
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLC-MS Orbitrap-based metabolomics using a novel hybrid zwitterionic hydrophilic interaction liquid chromatography and rigorous metabolite identification reveals doxorubicin-induced metabolic perturbations in breast cancer cells†
Salah Abdelrazig *ad,
Áine McCabeb,
Alia Yasinb,
Rajneil Chaudharya,
Michael A. Ochsenkühna,
David Scicchitanobc and
Shady A. Amin*ade
*ad,
Áine McCabeb,
Alia Yasinb,
Rajneil Chaudharya,
Michael A. Ochsenkühna,
David Scicchitanobc and
Shady A. Amin*ade
aMarine Microbiomics Lab, Biology Program, New York University Abu Dhabi (NYUAD), P.O. Box 129188, Abu Dhabi, United Arab Emirates. E-mail: salah.Abdelrazig@nyu.edu; samin@nyu.edu
bDivision of Science, New York University Abu Dhabi, P.O. Box 129188, Abu Dhabi, United Arab Emirates
cDepartment of Biology, New York University, 24 Waverly Place, 6th Floor, New York, New York 10003, USA
dMubadala ACCESS, New York University Abu Dhabi (NYUAD), P.O. Box 129188, Abu Dhabi, United Arab Emirates
eCenter for Genomics and Systems Biology, New York University Abu Dhabi (NYUAD), P.O. Box 129188, Abu Dhabi, United Arab Emirates
First published on 19th June 2025
Abstract
The identification of metabolites in biological samples presents a challenge in untargeted metabolomics, mainly due to limited databases and inadequate chromatography. Current LC columns suffer from high pH instability (silica-based), low efficiencies and pressure limitations (polymer-based), or inadequate retention of polar/semi-polar metabolites (reverse-phase). In this study, a comprehensive LC-MS workflow was developed to address these limitations using a novel zwitterionic HILIC (Z-HILIC), high-resolution MS, deep-scan data-dependent acquisition (DDA), and a large chemical library comprising 990 standards. The method performance was evaluated and compared with a widely-used ZIC-pHILIC method. Z-HILIC detected 707 (71%) of the standards compared to 543 (55%) standards with the ZIC-pHILIC showing enhanced resolution, sensitivity, selectivity and retention time (RT) distribution. In triple-negative Hs578T breast cancer cell extracts spiked with the standards, Z-HILIC annotated 79.1% of the detected standards versus 66.6% with ZIC-pHILIC, demonstrating improved sensitivity, stability, and reduced matrix effects for metabolite profiling. Deep-scan DDA of the spiked cell extracts increased the number of the identified metabolites using RT, m/z and MS/MS by more than 80% compared to standard DDA. The workflow was used to investigate the metabolic signature of doxorubicin-treated Hs578T cells (n = 15). The analysis resulted in identifying 173 metabolites, of which 26 metabolites and 20 metabolic pathways were significantly altered in doxorubicin treated cells compared to controls. These pathways were associated with oxidative stress, mitochondrial dysfunction, and impaired biosynthesis, consistent with prior knowledge about the action of doxorubicin. This comprehensive workflow promises to enhance metabolite profiling across diverse metabolomics studies.
1. Introduction
Untargeted metabolomics is an unbiased comprehensive analysis of metabolites that provides a powerful tool to study the metabolic perturbation and pathways within a biological or environmental system under different conditions.1 Liquid chromatography-mass spectrometry (LC-MS) using high-resolution instruments, such as Orbitrap MS, is considered the preferred choice for untargeted metabolomics due to its higher sensitivity, mass accuracy and wide metabolite coverage compared to nominal MS.1 The choice of chromatographic columns plays a pivotal role in the simultaneous separation of a broad spectrum of metabolites, particularly in complex biological and environmental samples. However, efficient chromatography alone is insufficient for a successful metabolomics study due to the challenges associated with metabolite identification. The lack of a universal LC-MS platform, limited reference standards, well-characterized databases, complex fragmentation, natural presence of isomers and the vast chemical diversity of metabolites are major challenges for accurate metabolite identification.2 For instance, using m/z of metabolite from experimental data against online databases annotates metabolites with low confidence (i.e. putative identification) due to the existence of a wide-range of isobaric and isomeric metabolites.3 This often leads to false identification and subsequently inaccurate biological interpretations.2 Therefore, the use of additional orthogonal features to m/z of metabolites such as retention time (RT) and/or MS/MS spectra, as recommended by the Metabolomics Standards Initiative (MSI),4 is essential to improve the confidence in confirming the identity of metabolites using authentic standards. That said, efficient chromatography, robust analytical LC-MS performance, the ability to acquire MS/MS spectra of metabolite features in biological samples, and access to large and well-curated metabolite standard databases are essential for reliable metabolite profiling and accurate identification.Chromatographic characteristics such as high sensitivity, resolution and adequate retention time distribution of metabolites are critical to obtain feature-rich profiles and aid metabolite identification in metabolomics studies. Sharp, well-resolved, and adequately distributed peaks of a wide-range of metabolites over the working RT range help to decrease ion suppression effects due to co-elution, and subsequently enhances the sensitivity of the MS. Reverse-phase ultra-high-performance LC (UHPLC) columns are capable of obtaining sharp peaks but they suffer from poor retention of polar and semi-polar metabolites and high pH instability (silica-based). In contrast, zwitterionic hydrophilic interaction liquid chromatography (HILIC) has demonstrated superior performance and enhanced retention of these metabolites.5,6 For instance, ZIC-pHILIC has been widely used in different metabolomics applications.6–10 However, current zwitterionic HILIC columns, including ZIC-pHILIC, are either unstable at high pH (silica-based) or suffer from low chromatographic efficiencies and pressure limitations (polymer-based). Recently, a novel zwitterionic HILIC column (Z-HILIC), designed for UHPLC with <3 μm particle sizes has been introduced. Z-HILIC and ZIC-pHILIC contain sulfobetaine zwitterionic ligands that enable analyte elution with a combination of hydrogen bonding, hydrophilic and electrostatic interactions.6 The key difference in Z-HILIC is that the sulfobetaine moiety is attached to an ethylene-bridged organic/inorganic particle hybrid (BEH). These differences in stationery phase composition improves the pH stability (pH 2–10) and pressure tolerance compared to silica-based and polymer-based HILIC columns, respectively.11 In addition, Z-HILIC features a high-performance surface coating designed to mitigate metal-analyte interactions to obtain improved peak shape and recovery for metal-sensitive analytes.11 Few studies characterized Z-HILIC under different chromatographic and pH conditions using a small set of metabolite standards.11–13 However, no comprehensive study, to the best of our knowledge, tested and compared the performance and sensitivity of the newly introduced column using large and diverse chemical classes of metabolites for LC-MS profiling.
The analytical capability of the MS system to obtain rich MS/MS spectra of metabolite features in samples is crucial to identify metabolites using spectral database of standards. Data-dependent acquisition (DDA) is widely used in metabolomics8,10,14–16 for metabolite identification. However, low numbers of MS/MS spectra are typically obtained due to the speed limitations of the MS data acquisition and its bias towards the most abundant peaks for fragmentation.15 In contrast, deep-scan DDA techniques have been introduced to increase the MS/MS fragmentation of metabolites.15 The combination of a metabolomics workflow that utilizes Z-HILIC and deep-scan DDA has the potential to improve metabolites separation efficiently, increase the number of acquired MS/MS spectra and subsequently enhance the detection and identification of metabolites.
Triple-negative breast cancer (TNBC) is an aggressive breast cancer subtype characterized by the lack of estrogen and progesterone receptors as well as human epidermal growth factor receptor 2, which makes it challenging to treat with targeted therapy.17 Hence, conventional chemotherapeutic agents and radiotherapy remain the primary treatment strategy for TNBC.18 Doxorubicin (Dox), a DNA intercalator and topoisomerase II inhibitor, is often used in combination with other chemotherapeutic agents for treating various breast cancers, including TNBC. However, the efficacy of Dox in TNBC can be compromised by the activation of chemoresistance-related molecular mechanisms,19 a poorly understood phenomenon partially driven by metabolic reprogramming or metabolic rewiring in cancer cells. TNBC undergoes metabolic reprogramming that sustains rapid cell proliferation, redox balance and metastasis to ensure survival under stress conditions, such as drug treatment.20 An in-depth understanding of these metabolic alterations can provide unique insights into the altered pathways that contribute to tumorigenesis, progression, and chemoresistance.20 Untargeted metabolomics provides a powerful tool to explore these metabolic changes in TNBC, and may improve our understanding of the metabolic landscape to pave the way for effective therapeutic intervention and ultimately the development of new therapies.
In this study, we develop and optimize an LC-MS untargeted metabolomics workflow using Z-HILIC and high-resolution hybrid Orbitrap MS combined with deep-scan DDA to investigate the metabolic changes in Hs578T cells (TNBC human cell line) treated with Dox. The chromatographic performance and metabolite coverage of the Z-HILIC was compared with ZIC-pHILIC for untargeted metabolomics using a comprehensive set of 990 metabolite standards. In addition, an in-house fragmentation database of the standards was curated and used with the deep-scan DDA (MS/MS) acquisition for precise and accurate identification of metabolites.
2. Experimental
2.1. Chemicals and LC-MS reagents
Analytical standards of 990 metabolites covering a wide range of primary and secondary metabolism were obtained from IROA technologies LLC (Chapel Hill, NC, USA). Each standard was supplied as dried weight of 5 μg in 96-well plates; full details of the standards are available in Table S1.† Methanol and acetonitrile used in this study were LC-MS grade and were purchased from Sigma Aldrich (Darmstadt, Germany) and Honeywell (Seelze, Germany), respectively. Ammonium carbonate (purity: 99.999%) and doxorubicin HCl were obtained from Sigma-Aldrich (Saint Louis, MO, USA). Ultrapure water (18.2 MΩ cm) was prepared using a water purification system (Milli-Q Integral 10, Merck Millipore, Burlington, VT, USA).2.2. Preparation of the standards for LC-MS
Batches of 8–12 standards (4 μg mL−1) were pooled in LC-MS grade solvents following the supplier's recommendation and used for the optimization, development and comparison of the LC-MS methods in the study. Full details including reconstitution solvents used for each standard are available in Table S1.†A mixture of all standards was also prepared by pooling all the batch standard solutions, drying and reconstituting them in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol:water (standard mixture) or spiking them in a pooled cell extract of Hs578T (spiked cell extract). The concentration of each standard in the standard mixture and the spiked cell extract was 4 μg mL−1.
:1 methanol:water (standard mixture) or spiking them in a pooled cell extract of Hs578T (spiked cell extract). The concentration of each standard in the standard mixture and the spiked cell extract was 4 μg mL−1.
2.3. LC-MS and LC-MS/MS for metabolite profiling and metabolite identification
Liquid chromatography was performed using Vanquish UHPLC system (Thermo Fisher Scientific, Waltham, MA, USA) on either (1) Z-HILIC column (4.6 × 150 mm, 2.5 μm, 95 Å) with Z-HILIC guard column (4.6 × 5 mm, 2.5 μm) (Atlantis Premier BEH Z-HILIC, Waters, Milford, MA, USA), or (2) ZIC-pHILIC column (4.6 × 150 mm, 5 μm) with ZIC-pHILIC guard column (2.1 × 20 mm, 5 μm) (Merck SeQuant, Watford, UK). Both columns were maintained at 45 °C with a flow rate of 300 μL min−1 of 20% 20 mM ammonium carbonate in water, pH 9.2 (A) and 80% acetonitrile (B). The gradient used for both methods was: 0–15 min: 20% to 95% A, 15–17 min: 95% to 20% A and 17–24 min 20% A. The injection volume was 5 μL and the samples were maintained at 4 °C during analysis. The chromatographic flow was diverted to waste before 1 min and after 23 min of the gradient, during which the heated ESI needle was washed with water (50 μL min−1) to minimize precipitation of the mobile phase buffer.Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) was used in switching ESI+ and ESI− acquisition modes for full LC-MS profiling and DDA to generate MS/MS spectra using Xcalibur and/or AcquireX acquisition softwares (Thermo Fisher Scientific, Waltham, MA, USA). The MS parameters for both ionization modes were: spray voltage 4.5 kV, sheath, auxiliary and sweep gas flow rate were: 40, 5 and 1 (arbitrary units), respectively. Ion transfer tube and vaporizer temperatures were 275 °C and 150 °C, respectively. Data were acquired in full scan mode with a resolution of 60000 from m/z 70–1400. DDA was performed by either Xcalibur or AcquireX deep-scan DDA (MS/MS) at a resolution of 15000 in separate ESI+ and ESI− modes and a normalized collision energies (CE) of 20, 30, 50 and/or a stepped CE of 20, 30 and 50. An inclusion list containing the m/z, RT and CE of the in-house database standards was used for a targeted fragmentation of the metabolite peaks in the study samples.
2.4. Evaluation of the performance of the developed LC-MS for metabolite profiling
The authentic standards (4 μg mL−1) were analyzed on both Z-HILIC and ZIC-pHILIC columns using LC-MS. The raw datasets from both methods were processed using Xcalibur 4.1 and TraceFinder 4.1 (Thermo Fisher Scientific, Waltham, MA, USA) for peak detection, retention times (RT) extraction and integration. The chromatographic performance of the two LC-MS methods were evaluated by assessing the quality and the retention profiles of the detected peaks of the standards. Different LC-MS properties, such as RT, peak quality (i.e., symmetry, tailing, full width at half-maximum (FWHM)), sensitivity (S/N) and selectivity were used for the assessment. Moreover, important characteristics for LC-MS metabolic profiling such as repeatability (RT, peak area), sensitivity and metabolite coverage were studied by analyzing the standard mixture (n = 3) and the spiked cell extract (n = 3) on both columns.2.5. Curation of in-house fragmentation database
The developed Z-HILIC method was used to create an in-house fragmentation database of the standards (standard database) using mzVault 2.3 (Thermo Fisher Scientific, Waltham, MA, USA). Each standard (4 μg mL−1) was analyzed using LC-MS and LC-MS/MS with a modified acquisition method for a targeted fragmentation using the RT and exact mass of the standard at CE of 20, 30 and 50 in both ESI modes. The quality of the acquired MS/MS spectra of each standard was then assessed based on the number of fragments and intensity using Xcalibur. The MS/MS spectrum of each standard containing 5–10 fragments with relatively high intensity compared to the rest of fragmentation spectra at a specific CE in either ESI+ or ESI− was selected for further processing. The selected spectra of the standards were then imported into mzVault 2.3 to curate the standard database. A comprehensive list of RT, exact mass and specific CE of each standard (standard inclusion list) was also created and used for targeted fragmentation as a part of the LC-MS workflow for metabolite identification.2.6. Evaluation of data-dependent acquisition methods for metabolite identification
Blanks, standard mixture, spiked cell extract and standard inclusion list were used to evaluate and compare deep-scan DDA using AcquireX (Thermo Fisher Scientific, Waltham, MA, USA) to the standard DDA in Xcalibur (Thermo Fisher Scientific, Waltham, MA, USA) for metabolite identification. AcquireX deep-scan DDA is an automated, iterative, inclusion and exclusion-based acquisition approach to maximize the MS/MS of low-abundance analytes by dynamically excluding previously fragmented ions across multiple injections.15 The list of the standards was divided into 4 inclusion lists and were used with Xcalibur DDA to analyse the standard mixture (n = 4) and the spiked cell extract (n = 4), while AcquireX used deep-scan DDA method with a single inclusion list of all the standards. AcquireX injections of the standard mixture (n = 4) and the spiked cell extract (n = 4) was automated to create an exclusion list of the blank peaks and continuously update the inclusion list of the standards after each injection.2.7. Untargeted metabolomics of Hs578T cell culture samples
000 × g, 4 °C for 10 min. The supernatants of the cell extracts were transferred and dried using a Genevac HT-12 Vacuum Evaporator (Genevac, Scientific Products, Ipswich, UK). The dried extract of each sample was then reconstituted in 100 μL of 1:1 methanol:water, centrifuged at 13000 × g, 4 °C for 5 min, and 60 μL of the supernatant was analyzed immediately with LC-MS and LC-MS/MS. Cell-free media were extracted following the same protocol and used as blanks. An equal volume from each extracted sample in the study (30 μL) was collected, pooled and vortexed (30 s) and used as pooled quality control (QC) sample or pooled cell extract. The pooled QC was used for metabolite identification and assessment of the instrument performance, while the pooled cell extract was used for method development and evaluating the performance of the chromatographic methods for metabolite profiling.Significantly altered metabolites in Dox 3 d extracts compared to controls were selected using variable importance in the projection (VIP) of the PLS-DA and adjusted p-values of Student's t-test with false discovery rate (Benjamini–Hochberg approach).22 Metabolites with VIP > 1.0 and adjusted p-value < 0.05 were considered significantly altered in Dox 3 d extracts compared to controls.
2.8. Metabolite enrichment and pathway analysis
Normalized abundance of the identified metabolites in Dox 3 d and control 3 d extracts were Pareto scaled and processed with MetaboAnalyst 6.023 for metabolite enrichment and pathway analysis.
3. Results and discussion
In this study, the chromatographic performance of Z-HILIC was evaluated and compared with an existing ZIC-pHILIC6–10 using a comprehensive set of 990 authentic standards of metabolites under the same LC-MS conditions. These standards comprised chemically-diverse classes of metabolites including organic acids, amino acids, amines, energy intermediates, mono-/di-saccharides, nucleotides, coenzymes, vitamins, phytochemicals and their derivatives (Table S1†).3.1. Evaluation of the chromatographic performance of Z-HILIC LC-MS
Metabolite standards were analyzed with Z-HILIC and ZIC-pHILIC LC-MS and the peak shape of each analyte was examined and evaluated (Fig. 1). Amino acids, organic acids and derivatives showed a comparable LC performance with both methods; however, sharper peaks were observed with Z-HILIC. Both methods showed comparable sensitivity and high retention towards the middle of the LC run of these metabolite classes. This is mainly due to enhanced electrostatic interactions at high pH (9.2) that contributed to the improved retention and peak quality of basic and acidic ionizable species, as previously reported.6 Nonetheless, amino acids and their derivatives, such as glutamine and taurine, showed improved, sharp (FWHM < 15 s) and symmetrical peaks using Z-HILIC compared to ZIC-pHILIC, a direct reflection of the UHPLC performance of the Z-HILIC and consistent with a previous column characterization study.24 Similar results were also observed with organic acids and derivatives such as succinate and guanidinosuccinate. For neutral analytes including mono- and di-saccharides such as glucose, sucrose and lactose, an identical retention but sharper peaks with increased sensitivity and peak symmetry were observed with Z-HILIC compared to ZIC-pHILIC. The retention of sugars was previously found not to be affected by modulating the pH strengths on ZIC-pHILIC, concluding that separation was achieved only by hydrophilic interactions.6 Thus, identical retention of sugars on both columns indicates similar separation mechanism. | ||
| Fig. 1 Comparison of representative chromatographic peaks of the standards covering various chemical classes analyzed with LC-MS using (a) Z-HILIC and (b) ZIC-pHILIC columns. The peak quality of all standards was assessed and compared for both methods as illustrated in Fig. 2. | ||
In contrast, some primary and secondary amines with dominant basic groups, such as 2-hydroxyphenethylamine and purine, showed distinct retention and highly improved peak shape and sensitivity on the Z-HILIC compared to poor chromatography on ZIC-pHILIC (Fig. 1). This may indicate that the electrostatic repulsion of the charged species on the surface of ZIC-pHILIC was stronger compared to Z-HILIC, a phenomenon previously shown to deteriorate peak shape and retention on ZIC-pHILIC.12 Nevertheless, 2-hydroxyphenethylamine was highly retained on ZIC-pHILIC signaling the possibility that the improved peak shape on Z-HILIC is related to the different percentage of ionizable species on the surface of the stationary phase and the mitigated metal-analyte interactions compared to ZIC-pHILIC, as previously discussed.11
Analytes with mixed functional groups such as pyrimidines, energy intermediates and derivatives, e.g., cytidine, deoxycytidine, ATP, ADP and AMP, showed higher peak quality and sensitivity on Z-HILIC but similar retention profiles, whereas some analytes were not detected on ZIC-pHILIC column such as ADP and AMP. Although these analytes were detected and quantified previously using ZIC-pHILIC,7,9 the working concentrations of standards (4 μg mL−1) used in this study was lower than previous studies, indicating that the use of Z-HILIC improved the LC-MS sensitivity for these analytes.
A holistic overview of the chromatographic performance of all standards in this study further confirmed the superiority of Z-HILIC relative to ZIC-pHILIC (Fig. 2). All detected standards on either column were categorized based on their observed peak shapes to enable direct comparison between the two methods (Fig. 2A). Z-HILIC LC-MS detected 707 standards (71.41%), while the ZIC-pHILIC LC-MS detected only 543 (54.85%) of all 990 standards. The undetected analytes were re-analyzed at higher concentrations 8–12 μg mL−1, however, they remained undetectable on both columns (data not included). Z-HILIC produced overall sharper peaks and less broad peaks (547 and 27, respectively) relative to ZIC-pHILIC (341 and 66, respectively) (Fig. 2B). Adequate distribution of the detected analytes along the RT axis minimizes co-elution of peaks and reduces interferences and ion suppression during electrospray ionization (ESI).25 Subsequently, this enhances the detection and sensitivity for LC-MS metabolite profiling. The distribution of eluted standards across the working RT range in each column (Z-HILIC: 3.42–22.96 min; ZIC-pHILIC: 3.54–19.55 min) exhibited a similar RT distribution pattern with 41% (Z-HILIC) and 35% (ZIC-pHILIC) of the analytes being well-retained and adequately distributed in RT region 9.00–15.50 min (Fig. 2C–E). Overall, an improved RT distribution pattern was observed with Z-HILIC compared to ZIC-pHILIC.
 | ||
| Fig. 2 Chromatographic performance of the Z-HILIC compared to ZIC-pHILIC for the LC-MS analysis of standards. (A) Top: examples of the coloring schemes of the heatmap below and how it corresponds to different peak shape qualities. Bottom: heatmap of all detected standards on both columns with corresponding peak shape quality observed using (a) Z-HILIC and (b) ZIC-pHILIC LC-MS. Dark blue box indicates a sharp peak (symmetrical peak, FWHM < 15 s and S/N > 3), light blue box indicates an adequate peak (symmetrical peak with FWHM = 15–30 s, S/N > 3), yellow box indicates a broad peak (asymmetrical peak with or without tailing, FWHM >30 s, S/N > 3), and red box indicates no detection. Numbers 1–11: 96-well plate ID and A1-H12: well ID (Table S1†). (B) Total numbers of standards observed in each peak quality category using both columns. (C) RT distribution showing the percentage of the retained peaks of standards at different RT range. (D) and (E) Histograms of the eluted peaks of the standards showing the number of the peaks detected over the course of the chromatographic run (RT 3.2–23 min). | ||
The ability to resolve isobaric metabolites in the RT dimension is crucial for accurate metabolite identification in untargeted metabolomics. Therefore, we investigated the co-elution of isobaric and isomeric analytes in our standards by combining all 990 metabolite standards into a single ‘standard mixture’. There were 256 metabolite standards with 2–10 isobaric/isomeric species detected using Z-HILIC and only 219 of these standards were detected using ZIC-pHILIC. 172 (67%) and 112 (51%) isobaric/isomeric analytes of the detected standards were resolved with Z-HILIC and ZIC-pHILIC LC-MS, respectively (Fig. 3), indicating that Z-HILIC outperformed ZIC-pHILIC in selectivity, providing better separation and reducing co-elution. Modulating the gradient, pH and compositions of the mobile phase buffer were found to improve selectivity and separation of charged isobaric and isomeric analytes on zwitterionic HILIC columns;6,7,9,12,13 however, such changes to separate the rest of the isobaric and isomeric analytes are beyond the scope of this study. Overall, these results demonstrate that the Z-HILIC method offers superior chromatographic performance and sensitivity compared to ZIC-pHILIC for metabolite profiling.
 | ||
| Fig. 3 The performance of the Z-HILIC and ZIC-pHILIC for the separation of isobaric and isomeric analytes in the metabolite standards. (A) and (B) Show the RT distribution and overlapping of different m/z groups of the detected isobaric and isomeric analytes in the standards using Z-HILIC and ZIC-pHILIC, respectively. Each horizonal line in the Y-axis represents a single m/z group of 2–10 isobaric and isomeric analytes. (C) The overall performance of the Z-HILIC compared to ZIC-pHILIC in detecting and resolving the isobaric and isomeric analytes. | ||
3.2. Performance of Z-HILIC LC-MS for metabolite profiling
Spiked cell extracts and standard mixtures were used as feature-rich samples of unknown and known analytes to evaluate the Z-HILIC LC-MS performance for metabolite profiling and assess the analyte/matrix effect. LC-MS for metabolite profiling aims to detect a wide range of metabolites with high precision and accuracy across samples in a given study. This requires efficient, robust and stable chromatography with high peak capacity, well-defined peaks and minimal co-elution/matrix interference.1,25 These characteristics are critical to enhance the detection of a large number of metabolite features and ensure reliable MS signals for peak alignment, deconvolution, identification and fold change calculations. Therefore, the extent of metabolite coverage, RT distribution, sensitivity, repeatability (i.e. within-batch variation for RT and peak areas), and sample matrix interference were assessed.Spiked cell extracts (n = 3), pure standard mixtures (n = 3) and blanks (n = 3) were analyzed in a single analytical run with Z-HILIC and ZIC-pHILIC and processed with Compound Discoverer for metabolite profiling. Spiked cell extracts generated 4314 features with Z-HILIC compared to 3297 features with ZIC-pHILIC, an improvement of 31% detection coverage (Fig. 4A). Similar RT distribution of these features were observed with both methods in RT region 3.2–14 min; however, more features were found to be highly retained with Z-HILIC in RT region 16–23 min. Thus, Z-HILIC provides an extended elution range and increased the spread of metabolites on the RT axis, a desirable characteristic for metabolite profiling, compared to ZIC-pHILIC and other columns12,13 (Fig. S1†). Of those features, 559 (Z-HILIC) and 471 (ZIC-pHILIC) metabolites were identified in the spiked cell extracts using RT and exact mass of the standards, a metabolite coverage increase of 79.1% with Z-HILIC compared to 66.6% with ZIC-pHILIC for all detected standards in this study, and indicates that the Z-HILIC is more sensitive and less prone to matrix effects. This is evident in the enhanced sensitivity observed in the average peak areas of all detected features and identified metabolites with Z-HILIC compared to ZIC-pHILIC (Fig. 4B), which leads to less influence by the matrix species as the ion-suppression in electrospray ionization is known to be more prominent against low abundance peaks.25
 | ||
| Fig. 4 Assessment of the metabolic profiling and metabolite identification using Z-HILIC LC-MS. Comparison between the metabolic profiles obtained with Z-HILIC and ZIC-pHILIC LC-MS showing (A) the number of the detected features and identified metabolites and their (B) average peak areas in the spiked cell extract and the standard mixture. (C) The number of the acquired MS/MS spectra and (D) identified metabolites using DDA (Xcalibur) compared to deep-scan DDA (AcquireX). | ||
Variability in the LC-MS analytical run (repeatability) was assessed by calculating relative standard deviations (RSD) of the RT and peak areas of the identified metabolites in the spiked cell extracts using both methods. Z-HILIC was more stable and less prone to RT shifts, with an average RSD of 0.79% and 85% of the metabolites falling within an RSD range of 2%. In contrast, ZIC-pHILIC showed a higher average RSD of 1.00%, with only 73% of the metabolites falling within RSD range of 2%. However, both methods were within the acceptable limit of ≥70% of the peaks having an RSD range of 2%.26 In addition, enhanced repeatability across peak areas of the identified metabolites was observed with Z-HILIC (RSD within 17.5%) compared to ZIC-pHILIC (RSD within 71.8%). Overall, the results showed that Z-HILIC analysis enabled detection of a wide range of metabolites with high sensitivity, precision and stability compared to ZIC-pHILIC and therefore, the former is more suited for LC-MS metabolite profiling.
3.3. Development of standard spectral database for metabolite identification
Broad chemical-diversity of metabolites in biological samples increases the possibility of false identification using m/z only; in addition, co-eluted isobaric/isomeric metabolites cannot be distinguished even with the use of authentic standards RT and m/z. MS/MS techniques generate high-energy spectra with a specific set of fragments for each metabolite that can be matched with an in-house or online standard spectral database for identification.1,27 Considering that, fragmentation in MS is structure-dependent and each metabolite has an optimum collision energy (CE) that produces informative MS/MS spectra essential for manual or automated metabolite identification. The attempt to match metabolite fragmentation spectra with reference spectral databases using different CE or tandem MS techniques is suboptimal and leads to false identification. Therefore, our spectral database of standards was curated with a specific CE for each standard to enable accurate matching of metabolite fragments with the reference spectra. The use of MS/MS along with (m/z, RT) in the database provided a confirmatory layer of confidence in metabolite identification in addition to the potential of identifying isobaric/isomeric metabolites with high accuracy. The developed Z-HILIC LC-MS method was used in DDA employing higher-energy collisional dissociation (HCD) to optimize the collision energy (CE) for each standard and curate an MS/MS spectral database for metabolite identification. Individual standards were analyzed using a modified acquisition method to target their exact mass and RT for fragmentation at different CE values of 20, 30 and 50 in both ESI modes. MS/MS spectra for each standard were then visually examined and evaluated to select fragment-rich spectra (5–10 fragments) with at least 10% relative abundance and the database was then populated with one specific CE value for each standard (Fig. S2†). As a result, an in-house spectral database of 707 metabolite standards with m/z, RT and MS/MS at specific CEs in ESI+ or ESI− modes (Table S2†) was curated using the mzVault spectral database (Thermo Fisher Scientific, Waltham, MA, USA). Therefore, our in-house database of standards enabled metabolite identification in the samples with high confidence (level 1, MSI classification).43.4. Evaluation of MS/MS acquisition techniques for metabolite identification
DDA is widely used in metabolomics to generate MS/MS spectra of the metabolites in the biological samples to aid identification and annotation using spectral database.8,10,14–16 However, low numbers of MS/MS spectra of the metabolite features in the samples were typically obtained with DDA as only the most abundant peaks are selected for fragmentation, leaving behind potentially important metabolites that are present at low abundances.15 AcquireX is an automated fragmentation acquisition software using different DDA techniques, such as deep-scan DDA, to increase MS/MS coverage of metabolite features in experimental samples. By injecting samples multiple times for fragmentation of unique peaks in a samples, while continuously excluding blank background peaks and previously fragmented peaks, more MS/MS spectra can be collected.15 Therefore, a metabolomics workflow was established using the developed Z-HILIC LC-MS profiling method and AcquireX deep-scan DDA. The workflow was used with the curated standard spectral database and Compound Discoverer for high-throughput identification of metabolites in the study. The spiked cell extracts and the standard mixture were analyzed with the AcquireX workflow using 3 consecutives MS/MS iterations for each CE in both modes and compared with a standard DDA (Xcalibur). Multiple injections led to a marked increase in the number of MS/MS spectra acquired, with a total of 608 fragmentation spectra obtained with deep-scan DDA (AcquireX) (Fig. 4C) compared to only 278 acquired with standard DDA (Xcalibur). This enhancement enabled identification of 80% more metabolites with 289 and 365 metabolites identified using MS/MS in the spiked cell extract and the standard mixture, respectively, compared to only 160 and 173 identified using standard DDA (Fig. 4D). These results indicate that the use of deep-scan DDA, compared to standard DDA, is more effective in identifying metabolites, especially in samples were matrix effects can compromise ion signals; this is higher than the previously reported 50% increase obtained using a different hybrid-Orbitrap MS.15 Therefore, the developed Z-HILIC LC-MS with AcquireX deep-scan DDA workflow was used for the analysis of the biological samples below.3.5. Untargeted metabolomics of Hs578T cell extracts exposed to doxorubicin (Dox)
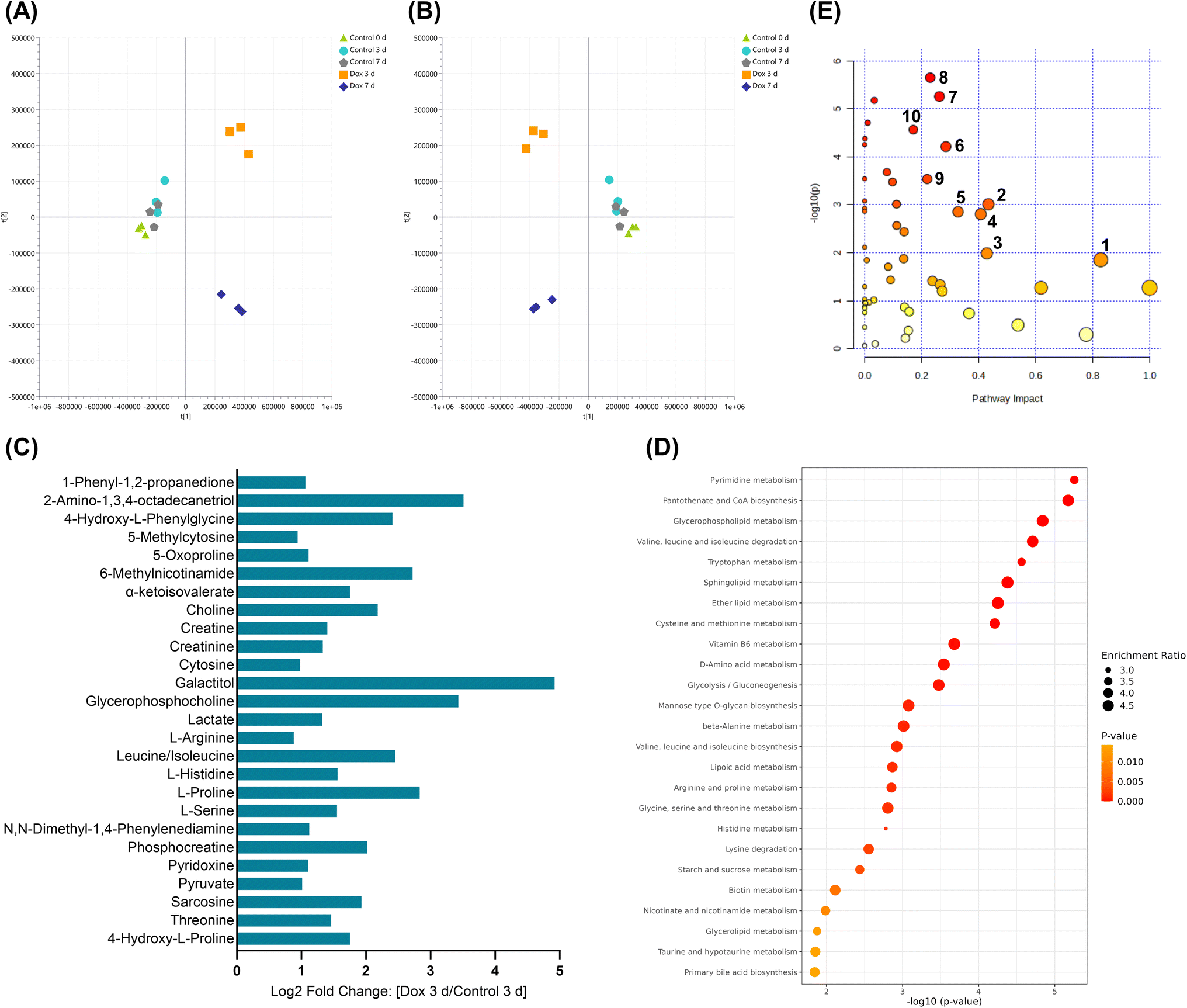 | ||
| Fig. 5 Significantly affected metabolic pathways in Hs578T cells exposed to doxorubicin (Dox) compared to controls using multivariate, enrichment and pathway analysis. (A) PCA and (B) PLS-DA score plots of the metabolic profiles of the study samples; cross validation values of the models were: R2X = 0.792, Q2 = 0.657 for PCA and R2X = 0.890, R2Y = 0.899, Q2 = 0.541 for PLS-DA. (C) Log2 fold change of the significantly affected metabolites identified in Dox 3 d compared to control 3 d. (D) Pathway enrichment analysis showing the top 25 enriched metabolic pathways in Dox 3 d compared to control 3 d. (E) Pathway analysis highlighting the top significantly altered metabolic pathways in Hs578T cells due to Dox treatment. Numbers correspond to (1) taurine and hypotaurine metabolism, (2) histidine metabolism, (3) nicotinate and nicotinamide metabolism, (4) glycine, serine and threonine metabolism, (5) arginine and proline metabolism, (6) cysteine and methionine metabolism, (7) pyrimidine metabolism, (8) glycerophospholipid metabolism, (9) pyruvate metabolism and (10) tryptophan metabolism. The value of −log10(p-value) represented by colour graduation from white (small), yellow, orange to red (large), whereas, the pathway impact represented by the circle size graduation: small (low impact) to large (high impact). | ||
The accumulation of lactate, pyruvate and suppression of the TCA cycle were previously linked to glycolysis as the main source of energy in cancer cells under oxidative stress.38 Our results similarly showed that lactate and pyruvate were significantly elevated in Hs578T cells treated with Dox. However, the increased levels of creatine, phosphocreatine and creatinine support a possible shift in central energy pathways, reflecting a potential Warburg effect reversal under Dox exposure toward oxidative phosphorylation.39 Pathway analysis identified alterations in the pantothenate and CoA biosynthesis, vitamin B6 and biotin metabolism, which may indicate a perturbed acetyl CoA production, TCA cycle and energy generation in Dox-treated cells, similar to a previous investigation in MCF-7 breast cancer cells.40 Dox is, therefore, limiting cell proliferation by depleting energy resources; however, such stress causes Hs578T cells to undergo metabolic reprogramming to be less reliant on glycolysis for energy production, thereby conferring resistance towards Dox.41
Cytosine and 5-methylcytosine were found significantly elevated in Dox-treated cells compared to control, suggesting disrupted DNA methylation, which impairs DNA repair and replication.32 Concurrently, altered levels of glycerophosphocholine, choline, and 2-amino-1,3,4-octadecanetriol due to Dox treatment indicate an impaired lipid metabolism, destabilizing the lipid bilayer and triggering apoptotic signaling.42 This observation aligns with the ability of Dox to activate cell death through apoptosis.43 In addition, disruption of biotin metabolism and lipoic acid metabolism in Hs578T cells by Dox correlates with the reported Dox-induced mitochondrial dysfunction in cancer cells, causing further oxidative stress and apoptosis.44,45
In summary, Dox was found to alter many metabolic pathways in Hs578T cells reflecting its multi-targeted modes of action and a global oxidative stress, mitochondrial dysfunction, and impaired biosynthesis response. These insights underscore the importance of understanding the metabolic vulnerabilities in TNBC to improve the efficacy of chemotherapeutic agents and may contribute in defining a new targeted therapy for TNBC.
4. Conclusion
A new LC-MS method using Z-HILIC column and a large number of metabolite standards was developed, evaluated for untargeted metabolomics studies and applied for metabolic characterization of TNBC cells treated with Dox. The method demonstrated higher sensitivity, selectivity, metabolite coverage and analytical stability compared to the widely used ZIC-pHILIC method.6–10 The use of the method with a comprehensive local spectral MS/MS database and RT along with MS/MS deep-scan acquisition method provides a high-throughput workflow for metabolic profiling and rigorous metabolite identification. The developed metabolomics workflow offers improved metabolite and pathway profiling related to Dox-induced metabolic alterations in TNBC cells and is suitable for different untargeted metabolomics applications.Data availability
Raw datasets for this article are available at MassIVE Repository (University of California, San Diego, CA, USA) at [https://doi.org/10.25345/C5VM4388P]. The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was carried out using the Core Technology Platforms Mass Spectrometry Facilities at New York University Abu Dhabi. This work was funded by a Tamkeen grant to S. A. A. (AD179), a Gordon and Betty Moore Foundation grant (GBMF9335) to S. A. A., a CGSB grant to S. A. A., a Mubadala ACCESS grant to S. A. A. and by a Tamkeen grant to D. A. S.References
- H. O. Doğan, Metabolomics: a review of liquid chromatography mass spectrometry-based methods and clinical applications, Turk. J. Biochem., 2024, 49(1), 1–14, DOI:10.1515/tjb-2023-0095.
- G. Theodoridis, H. Gika, D. Raftery, R. Goodacre, R. S. Plumb and I. D. Wilson, Ensuring Fact-Based Metabolite Identification in Liquid Chromatography-Mass Spectrometry-Based Metabolomics, Anal. Chem., 2023, 95(8), 3909–3916, DOI:10.1021/acs.analchem.2c05192.
- T. Kind and O. Fiehn, Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm, BMC Bioinf., 2006, 7, 234, DOI:10.1186/1471-2105-7-234FromNLM.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. M. Fan, O. Fiehn, R. Goodacre and J. L. Griffin, et al., Proposed minimum reporting standards for chemical analysis, Metabolomics, 2007, 3(3), 211–221, DOI:10.1007/s11306-007-0082-2.
- C. González Olmedo, L. Díaz Beltrán, V. Madrid García, J. L. Palacios Ferrer, A. Cano Jiménez, R. Urbano Cubero, J. Pérez Del Palacio, C. Díaz, F. Vicente and P. Sánchez Rovira, Assessment of Untargeted Metabolomics by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry to Define Breast Cancer Liquid Biopsy-Based Biomarkers in Plasma Samples, Int. J. Mol. Sci., 2024, 25(10), 5098, DOI:10.3390/ijms25105098.
- R. Zhang, D. G. Watson, L. Wang, G. D. Westrop, G. H. Coombs and T. Zhang, Evaluation of mobile phase characteristics on three zwitterionic columns in hydrophilic interaction liquid chromatography mode for liquid chromatography-high resolution mass spectrometry based untargeted metabolite profiling of Leishmania parasites, J. Chromatogr. A, 2014, 1362, 168–179, DOI:10.1016/j.chroma.2014.08.039.
- L. Safo, S. Abdelrazig, A. Grosse-Honebrink, T. Millat, A. M. Henstra, R. Norman, N. R. Thomas, K. Winzer, N. P. Minton and D. H. Kim, et al., Quantitative Bioreactor Monitoring of Intracellular Bacterial Metabolites in Clostridium autoethanogenum Using Liquid Chromatography-Isotope Dilution Mass Spectrometry, ACS Omega, 2021, 6(21), 13518–13526, DOI:10.1021/acsomega.0c05588FromNLM.
- D. M. Hijazi, L. A. Dahabiyeh, S. Abdelrazig, D. A. Alqudah and A. G. Al-Bakri, Micafungin effect on Pseudomonas aeruginosa metabolome, virulence and biofilm: potential quorum sensing inhibitor, AMB Express, 2023, 13(1), 20, DOI:10.1186/s13568-023-01523-0.
- S. Schatschneider, S. Abdelrazig, L. Safo, A. M. Henstra, T. Millat, D.-H. Kim, K. Winzer, N. P. Minton and D. A. Barrett, Quantitative Isotope-Dilution High-Resolution-Mass-Spectrometry Analysis of Multiple Intracellular Metabolites in Clostridium autoethanogenum with Uniformly 13C-Labeled Standards Derived from Spirulina, Anal. Chem., 2018, 90(7), 4470–4477, DOI:10.1021/acs.analchem.7b04758.
- S. Abdelrazig, L. Safo, G. Rance, M. Fay, E. Theodosiou, P. Topham, D.-H. Kim and A. Fernandez-Castane, Metabolic Characterisation of Magnetospirillum gryphiswaldense MSR-1 Using LC-MS-Based Metabolite Profiling, RSC Adv., 2020, 10(54), 32548–32560, 10.1039/D0RA05326K.
- T. H. Walter, B. A. Alden, K. Berthelette, J. A. Field, N. L. Lawrence, J. McLaughlin and A. V. Patel, Characterization of a highly stable zwitterionic hydrophilic interaction chromatography stationary phase based on hybrid organic–inorganic particles, J. Sep. Sci., 2022, 45(8), 1389–1399, DOI:10.1002/jssc.202100859.
- S. Girel, D. Guillarme, S. Fekete, S. Rudaz and V. González-Ruiz, Investigation of several chromatographic approaches for untargeted profiling of central carbon metabolism, J. Chromatogr. A, 2023, 1697, 463994, DOI:10.1016/j.chroma.2023.463994FromNLM.
- A. Lioupi, C. Virgiliou, T. H. Walter, K. M. Smith, P. Rainville, I. D. Wilson, G. Theodoridis and H. G. Gika, Application of a hybrid zwitterionic hydrophilic interaction liquid chromatography column in metabolic profiling studies, J. Chromatogr. A, 2022, 1672, 463013, DOI:10.1016/j.chroma.2022.463013.
- H. Abuzaid, S. Abdelrazig, L. Ferreira, H. M. Collins, D.-H. Kim, K.-H. Lim, T.-S. Kam, L. Turyanska and T. D. Bradshaw, Apoferritin-Encapsulated Jerantinine A for Transferrin Receptor Targeting and Enhanced Selectivity in Breast Cancer Therapy, ACS Omega, 2022, 7(25), 21473–21482, DOI:10.1021/acsomega.2c00997.
- B. Cooper and R. Yang, An assessment of AcquireX and Compound Discoverer software 3.3 for non-targeted metabolomics, Sci. Rep., 2024, 14(1), 4841, DOI:10.1038/s41598-024-55356-3.
- M. Kaczmarek, N. Zhang, L. Buzhansky, S. Gilead and E. Gazit, Optimization Strategies for Mass Spectrometry-Based Untargeted Metabolomics Analysis of Small Polar Molecules in Human Plasma, Metabolites, 2023, 13(8), 923, DOI:10.3390/metabo13080923.
- C. A. Hudis and L. Gianni, Triple-Negative Breast Cancer: An Unmet Medical Need, Oncologist, 2011, 16(S1), 1–11, DOI:10.1634/theoncologist.2011-S1-01.
- C. Braicu, R. Chiorean, A. Irimie, S. Chira, C. Tomuleasa, E. Neagoe, A. Paradiso, P. Achimas-Cadariu, V. Lazar and I. Berindan-Neagoe, Novel insight into triple-negative breast cancers, the emerging role of angiogenesis, and antiangiogenic therapy, Expert Rev. Mol. Med., 2016, 18, e18, DOI:10.1017/erm.2016.17FromCambridgeUniversityPressCambridgeCore.
- C. A. Ciocan-Cartita, A. Jurj, O. Zanoaga, R. Cojocneanu, L.-A. Pop, A. Moldovan, C. Moldovan, A. A. Zimta, L. Raduly and C. Pop-Bica, et al., New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells, J. Exp. Clin. Cancer Res., 2020, 39(1), 241, DOI:10.1186/s13046-020-01736-2.
- Z. Wang, Q. Jiang and C. Dong, Metabolic reprogramming in triple-negative breast cancer, Cancer Biol. Med., 2020, 17(1), 44–59, DOI:10.20892/j.issn.2095-3941.2019.0210FromNLM.
- C. A. Ciocan-Cartita, A. Jurj, O. Zanoaga, R. Cojocneanu, L. A. Pop, A. Moldovan, C. Moldovan, A. A. Zimta, L. Raduly and C. Pop-Bica, et al., New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells, J. Exp. Clin. Cancer Res., 2020, 39(1), 241, DOI:10.1186/s13046-020-01736-2FromNLM.
- Y. Benjamini and Y. Hochberg, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing, J. Roy. Stat. Soc. B, 1995, 57(1), 289–300, DOI:10.1111/j.2517-6161.1995.tb02031.x.
- Z. Pang, Y. Lu, G. Zhou, F. Hui, L. Xu, C. Viau, A. F. Spigelman, P. E. MacDonald, D. S. Wishart and S. Li, et al., MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation, Nucleic Acids Res., 2024, 52(W1), W398–W406, DOI:10.1093/nar/gkae253.
- A. Lioupi, C. Virgiliou, T. H. Walter, K. M. Smith, P. Rainville, I. D. Wilson, G. Theodoridis and H. G. Gika, Application of a hybrid zwitterionic hydrophilic interaction liquid chromatography column in metabolic profiling studies, J. Chromatogr. A, 2022, 1672, 463013, DOI:10.1016/j.chroma.2022.463013FromNLM.
- A. Furey, M. Moriarty, V. Bane, B. Kinsella and M. Lehane, Ion suppression; A critical review on causes, evaluation, prevention and applications, Talanta, 2013, 115, 104–122, DOI:10.1016/j.talanta.2013.03.048.
- O. Begou, H. G. Gika, G. A. Theodoridis and I. D. Wilson Quality Control and Validation Issues in LC-MS Metabolomics, in Metabolic Profiling: Methods and Protocols, ed. Theodoridis, G. A., Gika, H. G. and Wilson, I. D., Springer, New York, 2018, pp. 15–26 Search PubMed.
- S. Heiles, Advanced tandem mass spectrometry in metabolomics and lipidomics-methods and applications, Anal. Bioanal. Chem., 2021, 413(24), 5927–5948, DOI:10.1007/s00216-021-03425-1FromNLM.
- E. J. Want, I. D. Wilson, H. Gika, G. Theodoridis, R. S. Plumb, J. Shockcor, E. Holmes and J. K. Nicholson, Global metabolic profiling procedures for urine using UPLC-MS, Nat. Protoc., 2010, 5(6), 1005–1018, DOI:10.1038/nprot.2010.50.
- H. G. Gika, G. A. Theodoridis, J. E. Wingate and I. D. Wilson, Within-Day Reproducibility of an HPLC-MS-Based Method for Metabonomic Analysis: Application to Human Urine, J. Proteome Res., 2007, 6(8), 3291–3303, DOI:10.1021/pr070183p.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold, J. Trygg, C. Wikström and S. Wold, Multi- and Megavariate Data Analysis: Basic Principles and Applications, Umetrics AB, 2006 Search PubMed.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold, J. Trygg, C. Wikström and S. Wold, Multi- and Megavariate Data Analysis: Advanced Applications and Method Extensions, Umetrics AB, 2006 Search PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity, Pharmacol. Rev., 2004, 56(2), 185–229, DOI:10.1124/pr.56.2.6FromNLM.
- E. Xu, B. Ji, K. Jin and Y. Chen, Branched-chain amino acids catabolism and cancer progression: focus on therapeutic interventions, Front. Oncol., 2023, 13, 1220638, DOI:10.3389/fonc.2023.1220638FromNLM.
- S. Pan, M. Fan, Z. Liu, X. Li and H. Wang, Serine, glycine and one-carbon metabolism in cancer (Review), Int. J. Oncol., 2021, 58(2), 158–170, DOI:10.3892/ijo.2020.5158FromNLM.
- B. R. Rushing, A. Wiggs, S. Molina, M. Schroder and S. Sumner, Metabolomics Analysis Reveals Novel Targets of Chemosensitizing Polyphenols and Omega-3 Polyunsaturated Fatty Acids in Triple Negative Breast Cancer Cells, Int. J. Mol. Sci., 2023, 24(5), 4406, DOI:10.3390/ijms24054406.
- N. J. Lanning, J. P. Castle, S. J. Singh, A. N. Leon, E. A. Tovar, A. Sanghera, J. P. MacKeigan, F. V. Filipp and C. R. Graveel, Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities, Cancer Metabol., 2017, 5, 6, DOI:10.1186/s40170-017-0168-xFromNLM.
- B. R. Rushing, Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents, Int. J. Mol. Sci., 2023, 24(17), 13242 CrossRef CAS PubMed.
- C. N. Nagineni, S. Naz, R. Choudhuri, G. V. R. Chandramouli, M. C. Krishna, J. R. Brender, J. A. Cook and J. B. Mitchell, Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells, Int. J. Mol. Sci., 2021, 22(9), 4835 CrossRef CAS PubMed.
- Z. Tang, Z. Xu, X. Zhu and J. Zhang, New insights into molecules and pathways of cancer metabolism and therapeutic implications, Cancer Commun., 2021, 41(1), 16–36, DOI:10.1002/cac2.12112FromNLM.
- N. George, M. B. Joshi and K. Satyamoorthy, DNA damage-induced senescence is associated with metabolomic reprogramming in breast cancer cells, Biochimie, 2024, 216, 71–82, DOI:10.1016/j.biochi.2023.09.021.
- E. Varghese, S. M. Samuel, A. Líšková, M. Samec, P. Kubatka and D. Büsselberg, Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer, Cancers, 2020, 12(8), 2252, DOI:10.3390/cancers12082252.
- C. Zhang, N. Zhu, H. Li, Y. Gong, J. Gu, Y. Shi, D. Liao, W. Wang, A. Dai and L. Qin, New dawn for cancer cell death: emerging role of lipid metabolism, Mol. Metab., 2022, 63, 101529, DOI:10.1016/j.molmet.2022.101529.
- Z. Wang, Y. Wang, Z. Li, W. Xue, S. Hu and X. Kong, Lipid metabolism as a target for cancer drug resistance: progress and prospects, Front. Pharmacol, 2023, 14, 1274335, DOI:10.3389/fphar.2023.1274335.
- P. Jungsuwadee, M. Vore and D. K. S. Clair, Chemotherapy-Induced Oxidative Stress in Nontargeted Normal Tissues, in Oxidative Stress in Cancer Biology and Therapy, ed. Spitz, D. R., Dornfeld, K. J., Krishnan, K. and Gius, D., Humana Press, 2012, pp. 97–129 Search PubMed.
- J. P. J. Merlin, A. Crous and H. Abrahamse, Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes, Int. J. Mol. Sci., 2024, 25(19), 10796 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01044f |
| This journal is © The Royal Society of Chemistry 2025 |