
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two types of two-step mechanochromic luminescence of phenanthroimidazolylbenzothiadiazoles†
Shohei
Takahashi
,
Sayaka
Nagai
,
Masatoshi
Asami
and
Suguru
Ito
 *
*
Department of Chemistry and Life Science, Graduate School of Engineering Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: suguru-ito@ynu.ac.jp
First published on 1st June 2020
Abstract
The reversible color change of the solid-state emission upon exposure to mechanical stimuli is called mechanochromic luminescence (MCL). In spite of the recent growing interest in MCL materials, only a limited number of organic crystalline materials exhibit two-step MCL that responds to the mechanical stimuli of different intensities. Herein, we report two types of two-step MCL as well as bicolor MCL of phenanthroimidazolylbenzothiadiazole derivatives that bear a substituted phenyl group on the nitrogen atom of the phenanthroimidazole ring. One-way type two-step MCL was observed for a p-bromo-substituted derivative, whereas an o-bromo-substituted derivative displayed a rare back-and-forth type two-step MCL. The other derivatives including the m-bromo-substituted compound showed typical bicolor MCL. Specifically, three types of MCL behaviors are exhibited by changing the position of the bromo group. Single-crystal X-ray diffraction analyses revealed that the crystal system is altered by introducing different substituents. The mode of intermolecular interactions differs depending on the crystal system, which should account for the difference in emission wavelength and MCL properties of the crystalline samples. Based on powder X-ray diffraction and differential scanning calorimetry measurements, one-way type MCL can be rationalized in terms of the crystal-to-crystal transitions followed by crystal-to-amorphous transitions. The mechanism of the back-and-forth type MCL is proposed as the destruction of extended intermolecular interactions upon crushing the crystals followed by amorphization upon strong grinding. These results provide useful insights into the future development of multi-step MCL systems with potential applications in advanced mechano-sensing technologies.
Introduction
Stimuli-responsive luminescent materials1 that can switch the emission properties upon exposure to external stimuli such as light,2 heat,3 mechanical stress,4 and chemical vapor5 have attracted considerable interest because of their potential applications in sensors, security devices, data storage, etc. Mechanochromic luminescence (MCL) is a reversible emission-color change of solid-state materials in response to mechanical stimuli such as pressing, grinding, and scratching.1d,e,4,6 An increasing number of MCL materials have been reported owing to the wealth of their appealing potential as mechanosensors, rewritable memory devices, anti-counterfeiting inks, etc. Typical MCL materials exhibit bathochromically shifted bicolor MCL, whereby the emission wavelength switches in the bathochromic direction upon grinding (Fig. 1a). The mechanism for the bicolor MCL of organic crystalline dyes is usually based on phase transitions between crystalline and amorphous states.7 In some cases, the emission-color change is attributed to crystal-to-crystal phase transitions induced by mechanical stimuli.8 A significant challenge in the development of MCL materials is to achieve two-step MCL that can switch the emission color in a stepwise fashion between three states in response to mechanical stimuli of different intensities.8a,d,9 Two-step MCL materials are classified into one-way type and back-and-forth type. One-way type MCL dyes switch the emission color in one direction in response to crushing with low intensity followed by strong grinding (Fig. 1b). In the case of back-and-forth type MCL, the emission wavelength of the dye that shifted in one direction returns to the opposite direction upon increasing the intensity of mechanical stimuli (Fig. 1c). Most of the two-step MCL materials are one-way type,8a,d,9a,c–h and little is known about the back-and-forth type MCL.9b It remains a critical challenge to realize both one-way type and back-and-forth type MCL by organic crystalline dyes based on the same molecular scaffold. The development of such dyes should help to establish rational design guidelines for the organic crystalline dyes with two-step MCL properties.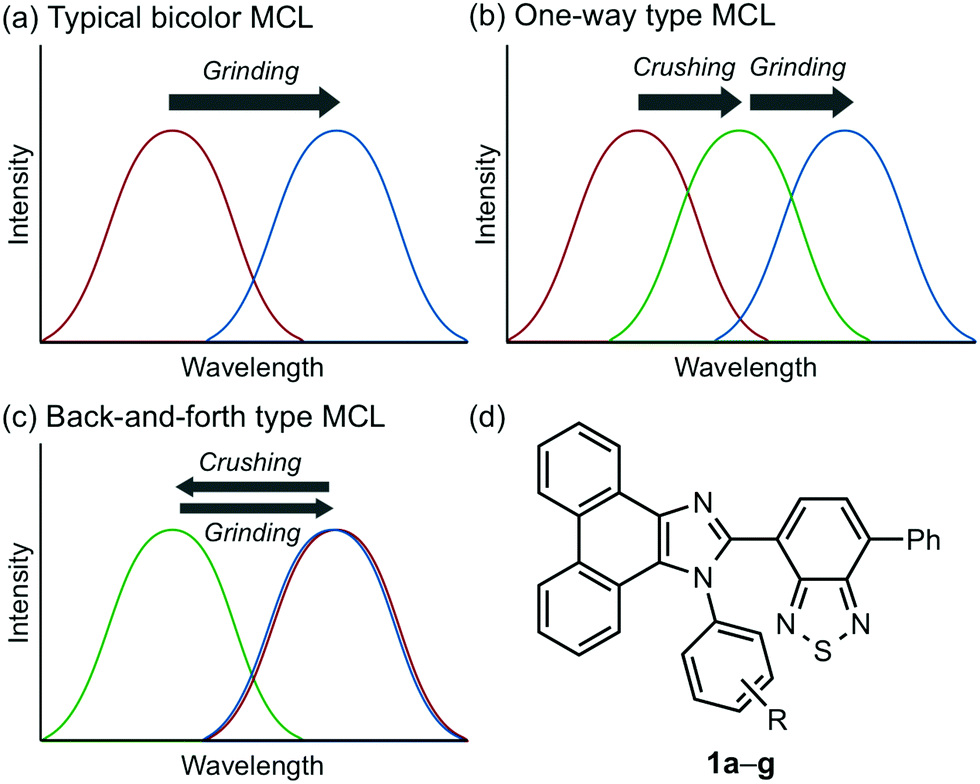 | ||
| Fig. 1 (a)–(c) Schematic representation of the changes in emission spectra in response to mechanical stimuli: crushing (weak intensity) and grinding (strong intensity). (a) Typical bathochromically shifted bicolor MCL. (b) One-way type two-step MCL. (c) Back-and-forth type two-step MCL. (d) Structural formula of phenanthroimidazolylbenzothiadiazoles 1a–g. | ||
We recently reported a series of crystalline organic MCL dyes composed of an electron-donating heteroaromatic ring and an electron-accepting benzothiadiazole ring.10 Among these, phenanthroimidazolylbenzothiadiazoles with different substituents on the phenyl group attached to the benzothiadiazole ring exhibited versatile MCL behaviors, including tricolor MCL as well as bathochromically and hypsochromically shifted bicolor MCL.10b Herein, we have synthesized a series of phenanthroimidazolylbenzothiadiazole derivatives 1a–g that bear different R substituents on the phenyl group at the nitrogen atom of the phenanthroimidazole ring (Fig. 1d). In contrast to our previous report, two types of two-step MCL based on different mechanisms have been achieved. Although p-substituted derivatives 1a–d (R = p-Me, p-OMe, p-F, and p-CF3) exhibit typical bathochromically shifted bicolor MCL, p-bromo-substituted 1e shows one-way type two-step MCL. Remarkably, the MCL behavior has been controlled by changing the position of the bromo group. Typical bicolor MCL is observed for m-bromo-substituted 1f, whereas o-bromo-substituted 1g exhibits a rare back-and-forth type two-step MCL. We propose that the formation of extended intermolecular interactions between electron-donating phenanthroimidazole and electron-accepting benzothiadiazole rings in the crystalline state should account for the mechanism of the back-and-forth type MCL of 1g.
Results and discussion
In accordance with a previously reported procedure with modifications,10b a mixture of 9,10-phenanthrenequinone, ammonium acetate (5.0 equiv.), 4-methylaniline (1.5 equiv.) and 4-formyl-7-phenyl-2,1,3-benzothiadiazole (1.0 equiv.) was refluxed in toluene for 8 h in the presence of anhydrous sodium sulfate (2.8 equiv.) to afford phenanthroimidazolylbenzothiadiazole derivative 1a (R = p-Me) in 57% yield (Scheme 1). In a similar manner, other derivatives 1b–d having electron-donating (1b: R = p-OMe) or electron-withdrawing (1c: R = p-F; 1d: R = p-CF3; 1e: R = p-Br) substituents on the p-position of the N-phenyl group were synthesized in 45–72% yields. Among these, 1d was synthesised in refluxing acetic acid to improve the reactivity of electron-deficient 4-trifluoromethylaniline. As described later in this paper, only 1e (R = p-Br) exhibited two-step MCL. Accordingly, m-bromo-substituted 1f (R = m-Br) and o-bromo-substituted 1g (R = o-Br) were also synthesized in refluxing acetic acid in 65% and 18% yield, respectively. The relatively low yield of 1g should be explained by the steric hindrance of the bromo group on the o-position of the N-phenyl group.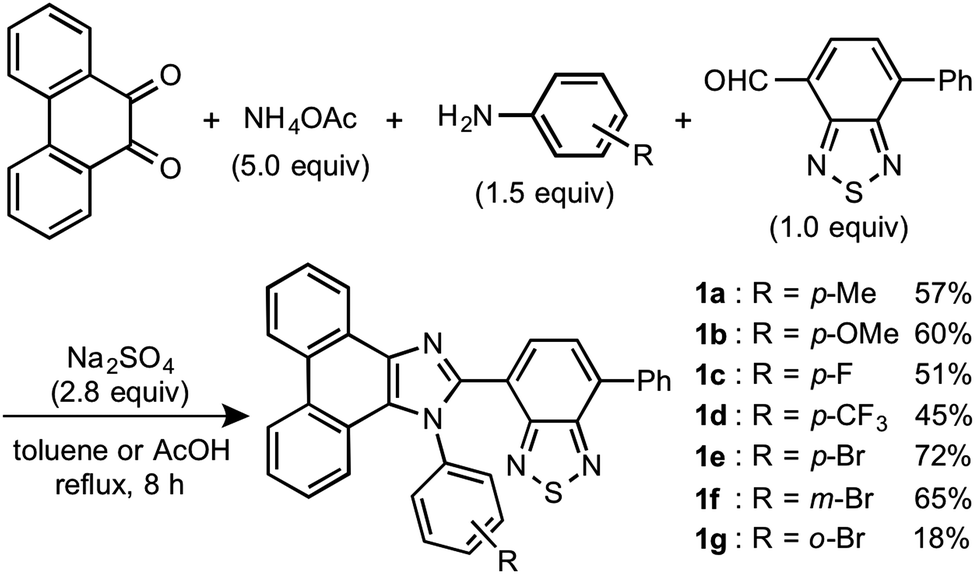 | ||
| Scheme 1 Synthesis of phenanthroimidazolylbenzothiadiazoles 1a–g. | ||
The absorption and emission properties of 1a–g were measured for their toluene solutions (1.0 × 10−5 M) (Fig. 2a and Fig. S1, ESI†). The maximum absorption band (λabs) of 1a (R = p-Me) in toluene was observed at 413 nm. Upon irradiation with UV light (365 nm), the toluene solution of 1a exhibited yellow-green emission (λem = 539 nm) with high fluorescence quantum yield (ΦF = 0.77) and a large Stokes shift of 126 nm. For the other derivatives 1b–g, the maximum absorption and emission bands were observed in the same regions as those of 1a (1b–g: λabs = 409–415 nm; λem = 538–543 nm). Accordingly, the electronic effect of the R substituent on the N-phenyl group should have negligible influence on the absorption and emission properties of 1a–g in solution. The emission properties of the crystalline samples of 1a–g, prepared by recrystallization from toluene, are summarized in Fig. 2b and Fig. S2 in the ESI.† Crystalline 1a showed green emission (λem = 523 nm) with high fluorescence quantum yield (ΦF = 0.70). The emission bands of crystalline 1b–g were observed over a wide range from blue to yellow depending on the R substituent (1b: λem = 490 nm, ΦF = 0.46; 1c: λem = 519 nm, ΦF = 0.55; 1d: λem = 498 nm, ΦF = 0.39; 1e: λem = 495 nm, ΦF = 0.56; 1f: λem = 503 nm, ΦF = 0.54; and 1g: λem = 564 nm, ΦF = 0.40). Compared to the maximum emission bands in toluene, the emission maxima of 1a–f hypsochromically shifted by 16–52 nm in the crystalline state. In contrast, the maximum emission wavelength of 1g in the crystalline state shifted by 26 nm in the bathochromic direction compared to that of its toluene solution. Since there were no significant changes in the emission wavelengths in toluene regardless of the differences in the electronic properties of the R substituent, the observation of the emission wavelength over a wide range in the crystalline state should be attributed to the effect of the packing structures of crystalline 1a–g.
 | ||
| Fig. 2 Photophysical properties of phenanthroimidazolyl-benzothiadiazoles 1a–g in toluene (a) and in the crystalline powdered form (b). λabs: maximum absorption bands (λabs > 350 nm); λem: maximum emission bands excited with UV light (365 nm); ΦF: absolute quantum yields measured using an integrating sphere. | ||
Single crystals of 1a–g except 1c and 1f could be obtained from vapor diffusion of hexane into a chloroform solution of 1a–g. Single-crystal X-ray diffraction analyses revealed their molecular structures in the crystalline state (Fig. S3–S7, ESI†). Calculated powder X-ray diffraction patterns of the single crystals of 1a, 1b, 1d, 1e, and 1g are consistent with those of their powdered samples prepared by recrystallization from toluene (Fig. S8, ESI†). Among these, the molecular conformation of 1d (R = p-CF3) is in good agreement with that of 1e (R = p-Br) owing to the similarity in their packing structures (Fig. S9c and d, ESI†). The other derivatives 1a (R = p-Me), 1b (R = p-OMe), and 1g (R = o-Br) showed different molecular conformations depending on the difference in their packing structures (Fig. 3 and Fig. S9a, b and e, ESI†). To reveal the mechanism that causes the difference in the emission wavelength of the crystalline samples, time-dependent density functional theory (TD-DFT) calculations of 1a, 1b, 1d, 1e, and 1g were carried out at the CAM-B3LYP/6-31G(d) level of theory using the molecular geometries obtained from the single-crystal X-ray diffraction analyses (Table 1 and Table S1, ESI†). The CAM-B3LYP approach was applied in this study as this method often provides better results in the TD-DFT calculations of charge-transfer systems than the typical B3LYP approach (Table S2, ESI†).11 The HOMO of 1a is mainly located on the phenanthroimidazole ring and some portion of the HOMO spreads to the benzothiadiazole ring (Fig. 4a). Meanwhile, the LUMO of 1a is localized on the benzothiadiazole ring. The calculated absorption wavelength of the isolated molecule of 1a is 363 nm with an oscillator strength (f) of 0.398, which is assignable to the intramolecular charge transfer from the HOMO to the LUMO. The HOMO and LUMO of 1b are distributed on similar locations to those of 1a, and the calculated absorption wavelength of 1b corresponding to the electronic transition from the HOMO to the LUMO is 352 nm. However, the oscillator strength of this transition is 0.037, which should be ascribed to the almost orthogonal dihedral angle (ϕ = 84.33°) between the phenanthroimidazole and benzothiadiazole rings (Fig. 3b). When the dihedral angle between the electron-donating and electron-accepting moieties of a D–A-type molecule is orthogonal, the intramolecular charge transfer should be prohibited due to the isolation of their orbitals.12 Therefore, the absorption of 1b should mainly be attributed to the local excitation from HOMO−2 to the LUMO at the phenylbenzothiadiazole ring (λabs,calcd = 331 nm, f = 0.317; Fig. 4b). The dihedral angles ϕ of 1d, 1e, and 1g are 64.92°, 65.01°, and 74.64°, respectively. Based on the calculated absorption wavelengths (1d: λabs,calcd = 360 nm, f = 0.377; 1e: λabs,calcd = 355 nm, f = 0.315; and 1g: λabs,calcd = 350 nm, f = 0.188), the absorption of 1d, 1e, and 1g should mainly originate from the HOMO → LUMO transitions as is described in the case of 1a (Fig. S10, ESI†). The optimized molecular structures of 1a, 1b, 1d, 1e, and 1g were also calculated at the CAM-B3LYP/6-31G(d) level of theory using their X-ray molecular structures as starting points. The optimized structures except for 1g that bears a sterically hindered o-bromo group exhibited similar dihedral angles between adjacent (hetero)aromatic groups (Fig. S11, ESI†). The calculated absorption wavelengths for the HOMO → LUMO transitions of these derivatives including 1g are in a narrower range (λabs,calcd = 367–374 nm, Table S3, ESI†) compared to those of the isolated molecules in crystalline states. These results indicate that the electronic effect of substituents on the N-phenyl group has negligible influence on the absorption wavelengths of these molecules, whereas the solid-state photophysical properties should be significantly affected by the intermolecular interactions in the crystalline state.
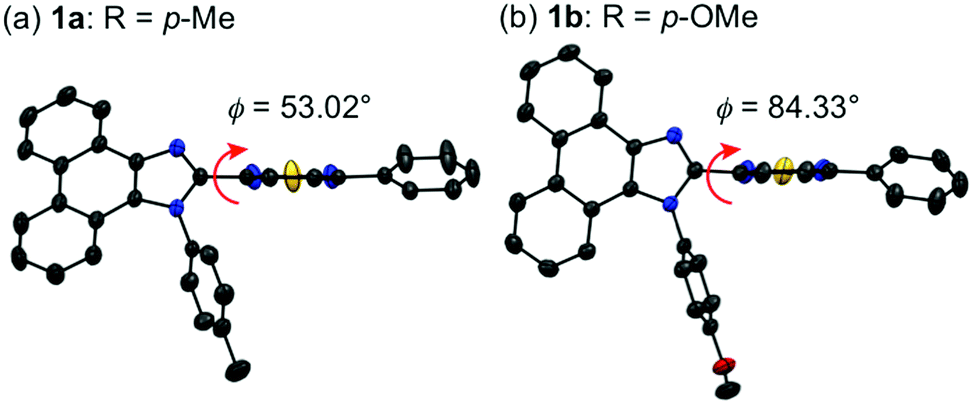 | ||
| Fig. 3 Molecular structures of 1a (a) and 1b (b) with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. Color code: gray = C; blue = N; red = O; and yellow = S. | ||
| Compd | Transition | λ abs,calcd (nm) | Oscillator strength |
|---|---|---|---|
| a Calculated at the CAM-B3LYP/6-31G(d) level of theory. | |||
| 1a | HOMO → LUMO | 363 | 0.398 |
| 1b | HOMO → LUMO | 352 | 0.037 |
| HOMO−2 → LUMO | 331 | 0.317 | |
| 1d | HOMO → LUMO | 360 | 0.377 |
| 1e | HOMO → LUMO | 355 | 0.315 |
| 1g | HOMO → LUMO | 350 | 0.188 |
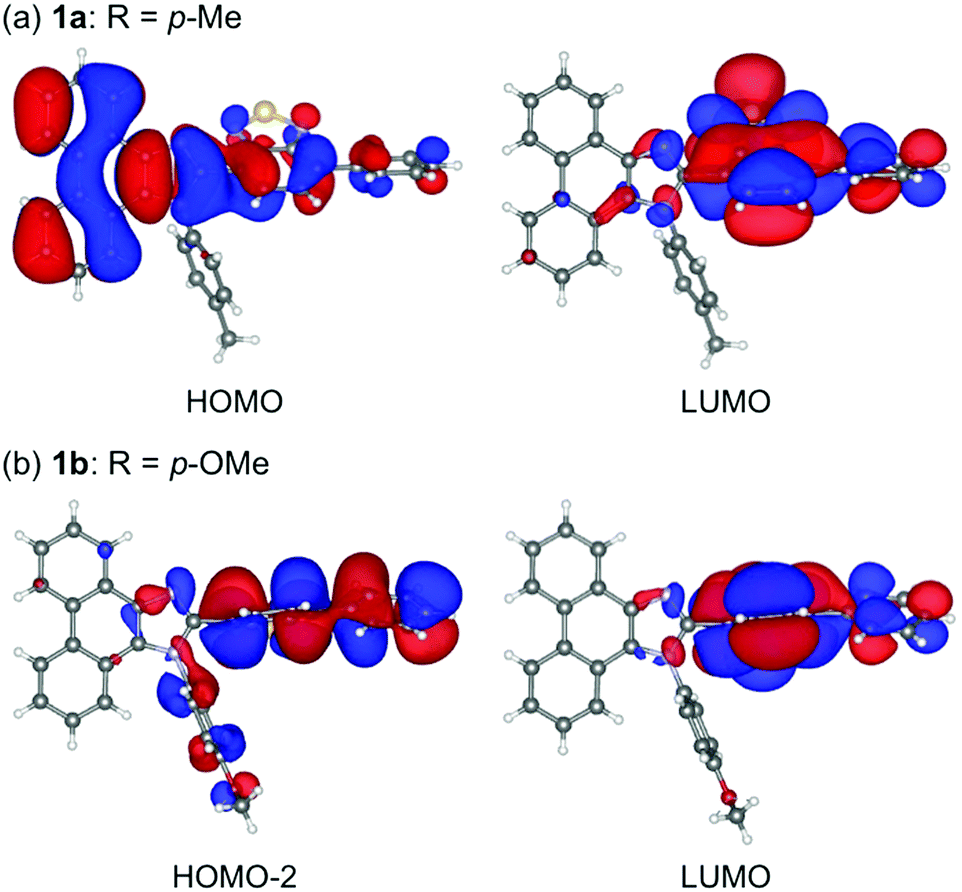 | ||
| Fig. 4 HOMO and LUMO of 1a (a) and HOMO−2 and LUMO of 1b (b) calculated at the CAM-B3LYP/6-31G(d) level.13 | ||
The absorption spectra of 1a, 1b, 1d, 1e and 1g in the crystalline state were obtained from the measurement of diffuse reflectance spectra. The absorption bands of 1a and 1b were observed at the longest- and shortest-wavelength regions, respectively (Fig. S13, ESI†). The order of the experimental absorption wavelengths is in good agreement with that of the calculated absorption bands since the major calculated absorption wavelengths of 1a and 1b exhibit the longest and shortest values, respectively (1a: λabs,calcd = 363 nm and 1b: λabs,calcd = 331 nm). On the other hand, the order of emission wavelengths in the crystalline state is inconsistent with that of absorption wavelengths. The o-bromo-substituted derivative 1g exhibited the longest emission wavelength in the crystalline state (λem = 564 nm).
The difference in the orders of the absorption and emission wavelengths of 1a–g should be explained in terms of the stabilizing effect of the excited state by intermolecular interactions in the crystal structures. The effect of intermolecular interactions with adjacent molecules on the maximum emission wavelengths is considered by focusing on the packing structures obtained from the single crystal X-ray structure analyses. In the crystal structure of 1a (monoclinic, C2/c), two adjacent molecules are stacked at the phenanthroimidazole moieties (Fig. 5a). The distance between the plane of one phenanthroimidazole ring and the center of the other phenanthroimidazole ring is 3.696 Å, which is in a typical π-stacking distance.14 Two molecules of 1b in the crystal structure (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) are stacked alternately in opposite directions at the phenylbenzothiadiazole moieties (Fig. 5b). The distance between the benzothiadiazole rings is 3.965 Å. Similarly, two adjacent molecules of 1d (triclinic, P) form antiparallel stacks at the phenylbenzothiadiazole moieties (Fig. 5c). Although a molecule of 1b is stacked from the other side of the N-phenyl group of another molecule, a molecule of 1d is located in the same side of the N-phenyl group of another molecule. The distance between the benzothiadiazole rings of stacked 1d is 3.672 Å, which is closer than that observed for 1b. The packing structure of 1e (triclinic, P) is identical to that of 1d, and intermolecular stacks of 1e are formed at the phenylbenzothiadiazole moieties (Fig. 5e).
) are stacked alternately in opposite directions at the phenylbenzothiadiazole moieties (Fig. 5b). The distance between the benzothiadiazole rings is 3.965 Å. Similarly, two adjacent molecules of 1d (triclinic, P) form antiparallel stacks at the phenylbenzothiadiazole moieties (Fig. 5c). Although a molecule of 1b is stacked from the other side of the N-phenyl group of another molecule, a molecule of 1d is located in the same side of the N-phenyl group of another molecule. The distance between the benzothiadiazole rings of stacked 1d is 3.672 Å, which is closer than that observed for 1b. The packing structure of 1e (triclinic, P) is identical to that of 1d, and intermolecular stacks of 1e are formed at the phenylbenzothiadiazole moieties (Fig. 5e).
 | ||
| Fig. 5 Molecular structures with an adjacent molecule of 1a, 1b, 1d, 1e and 1g with thermal ellipsoids at 50% probability. All hydrogen atoms are omitted for clarity. Color code: gray = C; blue = N; red = O; green = F; yellow = S; and orange = Br. | ||
Accordingly, the distance between the benzothiadiazole rings of 1e (3.678 Å) is almost the same as that of 1d. In the case of 1g (monoclinic, P21/n), the electron-rich phenanthroimidazole ring of a molecule is in close proximity to the electron-deficient benzothiadiazole ring of another molecule (Fig. 5d). The distance between the plane of the phenanthroimidazole ring and the center of the benzothiadiazole ring is 3.388 Å. Moreover, extended intermolecular interactions are observed between the donor and acceptor moieties of adjacent 1g molecules as shown in Fig. 6.
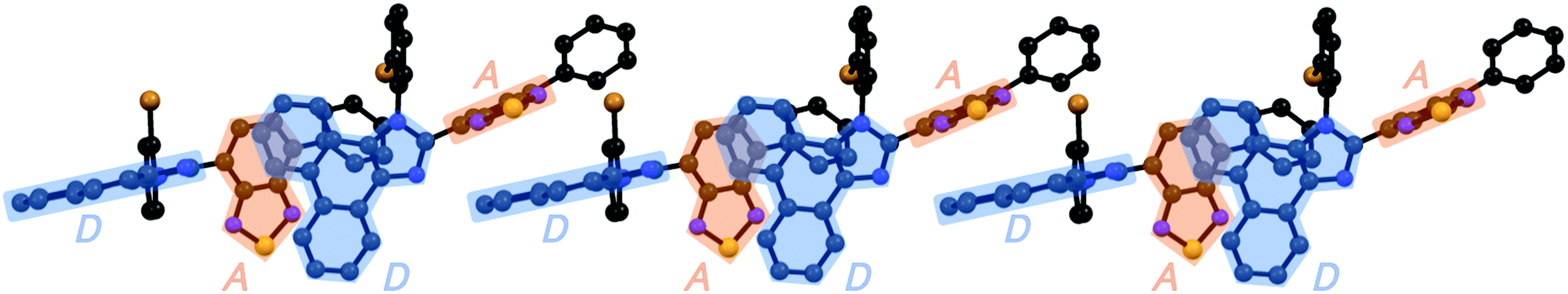 | ||
| Fig. 6 The extended intermolecular interactions in the crystalline state of 1g. All hydrogen atoms are omitted for clarity. Electron-donating phenanthroimidazole ring (D) and electron-accepting benzothiadiazole ring (A) are highlighted in blue and orange, respectively. Color code: gray = C; blue = N; yellow = S; and orange = Br. | ||
The absorption properties of the stacked dimers of 1a, 1b, 1d, 1e, and 1g observed in the X-ray diffraction analyses were calculated by the TD-DFT method at the CAM-B3LYP/6-31G(d) level of theory (Table 2 and Table S4, Fig. S12, ESI†). The calculated absorption wavelengths of the stacked 1a are 366 nm (f = 0.432) and 362 nm (f = 0.564). These transitions are mainly attributed to the HOMO → LUMO and HOMO → LUMO+1 transitions, respectively. The HOMOs of the stacked 1a are mainly localized on the phenanthroimidazole rings of the two molecules, whereas LUMO and LUMO+1 are localized on the benzothiadiazole rings of the two molecules. As almost no intermolecular overlap of molecular orbitals is observed in the stacked 1a and the calculated absorption wavelength of stacked 1a is similar to that of isolated 1a, the transitions of the stacked dimer should be attributed to the independent intramolecular charge-transfer transitions of each molecule. In the case of 1b, the absorption of the stacked dimer is attributed to the HOMO−1 to LUMO transition (λabs,calcd = 339 nm, f = 0.364). The HOMO−1 and LUMO are located on the phenanthroimidazole and phenylbenzothiadiazole rings of both molecules, respectively. Therefore, this transition should also exhibit intramolecular charge-transfer nature. The stacked 1d and 1e show the calculated absorption wavelengths of 356 nm (f = 0.862) and 350 nm (f = 0.738), respectively, based on the HOMO → LUMO transitions. These transitions should also have intramolecular charge-transfer nature, as the HOMO and LUMO are located on the phenanthroimidazole and phenylbenzothiadiazole rings of two molecules, respectively. Notably, a transition attributed to the intermolecular charge transfer is calculated for the stacked 1g. The calculated HOMO is mainly distributed on the one molecule of dimer 1g. The LUMO is located on the benzothiadiazole ring of the same molecule on which the HOMO is distributed. Meanwhile, LUMO+1 is distributed on the phenylbenzothiadiazole ring of the other molecule. The HOMO → LUMO+1 and HOMO → LUMO transitions are calculated at 357 nm (f = 0.007) and 356 nm (f = 0.218), respectively.
| Compd | Transition | λ abs,calcd (nm) | Oscillator strength |
|---|---|---|---|
| a Calculated at the CAM-B3LYP/6-31G(d) level of theory. | |||
| 1a | HOMO → LUMO | 366 | 0.432 |
| HOMO → LUMO+1 | 362 | 0.564 | |
| 1b | HOMO−1 → LUMO | 339 | 0.364 |
| 1d | HOMO → LUMO | 356 | 0.862 |
| 1e | HOMO → LUMO | 350 | 0.738 |
| 1g | HOMO → LUMO+1 | 357 | 0.007 |
| HOMO → LUMO | 356 | 0.218 | |
| HOMO−1 → LUMO+1 | 332 | 0.192 | |
Considering the effect of adjacent molecules for stabilizing the excited state of a molecule in the crystalline structure, an excited molecule of 1a should be more stabilized than those of 1b, 1d, and 1e. When a molecule of 1a in the crystalline state is excited based on an intramolecular charge transfer from the phenanthroimidazole ring to the benzothiadiazole ring, the positively charged phenanthroimidazole ring of the excited molecule of 1a should be stabilized by the adjacent electron-rich phenanthroimidazole ring of another molecule. Since the distance between adjacent molecules is relatively large (3.965 Å) and the twisted phenyl groups are stacked over the benzothiadiazole rings, a locally excited state of a molecule of 1b should not be efficiently stabilized by another molecule. In the cases of 1d and 1e, the negatively charged benzothiadiazole ring of an excited molecule is only partially overlapped with the phenyl group of another molecule, which would have little influence on the stabilization of the excited molecule. Therefore, it is logical that the maximum emission wavelength of crystalline 1a (523 nm) is longer than those of crystalline 1b (490 nm), 1d (498 nm), and 1e (495 nm). An excited molecule of 1g should be greatly stabilized by the extended intermolecular interactions in the crystalline state (Fig. 6), which should account for the longest emission wavelength (564 nm) of crystalline 1g. As described above, intermolecular interactions as well as molecular conformations should contribute to the determination of the emission wavelength of crystalline 1a–g.
With highly solid-state emissive crystalline samples of 1a–g in hand, their mechanoresponsive properties were subsequently evaluated by grinding with a spatula (Fig. 7). The emission color of crystalline 1a (R = p-Me) changed from green (λem = 523 nm, ΦF = 0.70) to orange (λem = 575 nm, ΦF = 0.76) upon grinding. The original green emission was restored by heating the ground sample to 180 °C. Powder X-ray diffraction (PXRD) analyses of 1a (Fig. 8a) showed that the intensity of the diffraction peaks observed for powdered crystalline 1a significantly decreased upon grinding, which indicates the loss of crystallinity. The intensity of the diffraction peaks was restored after heating the ground sample. In a differential scanning calorimetry (DSC) measurement of ground 1a (Fig. 8b), an exothermic peak that corresponds to the cold-crystallization transition (Tc = 131 °C) was observed followed by an endothermic peak that corresponds to the melting point of crystalline 1a (Tm = 226 °C). The PXRD and DSC analyses indicate that the mechanism for the MCL of 1a should be based on typical crystal-to-amorphous transitions. The bathochromic shift of the emission wavelength upon amorphization would be explained by the increased intermolecular interactions of 1a in the amorphous state. Similarly, 1b–d and 1f showed bathochromically shifted MCL between two colors (Fig. 7 and Fig. S14, ESI†). The emission color of crystalline 1b–d and 1f shifted in the bathochromic direction upon grinding, and the original emission color was recovered upon heating the ground samples. Based on the PXRD and DSC analyses of 1b–d and 1f, the MCL of these derivatives should also be attributed to the crystal-to-amorphous transitions (Fig. S15 and S16, ESI†).
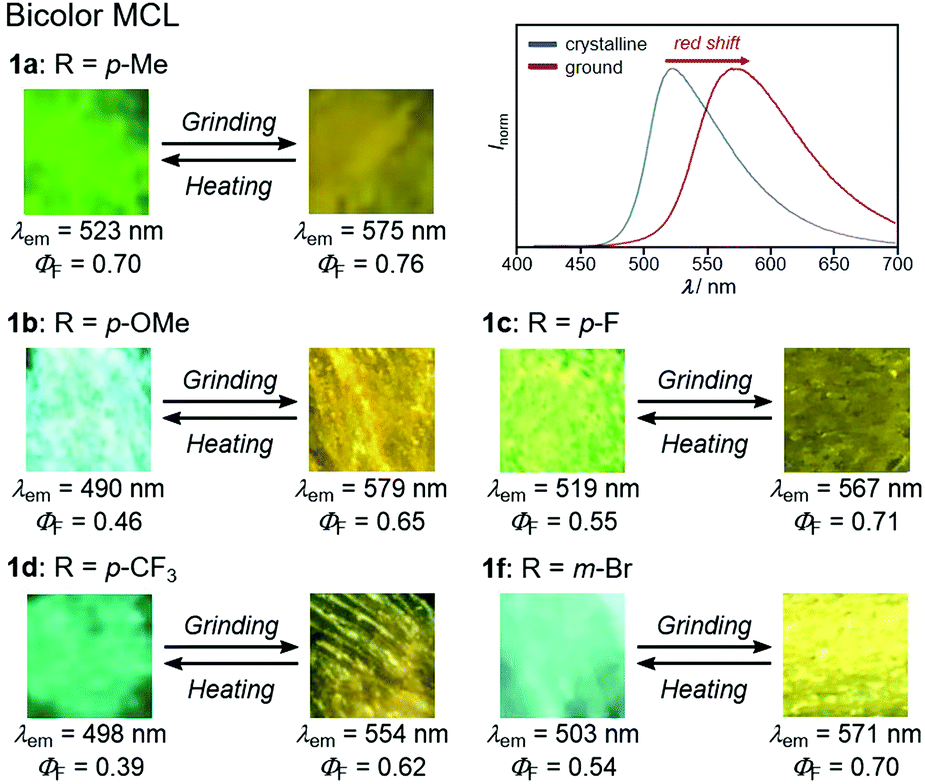 | ||
| Fig. 7 Photographs and fluorescence spectra for the bathochromically shifted bicolor MCL of 1a–d and 1f excited with UV light (365 nm). For the fluorescence spectra for the MCL of 1b–d and 1f, see Fig. S12 (ESI†). | ||
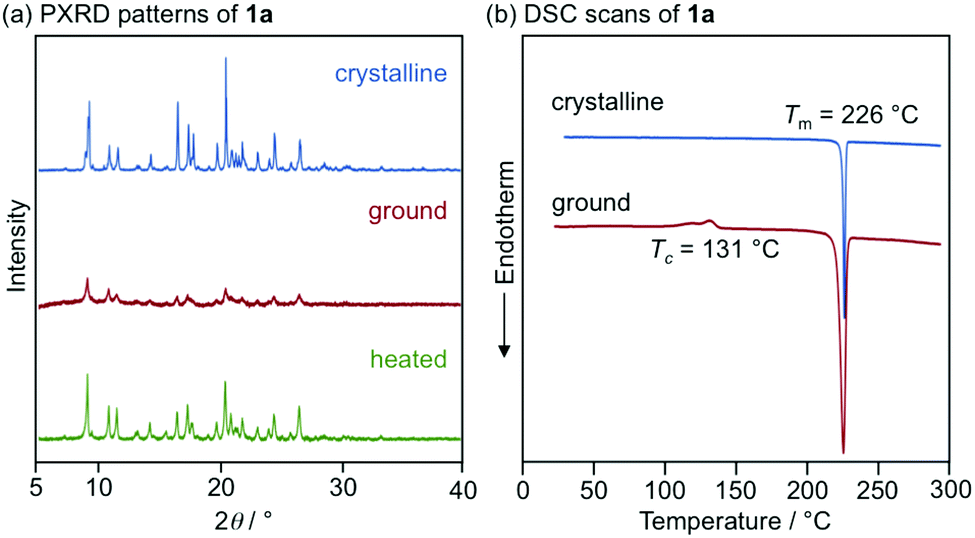 | ||
| Fig. 8 (a) PXRD patterns for the powdered crystalline (blue), ground (red), and heated (green) samples of 1a. (b) DSC scans for crystalline (blue) and ground (red) samples of 1a. Tm and Tc values are noted near the corresponding peaks. | ||
In sharp contrast to the bicolor MCL of 1a–d and 1f, the other derivatives 1e (R = p-Br) and 1g (R = o-Br) exhibited two-step MCL in response to the mechanical stimuli of different intensities (Fig. 9 and 10). The emission color of crystalline 1e bathochromically shifted from blue (λem = 495 nm, ΦF = 0.56) to green (λem = 518 nm, ΦF = 0.29) upon gently crushing into a fine powder by using a spatula (Fig. S17, ESI†). Upon strong grinding, the emission color of the crushed sample further shifted in the bathochromic direction to yellow (λem = 573 nm, ΦF = 0.57). The orange-emissive state recovered to the green-emissive state on heating to 120 °C. Heating the green-emissive state further at 180 °C restored the original blue-emissive state (Fig. 9a and Fig. S18a, ESI†). The PXRD analysis for the two-step MCL of 1e revealed that the diffraction pattern of crystalline 1e changed upon crushing. Upon strong grinding, the diffraction peaks of the crushed sample almost disappeared. When the ground sample was heated at 120 °C, the diffraction patterns were recovered to those of the crushed sample. After heating to a higher temperature (180 °C), the diffraction patterns of the original crystalline state were restored (Fig. 9b). The DSC thermogram of crystalline 1e showed only one endothermic peak that corresponds to the melting point (Tm = 238 °C). In the case of crushed 1e, an exothermic peak was observed at 142 °C. Moreover, two exothermic peaks were observed in the DSC thermogram of ground 1e at 109 °C and 142 °C. The first exothermic peak should correspond to the cold-crystallization transition (Tc = 109 °C) from the yellow-emissive amorphous state to the green-emissive crystalline state, whereas the second exothermic peak should be attributed to the phase transition (Tp = 142 °C) from the green-emissive crystalline state to another crystalline state that exhibits blue emission (Fig. 9c). These results suggest that the blue-emissive crystalline 1e changes to another crystalline state upon crushing along with the bathochromic shift of the emission color to green and the subsequent strong grinding of crushed samples induces the phase transition to the yellow-emissive amorphous state. The decrease in the ΦF value upon crushing (ΦF = 0.29) should potentially be attributed to the low crystallinity15 of the crushed state, which would be supported by the fact that the recovered blue-emissive state with low crystallinity, which was obtained by heating the crushed state, exhibited the decreased ΦF value (ΦF = 0.28) compared to that of the initial blue-emissive state with high crystallinity (ΦF = 0.56). On the other hand, the increase in the ΦF value after grinding (ΦF = 0.57) should be attributed to the suppression of non-radiative decay processes of the excited state.10b The increased intermolecular interactions upon amorphization should restrict molecular motions in the excited state.
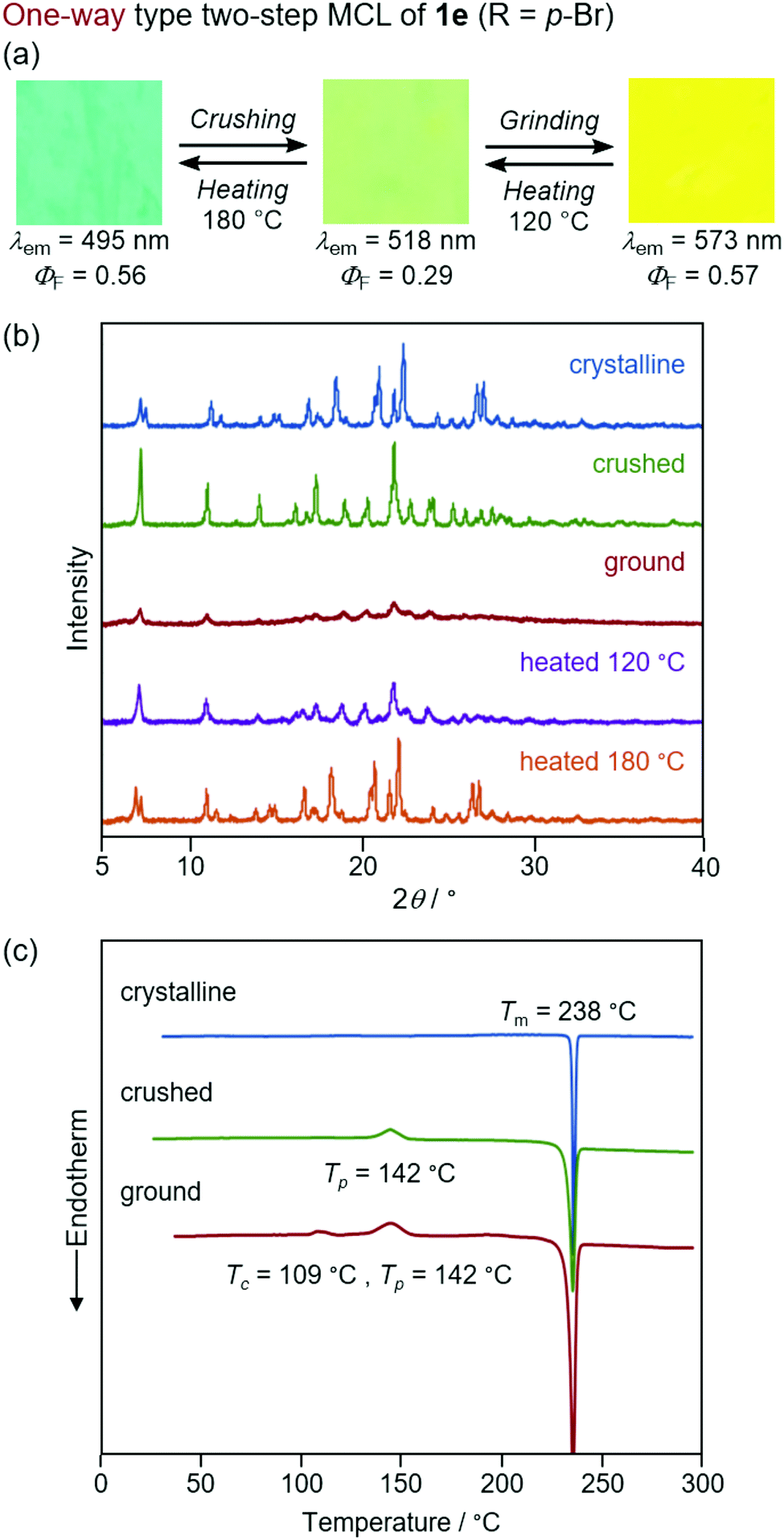 | ||
| Fig. 9 (a) Photographs for the two-step MCL of 1e excited with UV light (365 nm). (b) PXRD patterns for the crystalline (blue), crushed (green), ground (red), and heated (120 °C, purple and 180 °C, orange) samples of 1e. (c) DSC scans for the crystalline (blue), crushed (green), and ground (red) samples of 1e. Tm, Tp, and Tc values are noted near the corresponding peaks. | ||
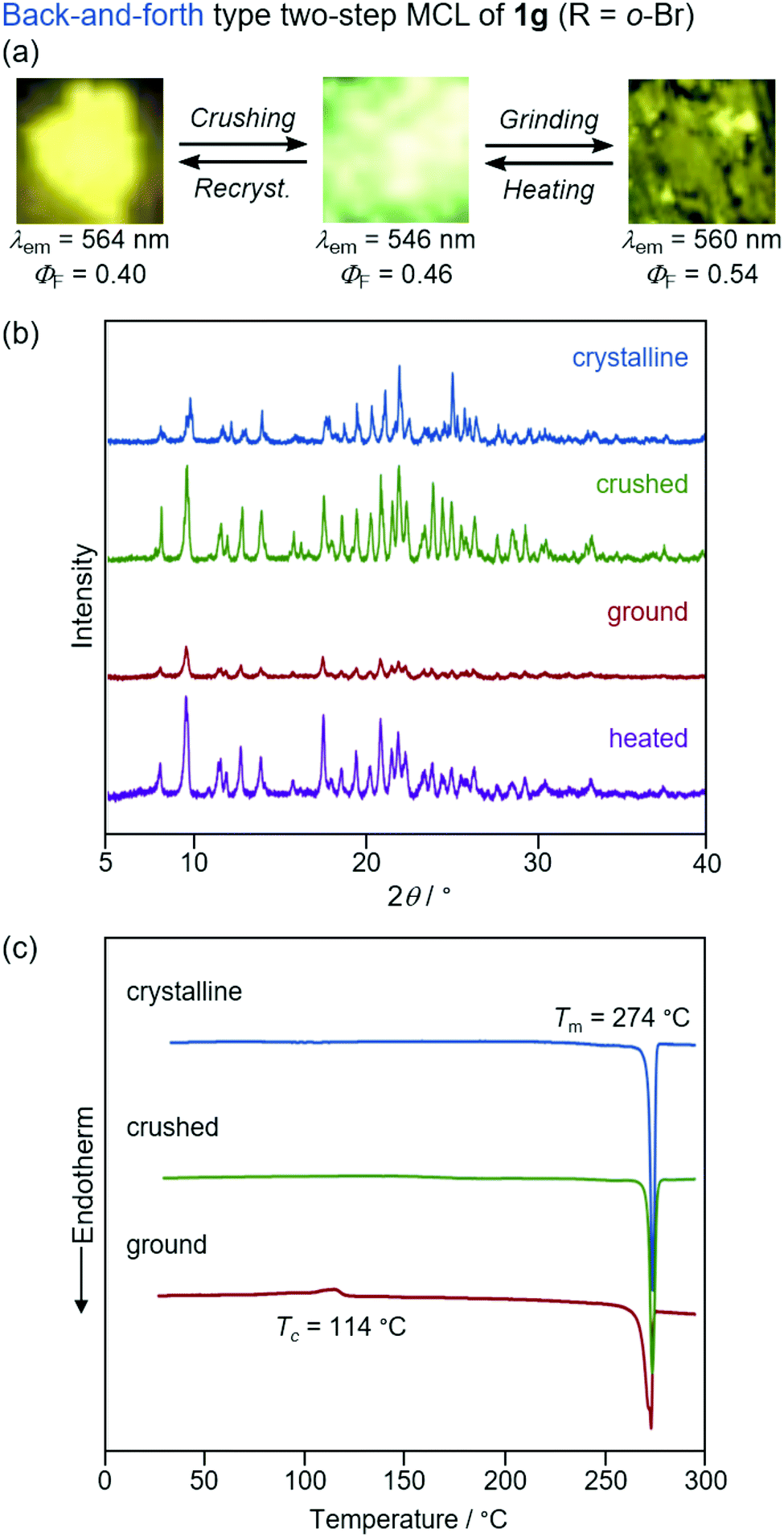 | ||
| Fig. 10 (a) Photographs for the two-step MCL of 1g excited with UV light (365 nm). (b) PXRD patterns for the crystalline (blue), crushed (green), ground (red), and heated (purple) samples of 1g. (c) DSC scans for the crystalline (blue), crushed (green), and ground (red) samples of 1g. Tm and Tc values are noted near the corresponding peaks. | ||
The yellow-emissive crystals of 1g (R = o-Br) exhibited a rare back-and-forth type two-step MCL, whereby the emission color shifts in the hypsochromic direction upon gentle crushing followed by a bathochromic shift upon strong grinding (Fig. 10). Upon crushing crystalline 1g with a spatula, the emission color shifted in the hypsochromic direction from yellow (λem = 564 nm, ΦF = 0.40) to yellow-green (λem = 546 nm, ΦF = 0.46). In response to the strong grinding, the emission color of the crushed sample shifted in the bathochromic direction to yellow (λem = 560 nm, ΦF = 0.54). After heating the ground 1g at 180 °C, the yellow-green-emissive state was recovered. However, the emission color of the yellow-green-emissive state was unchanged even after being treated with elevated temperature below its melting point. Recrystallization from a toluene solution is required to recover the original yellow-emissive crystals (Fig. 10a and Fig. S15b, ESI†). The PXRD analyses of the initial crystalline and the crushed samples of 1g showed that only the intensity ratio of the diffraction pattern changed after crushing without changing the position of the peaks (Fig. 10b). In the DSC diagram of the crushed sample, only one endothermic peak that corresponds to the melting point of the initial yellow-emissive crystals (Tm = 274 °C) was observed (Fig. 10c). This observation supports the lack of thermally induced phase transition from the crushed state to the yellow-emissive crystalline state. The PXRD and DSC analyses of crushed 1g suggest that the crushed samples are in the same crystal system as in the initial yellow-emissive state although the fracture surface of the crushed crystals should have defects. Accordingly, the hypsochromic shift in the emission wavelength upon crushing could potentially be rationalized by the formation of defects in the extended intermolecular interactions (Fig. 6) that should account for the intermolecular charge-transfer transitions and efficient stabilization of the excited state. The excitation spectrum of the yellow-green-emissive crushed state shifted in the hypsochromic direction compared to that of initial yellow-emissive crystals, which supports the significant disappearance of intermolecular charge-transfer transitions in the crushed state (Fig. S19, ESI†). The slight increase in the ΦF value after crushing would also be attributed to the destruction of the extended intermolecular interactions, which should cause non-radiative decays of the excited molecules during intermolecular energy-transfer processes. The PXRD analyses of the ground and the heated samples of 1g revealed that the intensity of the diffraction patterns significantly reduced after grinding and recovered after heating (Fig. 10b). The DSC measurement of ground 1g showed an exothermic peak at 114 °C that corresponds to Tc followed by an endothermic Tm peak at 274 °C (Fig. 10c). Therefore, the MCL between the crushed and the ground states should be based on the crystal-to-amorphous phase transitions. The bathochromically shifted emission in the ground state compared to that in the crushed state should be based on the intermolecular charge-transfer transitions in the amorphous state. The excitation spectrum of the amorphous state was observed in the same region as that of the initial yellow-emissive crystalline state (Fig. S19, ESI†). As discussed in the two-step MCL of 1e, the increased ΦF value of 1g in the amorphous state could potentially be explained by the increased intermolecular interactions, which should suppress non-radiative processes caused by molecular motions.
Notably, the amorphous samples of all derivatives 1a–g after grinding showed yellow emission (λem = 554–579 nm), although the crystalline samples of 1a–g exhibited blue to yellow emission (λem = 490–564 nm). In the amorphous states, these derivatives should exhibit similar molecular conformations and the degree of intermolecular interactions would not be affected by the difference in the R substituent. Accordingly, similar to the case of the emission from toluene solutions, the electronic and steric effects of the R substituent should have little influence on the emission color of the amorphous 1a–g. The introduction of different substituents on the N-phenyl group is only effective in controlling the emission color and MCL properties of crystalline samples.
Conclusions
In summary, phenanthroimidazolylbenzothiadiazoles 1a–g with different R substituents on the phenyl group at the nitrogen atom of the phenanthroimidazole ring were synthesized by a four-component condensation reaction. All derivatives exhibited yellow-green emission in toluene (λem = 538–543 nm), whereas the emission colors of the crystalline samples were observed over a wide range between blue and yellow (λem = 490–564 nm). Single-crystal X-ray diffraction analyses and TD-DFT calculations revealed that the intermolecular interactions as well as the molecular conformation should significantly contribute to the emission wavelengths of the crystalline samples. Among these derivatives, 1a–d (R = p-Me, p-OMe, p-F, and p-CF3) and 1f (R = m-Br) exhibited typical bathochromically shifted bicolor MCL. On the other hand, 1e (R = p-Br) and 1g (R = o-Br) displayed two-step MCL that can respond to the mechanical stimuli of different intensities. One-way type two-step MCL was observed for 1e, whereby the emission color shifts in the bathochromic direction in a stepwise manner. The MCL of 1g was a rare back-and-forth type, whereby the emission color once shifts in the hypsochromic direction and then in the bathochromic direction. According to the results of PXRD and DSC analyses, typical crystal-to-amorphous transitions should account for the emission-color change of the bicolor MCL of 1a–d and 1f. The emission-color change of crystalline 1e upon crushing could be caused by the phase transition between different crystal systems. In the case of 1g, the origin of the hypsochromic shift in the emission wavelength upon crushing crystalline samples could potentially be explained by the formation of defects on the fracture surface, which should hamper the stabilization of an excited molecule by intermolecular interactions. The second emission-color changes of 1e and 1g upon strong grinding were attributed to the crystal-to-amorphous transitions. Although the mechanism of the first steps of two-step MCL would be different, the formation of fragile crystals that can respond to gentle crushing stimuli is indispensable to achieve the stepwise emission-color change. The present study demonstrates that two-step MCL could be accomplished by slight modifications of the molecular structure. Particularly, bromo-substituted derivatives 1e–g exhibited different MCL behaviors depending on the position of the bromo group on the N-phenyl ring. The basic insights acquired in this study can be expected to provide future design principles for the rational synthesis of multi-step MCL dyes.Experimental
General
All air-sensitive experiments were carried out under an atmosphere of argon unless otherwise noted. IR spectra were recorded on a Nicolet iS10 FT-IR spectrometer. 1H and 13C NMR spectra were recorded on a Bruker DRX-500 spectrometer using tetramethylsilane as an internal standard. Fluorescence, excitation, and UV-vis absorption spectra were measured on a JASCO FP-8300 fluorescence spectrometer. For the measurement of UV-vis absorption spectra in toluene, a FUV-803 absorbance measurement cell block was used. The solid-state absorption spectra were obtained by measuring diffuse reflectance spectra using an FPA-810 powder sample cell block. The absolute fluorescence quantum yields were determined using a 100 mm ϕ integrating sphere JASCO ILF-835. A miniature fiber-optic spectrometer (FLAME-S-XR1-ES, Ocean Optics) was used for the measurements of MCL. PXRD measurements were performed on a Rigaku SmartLab system using CuKα radiation. Melting points were determined on a Stuart melting point apparatus SMP3 and are uncorrected. DSC data were recorded on a Shimadzu DSC-60 plus instrument (heating rate: 10 °C min−1). The high-resolution electrospray ionization (HRMS-ESI) mass spectra were recorded on a Hitachi Nano Frontier LD spectrometer. SEM images were recorded on a Keyence VE-8800 microscope. Silica gel 60 N (spherical, neutral, 63–210 mm) was used for column chromatography. The spectroscopic grade toluene for UV-vis absorption and fluorescence measurements was purchased from Wako Pure Chemical Industries, Ltd, and used as received.Synthesis of phenanthroimidazolylbenzothiadiazole derivatives 1a–f
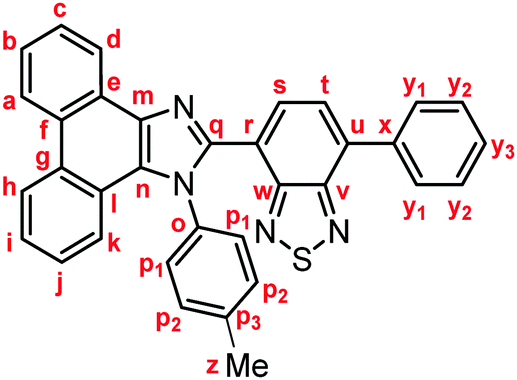
Representative experimental procedure (1a, Scheme 1). To a solution of 7-phenylbenzo[c][1,2,5]thiadiazole-4-carbaldehyde (120.1 mg, 0.50 mmol), 4-methylaniline (80.4 mg, 0.75 mmol), and anhydrous sodium sulfate (192.4 mg, 1.40 mmol) in toluene (5.0 mL) were added 9,10-phenanthrenequinone (104.1 mg, 0.50 mmol) and ammonium acetate (192.7 mg, 5.0 mmol). After the mixture was refluxed for 8 h, a saturated aqueous solution of NaHCO3 and dichloromethane were added to the mixture. The organic layer was separated, and the aqueous layer was extracted three times with dichloromethane. The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and filtered. After removal of the solvent under reduced pressure, the crude product was purified by column chromatography on silica gel (dichloromethane/toluene = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 4-phenyl-7-[1-(p-tolyl)-1H-phenanthro[9,10-d]imidazol-2-yl]benzo[c][1,2,5]thiadiazole (1a: 148.0 mg, 57%) as a yellow solid.
:1) to give 4-phenyl-7-[1-(p-tolyl)-1H-phenanthro[9,10-d]imidazol-2-yl]benzo[c][1,2,5]thiadiazole (1a: 148.0 mg, 57%) as a yellow solid.
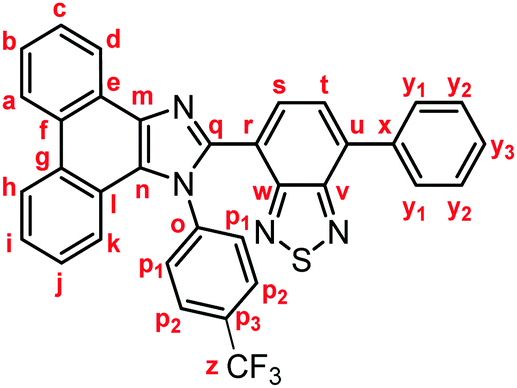
Yellow solid; M.p. 225.6–227.4 °C; IR (KBr): νmax 3057, 1513, 1474, 1451, 1378, 890, 858, 755, 729 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.87 (dd, J = 7.8, 1.2 Hz, 1H, Hd), 8.80 (d, J = 8.8 Hz, 1H, Hh), 8.73 (d, J = 8.2 Hz, 1H, Ha), 7.94–7.91 (m, 2H, Hy1), 7.79 (d, J = 7.3 Hz, 1H, Hs), 7.75–7.71 (m, 1H, Hc), 7.67 (d, J = 7.3 Hz, 1H, Ht), 7.68–7.64 (m, 1H, Hb), 7.57–7.51 (m, 3H, Hi, Hy2), 7.48–7.44 (m, 1H, Hy3), 7.42–7.40 (m, 2H, Hp1), 7.32–7.29 (m, 2H, Hj, Hk), 7.21–7.18 (m, 2H, Hp2), 2.39 (s, 3H, Hz); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.5 (Cw), 153.4 (Cv), 148.0 (Cq), 139.5 (Co), 137.7 (Cm), 136.9 (Cx), 135.4 (Cu), 135.3 (Cp3), 132.3 (Cs), 130.1 (Cp2), 129.5 (Cg), 129.3 (Cy1), 128.7 (Cy3), 128.62 (Cy2), 128.58 (Cp1), 128.3 (Cf), 128.2 (Cn), 127.31 (Ce), 127.25 (Cc), 127.1 (Ct), 126.3 (Cj), 125.6 (Cb), 125.1 (Ci), 124.1 (Ch), 123.3 (Cr), 123.1 (Ca), 123.0 (Cl), 122.9 (Cd), 121.1 (Ck), 21.3 (Cz); HRMS-ESI (m/z): [M + H]+ calcd for C34H23N4S, 519.1638; found, 519.1664. Crystal data for 1a (CCDC 1995249†): C34H22N4S, M = 518.63, monoclinic, a = 13.13381(11) Å, b = 16.14963(13) Å, c = 24.1369(2) Å, β = 94.3385(7)°, V = 5104.92(7) Å3, space group C2/c (no. 15), Z = 8, Dc = 1.350 g cm−3, F(000) = 2160.00, T = 223(1) K, μ(Cu-Kα) = 13.683 cm−1, 15
 141 reflections measured, 4685 independent (Rint = 0.0326). The final refinement converged to R1 = 0.0451 for I > 2.0σ(I), wR2 = 0.1217 for all data.
141 reflections measured, 4685 independent (Rint = 0.0326). The final refinement converged to R1 = 0.0451 for I > 2.0σ(I), wR2 = 0.1217 for all data.
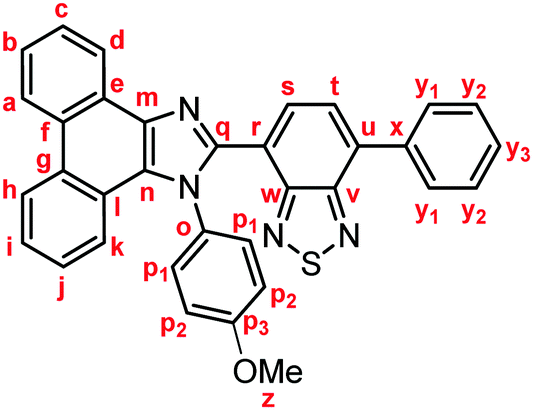
Yellow solid; M.p. 206.6–209.4 °C; IR (KBr): νmax 3057, 1512, 1453, 1253, 885, 837, 760, 730 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.87 (dd, J = 7.9, 1.2 Hz, 1H, Hd), 8.79 (d, J = 8.5 Hz, 1H, Hh), 8.72 (d, J = 8.5 Hz, 1H, Ha), 7.93–7.90 (m, 2H, Hy1), 7.78 (d, J = 7.3 Hz, 1H, Hs), 7.74–7.70 (m, 1H, Hc), 7.66 (d, J = 7.3 Hz, 1H, Ht), 7.67–7.63 (m, 1H, Hb), 7.55–7.51 (m, 3H, Hi, Hy2), 7.47–7.41 (m, 3H, Hy3, Hp1), 7.34–7.29 (m, 2H, Hj, Hk), 6.89–6.86 (m, 2H, Hp2), 3.80 (s, 3H, Hz); 13C NMR (126 MHz, CDCl3): δ (ppm) 160.0 (Co), 154.4 (Cw), 153.4 (Cv), 148.3 (Cq), 137.6 (Cm), 136.9 (Cx), 135.4 (Cu), 132.3 (Cs), 130.4 (Cp3), 130.0 (Cp1), 129.4 (Cg), 129.3 (Cy1), 128.7 (Cy3), 128.6 (Cy2) 128.3 (Cf, Cn), 127.30 (Ce), 127.26 (Cc), 127.1 (Ct), 126.3 (Cj), 125.6 (Cb), 125.1 (Ci), 124.1 (Ch), 123.3 (Cr), 123.09 (Ca), 123.04 (Cl), 122.9 (Cd), 121.0 (Ck), 114.5 (Cp2), 55.4 (Cz); HRMS-ESI (m/z): [M + H]+ calcd for C34H23N4OS, 535.1587; found, 535.1577. Crystal data for 1b (CCDC 1995250†): C34H22N4OS, M = 534.63, triclinic, a = 9.72015(13) Å, b = 10.26595(12) Å, c = 14.23572(16) Å, α = 100.4892(9)°, β = 96.5213(10)°, γ = 106.5436(11)°, V = 1318.19(3) Å3, space group P (no. 2), Z = 2, Dc = 1.347 g cm−3, F(000) = 556.00, T = 223(1) K, μ(Cu-Kα) = 13.711 cm−1, 14064 reflections measured, 4822 independent (Rint = 0.0292). The final refinement converged to R1 = 0.0448 for I > 2.0σ(I), wR2 = 0.1214 for all data.
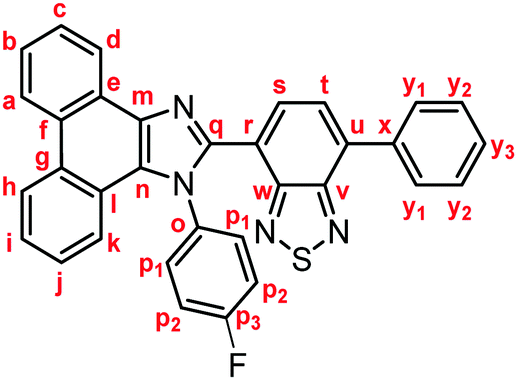
Yellow solid; M.p. 240.6–243.4 °C; IR (KBr): νmax 3040, 1513, 1469, 1453, 1224, 887, 851, 755, 727 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.86 (d, J = 7.6 Hz, 1H, Hd), 8.81 (d, J = 8.2 Hz, 1H, Hh), 8.73 (d, J = 8.2 Hz, 1H, Ha), 7.94–7.91 (m, 2H, Hy1), 7.85 (d, J = 7.3 Hz, 1H, Hs), 7.75–7.72 (m, 1H, Hc), 7.71 (d, J = 7.3 Hz, 1H, Ht), 7.69–7.65 (m, 1H, Hb), 7.59–7.52 (m, 5H, Hi, Hp1, Hy2), 7.48–7.45 (m, 1H, Hy3), 7.35–7.31 (m, 1H, Hj), 7.27 (d, J = 7.7 Hz, 1H, Hk), 7.11–7.07 (m, 2H, Hp2); 13C NMR (126 MHz, CDCl3): δ (ppm) 162.4 (d, 1JC–F = 250 Hz, Cp3), 153.9 (Cw), 153.1 (Cv), 147.9 (Cq), 137.5 (Cm), 136.5 (Cx), 135.5 (Cu), 133.7 (d, 4JC–F = 2.5 Hz, Co), 132.2 (Cs), 130.5 (d, 3JC–F = 8.8 Hz, Cp1), 129.3 (Cg), 129.1 (Cy1), 128.6 (Cy3), 128.4 (Cy2), 128.1 (Cf), 127.9 (Cn), 127.1 (Cc), 126.92 (Ce), 126.90 (Ct), 126.1 (Cj), 125.6 (Cb), 125.0 (Ci), 124.0 (Ch), 122.9 (Ca), 122.7 (Cr), 122.6 (Cd), 122.5 (Cl), 120.5 (Ck), 116.3 (d, 2JC–F = 22.7 Hz, Cp2); HRMS-ESI (m/z): [M + H]+ calcd for C33H20FN4S, 523.1387; found, 523.1409.
Yellow solid; M.p. 223.6–226.4 °C; IR (KBr): νmax 3073, 1613, 1518, 1474, 1451, 1373, 1326, 1168, 1125, 1068, 858, 850, 754, 728 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.86 (dd, J = 7.9, 1.3 Hz, 1H, Hd), 8.82 (d, J = 8.0 Hz, 1H, Hh), 8.74 (d, J = 8.2 Hz, 1H, Ha), 7.94–7.92 (m, 2H, Hy1), 7.88 (d, J = 7.3 Hz, 1H, Hs), 7.77–7.73 (m, 1H, Hc), 7.72 (d, J = 7.3 Hz, 1H, Ht), 7.71–7.66 (m, 5H, Hb, Hp1, Hp2), 7.59–7.53 (m, 3H, Hi, Hy2), 7.49–7.46 (m, 1H, Hy3), 7.35–7.32 (m, 1H, Hj), 7.22 (d, J = 8.2 Hz, 1H, Hk); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.0 (Cw), 153.3 (Cv), 148.0 (Cq), 141.2 (Co), 138.1 (Cm), 136.6 (Cx), 136.0 (Cu), 132.6 (Cs), 131.5 (q, 2JC–F = 33.0 Hz, Cp3), 129.6 (Cg), 129.5 (Cp1), 129.3 (Cy1), 128.9 (Cy3), 128.7 (Cy2), 128.4 (Cf), 127.9 (Cn), 127.5 (Cc), 127.2 (Ct), 127.1 (Ce), 126.51 (q, 3JC–F = 4.6 Hz, Cp2), 126.50 (Cj), 126.0 (Cb), 125.4 (Ci), 124.3 (Ch), 123.4 (q, 1JC–F = 274 Hz, Cz), 123.1 (Ca), 122.9 (Cd), 122.7 (Cr), 122.5 (Cl), 120.8 (Ck); HRMS-ESI (m/z): [M + H]+ calcd for C34H20F3N4S, 573.1355; found, 573.1337. Crystal data for 1d (CCDC 1995251†): C34H19F3N4S, M = 572.61, triclinic, a = 6.7391(3) Å, b = 13.8406(5) Å, c = 15.2075(5) Å, α = 113.486(3)°, β = 90.031(3)°, γ = 96.987(3)°, V = 1289.41(9) Å3, space group P (no. 2), Z = 2, Dc = 1.475 g cm−3, F(000) = 588.00, T = 223(1) K, μ(Cu-Kα) = 15.859 cm−1, 12728 reflections measured, 4685 independent (Rint = 0.1047). The final refinement converged to R1 = 0.0695 for I > 2.0σ(I), wR2 = 0.2109 for all data.
Yellow solid; M.p. 236.6–239.4 °C; IR (KBr): νmax 3056, 1519, 1489, 1474, 1374, 1008, 859, 833, 751, 729 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.85 (d, J = 6.9 Hz, 1H), 8.79 (d, J = 8.5 Hz, 1H), 8.72 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 7.3 Hz, 2H), 7.83 (d, J = 7.3 Hz, 1H), 7.74–7.71 (m, 1H), 7.70 (d, J = 7.3 Hz, 1H), 7.68–7.64 (m, 1H), 7.58–7.50 (m, 5H), 7.49–7.44 (m, 1H), 7.41 (d, J = 7.6 Hz, 2H), 7.36–7.32 (m, 1H), 7.29 (d, J = 7.3 Hz, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.2, 153.3, 148.0, 137.9, 137.0, 136.7, 135.9, 132.7 (2C), 132.5, 130.5 (2C), 129.5, 129.3 (2C), 128.8, 128.7 (2C), 128.4, 128.0, 127.4, 127.17, 127.13, 126.5, 125.9, 125.3, 124.3, 123.6, 123.1, 122.9 (2C), 122.7, 120.9; HRMS-ESI (m/z): [M + H]+ calcd for C33H20BrN4S, 583.0587; found, 583.0593. Crystal data for 1e (CCDC 1995252†): C33H19BrN4S, M = 583.50, triclinic, a = 6.63294(10) Å, b = 13.8905(2) Å, c = 15.0996(2) Å, α = 66.2986(14)°, β = 88.1740(12)°, γ = 83.1236(13)°, V = 1264.49(3) Å3, space group P (no. 2), Z = 2, Dc = 1.532 g cm−3, F(000) = 592.00, T = 223(1) K, μ(Cu-Kα) = 32.341 cm−1, 14175 reflections measured, 5135 independent (Rint = 0.0664). The final refinement converged to R1 = 0.0506 for I > 2.0σ(I), wR2 = 0.1436 for all data.
Yellow solid; M.p. 216.6–219.4 °C; IR (KBr): νmax 3064, 1573, 1478, 1365, 886, 855, 754, 727 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.81 (d, J = 7.8 Hz, 1H), 8.74 (d, J = 8.5 Hz, 1H), 8.72 (d, J = 8.5 Hz, 1H), 7.94 (m, 2H), 7.89 (d, J = 7.3 Hz, 1H), 7.80–7.79 (m, 1H), 7.76–7.72 (m, 2H), 7.70–7.66 (m, 1H), 7.59–7.53 (m, 4H), 7.49–7.46 (m, 2H), 7.35 (t, J = 7.5 Hz, 1H), 7.29–7.25 (m, 2H); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.1, 153.3, 148.0, 139.2, 137.8, 136.7, 135.9, 132.7, 132.5, 132.3, 130.5, 129.5, 129.3 (2C), 128.8, 128.6 (2C), 128.4, 128.0, 127.7, 127.4, 127.2, 127.1, 126.5, 125.9, 125.3, 124.2, 123.1, 122.9, 122.8, 122.63, 122.56, 120.9; HRMS-ESI (m/z): [M + H]+ calcd for C33H20BrN4S, 583.0587; found, 583.0575.
Yellow solid; M.p. 272.6–275.4 °C; IR (KBr): νmax 3058, 1522, 1483, 1318, 1223, 1166, 1075, 886, 858, 759, 727 cm−1; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.88 (dd, J = 7.9, 1.2 Hz, 1H), 8.81 (d, J = 8.2 Hz, 1H), 8.74 (d, J = 8.2 Hz, 1H), 7.93–7.92 (m, 3H), 7.79 (dd, J = 7.6, 1.9 Hz, 1H), 7.74 (t, J = 7.2 Hz, 1H), 7.71–7.65 (m, 3H), 7.58–7.51 (m, 3H), 7.48–7.44 (m, 1H), 7.39–7.29 (m, 3H), 7.10 (d, J = 8.1 Hz, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 154.4, 153.4, 147.5, 138.0, 137.4, 136.8, 135.7, 133.7 (2C), 132.3, 131.3 (2C), 129.4, 129.3 (2C), 128.8, 128.6 (2C), 128.4, 128.3, 127.7, 127.33, 127.29, 127.18, 126.6, 125.8, 125.3, 124.22, 124.15, 123.1, 123.0, 122.7, 120.2; HRMS-ESI (m/z): [M + H]+ calcd for C33H20BrN4S, 583.0587; found, 583.0592. Crystal data for 1g (CCDC 1995253†): C33H19BrN4S, M = 583.50, monoclinic, a = 10.39805(19) Å, b = 18.2187(4) Å, c = 13.3287(2) Å, β = 93.0879(17)°, V = 2521.31(8) Å3, space group P21/n (no. 14), Z = 4, Dc = 1.537 g cm−3, F(000) = 1184.00, T = 223(1) K, μ(Cu-Kα) = 32.439 cm−1, 17435 reflections measured, 4612 independent (Rint = 0.0499). The final refinement converged to R1 = 0.0540 for I > 2.0σ(I), wR2 = 0.1569 for all data.
Theoretical calculations
The theoretical calculations were performed using the Gaussian 16 program.16 The six lowest singlet–singlet transitions of 1a were calculated using time-dependent density functional theory (TD-DFT) calculations at the CAM-B3LYP/6-31G(d) level of theory. The molecular structure of 1a, obtained from the single-crystal X-ray diffraction analysis, was used as a starting point. Here, the long-range-corrected hybrid functional CAM-B3LYP was used, as CAM-B3LYP often provides better results in TD-DFT calculations than B3LYP, which is conventionally used in DFT calculations.11 The HOMO and LUMO energy levels of 1a are −6.46 eV and −1.18 eV, respectively. The calculated first excited state, which consists of the transition from the HOMO to the LUMO (0.634), is 363 nm (3.41 eV) with an oscillator strength of 0.398. The theoretical calculations of 1b, 1d, 1e, and 1g were carried out as described for 1a (Table S1, ESI†).Typical experimental procedure for the two-step MCL
Crystalline samples were prepared by recrystallization from hot toluene solutions. Several pieces of the crystalline samples were placed on a glass plate, and the fluorescence spectrum of the sample under irradiation with UV light (365 nm) was measured using the miniature fiber-optic spectrometer. The crystalline sample was manually crushed into a fine powder by using a spatula until the emission color of the sample no longer changes. The crushed sample was further ground manually with a spatula. After the measurement of the fluorescence spectrum, the ground sample on the glass plate was heated on a hot plate at 120 °C or 180 °C. The fluorescence spectrum of the heated sample was measured after cooling the samples to room temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number 18H04508 in Grant-in-Aid for Scientific Research on Innovative Areas “Soft Crystals: Area No. 2903”.Notes and references
- For recent reviews, see:
(a) E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517 RSC
; (b) R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2019, 1905227 Search PubMed
- For recent examples, see:
(a) Y.-J. Kong, Z.-P. Yan, S. Li, H.-F. Su, K. Li, Y.-X. Zheng and S.-Q. Zang, Angew. Chem., Int. Ed., 2020, 59, 5336 CrossRef CAS PubMed
- For recent examples, see:
(a) B. Mu, Y. Zhao, X. Li, X. Quan and W. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 9637 CrossRef CAS PubMed
- For recent examples, see:
(a) A. Ekbote, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2020, 8, 3589 RSC
- For recent examples, see:
(a) A. Kobayashi, S. Imada, Y. Shigeta, Y. Nagao, M. Yoshida and M. Kato, J. Mater. Chem. C, 2019, 7, 14923 RSC
- For seminal examples of MCL dyes, see:
(a) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS PubMed
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Métivier, C. Allain and I. Ciofini, Adv. Mater., 2018, 30, 1800817 CrossRef PubMed
- For examples, see:
(a) S. Li, M. Wu, Y. Kang, H.-W. Zheng, X.-J. Zheng, D.-C. Fang and L.-P. Jin, Inorg. Chem., 2019, 58, 4626 CrossRef CAS PubMed
- Little is known about MCL dyes that exhibit two-step MCL. For examples, see:
(a) C. Duan, Y. Zhou, G.-G. Shan, Y. Chen, W. Zhao, D. Yuan, L. Zeng, X. Huang and G. Niu, J. Mater. Chem. C, 2019, 7, 3471 RSC
-
(a) S. Ito, C. Nishimoto and S. Nagai, CrystEngComm, 2019, 21, 5699 RSC
-
(a) A. Arunkumar, S. Shanavas, R. Acevedo and P. M. Anbarasan, Struct. Chem., 2020, 31, 1029 CrossRef CAS
-
Polyimides: Fundamentals and Applications, ed. M. K. Ghosh and K. L. Mittal, Marcel Dekker, New York, NY, 1996 Search PubMed
- The structures are drawn by VESTA. See: K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2004, 108, 10200 CrossRef CAS
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, X-ray diffraction analyses, theoretical calculations, PXRD data, and DSC diagrams. CCDC 1995249 (1a), 1995250 (1b), 1995251 (1d), 1995252 (1e) and 1995253 (1g). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00198h |
| This journal is © The Royal Society of Chemistry 2020 |