
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating charge carrier interactions at solid electrolyte interfaces for enhanced micro-supercapacitor performance†
Abhirami Sukumaran,
Premkumar Jayaraman and
Helen Annal Therese *
*
Futuristic Energy Storage Technology Lab (FESTL), Department of Chemistry, SRM Institute of Science and Technology, Kattankulathur, Chennai 603203, India. E-mail: helena@srmist.edu.in
First published on 25th June 2025
Abstract
Manipulation of ionic charges in the solid electrolyte interface can result in unprecedented device characteristics for energy conversion and storage applications. An ultrathin gel polymer electrolyte film in a stacked device architecture has been proposed to offer a crucial method for exhibiting the characteristics of electric double layer capacitance (EDLC) at interfaces. A thorough understanding and control of ionic transport channels within the electrode/electrolyte interface are essential for the development of suitable design concepts and the fabrication of micro-scale energy storage devices. A stacked device with nanostructured thin gel polymer electrolyte (GPE) – polyvinyl alcohol (PVA) with lithium perchlorate (LiClO4), and additive lithium sulphate (Li2SO4) were studied for micro-supercapacitors (MSCs). The electrochemical properties of supercapacitor devices were studied to confirm how the anions and cations are separated at electrode/electrolyte interfaces utilizing an electromotive force. Significantly, the charge migration and separation of cations and anions at the electrode/electrolyte interfaces were also studied.
1. Introduction
Supercapacitors are advanced electrochemical energy storage devices that have attracted tremendous scientific interest in recent years. Supercapacitors are known to have potential advantages in meeting the demand for future electronic devices because of their enhanced charge storage mechanism, higher cycling stability, high-rate capability, and increased power density.1 As the demand for clean energy sources and energy storage, and conversion technologies increased, supercapacitors have emerged as a promising technology to store renewable energy resources. Supercapacitors are excellent recovery systems due to their shorter charging and discharging times and extended life cycles compared to conventional batteries. They can sustain millions of cycles of charge storage, but suffer from limited energy densities and low potential windows. Supercapacitors store electric energy in electric double layers between electrodes, unlike batteries that experience swelling.2Micro supercapacitors (MSC) are developing quickly with potential uses in microelectronics, on-chip energy storage, integrated sensors, portable and wearable electronics, and photonic devices with a focus on versatility, miniaturization, and shape-tuning capabilities.3–6 Despite having several names, supercapacitors still serve the same basic purpose of storing energy between an electrolyte and a solid electrode. Despite their advantages, supercapacitors face challenges like high self-discharge and low energy density, necessitating extensive research on electrode materials and electrolyte development to enhance their competitiveness.7–12 But in addition to the electrode materials, the performance of supercapacitors is also influenced by the properties of the electrolyte. The development of dendrites on the metal anode surface during repeated charge/discharge cycles in liquid electrolytes is a significant issue that affects the performance of supercapacitors.13 This can lead to short circuiting and the subsequent loss of capacity.
Numerous research teams have worked very hard in recent years to prevent dendritic growth. Several innovative methods have been suggested, such as incorporating an artificial solid-electrolyte interphase (SEI) or adding additives to the electrolyte solution. Structurally confined gel polymer electrolyte (GPE) offers several advantages over other electrode types in the creation of supercapacitors without altering the electrode's characteristics.14,15 Gel-based electrolytes are influenced by polymer-based electrolytes, which is used to overcome the electrolyte leakage issue and make it simple to fabricate supercapacitor devices.16–20 The polymer electrolytes based on polyvinyl alcohol (PVA) are some of the most commonly reported ones, with 5 to 67 Wh kg−1 as their stated energy densities.21–23 In general, polymer electrolytes have ionic conductivity lower than 10−3 S cm−1, which is lower than that of aqueous electrolytes (10−1 S cm−1).24 To boost its ionic conductivity and electrochemical activity, PVA is frequently employed as an electrolyte in combination with H2SO4, KOH, LiCl, TiO2, LiClO4, and H3PO4.25,26 Neutral GPEs were shown to have a larger operating potential window than basic and acidic GPEs. This is due to the fact that neutral salts have fewer hydrogen and hydroxide ions than acidic or alkaline substances.27
Krishnan et al. studied high-performance supercapacitors, revealing that changes in ionic charges at the solid-electrolyte border create new energy conversion and storage features.3 They demonstrated EDLC properties using a planar device with a thin film, revealing dynamic supercapacitance characteristics. J. Zhao et al. found that adding polysulfide to a carbonate electrolyte significantly increased the number of polyethylene oxide (PEO) polymers, inhibiting the growth of lithium dendrites, which enhanced the stability of lithium metal batteries during extended cycling. The polysulfide formed a durable solid electrolyte interphase film, suppressing lithium dendrite development and enhancing the battery's long cycling.28 F. Wan found that the Na2SO4 additive inhibits Zn dendrite and NVO nanobelt dissolution, resulting in high-performance aqueous ZIBs with NVO nanobelts as positive electrodes, resulting in negligible redox reactions, long cycle life, and high-capacity retention.29 X. Guo et al. found that adding lithium chloride (LiCl) stabilizes the Zn metal anode and inhibits dendrite growth. Li+ cations form a shield on the Zn surface, while Cl− anions reduce zinc polarization and aid in ion transport. Cells with LiCl electrolyte additive showed greater stability during cycling, decreased side reactions, and increased capacity.30 K. Krishnan et al. studied the ionic charges in the interfacial domination of ITO/polyvinyl alcohol–KOH, focusing on electrical double-layer capacitance (EDLC) characteristics. They found that planar MSC cells showed significant differences in volumetric capacitance and retention. The study also examined the distribution of ionic charges at interfaces with thin and thick PVI–KOH in planar device arrangements. Limiting the persistence of PVI–KOH to 28 nm improved capacitance retention, clarifying the confinement effect of MSC bias.31 Zhang et al. in the year of 2015 reported that Li2SO4 could be readily added to PVA aqueous solution in large quantities to address the issue of low ionic conductivity of polymer gel supercapacitors. LiCF3SO3, LiN(SO2CF3)2, LiPF6, LiBF4, and LiClO4 are among the various lithium salts that have also been investigated as conducting salts in PVA hydrogel.32,33 There are several studies investigating how the structural, electrochemical, and mechanical characteristics of gel electrolyte are affected by Li2SO4 and how it changes the ionic conductivity of the gel, enhancing ionic conduction via interactions with the polymer matrix, and raising the concentration of free lithium and sulfate ions as charge carriers.34–36
The use of Li2SO4 in PVA–LiClO4 gel electrolyte is a significant advancement in supercapacitor electrolyte design, addressing limitations of single-ion additive systems. Li2SO4 can provide Li+ cations and SO42− anions, enhancing bulk electrolyte properties and electrode–electrolyte interface dynamics. Traditional PVA gel electrolytes use single-ion additives for conductivity enhancement, achieving ionic conductivities up to 48 mS cm−1.37 ClO4− anions in these systems show limited interfacial activity, while H2SO4 containing PVA hydrogels show high conductivity but face challenges with hydrogen evolution reactions at high voltages.38 The main limitation of these additives is their monofunctional nature, focusing on enhancing cation mobility or anion effects but not simultaneously. However, when Li2SO4 is added to PVA–LiClO4 gel matrix, Li+ ions maintain high conductivity, while large divalent SO42− anions adsorb on electrode surfaces, modifying the electric double layer structure.39 Hybrid devices using Li2SO4 gel electrolytes showed higher areal capacitance and energy density compared to LiCl-based electrolytes, with 76.8% retention after 1000 cycles. The stability of Li2SO4 in aqueous environments addresses the corrosive nature of LiClO4 electrolytes. Thus PVA-based gel electrolytes incorporating a variety of electrolyte ions exhibit high conductivity and good chemical stability over a wide pH range.39,40 The dual-ion approach in SS‖PVA-LiClO4 + Li2SO4‖SS particularly benefits hybrid supercapacitors requiring balanced capacitive and faradaic performance, while its compatibility with industrial-scale PVA processing methods makes Li2SO4 as a transformative additive for next-generation energy storage systems.
Gel electrolytes for supercapacitors often suffer from low ionic conductivity and poor cycling stability due to lithium dendrite formation. This study addresses these issues by proposing a new gel electrolyte system in which Li2SO4 is introduced into a PVA–LiClO4 matrix, resulting in enhanced ionic conductivity and effective dendrite suppression on a stainless-steel substrate. Using a two-electrode cell design, electrochemical performances such as cyclic voltammetry (CV), galvanostatic charge–discharge cycles (GCD), electrochemical impedance spectroscopy (EIS), and ionic conductivity before and after cycles of stack MSCs were studied. This simple yet effective modification achieves an ultra-high volumetric capacitance of 11.8 F cm−3 and maintains stability over 2000 cycles, outperforming conventional PVA-based electrolytes. Li2SO4 additive enhanced the performance of the supercapacitor device by acting as a safer and more efficient gel electrolyte for high-performance supercapacitors by improving the overall volumetric capacitance and cycling stability of the stack MSC.
2. Materials and methods
2.1 Fabrication of gel polymer electrolyte – based stacked devices
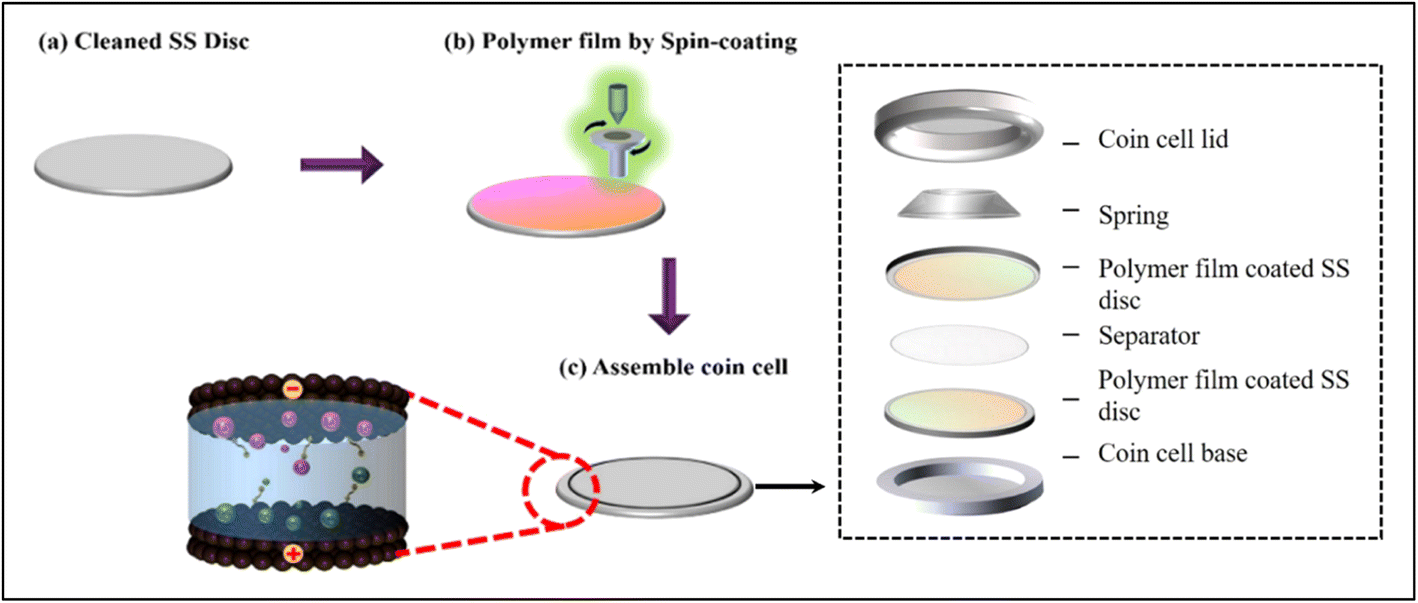 | ||
| Scheme 1 Schematic representation of device fabrication (a and b) and coin cell assembly (c). | ||
3. Characterizations
The electrochemical characteristics of the EDLC devices were measured with a two-electrode configuration. The cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and impedance were conducted using Bio-Logic BCS-810. In the CV measurements, the sweep rate varied from 5 to 100 mV s−1 and the potential window varied in the range of 0–1 V. In the GCD studies, the device was charged and discharged with a current of 0.5 and 1 microampere. By using CV curves, the volumetric capacitance (CV) was calculated. By using GCD profiles, the discharge capacitances of the devices were estimated. The surface morphology and thickness of the polymer film were observed using AFM.4. Results and discussions
The electrochemical performances of SS‖PVA-LiClO4‖SS stack MSC were studied in the potential window of 0–1.0 V. CV measurements of SS‖PVA-LiClO4‖SS at several sweep rates varying from 5–100 mV s−1 were plotted. CV plots of MSC (Fig. 1a) correspond to quasi-rectangular shapes, which indicates electric double layer behaviour. In addition, the quasi-rectangular behaviour of the CV curve was maintained throughout all sweep rates, indicating that the SS‖PVA-LiClO4‖SS has both excellent rate capability and optimal capacitive behaviour. As the sweep rate was increased from 10–100 mV s−1, the volumetric capacitance dropped from 5.8 F cm−3 until it reached 1.9 F cm−3 (Fig. 1c). The volumetric capacitance (CV) was estimated by the following relation:Here, ν is the scan rate, ΔV is the potential difference, i is the measured current in CV, and t is time. Using electrochemical impedance spectroscopy (EIS), the charge transfer properties of the SS‖PVA-LiClO4‖SS device were analysed. From the Nyquist plot (Fig. 1e), the increased ionic conductivity of the gel polymer electrolyte is further supported by the absence of a half circle pattern in the high frequency zone. The ionic conductivity (σ) of the thin film was measured using the following relationship:

where d is the distance between the electrodes, Rct is the resistance value obtained directly from EIS measurement, and A is the area of the electrode. According to the calculations, the pristine device has an ionic conductivity of about 7.6 × 10−7 S cm−1, and after EC studies, the ionic conductivity was found to be 7.6 × 10−7 S cm−1.

 | ||
| Fig. 1 (a) CV curves at different scan rate of device SS‖PVA-LiClO4‖SS (b) CV curves at different scan rate of device SS‖PVA-LiClO4 + Li2SO4‖SS, (c) scan rate dependent CVol of SS‖PVA-LiClO4‖SS, (d) scan rate dependent CV of SS‖PVA-LiClO4 + Li2SO4‖SS (e) EIS measurement of SS‖PVA-LiClO4‖SS before and after cycling (f) EIS measurement of SS‖PVA-LiClO4 + Li2SO4‖SS before and after cycling. | ||
Using a two-electrode cell design, we studied the electrochemical performance of SS‖PVA-LiClO4 + Li2SO4‖SS. Cyclic voltammetry (CV) measurements were performed in a potential window of 0–1.0 V at several scan rates (5–100 mV s−1). The cyclic voltammetry graphs (Fig. 1b) showed a quasi-rectangular shape across all sweep rates, representing the pseudocapacitive behaviour. The quasi-rectangular behaviour of the CV curve was maintained throughout the sweep rates, indicating the good capacitance behaviour of SS‖PVA-LiClO4 + Li2SO4‖SS and its rate capability. According to calculations, the maximum CV of the SS‖PVA-LiClO4 + Li2SO4‖SS is found to be 10.5 F cm−3 at 10 mV s−1. Fig. 1d shows a volumetric capacitance of a minimum value of 2.8 F cm−3 at 100 mV s−1 as the sweep rate was increased. Using electrochemical impedance spectroscopy (EIS), the charge transfer properties of additive-based SS‖PVA-LiClO4 + Li2SO4‖SS were analysed. Fig. 1f shows a Nyquist plot, where the low-frequency area is represented by a straight line with a small semicircle, which is typically associated with a better capacitance. In addition, the gel polymer electrolyte's increased ionic conductivity is inferred from the lack of a semicircle pattern in the high-frequency area.
The ionic conductivity of SS‖PVA-LiClO4 + Li2SO4‖SS is about 1.18 × 10−6 S cm−1, while the ionic conductivity of the device after EC tests increased to 1.1 × 10−6 S cm−1. After 1000 continuous CV cycling of SS‖PVA-LiClO4 + Li2SO4‖SS at 100 mV s−1, additional EIS measurements were taken to analyse the ion transport property of the device. Later in the CV experiment, the cell's conductivity returned, showing that the device maintains its performance over time. It is also important to know the amount of current that is carried by each mobile charged species than the overall conductivity of a polymer when the polymer contains more than one mobile charged species. So, by considering the ionic resistance (Ri) and the electronic resistance (Rele), tion (the mean transfer number) can be estimated from the equation given below:

Using the Vcell to tion relation, tion values were estimated to be 0.51 and 0.61 for SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS, respectively. This indicates that the gel polymer electrolyte of SS‖PVA-LiClO4 + Li2SO4‖SS enhances ionic transport efficiency, possibly resulting in superior electrochemical performance. SS‖PVA-LiClO4 + Li2SO4‖SS may provide enhanced performance, including greater capacitance, energy density, improved ionic mobility, and reduced hindrance within the polymer matrix when compared to SS‖PVA-LiClO4‖SS.
Comparison of SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS is shown in Fig. 2a. Due to the presence of the electrical double layer at the interface, the CV plots of the devices, which is thickness dependent, displayed a quasi-rectangular behaviour. Quasi-rectangular shape in the CV curve was maintained throughout all scan rates, indicating that the polymer- Li ion integrated devices have excellent capacitance properties and a high-rate capability. The CV plot was considered to measure the volumetric capacitance (CV) of both devices. The compared volumetric capacitance of the devices based on the role of the additive is displayed in Fig. 2b. CV studies were performed according to the device capacity to evaluate the cycle characteristics of the constructed stack MSC device. As in Fig. 2c, the cycling performance of the SS‖PVA-LiClO4‖SS stack MSC was inferior, with 67% of capacitance retention after 1000 cycles. It was found that at a specific characteristic nanoscale, the cycle performance and capacitance retention both improved by adding Li2SO4. Compared to the other device, the SS‖PVA-LiClO4 + Li2SO4‖SS demonstrated superior cycling performance, with 1000 cycles on continuous use and considerable capacitance retention of 90%.
 | ||
| Fig. 2 (a) Stability performance of device A (SS‖PVA-LiClO4‖SS) and B (SS‖PVA-LiClO4 + Li2SO4‖SS), (b) CV comparison of device A and B, (c) CV comparison at 10 mV s−1 scan rate of device SS‖PVA-LiClO4 + Li2SO4‖SS (A) and SS‖PVA-LiClO4 + Li2SO4‖SS (B). | ||
Similarly, using a two-electrode cell design, galvanostatic charge–discharge (GCD) characteristics of SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS stack MSC were studied at currents varying from 0.5 to 1 micro ampere in the potential window of 0–1.0 V as shown in Fig. 3a and b.
 | ||
| Fig. 3 (a) GCD 1st cycle at 0.5 microampere (b) 1stcycle at 1 microampere, (c) cyclic stability of SS‖PVA-LiClO4‖SS (d) cyclic stability SS‖PVA-LiClO4 + Li2SO4‖SS (e) comparison of CV retention of both devices (f) comparison with previous literature. | ||
By using GCD plots, the volumetric capacitances of SS‖PVA-LiClO4‖SS were found to be 6.821 and 3.76 F cm−3 at 0.5 to 1 microampere, respectively. The volumetric capacitances of SS‖PVA-LiClO4 + Li2SO4‖SS were found to be 11.8 and 4.43 F cm−3 at different currents of 0.5 and 1 microampere respectively, using the following equation:

In order to investigate the cycling performance of fabricated stack MSC devices, the GCD measurement was tested up to 2000 continuous cycles at a constant current of 1.0 μA (Fig. 3d). It infers that the electrolyte PVA-LiClO4 + Li2SO4 can give excellent device characteristics, which are comparable to the previously reported devices as mentioned in Table 1.
| SI no. | Electrolyte | Capacitance | Voltage | Capacitance retention/cycle life | Power density | Energy density | Ref. no. |
|---|---|---|---|---|---|---|---|
| 1 | PVA–KOH | 5.39 F cm−3 | 0–1 V | 98%/11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles 000 cycles |
300 mW cm−3 | 2.36 mW h cm−3 | 3 |
| 2 | PVI–KOH | 0.128 F cm−3 | 0–1 V | 92%/1400 cycles | 6.89 mW cm−3 | 0.056 mW h cm−3 | 41 |
| 3 | PAM (polyacrylamide)–LiCl | 0.341 F cm−3 | 0–0.6 V | 99.12%/30000 |
257.07 mW cm−3 | 17.07 mW h cm−3 | 42 |
| 4 | PVA + LiClO4+ Li2SO4 | 11.8 F cm−3 | 0–1 V | 83% after 2000 cycles | 6.58 mW cm−3 | 1.6 mW h cm−3 | This Work |
The stability of SS‖PVA-LiClO4‖SS started fading at 300 cycles (Fig. 3c), whereas SS‖PVA-LiClO4 + Li2SO4‖SS (Li2SO4 as additive) was stable even after 2000 cycles. The shape of the GCD profile remained almost unchanged (Fig. 3b) and exhibited good capacitance retention of 83% even after 2000 continuous cycles (Fig. 3e). The device with additive, showed excellent cycling characteristics, which indicate its potential for use in future energy-storage devices. Despite exhibiting a slightly lower coulombic efficiency than SS‖PVA-LiClO4‖SS (Fig. S1(b and d)),† the inclusion of Li2SO4 is advantageous overall, since it promotes pseudocapacitive redox reactions at the electrode/electrolyte interface, enhancing specific capacitance and cycling stability, hence enabling the device to maintain its performance over a greater number of cycles. Although these faradaic processes may induce minor side reactions that reduce coulombic efficiency per cycle,39,43,44 the overall energy storage and long-term dependability are significantly enhanced, making the Li2SO4-based device the superior option for high-performance supercapacitors.
Microscopic analysis to find the thickness of the thin film coating was conducted using AFM. The AFM was measured by spin coating the SS disc with the electrolyte and then masked the film edge with respect to the substrate then the step height of the edge was measured (Fig. 4b). From the corresponding depth profile (Fig. 4c), the thickness of the film was found to be 246.7 nm.
 | ||
| Fig. 4 (a) 2D AFM image of the thin film (b), 3D image (c) height profile (thickness) of the film. | ||
Open circuit potential (OCP), being the crucial parameter, represents the voltage across the terminals of supercapacitors when no current is flowing, indicating energy storage in the device. A higher OCP indicates a higher energy storage capacity of the supercapacitor. Here, the device SS‖PVA-LiClO4 + Li2SO4‖SS has a higher OCP than SS‖PVA-LiClO4‖SS (Fig. 5). It suggests that the device with the additive can store more energy than the device without the additive. This higher OCP leads to the storage of more power per unit volume compared to a device without an additive. SS‖PVA-LiClO4 + Li2SO4‖SS device has minimal voltage drop and maintains a stable OCV after the sweep, ensuring stability. Fig. 5 shows the time-dependent cell voltage of the SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS MSCs measured under a certain scan rate. The measurement was carried out in two steps, first, a positive cyclic sweep was performed at 0–1 V at certain sweep rates. Then, the voltage was measured as a function of time under an open circuit condition. In case of SS‖PVA-LiClO4 + Li2SO4‖SS MSC, a gradual increase in cell voltage for more than 300 s was observed because of the equilibration of ionic charges Li+ and ClO4−/SO42− at the electrode/electrolyte interface, whereas in SS‖PVA-LiClO4‖SS the cell voltage was dropping drastically. It was observed that the increase in scan rate increased the cell voltage for the device with additive, whereas the device without additive showed a decrease in cell voltage when the scan rate was increased due to the difference in ionic charge separated at the electrode/electrolyte interfaces.
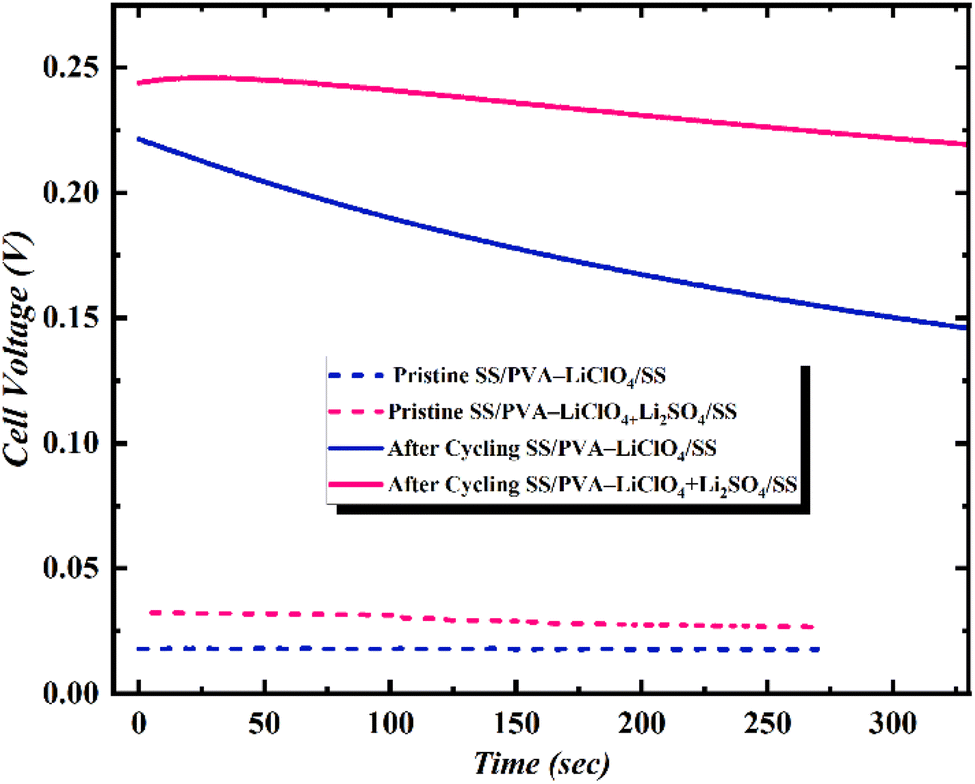 | ||
| Fig. 5 Time-dependent Vcell of SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS measured as a function of time under pristine state v/s after cycling. | ||
The cyclic voltammograms of both devices did not deviate from rectangular shapes even at higher scan rates. These results reveal that the electrolyte plays a crucial role in the capacitive property of both MSCs. The charge storage kinetics is analysed with the help of the power law as given by,
| i = avb |
Fig. 6(a & b) shows the dual logarithm plots for the cyclic voltametric current of both devices from 5 to 100 mV s−1 in CV. From the dual logarithm plots, the slope b for SS‖PVA-LiClO4‖SS and SS‖PVA-LiClO4 + Li2SO4‖SS were found to be 0.66 and 0.61, respectively. The increased ionic transference number in SS‖PVA-LiClO4 + Li2SO4‖SS (0.66) suggests that the system transitions towards a more diffusion-controlled process, which improves the ionic conductivity and electrode–electrolyte interaction. SO42− ions, due to their smaller size and more hydration compared to ClO4−, facilitate enhanced ion mobility in the electrolyte, hence promoting accelerated Li+ movement and boosting total ionic conductivity. The combination of monovalent (ClO4−) and divalent (SO42−) anions results in a more effective ion transport network, reducing ionic clustering or ion pairing. The interaction between Li+ and SO42− reduces ion aggregation and improves the dissociation of ionic species, hence decreasing the internal resistance. Li+ ions typically have a smaller hydration radius compared to SO42− ions. A smaller hydration radius allows Li+ to approach the electrode surface more closely, enabling faster adsorption which leads to lithium deposition in SS‖PVA-LiClO4‖SS device (Fig. 6c). When Li2SO4 is added SO42− ions may compete with Li+ for access to the electrode surface as it has higher charge density than ClO4−, which would potentially slow down lithium deposition at the surface as shown in Fig. 6d. SO42− is a divalent anion with a higher charge density and forms strong hydration shells this helps stabilize dissociated Li+ ions by competing with ClO4− or other anions for Li+, thus reducing ion pairing. This results in an optimized ionic atmosphere that enhances mobility without excessive pairing. SO42− ions adsorb at the electrode–electrolyte interface by weak electrostatic interactions that are driven by the applied electric field. This reduces degradation of the electrode and maintains consistent performance over many cycles. The additional Li+ ions increase the overall concentration of charge carriers in the electrolyte, thereby enhancing ionic conductivity within the PVA gel matrix.34 Here, a small amount of Li2SO4 is added, in which the sulfate ions participate in forming a passivation layer at the electrode surface. This layer inhibits the deposition of more Li+ ions at the electrode surface and further enhances the cycle stability of the device with Li2SO4 as an additive, which is evident from the GCD cycling which was stable even after 2000 continuous cycles.
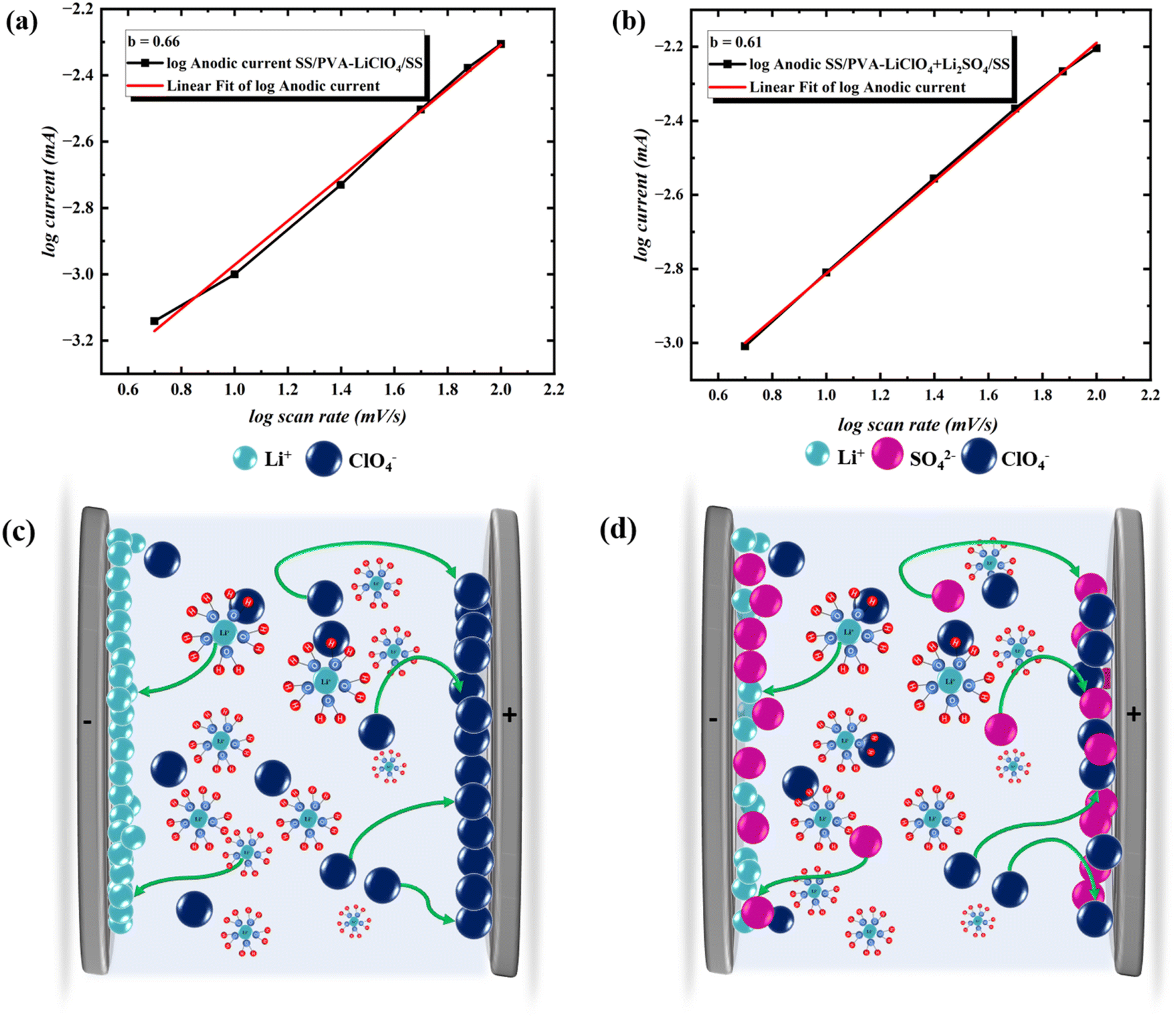 | ||
| Fig. 6 (a) Dual logarithm plot of current versus scan rate of CV in SS‖PVA-LiClO4‖SS, (b) dual logarithm plot of current versus scan rate of CV in SS‖PVA-LiClO4 + Li2SO4‖SS, (c) deposition of Li+ in the electrode surface, (d) inhibition of further Li+ deposition by SO42−. | ||
5. Conclusions
The volumetric capacitance and the stability of the device were enhanced by the addition of Li2SO4 into the PVA-gel electrolyte. Thus, SS‖PVA-LiClO4 + Li2SO4‖SS showed better ionic conductivity and the thin film device shows ultra-high volumetric capacitance 11 F cm−3 at 5 mV s−1 scan rate. From the GCD plots, the volumetric capacitances of SS‖PVA-LiClO4‖SS were found to be 6.8 F cm−3 and 3.76 F cm−3 at 0.5 μA and 1 μA respectively, whereas the volumetric capacitances of SS‖PVA-LiClO4 + Li2SO4‖SS were found to be 11.8 F cm−3 and 4.43 F cm−3 at currents ranging from 0.5 μA to 1 μA respectively. SS‖PVA-LiClO4 + Li2SO4‖SS showed a good cycling performance, which was stable for 2000 continuous cycles with 83% capacitance retention. The device remarkably achieved an energy density of 1.6 mWh cm−3 and a power density of 6.58 mW cm−3. The optimized PVA-LiClO4 + Li2SO4 gel electrolyte significantly inhibited the lithium deposition (dendrite development) on the surface of the SS disc during the charging and discharging process, which improved overall capacitance and cycle stability. In addition to the previously mentioned performance enhancements, our findings highlight the promise of Li2SO4 as a valuable additive in gel electrolytes for supercapacitor systems. The inclusion of Li2SO4 seems to enhance ionic conductivity and electrochemical stability. This study lays the groundwork for future investigations into the effective use of Li2SO4 in different electrolyte formulations and electrode materials, potentially leading to the improvement in energy density, power output, and device lifespan. In addition, understanding the mechanisms by which Li2SO4 inhibits dendrite formation could facilitate advancements in the creation of safer and more resilient supercapacitors and associated battery technologies in the future. In summary, our results enhance the development of gel electrolyte formulations, establishing a foundation for advanced supercapacitors that exhibit enhanced performance and stability.Data availability
The data that support the findings of this study are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the department of Physics & Nanotechnology for providing atomic force microscopy (AFM) Instrumental facility.References
- R. T. Yadlapalli, R. R. Alla, R. Kandipati and K. Anuradha, J. Energy Storage, 2022, 49, 104194 CrossRef.
- M. Czagány, S. Hompoth, A. K. Keshri, N. Pandit, I. Galambos, Z. Gácsi and P. Baumli, Materials, 2024, 17(3), 702 CrossRef PubMed.
- K. Krishnan, P. Jayaraman, B. Subramanian and M. Ulaganathan, J. Mater. Chem. A, 2018, 6(46), 23650–23658 RSC.
- X. Han, X. Wu and L. Zhao, et al., Microsyst. Nanoeng., 2024, 10, 107 CrossRef CAS PubMed.
- S. Ma, Y. Shi, Y. Zhang, L. Zheng, Q. Zhang and X. Xu, ACS Appl. Mater. Interfaces, 2019, 11(33), 29960–29969 CrossRef CAS PubMed.
- D. Li, S. Yang, X. Chen, L. Wei and W. Huang, Adv. Funct. Mater., 2021, 31(50), 2107484 CrossRef CAS.
- A. Afif, S. M. Rahman, A. T. Azad, J. Zaini, A. Islan and A. K. Azad, J. Energy Storage, 2019, 25, 100852 CrossRef.
- H. U. Lee, J.-H. Jin and S. W. Kim, J. Ind. Eng. Chem., 2019, 71, 184–190 CrossRef CAS.
- J.-H. Sung, S.-J. Kim, S.-H. Jeong, E.-H. Kim and K.-H. Lee, J. Power Sources, 2006, 162(2), 1467–1470 CrossRef CAS.
- Poonam, K. Sharma, A. Arora and S. K. Tripathi, J. Energy Storage, 2019, 21, 801–825 CrossRef.
- P. Lokhande, U. Chavan and A. Pandey, Electrochem. Energy Rev., 2019, 3(1), 155–186 CrossRef.
- A. Mendhe and H. S. Panda, Discov. Mater., 2023, 3(1), 65 Search PubMed.
- S. Samantaray, D. Mohanty, I. Hung, M. Moniruzzaman and S. K. Satpathy, J. Energy Storage, 2023, 72, 108352 CrossRef.
- U. S. Meda, L. Lal, M. Sushantha and P. Garg, J. Energy Storage, 2022, 47, 103564 CrossRef.
- S. Liu, W. Tian, J. Shen, Z. Wang, H. Pan, X. Kuang, C. Yang, S. Chen, X. Han, H. Quan and S. Zhu, Nat. Commun., 2025, 16(1), 57856 Search PubMed.
- S. S. Raut, L. K. Bommineedi, S. Pande and B. R. Sankapal, Synth. Met., 2021, 271, 116629 CrossRef CAS.
- K. Xiao, T. Yang, J. Liang, A. Rawal, H. Liu, R. Fang, R. Amal, H. Xu and D.-W. Wang, Nat. Commun., 2021, 12(1), 25817–25818 Search PubMed.
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7(1), 11782 CrossRef CAS PubMed.
- A. Railanmaa, M. Kujala, J. Keskinen, T. Kololuoma and D. Lupo, Appl. Phys. A, 2019, 125(3), 2461–2468 CrossRef.
- X. Ding, X. Xu, Z. He, Y. Liang, X. Wu and Z. Li, Carbon, 2023, 213, 118177 CrossRef CAS.
- H. N. Fard, G. B. Pour, M. N. Sarvi and P. Esmaili, Ionics, 2019, 25(7), 2951–2963 CrossRef CAS.
- J. O. Dennis, M. F. Shukur, O. A. Aldaghri, K. H. Ibnaouf, A. A. Adam, F. Usman, Y. M. Hassan, A. Alsadig, W. L. Danbature and B. A. Abdulkadir, Molecules, 2023, 28(4), 1781 CrossRef CAS PubMed.
- A. D. Shuaibu, S. S. Shah, A. S. Alzahrani and Md. A. Aziz, J. Energy Storage, 2025, 107, 114851 CrossRef.
- Y. Wang, C. Zhang, X. Qiao, A. N. Mansour and X. Zhou, J. Power Sources, 2019, 423, 18–25 CrossRef CAS.
- Y. Wang, X. Qiao, C. Zhang and X. Zhou, J. Energy Storage, 2019, 26, 100968 CrossRef.
- C.-S. Lim, K. H. Teoh, C.-W. Liew and S. Ramesh, Mater. Chem. Phys., 2014, 143(2), 661–667 CrossRef CAS.
- H. Dai, G. Zhang, D. Rawach, C. Fu, C. Wang, X. Liu, M. Dubois, C. Lai and S. Sun, Energy Storage Mater., 2021, 34, 320–355 CrossRef.
- J. Zhao, H. Yu, L. Ben, Y. Zhan, Y. Wu, X. Huang and Z. Zhou, J. Mater. Chem. A, 2018, 6(35), 16818–16823 RSC.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9(1), 4041 CrossRef PubMed.
- X. Guo, Z. Zhang, J. Li, N. Luo, G. Chai, T. S. Miller, F. Lai, P. R. Shearing, D. J. L. Brett, D. Han, Z. Weng, G. He and I. P. Parkin, ACS Energy Lett., 2021, 6(2), 395–403 CrossRef CAS.
- K. Krishnan, K. Selvakumar and S. Vijayaraghavan, RSC Adv., 2020, 10(73), 45019–45027 RSC.
- X. Zhang, L. Wang, J. Peng, P. Cao, X. Cai, J. Li and M. Zhai, Adv. Mater. Interfaces, 2015, 2(15), 1500267 CrossRef.
- Y. Ji, N. Liang, J. Xu, D. Zuo, D. Chen and H. Zhang, Cellulose, 2018, 26(2), 1055–1065 CrossRef.
- A. D. Shuaibu, S. S. Shah, A. S. Alzahrani and Md. A. Aziz, J. Energy Storage, 2024, 92, 112040 CrossRef.
- T. K. L. Nguyen and T.-N. Pham-Truong, Polymers, 2024, 16(17), 2506 CrossRef CAS PubMed.
- T. Lin, M. Shi, F. Huang, J. Peng, Q. Bai, J. Li and M. Zhai, ACS Appl. Mater. Interfaces, 2018, 10(35), 29684–29693 CrossRef CAS PubMed.
- N. R. Chodankar, D. P. Dubal, A. C. Lokhande and C. D. Lokhande, J. Colloid Interface Sci., 2015, 460, 370–376 CrossRef CAS PubMed.
- G. D'Altri, L. Yeasmin, V. Di Matteo, S. Scurti, A. Giovagnoli, M. F. Di Filippo, I. Gualandi, M. C. Cassani, D. Caretti, S. Panzavolta, E. Scavetta, M. Rea and B. Ballarin, ACS Omega, 2024, 9(6), 6391–6402 CrossRef PubMed.
- P.-L. Lan, I.-C. Ni, C.-I. Wu, C.-C. Hsu, I.-C. Cheng and J.-Z. Chen, Micromachines, 2023, 14(9), 1701 CrossRef PubMed.
- Z. Liu, J. Zhang, J. Liu, Y. Long, L. Fang, Q. Wang and T. Liu, J. Mater. Chem. A, 2020, 8(13), 6219–6228 RSC.
- K. Krishnan, S. Karuthapandi and S. Vijayaraghavan, RSC Adv., 2020, 10(73), 45019–45027 RSC.
- Q. Zhang, X. Jin, H. Li, R. Wang, F. Gao, T. Jiao, X. Cao and J. Ma, Nano Energy, 2024, 132, 110375 CrossRef.
- L. Guan, L. Guo, H. Yao, J. Cai, X. Dong, R. Wang, Z. Zhai, X. Chen, X. Wei, D. Li, X. Liu, S. Ji and F. Meng, Molecules, 2025, 30(8), 1764 CrossRef CAS PubMed.
- W. Qin, N. Zhou, C. Wu, M. Xie, H. Sun, Y. Guo and L. Pan, ACS Omega, 2020, 5(8), 3801–3808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02402a |
| This journal is © The Royal Society of Chemistry 2025 |