
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H activation-annulation on the N-heterocyclic carbene platform
Champak Dutta
 and
Joyanta Choudhury
*
and
Joyanta Choudhury
*
Organometallics and Smart Materials Laboratory, Indian Institute of Science, Education and Research Bhopal, Bhopal 462 066, India. E-mail: joyanta@iiserb.ac.in
First published on 6th August 2018
Abstract
Ring-fused cationic N-heterocycles are an important class of organic compounds recognized to be of significant interest in diverse research areas including bioactivity, materials chemistry, supramolecular chemistry, etc. Toward the synthesis of such molecules, recently unique chemistry has been explored utilizing a novel conjugative action of NHC ligands as a functionalizable directing group in rhodium(III)-catalyzed aromatic/heteroaromatic/non-aromatic C–H activation and subsequent annulation of various imidazolium salts with internal alkynes. This review highlights the initial development and underscores the potential of this chemistry.
 Champak Dutta | Champak Dutta obtained his B.Sc. from Dibrugarh University and M.Sc. from Tezpur University, Assam, India in 2011 and 2013 respectively. After that, in 2014 he joined the group of Dr Joyanta Choudhury at IISER Bhopal, India for his doctoral studies. He works on NHC-directed C–H activation-functionalization catalysis based on Rh and Co catalysts. |
 Joyanta Choudhury | Joyanta Choudhury obtained Ph.D. in 2006 from IIT Kharagpur, India working under Professor Sujit Roy on the development of well-defined Ir–Sn heterobimetallic complexes for aromatic C–H functionalization. He then moved to The Scripps Research Institute, Florida, USA for postdoctoral work with Professor Roy A. Periana in the field of alkane functionalization chemistry. In 2008, he received Marie Curie International Incoming Fellowship from the European Union and went to the Weizmann Institute of Science, Israel to work with Professor Milko E. van der Boom on coordination-driven molecular assemblies on solid surfaces. Currently, he is an Associate Professor at the Department of Chemistry, IISER Bhopal, India. His research focusses on (a) developing new reactivity with metal–NHC complexes and (b) small molecule activation-functionalization including CO2, H2, alkanes, arenes, and H2O to address renewable energy related problems. His research work on ‘Switchable Catalysis’ has been highlighted in the popular Chemistry magazine, ChemistryWorld. Recently, he has been featured as ‘Movers & Shakers’ by the industry-leading magazine ‘The Catalyst Review’, published by The Catalyst Group (TCG), PA, USA. |
1. Introduction
Their unique stereoelectronic properties and versatile metal–ligand bonding bestow N-heterocyclic carbenes (NHCs) with remarkable importance in various research fields including homogeneous catalysis, materials and medicinal chemistry.1–7 The NHC ligand acts as a strong σ-donor and considerably strong π-acceptor when bound to transition metals thereby affording desired stability and activity. From the frontier molecular orbitals (FMO) point of view, the sp2 hybridized orbital (HOMO) on the carbon atom acts as a σ-donor whereas the perpendicularly located vacant p-orbital (LUMO) acts as a π-acceptor from the metal bound to the carbon atom (Fig. 1). The robust bonding features impart the key criteria of excellent stability to metal–CNHC backbones so that tremendous catalytic and material applications have been possible with metal–NHC complexes, without any self-transformative side reactions.3–7 However, due to the same features, NHCs were found to be reluctant to function as a directing group and take part in powerful C–H functionalization catalysis leading to C–C and C-heteroatom bond-forming reactions.8 Very recently, this unique reactivity pattern on the NHC platform has been explored with the help of transition metals, especially Rh, to develop new catalytic protocols toward synthesizing various ring-fused cationic N-heterocyclic structural motifs. These and similar motifs are synthetically demanding due to their presence in various biologically active compounds and pharmaceuticals,9 and due to potential in organic light-emitting diode (OLED) applications.10 The main chemical strategy involves C–H activation/cyclometalation–insertion–annulation/functionalization sequence surrounding the metal–NHC templates accessible from desired quaternized ‘azolium’ salts as precursors/substrates (Fig. 2). While substituted imidazolium salts are a priority as substrate as they form building blocks for vastly known M–NHC motifs, internal alkynes have been found to be ideal inserting partner for coupling to the azolium substrates to furnish functionalized products (Fig. 2). The initial development and the potentiality of this chemistry have been highlighted in this article.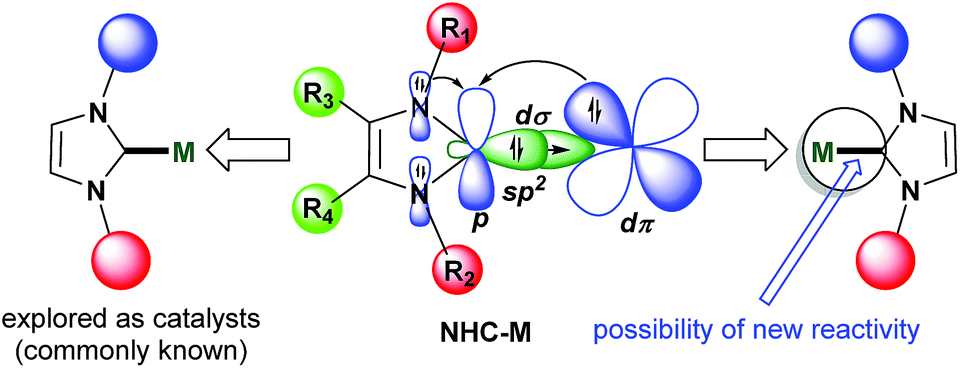 | ||
| Fig. 1 Scope for exploitation of reactivity in M–NHC motifs. | ||
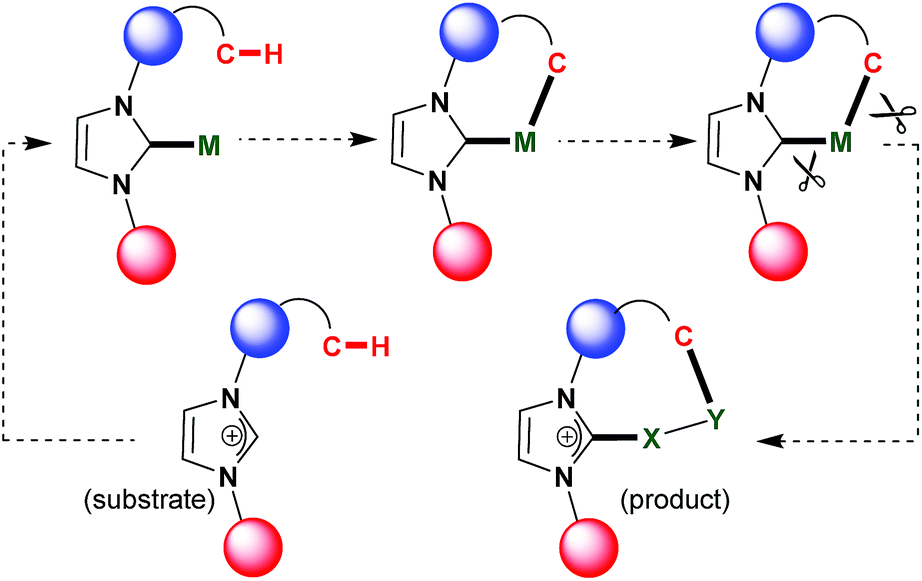 | ||
| Fig. 2 Strategy of C–H functionalization on M–NHC platform. | ||
2. Chemistry at Rh–NHC template
2.1 Background
For a long time Rh–NHC templates are being used as catalyst for various organic transformations.11–13 The idea of productive functionalization of Rh–NHC itself toward further conversion into useful end products was not realized until recently. In the context of C–H activation, initially, Peris and co-workers reported an Ir(III)–NHC complex which could act as catalyst for deuteration of organic molecules via C–H activation process.14a Similarly, Nolan and co-workers reported a unique double cyclometalation process as a result of double C–H activation in a Rh(III)–NHC complex.14b In due course of time, stoichiometric reactions involving insertion of internal alkyne to Rh–NHC complexes and reductive elimination of such species were also reported.14c A catalytic functionalization of NHC motifs was reported by the group of Cavell in 2008 catalysed by Ni(II) catalyst.15a They successfully exhibited catalytic intramolecular annulation of alkyl substituted imidazolium salts in presence of Ni(0)/Ni(II) redox couple as catalyst. Another catalytic intramolecular annulative transformation of alkyl substituted benzimidazoles reported by Bergman and Ellman involved Rh(I)–NHC moiety as an intermediate. In this work, the authors reported to form a Rh(I)–NHC–alkene complex that acted as resting state of the catalyst for intramolecular catalytic transformation of alkyl substituted benzimidazoles.162.2 Intermolecular monoannulation at non-chelating Rh–NHC template
The first idea of catalytic intermolecular transformation of Rh–NHC template was established by our group where we explored the unique directing group behaviour of NHC to activate aromatic sp2 C–H bond.17 Using Rh–NHC template as the platform we succeeded in functionalizing the Rh–NHC motif to useful organic products. The directing group (DG) ability of NHC was used in activating aromatic sp2 C–H bond of a suitably placed phenyl ring from NHC group with the help of Rh(III) catalyst (Scheme 1). This Rh(III)-catalyzed intermolecular aromatic C–H activation annulation involved coupling of the NHC backbone with an internal alkyne as electrophile. The DG assisted C–H activation is not a new strategy and has been in practice among synthetic chemists for a long time.8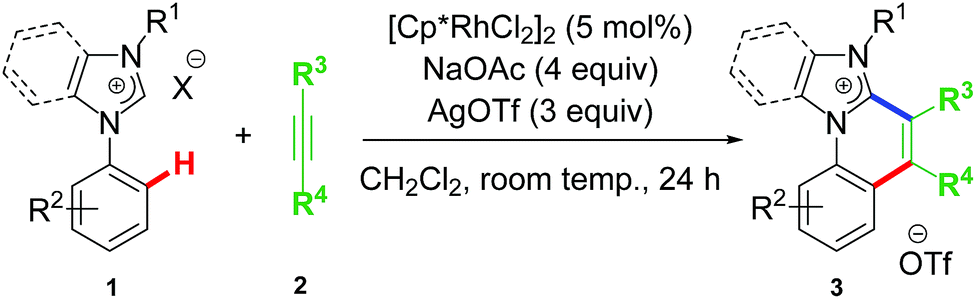 | ||
| Scheme 1 First report on catalytic transformation of Rh–NHC template. | ||
The Rh–NHC backbone was generated by using aryl imidazole salts. The annulation behaviour of Rh–NHC template was validated initially in a stoichiometric fashion. Using Ag transmetallation procedure, Rh–NHC complex was synthesised in situ and later treated with diphenylacetylene as a coupling partner to give the annulated organic compound 3a (Scheme 2a). The structural confirmation of 3a was made by various analytical techniques and single crystal X-ray diffraction (SC-XRD) analysis. Motivated by this information, a catalytic transformation was carried out using phenylimidazolium iodide; 1a as NHC precursor and diphenylacetylene to get the same product 3a′ (Scheme 2b). The successful catalytic reaction needed 5 mol% of [RhCp*Cl2]2 as catalyst, 4 eq. of NaOAc as base, 3 eq. of AgOTf as oxidant and dichloromethane as solvent under optimised conditions. This result also confirmed the successful reductive elimination from a Rh–NHC template without undergoing side reaction or decomposition. The annulation protocol was found to be operative for various substituted imidazolium substrates and internal alkynes as well. The end products of this annulation protocol were a newer class of cationic aromatic conjugated polyaromatic hydrocarbons which might have potential in organic material applications, pharmaceutics, imaging etc.9,10
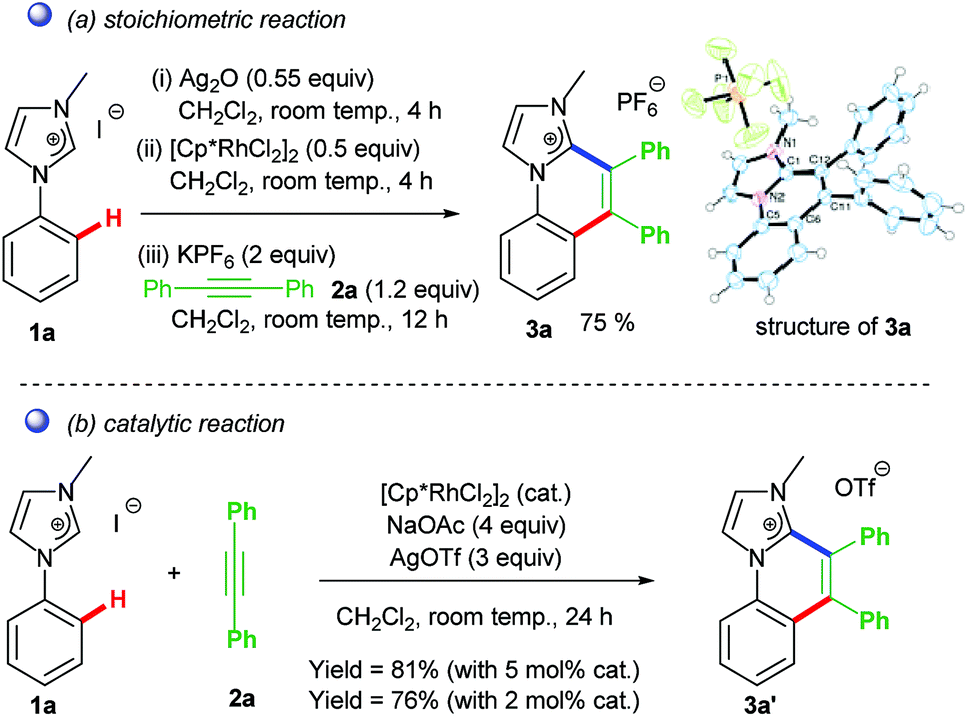 | ||
| Scheme 2 Stoichiometric/catalytic C–H activation–functionalization directed by NHC. | ||
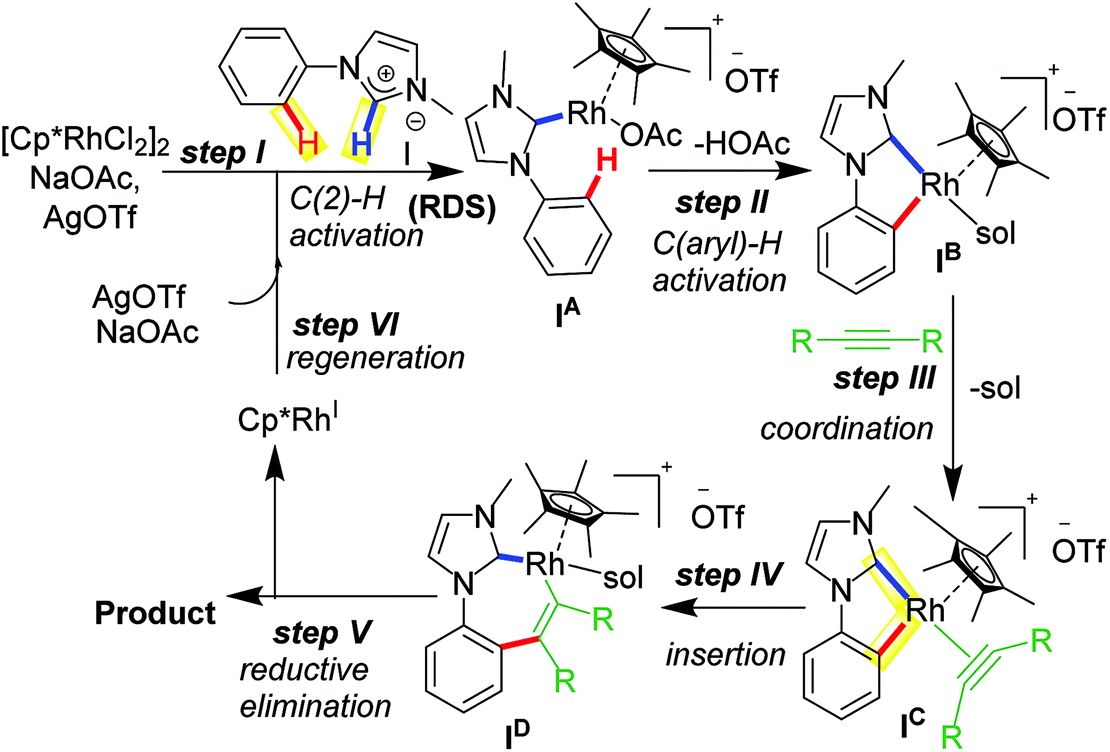
A widely accepted mechanism for C–H activation catalyzed by Rh(III) species is concerted metalation deprotonation (CMD) pathway assisted by a carboxylate base. Numerous experimental and theoretical reports on CMD-type mechanism validated for DG-assisted C–H activation-annulation are known.18 These investigations suggested involvement of carboxylate ligand acting as internal base to deprotonate the desired proton to undergo cyclometalation. We investigated the plausible mechanism of mono-annulation process for aromatic sp2 C–H bond activation/functionalization at Rh–NHC platform.19 After rigorous control studies supported by experimental and theoretical evidences and SC-XRD structural confirmations, a mechanism as shown in Scheme 3 was proposed. After initial formation of Rh–NHC coordinated species, the carboxylate (OAc) ligand at the Rh center acts as an internal base to abstract the proton from phenyl ring to undergo cyclometalation. An iodo-coordinated cyclometalated intermediate 4 (iodo version of IB) was synthesized and structural confirmation was done by SC-XRD technique. The intermediacy of 4 was confirmed by various control experiments under stoichiometric and catalytic conditions. To the cyclometalated species, the alkyne coordinates and inserts into Rh–CPh bond instead of Rh–CNHC bond. Thermodynamically viable path is the former, supported by theoretical calculations and later by X-ray structure confirmation of a seven-membered rhodacycle intermediate. The seven-membered intermediate undergoes reductive elimination to give desired product. Silver mirror in the reaction vessel was observed as well because of formation elemental Ag as a result of reductive elimination.20 From kinetic studies it was realized that imidazolium C2–H activation was the rate-determining step (rds) for this catalytic transformation.
 | ||
| Scheme 3 Plausible mechanism for C–H annulation at non-chelating Rh–NHC platform. | ||
2.3 NHC directed pyridine C–H activation/annulation at Rh–NHC platform
The success with NHC directed C–H activation/annulation prompted to extend its viability in more difficult C–H activation/annulation of pyridine ring. In 2015, our group reported similar catalytic annulative sp2 C–H activation/functionalization based on 4-pyridyl-Rh–NHC platform (Scheme 4).21 It is noteworthy that although several DG-assisted C–H functionalization were known at that time, pyridine functionalization catalysed by transition metal via similar strategy was limited mostly to amide directing group only.22,23 The Li group reported synthesis of quinolines via Rh(III) catalysed annulation strategy where DGs were amide, pyridine and imidazole as well.23 From theoretical standpoint, functionalization of pyridine C–H is more difficult because of high Lewis basicity of pyridine N atom that may coordinate to the metal strongly. This could be an inhibiting effect on the overall reaction. However, in the work reported by our group, the pendant pyridine-coordination enhanced the reactivity thereby providing a remote support. One significant change achieved in this work was reduction of catalyst loading to 2.5 mol% from earlier reported 5 mol% of [RhCp*Cl2]2.17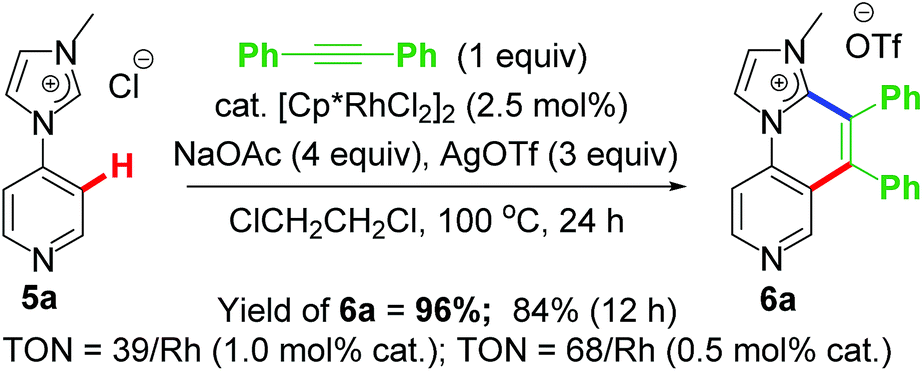 | ||
| Scheme 4 C–H activation/annulation at 4-pyridyl-Rh–NHC template. | ||
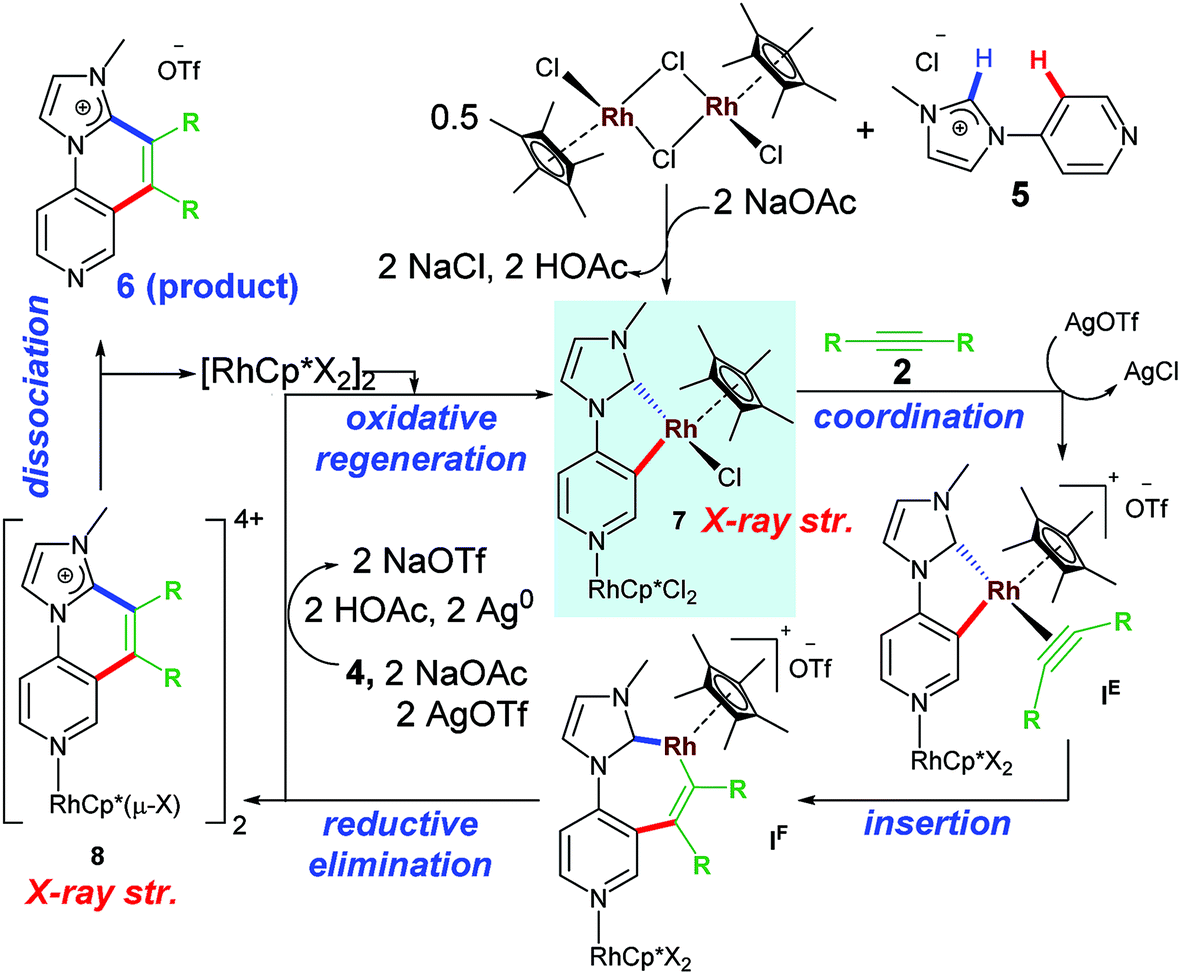
The plausible mechanistic pathway for this type of C–H activation/annulation could be depicted as similar to Scheme 5 above. However, presence of a coordinating N atom at 4-position could influence the reactivity in such cases. In fact, the first rhodacycle isolated from a stoichiometric reaction, contained a pendent Rh–N coordination at the pyridine ring. This particular work shed light upon the mechanistic path taken in terms of formation of both early and late stage intermediates. The free N at the 4-position of pyridine ring acts as a remote coordination site besides usual cyclometalation of Rh(III) to the NHC backbone. The pendant Rh(III) remains even after reductive elimination step leading to desired annulated product. Presumably, during work up stages the pendant centre was removed to furnish imidazo-[1,2-a][1,6] naphthyridinium salts as new products (Scheme 5).
 | ||
| Scheme 5 Plausible mechanism for NHC directed C–H activation/annulation of pyridyl C–H bond. | ||
2.4 Cascade annulation at non-chelating Rh–NHC motif
After the mono-annulation reaction of phenylimidazolium salts, the structural backbone of the products could, in principle, undergo another annulative functionalization in cascade if double equivalents of alkyne were used. This could confirm functionalization of Rh–aNHC template (aNHC = abnormal NHC) in an annulative pattern. This possibility was explored as well. Our group reported a double functionalization in cascade involving Rh–NHC template for the first time.24 In this particular work, we exploited the potential abnormal-NHC functionalization in addition to normal NHC-directed annulation. The double annulation resulted in nicely framed polyaromatic architectures of highly conjugated nature (Scheme 6).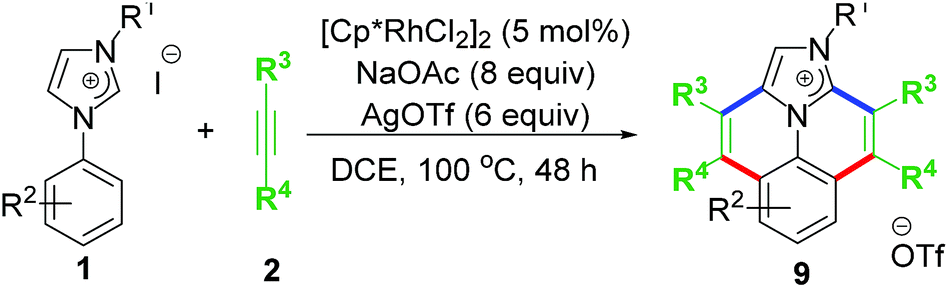 | ||
| Scheme 6 Cascade functionalization of Rh–nNHC and Rh–aNHC template. | ||
From mechanistic point of view, the first annulation step was expected to follow a similar mechanism as shown in Scheme 7. Later, to the product of first annulation, in presence of Rh(III) precursor and NaOAc, another activation and cyclometallation is possible. Plausibly, an abnormal Rh–NHC complex 10 acts as an intermediate in this step (Scheme 7). To the complex 10, another equiv. of internal alkyne 2 inserts to form 7-memebered inserted complex that may undergo reductive elimination. The end result of the reductive elimination is the desired bis-functionalized cationic annulated product.
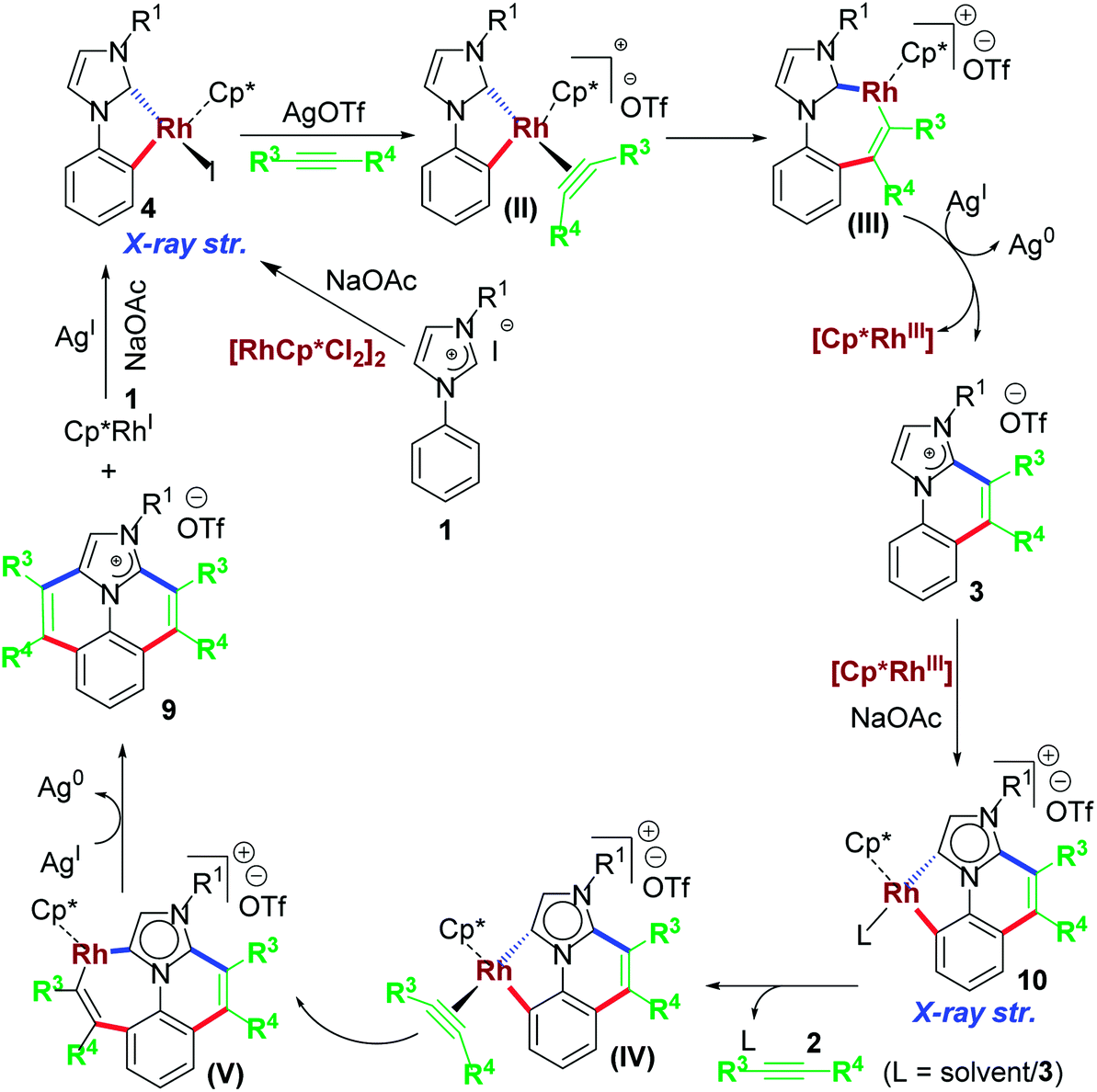 | ||
| Scheme 7 Proposed mechanism for cascade C–H activation/annulation. | ||
The elevated interest in functionalizing NHC-backbone was displayed by overlapping of almost similar reports by the Wang group on cascade annulation which involved Rh–NHC template and similar ligand backbone.25a They reported an annulation protocol involving Rh(III) as catalyst and Cu(II) salts as oxidant and base in t-AmOH as solvent at a reaction temperature of 80 °C (Scheme 8).
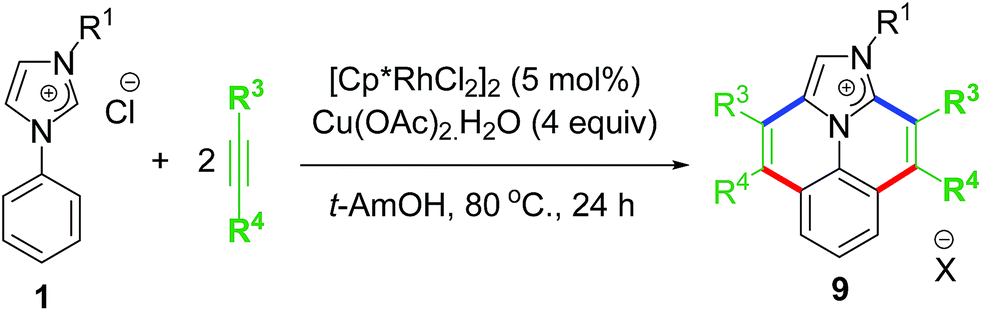 | ||
| Scheme 8 Wang's procedure of C–H activation-annulation at Rh–NHC platform. | ||
The Wang group later explored their annulation protocol in multiple C–H activations of arylimidazoles too, thereby resulting substituted benzo[ij]imidazo[2,1,5-de]quinolizine based polyheteroaromatic compounds as end products.25b Contemporarily, they also succeeded in replacing expensive Rh catalysts with comparably cheaper Ru(II), but higher reaction temperature of 130 °C and both Cu(OAc)2 and AgSbF6 were required as additives.25c Later, Li, Wang and other few groups extended this kind of annulation strategy to various other directing groups and coupling partners as well.26
2.5 Annulative transformation of non-aromatic sp2 C–H bond
After success with aromatic sp2 C–H activation/annulation, our group reported another successful functionalization of Rh–NHC template wherein the NHC backbone was based on non-aromatic imidazolium salts. In this work, we succeeded in activating various non-aromatic sp2 C–H bonds and coupling them with internal alkynes to form cationic annulated scaffolds (Scheme 9).27 Interestingly, no intramolecular C–H activation/annulation of vinylic group was observed under these reaction conditions. Two protocols using AgOTf or Cu(OAc)2 were achieved. The latter did not require any external base as it acted as internal base too. It was observed that under standard reaction conditions there was a competition between phenylic C–H bond and vinylic C–H bond leading to mixture of annulated products of both types.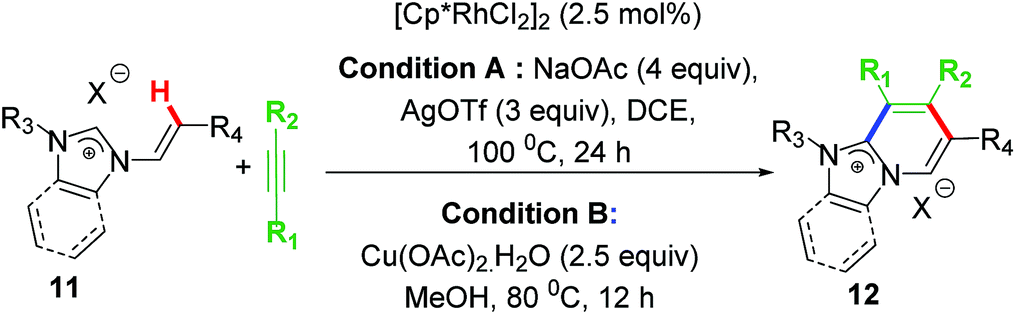 | ||
| Scheme 9 Functionalizing Rh(III)–NHC template for non-aromatic sp2 C–H activation. | ||
However, complete switching of reactivity toward pyridyl sp2 C–H activation over the vinylic sp2 C–H activation under the competitive situation was observed when 4-pyridyl substituent was used. These results suggested that the regioselectivity in the sp2 C–H activation could be guided by the electronic nature of the N-substituents within the NHC motifs. In agreement with the cascade annulation report earlier,24 excess alkyne provided a double sp2 C–H (aromatic as well as non-aromatic) activated product at higher temperature, wherein the second annulation was directed by an abnormal carbene generated from C(5)Im–H deprotonative metalation.
The mechanistic steps were presumed to follow a similar pathway as in monoannulation process (Scheme 10). In fact, isolation of rhodacycle intermediate 13 and stoichiometric and catalytic intermediacy test supported the proposed intermediates in the mechanism. Undoubtedly, this work motivated others to explore other annulative transformations and mechanistic investigations too.28
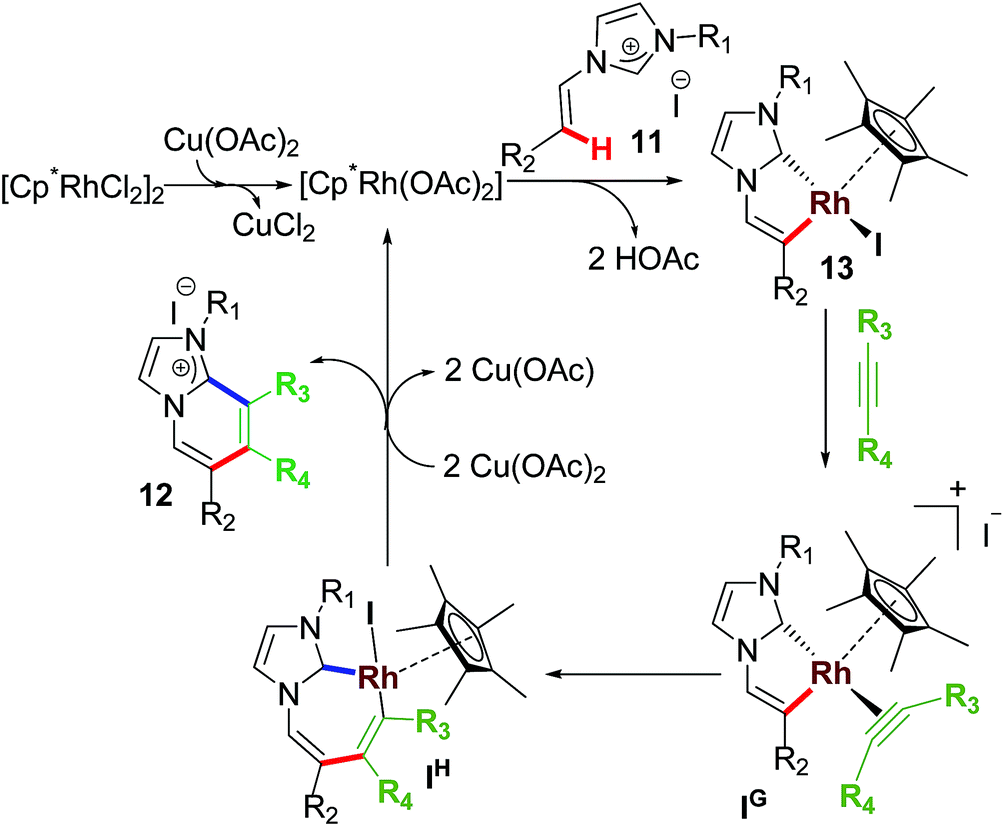 | ||
| Scheme 10 Proposed mechanism for non-aromatic sp2 C–H activation/annulation. | ||
2.6 Rollover annulation at chelating Rh–NHC template
Upon successful exhibition of directing group behaviour of NHC to activate aromatic17,19,21,24 and non-aromatic sp2 C–H bonds at Rh–NHC platform,27 we extrapolated our investigations to difficult-to-functionalise chelating organic molecules having strongly coordinating heteroatoms in suitable metal-chelating positions. Thus, in early 2016, our group reported an unprecedented ‘rollover’ C–H activation-annulation of such chelating molecules based on 2-pyridylimidazolium halide motifs as NHC backbone.29 In this work, we reported a ‘switchable rollover annulation’ protocol wherein Rh–NHC metallachelate underwent a rollover process to give C–C coupled annulated product instead of expected C–N coupled product without any ring opening process (path a, Scheme 11).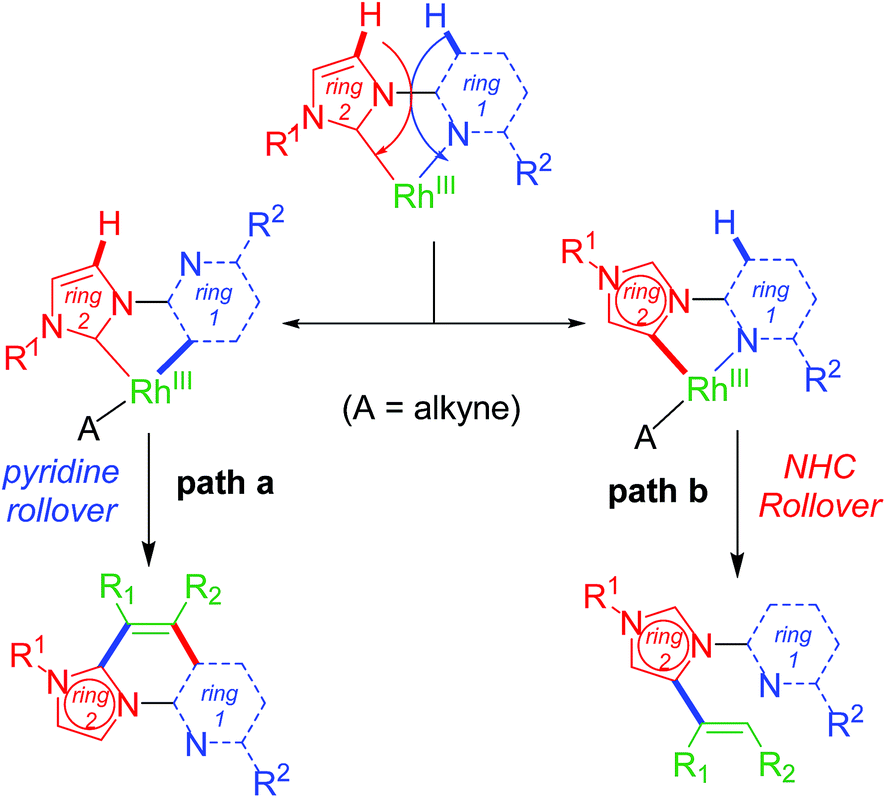 | ||
| Scheme 11 Bimodal ‘rollover’ process at chelated Rh–NHC platform. | ||
The ‘rollover’ process signifies initial formation of a metallachelate where the metal centre is bound to a heteroatom such as N or S.30 Then, under certain conditions and influencing factors, the ring opens followed by a rotation of the pivotal bond and finally formation of a new metallacycle wherein the connectivity of the metal is no more with the earlier heteroatom. This kind of rollover cyclometallation (ROCM) was known for various square planar complexes based on Pt metal, reported by the Zucca group (Scheme 12).31
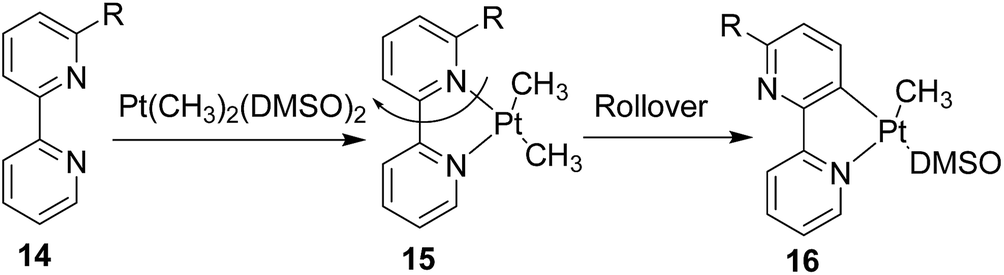 | ||
| Scheme 12 Steric and electronics induced stoichiometric rollover cyclometalation. | ||
Usually, the rollover process in these stoichiometric transformations was induced by trans effect of the substituent on square plane, anti to the heteroatom.30,31 Solvent-induced rollover was also observed in these cases. Cheng group investigated the ROCM process in both strong bidentate and tridentate heteroaryl ligands.32 In 2015 Miura and Satoh reported ROCM process for the synthesis of indolonaphthyridines from 2-pyridylindole derivatives, catalysed by Rh(III).33a Oro group also reported a similar rollover process in 2014, for 2-thienylpyridine C–H activation catalysed by a Rh(III)–NHC complex.33b
In addition to usual challenges associated to C–H activation/functionalization, chelate functionalization involves two major problems. The former being the facile formation of stable metallachelate resistant to ring opening process without undergoing decomplexation and the latter is desired and selective coupling with incoming electrophile to get functionalized products without undergoing decomposition. Owing to these facts, reports on ‘rollover’ C–H functionalization are extremely rare till date. However, the pyridine and thiophene motifs are in the suitable range of forming metallachelate of desired stability that may undergo ROCM process. Regardless, we succeeded in functionalization of strongly bonded Rh–NHC metallachelate which gave a newer class of cationic C–C coupled polyheteroaromatics (Scheme 13).
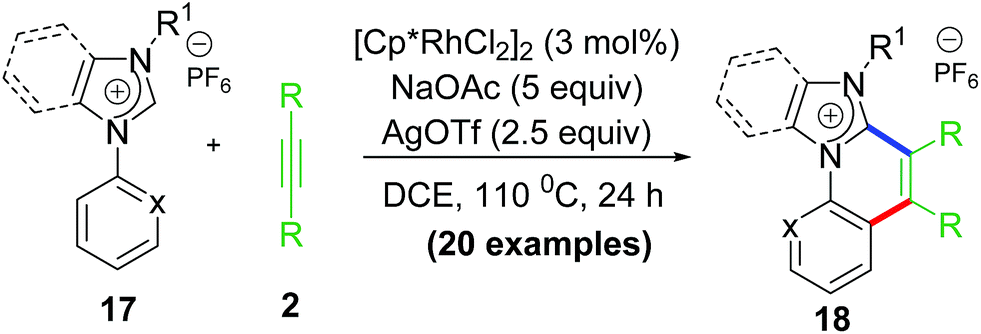 | ||
| Scheme 13 Rollover functionalization of Rh–NHC metallachelate. | ||
This type of rollover transformation giving annulated product is an outcome of pyridine rollover as apparent from path a in Scheme 11. The mechanistic investigations suggested that initially an Npy-Rh–NHC metallachelate 19 (Scheme 14) is formed. It was understood that in the above-mentioned rollover C–H functionalization strategies, stereoelectronic factors operating at the catalytic metal center play a pivotal role for the success to overcome high activation barrier rollover process to initiate the subsequent C–H bond cleavage step. Other factors such as ligand trans effect, intramolecular H-bonding, and water-assisted H-bonding were proposed as the driving force in this particular type rollover functionalization process.32,34
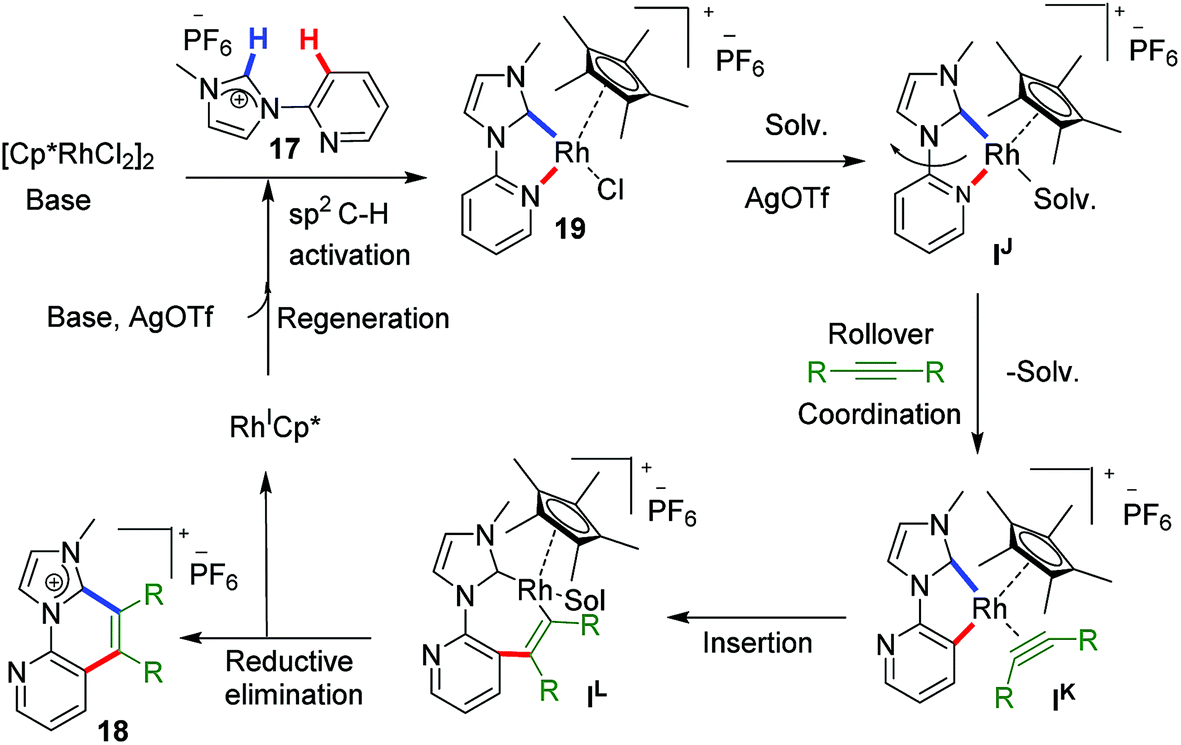 | ||
| Scheme 14 Plausible mechanism for ‘rollover’ C–H annulation at Rh–NHC platform. | ||
Under influence of these factors a pyridine rollover process is expected in complex 5 that may undergo alkyne insertion and via reductive elimination final annulated product is obtained. Presumably, the pyridine rollover in piano-stool type Rh–NHC complexes is driven by an induced bond weakening and hemi-lability of pyridine ligand attached to a highly electrophilic cationic, alkyne coordinated Rh(III) centre generated during the reaction. However, till date the exact step of the rollover process has not been confirmed for piano-stool type of complexes.
The path b in Scheme 11 suggests the possibility of a non-annulative transformation at Rh–NHC template. By virtue of NHC rollover of the rhodium metallachelate, alkenylation was observed under certain constraints (Scheme 15). The NHC ROCM process could possibly be due to the formation of stronger aNHC–Rh bonds with stronger σ-donation from aNHC site. The steric control generated from N-substituent of imidazolium ring and electronics of alkyne incorporated are also determining factors in such rollover process. A subtle steric and electronics balance between the normal and the abnormal NHC ligands dictates the direction of the rollover process. We observed NHC rollover when incoming alkyne is a very electron-deficient one such as dimethylacetylenedicarboxylate (DMAD) or when a very bulky group (such as adamantyl) is present within the imidazolium ring. Hence, the results were alkenylated products via non-annulative transformation at Rh–NHC template. Prior to this report, few similar Pd–NHC metal complex based non-annulative catalytic transformations were reported by Cavell15b and other groups during 2000–2008.15c,d
 | ||
| Scheme 15 NHC rollover functionalization of Rh–NHC metallachelate. | ||
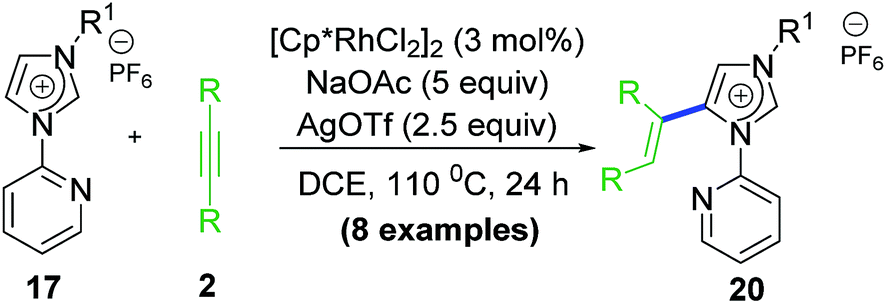
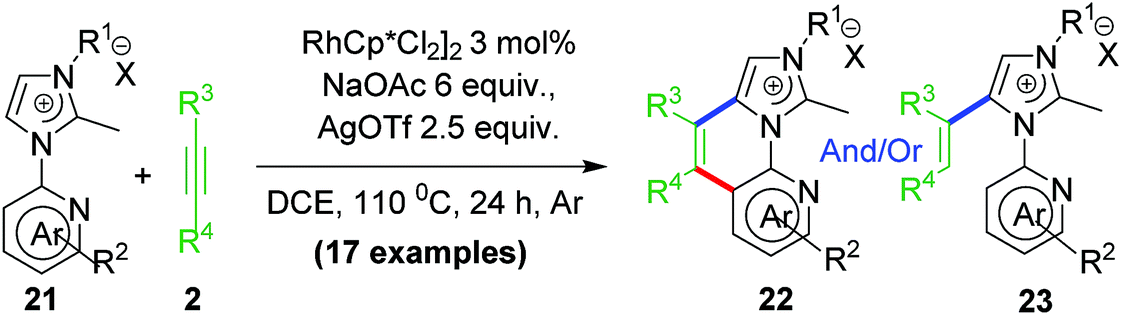
2.7 Rollover transformation at Rh–aNHC platform
NHC rollover is a less general process and controlled by steric and electronic factors to greater extent than pyridine rollover process. Also, NHC rollover was expected to be prominent for abnormal aNHC–Rh chelate than normal nNHC–Rh chelate. Based on these observations, our group investigated C–H activation–functionalization at Rh–aNHC platform too. A specially designed ligand via blocking the C2 position of the imidazolium backbone, provided the desired abnormal Rh–aNHC metallachelate template (Scheme 16). Functionalization of Rh–aNHC template is expected to be more difficult attributed to their enhanced stability as result of stronger σ-donation from NHC to metal centre which can terminate the reaction from undergoing functionalization or decomposition under reaction conditions.35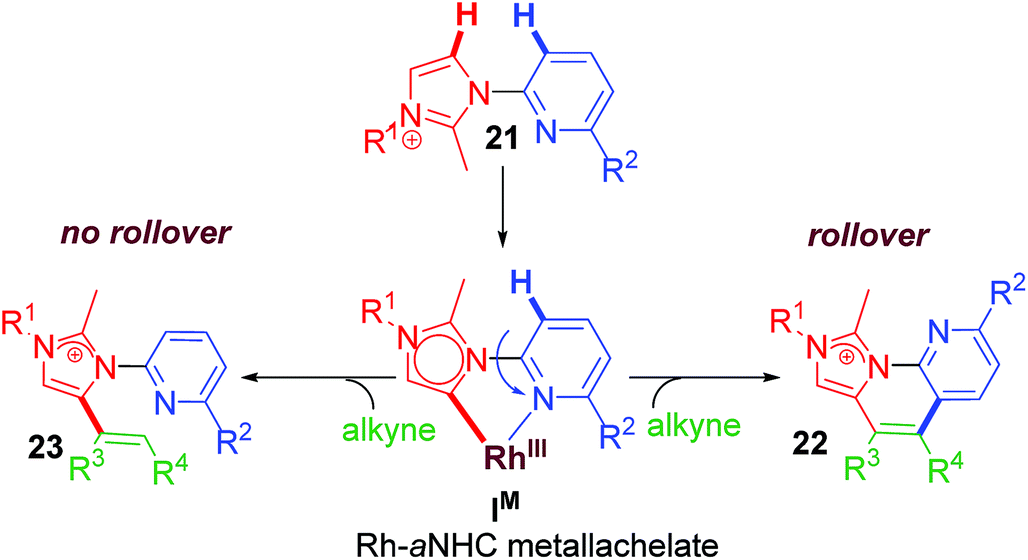 | ||
| Scheme 16 Stereoelectronically biased C–H functionalization at Rh–aNHC platform. | ||
In this context, C–H cyanation of arylimidazo[1,2-α]pyridines reported by Song and Hao group,34a C–H diamination of arylpyridines by Lu group,34b and rollover annulation at tridentate motifs by Chang group added significant value to the field of rollover functionalization of metallachelates.32b One recent report from Thiel group demonstrated ROCM process in the synthesis of Ir–Pt or Ir–Pd heterobimetallic complexes based on substituted pyrimidine as ligand backbone.36
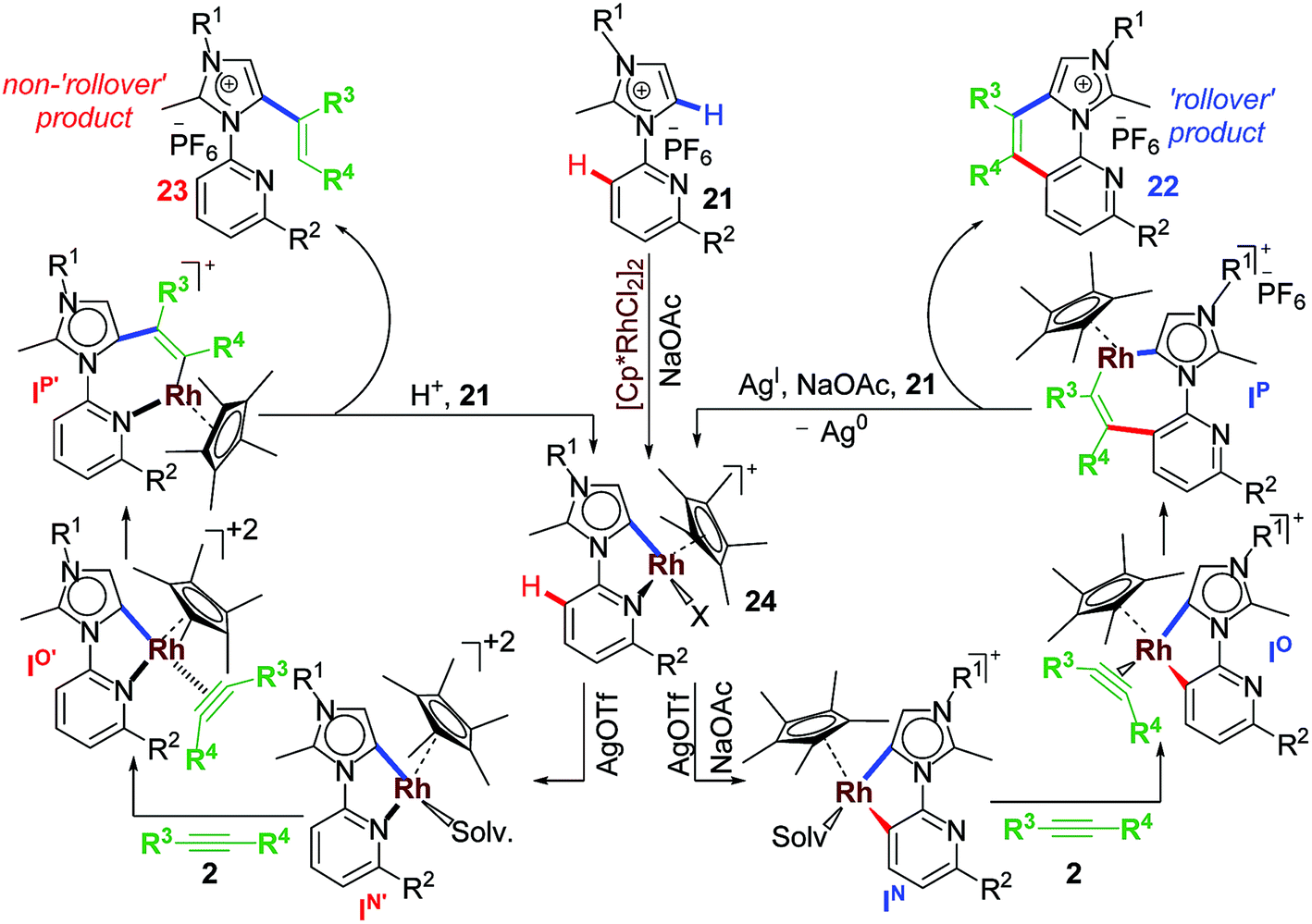
Following our established annulation protocol, we investigated functionalization of Rh–aNHC platform (Scheme 17).20 The expected transformation in case of Rh–aNHC template was alkenylation likewise in aNHC-directed rollover for chelators, without undergoing C–N coupling. However, it was observed that even in case of Rh–aNHC template bimodal functionalization leading to both alkenylation and annulation was possible. The usual pathway for this was alkenylation whereas under certain stereoelectronic constraints, annulation was also possible via a pyridine rollover process. When the adjacent 2-position of the pyridyl ring is substituted (R2 in Scheme 17) by electron donating group, rollover annulation was observed for electron-deficient alkynes such as diphenyl acetylene. No rollover product was obtained for electron-rich alkyne such as 4-octyne. On the other hand, with substantial steric bias at 2-position led to rollover exclusively. The steric bias involved at pyridyl ring led to exclusive alkenylation as a result of non-rollover. Therefore, it was evident that the reaction path was never controlled by one factor; rather it was guided by a collective contribution from sterics and electronics of substituents and incoming electrophile as well.
 | ||
| Scheme 17 Bimodal C–H activation/annulation at Rh–aNHC template. | ||
Preliminary mechanistic investigations led to synthesis, isolation and structural confirmation of various intermediates. Owing to experimental challenges involved in the ‘rollover’ process, full proof investigations have not yet been completed. It was observed that ‘rollover’ in sterically-biased Rh–aNHC platform is controlled by substituents, electronics of the incoming electrophile and the base involved (Scheme 19). When R2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H in the metallachelate 24 (Scheme 18) no rollover was observed and alkenylation path was followed. The main difference in alkenylation and annulation is that in the former the last step is a protodemetalation step while for annulation it is a reductive elimination step.
H in the metallachelate 24 (Scheme 18) no rollover was observed and alkenylation path was followed. The main difference in alkenylation and annulation is that in the former the last step is a protodemetalation step while for annulation it is a reductive elimination step.
 | ||
| Scheme 18 Plausible mechanism for ‘rollover’ C–H annulation at Rh–aNHC platform. | ||
2.8 C–C vs. C–N ring-extension annulation
Based on the earlier works, our group recently reported the switching of reaction pathway controlled by anion and solvents giving dichotomous end products.37 However, the substrates used in this work were based on aryl imidazoles rather than imidazolium salts (Scheme 19). The anion attached to the common rhodacycle intermediate along with the nature of the reaction solvent played a crucial role in dictating chemoselectivity in this divergent annulation protocol catalyzed by Rh(III). A strongly coordinating anion such as acetate (from Cu(OAc)2·H2O) forms a neutral rhodacycle intermediate which prefers non-polar solvents for stabilization. As a result of this, it undergoes selective C–C annulation (path a; Scheme 19). On the other hand, a weakly/non coordinating anion such as BF4 or OTf (from Cu(BF4)2·6H2O; AgOTf) prefers formation of cationic rhodacycle stabilized better by polar solvents. Eventually it leads to C–N coupled product involving a ring-extension process (path b, Scheme 19). These observations added further to the mechanistic understanding of catalytic rollover process involving piano-stool chelating type of metallacycles.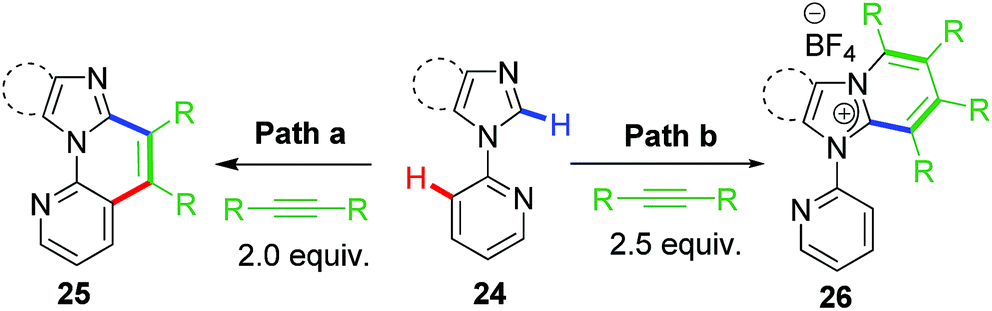 | ||
| Scheme 19 Switching of reaction pathway from C–C rollover to C–N ring-extension annulation. | ||
3. Conclusion and outlook
The chemistry of C–H activation-annulation at the metal–NHC platform described herein shows a new direction of applications in the versatile field of transition-metal-catalyzed C–H functionalization. It was suggested that productive transformation of in situ generated metal–NHC templates could be a potential strategy for derivatizing a large number of azolium-containing organic substrates to the valuable products. Aromatic, heteroaromatic and non-aromatic C–H bonds were shown to be possible to functionalize via these protocols. In terms of reaction pathway, one can control the same by several factors including electronic and steric on the substrates, solvents, base, temperature etc. Interestingly, the end-products represent a newer class of cationic annulated polyazaheteroaromatic compounds with extensive conjugation. These compounds exhibit variable fluorescence emission in organic as well as aqueous solutions (Fig. 3). These may find potential applications in the field of organic materials, cellular imaging etc. | ||
| Fig. 3 Images of selected synthesized annulated products under UV light. | ||
More importantly, the one-pot synthesis of these polyheteroaromatics achieved by the current annulation protocol is an attractive route in comparison to traditionally-practiced multistep tedious organic synthetic protocols. However, these protocols are not free of drawbacks. The major drawback is the use of metal-based oxidants, such as either Ag salts or Cu salts. From synthetic point of view, it is a limitation that has to be resolved. Investigations can be focused toward application of cheaper non-metal oxidants including aerobic conditions.
On the other hand, the mechanistic picture of the highly attractive rollover C–H functionalization protocols is not clear, particularly due to various thermodynamic and kinetic restrictions involved in arresting the crucial organometallic intermediates. An extensive research direction consisting of experimental and computational approaches can address the mechanistic issues and thereby establish the required understanding and knowledge-base which may help in designing newer methods as well as broadening the reaction scope. Lastly and most importantly, there is a significant scope for future research involving these new synthetic reactions toward the development of cheaper first-row transition metal-based catalysts to replace the expensive Rh-based ones. Recently, there started a trend toward this direction, although the implication in the NHC-directed/assisted C–H activation-annulation is yet to be achieved.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge generous funding from DST-SERB (grant no. EMR/2016/003002) and IISER Bhopal. C. D. thanks doctoral fellowship from IISER Bhopal. The authors also thank the other past and present co-workers in the group whose works have been described herein.Notes and references
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- (a) H.-W. Wanzlick and H.-J. Schonherr, Angew. Chem., Int. Ed., 1968, 7, 141 CrossRef; (b) K. Ofele, J. Organomet. Chem., 1968, 12, 42 CrossRef.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef PubMed; (c) T. Droge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed; (d) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CrossRef PubMed; (e) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef PubMed.
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef; (c) T. Zou, C. Lok, P. Wan, Z. Zhang, S. Fung and C. Che, Curr. Opin. Chem. Biol., 2018, 43, 30 CrossRef PubMed; (d) M. Elie, J.-L. Renaud and S. Gaillard, Polyhedron, 2018, 140, 158 CrossRef; (e) L. Oehninger, R. Rubbiania and I. Ott, Dalton Trans., 2013, 42, 3269 RSC; (f) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859 CrossRef PubMed; (g) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599 CrossRef PubMed.
- N. Kuhn and A. Al-Sheikh, Coord. Chem. Rev., 2005, 249, 829 CrossRef.
- (a) E. Peris, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.6b00695; (b) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef; (c) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC.
- (a) M. C. Jahnke and E. Hahn in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, S. Diez-Gonzalez, RSC, 2011, vol. 6, ch. 1, pp. 1–45 Search PubMed; (b) R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970 CrossRef PubMed.
- For reviews, see (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef PubMed; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef PubMed; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef PubMed; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed.
- For representative examples, see (a) P. E. Vivas-Mejía, O. Cox and F. A. González, Mol. Cell. Biochem., 1998, 178, 203 CrossRef; (b) M. Hranjec, I. Piantanida, M. Kralj, L. Suman, K. Pavelic and G. Karminski-Zamola, J. Med. Chem., 2008, 51, 4899 CrossRef PubMed.
- (a) H. H. Chou and C. H. Cheng, Adv. Mater., 2010, 22, 2468 CrossRef PubMed; (b) Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef PubMed; (c) B. Park, Y. H. Huh, H. G. Jeon, C. H. Park, T. K. Kang, B. H. Kim and J. Park, J. Appl. Phys., 2010, 108, 094506 CrossRef.
- A. C. Chen, L. Ren, A. Decken and C. M. Crudden, Organometallics, 2000, 19, 3459 CrossRef.
- (a) J. M. Praetorius and C. M. Crudden, Dalton Trans., 2008, 4079 RSC; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef PubMed.
- (a) D. Janssen-Muller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845 RSC; (b) M. S. Jeletic, M. T. Jan, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2009, 2764 RSC.
- (a) R. Corberan, M. Sanau and E. Peris, J. Am. Chem. Soc., 2006, 128, 3974 CrossRef PubMed; (b) R. Dorta, E. D. Stevens and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5054 CrossRef PubMed; (c) R. Sun, S. Zhang, X. Chu and B. Zhu, Organometallics, 2017, 36, 1133 CrossRef.
- (a) A. T. Normand, S. K. Yen, H. V. Huynh, T. S. A. Hor and K. J. Cavell, Organometallics, 2008, 27, 3153 CrossRef; For non-annulative NHC-alkyl/aryl reductive elimination, see (b) D. S. McGuinness and K. J. Cavell, Organometallics, 2000, 19, 4918 CrossRef; (c) W. J. Marshall and V. V. Grushin, Organometallics, 2003, 22, 1591 CrossRef; (d) A. T. Normand, A. Stasch, L.-L. Ooi and K. J. Cavell, Organometallics, 2008, 27, 6507 CrossRef.
- K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 3202 CrossRef PubMed.
- D. Ghorai and J. Choudhury, Chem. Commun., 2014, 50, 15159 RSC.
- (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (b) A. P. Walsh and W. D. Jones, Organometallics, 2015, 34, 3400 CrossRef; (c) D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin and K. Singh, J. Am. Chem. Soc., 2015, 137, 9659 CrossRef PubMed.
- R. Thenarukandiyil, S. K. Gupta and J. Choudhury, ACS Catal., 2016, 6, 5132 CrossRef.
- C. Dutta, D. Ghorai and J. Choudhury, ACS Omega, 2018, 3, 1614 CrossRef.
- R. Thenarukandiyil and J. Choudhury, Organometallics, 2015, 34, 1890 CrossRef.
- H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef PubMed.
- G. Song, X. Gong and X. Li, J. Org. Chem., 2011, 76, 7583 CrossRef PubMed.
- D. Ghorai and J. Choudhury, ACS Catal., 2015, 5, 2692 CrossRef.
- (a) Q. Ge, B. Li, H. Songa and B. Wang, Org. Biomol. Chem., 2015, 13, 7695 RSC; (b) Q. Ge, B. Li and B. Wang, Org. Biomol. Chem., 2016, 14, 1814 RSC; (c) R. Li, Y. Hu, R. Liu, R. Hu, B. Li and B. Wang, Adv. Synth. Catal., 2015, 357, 3885 CrossRef.
- (a) L. Li, H. Wang, X. Yang, L. Kong, F. Wang and X. Li, J. Org. Chem., 2016, 81, 12038 CrossRef PubMed; (b) Q. Ge, Y. Hu, B. Li and B. Wang, Org. Lett., 2016, 18, 2483 CrossRef PubMed; (c) J. M. Villar, J. Suárez, J. A. Varela and C. Saá, Org. Lett., 2017, 19, 1702 CrossRef PubMed; (d) Y. R. Han, S.-H. Shim, D.-S. Kim and C.-H. Jun, Org. Lett., 2017, 19, 2941 CrossRef PubMed; (e) D. Sucunza, A. M. Cuadro, J. Alvarez-Builla and J. J. Vaquero, J. Org. Chem., 2016, 81, 10126 CrossRef PubMed.
- R. Thenarukandiyil, H. Thrikkykkal and J. Choudhury, Organometallics, 2016, 35, 3007 CrossRef.
- P. Xie, M. Jia, X. Xu, F. Chen and Y. Xia, Asian J. Org. Chem., 2018, 7, 586 CrossRef.
- D. Ghorai, C. Dutta and J. Choudhury, ACS Catal., 2016, 6, 709 CrossRef.
- B. Butschke and H. Schwarz, Chem. Sci., 2012, 3, 308 RSC.
- (a) L. Maidich, G. Dettori, S. Stoccoro, M. A. Cinellu, J. P. Rourke and A. Zucca, Organometallics, 2015, 34, 817 CrossRef; (b) F. Cocco, A. Zucca, S. Stoccoro, M. Serratrice, A. Guerri and M. A. Cinellu, Organometallics, 2014, 33, 3414 CrossRef; (c) M. E. Moustafa, P. D. Boyle and R. J. Puddephatt, Organometallics, 2014, 33, 5402 CrossRef; (d) A. Zucca, L. Maidich, L. Canu, G. L. Petretto, S. Stoccoro, S. M. A. Cinellu, G. J. Clarkson and J. P. Rourke, Chem.–Eur. J., 2014, 20, 5501 CrossRef PubMed; (e) L. Maidich, G. Zuri, S. Stoccoro, M. A. Cinellu, M. Masia and A. Zucca, Organometallics, 2013, 32, 438 CrossRef; (f) A. Zucca, D. Cordeschi, L. Maidich, M. I. Pilo, E. Masolo, S. Stoccoro, M. A. Cinellu and S. Galli, Inorg. Chem., 2013, 52, 7717 CrossRef PubMed.
- (a) J. Kwak, Y. Ohk, Y. , Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778 CrossRef PubMed; (b) S. Y. Hong, J. Kwak and S. Chang, Chem. Commun., 2016, 52, 3159 RSC.
- (a) R. Morioka, K. Nobushige, T. Satoh, K. Hirano and M. Miura, Org. Lett., 2015, 17, 3130 CrossRef PubMed; (b) L. Rubio-Perez, M. Iglesius, R. Castarlenas, V. Polo, J. J. Perez-Torrente and L. A. Oro, ChemCatChem, 2014, 6, 3192 CrossRef.
- (a) X. Zhu, X.-J. Shen, Z.-Y. Tian, S. Lu, L.-L. Tian, W.-B. Liu, B. Song and X.-Q. Hao, J. Org. Chem., 2017, 82, 6022 CrossRef PubMed; (b) M. A. Ali, X. Yao, G. Li and H. Lu, Org. Lett., 2016, 18, 1386 CrossRef PubMed; (c) H. J. Kim, M. J. Ajitha, Y. Lee, J. Ryu, J. Kim, Y. Lee, Y. Jung and S. Chang, J. Am. Chem. Soc., 2014, 136, 1132 CrossRef PubMed; (d) Z. Qi, S. Yu and X. Li, J. Org. Chem., 2015, 80, 3471 CrossRef PubMed.
- (a) A. R. Chianese, A. Kovacevic, B. M. Zeglis, J. W. Faller and R. H. Crabtree, Organometallics, 2004, 23, 2461 CrossRef; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef PubMed.
- F. Schön, M. Leist, A. Neuba, J. Lang, C. Braun, Y. Sun, G. Niedner-Schatteburg, S. Bräse and R. W. Thiel, Chem. Commun., 2017, 53, 12016 RSC.
- R. Thenarukandiyil, C. Dutta and J. Choudhury, Chem.–Eur. J., 2017, 23, 15529 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |