
Visible-light-mediated α-phosphorylation of N-aryl tertiary amines through the formation of electron-donor–acceptor complexes: synthetic and mechanistic studies†
Valentin
Quint
a,
Nourhène
Chouchène
b,
Moheddine
Askri
b,
Jacques
Lalevée
 c,
Annie-Claude
Gaumont
a and
Sami
Lakhdar
*a
c,
Annie-Claude
Gaumont
a and
Sami
Lakhdar
*a
aNormandie Univ., ENSICAEN, Unicaen, CNRS, LCMT, 14000 Caen, France. E-mail: sami.lakhdar@ensicaen.fr
bUniversité de Monastir, Faculté des Sciences de Monastir, avenue de l'environnement, Monastir 5000, Tunisia
cInstitut de Science des Matériaux de Mulhouse IS2 M – UMR CNRS 7361 – UHA, 15, rue Jean Starcky, 68057 Mulhouse Cedex, France
First published on 12th November 2018
Abstract
This work describes an efficient α-phosphorylation of a wide variety of N-aryl tertiary amines under mild conditions. It consists of a simple combination of amines (electron donors) with N-ethoxy-2-methylpyridinium tetrafluoroborate (electron acceptor) in the presence of P(V)–H compounds under visible light irradiation. Physical organic investigations support the formation of an electron-donor–acceptor complex (amine/pyridinium ion) as a key intermediate.
Introduction
The direct and efficient α-C(sp3)–H functionalization of tertiary amines into carbon–phosphorus bonds is a challenging organic transformation that has attracted much attention during the last few decades.1 This is obviously due to the potential biological activities of α-aminophosphonates and α-aminophosphonic acids in numerous natural and pharmaceutical compounds.2Based on the use of the well-known cross-dehydrogenative coupling (CDC) technology3 between an sp3 C–H bond adjacent to a nitrogen atom of N-aryl amines 1 and an H–P bond, various approaches have been reported for the synthesis of α-phosphorylated amines 3. As depicted in Scheme 1 (upper part), these methods rely on the use of transition metal catalysts (method a)1a–c,1i and organic- or organometallic-based photocatalysts (method b)1d–j. It should be mentioned that Zeitler et al. have recently reported a visible light photocatalyst free approach for the α-functionalization of tertiary amines, which has been successfully applied for carbon–carbon as well as carbon–phosphorus bonds (method c).1k Detailed mechanistic studies of these different approaches revealed the intermediacy of an iminium ion, which can then be intercepted with the phosphorus nucleophiles to furnish the desired α-phosphonated amines.
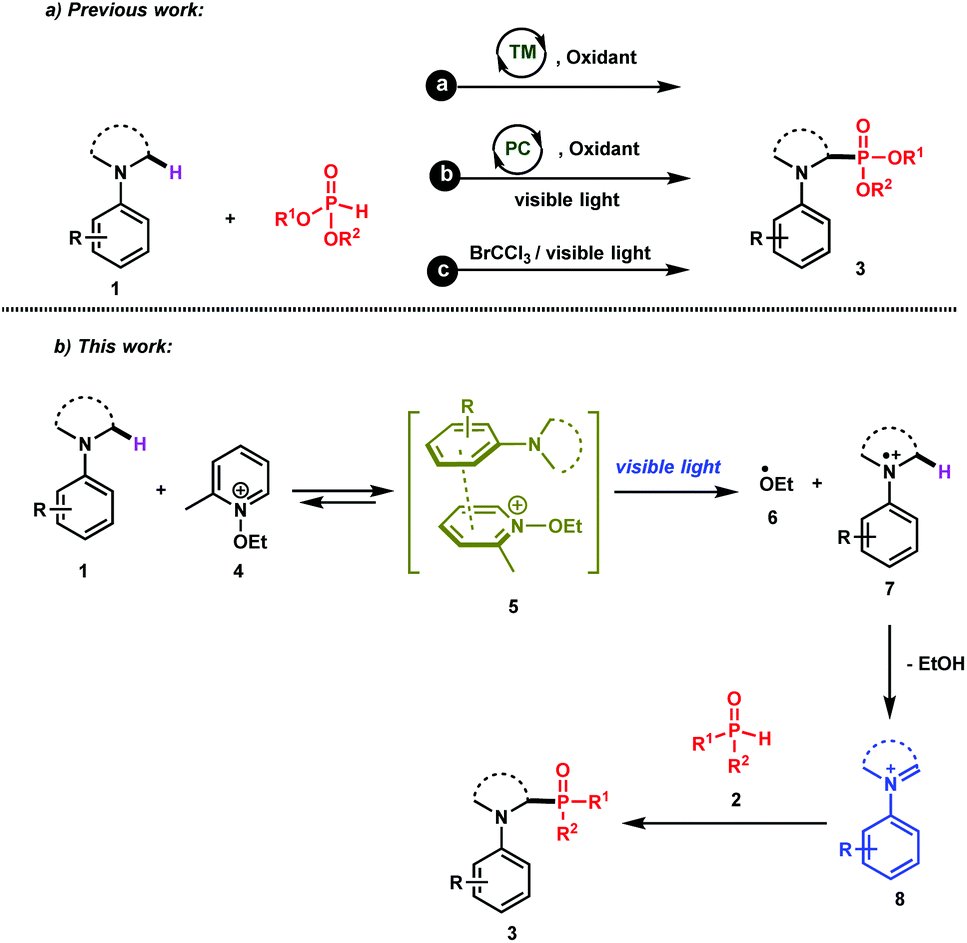 | ||
| Scheme 1 (a) Common approaches for the phosphorylation of tertiary amines. (b) Our working hypothesis for the generation of iminium ions through the association of N-aryl tertiary amines with N-ethoxy-2-methylpyridinium tetrafluoroborate under visible light irradiation. TM: Transition metal catalysts; PC: photocatalysts. | ||
While these methods are efficient, they have mainly been restricted to tetrahydroisoquinolines (THIQ) as tertiary amines and to H-phosphonates as phosphorus nucleophiles. For instance, secondary phosphine oxides 2 (R1R2P(O)H) are not compatible with many of the reaction conditions outlined above due to their low stability in the presence of oxidants.4 In order to solve this synthetic issue and to provide an efficient approach for the formation of C–P bonds between N-aryl tertiary amines and secondary phosphine oxides, we hypothesized the working reaction mechanism depicted in Scheme 1. It consists of the association of an N-aryl amine 1 with N-ethoxy-2-methylpyridinium tetrafluoroborate 4,5 which is a well-known electron-acceptor cation, to form an electron-donor–acceptor (EDA) complex 5.6
Upon visible light irradiation, a single electron transfer (SET) between both components should take place to give rise to the formation of the ethoxy radical (EtO˙, 6) along with the tertiary amine radical cation 7 (Scheme 1). Because of the very weak bond dissociation energy of the α C–H bond of the radical cation (BDE ≈ 42 kcal mol−1),7 a fast hydrogen atom abstraction (HAT) occurs with EtO˙ to form the corresponding iminium ion 8, which can be intercepted by secondary phosphine oxides (Scheme 1).
Results and discussion
To test this hypothesis, we started our investigation by combining N,N-dimethylaniline 1a (1 equiv.) with diphenylphosphine oxide 2a (1 equiv.) in the presence of N-ethoxy-2-methylpyridinium tetrafluoroborate 4 (1 equiv.) as an oxidant and NaHCO3 (1.2 equiv.) as a base in DMF under blue light irradiation. Under these conditions, a good conversion of 3a has been attained by 31P NMR after 24 h (Table 1, entry 1, 78%). Using sodium acetate as a base (entry 2), or carrying out the reaction in the absence of a base (entry 3), gave modest 63% and 47% conversions, respectively, thus showing the efficiency of NaHCO3 as a base in the reaction.|
|
||
|---|---|---|
| Entry | Solvent | Yield of 3ab, % |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), 4 (0.5 mmol), NaHCO3 (0.6 mmol), solvent (4 mL), 24 h. b NMR yields are determined from 31P NMR spectroscopy, using tri-n-octylphosphine oxide as an internal standard. c Base: NaOAc. d No base. e Irradiation with white LEDs. f 1a (0.5 mmol), 2a (1 mmol), 4 (0.6 mmol), NaHCO3 (0.6 mmol), solvent (4 mL), 24 h. g 1a (1 mmol), 2a (0.5 mmol), 4 (0.6 mmol), NaHCO3 (0.6 mmol), solvent (4 mL), 24 h. h The reaction is protected from external light with foil and heated at 40 °C to reproduce the heat caused by the LED lamp. i No oxidant. | ||
| 1 | DMF | 78 |
| 2 | DMF | 63c |
| 3 | DMF | 47d |
| 4 | CH3CN | 66 |
| 5 | DMSO | 75 |
| 6 | MeOH | 57 |
| 7 | DMF | 74e |
| 8 | DMF | 93f |
| 9 | DMF | 82g |
| 10 | DMF | 0h |
| 11 | DMF | 0i |
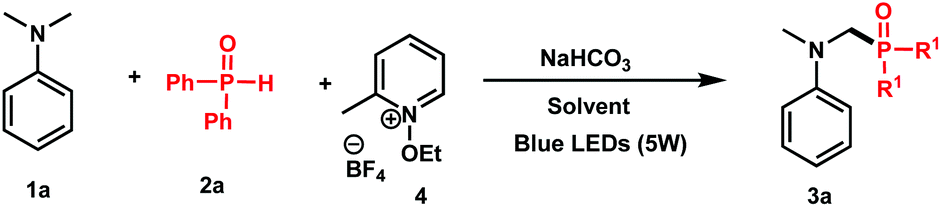
Although good results have been observed with acetonitrile (entry 4, 66%) and DMSO (entry 5, 75%), the conversion dropped in polar and protic solvents such as MeOH (entry 6, 57%). We furthermore tested the stoichiometry of the reactants and observed that using two equivalents of 2a with respect to 1a furnished 93% of 3a (entry 8), whereas 82% of conversion was observed when an excess of 1a was used (entry 9). We finally confirmed the necessity of light (entry 10) and oxidant (entry 11) as no reaction took place in their absence.
With the optimized reaction conditions in hand, we next explored the scope of the process by using different N-aryl amines and secondary phosphine oxides.
As shown in Fig. 1, various ring-substituted N,N-dimethylanilines carrying electron donor substituents 1b,d can be phosphorylated in excellent yields (81–87%). The reaction also proceeds well with N,N-dimethylanilines having fluoro or bromo groups at the para position of the aromatic ring 1e (71%) and 1f (68%), respectively. Interestingly, apart from the N,N-dimethylanilines, the phosphorylation worked efficiently with N,N-diethylaniline 1g (76%) and THIQ 1h (91%).
 | ||
| Fig. 1 Scope of the reaction. Reaction conditions: 1 (0.5 mmol), 2 (1 mmol), 4 (0.6 mmol), NaHCO3 (0.6 mmol), and DMF (4 mL) were irradiated with blue LEDs (5 W) for 24 h. | ||
Diarylphosphine oxides bearing different groups such as t-Bu (2i), OMe (2j), and CF3 (2k) at the para position of the aromatic rings were all applicable to the coupling with 72–93% yields. The reaction of sterically hindered di(o-tolyl)phosphine (2l) with 1a successfully afforded the desired adduct 3l in 74% yield. Finally, the reaction of 1a with H-diethylphosphite gave the corresponding phosphonated adduct 3m in 77% yield.
Having investigated the scope of the photoreaction with different N-aryl amines and secondary phosphine oxides, we next turned our attention to study the mechanism of this phosphorylation reaction. As shown in Fig. 2, while the UV-Vis spectra of 1a (blue) and 4 (red) in DMF are silent in the visible region, the spectrum of a mixture of both components (green) shows a bathochromic displacement above 400 nm. This new intermediate has been assigned to the formation of an EDA complex 5.6 We next employed Job's method of continuous variations to determine the molar donor/acceptor ratio, which has been found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in DMF at 20 °C (Fig. 3). Furthermore, an association constant KEDA of 2.7 M−1 has been determined photometrically in DMF by using the Benesi–Hildebrand method (see the ESI†). This constant is in good agreement with those reported by Melchiorre for the EDA complexes formed upon the association of substituted 1H-indoles with electron accepting benzyl and phenacyl bromides.6f
:1 in DMF at 20 °C (Fig. 3). Furthermore, an association constant KEDA of 2.7 M−1 has been determined photometrically in DMF by using the Benesi–Hildebrand method (see the ESI†). This constant is in good agreement with those reported by Melchiorre for the EDA complexes formed upon the association of substituted 1H-indoles with electron accepting benzyl and phenacyl bromides.6f
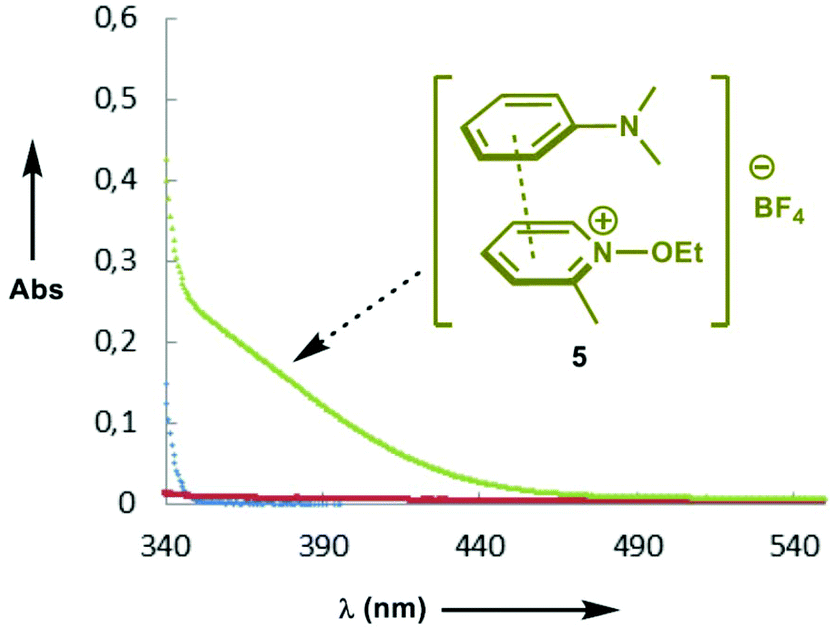 | ||
| Fig. 2 UV-Vis spectra of N-ethoxy-2-methylpyridinium tetrafluoroborate 4 ([4] = 5 × 10−2 M) (red); N,N-dimethylaniline 1a ([1a] = 5 × 10−2 M) (blue); a mixture of 1a and 4 (green) in DMF at 20 °C. | ||
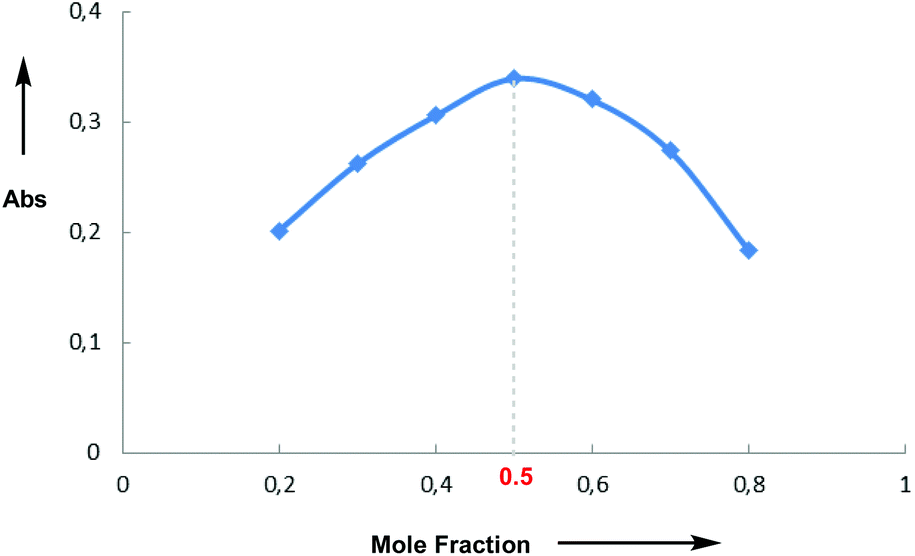 | ||
| Fig. 3 Job's plot for the association of 1a and 4 in DMF at 20 °C. | ||
Light on/off experiments of the reaction of 1a with 2a under the optimized reaction conditions have been conducted and revealed an interruption of the reaction progress in the absence of light, and recuperation of reactivity upon further illumination (see Fig. S5, ESI†). Furthermore, a quantum yield (Φ) of 0.28 was determined, indicating that a radical chain process is presumably not operative.8
To characterize the nature of the radical species involved in this reaction, a spin trap EPR experiment was undertaken in the presence of N-tert-butyl-α-phenylnitrone (PBN) 9 as a spin trap. Interestingly, the ethoxy-PBN adduct 10 was detected and the determined hyperfine coupling constants (aN = 13.7 G and aH = 1.9 G) were found to be in good agreement with those previously reported in the literature (Fig. 4).5
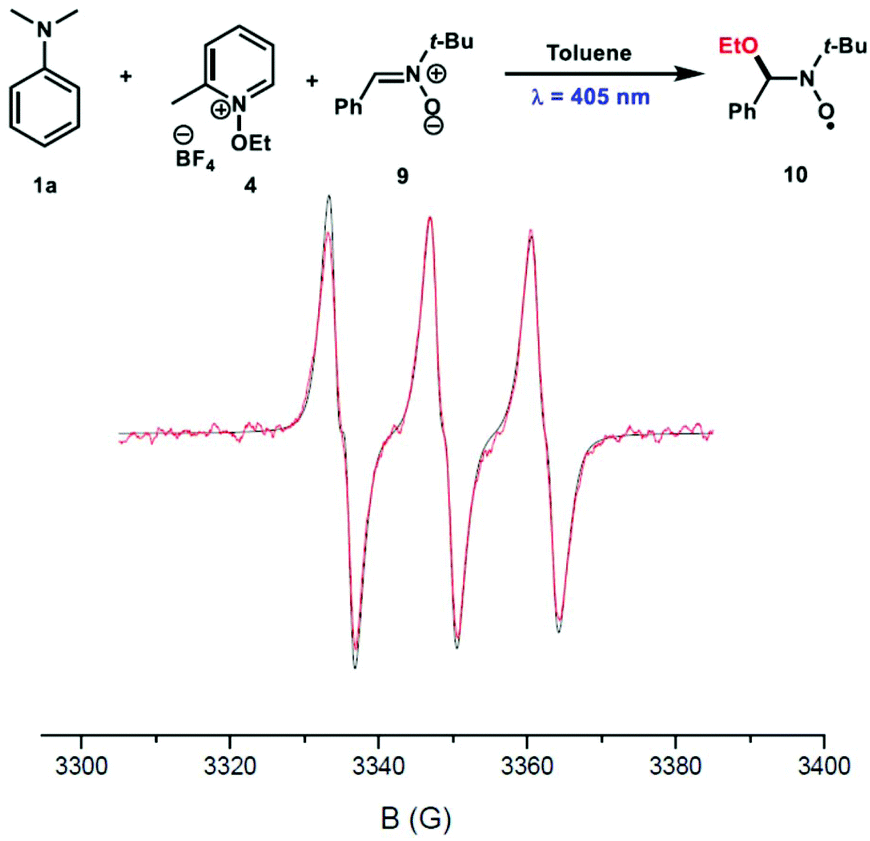 | ||
| Fig. 4 EPR spectra of spin adduct 10 generated in toluene from the reaction of N,N-dimethylaniline 1a (1 × 10−2 M) with N-ethoxy-2-methylpyridinium tetrafluoroborate 4 (1 × 10−2 M) in the presence of PBN 9 (1 × 10−2 M) at 20 °C. | ||
Finally, in order to elucidate the intermediacy of the iminium ion 8 derived from N,N-dimethylaniline 1a, we recorded the 1H NMR spectrum of the reaction of 1a with 4 in CD3CN. However, because of its high electrophilicity,9 it reacted with the generated ethanol to form the adduct 11 (Scheme 2). Interestingly, when THIQ (1h) was used instead of 1a, the corresponding iminium ion was detected by NMR spectroscopy.
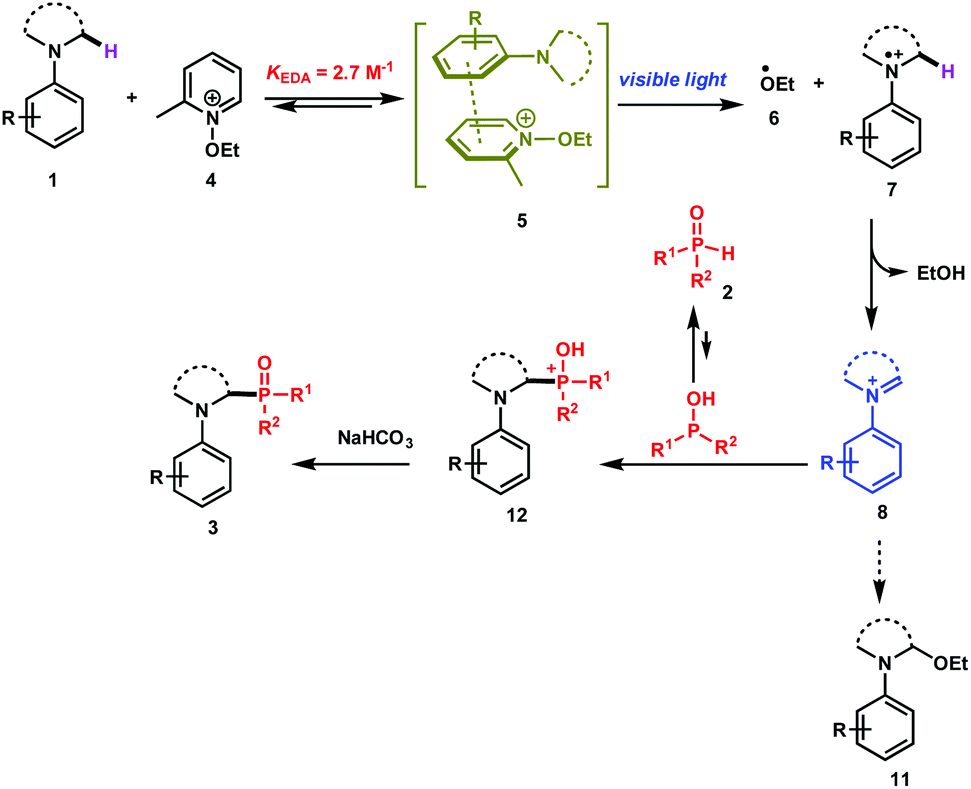 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Based on these physical organic investigations, a reaction mechanism is proposed (Scheme 2). The reaction starts with the association of the amine (electron donor) 1 with the pyridinium salt 4 to form an EDA complex 5. After visible light irradiation, a single electron transfer takes place to generate the ethoxy radical (6) and the aminium radical cation 7. A fast hydrogen atom abstraction takes place to generate the iminium ion 8 that reacts with the diarylphosphine oxide (P(III) tautomer) to form the phosphonium ion 12. The deprotonation of the latter with a base yields the desired product 3.
Conclusion
In conclusion, we have developed a metal-free, visible-light-induced method for the phosphorylation of N-aryl amines 1 with secondary phosphine oxides 2. The reaction proceeds under mild, catalyst-free conditions. The key to the success of the reaction was the formation of EDA complexes between the amines 1 and the electron-deficient pyridinium 4. The proposed reaction mechanism was supported by detailed mechanistic investigations, including spectrophotometry, NMR spectroscopy and EPR spectroscopy. We anticipate that this operationally simple approach should be amenable with other nucleophiles to synthesize various α-functionalized amines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the CNRS, Normandie Université, INSA Rouen, and Labex Synorg (ANR-11-LABX-0029) for financial support. V. Q. is grateful to the “Ministère de l'enseignement supérieur et de la recherche” for a fellowship. N. Ch. thanks the University of Monastir for a scholarship.Notes and references
- For selected recent examples, see:
(a) O. Baslé and C.-J. Li, Chem. Commun., 2009, 4124 RSC
; (b) W. Han and A. R. Ofial, Chem. Commun., 2009, 6023 RSC
-
(a) S. C. Fields, Tetrahedron, 1999, 55, 12237 CrossRef CAS
- For seminal reviews, see:
(a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS
- J. Xu, P. Zhang, X. Li, Y. Gao, J. Wu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2014, 356, 3331 CrossRef CAS
- V. Quint, F. Morlet-Savary, J. F. Lohier, J. Lalevée, A. C. Gaumont and S. Lakhdar, J. Am. Chem. Soc., 2016, 138, 7436 CrossRef CAS
- For a seminal work on the formation of EDA complexes between N-heteroatom-substituted pyridinium cations and arenes, see:
(a) K. Y. Lee and J. K. Kochi, J. Chem. Soc., Perkin Trans. 2, 1992, 1011 RSC
- H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860 CrossRef CAS
-
(a) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC
- The electrophilicity of the methyl(phenyl)methyleneammonium ion has been estimated to be −5.17; see: H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qo00985f |
| This journal is © the Partner Organisations 2019 |