
Enhancement of the oxygen reduction reaction electrocatalytic activity of metallo-corroles using contracted cobalt(III) CF3-corrole incorporated in a high surface area carbon support†
Hilah C.
Honig
,
Ariel
Friedman
,
Noam
Zion
and
Lior
Elbaz
 *
*
Chemistry Department, Bar-Ilan University, Ramat-Gan, Israel. E-mail: lior.elbaz@biu.ac.il
First published on 23rd June 2020
Abstract
Molecular oxygen reduction reaction catalysts based on metallo-corrole with the smallest meso-substituent reported to-date, Co(III)CF3-corrole, was synthesized and compared to the well-studied Co(III)tpfcorrole when adsorbed on a high surface area carbon support. This study shows the improved electrocatalytic performance with the new corrole, attributed to its unique compact structure, which enables surface interactions in favor of lowering the reaction overpotential by 70 mV.
The world's leading economies have come to realize that fossil fuels are not an everlasting resource, and that energy-related carbon dioxide emissions must be dropped significantly.1 Among the various technologies available, low temperature fuel cells (LTFCs), namely: proton exchange membrane and alkaline electrolyte membrane fuel cells, seem to have the most promise for high power density applications such as transportation, backup and main power.2 In order to increase the fuel cell's reaction rate, and lower the activation energy, electrocatalysts are needed.3 The relatively sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode results in a significant energy loss, and requires large amounts of catalysts.4
The most common ORR catalysts today are platinum-group metal (PGM) catalysts, which are scarce and account for the relatively high cost of LTFCs.4,5 Moreover, PGMs are considered to be non-selective catalysts, and tend to accelerate the degradation of carbon-based electrodes.6 For these reasons, the search for alternative, PGM-free catalysts has been increasing during the past couple of decades.3 The work on this class of catalysts has been inspired by biological systems that reduce oxygen very efficiently, using transition metal complexes.7,8 While the state-of-the-art PGM-free ORR catalysts are heat-treated mixtures of a first-row transition metal, nitrogen and carbon precursors (MNCs), their undefined structure limits their further improvement. Hence, the need for well-defined molecular catalysts is clear. We recently reported on the very promising activity of a family of molecular PGM-free ORR catalysts based on metallocorroles (Fig. 1).6,9–17
 | ||
| Fig. 1 The structure of Cobalt (III) corrole. CF3-corrole R = CF3 (trifluoromethyl), tpf-corrole R = C6F5 (tris-pentafluoro phenyl). | ||
In the past few years, we have studied the parameters that affect the ORR electrocatalytic activity of metallo-corroles.18 We concluded that in terms of overpotential and selectivity, the most active metal center is Co(III). In order to further lower the ORR overpotential, a wide array of substituents were studied, both at the beta and meso positions on the corrole ring, having different electronegativities, making them anywhere from very strong electron-donating to very strong electron-withdrawing substituents.10,11,18 Although electron-withdrawing substituents lower the overpotential when used as is, after their incorporation in high surface area carbon, the effect of the nature of the substituent diminishes, and all of the studied metallo-corroles catalyze the ORR at the same potential. These findings led us to postulate that the metallo-corrole's substituents do not affect the ORR overpotential when these complexes are adsorbed on high surface area carbon supports. We now report on the activity of the smallest corrole structure reported to date, which enables the removal of the complexity and the bulkiness of other metallo-corroles, and is expected to be one of the most electrocatalytically active corroles.
In this work, a contracted cobalt-corrole containing only the CF3 substituent on its meso-position was synthesized (see details in the SI†), and its ORR electrocatalytic activity was compared to that of Co(III) tri-(pentafluorophenyl)corrole (Co(III) tpf-corrole) (Fig. 1). It is expected that the compactness of this new corrole will increase its chance of forming favorable surface interactions with the carbon supports, as was previously shown with other catalysts,19,20 while maintaining the already high activity associated with this family of molecular catalysts, and will allow the increase of the activity and catalytic site density of these PGM-free molecular catalysts; one of the biggest challenges in the field today.21–23 Previous work with Co(III) CF3-corrole showed that it tends to form a very stable axial coordination on the metal center, whereas more elaborate structures show limited-to-no stability.24–26
Both substituents (CF3 and tpf) are considered very electro-negative, and stabilize the corrole's ring.11 And both significantly contribute to the electron-withdrawing tendency of the ring, which is important for lowering the ORR overpotential with transition metal complexes.27,28 The ORR activity of these corroles when adsorbed on a high surface area carbon support, black pearls 2000 (BP2000), was studied in alkaline and acidic environments. Both exhibit very high activity in the alkaline environment, on par with the state-of-the-art molecular catalysts (Fig. 2). While in the acidic environment, they show a much lower activity (Fig. S2, ESI†). Hence, in this work, we focused on the performance in an alkaline environment.
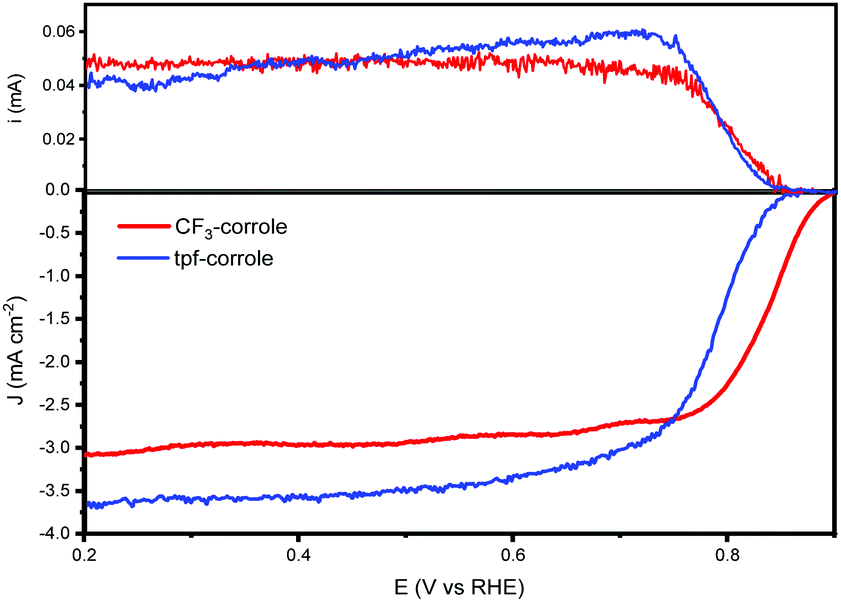 | ||
| Fig. 2 RRDE of CF3-corrole and tpf-corrole in 0.1 M KOH @ 900 rpm. | ||
As shown by the RRDE results (Fig. 2), the ORR activity of the CF3-substituted corrole outperforms that of tpf-corrole in terms of reaction potentials. The onset and half-wave potentials for ORR with the CF3-corrole are 0.89 and 0.83 V vs. RHE, respectively. The tpf-corrole has an onset potential of 0.85 V vs. RHE and a half-wave potential of 0.76 V vs. RHE. In contrast to other meso-substituted Co(III)-corroles that were studied in the past, which did not show a significant difference in their ORR potentials (Fig. S3, ESI†), the CF3-substituted corrole's half-wave potential was significantly shifted to a lower overpotential by 70 mV, hinting at some new effect that was not observed before.10
In order to understand the cause for the shift in ORR potentials, X-ray photoelectron spectroscopy (XPS) was used. Fig. 3 presents a summary of the XPS spectra of both corroles. The spectra before incorporation in BP2000 show one main O1s peak at 531.28 eV and 532.88 eV, for the CF3- and tpf-corrole, respectively. These peaks can be assigned to a combination of various Co–O species, most probably an environmental oxygen adduct on the cobalt metal center in the corrole (Co–O2).29 This observation was confirmed by the Co2p spectra (Fig. 3, left). After the adsorption on BP2000, the O1s peak of pristine tpf-corrole shifted by 0.3 eV to 533.18 eV, while the peak of the CF3-corrole was much more significantly shifted, by 2.0 eV to 533.28 eV. The fact that the peak of both pristine corroles shifted implies surface interactions with BP2000. It seems that these surface interactions are through the oxygen of the quinone-like moieties on the BP2000 surface, similar to previous observations with cobalt–porphyrins.20,30 After the incorporation in BP2000, the Co2p peak of CF3-corrole, at 780.08 eV, becomes significantly more dominant, implying the formation of a strong bond to the surface of BP2000 through the oxygen moieties. This is in agreement with the O1s spectra. In contrast, the tpf-corrole shows much weaker Co–O bonds after its incorporation in BP2000. These results are also in agreement with the stronger tendency of the CF3-corrole for binding axial ligands.24–26 Further computational study is needed in order to elucidate how these surface interactions affect the electronic configuration of the corroles, which in turn will help understand the shift in ORR potentials. It is important to emphasize that the CF3-corrole is much more compact than the tpf-corrole, and has a less elaborate Π system, which may also help in facilitating the formation of the strong surface interactions through the surface oxygen moieties on the carbon support.
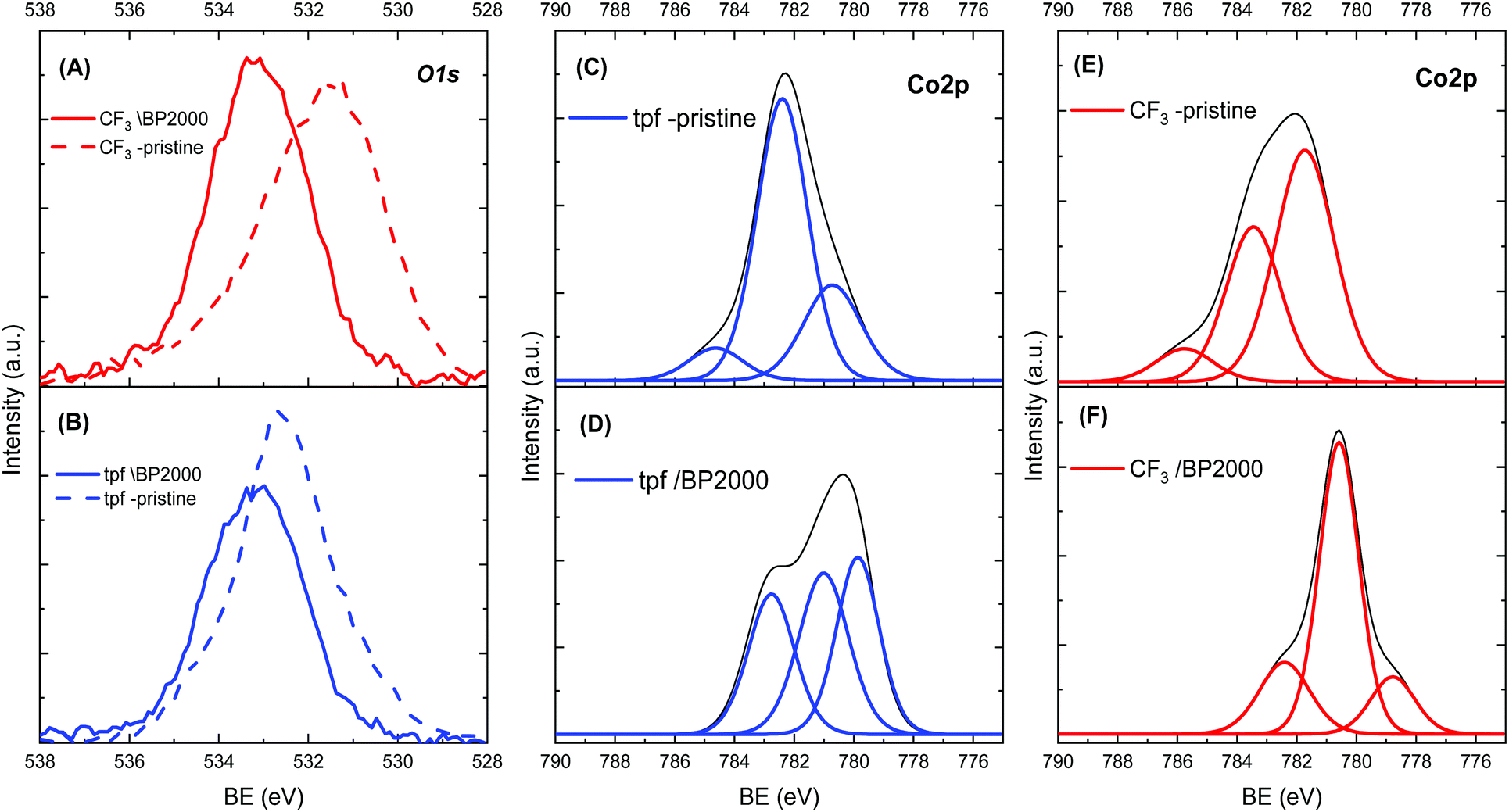 | ||
| Fig. 3 XPS spectra of (A) O1s of the Co(III)-CF3-corrole before and after incorporation in BP2000, (B) O1s of the Co(III)-tpf-corrole before and after incorporation in BP2000, (C) Co2p spectra of Co(III)-tpf-corrole, (D) Co2p of the Co(III)-tpf-corrole after incorporation in BP2000, (E) Co2p spectra of Co(III)-CF3-corrole, and (F) Co2p of the Co(III)-CF3-corrole after incorporation in BP2000. | ||
Apart from the ORR overpotential, the reaction mechanism was also elucidated from the RRDE results. The ORR pathway can either be a direct 4e−, consecutive 2 + 2e−, or 2e− mechanism. The number of electrons (n) transferred during the ORR was calculated using the Levich equation:31
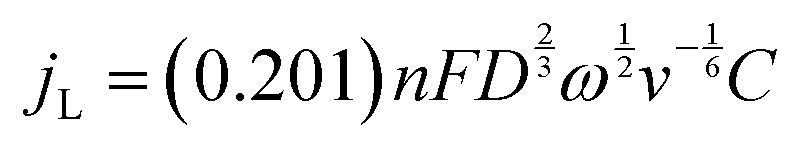
The number of transferred electrons at 0.4 V vs. RHE was calculated and found to be 3.06 and 3.67 for CF3-corrole and tpf-corrole, respectively. The tpf-corrole has a higher tendency for the 4e− pathway, but both seem to follow the 2 + 2e− pathway, since relatively high levels of peroxide anion were detected at the ring electrode (25–40%), as shown in Fig. 2.
To shed more light on the ORR mechanism with the CF3-corrole, the amount of HO2− formed during the reaction was quantified, and the number of electrons transferred during the ORR was calculated from it according to the following equations:34
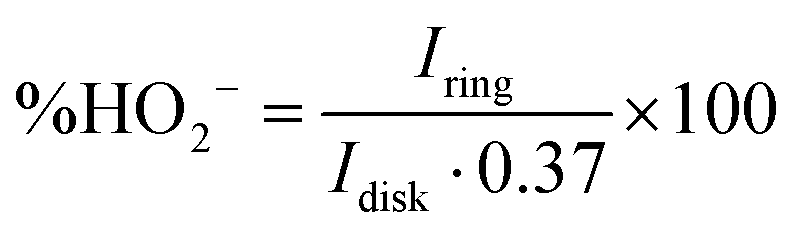 | (1) |
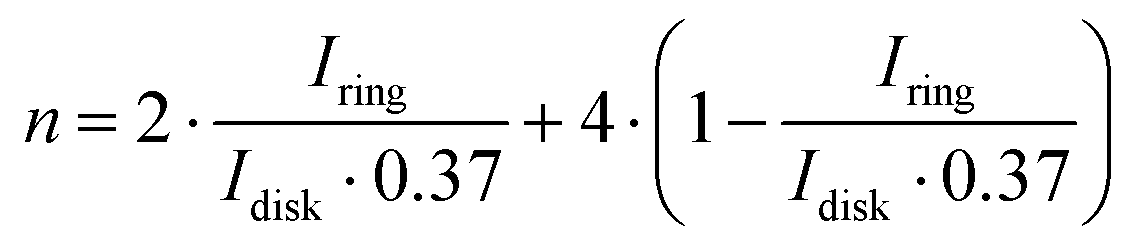 | (2) |
The results of these calculations over a wide range of voltages are presented in Fig. 4. They support the conclusions from the calculation using the Levich equation, and confirm that the ORR follows the 2 + 2e− reduction pathway with both corroles.35 The difference between the electron number calculated from the Levich equation and eqn (2) can be explained by the electrode roughness, which may have an effect on the surface area of the electrode, which is used in the Levich equation.36 Another possible reason might be related to the HO2− that is stranded in the carbon-pores and possibly continues its reduction to H2O through a disporportionation reaction. While the CF3-corrole maintains a relatively stable n value, the n value of the tpf-corrole increases significantly with the increase of the overpotential. This suggests that the tpf-corrole is a bit more active in the reduction of peroxide anion to hydroxide at high overpotentials.
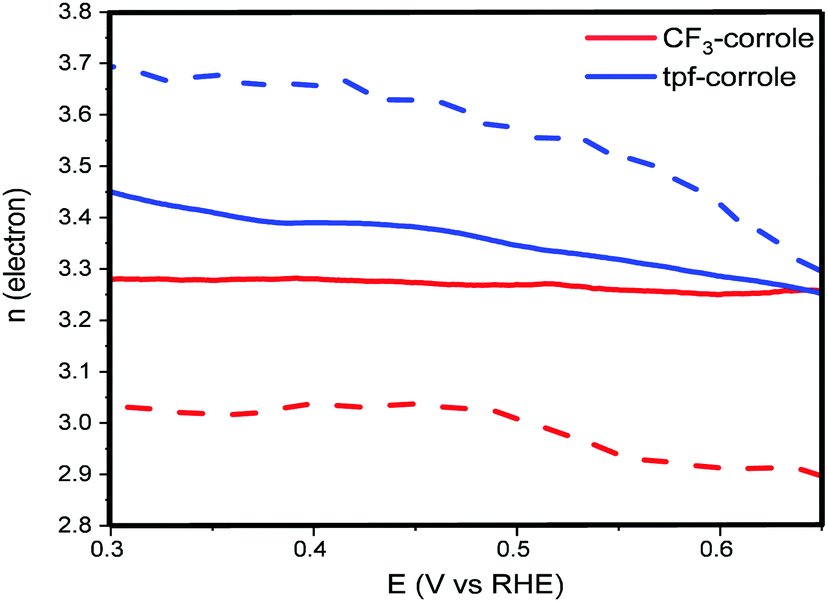 | ||
| Fig. 4 n electron obtained from eqn (2) (solid line) and Levich equation (dashed line). | ||
In pervious papers, we compared different structural aspects of metallocorrole to elucidate their effect on the ORR electrocatalysis and concluded that once adsorbed in high surface area carbon supports, the corrole substituent has no effect on the ORR overpotential. In order to prove this conclusion, we studied the most compact corrole available. Herein, we compared the well-studied Co(III) tpf-corrole and the contracted structure Co(III) CF3-corrole. We found that in contrast to the previous conclusion, lower overpotentials can be reached by using a less elaborate structure, which allows the formation of strong surface interactions with the support material. The new, compact Co(III) CF3-corrole shows a significant improvement in terms of overpotential. The Co–O binding energy differences, before and after the corroles were adsorbed on BP2000, hint at surface interactions between the corroles and the support. There is a correlation between the larger shift in the XPS spectra and the shift in the ORR overpotentials. Stronger surface interaction translated to higher ORR potentials. It seems that the surface interactions are facilitated through quinone-like moieties on the BP2000 surface. The reaction mechanism of both corroles is a consecutive 2 + 2e− reduction, where the tpf-corrole seems to be a better catalyst for peroxide anion reduction. The unique interactions observed with the Co(III) CF3-corrole incorporated in BP2000 advocate further investigation of this phenomena using in-depth computational study, in order to elucidate the effect of the surface interactions on the electron density and the Co(III) CF3-corrole's affinity to oxygen.
The authors would like to thank the Israeli Ministry of Energy and The Fuel Choices and Smart Mobility Initiative at the Israeli Prime Minister's Office for funding this work. One of the authors, A. F., would like to thank the Israeli Ministry of Energy for his fellowship. This work was conducted in the framework of the Israeli Fuel Cells Consortium (part of Israel National Center for Electrochemical Propulsion).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Moliner, M. J. Lázaro and I. Suelves, Int. J. Hydrogen Energy, 2016, 41, 19500–19508 CrossRef CAS.
- J. Zhang, PEM fuel cell electrocatalysts and catalyst layers: fundamentals and applications, Springer Science & Business Media, 2008 Search PubMed.
- L. Elbaz, G. Wu and P. Zelenay, in Electrocatalysis in Fuel Cells, ed. M. Shao, Springer, London, 2013, ch. 8, vol. 9, pp. 213–246 Search PubMed.
- V. Mehta and J. S. Cooper, J. Power Sources, 2003, 114, 32–53 CrossRef CAS.
- J. Van Mierlo, G. Maggetto and P. Lataire, Energy Convers. Manage., 2006, 47, 2748–2760 CrossRef CAS.
- O. Lori and L. Elbaz, Catalysts, 2015, 5, 1445–1464 CrossRef CAS.
- R. Jasinski, Nature, 1964, 201, 1212 CrossRef CAS.
- R. Othman, A. L. Dicks and Z. Zhu, Int. J. Hydrogen Energy, 2012, 37, 357–372 CrossRef CAS.
- N. Zion, A. Friedman, N. Levy and L. Elbaz, Adv. Mater., 2018, 30, 1800406 CrossRef PubMed.
- N. Levy, O. Lori, S. Gonen, M. Mizrahi, S. Ruthstein and L. Elbaz, Carbon, 2020, 158, 238–243 CrossRef CAS.
- J. S. Shpilman, A. Friedman, N. Zion, N. Levy, D. T. Major and L. Elbaz, J. Phys. Chem. C, 2019, 123, 30129–30136 CrossRef CAS.
- A. Friedman, I. Saltsman, Z. Gross and L. Elbaz, Electrochim. Acta, 2019, 310, 13–19 CrossRef CAS.
- M. Kosa, N. Levy, L. Elbaz and D. T. Major, J. Phys. Chem. C, 2018, 122, 17686–17694 CrossRef CAS.
- A. Friedman, L. Landau, S. Gonen, Z. Gross and L. Elbaz, ACS Catal., 2018, 8, 5024–5031 CrossRef CAS.
- N. Levy, A. Mahammed, A. Friedman, B. Gavriel, Z. Gross and L. Elbaz, ChemCatChem, 2016, 8, 2832–2837 CrossRef CAS.
- H. C. Honig, C. B. Krishnamurthy, I. Borge-Durán, M. Tasior, D. T. Gryko, I. Grinberg and L. Elbaz, J. Phys. Chem. C, 2019, 123, 26351–26357 CrossRef CAS.
- N. Levy, A. Mahammed, M. Kosa, D. T. Major, Z. Gross and L. Elbaz, Angew. Chem., Int. Ed., 2015, 54, 14080–14084 CrossRef CAS PubMed.
- N. Levy, J. S. Shpilman, H. C. Honig, D. T. Major and L. Elbaz, Chem. Commun., 2017, 53, 12942–12945 RSC.
- R. Z. Snitkoff, N. Levy, I. Ozery, S. Ruthstein and L. Elbaz, Carbon, 2019, 143, 223–229 CrossRef CAS.
- L. Elbaz, E. Korin, L. Soifer and A. Bettelheim, J. Phys. Chem. Lett., 2009, 1, 398–401 CrossRef.
- N. Zion, D. A. Cullen, P. Zelenay and L. Elbaz, Angew. Chem., Int. Ed., 2020, 59, 2483–2489 CrossRef CAS PubMed.
- N. D. Leonard, S. Wagner, F. Luo, J. Steinberg, W. Ju, N. Weidler, H. Wang, U. I. Kramm and P. Strasser, ACS Catal., 2018, 8, 1640–1647 CrossRef CAS.
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 13285 CrossRef CAS PubMed.
- Q.-C. Chen, M. Soll, A. Mizrahi, I. Saltsman, N. Fridman, M. Saphier and Z. Gross, Angew. Chem., Int. Ed., 2018, 57, 1006–1010 CrossRef CAS PubMed.
- A. Mahammed, I. Giladi, I. Goldberg and Z. Gross, Chem. – Eur. J., 2001, 7, 4259–4265 CrossRef CAS.
- V. A. Adamian, F. D’Souza, S. Licoccia, M. L. Di Vona, E. Tassoni, R. Paolesse, T. Boschi and K. M. Kadish, Inorg. Chem., 1995, 34, 532–540 CrossRef CAS.
- K. Sudhakar, A. Mahammed, N. Fridman and Z. Gross, Dalton Trans., 2019, 48, 4798–4810 RSC.
- K. M. Kadish and M. M. Morrison, Bioinorg. Chem., 1977, 7, 107–115 CrossRef CAS PubMed.
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263–275 CrossRef CAS.
- L. Elbaz, E. Korin, L. Soifer and A. Bettelheim, J. Electrochem. Soc., 2010, 157, B27–B31 CrossRef CAS.
- J. Nikolic, E. Expósito, J. Iniesta, J. González-Garcia and V. Montiel, J. Chem. Educ., 2000, 77, 1191 CrossRef CAS.
- R. Davis, The Solubility and Diffusion Coefficient of Oxygen in Potassium Hydroxide Solutions, Electrochim. Acta, 1967, 12, 287–297 CrossRef CAS.
- W. Robert, Handbook of chemistry and physics, CRC Press, 1988 Search PubMed.
- A. J. Bard, L. R. Faulkner, J. Leddy and C. G. Zoski, Electrochemical methods: fundamentals and applications, Wiley, New York, 1980 Search PubMed.
- K. M. Kadish, L. Frémond, Z. Ou, J. Shao, C. Shi, F. C. Anson, F. Burdet, C. P. Gros, J.-M. Barbe and R. Guilard, J. Am. Chem. Soc., 2005, 127, 5625–5631 CrossRef CAS PubMed.
- A. J. Bard, Integrated chemical systems: a chemical approach to nanotechnology, Wiley, New York, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and XPS data. See DOI: 10.1039/d0cc03122d |
| This journal is © The Royal Society of Chemistry 2020 |