DOI:
10.1039/D0NH00077A
(Communication)
Nanoscale Horiz., 2020,
5, 1213-1225
A brain tumor-homing tetra-peptide delivers a nano-therapeutic for more effective treatment of a mouse model of glioblastoma†
Received
4th February 2020
, Accepted 2nd June 2020
First published on 2nd June 2020
Abstract
Organ-specific cell-penetrating peptides (CPPs) are a class of molecules that can be highly effective at delivering therapeutic cargoes, and they are currently of great interest in cancer treatment strategies. Herein, we describe a new CPP (amino acid sequence serine-isoleucine-tyrosine-valine, or SIWV) that homes to glioblastoma multiforme (GBM) brain tumor tissues with remarkable specificity in vitro and in vivo. The SIWV sequence was identified from an isoform of annexin-A3 (AA3H), a membrane-interacting human protein. The mechanism of intracellular permeation is proposed to follow a caveolin-mediated endocytotic pathway, based on in vitro and in vivo receptor inhibition and genetic knockdown studies. Feasibility as a targeting agent for therapeutics is demonstrated in a GBM xenograft mouse model, where porous silicon nanoparticles (pSiNPs) containing the clinically relevant anticancer drug SN-38 are grafted with SIWV via a poly-(ethylene glycol) (PEG) linker. The formulation shows enhanced in vivo targeting ability relative to a formulation employing a scrambled control peptide, and significant (P < 0.05) therapeutic efficacy relative to free SN-38 in the GBM xenograft animal model.
New concepts
Glioblastoma multiforme (GBM) is the most aggressive brain cancer with a 5 year survival rate less than 5%. Traditionally, chemotherapy has been used as a treatment method, but the low organ specificity and penetration yield through blood–brain barrier of drugs induce many side-effects. To date, many drug delivery systems with cell-penetrating peptide have been researched. However, previous CPPs are still limited because of low organ specificity, non-specific cellular uptake, and rapid clearance into the kidney, spleen, and liver due to their intrinsic cationic nature. To solve these problems, we report a new cell-penetrating tetra-peptide, which shows a significant homing ability to brain cancer cells and tissues in vitro and in vivo, and which have successfully demonstrated delivery of a nano-therapeutic. The peptide, with the sequence SIWV, showed a mechanism of caveolin-mediated endocytosis with negligible cytotoxicity and immunogenicity. Moreover, porous silicon nanoparticles with SIWV showed that the GBM selective homing capability could yield significant improvements in both selectivity and efficacy for the treatment in a GBM mouse model. This work demonstrates that the hybridization of biocompatible GBM-homing tetra-peptide and biodegradable nano-carrier provides a more feasible pathway to clinical translation.
|
1. Introduction
Cell-penetrating peptides (CPPs) are short peptides that facilitate cellular intake of various substances; small/large organic molecules, inorganic materials, and nanosized particles.1–3 Typically identified from viruses, artificial synthetic sequences, or phage display selections, CPPs are widely used as a tool in basic research and they are of translational interest as components of a number of drug delivery systems (DDS) currently under development.4–6 At present, CPPs are generally limited by: (i) low organ specificity; (ii) non-specific cellular uptake; and (iii) rapid clearance into the kidney, spleen, and liver due to their intrinsic cationic nature (e.g., TAT, Antp, R8).3,7–9 In order to overcome these limitations, researchers have focused on hydrophobic and non-ionic CPPs that show high affinity for membrane receptors that are over-expressed on the specific cells of interest. For example, the anti-HER2/neu peptide (AHNP) can target HER2/neu positive breast cancer cells,10,11 and the DV3 can target CXC chemokine receptor 4 (CXCR4) that is overexpressed in various cancer cells.12,13 Cell specificity can be further increased by linking non-CPP targeting peptide sequences (e.g., MMPs) that have their own cellular receptor specificity.
In general, CPPs enter cells through the permeation pathway using specific molecules such as heparan sulfate, clathrin, caveolin, and phospholipids, which are expressed on the cell membrane.14–16 The efficiency of CPPs depends on these factors, and the effect is more dramatic for the hydrophobic CPPs. In this regard, the design strategy of new CPPs should be established depending on the target cells and tissues.
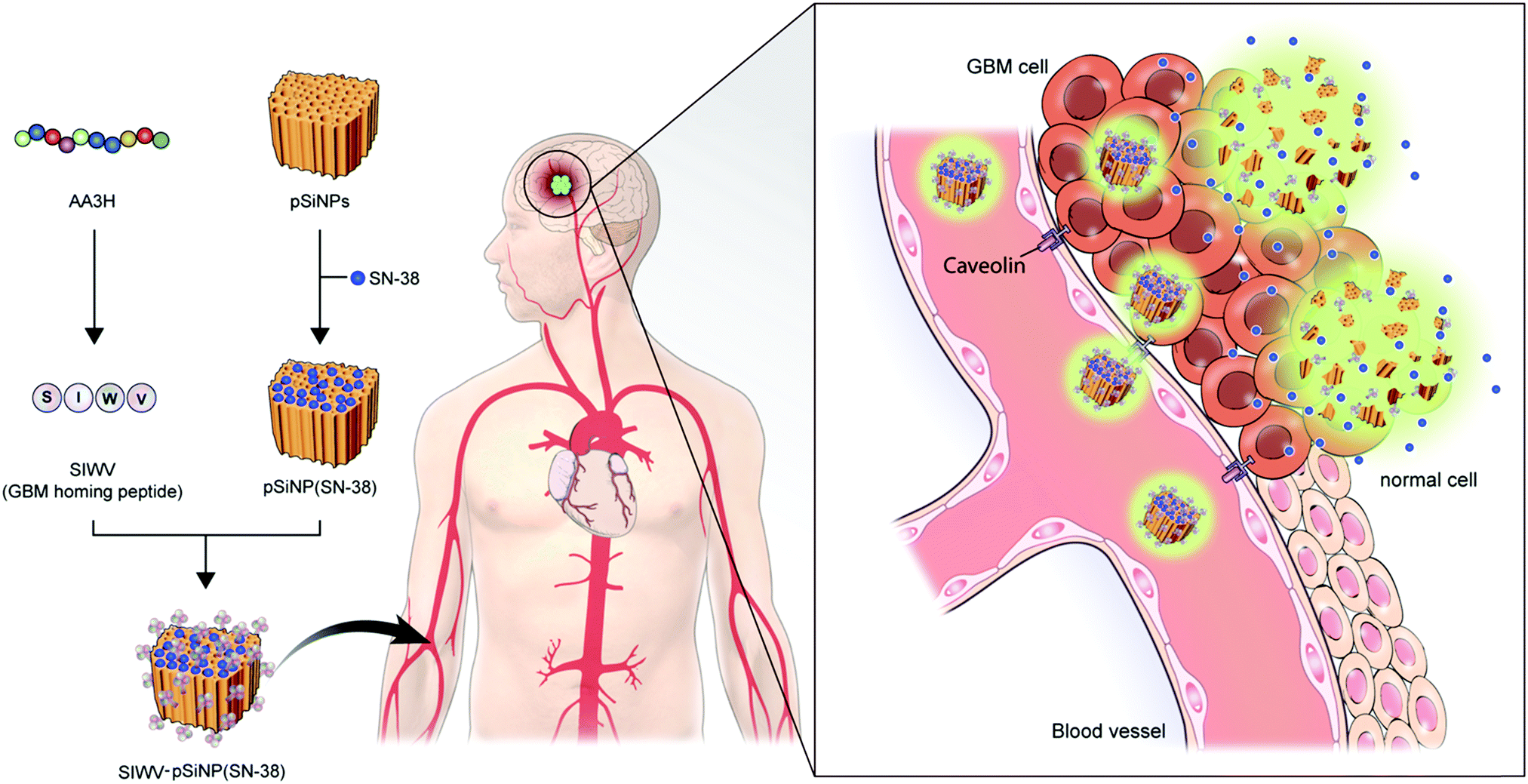
In this report, we report a new brain tumor-homing caveolin-mediated endocytotic CPP and its therapeutic applications with hybridization of porous silicon nanoparticles (pSiNPs) for brain tumor, in particular, glioblastoma multiforme (GBM) (Fig. 1). We systematically analyzed the essential elements of non-amphipathic CPP; AA3H, a 10-amino acid peptide (MASIWVGHRG) derived from the human annexin-A3 functions, with Ala-substitution mutagenesis of the amino acid residues.17 The discovered key part, name SIWV, consists of 4-amino acid of CPP sequence (SIWV), and it showed superior cell penetration efficacy with caveolae-dependent mechanism. This property has demonstrated for the specific delivery of therapeutic nano-carrier, pSiNPs, to the caveolae abundant cells and organs such as the brain, specifically GBM in this study. GBM is the most aggressive cancer that develops within the brain, and average survival length following diagnosis is 12–15 months, with a five-year survival rate of less than 5%.18,19 Palliative therapy, such as chemotherapy has proven to be the most successful, but the low organ specificity and penetration yield through blood–brain barrier of drugs induce many side-effects.20–22 In this vein, nanoparticle-based drug delivery systems (DDS) have been highlighted in order to address these issues. Among the known nano-materials, pSiNPs have been widely applied in preclinical research as a promising candidate for clinical translation because of its low toxicity, intrinsic photoluminescence property, high loading efficacy, and accessibility as a controlled-release system.23–34 This study demonstrated a successful targeting of GBM in vivo using SIWV-pSiNPs hybrid material, which represents abilities of delivering multiple agents for both therapeutic and diagnostic materials.
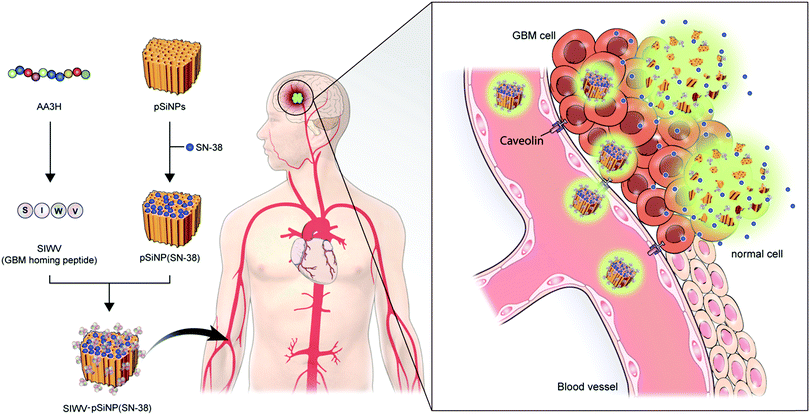 |
| | Fig. 1 Schematic of the approach and motivation for this work. The four-peptide sequence SIWV was identified from the larger AA3H cell-penetrating peptide as the essential construct needed to bind to and penetrate human glioblastoma multiforme (GBM) cells. The peptide appears to target and penetrate these cells via a caveolin-mediated endocytosis pathway. When attached to a porous nanoparticle delivery vehicle carrying the anti-cancer agent SN-38, the peptide effectively homes the nanoparticles to a mouse model of human GBM. | |
2. Results and discussion
Discovery of tetra-CPPs
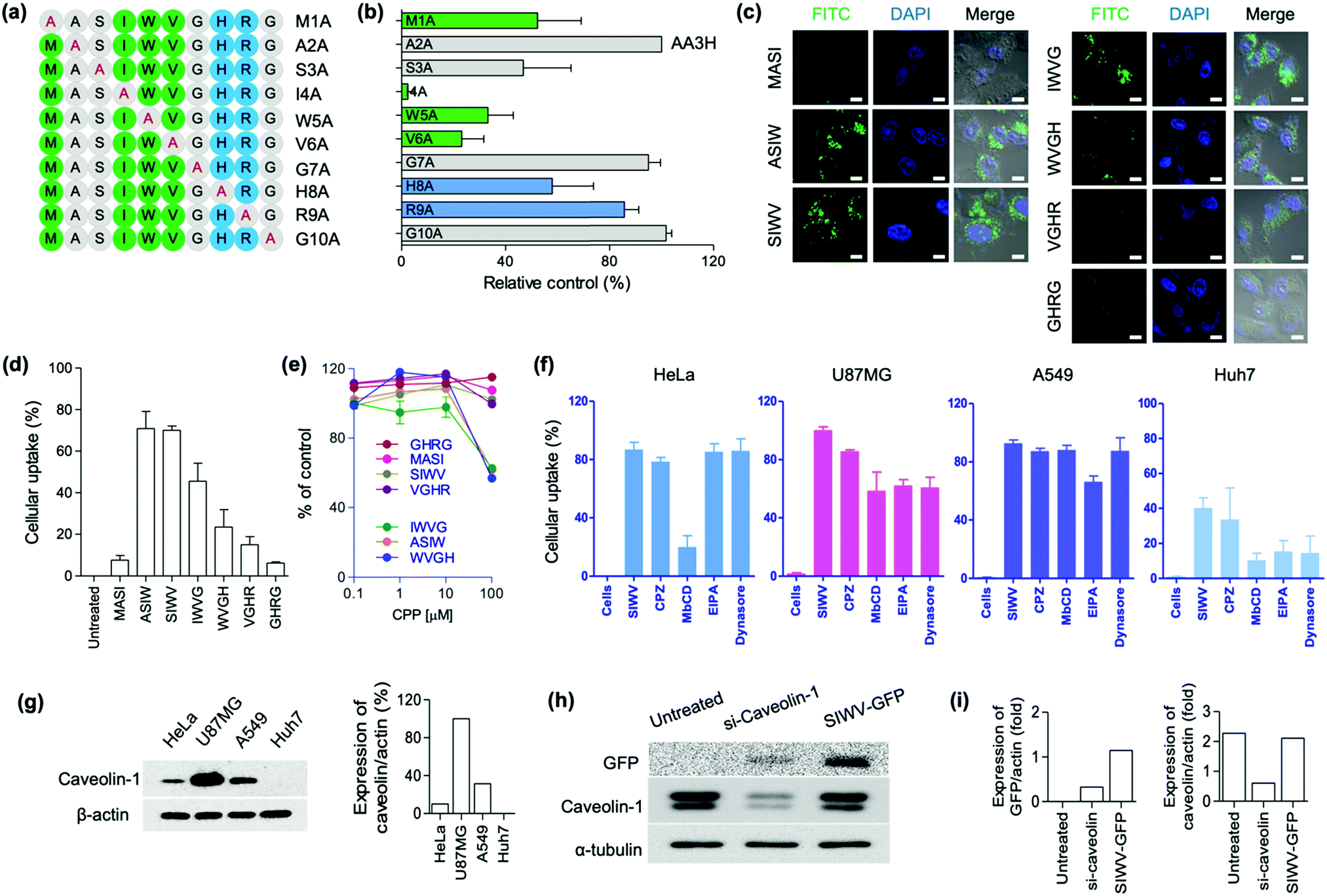
Recently, we focused on the development of new CPPs for biomedical applications. Up to date, the shortest lengths of CPPs are the five,35 and the development of short and effective CPPs have been demanded in terms of large-scale preparation and clinical translation. We previously reported AA3H-CPP as an endogenous membrane binding protein.17 During this research process, we discovered a new CPP, capable of brain tumor-homing via caveolin-mediated endocytotic cell-penetration. Multiple derivatives of the AA3H-CPP peptide were synthesized by solid-phase synthesis, and then the essential elements were systematically examined using alanine (A) scanning. The AA3H-CPP has 10 amino acids (MASIWVGHRG), and each non-alanine residue was substituted with alanine, generating M1A, A2A, S3A, I4A, W5A, V6A, G7A, H8A, R9A, and G10A (Fig. 2a); M1A indicates A substitution at first M point, A2A indicates A substitution at second A point, etc. To determine the uptake efficiency of each peptide derivatives, N-terminus fluorescein isothiocyanate (FITC)-labelled peptide was prepared and then treated to the cells (HeLa cells, 1 μM peptide, 4 h incubation). The uptake of variants into the cells was measured using flow cytometry, and the results represented that Ser (S), Ile (I), Trp (W), and Val (V) were important residues for cell penetration (Fig. 2b, relatively low control of S3A, I4A, W5A, and V6A). This result represents that the overall hydrophobic nature (I, W, V) of the AA3H is essential for the cell-penetration property, in contrast to the cationic properties of other well-known protein transduction domains (PTDs), such as TAT and R9. Like Ile, Val and Trp are hydrophobic amino acids that could be involved in the binding/recognition of hydrophobic ligands, such as lipids. Based on this result, we produced all possible truncated versions of the peptide each containing only four amino acids, sequentially from the M to G of AA3H-CPP. Again, we prepared N-terminus FITC-labelled peptides and then treated to the cells (HeLa cells, 1 μM peptide, 4 h incubation). In the confocal laser scanning microscopy (CLSM) imaging (Fig. 2c) and flow cytometry (Fig. 2d), ASIW, SIWV, IWVG, and WVGH showed high penetration efficiency. In the cytotoxicity analysis of peptides, we found that the ASIW, IWVG, and WVGH were toxic at 100 μM, whereas SIWV was nontoxic, even at high concentration levels (Fig. 2e). In order to measure aggregation factor of SIWV in serum, we incubated 100 μM of SIWV in human serum. In the dynamic light scattering (DLS) analysis, there was no significant aggregates in human serum up to 60 min at 37 °C.
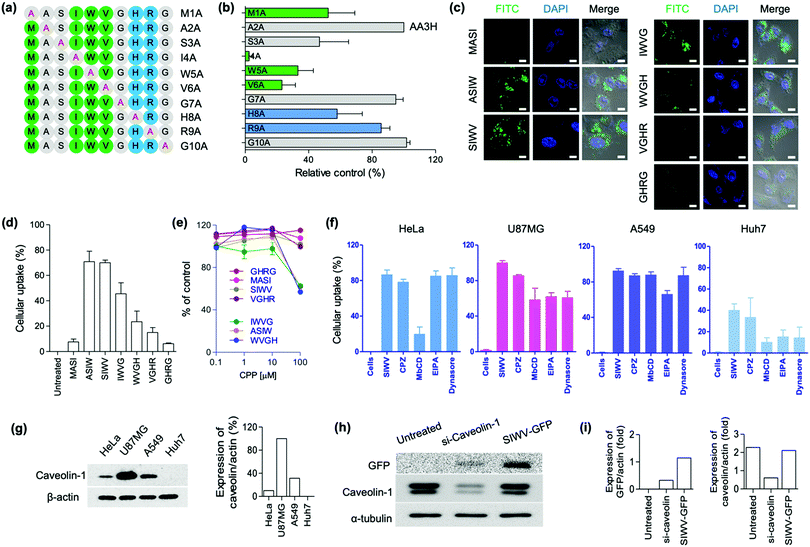 |
| | Fig. 2 Screening results aimed at identifying the essential peptide fragment in the known cell-penetrating peptide AA3H that is responsible for its ability to target and penetrate cells. (a) Alanine substitution scheme used to generate the series of AA3H-derived cell-penetrating peptide (CPP) candidates. Green: hydrophobic, uncharged residues (M, I, W, V), Blue: basic residues (R, H), Gray: other residues. Letter and number codes assigned to each sequence are given on the right. (b) Fluorescence intensity plots of the AA3H-derived CPPs from (a), showing their accumulation in HeLa cells determined by flow cytometry analysis. Each value given is a relative percentage in comparison to the original AA3H (A2A) sequence. Bars represent the means ± SDs of individual experiments performed in triplicate. Candidate sequences were each present at concentration of 1 μM. (c) Confocal microscope images of four-peptide fragments truncated from the AA3H peptide, FITC-labeled and administered to HeLa cells at a dose of 1 μM. Nuclei were stained with DAPI. Scale bars are 100 μm. (d) Flow cytometry data, presented as percentage cellular uptake based on fluorescence intensity values, for HeLa cells incubated with the FITC-labeled truncated peptides from (c). Cells were incubated with peptides of 5 μM concentration. (e) Viability of HeLa cells treated with truncated CPPs (0–100 μM). (f) Cellular uptake analysis of TAMRA-labeled SIWV (5 μM) in four cell lines (as indicated) when co-incubated with the inhibitors CPZ, MβCD, EIPA, and Dynasore. Uptake was quantified by fluorescence intensity from flow cytometry data. (g) Electrophoresis gels identifying the endogenous caveolin and β-actin expression levels in each cell. Bar graph: percentage of caveolin relative to actin. (h) Caveolin expression level analysis using SIWV-GFP in U87MG cells that had been transfected with si-Caveolin (siRNA knocking down caveolin expression). Untreated; U87MG cells with no treatment as a control, si-Caveolin-1; si-Caveolin-1 treated U87MG cells, and SIWV-GFP; SIWV-GFP treated U87MG cells. (i) Bar graph for the level of expressed GFP or caveolin with siRNA against actin. | |
Identification of tetra-CPPs
Based on these results, we selected four truncated CPPs (ASIW, SIWV, IWVG, and WVGH) in order to study the kinetic profiles of the penetration of truncated CPPs. FITC-labeled truncated CPPs were treated to HeLa cells for kinetic profiling; with different concentrations and incubation times, and the mean fluorescence intensities were then compared with the original AA3H-CPP, using fluorescence activated cell sorting (FACS) (Fig. S1, ESI†). We found that the penetration kinetics of truncated CPPs and AA3H-CPP produced similar patterns. We then quantified uptake of peptide into the cells as a function of CPP concentration (1, 5, 10, 15, and 20 μM), and optimized the incubation time for uptake at 2, 4, 8, and 18 h. The uptake rate of CPPs were slowly increased up to a concentration of 5 μM; however, at concentrations of over 5 μM, there were no significant differences in penetration properties (Fig. S1a, ESI†). With regard to the penetration time, the uptake of truncated CPPs to the cells was the highest at 4 h, while AA3H exhibited a steady increase of penetration over the incubation period (Fig. S1b, ESI†). These results suggested that the optimal uptake time and concentration of the tetra-peptides are 4 h and 5 μM, respectively. In the time-dependent flow cytometry analysis, the order of uptake efficiency was SIWV > ASIW > WVGH > IWVG, at 4 h incubation point (Fig. S1b, ESI†).
Homing ability and mechanism of SIWV
After identification of tetra-CPPs, we focused on SIWV-CPP which showed low cytotoxicity with high cell penetration ability. Firstly, we examined its mechanism of cell penetration and homing ability. Most CPPs penetrate cells via endocytosis, e.g., clathrin- or caveolae-mediated endocytosis or macro-pinocytosis.36,37 Prior to internalization of SIWV-CPP into cells, it is essential that the peptide interact with the cellular membrane surface. Cell surface-associated heparan sulfate is a regulatory polysaccharide that mediates a wide variety of cellular interactions, including constitutive as well as ligand-induced endocytosis.38 In order to assess if membranous heparan sulfate played a role in cellular uptake of SIWV-CPP, we performed a competition assay. When HeLa cells were treated with heparan sulfates (30 μg mL−1) for 30 min before adding SIWV, no change in the penetration ability of SIWV was observed, suggesting that SIWV did not depend on interaction with heparan sulfate in order to penetrate cells (Fig. S2a, ESI†). Additionally, we tested whether SIWV penetration was energy-dependent or -independent by treating the HeLa cells with SIWV at 4 °C for 2 h, which would slow cellular metabolism. The results indicated that SIWV penetration was energy-dependent pathway of SIWV penetration (Fig. S2b, ESI†). We then checked the cell penetration properties of SIWV-CPP towards different cell lines; human-derived brain carcinoma cells (U87MG cells), lung carcinoma cells (A549 cells), and liver carcinoma cells (Huh7 cells) (Fig. 2f). Interestingly, the highest cell penetration efficiency of SIWV was observed with U87MG cells, similar to the expression pattern of caveolin (Fig. 2g). Next, we treated cells with various inhibitors of endocytosis, including chlorpromazine (CPZ) to inhibit clathrin,39 methyl-beta-cyclodextrin (MβCD) to disrupt caveolae/lipid rafts,40 ethyl isopropyl amiloride (EIPA) to inhibit micropinocytosis,41 and dynasore to block dynamin in the cells (Fig. 2f).42 In HeLa cells, 87 ± 5.1% of SIWV was internalized; however, when cells were treated with MβCD before adding SIWV, the penetration ability decreased to 33 ± 8.0%. These results suggest that SIWV was internalized via a caveolin/lipid raft-dependent mechanism, but clathrin and dynamin were not involved in this process. In U87MG cells, SIWV showed 100% penetration into cells, and this penetration ability decreased to 86 ± 1.2%, 58 ± 13%, 61 ± 4.3%, and 61 ± 7.2% by using CPZ, MβCD, EIPA, and dynasore as inhibitors. These results suggest that SIWV was internalized via multiple pathways, primarily the caveolae/lipid raft-dependent pathways associated with dynamin. To investigate the synergistic effects of inhibitors, cells were simultaneously treated with MβCD and dynasore comparing the intracellular permeability of SIWV. As expected, simultaneous treatment with two inhibitors reduced the intracellular penetrability of SIWV to a greater extent than either singular inhibitor (Fig. S3, ESI†). These results represented the caveolae-dependent intracellular uptake mechanism of SIWV. In A549 cells, SIWV showed 93 ± 2.5% penetration, and the uptake was decreased to 74 ± 4.2% after treatment with the EIPA inhibitor, suggesting that SIWV was internalized via micropinocytosis. In Huh7 cells, SIWV showed a low cell permeability, which was decreased further with the use of MβCD, EIPA, and dynasore inhibitors. To understand the different cell permeabilities of SIWV, the caveolin expression level of cells was examined. The different amount of caveolin protein was observed in each of the four cell types, with the highest expression in U87MG cells, followed by A549, HeLa, and lastly the Huh7 cells. The amount of caveolin protein was expressed in each cell line and it was proportional to the cell permeation efficiency of SIWV (Fig. 2g). This finding serves as strong evidence for the cell penetration mechanism of SIWV, which is absorbed into cells via caveolin. The cell penetration mechanism of SIWV was also confirmed in U87MG cells where expression of caveolin protein was suppressed by siRNA-caveolin transfection (Fig. 2h). In this set, we observed dramatically decreased uptake of SIWV-GFP (Fig. 2i).
Localization of SIWV in U87MG cells
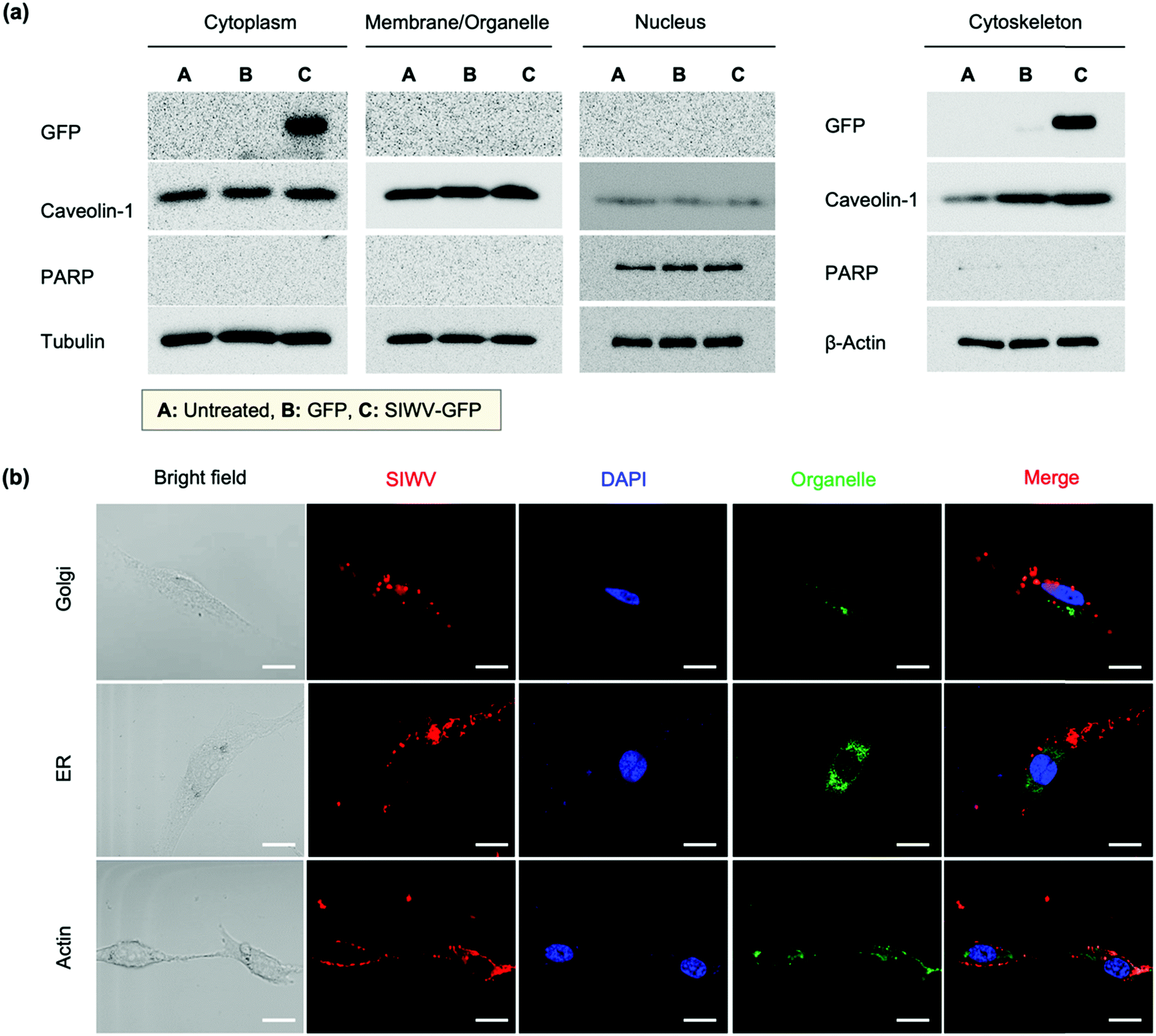
In order to understand the cellular localization of the SIWV peptide in U87MG cells, we performed cell extract analysis, subcellular fractionation, western blotting, and GFP fusion protein analysis. The GFP-labeled SIWV was prepared by chemical conjugation at the C-terminus, cloning, and purification on a His-tagged resin (Fig. S4a, ESI†). U87MG cells were then treated with the purified proteins at 2 μM and 10 μM concentration, and the results were evaluated using anti-GFP antibodies. Free GFP (with no SIWV peptide attached) showed no uptake into the cells, while SIWV-GFP fusion protein entered the cells when used at a concentration of 10 μM (Fig. S4b, ESI†). The cell lysates were then fractionated into cytoplasm, organelle membrane, nucleus, and cytoskeletal matrix components to assess the fate of the SIWV-GFP fusion protein. The SIWV-GFP fusion protein was mainly localized in the cytoplasm and cytoskeletal matrix, with negligible partitioning to the nucleus or organelle membranes (Fig. 3a). This result suggests that the pathway for SIWV penetration involves the cytoskeletal matrix, e.g., tubulin and actin. Localization of SIWV in the cellular organelles (e.g., golgi, endoplasmic reticulum (ER), nuclei, actin, etc.) was evaluated using a co-localization assay that employed known staining dyes and confocal microscopy (Fig. 3b). This data supports no specific localization of SIWV in the golgi or nucleus.
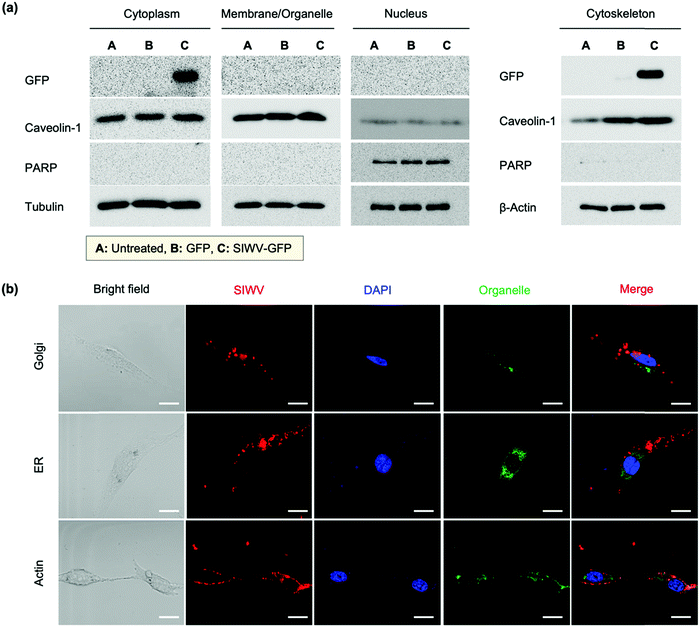 |
| | Fig. 3 Cellular localization analysis of SIWV in U87MG cells. (a) Results of a cellular fractionation study tracking the fate of the SIWV-GFP fusion protein in subcellular fractions (cytoplasm, membrane/organelle, nucleus, and cytoskeleton, as indicated) of U87MG cells. Fractions were detected using anti-GFP, anti-caveolin-1, anti-PARP, anti-tubulin, and anti-β-actin antibodies. Columns labeled A, B, and C are for untreated cells, cells treated with free GFP, and cells treated with the SIWV-GFP fusion protein, respectively. (b) Confocal microscope images evaluating co-localization of TAMRA-labeled SIWV (red) with suborganelle tracers in U87MG cells. Green (490 nm/525 nm ± 20 nm): golgi, ER, and actin. Blue (DAPI): nuclei. See the details about the staining reagents in the Methods section of the SI. Excitation wavelength and detection channels used: Blue (350 nm/461 nm ± 20 nm), Green (490 nm/525 nm ± 20 nm), and Red (555 nm/584 nm ± 20 nm). Scale bar: 20 μm. | |
In vivo distribution of SIWV in healthy and GBM model mice
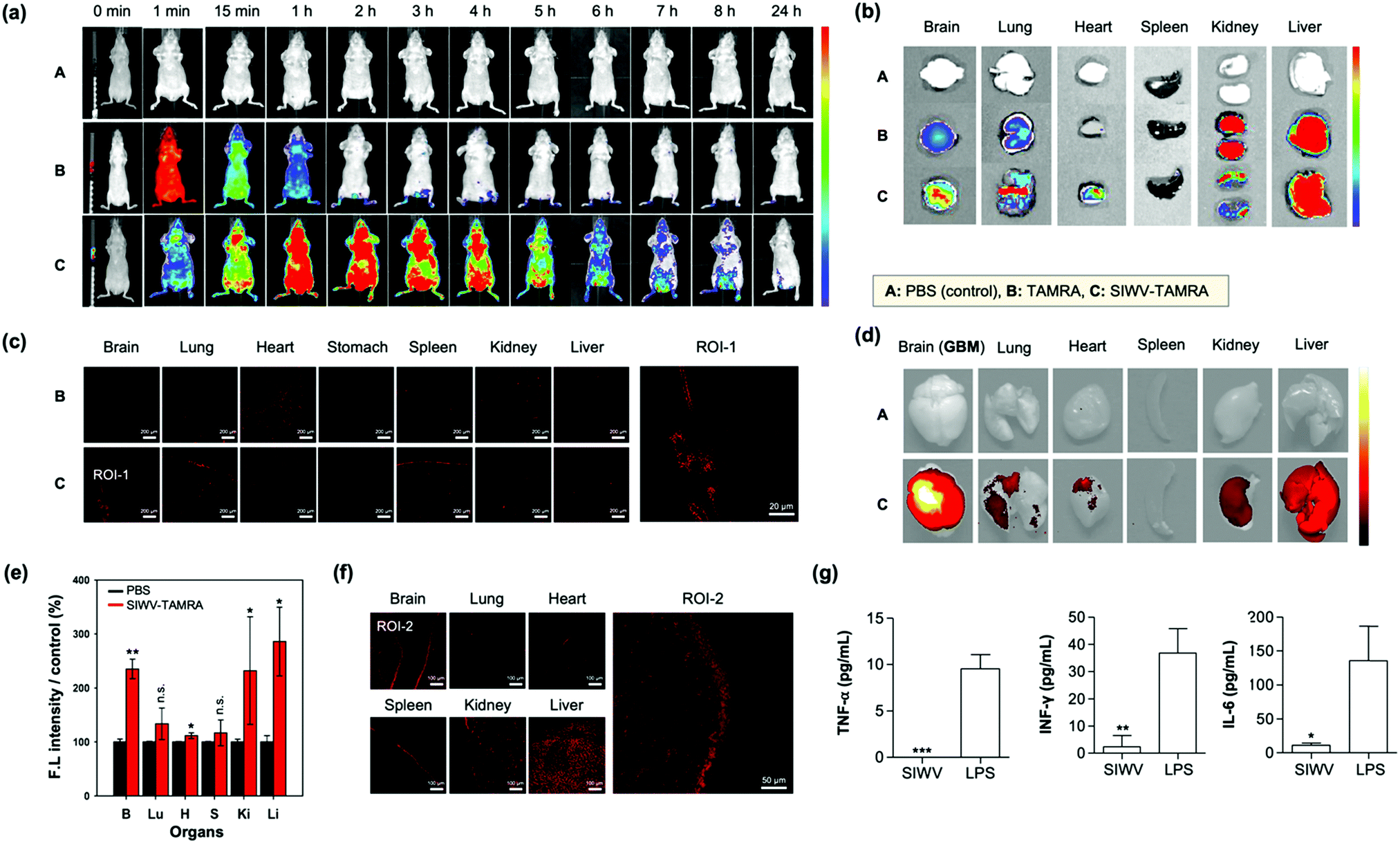
For the in vivo distribution assay, TAMRA (tetramethyl-rhodamine)-labelled SIWV was prepared. SIWV-TAMRA was intravenously (i.v.) injected into healthy nude mice and monitored over time using fluorescence in vivo imaging (Fig. 4a). When TAMRA was injected, it spread throughout the mouse in less than 1 min, and the fluorescence signal then disappeared within 2 h. In the case of SIWV-TAMRA, a strong signal was observed at 1 h, in the most parts of mouse, including the brain, and the signal became negligible after 24 h. This demonstrated that the peptide was effectively cleared. At 3 h circulation point after injection, the mice were dissected, and the fluorescence intensity of each organ was analyzed. Factors that could interfere with the fluorescence intensity were eliminated by perfusing of the mice. The fluorescence intensities of SIWV-TAMRA were monitored in the brain, lung, heart, and liver, and the signals were stronger than the corresponding signals from animals injected with free TAMRA, particularly in brain (Fig. 4b). In order to confirm that SIWV penetrated the tissues rather than just appearing on the tissue surface, frozen tissue sections were prepared and analyzed by confocal microscopy (Fig. 4c). These images confirmed that the peptide permeated the tissues.
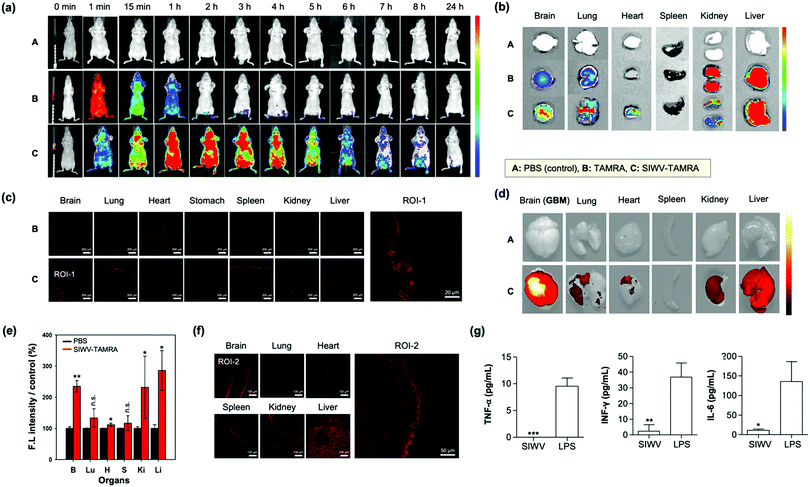 |
| | Fig. 4 Biodistribution of SIWV in mice. (a) IVIS image of TAMRA-labeled SIWV in healthy mice over time (0–24 h). A: PBS control, B: TAMRA, C: SIWV-TAMRA. (b) IVIS images of sacrificed organs of healthy mice (brain, lung, heart, stomach, spleen, kidney, and liver) after 3 h circulation of SIWV-TAMRA (i.v. injection). (c) Confocal microscope images of cryo-sectioned tissues from panel (b). Scale bar: 20 μm. (d) IVIS images of sacrificed organs of GBM mice after 3 h circulation of SIWV-TAMRA (i.v. injection). (e) Relative fluorescence intensity measured from each organ in panel (d). PBS injection was used as a control. Bars represent the means ± S.E.M. of individual experiments performed in triplicate, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, compared with PBS control of each organ (n.s. = non-significant). (f) Confocal microscope images of cryo-sectioned organs from panel (d). (g) Immunogenicity of SIWV in mouse serum, quantified as the concentration of inflammatory markers TNF-α, IFN-γ, and IL-6 measured in the sera from healthy mice injected with SIWV (5 μg mL−1) or with lipopolysaccharide (LPS, 10 μg mL−1) as a positive control. Bars represent the means ± SDs of individual experiments performed in triplicate, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 compared with LPS positive control. Excitation wavelength and detection channel for IVIS: 541 nm/580 ± 20 nm. Excitation wavelength and detection channel for confocal microscope: 543 nm/580 ± 20 nm. | |
The high organ specificity of SIWV to the brain encourages further study for brain related disease likes GBM because of its high cell permeability towards glioblastoma cell line, U87MG. Firstly, we prepared GBM xenograft mouse model via intracranial cancer cell implantation (U87MG cells, unilateral injection) (Fig. S5, ESI†). After 2 weeks of U87MG implantation, SIWV-TAMRA was injected into GBM xenograft mice through a tail vein. At 3 h circulation point after injection, the mice were sacrificed, and the fluorescence intensity of the harvested the organs (brain, lung, heart, spleen, kidney, and liver) were measured using an IVIS imaging system (Fig. 4d). The fluorescence intensities of SIWV-TAMRA were monitored in the brain, lung, heart, kidney, and liver, which were higher than the PBS control (Fig. 4e). Specifically, the signal within the brain (GBM site) was more than 2-fold compared to PBS. The undesired kidney and liver accumulation were noticeable within both healthy and GBM mouse models, but it was sufficiently higher within the GBM site. In order to confirm that the SIWV could be penetrated and accumulated into the brain tissue, frozen tissue sections were prepared and analyzed using confocal microscopy (Fig. 4f). These results indicated that the SIWV permeated into the brain tissue in the GBM mouse model. The liver accumulation factor was solved in the nano-formulation (see below). The SIWV accumulation into lung tissue was also confirmed, not only in the IVIS image but also in the frozen samples. In the IVIS image, it was confirmed to be permeable to the lung airway, but the efficiency of the penetrating tissue was low. Future studies are expected because the pattern of permeation of SIWV to lung tissue and pulmonary airway is expected to vary depending on the dose, time, and route of administration.
Immunogenecity of SIWV in serum
We next evaluated the immunogenicity of SIWV in serum. Generally, viral vectors have been used for the treatment of diseases, e.g., in gene therapy.43 But these are generally associated with serious problems, including immunogenicity and other side effects. So, small peptide-based CPPs, as non-viral carriers, have strong merits. In order to check the immunogenicity side effects, cytokine level in the blood were analyzed after injecting (i.v.) of SIWV into healthy mice. Inflammatory cytokines were produced by external stimuli, highlighted by Th1 cells in the blood, so we identified the immune system-related cytokines (e.g., TNF-α, IFN-γ, and IL-6) in the serum sample (Fig. 4g). The normal amount of TNF-α (7.08 pg mL−1), INF-γ (6.23 pg mL−1), and IL-6 (9.18 pg mL−1) amounts in a healthy mouse serum was confirmed before SIWV treatment. As a positive control for the immunogenicity assay, we treated the mouse with lipopolysaccharide (LPS), which is known to induce an immune response in macrophages. The SIWV treated mouse serum showed very a low level of TNF-α, while LPS induced its secretion more than 10 pg mL−1. The expression of INF-γ was greater than 40 pg mL−1 in the LPS-treated group, while SIWV group showed negligible values (2 pg mL−1). IL-6, another type of cytokine, was expressed at 150 pg mL−1 or more in the LPS group, while only 7 pg mL−1 in the SIWV group. From this data, we conclude that SIWV did not induce an inflammatory response in the animal model. SIWV may have potential as a drug delivery system to selectively transfer substances, not only at the cellular level, but also in disease sites.
Preparation of nano-formulation with SIWV and pSiNPs
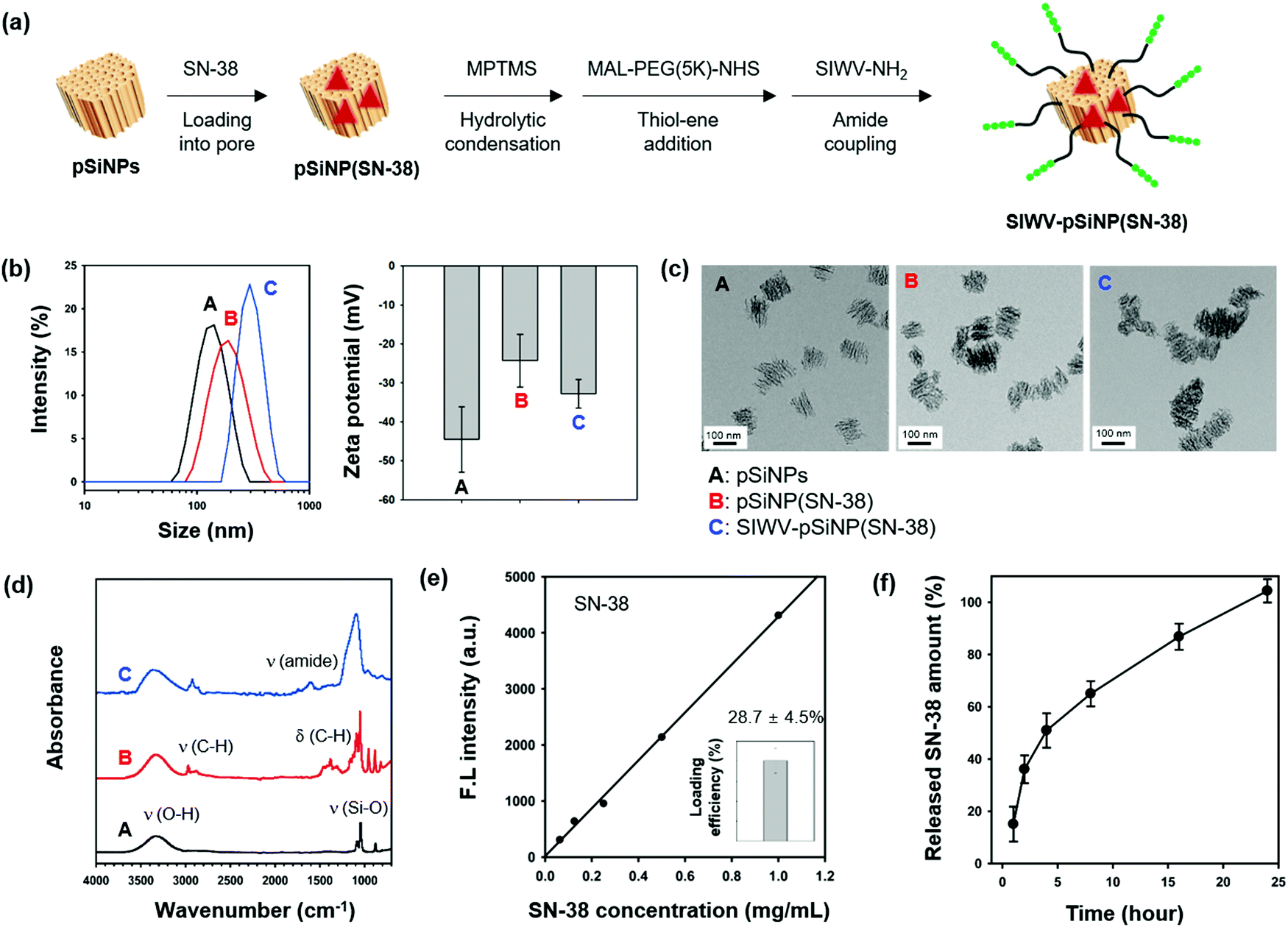
The ability of the SIWV peptide to act as a targeting moiety to selectively deliver a nanoparticle/therapeutic construct to GBM was then investigated. For these experiments we used porous silicon nanoparticles (pSiNPs) and the drug; 7-ethyl-10-hydroxy-camptothecin (SN-38), an FDA-approved anti-neoplastic agent.44 The pSiNPs were prepared by electrochemical etching of p++-type crystalline silicon wafers in a hydrofluoric acid (HF)-containing electrolyte, followed by ultrasonic fracture of the resulting porous silicon layers as previously described.45 As a drug delivery system, pSiNPs have been shown to exhibit several beneficial properties: (i) high loading efficiency, (ii) low cytotoxicity, (iii) non-toxic dissolution products, and (iv) high tissue specificity when conjugated to a targeting moiety. For the present work, the SN-38 antineoplastic agent was loaded into the pores using a calcium silicate sealing/condenser chemistry.46 The SN-38-loaded pSiNP formulation (named pSiNPs(SN-38)) was modified with the SIWV peptide via a poly-(ethyleneglycol) (PEG, 5 kDa) linker (Fig. 5a and Fig. S6, ESI†). Prior to drug loading and targeting peptide conjugation, the as-prepared pSiNPs displayed an average hydrodynamic diameter of 139.7 ± 18.8 nm (polydispersity index; PDI: 0.206) and zeta potential of −44.5 ± 8.39 mV, measured by dynamic light scattering (DLS, Fig. 5b and Table S1, ESI†). The SN-38-loaded particle, pSiNPs(SN-38), showed a slight increase in size (196.3 ± 32.2 nm, PDI: 0.103) with a lowering of the negative surface charge (to −24.3 ± 6.73 mV). Conjugation of SIWV peptide to the drug-loaded particle, SIWV-pSiNP(SN-38), further increased the size (to 306.2 ± 32.7 nm, PDI: 0.217) and it increased the negative charge (−34.8 ± 5.91 mV). The transmission electron microscope (TEM) images of pSiNPs prior to loading indicated a relatively homogeneous particle size. The size uniformity and general overall appearance of the porous nanostructure were both retained after the drug loading and peptide conjugation processes (Fig. 5c).
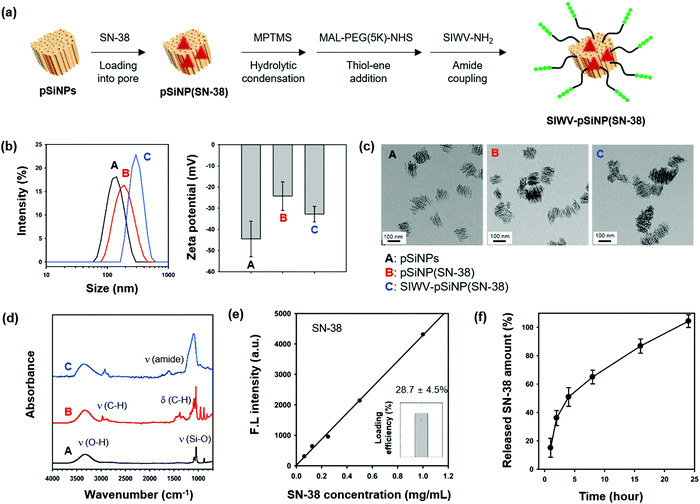 |
| | Fig. 5 Preparation and characterization of nano-carrier formulation. (a) Scheme depicting the preparation of the nano-carrier SIWV-pSiNP(SN-38). (b) Mean hydrodynamic diameter (intensity distribution) measured by dynamic light scattering (DLS), and zeta-potential value. Each mean and standard deviation was calculated in triplicate. (c) Transmission electron microscope (TEM) images of particles (scale bar = 100 nm). (d) Attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectra of the particles. Symbols: ν = stretching, δ = bending. The pSiNPs displayed bands associated with the Si–O–H functionality; broad band for ν (O–H) at 3550–3200 cm−1, ν (Si–O) at 1065 cm−1. The pSiNP(SN-38) showed strong bands at 2950–2850 cm−1, and 1480–1350 cm−1 associated with ν (C–H), and δ (C–H), respectively, which derived from the loaded SN-38. The SIWV-pSiNP(SN-38) construct showed a bending mode assigned to the amide bond at 1650 cm−1. (e) Loading efficiency of SIWV-pSiNP(SN-38). Loaded SN-38 content was analyzed by fluorescence intensity from the standard curve of SN-38 (λexi/emi: 367/561 nm). (f) Release profile depicting percent of SN-38 from SIWV-pSiNP(SN-38) released into the PBS solution as a function of time at 37 °C. | |
The attenuated total reflectance Fourier-transform infrared spectrum (ATR-FTIR) of the as-prepared pSiNPs (Fig. 5d) showed two major bands corresponding to ν (O–H) stretching at 3550–3200 cm−1 and ν (Si–O) stretching at 1065 cm−1. Upon drug loading, the pSiNP(SN-38) formulation displayed bands in the regions 2950–2860 cm−1 and 1480–1350 cm−1 associated with (C–H) stretching and bending vibrations, respectively, confirming the successful loading of SN-38 into the pSiNPs. The SIWV-pSiNPs(SN-38) construct showed strong bands associated with the ν (amide) of the SIWV peptide at 1650 cm−1. The final formulation, SIWV-pSiNP(SN-38), showed high loading efficiency of the SN-38 drug (28.7 ± 4.5% by mass) (Fig. 5e). The loading of SN-38 within the pore of pSiNPs, not stacked on the surface, was verified by incubation of SIWV-pSiNP(SN-38) within DMSO for 24 h at 37 °C and measurement of emission changes of the solution after spin-down the particle. In this condition, the stacked SN-38 on the surface of pSiNPs is rapidly washed away, but the encapsulated SN-38 is slowly released out by slow degradation of pSiNPs in the DMSO. No significant fluorescence intensity changes of solution were observed within 1 h, and the emission signal of SN-38 has appeared from 2 h incubation as pSiNPs degradation begins. From these results, we conclude that SN-38 is loaded in interior pores of pSiNPs, not surfaces of it (Fig. S7, ESI†). In order to evaluate the temporal drug release profile from the SIWV-pSiNP(SN-38) in vitro, we dispersed SIWV-pSiNP(SN-38) in phosphate-buffered saline (PBS, pH 7.4) for 24 h and measured the fluorescence intensity of the released SN-38 at regular time intervals (Fig. 5f). The majority of the drug load (50%) was released within 4 h and the remaining drug was released over the next 24 h.
Cellular uptake mechanism of nano-formulation
We next examined the cellular uptake mechanism of nano-formulation. We conducted an experiment that cells are internalized by different mechanisms depending on cargo size. We used four types of intracellular migration inhibitors in establishing the cellular uptake mechanism of SIWV-functionalized pSiNPs. In this experiment, the SIWV-pSiNP showed a similar mechanism of SIWV itself. In U87MG cells, the internalization of SIWV-pSiNP was reduced by 74% by MβCD inhibitor, 48% by EIPA, and 15% by dynasore. Huh-7 cells showed a similar trend, with MβCD 52%, EIPA 17%, but dynasore not inhibited. Mainly internalized primarily in caveolae/lipid-raft and concomitantly through macro-pinocytosis. Also, we confirmed that the SIWV-pSiNP has more effective cell permeability (1.2 times) in U87MG cells than in Huh-7 cells. These results showed that even though cargo was attached, the characteristics of SIWV's cell internalization is retained (Fig. S8, ESI†).
Evaluation of cellular toxicity of the nano-formulation in vitro
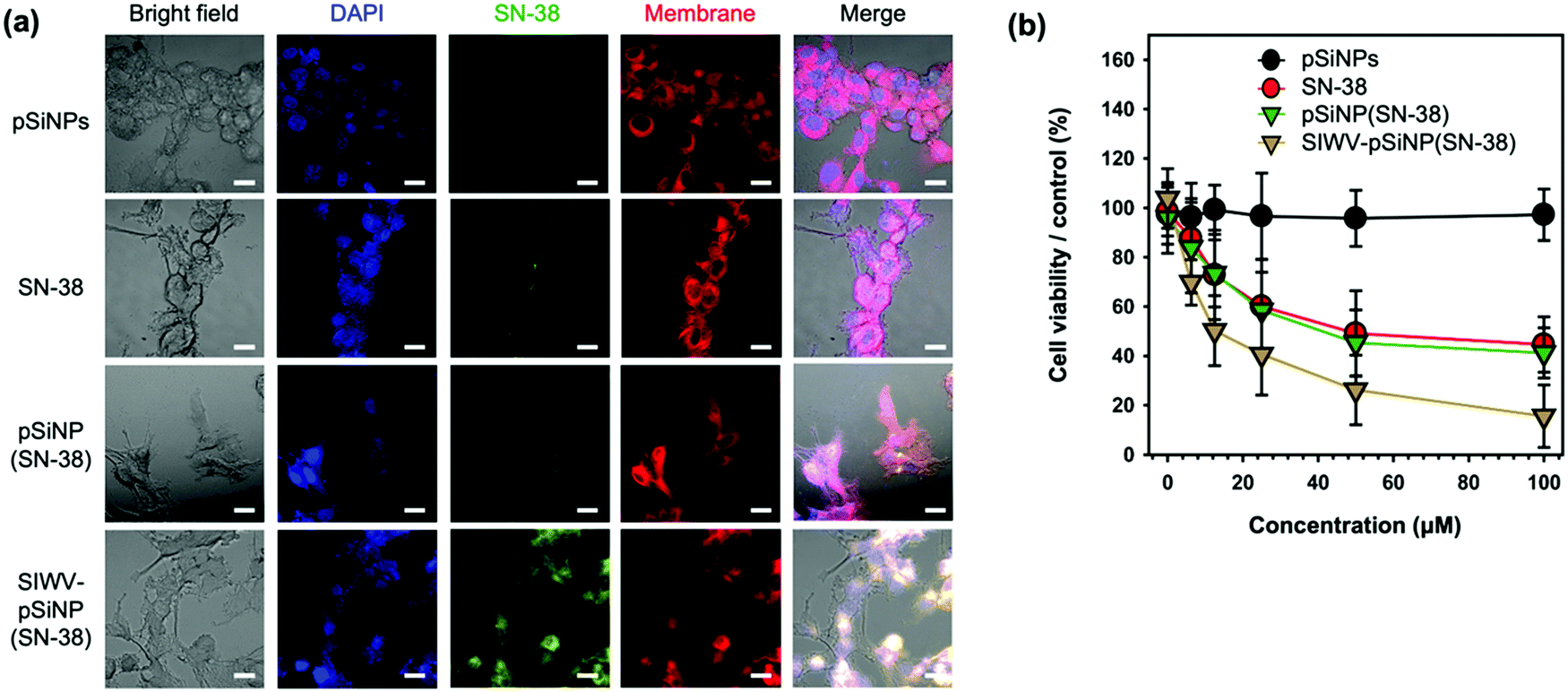
In order to investigate intracellular localization of the nano-formulation, U87MG cells were incubated with empty pSiNPs, free SN-38, pSiNP(SN-38), and SIWV-pSiNP(SN-38) for 6 h and analyzed using confocal laser scanning microscopy (CLSM) (Fig. 6a). The fluorescence images revealed that free SN-38 and non-targeted pSiNPs containing SN-38 payload (with no targeting group attached) were not internalized in the cells within the 6 h observation period, whereas the SIWV-pSiNP(SN-38) construct successfully delivered and released SN-38 inside the U87MG cells under the given conditions. In the co-stained images with DAPI (blue) and cell membrane (red) dyes, the green emitting SN-38 signal was mainly observed in the cytoplasm. To evaluate the cytotoxicity of the nano-formulations toward U87MG cells, we performed a cell viability assay, evaluating concentrations of SN-38 between 0 and 100 μM (Fig. 6b). The empty control pSiNPs showed no toxicity under these conditions, whereas cytotoxicity was observed to occur in a dose-dependent manner for free SN-38, SN-38-loaded pSiNPs, and SIWV-pSiNP(SN-38). At low SN-38 concentration (6.5 μM), the cytotoxicity of U87MG cells treated with SIWV-pSiNP(SN-38) was significantly higher (31.2%) than that observed in cells treated with free SN-38 (12.4%) or with the non-targeted SN-38-loaded pSiNPs (16.2%). At high SN-38 concentrations (100 μM), the cytotoxicity of SIWV-pSiNP(SN-38) was the highest (84.4%) compared to free SN-38 (55.4%) and non-targeted SN-38-loaded pSiNPs (58.8%). These results support the hypothesis that SIWV provides high cell penetration and efficient drug release in U87MG cells.
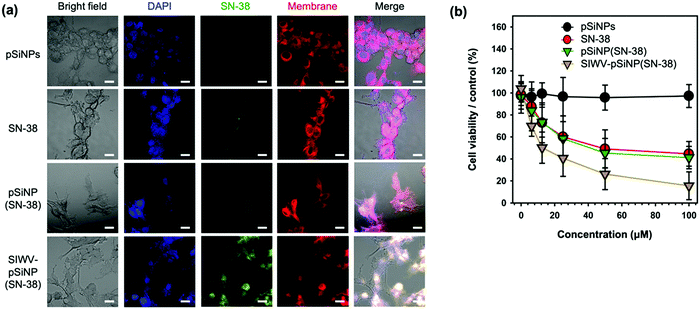 |
| | Fig. 6 Cytotoxicity analysis of SIWV-pSiNP(SN-38). (a) Confocal microscope images of U87MG cells treated with pSiNPs, SN-38, pSiNP(SN-38), and SIWV-pSiNP(SN-38). Excitation wavelength and detection channel for CLSM: Blue (DAPI, 405 nm/430 nm ± 20 nm), Green (SN-38, 405 nm/525 nm ± 20 nm), and Red (Cell-Membrane Mask, 640 nm/670 nm ± 20 nm). Scale bar: 20 μm. (b) Cytotoxicity of each samples (0–100 μM) towards U87MG cells, quantified by CCK-8 assay. | |
Evaluation of anticancer efficacy of the nano-formulation in vivo
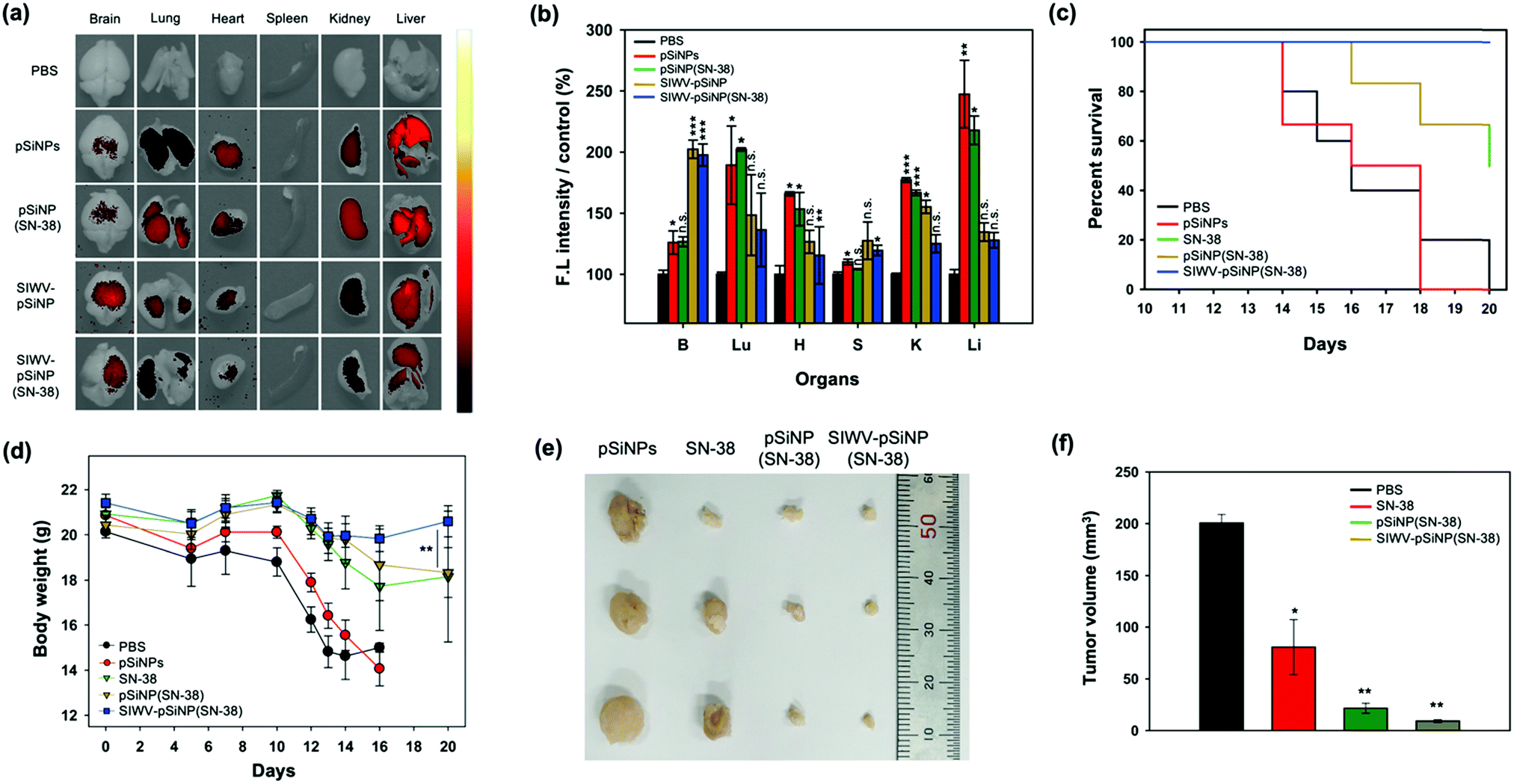
Next, we checked the bio-distribution of the nano-formulations, comparing the SIWV-targeted, SN-38-loaded nanoparticles with PBS buffer, empty pSiNPs, non-targeted SN-38-loaded pSiNPs, and SIWV-targeted empty pSiNPs (Fig. 7a and b). All pSiNP formulations were intravenously injected into GBM model mice at a dose of 40 mg kg−1. After 2 h of circulation, the mice were sacrificed, perfused, and the internal organs were removed and analysed by following the intrinsic photoluminescence (PL)23 of pSiNPs. In the control set, the PBS-treated set showed a negligible tissue autofluorescence signal, and non-targeted constructs (pSiNPs, pSiNPs(SN-38)) showed signals within the kidney and liver. The targeted constructs (SIWV-pSiNPs, SIWV-pSiNPs(SN-38)) showed intensive signals within the brain, and interestingly liver accumulation was dramatically decreased in comparison to a SIWV-TAMRA formulation (Fig. 4d). These results suggest (i) higher accumulation of the nanoparticle in the brain due to the targeting ability of SIWV, (ii) lower macrophage uptake of the nano-formulation relative to free SIWV, and/or (iii) longer blood circulation of the nano-formulation in the body. Bio-distribution analysis of SIWV-pSINP(SN-38) in healthy mice also revealed a significant accumulation in the brain (Fig. S9, ESI†).
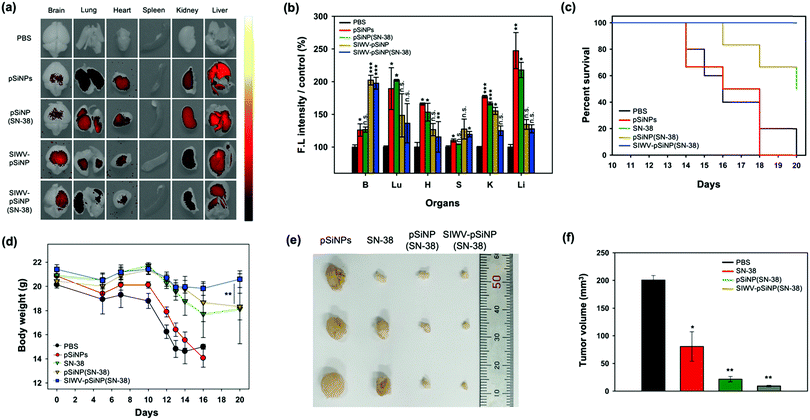 |
| | Fig. 7 Anticancer efficacy analysis of SIWV-pSiNP(SN-38). (a) IVIS images of sacrificed organ of GBM mice after 2 h circulation of PBS, pSiNPs, pSiNP(SN-38), SIWV-pSiNP, and SIWV-pSiNP(SN-38) (injected with 40 mg kg−1, i.v.). Excitation wavelength and detection channel for IVIS: 410 nm/830 ± 20 nm. (b) Relative fluorescence intensity plot of each organs from panel (a). Bars represent the means ± S.E.M. of individual experiments performed in triplicate, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, compared with PBS control of brain. (c) Therapeutic efficacy test in GBM mice. Each mouse was treated with PBS (control, n = 5), pSiNPs, free SN-38, pSiNP(SN-38), and SIWV-pSiNP(SN-38). (n = 6, injected with 20 mg kg−1, i.v.). The survival rate of each mouse was observed for 20 days, after U87MG inoculation. (d) Changes in body weight of the mice after injection of PBS, empty pSiNPs, free SN-38, pSiNP(SN-38), and SIWV-pSiNP(SN-38). The mice were weighed on the 14th, 15th, 16th, and 18th days after the inoculation. Bars represent the means ± S.E.M., *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, compared with the group after treatment with pSiNP(SN-38). (e) Representative photography images and (f) variation in harvested GBM tissue volume after treatment with PBS control, free SN-38, pSiNP(SN-38), and SIWV-pSiNP(SN-38) (n = 3). Bars represent the means ± S.E.M. of individual experiments performed in triplicate, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, compared with PBS control. The tumor tissue was excised on 20th day after the inoculation. | |
Finally, we evaluated the antitumor efficacy of the optimized nano-formulation in GBM xenograft model mice. Mice were implanted on day 0 with U87MG cells to induce the formation of GBM tumors. The GBM-bearing-mice were then administered PBS, SN-38, pSiNPs, pSiNP(SN-38), and SIWV-pSiNP(SN-38) at doses of 20 mg kg−1via the tail vein on day 14, 15, 16, and 18. The survival rate and the body weight of the animals were monitored (Fig. 7c and d). The mice treated with SIWV-pSiNP(SN-38) exhibited a significantly improved survival rate of over 20 days relative to PBS, empty pSiNP, free SN-38, and non-targeted, SN-38-loaded pSINP control groups (0, 0, 50, and 66.6% survival after 20 days, respectively). The survival curves showed that the SIWV-pSiNP(SN-38) treatment significantly prolonged the survival of the mice (Fig. 7c). The body weight of the PBS and pSiNPs treated mice rapidly decreased from 10 days after xenograft, and the body weight of SN-38 and pSiNP(SN-38)-treated mice slowly decreased over the observation period. However, the SIWV-pSiNP(SN-38) treated set showed no significant body weight loss over 20 days, consistent with an effective reduction in disease progression in the GBM implanted mice (Fig. 7d). Tumors in the SIWV-pSiNP(SN-38) treated group significantly decreased in size (Fig. 7e). Average tumor volume of the SIWV-pSiNP(SN-38)-treated group was 9.2 ± 2.2 mm3, whereas the tumor volume of PBS, SN-38, and pSiNP(SN-38)-treated groups was 200.5 ± 14.6 mm3, 80.6 ± 46.1 mm3, and 21.6 ± 8.3 mm3, respectively (Fig. 7f). Taken together, the results indicate that a GBM-homing peptide can effectively direct a nanoparticle-drug construct to the brain, more effectively treating a mouse model of GBM relative to the free anticancer drug, thus substantially reducing the possibility of severe side effects derived from low organ-specificity and poor drug solubility.
3. Conclusions
This study identified a sequence consisting of four residues from the substructure of the AA3H cell-penetrating peptide that is essential for cellular internalization, using Ala substitution assays. The peptide, with the sequence SIWV, showed negligible cytotoxicity and immunogenicity based on in vitro and in vivo assays. The SIWV peptide exhibited significant homing to the U87MG human glioblastoma (GBM) brain cancer cell line, and investigation of the mechanism of cell permeation in several cell lines indicated that the peptide undergoes caveolin-mediated endocytosis. The practical applicability of the SIWV peptide was demonstrated using a porous silicon nanoparticle (pSiNPs) carrier delivering the anticancer drug SN-38. While SN-38 is a promising therapeutic for treatment of GBM, it displays substantial adverse side effects due to a poor biodistribution profile. Biodistribution analysis and in vivo efficacy data showed that the selective homing capability of SIWV, when combined with the drug carrying capability of the pSiNPs, can yield significant improvements in both selectivity and efficacy for treatment of a mouse model of GBM.
4. Materials and methods
Peptide preparation
Peptides were prepared using the standard Fmoc-based solid-phase peptide synthesis method on Rink-amide resin (0.7 mmol g−1). For the coupling reactions, Fmoc-protected amino acids (5 equiv.), HOBt (5 equiv.), HBTU (5 equiv.), and DIPEA (5 equiv.) in DMF were added to the solution of resin in DMF (0.5 mL). We achieved de-protection of Fmoc by incubating the resin with 20% (v/v) piperidine in DMF (0.5 mL) for 30 min at room temperature. After labeling the N-terminus of the peptides with FITC, and the C-terminus of the truncated peptides with biotin, peptides were de-protected and cleaved from the resin by incubating in de-protection solution (1 mL, 95% [v/v] TFA, 2.5% TIS, and 2.5% H2O) for 2 h at room temperature. After cleavage of the peptides from the resin, the peptides were precipitated by addition of diethyl ether (5 mL) and centrifugation for 5 min at 1500g. The peptides were then purified on a C-18 reverse-phase high-performance liquid chromatography column using acetonitrile and water containing 0.1% TFA as the eluents. All the purified peptides were characterized using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (Kangwon National University, Rep. of Korea). Fluorescent-labeled (TAMRA, FITC) truncated CPPs were synthesized by Peptron (Daejeon, Korea, Rep. of Korea). High purity (>98%) peptides were obtained from Peptron. All peptides were reconstituted in high purity DMSO (10 mM) and stored at −70 °C.
Cellular uptake analysis of tetra-CPPs
To assess the uptake of CPP candidates by HeLa cells, the cells were seeded onto 6-well plates (1.0 × 106 cells per well). After 24 h, the cells were washed twice with serum-free DMEM and incubated with CPPs in serum-containing DMEM at the indicated concentrations (0.1, 1, 10, or 100 μM) at 37 °C. The cells were then washed twice with PBS, incubated in trypsin-EDTA solution (0.01% trypsin) for 10 min at 37 °C to digest extracellular proteins, then washed twice with ice-cold PBS, and finally resuspended in 500 μL ice-cold PBS for flow cytometry. For treatment with endocytosis inhibitors, the cells were pre-incubated with various inhibitors (30 μg heparan, 50 μM CPZ, 100 μM CQ, 1 mM MβCD, 30 μg dynasore, or 50 μM EIPA) for 30 min in serum-containing medium, and SIWV (5 μM) was then added. Samples for flow cytometry analysis were prepared as described above. Analysis of peptide internalization was performed with a GUAVA flow cytometry system (Merck Millipore, Darmstadt, Germany), following the manufacturer's instructions, using the FL-1 channel to measure fluorescence in GuavaSoft 2.6. The results are presented as the relative percentage of untreated HeLa cells. Data are presented as mean ± standard deviation (SD). To visualize the cellular uptake of peptides under confocal fluorescence microscopy, HeLa cells were seeded onto clear-bottomed 35 mm plates (SPL, Gyeonggi-Do, Rep. of Korea) and then incubated with truncated CPPs (2 μM) for 4 h. The Golgi were stained with CellLight reagent Golgi-GFP, endoplasmic reticulum (ER) were stained with CellLight reagent ER-GFP, actin was stained with CellLight Actin-GFP, and nuclei of truncated CPP-treated cells were stained with DAPI (Thermo Fisher). After washing with PBS twice, the intracellular fluorescence of HeLa cells was imaged using a confocal laser scanning microscope (LSM Zeiss 700, Jena, Germany).
Preparation of the porous silicon nanoparticles (pSiNPs)
Porous silicon nanoparticles (pSiNPs) were fabricated by electrochemical etching; galvanostatic anodization of heavily boron-doped p++-type single-crystal silicon wafers (polished on the (100) face, Virginia Semiconductor, Inc. (USA)) in an aqueous ethanolic hydrofluoric acid electrolyte consisting of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) of 48% aqueous HF:absolute ethanol. CAUTION: HF is highly toxic and proper care should be exerted to avoid contact with skin or lungs. Prior to the preparation of the pSi layers, the silicon wafer was anodized to generate a thin porous layer, commonly referred to as a “sacrificial layer”, in the HF-containing electrolyte, and the resulting porous layer was then dissolved by treatment with aqueous potassium hydroxide (KOH, 2 M). The etching waveform consisted of a lower current density of 46 mA cm−2 and was applied for 1.8 s, followed by a higher current density pulse of 334 mA cm−2 applied for 0.4 s. This waveform was repeated for 150 cycles, generating a porous silicon (pSi) film with “perforations” repeating approximately every 200 nm through the porous layer. The pSi film was removed from the silicon substrate by applying a current density of 3.1 mA cm−2 for 300 s in a solution containing 1:12 (v:v) of 48% aqueous HF:ethanol. The collected pSi film was placed in deionized water (DI H2O, 6 mL) in a sealed plastic vial and fractured into nanoparticles in an ultrasonic bath (VWR, PA, USA) for 24 h and then filtered through a 0.22 μm syringe filter (Millipore, Millex syringe filter unit, 220 nm model #SLGP033RS). The pSiNPs were then further incubated in deionized water for 10 days at room temperature (25 °C) to form oxidized silicon surfaces (Si–OH). The resulting pSiNPs were then collected using centrifugation (14000 rpm, 30 min) and washed 3 times with ethanol.
:1 (v:v) of 48% aqueous HF:absolute ethanol. CAUTION: HF is highly toxic and proper care should be exerted to avoid contact with skin or lungs. Prior to the preparation of the pSi layers, the silicon wafer was anodized to generate a thin porous layer, commonly referred to as a “sacrificial layer”, in the HF-containing electrolyte, and the resulting porous layer was then dissolved by treatment with aqueous potassium hydroxide (KOH, 2 M). The etching waveform consisted of a lower current density of 46 mA cm−2 and was applied for 1.8 s, followed by a higher current density pulse of 334 mA cm−2 applied for 0.4 s. This waveform was repeated for 150 cycles, generating a porous silicon (pSi) film with “perforations” repeating approximately every 200 nm through the porous layer. The pSi film was removed from the silicon substrate by applying a current density of 3.1 mA cm−2 for 300 s in a solution containing 1:12 (v:v) of 48% aqueous HF:ethanol. The collected pSi film was placed in deionized water (DI H2O, 6 mL) in a sealed plastic vial and fractured into nanoparticles in an ultrasonic bath (VWR, PA, USA) for 24 h and then filtered through a 0.22 μm syringe filter (Millipore, Millex syringe filter unit, 220 nm model #SLGP033RS). The pSiNPs were then further incubated in deionized water for 10 days at room temperature (25 °C) to form oxidized silicon surfaces (Si–OH). The resulting pSiNPs were then collected using centrifugation (14000 rpm, 30 min) and washed 3 times with ethanol.
Preparation of pSiNP(SN-38)
pSiNPs (∼1 mg) and SN-38 stock solution (20 mg mL−1 DMSO, 50 μL) were dispersed in ethanol (1.45 mL) and then mixed using a vortex mixer at room temperature (25 °C) for 24 h. The resulting pSiNPs were then washed 3 times with ethanol by centrifugation (14000 rpm, 15 min), in order to remove the remaining free SN-38. To analyze the SN-38 loading efficiency, supernatants from each centrifugation step were collected and analyzed by measurement of fluorescence intensity using a spectro-fluorophotometer (SHIMADZU CORP., Kyoto, Japan) and fit to a standard curve, using a 1 cm standard quartz cell (internal volume of 1 mL, Hellma Analytics, Germany).
Preparation of SIWV-pSiNP(SN-38)
The pSiNP(SN-38) construct (∼1 mg) and (3-mercaptopropyl)-triethoxysilane (MPTES, 20 μL) was dispersed in ethanol (1 mL), and then mixed using a vortex mixer at room temperature (25 °C) for 4 h. The thiol-terminated nanoparticles were then washed 3 times with ethanol using centrifugation (14000 rpm, 15 min), in order to remove the remaining MPTES. The thiolated nanoparticles were resuspended in ethanol (800 μL), and a maleimide (MAL)-PEG-N-hydroxy-succinimide (NHS) stock solution (5 mg mL−1 in ethanol, 200 μL) was added. The mixture was vortexed at room temperature (25 °C) for 2 h. The resulting particles were washed 3 times with ethanol using centrifugation (14000 rpm, 15 min) to remove the remaining unreacted MAL-PEG-NHS. For the peptide-conjugated formulations, SIWV peptide stock solution (2 mg mL−1 in DI H2O, 100 μL,) was added to 100 μL of nanoparticles in ethanol, incubated at 4 °C for 4 h, purified three times by centrifugation, dispersed in 10 μL DI H2O and stored at 4 °C before use.
Animals
5- or 6-week-old female BALB/c nu/nu mice or 7-week-old ICR mice (Taconic, provided by Daehan Biolink Co., Ltd, Eumseong, Rep. of Korea) were housed at an ambient temperature of 23 ± 1 °C and relative humidity of 60 ± 10% under a 12 h light/dark cycle and were allowed free access to food and water. All of the experiments performed with mice were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publications No. 80-23, revised 1996) and protocols approved by the Institutional Animal Care and Use Committee of Kyung Hee University for each experiment (KHUASP(SE)-19-002 and KHUASP(SE)-19-003) and institutional guidelines (assigned No. 2015-020).
Intracranial xenograft (tumor implantation)
BALB/c nu/nu mice were anesthetized with tribromoethanol and mounted in a stereotaxic apparatus [myNeuroLab (St. Louis, MO, USA)]. Each mouse received a unilateral injection of 1.0 × 106 U87MG cells, according to the following coordinates: anteroposterior: −3.0 mm from bregma; mediolateral: 1.8 mm from bregma; and dorsoventral: −3.0 mm from the skull bregma.47 The flow of the injections was controlled using an electronic pump at 1 μL min−1 for 5 min and followed by 2 min with the needle to the injection site in order to avoid reflux. After surgery, the mice were allowed to recover from anesthesia in a temperature-controlled chamber and then placed in individual cages. Animals were weighed every 2–3 days until the occurrence of the visible symptoms of impending death such as hunched posture, reduced mobility, and weight loss.48
In vivo distribution of SIWV in GBM model mice
2 weeks after implantation, mice were randomly divided into 2 groups (n = 3 per group) as follows: (1) control group (injected with PBS, i.v.), (2) SIWV-TAMRA group (injected intravenously with 1 mM SIWV-TAMRA in 0.2 mL). 1 h post-injection, the mice were anesthetized with tribromoethanol, perfused with PBS and then fixed with 4% PFA. The brain, lung, heart, spleen, kidney, and liver organs were harvested and immersed, in order to analyze the fluorescence intensity, using a VISQUE inVivo Smart LF luminescence and fluorescence animal imaging system (Vieworks Co., Ltd, Rep. of Korea). SIWV-TAMRA: PE excitation (530–570 nm) and the PE emission filter (575–640 nm) was used for image acquisition.
In vivo distribution of SIWV-pSiNP(SN-38) in GBM mice
2 weeks after implantation, the mice were randomly divided into 4 groups (n = 3 per group) as follows: (1) control group (injected with PBS, i.v.), (2) pSiNPs group (injected with 40 mg kg−1 empty pSiNPs, i.v.), (3) pSiNP(SN-38) group (injected with 40 mg kg−1 pSiNP(SN-38), i.v.) (4) SIWV-pSiNP group (injected with 40 mg kg−1 SIWV-pSiNP, i.v.), (5) SIWV-pSiNP(SN-38) group (injected with 40 mg kg−1 SIWV-pSiNP(SN-38), i.v.). 2 h post-injection, the mice were anesthetized with tribromoethanol, perfused with PBS and then fixed with 4% PFA. The brain, lung, heart, spleen, kidney, and liver organs were removed and immersed in buffer solution at pH 12 for 1 h and the fluorescence intensity was quantified using a VISQUE inVivo Smart LF luminescence and fluorescence animal imaging system, employing a GFP excitation filter (390–490 nm), 3 s exposure time, and an ICG (810–860 nm) emission filter.
Therapeutic efficacy of SIWV-pSiNP(SN-38)
After innoculation with U87MG cells, the mice were divided into 5 groups (n = 6 per group) to investigate the therapeutic efficacy of the nano-formulation of SIWV as follows: (1) control group (PBS, i.v.), (2) pSiNPs group (injected with empty pSiNPs, i.v.), (3) SN-38 group (injected with free SN-38, i.v.), (4) pSiNP(SN-38) group (injected with pSiNP(SN-38), i.v.) (5) SIWV-pSiNP(SN-38) group (injected with SIWV-pSiNP(SN-38), i.v.). Mice were treated on the 14th, 15th, 16th, and 18th days after the inoculation and weighed every 2–3 days. Mice were sacrificed on the 20th day and the tumor tissue was harvested for volume measurement. The tumor volume was measured using a caliper and calculated as V = (4/3) × π × (L/2) × (W/2) × (D/2), where L is the length, W is the width, and D is the depth of the tumor. Photographic images were captured using a digital camera.
Conflicts of interest
The authors declare the following competing financial interest(s): the authors are listed as inventors on a pending patent application related to the technology described in this work. D. K. is a scientific founder of N Therapeutics, Inc., a member of the Board of Directors, and has an equity interest in the company. M. J. S. holds an appointment as a “High-Level Talent” at the Key Laboratory of Organosilicon Chemistry and Material Technology of Hangzhou Normal University, China, he is a Guest Professor at Zhejiang University, China, and he is a scientific founder of Spinnaker Biosciences, Inc., a member of the Board of Directors, and has an equity interest in the company. Although one or more of the grants that supported this research has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Spinnaker Biosciences, Inc., the research findings included in this particular publication may not necessarily relate to the interests of Spinnaker Biosciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2013-R1A6A3A01064606, 2015-R1A6A3A04059360). We also had financial support from the Bio & Medical Technology Development Program of the NRF of Korea, funded by the Ministry of Science & ICT (2018-M3A9H3021707), and Basic Science Research Program through the NRF of Korea, funded by the Ministry of Education (2018-R1A6A1-A03025124, 2018-R1D1A1-B07043383), the U.S. National Institutes of Health, through contract #1R01CA214550-01, and by the Medical Research Center Program through the NRF of Korea, funded by the Ministry of Science and ICT (2017-R1A5A2014768).
References
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed.
- E. L. Snyder and S. F. Dowdy, Pharm. Res., 2004, 21, 389–393 CrossRef CAS PubMed.
- H. Derakhshankhah and S. Jafari, Biomed. Pharmacother., 2018, 108, 1090–1096 CrossRef CAS PubMed.
- S. Silva, A. J. Almeida and N. Vale, Biomolecules, 2019, 9, 22 CrossRef PubMed.
- M. Arite, S. Dikran, H. Uwe and M. Walter, Curr. Pharm. Biotechnol., 2014, 15, 200–209 Search PubMed.
- S. Jafari, S. Maleki Dizaj and K. Adibkia, BioImpacts, 2015, 5, 103–111 CrossRef CAS PubMed.
- J. Habault and J.-L. Poyet, Molecules, 2019, 24, 927 CrossRef PubMed.
- D. Sarko, B. Beijer, R. Garcia Boy, E.-M. Nothelfer, K. Leotta, M. Eisenhut, A. Altmann, U. Haberkorn and W. Mier, Mol. Pharmaceutics, 2010, 7, 2224–2231 CrossRef CAS PubMed.
- W. B. Kauffman, S. Guha and W. C. Wimley, Nat. Commun., 2018, 9, 2568 CrossRef PubMed.
- H. Ding, P. R. Gangalum, A. Galstyan, I. Fox, R. Patil, P. Hubbard, R. Murali, J. Y. Ljubimova and E. Holler, Nanomedicine, 2017, 13, 631–639 CrossRef CAS PubMed.
- Q. Mu, F. M. Kievit, R. J. Kant, G. Lin, M. Jeon and M. Zhang, Nanoscale, 2015, 7, 18010–18014 RSC.
- E. L. Snyder, C. C. Saenz, C. Denicourt, B. R. Meade, X.-S. Cui, I. M. Kaplan and S. F. Dowdy, Cancer Res., 2005, 65, 10646–10650 CrossRef CAS PubMed.
- Y. Wang, Y. Xie and D. Oupický, Curr. Pharmacol. Rep., 2016, 2, 1–10 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, U. Langel, S. Futaki and A. Gräslund, J. Biophys., 2011, 2011, 414729 Search PubMed.
- C.-J. Chen, K.-C. Tsai, P.-H. Kuo, P.-L. Chang, W.-C. Wang, Y.-J. Chuang and M. D.-T. Chang, BioMed Res. Int., 2015, 237969 Search PubMed.
- G. Ter-Avetisyan, G. Tünnemann, D. Nowak, M. Nitschke, A. Herrmann, M. Drab and M. C. Cardoso, J. Biol. Chem., 2009, 284, 3370–3378 CrossRef CAS.
- H. Y. Kim, S. Y. Yum, G. Jang and D.-R. Ahn, Sci. Rep., 2015, 5, 11719 CrossRef CAS PubMed.
- M. Weller, W. Wick, K. Aldape, M. Brada, M. Berger, S. M. Pfister, R. Nishikawa, M. Rosenthal, P. Y. Wen, R. Stupp and G. Reifenberger, Nat. Rev. Dis. Primers, 2015, 1, 15017 CrossRef.
- R. M. deSouza, H. Shaweis, C. Han, V. Sivasubramaniam, L. Brazil, R. Beaney, G. Sadler, S. Al-Sarraj, T. Hampton, J. Logan, V. Hurwitz, R. Bhangoo, R. Gullan and K. Ashkan, Br. J. Cancer, 2016, 114, 146–150 CrossRef CAS PubMed.
- K. Aldape, K. M. Brindle, L. Chesler, R. Chopra, A. Gajjar, M. R. Gilbert, N. Gottardo, D. H. Gutmann, D. Hargrave, E. C. Holland, D. T. W. Jones, J. A. Joyce, P. Kearns, M. W. Kieran, I. K. Mellinghoff, M. Merchant, S. M. Pfister, S. M. Pollard, V. Ramaswamy, J. N. Rich, G. W. Robinson, D. H. Rowitch, J. H. Sampson, M. D. Taylor, P. Workman and R. J. Gilbertson, Nat. Rev. Clin. Oncol., 2019, 16, 509–520 CrossRef CAS.
- A. Shergalis, A. Bankhead, U. Luesakul, N. Muangsin and N. Neamati, Pharmacol. Rev., 2018, 70, 412–445 CrossRef CAS.
-
T. Walbert and K. Chasteen, in Current Understanding and Treatment of Gliomas, ed. J. Raizer and A. Parsa, Springer International Publishing, Cham, 2015, pp. 171–184 DOI:10.1007/978-3-319-12048-5_11.
- J.-H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS.
- D. Kim, J. Kang, T. Wang, H. G. Ryu, J. M. Zuidema, J. Joo, M. Kim, Y. Huh, J. Jung, K. H. Ahn, K. H. Kim and M. J. Sailor, Adv. Mater., 2017, 29, 1703309 CrossRef.
- L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326 CrossRef PubMed.
- B. Kim, S. Sun, J. A. Varner, S. B. Howell, E. Ruoslahti and M. J. Sailor, Adv. Mater., 2019, 31, 1902952 CrossRef PubMed.
- E. J. Kwon, M. Skalak, A. Bertucci, G. Braun, F. Ricci, E. Ruoslahti, M. J. Sailor and S. N. Bhatia, Adv. Mater., 2017, 29, 1701527 CrossRef.
-
H. A. Santos, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2016, pp. 1–15 DOI:10.1007/978-3-319-04508-5_128-1.
-
J. Salonen, in Handbook of Porous Silicon, ed. L. Canham, Springer International Publishing, Cham, 2017, pp. 1–14 DOI:10.1007/978-3-319-04508-5_91-2.
- Y. Jin, D. Kim, H. Roh, S. Kim, S. Hussain, J. Kang, C.-G. Pack, J. K. Kim, S.-J. Myung, E. Ruoslahti, M. J. Sailor, S. C. Kim and J. Joo, Adv. Mater., 2018, 30, 1802878 CrossRef.
- C.-F. Wang, M. P. Sarparanta, E. M. Mäkilä, M. L. K. Hyvönen, P. M. Laakkonen, J. J. Salonen, J. T. Hirvonen, A. J. Airaksinen and H. A. Santos, Biomaterials, 2015, 48, 108–118 CrossRef CAS.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z.-G. She, D. Kim, A. P. Mann, T. Mölder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef CAS PubMed.
- J. Liu, F. Erogbogbo, K.-T. Yong, L. Ye, J. Liu, R. Hu, H. Chen, Y. Hu, Y. Yang and J. Yang, ACS Nano, 2013, 7, 7303–7310 CrossRef CAS PubMed.
- D. Warther, Y. Xiao, F. Li, Y. Wang, K. Huffman, W. R. Freeman, M. Sailor and L. Cheng, Drug Delivery, 2018, 25, 1537–1545 CrossRef CAS PubMed.
- B. R. Liu, Y.-W. Huang, R. S. Aronstam and H.-J. Lee, PLoS One, 2016, 11, e0150439 CrossRef PubMed.
- H. Räägel, P. Säälik and M. Pooga, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 2240–2248 CrossRef PubMed.
- C. Palm-Apergi, P. Lönn and S. F. Dowdy, Mol. Ther., 2012, 20, 695–697 CrossRef CAS.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Cold Spring Harbor Perspect. Biol., 2011, 3, a004952 Search PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- S. Guo, X. Zhang, M. Zheng, X. Zhang, C. Min, Z. Wang, S. H. Cheon, M.-H. Oak, S.-Y. Nah and K.-M. Kim, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 2101–2110 CrossRef CAS PubMed.
- S. Kälin, B. Amstutz, M. Gastaldelli, N. Wolfrum, K. Boucke, M. Havenga, F. DiGennaro, N. Liska, S. Hemmi and U. F. Greber, J. Virol., 2010, 84, 5336–5350 CrossRef PubMed.
-
T. Kirchhausen, E. Macia and H. E. Pelish, Methods in Enzymology, Academic Press, 2008, vol. 438, pp. 77–93 Search PubMed.
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef PubMed.
- M. Ramesh, P. Ahlawat and N. R. Srinivas, Biomed. Chromatogr., 2010, 24, 104–123 CrossRef PubMed.
- Z. Qin, J. Joo, L. Gu and M. J. Sailor, Part. Part. Syst. Charact., 2014, 31, 252–256 CrossRef CAS.
- J. Kang, J. Joo, E. J. Kwon, M. Skalak, S. Hussain, Z.-G. She, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2016, 28, 7962–7969 CrossRef CAS PubMed.
-
K. Franklin and G. Paxinos, Paxinos and Franklin's The mouse brain in stereotaxic coordinates, Elsevier, 2013 Search PubMed.
- D. M. Lussier, E. C. Woolf, J. L. Johnson, K. S. Brooks, J. N. Blattman and A. C. Scheck, BMC Cancer, 2016, 16, 310 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00077a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Dae-Ro
Ahn
b,
Dae-Ro
Ahn