
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo drug design and biological evaluation of coumarin–pyrimidine co-drug derivatives as diabetic inhibitors: expanding a multi-algorithm approach with the integration of machine learning in pharmaceutical research†
Maryam Rashida,
Warda Sarwara,
Shagufta Parveena,
Nida Naza,
Fatima Ridaa,
Eman Nawaza,
Nusrat Shafiq *a,
Dalal Hussien M. Alkhalifahb,
Wael N. Hozzeinc,
Mohamed Mohanyd and
Aniqa Moveeda
*a,
Dalal Hussien M. Alkhalifahb,
Wael N. Hozzeinc,
Mohamed Mohanyd and
Aniqa Moveeda
aSynthetic & Natural Product Discovery Lab, Department of Chemistry, Government College Women University Faisalabad, 38000, Pakistan. E-mail: Smilejust9@yahoo.com; shagufta_organic@yahoo.com; wardasarwar183@gmail.com; naznidagcuf@gmail.com; ridafatima0310@gmail.com; nawazeman86@gmail.com; aniqamaveed713@gmail.com; dr.nusratshafiq@gcwuf.edu.pk; gqumarin@gmail.com
bDepartment of Biology, College of Science, Princess Nourah Bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia. E-mail: DHALkalifah@pnu.edu.sa
cBotany and Microbiology Department, Faculty of Science, Beni-Suef University, Beni-Suef, Egypt. E-mail: hozzein29@yahoo.com
dDepartment of Pharmacology and Toxicology, College of Pharmacy, King Saud University, P. O. Box 55760, Riyadh 11451, Saudi Arabia. E-mail: mmohany@ksu.edu.sa
First published on 11th July 2025
Abstract
Optimization of hit compounds was carried out using the density functional theory. We introduce the coumarin-based co-drug by linkages (amide and oxime) of coumarins with pyrimidines to improve the pharmacokinetic activity of coumarin owing to the synergic effect of both collectively. Pharmacokinetic studies, including drug likeness, were performed using SWISSADMET, and toxicity was identified using a web-based server. Screened lead molecule W6 (C) was synthesized and characterized by NMR spectroscopy, and its anti-diabetic activity was evaluated by in vitro α-amylase inhibition. This study identified W6 as an active, new and more effective candidate for diabetes in drug discovery.
1 Introduction
Diabetes mellitus is a metabolic disorder in which the human body is unable to maintain blood sugar levels. In modern times, the frequency of diabetes is increasing rapidly, and it has become an alarming situation for human health worldwide. Diabetes can cause prolonged complications owing to the deprivation of different blood vessels and organs. In the 21st century, diabetes mellitus is a major chronic disease. It is estimated that in 2015, 415 million people faced this disease, but the ratio has increased rapidly and will reach 642 million by 2040. According to the World Health Organization forecast, in 2030, diabetes will become the 7th leading cause of death worldwide.1–3The two primary types of diabetes are type I diabetes and type II diabetes. Type I diabetes mainly depends on insulin and is most commonly diagnosed in adults and children. In type I diabetes, the immune system destroys the insulin-producing beta cells in the pancreases and leads to the loss of the peptide hormone, which inhibits the transfer of blood glucose to different body tissues, such as muscle and liver. Type II diabetes is defined as an abnormality of the blood glucose level.2 In hyperglycemia, blood glucose levels are high, which creates disturbances in the metabolism of protein and carbohydrates. A high level of glucose in the blood can damage the kidneys and eyes and cause different cardiovascular diseases.1
Several anti-diabetic drugs are available in the market to target different anti-diabetic agents. It is necessary to target those anti-diabetic agents that show better pharmacological activity. In this study, we target four anti-diabetic agents (alpha amylase, aldose reductase, DPP-IV, and glycogen phosphorylase) in anti-diabetic drug design. The fundamental function of an alpha amylase inhibitor is to prohibit the consumption of carbohydrates in the small intestine, and it can be very helpful in curing diabetes.4 One of the dominant approaches is to inhibit DPP-IV for the treatment of diabetes. DPP-IV is a serine protease enzyme that is most commonly present in the kidneys and intestines. The function of DPP-IV is glucose metabolism, and it removes incretin from the body. The function of aldose reductase enzymes is to convert glucose to sorbitol through the polyol pathway. However, for the treatment of diabetes, the main approach is to inhibit these enzymes. The main function of phosphorylase enzymes is glycogen metabolism. In the case of type II diabetes, the amount of glucose is high in the liver. The main approach for the treatment of diabetes is to inhibit excess glucose production in the liver through glycogenesis.5
In ancient times, humans used natural products,6 such as plants, to treat different diseases. In recent years, natural products have played an important role in drug discovery. It is estimated that 25 percent of the drug was isolated primarily from the plants. Plants are a rich source of secondary metabolites.7 Antidiabetic drugs that are derived from natural sources, such as plants, have fewer side effects.8 It is a great challenge for scientists to identify drugs from natural sources that have fewer side effects.9
Coumarin, a natural product, is an attractive candidate for researchers owing to its fewer toxic effects, being more stable, high efficiency, and the fact that coumarin is a low molecular weight compound. Coumarin belongs to the class of benzopyrone, which consists of a benzene ring connected to the pyrone ring. The fundamental application of coumarin is its widespread use as a food additive, in cosmetics and the perfume industry, as well as in electrophoresis, hair spray, detergents, plastics, toothpaste, and food and beverages. Coumarin was first introduced to the market as an additive in 1868.10
The main purpose of this study is to introduce coumarin derivatives as a prodrug (mutual co-drug). A series of naturally occurring coumarin derivatives isolated from natural sources is identified. Coumarin-based co-drugs (mutual prodrugs) were introduced by linkages (amide, oxime) of coumarin derivatives with previously synthesized pyrimidine derivatives11 by rational drug design. The advantage of a co-drug is that the absorption rate of the co-drug is better, and it can easily reach the target site.
Nowadays, in silico virtual screening methodologies play an important role in drug discovery. Molecular docking was performed to screen the compound. Molecular docking is a powerful tool for identifying the interaction of a drug with macromolecules, such as proteins. For virtual screening, molecular docking is the most widely used.12 In computational studies, virtual screening plays a significant role. The fundamental purpose of virtual screening is to identify the chemical structures that show the best affinity for the targeted protein. Docking can be proposed to screen thousands of libraries in the compound. After screening the compound, optimization of the compound was done by DFT (density functional theory). Computational studies, such as DFT of screened compounds, give us information about the reactivity of the compound, geometry optimization and stability of the compound. The DFT of the screened compound was performed using Gaussian 09 software. The fundamental function of DFT is to determine the molecular structure of the compound and its activity by calculating different reactivity descriptors. By virtue of the information, the synthesis of the target can be identified as either possible or not. Pharmacokinetic studies, including drug likeness and toxicity, were performed using a web-based server. The ADMET study describes pharmacokinetic properties, such as the absorption, distribution, metabolism, excretion, and toxicity of the compound.
2 Materials and methods
We explain here how the process unfolded in this study to provide a clear overview. At the outset, a set of known active coumarins and pyrimidines was assembled from the published literature. To assess whether these compounds interacted well with diabetic-related targets (α-amylase, DPP-IV, aldose reductase, and glycogen phosphorylase), they were used in molecular docking. These hits showing the highest performance were examined using Density Functional Theory (DFT) to check their electronic and chemical stability. Using SwissADME and Protox-II, the researchers carried out secondary checks on pharmacokinetics and toxicity to evaluate the drug-likeness of the synthetic compounds. W6, the main compound, was made using several chemical procedures, identified with NMR and IR spectroscopy, and its ability to reduce diabetes was confirmed by testing with an α-amylase inhibition assay. Because various experimental and computational techniques were integrated, a favorable antidiabetic drug with minimal toxicity was discovered.2.1. Software design for computational studies
Computational and virtual screening tools and software, such as ChemDraw Professional (Version 19.1.0.8, PerkinElmer, Waltham, WIA, USA), Gaussian 09W, Auto Dock Tools (ADT), BIOVIA Discovery Studio 2020 (v20.1.0.19295), the PyMOL molecules graphic system, version 2.4.1 (Schrodinger, LLC), and Open Babel 2.4.1, were utilized. Optimization of the compound was carried out using the basis set 6311G and B3LYP functions in Gaussian 09. A pharmacokinetic study was performed using a web-based server, such as Swiss ADMET (https://www.swissadme.ch/).2.2. Rational drug designing
The application of rational drug design has gained the interest of medicinal chemists in drug discovery and medicinal chemistry owing to its ability to use theoretical and experimental knowledge of the underlying system. Rational drug design has contributed to the development of features and advancements in biochemistry, medicinal chemistry, molecular sciences, and pharmacokinetics of drugs. Based on rational drug design, knowledge of the system is utilized, which reduces the cost of human power, time consumption and experimental expenses in the lab.13 Therefore, based on the use of structure- and core-based designs, a series of coumarin and pyrimidine analogues was selected. Their inhibition potential was screened against type II diabetes using a multi-disciplinary approach of computational tools with the incorporation of in vitro antidiabetic analysis. For this purpose, a dataset of coumarin from the literature and pyrimidine previously synthesized14 were used to screen hit-to-lead optimized compounds. Lead compounds were further subjected to design using a core-hopping approach.2.3. Preparation of the library
A library of 27 coumarin-based compounds was taken from the PubChem database, Google Scholar, DNP, and previously available literature. These coumarins were selected based on their in vitro activity, and pyrimidine derivatives used for co-drug were selected from the previously synthesized15,16 and in vitro and in silico optimized series.14 These selected compounds were collected in library form as a dataset (Table S1 (ESI)†).2.4. Molecular docking analysis for diabetes inhibition of coumarin–pyrimidine co-drugs
2.5. MM-GBSA
The determination of the free binding energy of the protein–ligand complex is performed using the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method. This is done by employing the prime module of the Schrödinger suite 2022, which incorporates the OPLS4 force field and the VSGB (Variable Dielectric Surface Generalized Born) solvation model to perform the calculations.18 Following the energy terms and equation systems presented below, an automatic calculation was performed to determine the complex energies.| ΔGbinding = Gcomplex − (Greceptor + Gligand), |
| ΔGbinding = ΔEMM + ΔGGB + ΔGSA. |
| ΔGbinding = ΔHbinding − TΔS, |
2.6. Molecular dynamics simulation
MD simulations evaluated the dynamic characteristics along with flexibility patterns in protein–ligand complexes. The iMODS server functions as an efficient tool for analyzing macromolecular complex intrinsic motions through Normal Mode Analysis (NMA). The key parameters included both deformability plots and B-factor analysis along with eigenvalues, covariance matrices and elastic network models. The computed eigenvalues describe how stiff different portions of the complex structures were, while deformability features together with B-factor analyses show where system flexibility occurs. The computed results validated both the structural stability and binding potential of the examined complexes when operating in dynamic environments.IMODS is an online server used for the molecular dynamics' simulation of anti-diabetic protein–ligand complexes, such as 3RX2–W6, 3BAJ–W6, 1GPY–W6, and 4J3J–W6. NMA was carried out using the iMODS server to analyze the mobility and deformability of the complexes. Owing to this analysis, we discovered how the complexes move and flex as units, which is an important factor in understanding their ability to stay together and join others. Homologous molecules of the complexes could be processed by IMOD, but there is no need for 100% sequencing of the complexes. Simulation by IMOD plays a crucial role in drug designing strategies, as it gives knowledge about the flexibility of the target protein–ligand complex.
2.7. Density functional theory calculation
The computational studies used density functional theory and the Becke three-parameter Lee–Yang–Parr (B3LYP) functional with a 6-311G basis set to analyze the systems through the Gaussian 09W software package. The optimization of all molecules occurred in a gas phase scenario using an unrestricted optimization method. The structures reached true minima through frequency calculations, which showed no imaginary frequencies when performed at the same theoretical level. The optimized molecular structures yielded essential electronic values of HOMO and LUMO orbital energies and ΔE, μ, η, and ω that helped analyze the stability and reactivity features of these molecules.The basic function of density functional theory is to calculate the structural parameters and orbital interactions of the chemical compound.20 In recent years, density functional theory has played a significant role in determining the reactivity of organic compounds. DFT (density functional theory) calculation was carried out using a basis set 6311G and the B3LYP function. Optimization of the compound was carried out using density functional theory. Using Gaussian 09, we can determine the quantum chemical parameters, such as envision the dipole moment, electronic energy, molecular electrostatic potential and energies of HOMO and LUMO.21 The reactivity of the compound was determined by measuring the energy gap present between the HOMO and LUMO. Frontier molecular orbitals are the most important parameters for calculating the other chemical reactivity descriptors. However, the stability of the compound correlates with its hardness, and the chemical softness of the compound determines the range of its reactivity.22 However, the electrophilicity index indicates the flow of electrons between the donor and the acceptor. The compound has a higher value of electrophilic index. This indicates that the compound is more electrophilic in nature.23 The compound has the highest value of the electrophilicity index, indicating that the compound has a higher binding affinity with organic molecules.24 The compound with the higher value of chemical potential represents a moving charge from this compound to another compound.25 The high dipole moment of the compound determines the binding site within the particular selected protein.26 Molecular electrostatic potential is a fundamental tool for determining the reactivity and behavior of a compound. However, the MEP scale represents the electrophilic and nucleophilic regions. The red color indicates the electron rich region, and the blue color represents the electron deficient region. Molecular electrostatic potential indicates the size of the molecules. Molecular electrostatic potential represents the electron density in the compound.27 The molecular electrostatic potential is determined using a basis set 6311G.28
The synthetic reactants and the complex product formed were computationally optimized using Gaussian 09W and GaussView 6.0, with a hybrid functional of B3LYP and a basis set of 6311-G(d,p).29 For parallel validation of the experimental details, calculations were carried out using the IEFPCM approach.30
2.8. Drug-likeness activity of the selected compound
The drug-like property of the compound is determined by an ADMET study. Using ChemDraw, SMILES representations of the coumarin co-drug derivatives were generated and then processed on the SwissADME website to assess pharmacokinetic properties.31 The Lipinski rule of 5 was applied to determine the drug-like behavior of the compound. The toxicity of the compound was determined using the online server PROTOX-II.2.9. Synthesis of target lead molecule
 | ||
| Scheme 1 (A) Synthesis of the precursor pyrimidine (A) for C-drug formation; (B) synthesis of the precursor coumarin (B) for co-drug formation; and (C) synthesis of the target lead molecule (C) for co-drug formation. | ||
2.10. Spectroscopic characterization
The synthesized target lead was analyzed for structure confirmation by 1H- and 13C-NMR spectroscopy (see ESI† for spectral detail).2.11. Biological evaluation as in vitro anti-diabetic leads
The synthesized lead co-complex W6 (C) was subjected to bio-assay to screen its in vitro anti-diabetic potential using the method by Uzma Arshad et al.14,32 According to this method, the test compounds were incubated together with the α-amylase enzyme and starch under set conditions. Next, the amount of reducing sugars was determined by measuring their reaction with the 3,5-dinitrosalicylic acid (DNSA) reagent, most often at a wavelength of 540 nm, using a spectrophotometer. All patient outcomes were compared to those of the standard drug acarbose. To learn about their potential as anti-diabetic drugs, the amount of α-amylase inhibition by each test compound was measured against the control. The activity was performed using α-amylase as the enzyme and starch as the substrate in addition to acarbose as a reference drug. The percentage inhibition was calculated using the following formula:
3 Results and discussion
3.1. Scaffold-based drug design
Based on the core-hopping approach (Fig. 1), the selected series of compounds was screened using different computational tools. Consequently, one lead from pyrimidine and one from coumarin were further selected. These leads from both series were further subjected to synthesis, characterized for structure elucidation and further validated for their inhibitory potential by in vitro antidiabetic bio-assay. | ||
| Fig. 1 Core-hopping and rational drug design-based approach used for the synthesis of C-drugs. | ||
3.2. Virtual screening of the compound based on the scoring function
Using a structure- and activity-based approach, coumarin-based compounds were downloaded from the PubChem database, which showed high IC50 values via in vitro and in vivo activities. A virtual screening of the compounds was performed to evaluate their biological potential as anti-diabetic agents. For this, a molecular docking approach was employed. Molecular docking of the compounds provided variable binding affinity values of the compounds. The screening of the compounds was achieved based on the scoring function of AutoDock software. Docking of the 27 naturally occurring coumarin compound-based pyrimidine hybrids was performed against target proteins, such as α-amylase with PDB-ID (3BAJ), DPP-IV with PDB-ID (4J3J), aldose reductase PDB-ID (3RX2), and glycogen phosphorylase PDB-ID (1GPY), which are related to diabetes. As hit compounds, they exhibited good binding affinity values when docked with AutoDock tools. Thus, a scoring function filter was applied, which provided the compounds with the best anti-diabetic potential with binding affinity values ranging from −10.1 to −9.0 kcal mol−1.Molecular docking studies of the compound with PDB ID 3BAJ showed a good binding affinity ranging from −9.7 to −7.0 kcal mol−1 compared to standard drugs. Acarbose showed a binding affinity of −6.9 kcal mol−1. All the compounds showed a favorable interaction against the target enzyme.33 The top five compounds showed a good binding score to the alpha amylase inhibitor shown in Table S3 (ESI).† Hydrogen bonding between the ligand and the targeted protein is depicted in Table S3 (ESI).† The compound that showed the highest binding score, 4-methyl umbelliferon–pyrimidine co-drug (W6), showed hydrogen bonding interaction through amino acid residues, such as GLN63, HIS201, HIS305, HIS305, TRP59, and GLU233. It forms a six-hydrogen bond. Other Van der Wall interactions have been observed via pi-anion, pi-alkyl, alkyl, and pi-sulfur. In the 4-methyl umbelliferon–pyrimidine co-drug (W6), electrostatic interaction was shown by ASP300, ASP197, and ASP300 amino acid residues, while hydrophobic interaction was shown by LYS200, ILE235, and HIS201 amino acid residues (Fig. 2).34 None of the 27 compounds that target alpha amylase for anti-diabetic activity show any unfavorable interactions. Docking studies of 27 compounds with PDB-ID 4J3J for DPP-IV inhibition were performed, and only four compounds showed good binding affinity ranging from −9.5 to −9.1 kcal mol−1. Sitagliptin, a standard drug, had a binding affinity of 8.7 kcal mol−1. All the hit compounds formed 15 hydrogen bonds. More than 15 hydrophobic interactions were observed between the hit compound 8-methoxy psoralen–pyrimidine complex (W24) and the amino acid residues LEU162, ILE235, HIS305, LEU162, LYS200, and ALA98. When the number of hydrogen bonds decreased, it increased the hydrophobicity of the compound and indicated the stability of the compound (Fig. S1 and S2: ESI†).35 Four hit compounds showed excellent binding affinity ranging from −10.1 to −9.0 kcal mol−1 compared to the standard drugs when docked with aldose reductase PDB ID 3RX2 (Table S1: ESI†). Epalrestat showed a binding affinity of −8.1 kcal mol−1.36 The hit compound (W1) showed 12 hydrogen bonding interactions. The hit compound (W1) showed hydrophobic interactions via amino acid residues Val47, leu300, Phe122, TRP219, Phe121, and others (Fig. 2).
 | ||
| Fig. 2 Interaction of W6 with 3BAJ (a) protein in complex with ligand; (b) 3D-protein–ligand interaction, (c) 2D-protein–ligand interaction, and (d) hydrophobic interaction. | ||
Docking of 27 compounds showed binding affinity ranging from −9.5 to −8.8 kcal mol−1 when docked with glycogen phosphorylase protein 1GPY. The hit compound (W19) showed higher binding affinity as well as four hydrogen bonds with binding protein 1GPY (Fig. S3: ESI†). However, metformin, which was used as a standard, showed a binding affinity of −5.7 kcal mol−1.37
3.3. MM-GBSA analysis
The performance accuracy of targeted proteins with selected lead ligands (selected from 27 studied complex compounds) was measured by the prime MM-GBSA calculation of receptor–ligand complexes.38 The molecular mechanical energy produced by the receptor–ligand interaction was considered in the MM calculation. The effectiveness of this method has been demonstrated for a wide range of protein–protein complexes.39,40 Molecular docking was also evaluated using MMGBSA free limiting energy, linked to the 1GPY, 3BAG, 3RX2 and 4J3J targets. Checking the expected low-energy poses is a surefire way to verify docking's precision. The Glide scores were quite close to the binding mode obtained experimentally using X-ray crystallography. Docking ligands into the binding pocket yields MM-GBSA free energy estimates.18 The free binding energies of the targeted complex with the respective receptors are presented in Table 1. The linear relationship between binding free energy for different parameters of the complex in the solvent (water) is represented in Fig. 3.| PDB ID | Ligand | ΔGbind | ΔGbind Coulomb | ΔGbind covalent | ΔGbind H-bond | ΔGbind lipo | ΔGbind vdW |
|---|---|---|---|---|---|---|---|
| 1GPY | W1 | −85.89 | −19.92 | 0 | −1.89 | −28.67 | −34.61 |
| W14 | −63.85 | −14.76 | 0 | −3.51 | −13.1 | −46.4 | |
| W19 | −69.48 | −41.7 | 0 | −4.54 | −13.97 | −44.79 | |
| Standard (metformin) | −37.65 | −33.1 | 0 | −3.34 | −3.12 | −24.12 | |
| 3BAJ | Standard (acarbose) | −37.68 | −23.28 | 17.03 | −7.15 | −27.5 | −74.18 |
| W24 | −46.54 | −20 | 9.12 | −2.29 | −19.95 | −63.37 | |
| W6 | −24.94 | −16.12 | −0.02 | −1.96 | −15.43 | −41.42 | |
| W3 | −12.43 | −9.28 | −7.07 | 0.1 | −18.26 | −59.81 | |
| W19 | −28.43 | −18.63 | 3.58 | −0.5 | −12.09 | −55.54 | |
| W23 | −28.4 | −17.09 | 4.6 | 0.02 | −16.85 | −50.1 | |
| 3RX2 | Standard (epalrestat) | −30.25 | −6.41 | 14.83 | −0.3 | −11.1 | −44.93 |
| W1 | −39.06 | −32.59 | 9.12 | −3.11 | −10.56 | −32.44 | |
| W3 | −39.89 | −1.57 | 4.8 | −1.54 | −17.31 | −51.74 | |
| W27 | −58.65 | −14.84 | 6.3 | −0.94 | −13.83 | −58.29 | |
| W23 | −31.23 | −6.96 | 6.27 | −2.32 | −14.14 | −42.46 | |
| 4J3J | Standard (sitagliptin) | −21.25 | −13.75 | 9.75 | −2.66 | −20.81 | −47.04 |
| W1 | −33.57 | −21.44 | 14.42 | −2.37 | −30.24 | −46.2 | |
| W3 | −33.32 | −9.83 | 11.54 | −2.51 | −26.51 | −47.32 | |
| W24 | −42.89 | −18.18 | 8.02 | −1.13 | −20.4 | −64.13 | |
| W18 | −38.54 | −17.61 | 16.57 | −1.96 | −18.39 | −55.16 | |
| W19 | −42.46 | −4.3 | 10.1 | −2.4 | −15.89 | −39.87 |
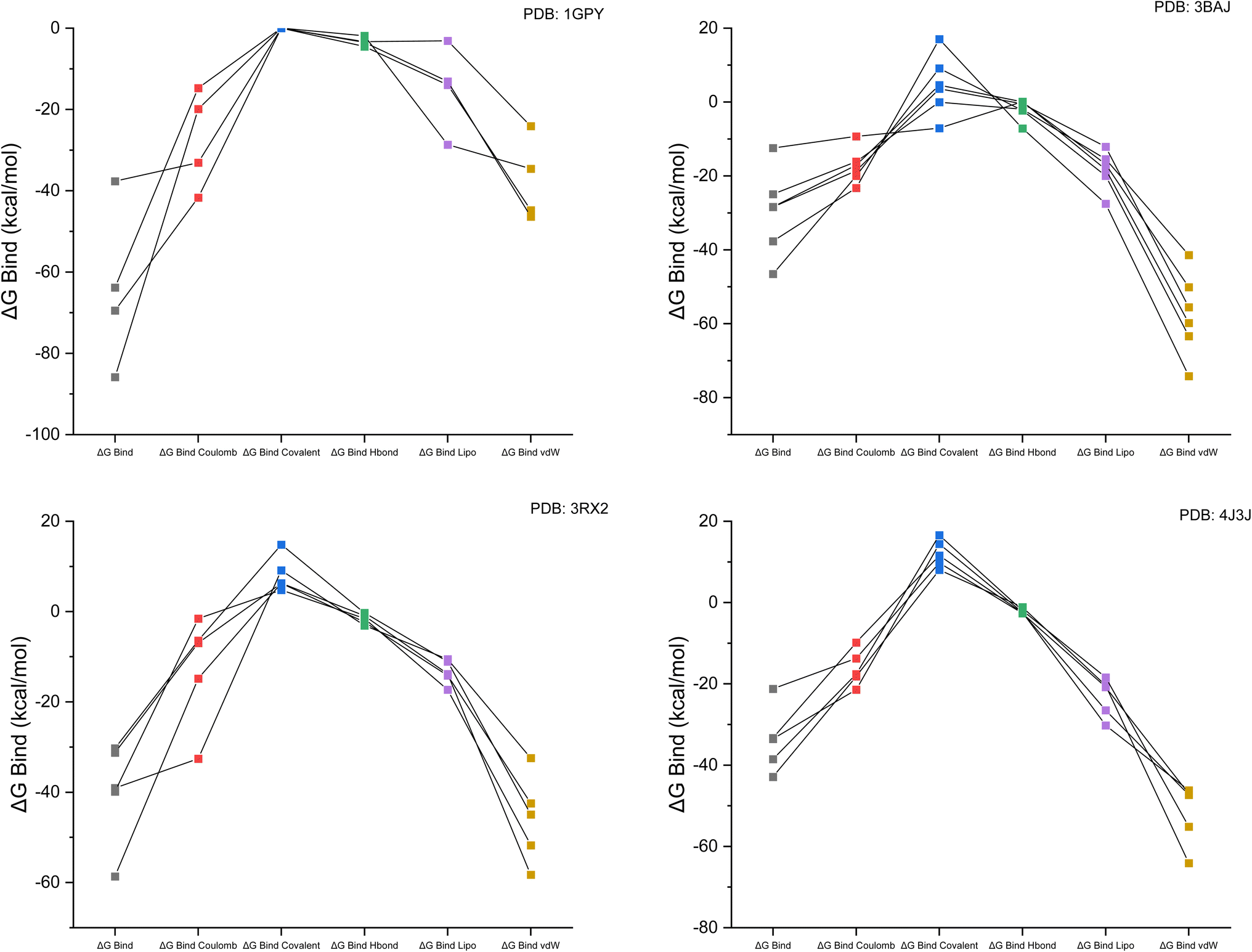 | ||
| Fig. 3 Linear graphical representation of the binding free energy for Coulomb, covalent, H-bond, lipophilic and van der Waals forces for each receptor with the targeted ligand complex. | ||
3.4. Re-docking
An RMSD value was calculated to validate the docking method. Re-docking of the native and docked ligand was carried out for docking validation. Docking validation for antidiabetic activity was performed with PDB-ID 3BAJ for alpha amylase, 4J3J for DPP-IV, 3RX2 for aldose reductase, and 1GPY for glycogen phosphorylase, with an RMSD value less than 2 Å. The redocking best pose of the native and docked ligands is shown in Fig. 4. | ||
| Fig. 4 Different redocking poses for the anti-diabetic activity. | ||
3.5. Molecular dynamics simulation
All 4 complexes (3RX2–W6, 3BAJ–W6, 1GPY–W6, and 4J3J–W6) undergo molecular dynamics simulation. The results from the iMODs summarize the NMA (normal mode analysis) for different antidiabetic docked complexes. This study was performed to regulate the actions of the proteins. The mobility of the docked complexes was checked using deformability and B-factor graphs. The deformability and B-factor graphs of complexes 3RX2–W6, 3BAJ–W6, 3RX2–W6, and 4J3J–W6 are shown in Fig. S4 (ESI).† The eigenvalue of the complexes was linked to the normal mode of the complexes. The lower the eigenvalue, the higher the mobility of the complexes. The eigenvalues of all the complexes are presented in Table S4 (ESI).†Complexes 1GPY–W6 and 3BAJ–W6 have lower eigenvalues, showing the greatest mobility of these complexes from the other two, as shown in Fig. S5 (ESI).† Complex 1GPY–W6 has the lowest eigenvalue and great mobility compared to the other complexes. The RMSF (root mean square fluctuation) of lead complex 1GPY–W6 was calculated by the iMODs, which showed the minimum fluctuation of 1.01 × 10−1 at residue no 128 and the maximum fluctuation of 9.87 × 10−2 at residue no. 718, as shown in Fig. 5.
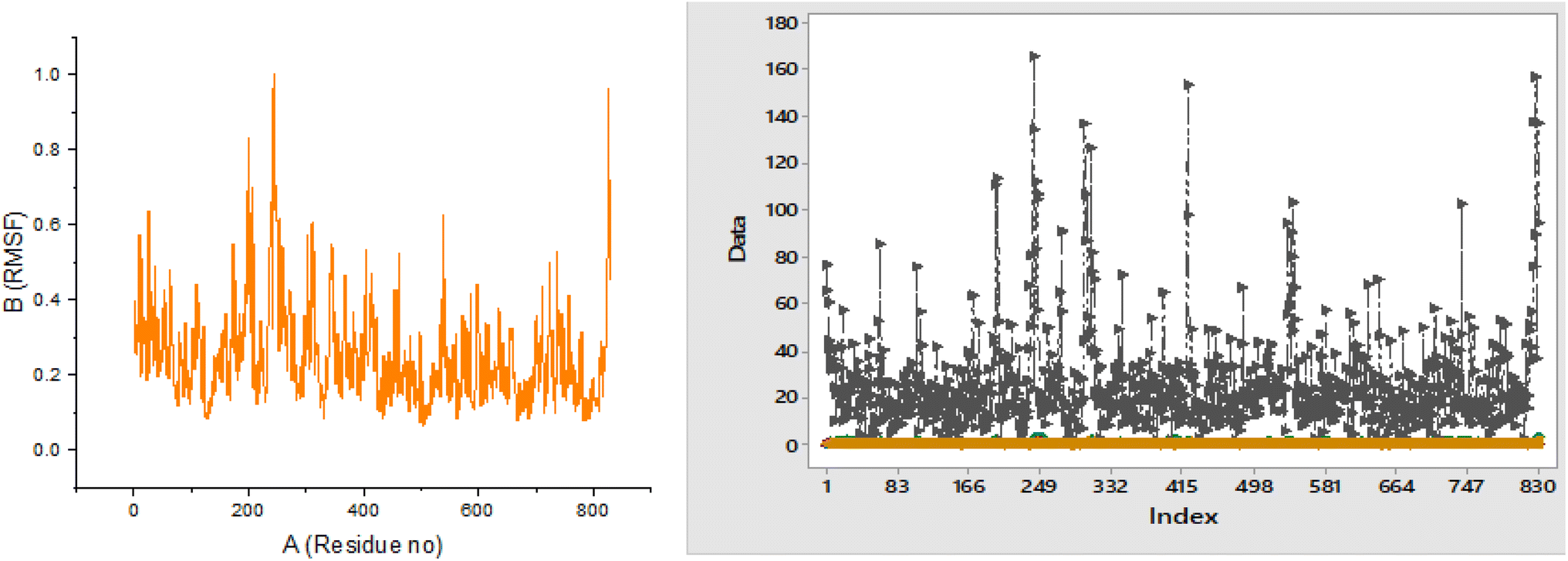 | ||
| Fig. 5 RMSF and RMSF overlapping of the complex 1GPY–W6. | ||
According to the literature, the RMSD value for a good docked complex should be less than 2 Å, while the complex 1GPY–W6 has an RMSD value of 0.25 Å, which is very good along with the radius of gyration that is also calculated by iMODs (Fig. 6).
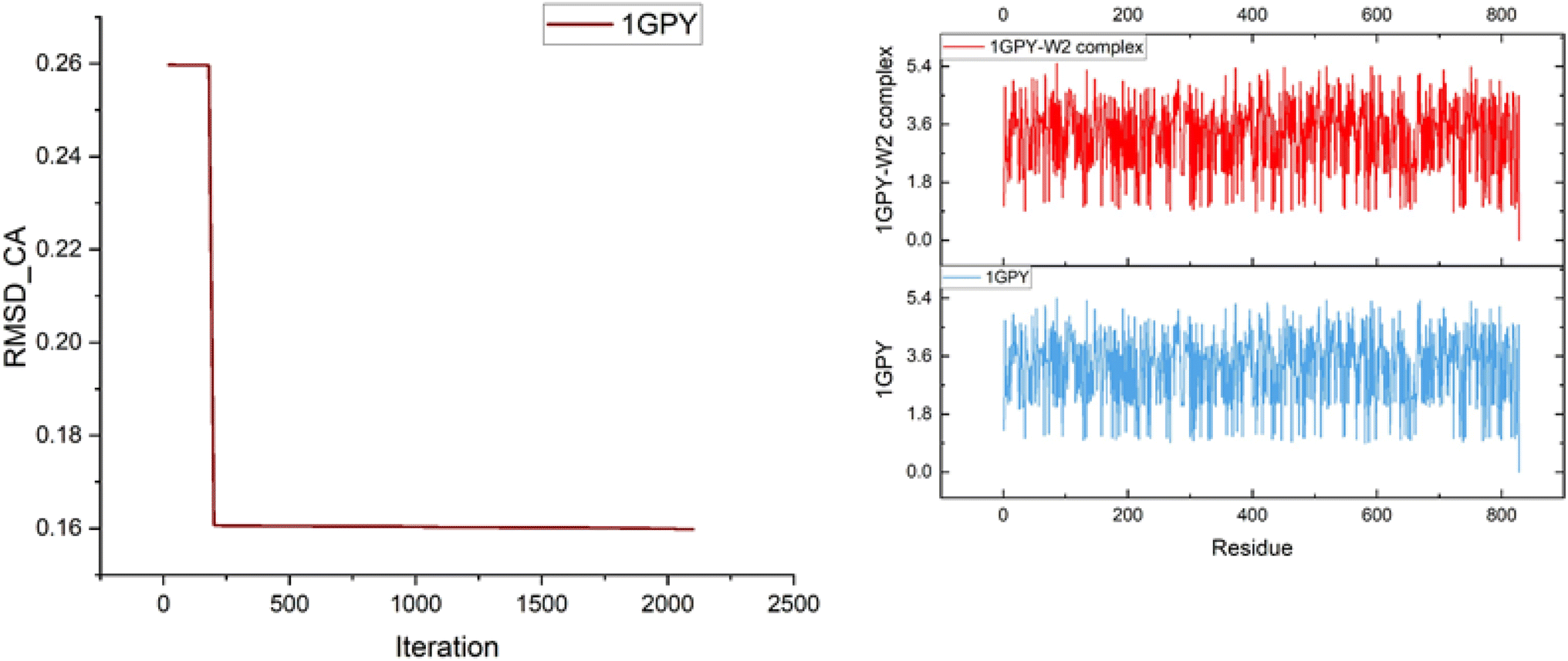 | ||
| Fig. 6 RMSD and radius of gyration. | ||
3.6. DFT studies
Optimization of the screened compounds was carried out using DFT (Fig. 7). DFT was performed using a basis set of 6-311G.41 Frontier molecular orbitals can be classified as HOMO (highest occupied molecular orbitals) and LUMO (lowest unoccupied molecular orbitals) (Fig. 7). The frontier molecular orbitals are very significant in determining the reactivity and stability of the compound.42 The compound with the highest HOMO value has the strongest ability to donate electrons. The compound has the lowest value of HOMO, and it has less ability to donate electrons, making it the least effective (Table 2).43 | ||
| Fig. 7 (a) Optimized structure of the lead W6; (b) MEP mapped of the lead W6; (c) LUMO frontier molecular orbitals; and (d) HOMO frontier molecular orbital. | ||
| Compound | EHOMO (eV) | ELUMO (eV) | Energy gap (ΔE) |
|---|---|---|---|
| W1 | −0.23 | −0.06 | 0.17 |
| W6 | −0.21 | −0.07 | 0.14 |
| W14 | −0.23 | −0.09 | 0.14 |
| W23 | −0.23 | −0.10 | 0.13 |
| W18 | −0.23 | −0.11 | 0.12 |
| W19 | −0.23 | −0.11 | 0.13 |
| W27 | −0.21 | −0.10 | 0.12 |
| W3 | −0.21 | −0.06 | 0.15 |
| W24 | −0.23 | −0.10 | 0.13 |
Frontier molecular orbitals describe the electrophilic and nucleophilic characteristics of the compound. The compound with a higher energy of HOMO indicates that it is a good nucleophile, and the compound with a lower energy of LUMO indicates that it is a good electrophile.44 Compound W23 has the highest value of HOMO (−0.23429 eV), indicating that it is a good nucleophile and has a greater ability to donate electrons. The order of ranking of the nucleophilic character of the compounds is as follows:
| W23 > W19 > W18 > W14 > W1 > W24 > W27 > W6 > W3. |
Compound W8 (−0.05721 eV) has a lower LUMO energy. It was found to have a good electrophile, meaning that it has a higher ability to accept electrons. The order of ranking of the electrophilic behavior of the compounds is as follows:
| W3 > W1 > W6 > W14 > W27 > W24 > W23 > W19 > W18. |
The most important parameter is the energy gap, which is related to the stability of the compound. Compound W7 has the lowest energy gap, which indicates the stability of the molecules. A small energy gap indicates that the compound is soft and more reactive. The ranking of the compound having the smallest energy gap and increasing the stability of the compound is given below:
| W27 > W18 > W19 > W24 > W23 > W14 > W6 > W3 > W1. |
Compound W1 has a larger energy gap, which represents the hardness and less reactivity of the compound. The order of the decreasing reactivity of the compound is as follows:45
| W1 > W3 > W6 > W14 > W23 > W24 > W19 > W18 > W27. |
Chemical reactivity descriptors, such as chemical hardness and chemical softness values, quantify resistance to charge transfer. The electrophilicity index provides information about the binding affinity of compounds with organic molecules. The values of the chemical reactivity descriptor are shown in Table 3. The first component of the chemical reactivity descriptor is the electrophilicity index. Compound W1 has the highest value of the electrophilicity index. This means that the compound has good binding affinity to the targeted protein. The order of compounds with good binding affinity is as follows:
| W1 > W18 > W19 > W23 > W24 > W27 > W14 > W6 > W3. |
| Compound | Electronic energy | Dipole moment | Chemical potent | Ionization potential | Chemical hardness | Chemical softness | Electrophilicity index | Electronegativity | Electron affinity |
|---|---|---|---|---|---|---|---|---|---|
| W1 | −1705.56 | 2.25 | −0.53 | 0.23 | 0.47 | 1.07 | 0.94 | 0.53 | 0.06 |
| W6 | −1912.017 | 5.29 | −0.53 | 0.21 | 0.47 | 1.07 | 0.31 | 0.53 | 0.07 |
| W14 | −1946.73 | 6.40 | −0.55 | 0.23 | 0.45 | 1.10 | 0.33 | 0.55 | 0.09 |
| W23 | −2024.13 | 11.66 | −0.55 | 0.23 | 0.45 | 1.11 | 0.34 | 0.55 | 0.10 |
| W18 | −8592175.55 | 3.55 | −0.55 | 0.23 | 0.45 | 1.12 | 0.34 | 0.55 | 0.55 |
| W19 | −2061.04 | 5.90 | −0.56 | 0.23 | 0.45 | 1.12 | 0.34 | 0.55 | 0.11 |
| W27 | −2393.91 | 5.49 | −0.55 | 0.21 | 0.45 | 1.11 | 0.33 | 0.55 | 0.10 |
| W3 | −1858.37 | 4.318 | −0.53 | 0.21 | 0.47 | 1.06 | 0.30 | 0.53 | 0.06 |
| W24 | −2175.54 | 5.88 | −0.55 | 0.23 | 0.45 | 1.11 | 0.34 | 0.55 | 0.10 |
The chemical hardness and chemical softness of the compound are interrelated with the energy gap. The compound with a small energy gap indicates that the compound is reactive. However, hard molecules have a large energy gap and are the least reactive. Biologically, the activity of a compound increases when the compound has a small energy gap and the molecules are soft.
| W27 > W18 > W19 > W24 > W23 > W14 > W6 > W3 > W1 |
The compound has a small value of electronegativity, which determines the delocalization of electrons on the entire compound. This makes it easier for the compound to correlate with the biological system by giving electrons. The ranking of the compound based on the smaller value of electronegativity is as follows:
| W3 > W1 > W6 > W14 > W27 > W24 > W23 > W19 > W18. |
The compound has the highest dipole moment, and it identifies the binding pose of the compound with the target protein. The order of the compound with the highest dipole moment is as follows:
| W23 > W14 > W19 > W24 > W27 > W6 > W3 > W18 > W1. |
Through DFT evaluations, we obtained essential data about the electronic structure and reactivity patterns in their target substances before conducting synthetic cycles. The compounds maintained effective HOMO–LUMO energy gaps that suggested optimal reactive qualities while ensuring chemical stability. Higher chemical hardness values showed resistance to deformation, thus predicting a favorable synthetic process. Electrophilicity displayed by the molecules confirmed their biological interaction ability while showing limited reactive potential, thus reducing nonspecific binding risks. Computational predictions built on quantum mechanics technologies allowed us to choose compounds for synthesis, which later served as the basis for molecular dynamics testing to establish the validity of our computational screening process.
| Targets | Experimental λmax (nm) | Theoretical λmax (nm) | Excitation energies (eV) | Oscillator strength (f) |
|---|---|---|---|---|
| Compound A | 325 | 310.37 | 3.9947 | 0.4399 |
| Compound B | 285.58 | 303.12 | 4.0903 | 0.3937 |
| Compound C | 284 | 389.65 | 3.1819 | 0.0114 |
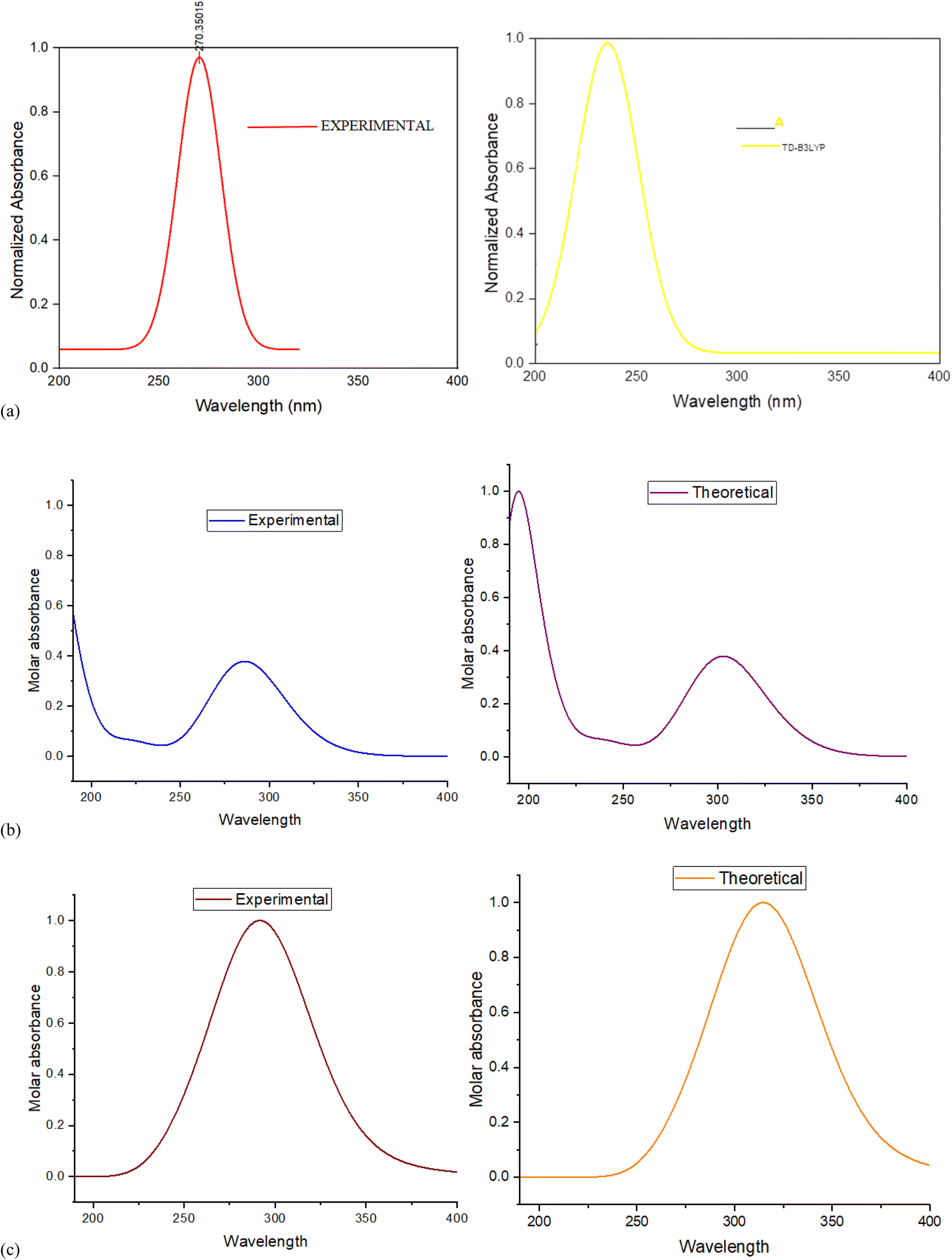 | ||
| Fig. 8 (a) Plot of λmax (nm) and molar absorbance of compound A. (b) Plot of λmax (nm) and molar absorbance of compound B. (c) Plot of λmax (nm) and molar absorbance of compound C. | ||
 | ||
| Fig. 9 (a) Experimental vibrational spectra and theoretical vibrational spectra of compound A. (b) Experimental vibrational spectra and theoretical vibrational spectra of compound B. (c) Experimental vibrational spectra and theoretical vibrational spectra of compound C. | ||
3.6.2.1. Aromatic ring C–H vibrations. In aromatic systems, the C–H of the benzene ring shows absorption at 3100–3000 cm−1, as reported in the literature. In the theoretical study, compound C shows the C–H stretching of the benzene ring at 3100 cm−1, but there is no peak observed in the experimental study. Compound A observed C–H stretching at 3106.81 cm−1, and experimentally, this stretching appeared at 3163 owing to the presence of the alcoholic functional group moiety.
3.6.2.2. Carbonyl group (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O) vibrations of ester linkage. In the literature, the ester-linked carbonyl functional group C
O) vibrations of ester linkage. In the literature, the ester-linked carbonyl functional group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration range is 1750–1700 cm−1. The main target compound (C) showed CO stretching at 1700 cm−1 in the theoretical study, which indicates the CO moiety of ester linkage, but in the experimental study, this stretching was observed at 1752 cm−1. The theoretical value of the same vibration for compound A appeared at 1705 cm−1, but in the experimental study, no peak was observed in this range. In the vibrational analysis spectrum of compound B, no peak was observed in the theoretical and experimental calculations, which indicated that no ester linkage functional group was present in the particular compound.
O stretching vibration range is 1750–1700 cm−1. The main target compound (C) showed CO stretching at 1700 cm−1 in the theoretical study, which indicates the CO moiety of ester linkage, but in the experimental study, this stretching was observed at 1752 cm−1. The theoretical value of the same vibration for compound A appeared at 1705 cm−1, but in the experimental study, no peak was observed in this range. In the vibrational analysis spectrum of compound B, no peak was observed in the theoretical and experimental calculations, which indicated that no ester linkage functional group was present in the particular compound.
3.6.2.3. Vibrational analysis of C–F. To assess the interaction of aromatic rings with halogen atoms from a functional group perspective, the stretching vibration for F–C is quite important. The detected infrared for this stretching is in the range of 1100–1300 cm−1. The C–F stretching for compound C is stretched at 1250 cm−1 theoretically and experimentally; it was observed in the range of 1135 cm−1. The theoretical and actual values of compound A for C–F stretching vibration were observed at 1127 cm−1 and 1135 cm−1, respectively. From the theoretical and experimental results, no peak appeared for compound B of the same stretching, indicating that no halogen group was present in the B precursor compound.
3.6.2.4. Vibrational analysis of the C–OH and alcoholic OH groups. The O–H and C–O stretching vibrations have characteristic bands in the range of 3650–3400 cm−1 and 1300–1000 cm−1, respectively, as reported in the literature. For target compound C, the O–H stretching appeared at 3327 cm−1 in the experimental results, but theoretically, no peak was observed in this range. The intense peak of the C–O stretching for compound C was observed at 1248 cm−1 theoretically and in the actual study; this vibration stretching appeared at 1018 cm−1. In compound A, no peak was observed theoretically and experimentally in this range, clearly indicating that the alcoholic group was not present in compound A. Compound B showed the peak for O–H and C–O stretching at 3808 cm−1 and 1026 cm−1 theoretically, respectively.
3.6.2.5. Vibration analysis of C–N and N–H stretching. To identify the nitrogen group in the compound, N–H stretching in the infrared spectrum is very important. The N–H and C–N stretching of aromatic compounds are observed in the range of 3500–3100 cm−1 and 1250–1450 cm−1, respectively, in the literature. No peak was observed in the range of N–H stretching for compound C, clearly indicating that no hydrogen was attached to the nitrogen atom in compound C. The C–N vibration stretching for compound C is observed at 1420 cm−1 in the theoretical study, while in the experimental study, the peak of the same stretching is observed at 1400 cm−1. Compound A showed N–H stretching at 3163 cm−1 in the experimental analysis, but in the theoretical analysis, this stretching was observed at 3106.81 cm−1. The C–N vibration peak for compound A was observed at 1216 cm−1 and 1445 cm−1 by experimental and theoretical analyses, respectively. Further, no peak appeared for compound B in the range of both stretching vibrations C–N and N–H, indicating that no nitrogen group was present in compound B.
The highest theoretical chemical shift value of 13C-NMR of co-drug C is 202.83 ppm, and the peak of this carbon was observed at 174.9 ppm. The irregularity in both peaks is due to the dependence on the surrounding functional groups present. The theoretical and experimental chemical shift values are close to each other, validating the experimental peaks, but the peak of C-20 appeared at δ 77.49 ppm in the GIAO method, but in the actual study, the peak of this carbon appeared at δ 98.6 ppm because of the presence of the pyran ring. The chemical shift values of 1H NMR were found to agree with the experimental values (Table 5 and Fig. 10).
| Carbon | Theoretical | Experimental | Proton | Theoretical | Experimental |
|---|---|---|---|---|---|
| Compound A | |||||
| 5-C | 1.38 | 14.46 | 26-H | 8.23 | 10.43 |
| 15-C | 11.31 | 17.67 | 27-H | 7.99 | 9.75 |
| 10-C | 12.38 | 53.99 | 25-H | 7.83 | 7.44 |
| 8-C | 30.07 | 60.18 | 24-H | 7.75 | 7.08 |
| 3-C | 31.46 | 100.74 | 22-H | 7.46 | 7.15 |
| 12-C | 47.05 | 122.87 | 23-H | 6.75 | 6.97 |
| 13-C | 55.15 | 113.44 | 21-H | 5.64 | 5.21 |
| 9-C | 63.42 | 113.73 | 34-H | 4.73 | 4.05 |
| 11-C | 63.70 | 131.27 | 35-H | 4.58 | 2.50 |
| 2-C | 75.73 | 164.21 | 29-H | 3.64 | 1.12 |
| 20-C | 117.13 | 146.51 | 28-H | 3.17 | |
| 1-C | 121.13 | 160.97 | 30-H | 2.13 | |
| 18-C | 160.68 | 174.61 | 33-H | 2.06 | |
| 19-C | 168.93 | 165.43 | 32-H | 2.04 | |
| 31-H | 1.83 | ||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Compound B | |||||
| 1-C | 184.11 | 161.1 | 15-H | 8.33 | 10.5 |
| 9-C | 180.60 | 160.2 | 16-H | 7.55 | 7.57 |
| 5-C | 179.85 | 154.8 | 14-H | 7.47 | 6.79 |
| 10-C | 178.98 | 153.4 | 18-H | 6.68 | 6.68 |
| 3-C | 148.54 | 126.5 | 17-H | 5.60 | 6.10 |
| 4-C | 134.89 | 112.8 | 20-H | 3.12 | 2.99 |
| 2-C | 131.46 | 110.2 | |||
| 11-C | 130.41 | 112.0 | |||
| 6-C | 122.08 | 102.1 | |||
| 13-C | 37.26 | 18.0 | |||
|
|||||
| Compound C | |||||
| 15-C | 68.8 | 69.2 | 47-H | 7.542587 | 9.24 |
| 28-C | 188.78 | 163.2 | 48-H | 7.51 | 7.78 |
| 23-C | 187.15 | 165.2 | 45-H | 7.25 | 7.39 |
| 3-C | 182.55 | 147.6 | 43-H | 7.23 | 6.99 |
| 1-C | 176.83 | 147.6 | 36-H | 7.23 | 9.24 |
| 21-C | 169.01 | 163.2 | 46-H | 7.16 | 7.09 |
| 18-C | 167.65 | 163.2 | 34-H | 6.48 | 5.15 |
| 7-C | 160.07 | 160.8 | 44-H | 6.46 | 7.36 |
| 25-C | 153.03 | 160.8 | 35-H | 6.45 | 7.05 |
| 5-C | 146.98 | 113.0 | 37-H | 4.73 | 6.99 |
| 26-C | 145.29 | 130.5 | 38-H | 4.54 | 4.03 |
| 22-C | 138.08 | 122.2 | 55-H | 4.10 | 4.00 |
| 8-C | 137.54 | 98.6 | 51-H | 2.38 | 2.24 |
| 24-C | 136.35 | 114.1 | 41-H | 2.05 | 2.25 |
| 6-C | 134.35 | 130.4 | 39-H | 1.86 | 1.09 |
| 4-C | 127.76 | 98.6 | |||
| 9-C | 127.28 | 98.6 | |||
| 19-C | 126.70 | 122.1 | |||
| 2-C | 121.73 | 122.0 | |||
| 33-C | 82.90 | 98.6 | |||
| 20-C | 77.49 | 98.6 | |||
| 31-C | 37.49 | 40.1 | |||
| 12-C | 35.70 | 14.0 | |||
| 32-C | 31.10 | 17.7 | |||
 | ||
| Fig. 10 (a) Theoretical 1H-NMR spectra of compound A; (b) theoretical 13C-NMR spectra of compound A; (c) theoretical 1H-NMR spectra of compound B; (d) theoretical 13C-NMR spectra of compound B; (e) theoretical 1H-NMR spectra of compound C; and (f) theoretical 13C-NMR spectra of compound C. | ||
In compound B, the highest chemical shift value is at a chemical shift of 184.11 ppm in the theoretical study, and while in the experimental calculation, this carbon shows a peak at 161.1 ppm. In GIAO, the methyl carbon exhibits a peak at 37.26 ppm; however, in the experimental investigation, this carbon demonstrated a peak at 18.0 ppm. In compound A, the highest peak appeared at 174.61 ppm in the experimental study, while in the GIAO calculation, 168.93 ppm was the highest nuclear chemical shift value. There are no anomalies or differences between the theoretical and experimental values of the nuclear shielding of 13C and 1H for the compounds B and A.
3.7. ADMET studies
A pharmacokinetic study of the hit compound was conducted. The ADMET study consists of different parameters, such as adsorption (A), distribution (D), metabolism (M), excretion (E) and toxicity (T). The toxicity of the lead compound was determined.The compound with a Lipinski rule of five with 0 violations indicated that the compound was orally active in the form of a drug in the human body. The compound with absorption percentage in the range of 84.9–100% indicates nontoxic.50 The ADMET study revealed that W6, W23 and W14 fully obey the Lipinski rule with 0 (zero) violations. This indicates the drug-like activity of the compound. However, the other compound showed 1 violation.
The molecular weight of the compound should be less than 500. The W6, W23 and W14 have low molecular weight compared to other compounds. However, the low molecular weight of the compound increases the absorption rate of the compound.51
Compounds W6, W23 and W14 have small TPSA values. If the compound has a small TPSA value, it indicates that the compound can be easily absorbed. Except for one compound, all other compounds had a TPSA (total polar surface area) value less than 140. This indicates that these compounds passively pass through the cellular path.52 If the compound has more than 5 hydrogen bond donors and more than 10 hydrogen bond acceptors, it does not obey the Lipinski rule. All the compounds have a number of hydrogen bond donors of less than 5, and a hydrogen bond acceptor of less than 10. The medicinal properties of these compounds indicate that they show good bioavailability. If the value of consensus logPo/w is smaller than 5, it demonstrates that a suitable quantity of drug could be reached and shows aqueous solubility inside the body. All the lead compounds have a value of less than 5.53 The toxicity of the hit compound was determined using the online server. Out of the three compounds, only 1 compound showed the least toxic behavior owing to inactive carcinogenicity and immune-toxicity. The LD50 dose of these compounds is shown in Table 6, which is calculated from Protox II. The results indicate that a high dose of this compound was safe.
| Properties | Parameter | W6 | W23 | W24 | W19 | W14 | W27 | W18 | W1 | W3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Drug-likeness | Lipinski rule | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 |
| Ghose rule | No | No | No | No | Yes | No | No | No | No | |
| Veber rule | Yes | Yes | No | Yes | Yes | No | No | Yes | Yes | |
| Egan rule | Yes | Yes | No | No | No | No | No | Yes | Yes | |
| Mugge rule | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | |
| Physico-chemical properties | Molecular weight | 470.51 g mol−1 | 496.51 g mol−1 | 536.53 g mol−1 | 506.50 g mol−1 | 470.47 g mol−1 | 606.6 g mol−1 | 536.53 g mol−1 | 500.52 g mol−1 | 536.55 g mol−1 |
| No of H-bond donors | 3 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 3 | |
| No of H-bond acceptors | 6 | 7 | 8 | 7 | 7 | 9 | 8 | 8 | 8 | |
| TPSA | 123.35 | 130.17 | 143.31 | 134.08 | 137.26 | 160.38 | 143 | 88.10 | 106.56 | |
| Lipophilicity of the compound | Consensus logPo/w |
3.24 | 3.53 | 3.82 | 3.63 | 2.73 | 4.40 | 3.86 | 4.06 | 3.80 |
| logPo/w (XLOGP3) |
3.22 | 3.87 | 4.27 | 4.30 | 3.17 | 5.07 | 4.27 | 4.24 | 3.95 | |
| logPo/w (WLOGP) |
3.08 | 3.18 | 3.93 | 3.92 | 2.10 | 4.53 | 3.93 | 4.23 | 3.79 | |
| Solubility | logS (ESOL) |
Moderate soluble | Moderate soluble | Moderate soluble | Moderate soluble | Moderate soluble | Poorly soluble | Moderate soluble | Moderate soluble | Moderate soluble |
| logS (Ali) |
−5.79 | −6.30 | −6.99 | −6.83 | −5.72 | −8.18 | −6.99 | −5.80 | −5.89 | |
| Pharmacokinetics | GI absorption | High | High | Low | Low | High | Low | Low | High | High |
| BBB permeant | No | No | No | No | No | No | No | No | No | |
| CYP1A2 inhibitor | No | Yes | Yes | Yes | No | Yes | Yes | No | No | |
| Medicinal properties | Synthetic score | 4.56 | 4.76 | 4.76 | 4.62 | 4.57 | 5.25 | 4.80 | 5.29 | 5.55 |
| Lead likeness | No | No | No | No | No | No | No | No | No | |
| Toxicity | Immunotoxicity | Inactive | Active | Inactive | ||||||
| Cytotoxicity | Inactive | Inactive | Inactive | |||||||
| Mutagenicity | Inactive | Inactive | Inactive | |||||||
| Carcinogenicity | Inactive | Inactive | Inactive | |||||||
| LD50 mg kg−1 | 1000 mg kg−1 | 1190 mg kg−1 | 785 mg kg−1 |
3.8. Chemistry of synthesized target molecules
The 1H- and 13C-NMR spectra of pyrimidine molecule A were found in accordance with the literature.14 Coumarin molecule B was elucidated by its 1H- and 13C-NMR spectrum analyses. 1H-NMR of B displayed four protons in the aromatic region with a chemical shift value of 6.10 ppm as a singlet at position-3. A signal at δ 6.68 ppm was observed with a coupling constant of 2.5 and 6.0 Hz, which confirmed it at position-8 as ortho- and meta-coupled to another proton in close vicinity. For a doublet signal at δ 6.79 ppm with coupling constant 9.0, 2.5 Hz was observed at position-6, whose coupling confirmed it as meta- and para- to other protons. A proton at δ 7.57 ppm with a coupling constant of J = 9.0 Hz confirmed it at position-5. Among protons, a singlet at δ 10.5 ppm was found in the spectrum, which confirmed a hydroxyl at position-7, and a methyl signal was found at δ 2.99 ppm as a singlet at position-4. The 13C-NMR data were found in accordance with 1H-NMR data of molecule B with a distinguished peak at δ 160.2 for carbonyl, 161.1 for a carbon with a hydroxyl group and 18.0 for a methyl group. All methine and quarterly carbons were also confirmed by their respective signals in 13C-NMR spectra.Lead molecule C (W6) was obtained as a white crystalline solid. The 1H- and 13C-NMR spectra showed characteristic peaks that help in identifying and confirming its structure. The 1H-NMR spectra of W6 displayed a characteristic peak at δ 9.24 ppm for a single amine, while the absence of another peak at δ 10.43 confirmed the formation of a junction of pyrimidine and coumarin. The presence of a characteristic peak at δ 130.4 and the absence of a peak at δ 160.2 for the carbonyl group also confirmed complex formation. These distinguished and characteristic peaks confirmed the formation of our target molecule. All other peaks of coumarin and pyrimidine rings in the spectra of W6 were found in accordance with the literature.

3.9. Antidiabetic evaluation of target molecule W6 (C)
Anti-diabetic results of the target molecule W6 (C) showed excellent inhibition potential with an IC50 value of 30.58 (Fig. 11). However, in comparison, acarbose was used as a standard and found to exhibit 61.40% inhibition. At various concentrations, W6 was screened for its potential against diabetes type II and was found to be more active with an IC50 value of 30.58 μM. In vitro antidiabetic activity was found in close co-relation with binding affinity capacity calculated by molecular docking. | ||
| Fig. 11 Graphical representation of anti-diabetic activity and estimation of the IC50 value by non-regression analysis in GraphPad Prism. | ||
4 Conclusion
Coumarin and its analogues have different biological activities. Owing to their wide range of biological applications, coumarin and its analogues are of great interest to researchers because of their less toxic behavior. The main focus of this study was to identify targets as antidiabetic agents using coumarin derivatives and their analogues by interacting with pyrimidine compounds via amide and oxime linkages. A computational study was performed, and optimization of the molecular geometry of 27 compounds was performed by Density Functional Theory (DFT) using a basis set 6311G with the function B3LYP. Virtual screening of these compounds was carried out by molecular docking using the Auto-Dock tool. Structure-based virtual screening provided us with nine hit compounds with oxygen oxygen-centered furan rings and methoxy derivatives. These hit compounds showed good binding affinity for antidiabetic protein active sites. ADMET studies of hit compounds were performed, which filtered compounds that did not fall into the criterion of drug likeness. Only 1 compound, W6 (C), fully obeys the Lipinski rule and is the least toxic, indicating drug-like behavior. Based on these screening results, lead molecule W6 was synthesized, characterized for structure and evaluated for antidiabetic potential by in vitro analysis. The in silico and in vitro results were found to be in close co-relation with each other, indicating that W6 (C) was a new effective candidate for diabetes inhibition.Data availability
Data will be made available upon request.Author contributions
Nusrat Shafiq: conceptualization, paper drafting, writing, supervision, molecular docking, NMR elucidation and funding; Warda Sarwar: library preparation, drafting, molecular docking, ADMET, and DFT; Maryam Rashid: supervision and editing; Nida NAz: synthesis of starting material and target molecule; Fatima Rida: MM-GBSA properties; Aniqa Moveed: DFT analysis; Dalal Hussien M. Alkhalifah: editing and validation; Mohamed Mohany: funding acquisition; Wael N. Hozzein: visualization; Shagufta Parveen: evaluation of results, data analysis, and literature; Eman Nawaz: editing, evaluation, and figures resolution.Conflicts of interest
There is no conflict of interest.Acknowledgements
The authors thank the Government College Women University for technical support and HEC Pakistan (Grant # TDF03-172). They also acknowledge the support from Princess Nourah Bint Abdulrahman University Researchers Supporting Project Number (PNURSP2025R15), Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia.References
- A. Donihi and M. Korytkowski, Br. Med. J., 2019, 365(l1114), 1–15 Search PubMed.
- U. Galicia-Garcia, A. Benito-Vicente, S. Jebari, A. Larrea-Sebal, H. Siddiqi, K. B. Uribe, H. Ostolaza and C. Martín, Int. J. Mol. Sci., 2020, 21(17), 6275 CrossRef CAS PubMed.
- K. Kaul, J. M. Tarr, S. I. Ahmad, E. M. Kohner and R. Chibber, Diabetes: An Old Disease, a New Insight, 2013, 1–11 CAS.
- R. Henry, T. Wiest-Kent, L. Scheaffer, O. Kolterman and J. Olefsky, Diabetes, 1986, 35(2), 155–164 CrossRef CAS PubMed.
- J. Jendle, M. Nauck, D. Matthews, A. Frid, K. Hermansen, M. Düring, M. Zdravkovic, B. Strauss, A. Garber and LEAD-2 and L. S. Groups, Diabetes Obes. Metabol., 2009, 11, 1163–1172 CrossRef CAS PubMed.
- H. Yuan, Q. Ma, L. Ye and G. Piao, Molecules, 2016, 21, 5–59 Search PubMed.
- A. Zhang, H. Sun and X. Wang, Eur. J. Med. Chem., 2013, 63, 570–577 CrossRef CAS PubMed.
- M. Verma, S. J. Gupta, A. Chaudhary and V. K. Garg, Bioorg. Chem., 2017, 70, 267–283 CrossRef CAS PubMed.
- N. E. Thomford, D. A. Senthebane, A. Rowe, D. Munro, P. Seele, A. Maroyi and K. Dzobo, Int. J. Mol. Sci., 2018, 19, 15–78 CrossRef PubMed.
- T. M. Costa, L. B. B. Tavares and D. de Oliveira, Appl. Microbiol. Biotechnol., 2016, 100, 6571–6584 CrossRef CAS PubMed.
- U. Arshad, S. Ahmed, N. Shafiq, Z. Ahmad, A. Hassan, N. Akhtar, S. Parveen and T. Mehmood, Molecules, 2021, 26, 1–24 Search PubMed.
- Z. Li, J. Gu, H. Zhuang, L. Kang, X. Zhao and Q. Guo, Appl. Soft Comput., 2015, 26, 299–302 CrossRef.
- T. Mavromoustakos, S. Durdagi, C. Koukoulitsa, M. Simcic, M. G. Papadopoulos, M. Hodoscek and S. G. Grdadolnik, Curr. Med. Chem., 2011, 18, 2517–2530 CrossRef CAS PubMed.
- U. Arshad, S. Ahmed, N. Shafiq, Z. Ahmad, A. Hassan, N. Akhtar, S. Parveen and T. Mehmood, Molecules, 2021, 26, 4424–4454 CrossRef CAS PubMed.
- U. Arshad, N. Shafiq, S. Parveen and M. Rashid, Future Med. Chem., 2024, 16, 1949–1969 CrossRef PubMed.
- U. Arshad, N. Shafiq, M. Rashid and S. Parveen, J. Mol. Struct., 2024, 15, 138670 CrossRef.
- S. Naseem, M. Khalid, M. N. Tahir, M. A. Halim, A. A. Braga, M. M. Naseer and Z. Shafiq, J. Mol. Struct., 2017, 1143, 235–244 CrossRef CAS.
- R. Kalirajan, A. Pandiselvi, B. Gowramma and P. Balachandran, Curr. Drug Res. Rev., 2019, 11, 118–128 CrossRef CAS PubMed.
- F. Chen, H. Liu, H. Sun, P. Pan, Y. Li, D. Li and T. Hou, Phys. Chem. Chem. Phys., 2016, 18, 22129–22139 RSC.
- D. Suresh, M. Amalanathan, S. Sebastian, D. Sajan, I. H. Joe, V. B. Jothy and I. Nemec, Spectrochim. Acta, Part A, 2013, 115, 595–602 CrossRef CAS PubMed.
- M. Hagar, H. Ahmed and O. Alhaddadd, Crystals, 2018, 8, 3–59 CrossRef.
- R. Parthasarathi, V. Subramanian, D. R. Roy and P. Chattaraj, Bioorg. Med. Chem., 2004, 12, 5533–5543 CrossRef CAS PubMed.
- A. K. Mishra and S. P. Tewari, Emerg. Mater. Res., 2019, 8, 651–662 Search PubMed.
- M. A. Mumit, T. K. Pal, M. A. Alam, M. A.-A.-A.-A. Islam, S. Paul and M. C. Sheikh, J. Mol. Struct., 2020, 1220, 128–715 CrossRef PubMed.
- A. M. Fahim, M. A. Shalaby and M. A. Ibrahim, J. Mol. Struct., 2019, 1194, 211–226 CrossRef CAS.
- M. Hagar, H. A. Ahmed, G. Aljohani and O. A. Alhaddad, Int. J. Mol. Sci., 2020, 21, 39–22 Search PubMed.
- M. Sheikhi, S. Shahab, M. Khaleghian and R. Kumar, Appl. Surf. Sci., 2018, 434, 504–513 CrossRef CAS.
- N. Sheela, S. Muthu and S. Sampathkrishnan, Spectrochim. Acta, Part A, 2014, 120, 237–251 CrossRef CAS PubMed.
- M. Almandoz, M. I. Sancho, P. R. Duchowicz and S. E. Blanco, Spectrochim. Acta, Part A, 2014, 129, 52–60 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct.: THEOCHEM, 1999, 464, 211–226 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- N. Shafiq, N. Shahzad, F. Rida, Z. Ahmad, H. A. Nazir, U. Arshad, G. Zareen, N. Attiq, S. Parveen, M. Rashid and B. Ali, Future Med. Chem., 2023, 15, 1–22 CrossRef PubMed.
- F. Rahim, S. Tariq, M. Taha, H. Ullah, K. Zaman, I. Uddin, A. Wadood, A. A. Khan, A. U. Rehman and N. Uddin, Bioorg. Chem., 2019, 92, 103–284 CrossRef PubMed.
- A. Aispuro-Pérez, J. López-Ávalos, F. García-Páez, J. Montes-Avila, L. A. Picos-Corrales, A. Ochoa-Terán, P. Bastidas, S. Montaño, L. Calderón-Zamora and U. Osuna-Martínez, Bioorg. Chem., 2020, 94, 103–491 CrossRef PubMed.
- Y. Shu, W. Xue, X. Xu, Z. Jia, X. Yao, S. Liu and L. Liu, Food Chem., 2015, 173, 31–37 CrossRef CAS PubMed.
- Y. Demir, P. Taslimi, Ü. M. Koçyiğit, M. Akkuş, M. S. Özaslan, H. E. Duran, Y. Budak, B. Tüzün, M. B. Gürdere and M. Ceylan, Arch. Pharmazie, 2020, 353, 200–118 Search PubMed.
- N. M. H. El-Ameen, M. M. E. Taha, S. I. Abdelwahab, A. Khalid, F. Elfatih, M. A. Kamel and B. Y. Sheikh, Pharmacogn. J., 2015, 7, 406–410 CrossRef CAS.
- E. Wang, W. Fu, D. Jiang, H. Sun, J. Wang, X. Zhang, G. Weng, H. Liu, P. Tao and T. Hou, J. Chem. Inf. Model., 2021, 61, 2844–2856 CrossRef CAS PubMed.
- W. Wang and P. A. Kollman, J. Mol. Biol., 2000, 303, 567–582 CrossRef CAS PubMed.
- T. Hou, K. Chen, W. A. McLaughlin, B. Lu and W. Wang, PLoS Comput. Biol., 2006, 2, e1 CrossRef PubMed.
- U. Arshad, S. Ahmed, N. Shafiq, Z. Ahmad, A. Hassan, N. Akhtar, S. Parveen and T. Mehmood, Molecules, 2021, 26, 44–24 Search PubMed.
- O. Noureddine, N. Issaoui and O. Al-Dossary, J. King Saud Univ., Sci., 2021, 33, 101248 CrossRef PubMed.
- H. Boulebd, Free Radic. Res., 2019, 53, 1125–1134 CrossRef CAS PubMed.
- N. Uludağ, G. Serdaroğlu and A. Yinanc, J. Mol. Struct., 2018, 1161, 152–168 CrossRef.
- M. Marinescu, D. G. Tudorache, G. I. Marton, C.-M. Zalaru, M. Popa, M.-C. Chifiriuc, C.-E. Stavarache and C. Constantinescu, J. Mol. Struct., 2017, 1130, 463–471 CrossRef CAS.
- N. Obi-Egbedi, I. Obot and M. I. El-Khaiary, J. Mol. Struct., 2011, 1002, 86–96 CrossRef CAS.
- S. D. Kanmazalp, M. Macit and N. Dege, J. Mol. Struct., 2019, 1179, 181–191 CrossRef CAS.
- V. Barone and A. Polimeno, Chem. Soc. Rev., 2007, 36, 1724–1731 RSC.
- B. Tirri, G. Mazzone, A. Ottochian, J. Gomar, U. Raucci, C. Adamo and I. Ciofini, J. Comput. Chem., 2021, 42, 1054–1063 CrossRef CAS PubMed.
- S. V. Tiwari, J. A. Seijas, M. P. Vazquez-Tato, A. P. Sarkate, K. S. Karnik and A. P. G. Nikalje, Molecules, 2017, 22, 11–72 Search PubMed.
- C. M. Nisha, A. Kumar, P. Nair, N. Gupta, C. Silakari, T. Tripathi and A. Kumar, Adv. Bioinf., 2016, 2016, 1–6 Search PubMed.
- R. Liu, H. Sun and S.-S. So, J. Chem. Inf. Comput. Sci., 2001, 41, 1623–1632 CrossRef CAS PubMed.
- S. B. Olasupo, A. Uzairu, G. Shallangwa and S. Uba, J. Iran. Chem. Soc., 2020, 17, 1953–1966 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01835h |
| This journal is © The Royal Society of Chemistry 2025 |