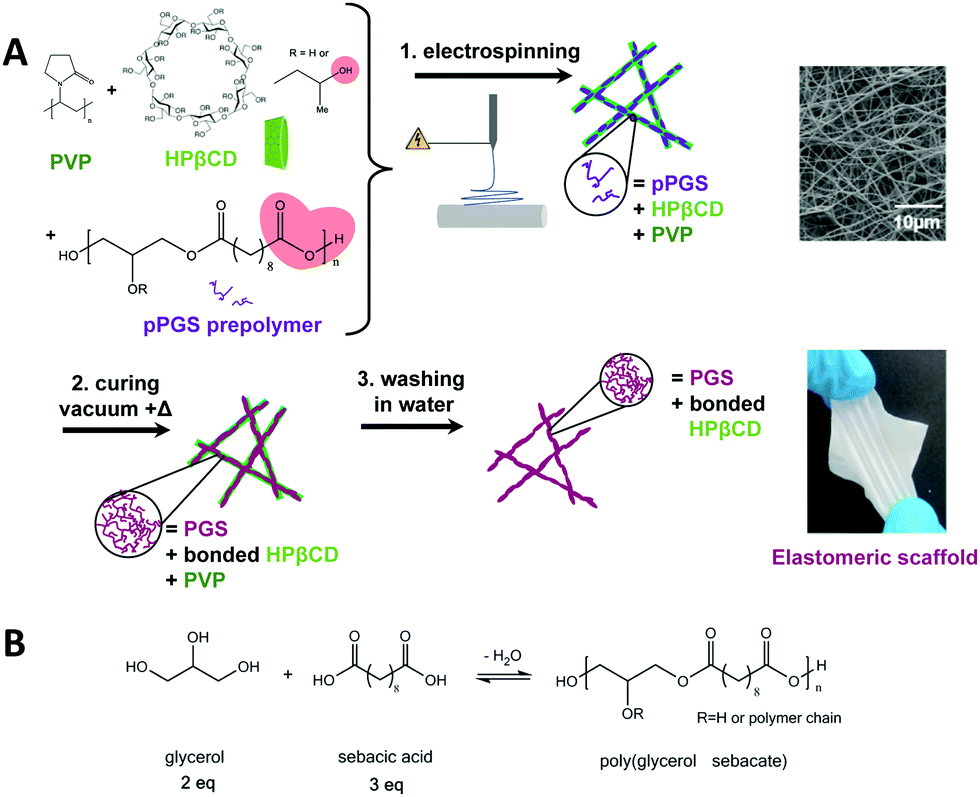 | ||
| Fig. 1 Strategy for the elaboration of the elastomeric PGS-based fibers. During thermal treatment, pPGS is crosslinked and reaction of –OH groups of HPβCD with –COOH groups of pPGS (in red) occurs (A). Chemical scheme of pPGS synthesis by polycondensation of glycerol and sebacic acid (B). | ||
pPGS was then electrospun with the carrier material. Preliminary experiments have shown that PVP alone is not a good carrier for PGS as it does not allow maintaining the fibrous structure of the scaffold during thermal treatment (Fig. S2 and S3, ESI†). HPβCD, with a high degradation temperature (278 °C), is thus required to maintain the fibrous structure during curing and to stabilize the electrospinning process.
Effect of cyclodextrin and PVP contents on the fiber morphology
In order to prepare elastomeric mats and investigate their properties depending on the crosslinking parameters, the material content in the PGS/PVP/HPβCD fibers was first optimized. Consistent with the literature,23 the amount of pPGS in the as-spun dry fibers was fixed at 50%. Indeed, a high enough percentage of pPGS is required to have continuous fibers after curing and washing, and a sufficient amount of carrier polymer is needed to retain the fiber shape during thermal treatment. The objective was thus to find a formulation able to reach an amount of 50% of pPGS with the help of PVP and HPβCD as carrier materials while preserving the fibrous morphology of the final scaffold.Electrospinning of formulations with less than 1.95% of HPβCD in the solution (CD15) was not stable due to periodic breaking of the jet (several times a minute) leading to solution projections on the collector. It did not allow the formation of continuous fibers, as shown for CD0 in Fig. 2. For CD5 and CD10, fibers could be seen on the collector but they tended to stick together and form an almost continuous film (Fig. S4, ESI†). PVP alone was not able to stabilize the jet and form solid enough fibers with such a high proportion of pPGS. A minimum of 15% of HPβCD was necessary to obtain membranes made of continuous fibers. Moreover, an increase of the HPβCD content combined with a decrease of PVP content resulted in a slight decrease of the fiber diameters, from 511 ± 136 nm for CD15, 382 ± 144 nm for CD20 to 318 ± 143 nm for CD25, the decrease of the carrier polymer quantity leading to a lower solution viscosity.
 | ||
| Fig. 2 Influence of the proportion of HPβCD on the PGS/HPβCD/PVP fiber morphology before and after curing for various formulations. CD0 contains 50% of PGS and 50% of PVP; CD15 contains 50% of PGS, 15% of HPβCD and 35% of PVP; CD20 contains 50% of PGS, 20% of HPβCD and 30% of PVP. The morphology is shown after the following curing steps: 24 h at 120 °C followed by 48 h at 140 °C (Cshort) or 24 h at 120 °C, 48 h at 140 °C and 24 h at 170 °C (Clong). | ||
These materials were cured to crosslink the PGS. Fig. 2 shows that 15% of HPβCD was not enough to avoid the melting of the fibers. Finally, with 20% of HPβCD and more, it was possible to obtain fibers with crosslinked PGS and only few melted areas, mostly at the contact points between fibers and around the few beads.
Fabrication of elastomeric mats
In this work, the mechanical properties of the mats prepared from pPGS/HPβCD/PVP and pPGS/PVA blends are compared with an identical pPGS weight amount of 50%. For the strategy based on the use of HPβCD and PVP, contents of 25 wt% of HPβCD and 25 wt% of PVP were taken. This condition, with a solid content of 13%, allows the fabrication of regular fibers as shown above. Moreover, it was assumed that a higher content in cyclodextrin would increase the modulus of the final materials as HPβCD can create multiple crosslinking points due to its large number of –OH groups. The selected formulation led to the fabrication of thin fibers (CD25 having a mean diameter of 318 ± 143 nm) comprising some beads (Fig. 3). In the case of 50/50 pPGS/PVA fibers, regular fibers were obtained from solutions containing a total concentration of solid of 6.4 wt%, while other values did not allow the formation of continuous fibers. It led to regular fibers being almost 5 times thicker than CD25 (PVA50 mean diameter: 1402 ± 503 nm, Fig. 3B). CD25 fibers are less regular, but in any case, the fiber morphology is more or less modified by the curing step. In addition, the preparation of PVA50 fibers requires the use of a costly and toxic solvent, HFIP, which is not the case for CD25. | ||
| Fig. 3 Morphology of the fibers as spun and after post-treatments. Low magnification SEM pictures (A), diameter distribution of diameters of as-spun fibers (B) and high magnification pictures of cured PVA50-washed fibers (C). CD25 contains 50% of PGS, 25% of HPβCD and 25% of PVP; PVA contains 50% of PGS and 50% of PVA. The morphology is shown here after Clong (24 h at 120 °C, 48 h at 140 °C and 24 h at 170 °C) and after 24 h washing in water. | ||
The mats were then crosslinked under vacuum following two different heating settings: Cshort (24 h at 120 °C followed by 48 h at 140 °C) or Clong (24 h at 120 °C followed by 48 h at 140 °C and 24 h at 170 °C). The influence of the curing parameters on the morphology and the mechanical properties of the final scaffolds was investigated. After curing, the crosslinked PGS/HPβCD/PVP fibers are fused together at the contact points and some melted spots are also visible. Fig. 3 shows the result after Clong, but the same observations can be noted after Cshort curing conditions (Fig. 2 and Fig. S5, ESI†). The integrity of the PVA/PGS fibers is better preserved after curing, even if fibers are also merged at contact points.
After the washing step in water, cured CD25 partially lost their fibrous morphology (Fig. 3A). In particular, numerous continuous areas are observed on cured CD25 crosslinked under Cshort conditions, leading to a high density of less porous areas. Under Clong crosslinking conditions, a higher porosity is observed. From these results, it can be concluded that if PGS is not enough crosslinked, some of the material can be dissolved or swollen out of the fibers and redeposit around them. In the case of washed crosslinked PVA50 fibers, the fibrous structure is well preserved thanks to the fact that the PVA50 fiber diameter is much larger than that of CD25 fibers (Fig. 3B). This result can also be justified by strong interactions between PGS and PVA as –OH groups of PVA can react with –COOH groups of PGS. Yet, cured PVA50 fibers are slightly attached together by melted materials too. Besides, it can be observed that PVA left a noticeable roughness or even small pores at the surface of the fibers after its removal (Fig. 3C).
In order to check the efficiency of the washing step, the membranes were weighed before and after washing and drying. In all cases, 65% to 80% of the initial mass remains after washing showing that pure PGS membranes are not obtained. However, the removal seems to be more efficient after the Cshort curing than after Clong: 68% of CD25-Cshort remains after washing versus 72% for CD25-Clong. The same tendency is observed in the case of PVA50: 75% of the initial weight remains after washing for PVA50-Cshort whereas a percentage of 78% is measured for PVA50-Clong. Thus, the Clong procedure leads to more “entrapment” of the carrier material in the fiber matrix. This “entrapment” can be the effect of covalent bonding, especially between PVA or HPβCD and PGS, or the effect of an entanglement of the carrier polymer (PVA for PVA50 or PVP for CD25) in the PGS network, both effects being increased for a higher reaction time or temperature and the resulting higher crosslinking density. It can be noted that the removal is more efficient for CD25 than for PVA50, demonstrating stronger interactions between PVA and PGS.
In order to obtain more insight into the efficiency of the fiber washing step, ATR-FTIR characterization was carried out. The spectra of cured CD25 and cured PVA50 before and after washing were compared to pPGS, PVP, HPβCD (Fig. 4A–C and Fig. S6, S7, ESI†) and PVA spectra (Fig. 4D–F and Fig. S8, S9, ESI†). Two areas of interest are considered. In the first one, corresponding to the elongation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (1650–1730 cm−1), the pPGS spectrum shows characteristic bands for acids at 1710 cm−1 and for esters at 1730 cm−1. In the fibers after curing, the shoulder at 1710 cm−1 is not visible anymore (Fig. 4B), confirming the esterification. The PGS characteristic bands partially overlap the PVP band arising from the amide CO bond (1650 cm−1) in the CD25 sample (Fig. 4B). In the same way, the CO band of residual acetate groups of PVA (1724 cm−1) is overlapped with the CO bands of PGS in PVA50 (Fig. 4E). The second interesting area includes the C–O of esters bonds (1160 cm−1), C–O in HPβCD (1027 cm−1), C–N in PVP (1287 cm−1) and C–C in PVA (845 cm−1). Because of the overlapping of the bands, it was difficult to quantify the presence of each component, but the evolution of intensities was investigated. In CD25-Clong, the intensity of the band at 1027 cm−1 clearly decreases relatively to PGS bands after washing (Fig. 4C). Indeed, the ratio of intensities of C–O in HPβCD over C–O in PGS decreases from 1.2 before washing to 0.7 after washing, showing that cyclodextrin was partially lost during the washing step. However, the PVP band at 1287 cm−1 does not clearly decrease relatively to PGS band: the ratio of intensities remains stable at 0.6. Surprisingly, it seems that HPβCD was easier to wash away, despite the fact that cyclodextrin can be covalently bonded with PGS. Indeed, all HPβCD molecules are not bonded to PGS, because there are 3 to 4 times more –OH groups (primary and secondary alcohols) from cyclodextrin than –COOH groups from PGS. Moreover, it can be assumed that the long PVP macromolecules are actually trapped in the PGS network. In PVA50, the band at 845 cm−1 decreased relatively to PGS band after washing (Fig. 4F): the ratio of intensities of C–C from PVA over C–O from PGS decreased from 0.7 to 0.4. This result highlights the partial removal of the PVA. The remaining PVA is bonded to the PGS or trapped in the network too.
O bonds (1650–1730 cm−1), the pPGS spectrum shows characteristic bands for acids at 1710 cm−1 and for esters at 1730 cm−1. In the fibers after curing, the shoulder at 1710 cm−1 is not visible anymore (Fig. 4B), confirming the esterification. The PGS characteristic bands partially overlap the PVP band arising from the amide CO bond (1650 cm−1) in the CD25 sample (Fig. 4B). In the same way, the CO band of residual acetate groups of PVA (1724 cm−1) is overlapped with the CO bands of PGS in PVA50 (Fig. 4E). The second interesting area includes the C–O of esters bonds (1160 cm−1), C–O in HPβCD (1027 cm−1), C–N in PVP (1287 cm−1) and C–C in PVA (845 cm−1). Because of the overlapping of the bands, it was difficult to quantify the presence of each component, but the evolution of intensities was investigated. In CD25-Clong, the intensity of the band at 1027 cm−1 clearly decreases relatively to PGS bands after washing (Fig. 4C). Indeed, the ratio of intensities of C–O in HPβCD over C–O in PGS decreases from 1.2 before washing to 0.7 after washing, showing that cyclodextrin was partially lost during the washing step. However, the PVP band at 1287 cm−1 does not clearly decrease relatively to PGS band: the ratio of intensities remains stable at 0.6. Surprisingly, it seems that HPβCD was easier to wash away, despite the fact that cyclodextrin can be covalently bonded with PGS. Indeed, all HPβCD molecules are not bonded to PGS, because there are 3 to 4 times more –OH groups (primary and secondary alcohols) from cyclodextrin than –COOH groups from PGS. Moreover, it can be assumed that the long PVP macromolecules are actually trapped in the PGS network. In PVA50, the band at 845 cm−1 decreased relatively to PGS band after washing (Fig. 4F): the ratio of intensities of C–C from PVA over C–O from PGS decreased from 0.7 to 0.4. This result highlights the partial removal of the PVA. The remaining PVA is bonded to the PGS or trapped in the network too.
 | ||
| Fig. 4 ATR-FTIR spectra of CD25 (A–C) and PVA50 (D and E) after curing and after purification compared to HPβCD, PVP, PVA and corresponding pPGS spectra. (A and D) Full spectra. (B and E) CO bands, absorbance normalized to the 1730 cm−1 band. (C and F) C–O, C–C and C–N bands, absorbance normalized to the 1160 cm−1 band. | ||
 | ||
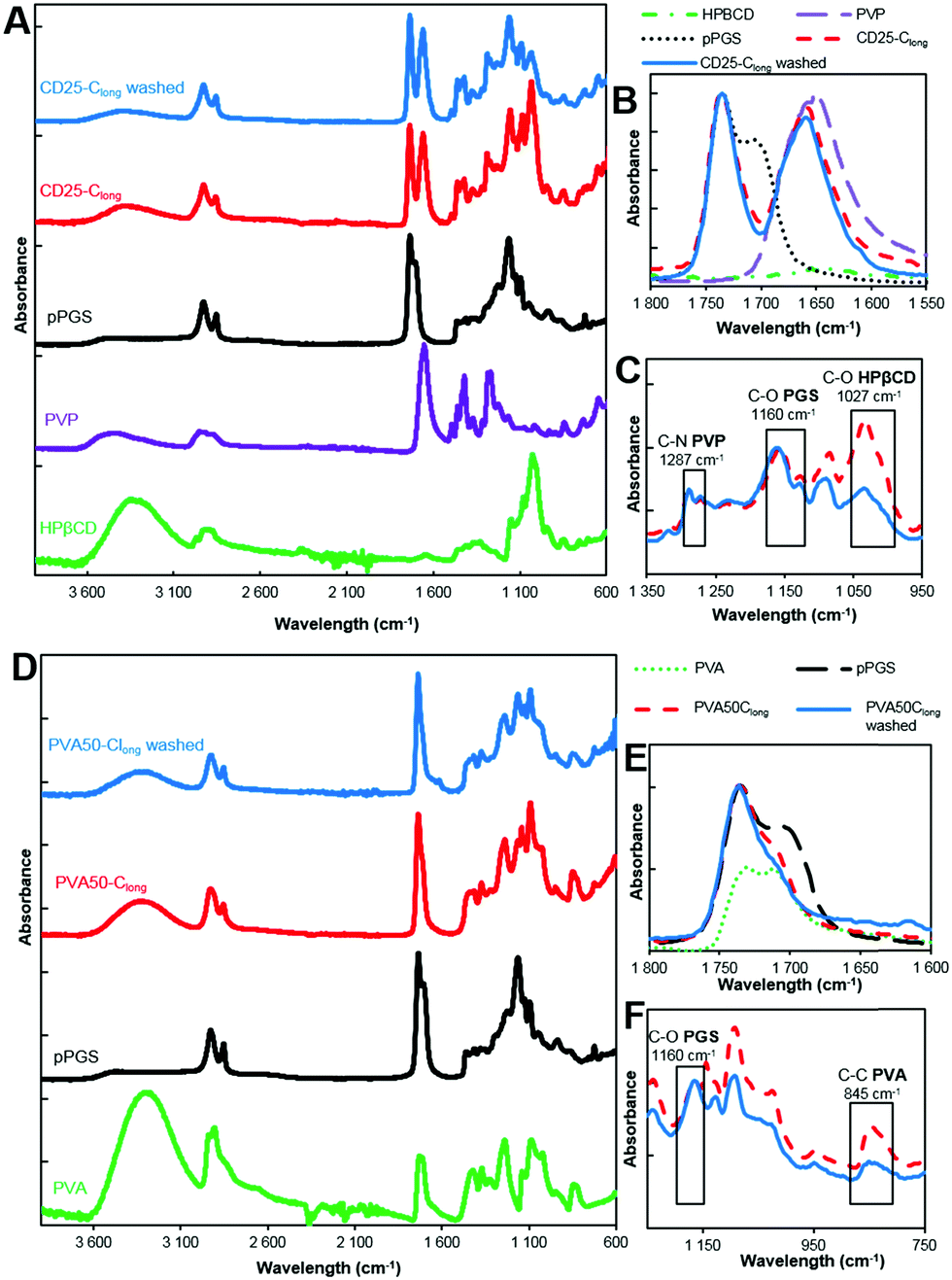
| Fig. 5 Influence of curing and washing on the mechanical properties of CD25 and PVA50. (A) Typical stress–strain curves. (B) Evolution of modulus, strain at break and UTS along curing and washing. | ||
The comparison of the results of the as-spun and cured samples clearly shows that the curing step increases the modulus and the stress at break. Two explanations can be given about this observation. First, the curing step leads to the crosslinked elastomer network which obviously enhances the overall strength. However, the maximal strain is not significantly improved by cross-linking because heating the fibers leads to their fusion at the contact points and therefore to strong bonding between them. The relative movement of fibers along each other is thus inhibited, which is a reason for the increase of the modulus and UTS.
The washing step decreases the Young's modulus and increases the strain at break. This result highlights the fact that the rigid carrier materials were at least partially removed, allowing the elastomer to have more impact on the overall mechanical properties of the scaffolds. After washing, it can be seen that the longest curing step leads to a higher modulus and UTS and lower strain at break. Indeed, Clong pushes further the reaction to a higher crosslinking density. Moreover, it has been shown by weighing that the rigid carrier materials are less efficiently washed away after Clong than after Cshort curing step, what could also explain the modulus increase.
In addition, it should be noted that cured and washed PVA50 and CD25 have different mechanical behaviors. PVA50 stress–strain curves show a clear first elastic domain with high modulus, before a region of irreversible deformations with lower modulus until breaking at higher strains. For cured and washed CD25, the first high modulus-domain is drastically reduced, and almost invisible. This difference can be explained by the structure and composition of the mats. PVA50 and CD25 can be compared to point-bonded PVC/TPU-based structures described by Lee et al.34 PVA50 still contains a large amount of rigid PVA but the density of bonding between its fibers is low; this case corresponds to fibers with a high PVC/TPU ratio. When the mat is stretched, these bonds hinder the slipping of the fibers, while the PVA phase endures the load until a critical stress is reached. Then, the elastomeric PGS phase governs the behavior. CD25 samples are more similar to PVC/TPU fibers with high TPU content with the density of bonding points being very high.
Moreover, it can be considered that the PGS content is higher because it was shown that the mass loss after washing is higher for CD25 than for PVA50. In addition to that, a fraction of the remaining carrier material of CD25 is cyclodextrin, which is included in the elastomeric network as crosslink points and influences the elastomeric behavior. So, the mat is a quasi-continuous elastomeric phase, and behaves almost as such.
The values given in Fig. 5B indicate that PVA50 has a higher modulus and UTS than CD25 regardless of the post-treatments. After curing and washing, PVA50 has a Young's modulus higher than 50 MPa for a strain at break close to 150% and an UTS close to 5 MPa. The washed CD25-Cshort and Clong have Young's moduli of 2 and 6 MPa respectively and UTS below 2 MPa. After washing, the average strain at break reached 390% for Cshort and 111% for Clong. In summary, the use of PVA as the carrier polymer leads to stiffer materials than in the case of the use of HPβCD and PVP.
From Fig. 5, it can be noted that most of the samples do not show a linear elastic behavior for strains higher than 3%. Cyclic mechanical tests were thus conducted to study how the materials recover after several cycles of deformation. Fig. 6 represents some typical stress–strain curves obtained with samples over 3 cycles of deformation varying from 0 to 15%. The maximal strain value of 15% was chosen to mimic the strain underwent by the heart tissue.32
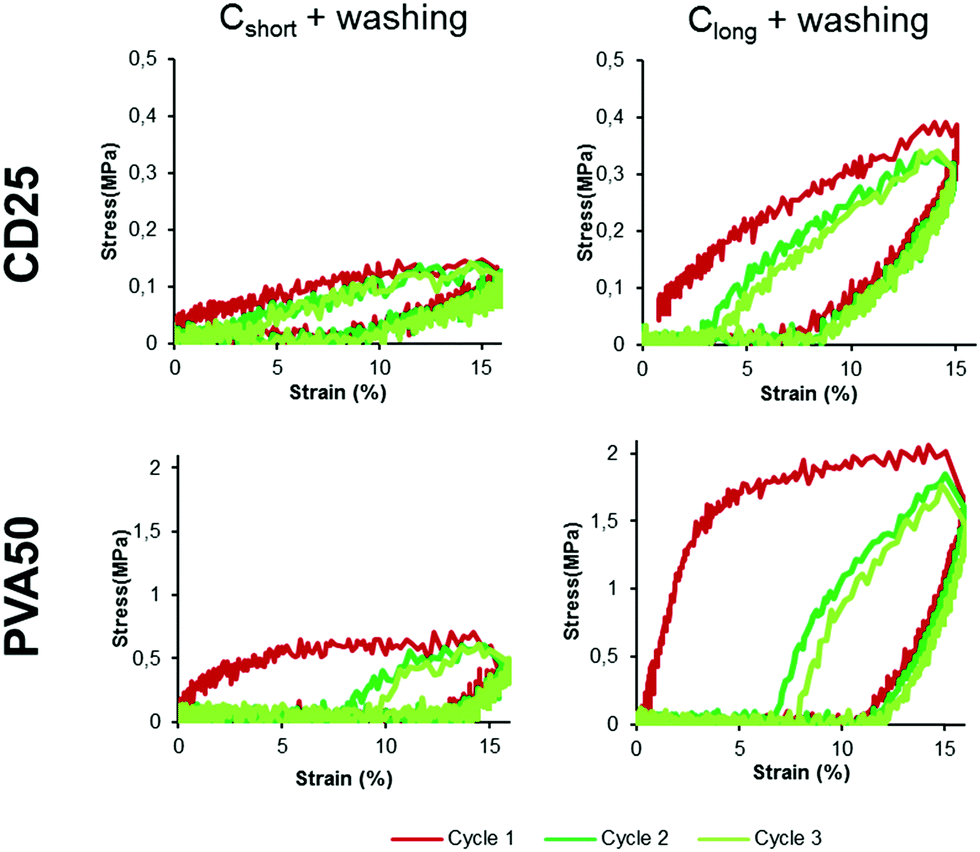 | ||
| Fig. 6 Stress–strain curves obtained for cured and washed CD25 and PVA50 between 0 and 15% of strain for the three first cycles. | ||
For all samples, during the first cycle, the stretching part of the curve is very different from the releasing part of the curve. But after the first deformation and release, all the following cycles overlap. This is visible in Fig. 6 for cycles 2 and 3 (10 cycles can be seen for all samples in Fig. S12 and S13, ESI†). This overlapping illustrates a lower loss of energy than after the first cycle, suggesting that the membranes underwent an irreversible deformation during this first stretching-release. This can be justified be the relative movement of the fibers along each other.
The energy recovery after stretching and release back to 0% of strain was calculated as the ratio of the area under the stress–strain curve during release over the area under the curve during stretching. The calculated values confirm the poor recovery during the first cycle (Fig. 7 in grey). PVA50 recovery is close to 10% after Cshort and Clong whereas the recovery of CD25 is slightly higher with values above 20%. An average value was calculated for cycle 2 to 10 as they are similar. The recovery reaches then nearly 40% for CD25 and 50% for PVA50 for samples after Cshort. After Clong, the recovery is worse for PVA50 with 33% but stable for CD25.
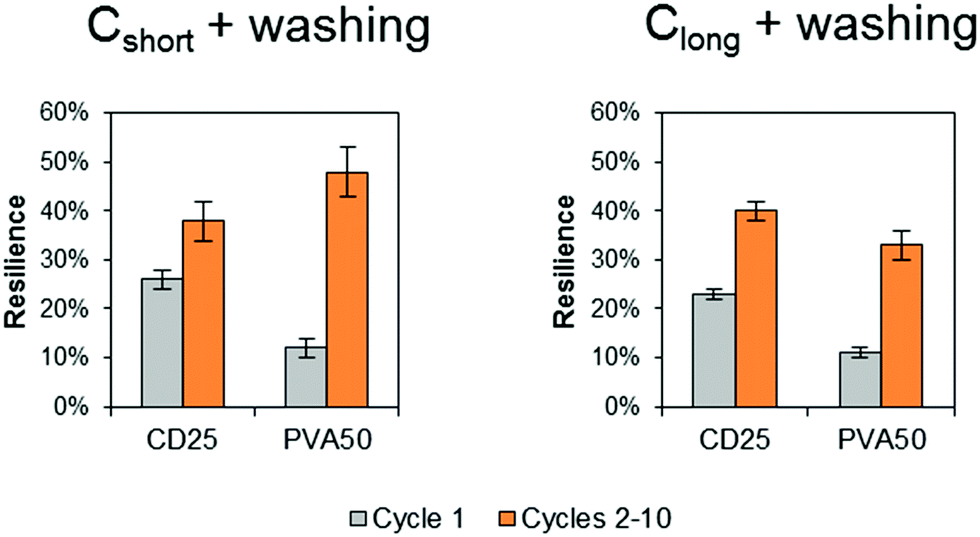 | ||
| Fig. 7 Energy recovery after the first cycle and mean energy recovery during cycles 2 to 10. The given value is the average value for 3 samples with the standard deviation. | ||
Finally, both materials show similar behavior under repeated cycles except during the first extension of the material: PVA50 underwent more important irreversible structural modifications. The morphology of the fibrous samples can help to justify this result. Indeed, as PVA50's fibers are better defined and include a lower density of fixed contact points, they can move with higher amplitudes, these displacements being permanent. In contrast, the washed CD25-Cshort sample comprises a high density of fixed contact points and shows thus less irreversible deformations.
| Fiber composition (%PGS) | Electrospinning solvent | Carrier polymer removal | Elastic modulus (MPa) | Strain at break (%) | UTS (MPa) | State | Ref. |
|---|---|---|---|---|---|---|---|
| pPGS/Zein (20%) | Acetic acid | No | 6–14 | 5–7 | 0.3–13 | Dry | 12 |
| PGS/PLA (30%) | DMF:DCM | No | 55–278 | 50–120 | 0.2–7 | Dry | 18 |
| PGS/PLLA (50%) | DMF:DCM | No | 7–336 | 13–66 | 1–7 | Dry | 19 |
| pPGS/gelatin (77%) acrylate (UV) | HFIP | No | 10 | 20–40 | 0.65 | Dry | 14 |
| pPGS/gelatin (30–60%) EDC-NHS | Acetic acid | No | 9–14 | 31–34 | 1.5–1.6 | Dry | 13 |
| pPGS/collagen (32%) glutaraldehyde | HFIP | No | 4.2 | 84 | 2 | Dry | 15 |
| PGS/PVA (>90%) | HFIP | Yes (water) | 50 | 150 | 4–6.5 | Dry | This article |
| PGS/HPβCD/PVP (>90%) | DMF:ethanol | Yes (water) | 2–6 | 110–390 | 0.6–2 | Dry | This article |
| PGS/gelatin (30–60%) EDC-NHS | Acetic acid | No | 0.15–0.30 | 90–180 | 0.3 | Wet | 13 |
| PGS/gelatin (77%) acrylate (UV) | HFIP | No | 0.1–1 | 30–45 | 0.03 | Wet | 14 |
| PGS/PVA (>90%) | HFIP | Yes (water/ethanol) | 0.1–0.8 | 200–750 | 0.8–1.2 | Wet | 23 |
| PGS/PVA (>90%) | HFIP | Yes (water) | 0.15 | 70 | 0.1 | Wet | This article |
| PGS/HPβCD/PVP (>90%) | DMF:ethanol | Yes (water) | 0.12–0.17 | 50–70 | 0.08–0.09 | Wet | This article |
Mechanical properties were also measured on hydrated scaffolds (Fig. 8). The results show a clear decrease in modulus, UTS and strain at break for all samples after hydration. Indeed, the material swells in water and behaves as a hydrogel. It results in a wider elastic domain, the linear part reaching several tens of %. For all samples, the moduli of hydrated scaffolds are close to 100–200 kPa (Fig. 8B), which is more suitable for soft tissue than the same dry samples. The maximal strains are lower than those in the dry state but still high enough for cardiac regeneration as the typical strain underwent by the heart tissue is 15%. Cyclic tensile tests show the reversibility of the deformation in the elastic domain below 15% (Fig. 8C).
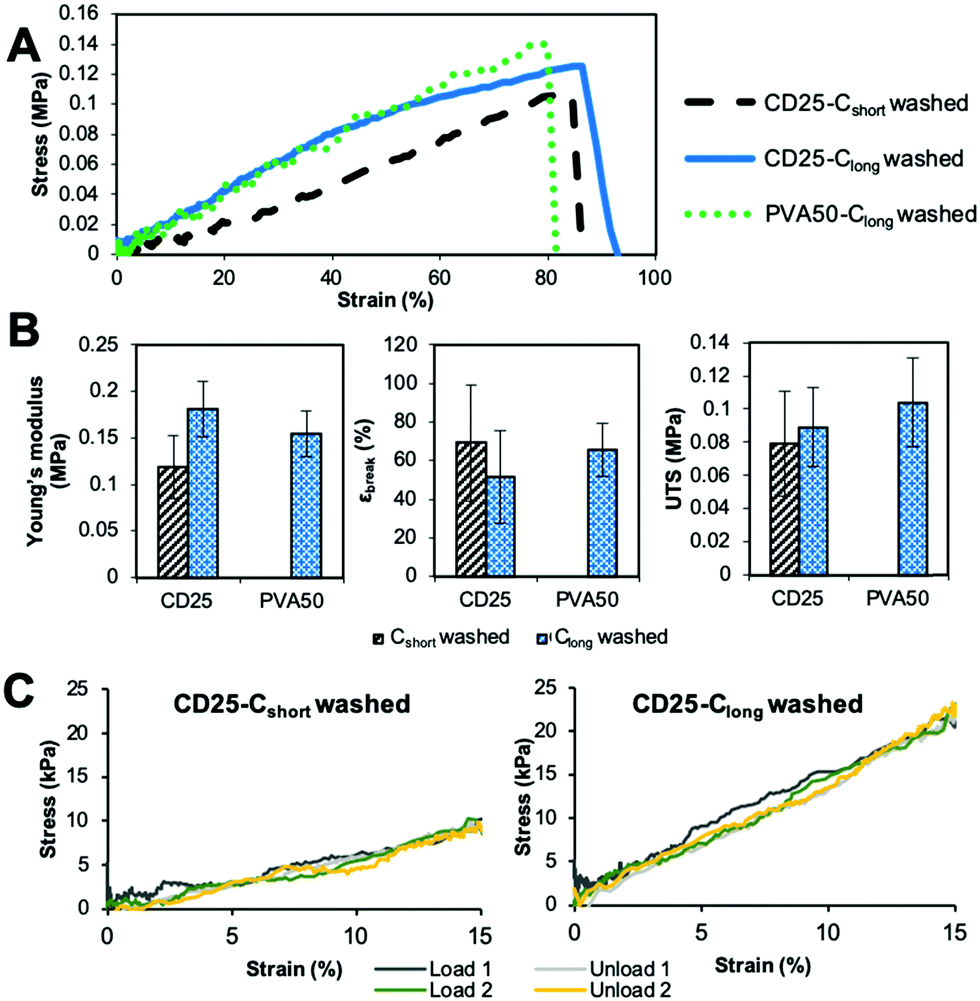 | ||
| Fig. 8 Mechanical behavior of the scaffolds in the wet state. (A) Typical stress–strain curves, (B) evolution of modulus, strain at break and UTS with curing and washing. The given value is the average value for 3 samples with the standard deviation. (C) Stress–strain curves obtained for cured and washed CD25 samples between 0 and 15% strain. | ||
Moreover, the mechanical properties in dry and wet states of our scaffolds were compared with those reported in the literature (Table 2). They fall in the same range as the other developed PGS containing fibers. What is important to note is that our PGS/HPβCD/PVP system and the PGS/PVA one, first developed by Jeffries et al.,23 are the only systems for which the pPGS is cured after electrospinning and the carrier polymer is washed, leaving almost pure PGS fibers, and without chemical crosslinking with toxic agents such as acrylate, EDC-NHS or glutaraldehyde.
Conclusions
In this work, PGS-based membranes were successfully prepared using a blend of PVP and HPβCD as carrier molecules. Composite fibers with 50% of pPGS, 25% of PVP and 25% of HPβCD were electrospun, cured and washed. For comparison, fibers with 50% of pPGS and 50% of PVA were also electrospun and post-treated in the same way. The fibers obtained from pPGS/PVP/HPβCD are almost 5 times thinner than those obtained from pPGS/PVA. In both cases, the washing step did not lead to pure PGS fibers. Washing was however slightly more efficient for materials prepared with PVP and HPβCD. Mechanical characterization in the dry state showed that both types of membranes can be stretched at strains larger than 100% after washing but the moduli of pPGS/PVP/HPβCD scaffolds are lower than those prepared with PVA. In addition, cyclic mechanical tests showed that samples prepared with PVP/HPβCD have a good resilience, making them good candidates for soft tissue engineering. In the wet state, the moduli and tensile strength decrease, reaching values suitable for soft tissue engineering and the elastic domains increase allowing real elastic deformation of the scaffolds. Finally, the presence of cyclodextrin in the material should offer the possibility to encapsulate and deliver molecules of therapeutic interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the University of Strasbourg and CNRS. This work was supported by the Agence Nationale de la Recherche (MimHeart project ANR-15-CE08-0010-02) CFB has received funding from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, process no. 2014/17939-0 and process no. 2016/04418-8) and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. The authors also thank Christophe Mélart for technical help.References
- Q.-Z. Chen, S. E. Harding, N. N. Ali, A. R. Lyon and A. R. Boccaccini, Materials Science and Engineering: R: Reports, 2008, 59, 1–37 CrossRef.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS.
- Q.-Z. Chen, A. Bismarck, U. Hansen, S. Junaid, M. Q. Tran, S. E. Harding, N. N. Ali and A. R. Boccaccini, Biomaterials, 2008, 29, 47–57 CrossRef CAS.
- Q.-Z. Chen, H. Ishii, G. A. Thouas, A. R. Lyon, J. S. Wright, J. J. Blaker, W. Chrzanowski, A. R. Boccaccini, N. N. Ali, J. C. Knowles and S. E. Harding, Biomaterials, 2010, 31, 3885–3893 CrossRef CAS.
- G. C. Engelmayr Jr, M. Cheng, C. J. Bettinger, J. T. Borenstein, R. Langer and L. E. Freed, Nat. Mater., 2008, 7, 1003–1010 CrossRef.
- S. Sell, C. Barnes, M. Smith, M. McClure, P. Madurantakam, J. Grant, M. McManus and G. Bowlin, Polym. Int., 2007, 56, 1349–1360 CrossRef CAS.
- U. Boudriot, R. Dersch, A. Greiner and J. H. Wendorff, Artif. Organs, 2006, 30, 785–792 CrossRef CAS.
- J. Lannutti, D. Reneker, T. Ma, D. Tomasko and D. Farson, Mater. Sci. Eng., C, 2007, 27, 504–509 CrossRef CAS.
- S. Agarwal, J. H. Wendorff and A. Greiner, Adv. Mater., 2009, 21, 3343–3351 CrossRef CAS.
- F. Yi and D. A. LaVan, Macromol. Biosci., 2008, 8, 803–806 CrossRef CAS.
- D. Dippold, M. Tallawi, S. Tansaz, J. A. Roether and A. R. Boccaccini, Eur. Polym. J., 2016, 75, 504–513 CrossRef CAS.
- M. Kharaziha, M. Nikkhah, S.-R. Shin, N. Annabi, N. Masoumi, A. K. Gaharwar, G. Camci-Unal and A. Khademhosseini, Biomaterials, 2013, 34, 6355–6366 CrossRef CAS.
- J. L. Ifkovits, J. J. Devlin, G. Eng, T. P. Martens, G. Vunjak-Novakovic and J. A. Burdick, ACS Appl. Mater. Interfaces, 2009, 1, 1878–1886 CrossRef CAS.
- R. Ravichandran, J. R. Venugopal, S. Sundarrajan, S. Mukherjee and S. Ramakrishna, World J. Cardiol., 2013, 5, 28–41 CrossRef.
- R. Rai, M. Tallawi, C. Frati, A. Falco, A. Gervasi, F. Quaini, J. A. Roether, T. Hochburger, D. W. Schubert, L. Seik, N. Barbani, L. Lazzeri, E. Rosellini and A. R. Boccaccini, Adv. Healthcare Mater., 2015, 4, 2012–2025 CrossRef CAS.
- M. Tallawi, D. Dippold, R. Rai, D. D’Atri, J. A. Roether, D. W. Schubert, E. Rosellini, F. B. Engel and A. R. Boccaccini, Mater. Sci. Eng., C, 2016, 69, 569–576 CrossRef CAS.
- F. Flaig, H. Ragot, A. Simon, G. Revet, M. Kitsara, L. Kitasato, A. Hébraud, O. Agbulut and G. Schlatter, ACS Biomater. Sci. Eng., 2020, 6, 2388–2400 CrossRef CAS.
- Y. Yan, V. Sencadas, T. Jin, X. Huang, J. Chen, D. Wei and Z. Jiang, J. Colloid Interface Sci., 2017, 508, 87–94 CrossRef CAS.
- Z.-R. You, M.-H. Hu, H.-Y. Tuan-Mu and J.-J. Hu, J. Mech. Behav. Biomed. Mater., 2016, 63, 220–231 CrossRef CAS.
- H.-J. Wu, M.-H. Hu, H.-Y. Tuan-Mu and J.-J. Hu, Mater. Sci. Eng., C, 2019, 100, 30–37 CrossRef CAS.
- B. Xu, Y. Li, C. Zhu, W. D. Cook, J. Forsythe and Q. Chen, Eur. Polym. J., 2015, 64, 79–92 CrossRef CAS.
- E. M. Jeffries, R. A. Allen, J. Gao, M. Pesce and Y. Wang, Acta Biomater., 2015, 18, 30–39 CrossRef CAS.
- A. Keirouz, G. Fortunato, M. Zhang, A. Callanan and N. Radacsi, Med. Eng. Phys., 2019, 71, 56–67 CrossRef.
- Y. Li, W. D. Cook, C. Moorhoff, W.-C. Huang and Q.-Z. Chen, Polym. Int., 2012, 62, 534–547 CrossRef.
- T. Uyar, A. Balan, L. Toppare and F. Besenbacher, Polymer, 2009, 50, 475–480 CrossRef CAS.
- F. Kayaci and T. Uyar, Carbohydr. Polym., 2012, 90, 558–568 CrossRef CAS.
- M. Séon-Lutz, A.-C. Couffin, S. Vignoud, G. Schlatter and A. Hébraud, Carbohydr. Polym., 2019, 207, 276–287 CrossRef.
- F. J. Otero-Espinar, J. J. Torres-Labandeira, C. Alvarez-Lorenzo and J. Blanco-Méndez, J. Drug Delivery Sci. Technol., 2010, 20, 289–301 CrossRef CAS.
- X. Li, A. T.-L. Hong, N. Naskar and H.-J. Chung, Biomacromolecules, 2015, 16, 1525–1533 CrossRef CAS.
- G. Coativy, M. Misra and A. K. Mohanty, ACS Sustainable Chem. Eng., 2016, 4, 2142–2149 CrossRef CAS.
- C. Zhu, A. E. Rodda, V. X. Truong, Y. Shi, K. Zhou, J. M. Haynes, B. Wang, W. D. Cook and J. S. Forsythe, ACS Biomater. Sci. Eng., 2018, 4, 2494–2504 CrossRef CAS.
- A. I. Veress, G. T. Gullberg and J. A. Weiss, J. Biomech. Eng., 2005, 127, 1195–1207 CrossRef.
- K. H. Lee, H. Y. Kim, Y. J. Ryu, K. W. Kim and S. W. Choi, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1256–1262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00673d |
| This journal is © The Royal Society of Chemistry 2021 |