
Photoresponsive lanthanide luminescent materials
Lingna
Su
a,
Xiao
Liu
*a,
Qingyu
Niu
a and
Zhiqiang
Li
 *ab
*ab
aSchool of Chemical Engineering and Technology, Hebei University of Technology, GuangRong Dao 8, Hongqiao District, Tianjin 300130, P. R. China. E-mail: 2023925@hebut.edu.cn; zhiqiangli@hebut.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
First published on 6th June 2024
Abstract
Lanthanide luminescent materials have attracted great attention in the last few decades due to their intriguing optical properties, such as large Stokes shift, long excited state lifetimes, high quantum efficiencies, abundant emission spectra, and high color purity. Stimuli-responsive materials are smart materials that can sense and respond to surrounding stimuli. The combination of stimuli-response with the unique luminescent properties of lanthanides has great potential for numerous applications. Compared to other stimuli, light irradiation offers many advantages, such as cleanliness, easy availability, high efficiency, and remoteness. This review focuses on the construction and response mechanism of photoresponsive lanthanide luminescent materials. Furthermore, various important applications in luminescent actuators, photoswitches, intelligent anticounterfeiting, and theranostics are also highlighted in detail.
 Lingna Su | Lingna Su is a master's student at Hebei University of Technology. She received her bachelor's degree from Datong University. Her research interests are focused on constructing lanthanide luminescent materials. |
 Xiao Liu | Xiao Liu received her PhD degree under the supervision of Prof. Zhiqiang Li at Hebei University of Technology in 2023. Then, she joined Hebei University of Technology as a post-doctoral researcher. Her research interests focus on the design and synthesis of lanthanide luminescent materials. |
 Qingyu Niu | Qingyu Niu is a graduate student under the supervision of Prof. Zhiqiang Li at Hebei University of Technology. His current research focuses on the design and synthesis of supramolecular luminescent materials. |
 Zhiqiang Li | Zhiqiang Li received his BS at Inner Mongolia University in 2009 and his PhD at Nankai University under the guidance of Prof. Yu Liu in 2014. From 2017 to 2019, he worked as a Postdoctoral Research Fellow under the supervision of Prof. Banglin Chen at the University of Texas at San Antonio. Currently, he is a Full Professor of School of Chemical Engineering and Technology, Hebei University of Technology. His current research interest is the self-assembly of lanthanide-containing hybrid luminescent materials. |
1. Lanthanide luminescent materials
Lanthanide elements located in the sixth period and IIIB group in the periodic table consist of 15 elements from lanthanum (atomic number 57) to lutetium (atomic number 71), and their unique electronic configuration is [Xe] 4fn−15d0−16s2 (n = 1–15).1–4 The lanthanide elements have similar chemical properties due to their nearly identical outermost electron arrangements. The valence states of lanthanide elements can range from +2 to +4, with +3 being the most common, characterized by the electron configurations [Xe]4f0–14, which indeed produce a diverse range of electronic levels.5–7 Therefore, lanthanide materials display distinctive optical, magnetic, and electrical properties, showing a wide range of applications, including but not limited to photonic devices, renewable energy, biomedical devices, industrial products, and display devices.8–11As an important branch, the luminescence of lanthanide (Ln3+) ions originated from transitions inside the 4f shell, thus intraconfigurational f–f transitions have become a research hot spot.12,13 Different Ln3+ ions emit luminescence of various colors because of the different energy gaps of radiative transitions. For instance, Eu3+ emits red light, Tb3+ emits green light, Sm3+ emits orange light, Tm3+ emits blue light, and Dy3+ emits white or near-white light. Additionally, Pr3+, Nd3+, Dy3+, Ho3+, Er3+, Tm3+, and Yb3+ all emit radiation in near-infrared (NIR) light.14–16 The emission spectra of Ln3+ ions cover a wide range, including the ultraviolet (UV), visible (400–800 nm), and NIR (800–1700 nm) regions, which are widely recognized as valuable emitting sources.17,18 Due to the shielding effect of the partially filled 4f shell by the closed 5s2 and 5p6 shells, the emission wavelengths of Ln3+ ions are hardly affected by ligands in the first and second coordination sphere, which in particular results in narrow emission bands and long excited lifetimes.19
However, all Ln3+ ions have low absorption cross-sections (absorption coefficients ε, <10 L mol−1 cm−1), so only a limited amount of radiation is absorbed through direct excitation in the 4f energy level, resulting in weak luminescence.20 In order to solve this problem, coordinating Ln3+ ions with organic ligands offers a viable approach, which can resolve the weak light absorption caused by the forbidden f–f transitions. Upon exposure to external UV light, the organic ligands absorb energy and transition from the ground state (S0) to the excited singlet state (S1), then transfer the energy to the excited triplet state (T1) through the intersystem crossing (ISC). In addition, the energy is transferred from the triplet state to the excited state of the central Ln3+ ions via nonradiative transitions (Non-rad.). Finally, Ln3+ ions emit their characteristic luminescence through radiative transitions from the excited state to the ground state (Fig. 1).21 It is important to note that not all organic ligands can sensitize Ln3+ ions, which relies on the energy gap of the lowest excited triplet state T1 and the 4f excited state of the Ln3+ ions. Effective transfer can only occur if the energy gap between is in the appropriate range. Commonly used organic ligands include β-diketones (2-thiophenyltrifluoroacetone, hexafluoroacetylacetone, and acetylacetone), aromatic carboxylic acids (terephthalic acid, pyridine-2,6-dicarboxylic acid, and 1,3,5-benzenetricarboxylic acid), heterocyclic compounds (2,2′-bipyridine, 1,10-phenanthroline, and terpyridyl), macrocyclic ligands (macrocyclic polyether and macrocyclic polyketone), and compounds containing phosphoryl groups (triphenylphosphine oxide) (Fig. 2).22–27 Luminescence from lanthanide materials with narrow-band, large Stokes shift and longer decay time,28,29 is obtained by the so-called “antenna effect”,30,31 showing various optical applications.
 | ||
| Fig. 1 Intramolecular energy transfer mechanism for the lanthanide materials. | ||
 | ||
| Fig. 2 Chemical structure of common organic ligands: 2-thiophenyltrifluoroacetone (tta), hexafluoroacetylacetone (hfac), acetylacetone (acac), terephthalic acid (TPA), pyridine-2,6-dicarboxylic acid (DPA), 1,3,5-benzenetricarboxylic acid (BTC), 2,2′-bipyridine (Bpy), 1,10-phenanthroline (Phen), terpyridyl (Tpy), 2,2′,2′′-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triaceticacid (DOTA), and triphenylphosphine oxide (Ph3PO). | ||
2. Stimuli-responsive lanthanide luminescent materials
Stimuli-responsive materials are smart materials that undergo chemical and physical changes in response to external stimuli.32–34 External stimuli can be classified into physical stimuli, such as light,35 electricity,36 magnetism,37 mechanical force,38 humidity,39 and temperature,40 and chemical stimuli,41 such as pH,42 redox,43 and ions.44 These interactive, adaptive, and self-regulating features make them attractive as smart and multifunctional materials in various fields. Stimuli-responsive lanthanide luminescent materials,45,46 which achieve structure or luminescence signal changes in response to external stimuli, are one of the most important subclasses of lanthanide luminescent materials, showing potential applications in the fields of medical diagnosis,47,48 chemical sensors,49,50 biomimetic soft robotics,51,52 data storage,53 and security technologies.54 The response mechanism of various stimuli-responsive materials can be categorized as energy transfer and competitive coordination, etc. Energy transfer encompasses interactions between the two Ln3+ ions, as well as between the ligand and the Ln3+ ion. Competitive coordination encompasses competition between the water molecule and the organic ligand, as well as competition between other metal ions and the Ln3+ ion. Next, some representative examples of stimuli-responsive lanthanide luminescent materials will be discussed.2.1. Energy transfer
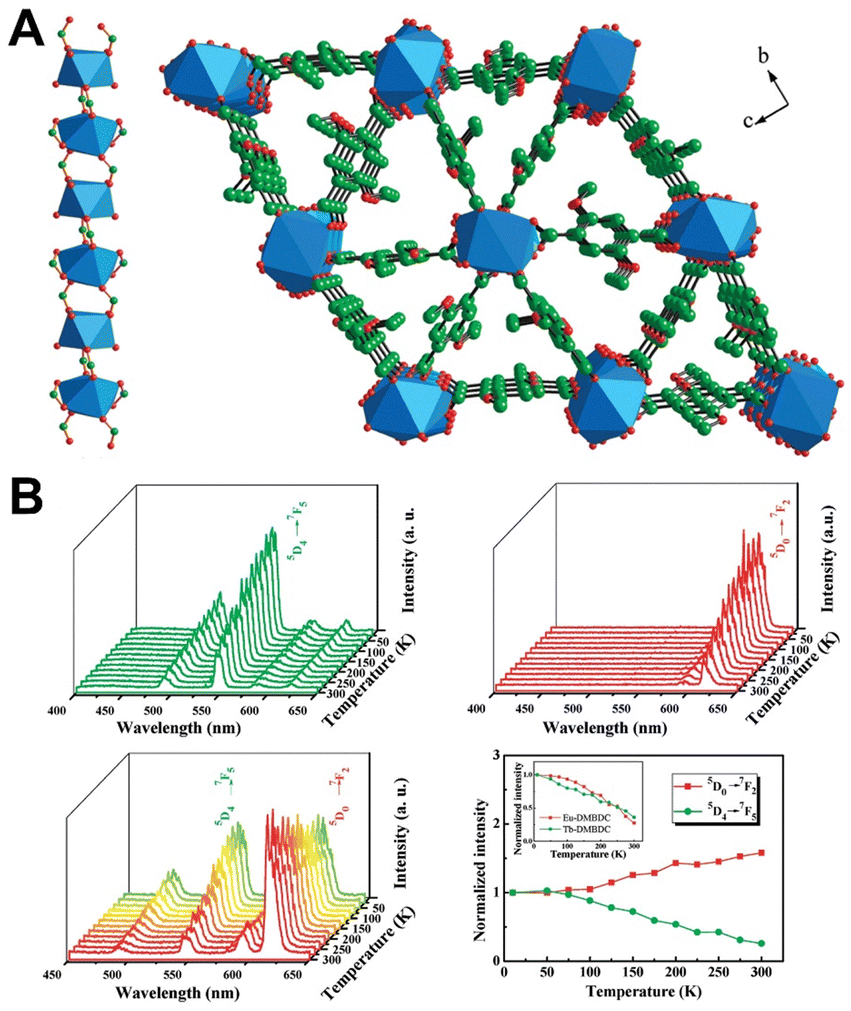 | ||
| Fig. 3 (A) Structure of Tb-DMBDC. (B) Emission spectra of Tb-DMBDC, Eu-DMBDC, and Eu0.0069Tb0.9931-DMBDC recorded between 10 and 300 K, and the temperature dependence of integrated intensities of Eu0.0069Tb0.9931-DMBDC. Reproduced with permission from ref. 58. Copyright: 2012, American Chemical Society. | ||
 | ||
| Fig. 4 (A) Protonation and deprotonation of Eu@UiO-67-bpydc. (B) The relationship between the emission intensity at 615 nm and the pH value. Reproduced with permission from ref. 42 Copyright: 2018, Elsevier. | ||
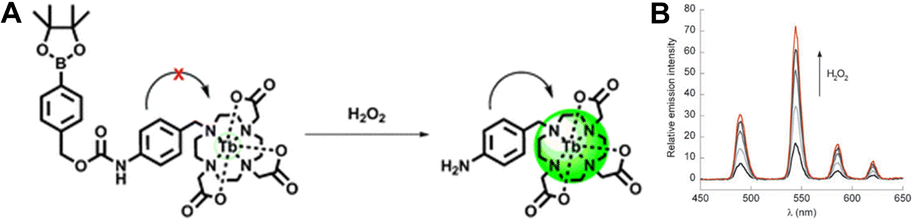 | ||
| Fig. 5 (A) Chemical structure and response mechanism of luminescent probes. (B) Emission spectra at different concentrations of H2O2. Reproduced with permission from ref. 63 Copyright: 2010, the Royal Society of Chemistry. | ||
2.2. Competitive coordination
 | ||
| Fig. 6 (A) Structure of [Eu(H2O)2(mpca)2Eu(H2O)6M(CN)8]·nH2O. (B) Emission spectra at different humidity. Inset: Emission intensity at 617 nm as a function of humidity. Reproduced with permission from ref. 39 Copyright: 2017, the Royal Society of Chemistry. | ||
 | ||
| Fig. 7 (A) Schematic illustration and chemical structure of luminescent hydrogels with Eu3+ as the cross-linking points. (B) Fe3+-induced luminescence quenching and gel–sol transition under UV light (upper) and daylight (lower). Reproduced with permission from ref. 67. Copyright: 2018, Wiley-VCH. | ||
3. Photoresponsive lanthanide luminescent materials
Despite many advances in stimuli-responsive lanthanide luminescent materials, there are still some challenges. First, some invasive stimuli can cause irreversible luminescence quenching. For example, the addition of transition metal ions (Fe3+, Fe2+, Cu2+, and so on) triggers the gel–sol transition, but the luminescence signal also irreversibly disappears during this process because the organic ligand coordinates with these ions.67 Luminescence on–off switching can be achieved to some extent by pH stimulation, which is based on the protonation or deprotonation of organic ligands. However, its fatigue resistance can only be maintained for a few cycles.68–70 An excess of alkali may cause Ln3+ ions to precipitate as hydroxides or oxides, resulting in irreversible luminescence quenching.71 Second, certain stimuli have the potential to harm biological tissues in biomedical applications, such as tumor diagnosis and targeted drug delivery. For instance, a pH-responsive anticancer system may release drugs in the acidic endolysosomes of normal cells, causing side effects.72,73 Direct heating can cause thermal damage to the physiological functions of normal tissues, whereas photothermal conversion can accurately and precisely stimulate specific locations on command.74 Third, obtaining certain stimuli, such as strong magnetic and electric fields, may be challenging in reality. Forth, the redox process may produce by-products and alter the valence state of Ln3+ ions.75,76In contrast, light irradiation has emerged as an ideal stimulus because of its high spatial/temporal resolution, controllability, rapidness, cleanliness, and handiness.77,78 Researchers have combined photoresponse with lanthanide luminescent materials by introducing various photoresponsive molecules into lanthanide systems.79–82 This has resulted in the development of a range of functional photoresponsive lanthanide luminescent materials including in luminescent actuators, photoswitches, intelligent anticounterfeiting, and theranostics. Common photoresponsive molecules can be divided into different types (Fig. 8).
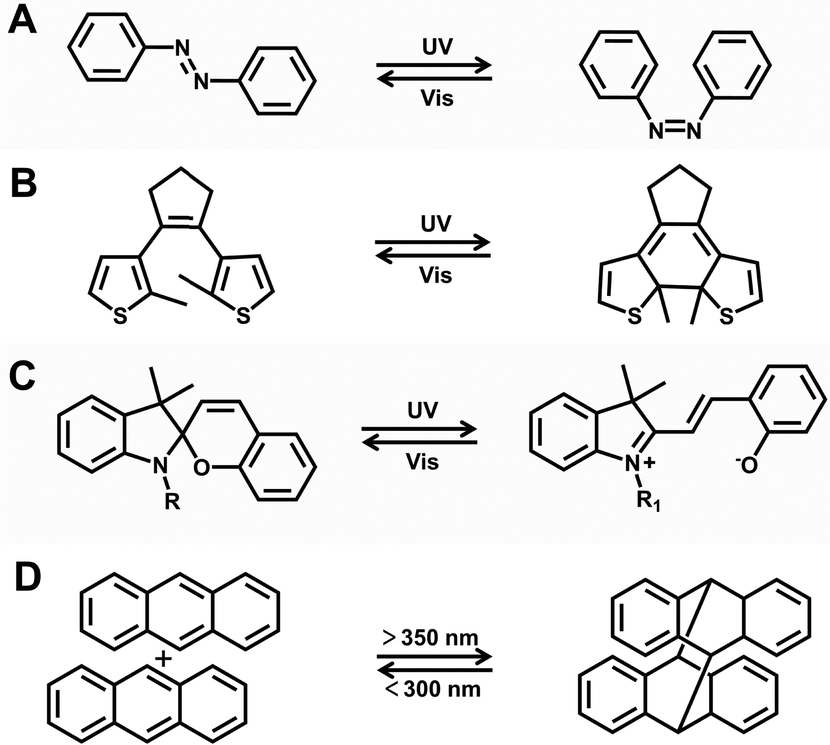 | ||
| Fig. 8 Common photoresponsive molecules. (A) Azobenzene. (B) Diarylethenes. (C) Spiropyran. (D) Anthracene. | ||
3.1. Photoresponsive molecules and mechanisms
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond to produce cis(Z)-azobenzene. When irradiated with visible light, it isomerizes back to E-azobenzene (Fig. 8A).83,84
N bond to produce cis(Z)-azobenzene. When irradiated with visible light, it isomerizes back to E-azobenzene (Fig. 8A).83,84
Similar to DTEs, spiropyran (SP) and its derivatives have also received significant attention due to their distinctive photochromic properties. When exposed to UV light, SP can be converted into cyanine (MC), resulting in a color change from colorless to blue-violet (with new absorption peaks appearing in the absorption spectrum at 550–600 nm) and a change from non-fluorescence to red fluorescence (with new emission peaks appearing at 620–750 nm). When exposed to visible light, MC quickly restores to the ring-closure SP, causing the color and fluorescence to disappear simultaneously (Fig. 8C).89–91
3.2. Functionalities of photoresponsive molecules
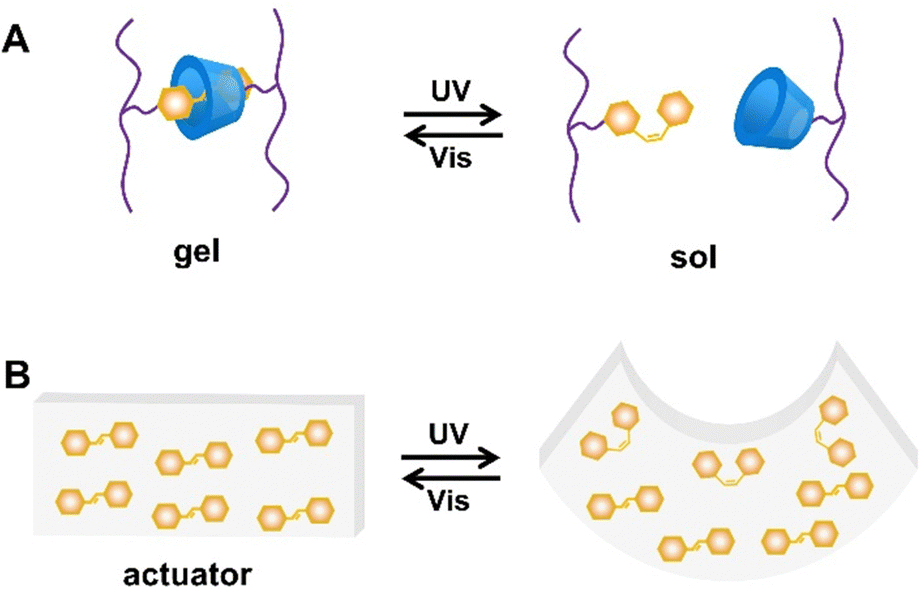 | ||
| Fig. 9 Schematic illustration of (A) sol–gel transition and (B) an actuator based on trans/cis isomerization of azobenzene under UV and visible light irradiation alternately. | ||
In addition, liquid crystal systems are often prepared by introducing azobenzene to achieve photoresponsive deformation. E-azobenzene is a nearly rod-shaped molecule, while Z-azobenzene displays a curved “V” shape. The long rod-shaped E-azobenzene can exist as a liquid crystal radical in azobenzene-containing liquid crystal systems. After irradiating with UV light, the orderly arrangement of liquid crystal motifs can be disrupted and cause the entire liquid crystal network to macroscopically deform (Fig. 9B).
Harada et al. prepared supramolecular hydrogels by using α-CD modified curdlan as the backbone, which were further cross-linked with azobenzene as the guest (Fig. 10).96 The hydrogels underwent a reversible sol–gel transition by controlling the trans/cis isomerization of azobenzene through UV/visible light irradiation alternately to modulate the dissociation and formation of α-CD/azobenzene inclusion complexes, providing a reference for the design of photoresponsive lanthanide luminescent hydrogels.
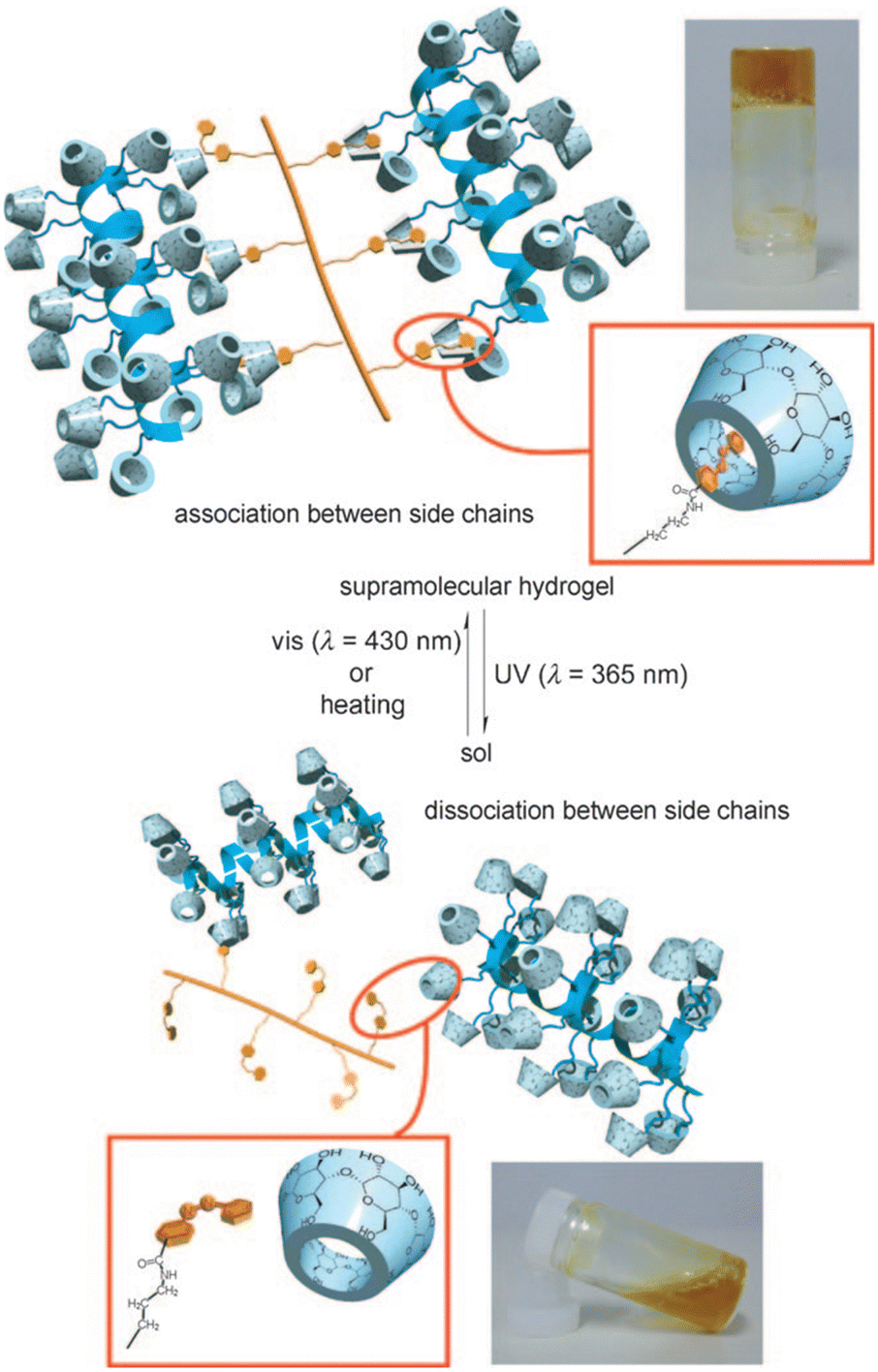 | ||
| Fig. 10 Schematic illustration of sol–gel transition based on α-CD and azobenzene with UV (365 nm) and visible (430 nm) light. Reproduced with permission from ref. 96. Copyright: 2010, Wiley-VCH. | ||
 | ||
| Fig. 11 Schematic illustration of luminescence on–off switching based on DTEs and lanthanide luminescent materials. | ||
Liu et al. designed a host–guest system assembled by DTEs (guest molecule) and terpyridinyldibenzo-24-crown-8 as well as Eu3+ ions (host molecule) through noncovalent interaction (Fig. 12).99 The UV-Vis absorption spectra of the ring-closure form of guest molecules and the luminescent emission spectra of the host molecules Eu3+/terpyridinyldibenzo-24-crown-8 overlap perfectly. Furthermore, the distance between host and guest molecules is narrowed due to the host–guest interaction, which quenches the luminescence of Eu3+ through the FRET process. Luminescence on–off switching of the Eu3+ can be effectively controlled by alternating external UV and visible light, which provided a novel design for multistage driven molecular machines and logic gates.
 | ||
| Fig. 12 (A) Schematic illustration of the luminescence on–off switching based on DTEs and Eu3+ complex. (B) Emission spectra at different times under 300 nm UV light irradiation. Insets: Emission intensity at 619 nm at different times (upper); photographs of emission color under 254 nm UV light (lower). Reproduced with permission from ref. 99. Copyright: 2013, American Chemical Society. | ||
SP is frequently added to lanthanide luminescent systems. The luminescence of MC can be enhanced by quenching the luminescence of Ln3+ ions through FRET, due to the overlap between the UV-Vis absorption spectrum of MC and the emission spectrum of Ln3+ ions (Fig. 13). Efficient FRET between Ln3+ ions and SP can be used to create luminescent materials with multiple colors, providing a new idea for preparing anti-counterfeiting materials with dynamic photochromic performance.
 | ||
| Fig. 13 Schematic illustration of multicolor luminescence based on spiropyran and lanthanide luminescent materials. | ||
Lee et al. reported a core–shell structure of lanthanide-doped upconversion nanoparticles (Ln-UCNPs) (β-NaYF4:Nd3+/Yb3+/Tm3+@NaYF4:Nd3+@NaYF4@NaYF4:Yb3+/Er3+) that produced UV (347–475 nm) and visible (545 nm) emission upon excitation with NIR light at 808 and 980 nm, respectively, and then the Ln-UCNPs were further assembled with SP (Fig. 14).100 The maximum absorption peaks of SP and MC are located at 342 nm and 560 nm, respectively, which have good overlap with the UV emission of Ln-UCNPs under 808 nm excitation and the green emission under 980 nm excitation. Therefore, this strategy achieves two-way photoswitching using dual wavelengths of NIR light.
 | ||
| Fig. 14 (A) Schematic illustration of photoswitching of Ln-UCNPs/SP under 808 nm and 980 nm NIR light irradiation. (B) Emission spectra of Ln-UCNPs and the UV-Vis absorption spectra of SP and MC under 808 nm and 980 nm irradiation alternately. Reproduced with permission from ref. 100. Copyright: 2014, Wiley-VCH. | ||
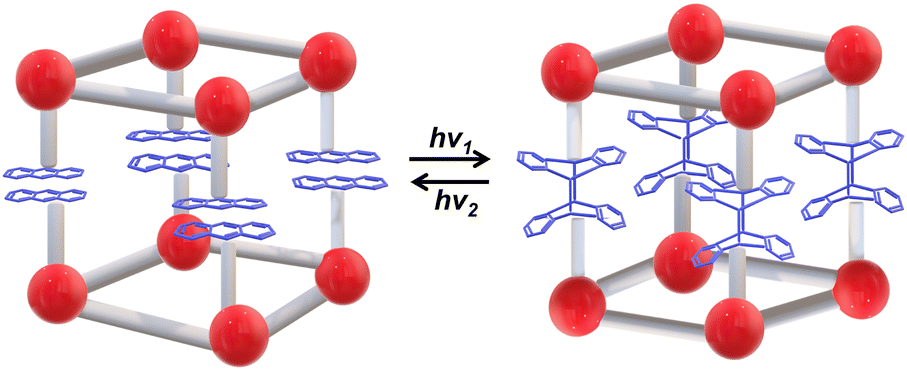 | ||
| Fig. 15 Schematic illustration of the photoresponsive SC–SC structural transition based on anthracene-containing LnMOFs. | ||
Zheng et al. synthesized a mononuclear compound LnIII(depma)(NO3)3(hmpa)2 (Ln = Dy, Gd; depma = 9-diethylphosphono-methylanthracene; hmpa = hexamethylphosphoramide) using Ln3+ ions and anthracene derivatives (Fig. 16).102 The face-to-face anthracene group underwent a [4+4]-cycloaddition reaction after irradiation with 365 nm UV light without loss of crystallinity, and mononuclear compounds transformed into LnIII2(depma2)(NO3)6(hmpa)4 dimers. Dy-containing single compounds change from single-ion to a single-molecule magnet with the luminescence color changes from yellowish-green to bluish-white, which opens up new possibilities for designing intelligent opto-magnetic devices.
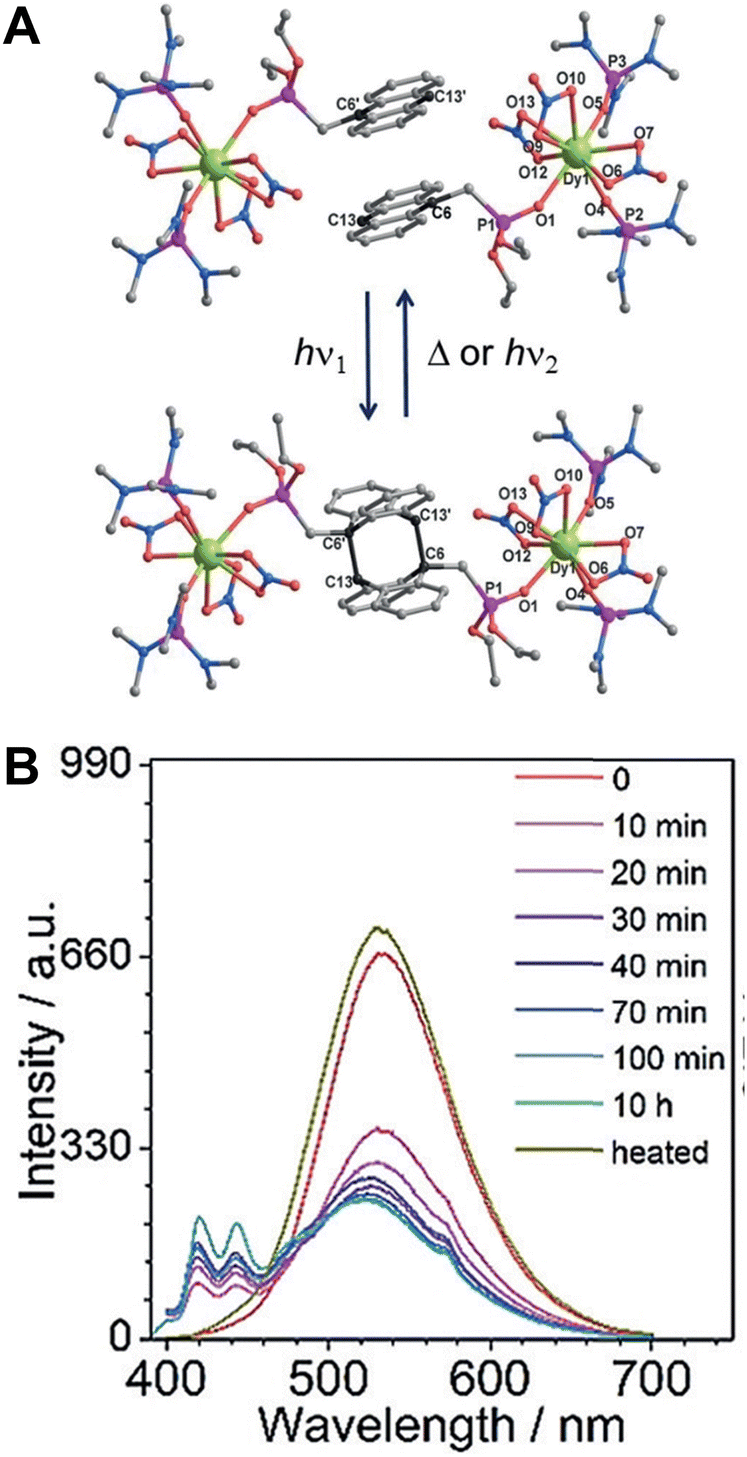 | ||
| Fig. 16 (A) Structure of DyIII(depma)(NO3)3(hmpa)2 and dimers. (B) Emission spectra of DyIII(depma)(NO3)3(hmpa)2 illuminated with 365 nm UV light at different times. Reproduced with permission from ref. 102. Copyright: 2018, Wiley-VCH. | ||
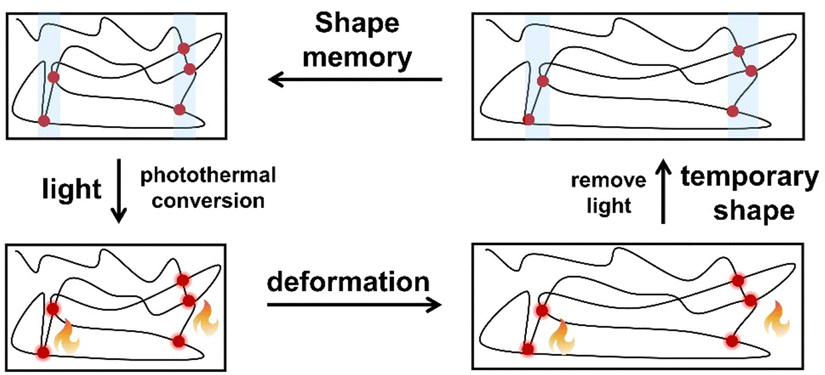 | ||
| Fig. 17 Mechanism of photoresponsive SMPs: the absorption of light by Ln-UCNPs or lanthanide complexes inducing photothermal conversion and phase separation; the material can be deformed; removing light during deformation causes the material to be locked in a temporary shape; restore the material to its permanent shape with additional light irradiation and removal. | ||
Rowan et al. prepared metal-supramolecular polymer films with photoresponsive shape memory properties (Fig. 18).105 Poly(butadiene) was first terminated by 4-hydroxy-2,6-bis(10-methylbenzimidazoyl)pyridine, and then cross-linked with pentaerythritol tetrakis(3-mercaptopropionate). Eu(NTf2)3 was also added during photopolymerization to prepare metal-supramolecular polymers. The Eu3+ complexes absorbed the UV light, and converted it into luminescence and localized heating. This caused softening of the hard phases and increased decomplexation of the Eu3+ complexes. As a result, the materials could be deformed under UV light. Upon removal of light, the materials cooled and the metal–ligand interactions rejoined, locking them in a temporary deformed state. After further UV light irradiation, the strain was released, and the material returned to its original shape.
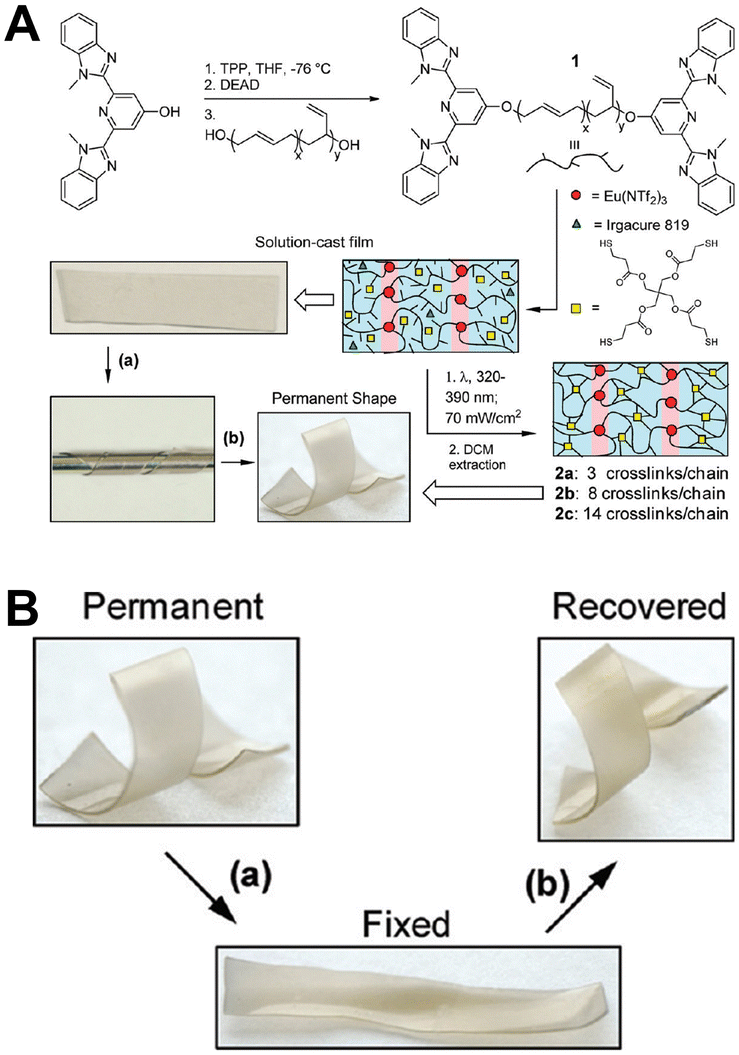 | ||
| Fig. 18 (A) Chemical structure and the synthetic process of metal-supramolecular polymer films. (B) Pictures of the shape memory behavior. Reproduced with permission from ref. 105. Copyright: 2011, American Chemical Society. | ||
4. Application of photoresponsive lanthanide luminescent materials
4.1. Luminescent actuators
An actuator is a material that can change its volume, shape, or position under external stimulation. These materials have attracted attention due to their potential in bionic domains, and have been used to design grippers, walkers, artificial muscles, and valves.106–109 The integration of a photoresponse in lanthanide luminescent systems enables the realization of their photoresponsive sol–gel transition and actuation.Capobianco et al. used Ln-UCNPs that respond to NIR light to induce a sol–gel transition in supramolecular hydrogels based on azobenzene and α-CD (Fig. 19).110 The high upconversion efficiency of Ln-UCNPs in the ultraviolet region could convert low-energy photons into high-energy UV light. This, in turn, induces the isomerization of azobenzene in the supramolecular hydrogel, leading to the dissociation of an azobenzene/α-CD inclusion complex. Therefore, the gel–sol transition was realized in this system. NIR light has excellent penetration and low cytotoxicity in biological tissues. After implantation of the hydrogel into the surgical site, the NIR light triggers the release of the guest, which can be utilized for the development of efficient and controllable drug delivery systems.
 | ||
| Fig. 19 (A) Schematic illustration of the interactions of Ln-UCNPs, α-CD and azobenzene within the hydrogel matrix. (B) Digital photographs of gels illuminated with 980 nm NIR laser for different times. Reproduced with permission from ref. 110. Copyright: 2018, the Royal Society of Chemistry. | ||
In 2018, we modified α-CD with 2,6-pyridine dicarboxylic acid (DPA) via a polyethylene glycol polymer chain, and then Ln3+ ions were added to coordinate with PDA forming a lanthanide complex. Subsequently, the guanidinium-modified azobenzene was incorporated into the lanthanide complex periphery through molecular recognition of azobenzene and α-CD, resulting in host–guest inclusion (Fig. 20).111 Finally, a hybrid supramolecular luminescent hydrogel was constructed by the electrostatic force driving between guanidinium salts and LAPONITE® (LAP) nanosheets to form a three-dimensional network. Under UV light irradiation, E-azobenzene isomerizes to Z-azobenzene, which has greater spatial resistance, causing it to detach from the cavity of α-CD. This process cut off the cross-linking between the coordination material and the surrounding LAP nanosheets, resulting in gel–sol transition. Upon further irradiation with visible light, the Z-azobenzene isomerized back to E-azobenzene. As a result, the azobenzene and α-CD once again formed the inclusion compound and re-cross-linked the surrounding LAP, thus initiating the transformation process from sol to gel once again. Furthermore, since the photoisomerization wavelength of azobenzene did not conflict with the excitation/emission wavelengths of lanthanide complex, the remote light-controlled gel–sol transition thus does not affect the luminescent performance of supramolecular hybrid hydrogels. In addition, the luminescence color of the hybrid hydrogel was regulated by adjusting the molar ratio of Ln3+ ions (Eu3+/Tb3+).
 | ||
| Fig. 20 (A) Schematic illustration of the sol–gel phase transition of photoresponsive hybrid luminescent supramolecular hydrogels. (B) Photographs of luminescent hydrogels with sol–gel phase transition under 254 nm UV light irradiation. Reproduced with permission from ref. 111. Copyright: 2018, Wiley-VCH. | ||
4.2. Photoswitches
Photoswitching materials are able to interconvert between two different light output states after being exposed to light. This property makes them a promising candidate in various fields, such as optoelectronic switching devices, molecular data processing and storage, chemical sensing, and super-resolution fluorescence microscopy.112,113 Introducing photoresponsive molecules into lanthanide luminescent materials is an effective way to achieve luminescence on–off switching by regulating their conformation.Liu et al. reported a multi-component nanosystem and explored its function as a lanthanide supramolecular photoswitch (Fig. 21A).114 The neighbouring azobenzene units’ disordered motion suppressed the lanthanide complex's luminescence. α-CD was added to immobilize the E-azobenzene's conformation, allowing it to display the characteristic emission of Ln3+ ions in both aqueous solution and hydrogel. The photoluminescence of the supramolecular lanthanide system can be modulated by alternately irradiating with UV and visible light, causing reversible association and dissociation of the α-CD/azobenzene host–guest inclusion complex. Recently, they also designed a full-color lanthanide supramolecular photoswitches, which were assembled using pyridine-2,6-dicarboxylic acid (DPA)-modified pillar[5]arene, Ln3+ ions, and diethylene derivatives through noncovalent interactions (Fig. 21B).115 The supramolecular system exhibited strong luminescence emission in both organic solvents and aqueous solution due to the appropriate energy gap and strong coordination between DPA and Ln3+ ions. By adjusting the molar ratio of Eu3+, Tb3+, and host materials, the supramolecular system produced full-color emission, including white luminescence. The system's on–off luminescence can be controlled by the FRET between Ln3+ ions and DTEs through the ring-closure/ring-opening isomerization of diethylene. This allows for on-demand luminescence color tuning of advanced photoresponsive materials.
 | ||
| Fig. 21 Schematic illustration and molecular structures of lanthanide supramolecular photoswitches assembled by Ln3+ ions. (A) DPA-modified azobenzene and α-CD. Reproduced with permission from ref. 114. Copyright: 2022, Wiley-VCH. (B) DPA-modified pillar[5]arene and DTEs. Reproduced with permission from ref. 115. Copyright: 2023, the Royal Society of Chemistry. | ||
We reported an approach to construct photoresponsive lanthanide metal–organic frameworks (Ln-MOFs) by loading solid-state DTEs into MOFs via host–guest interactions, which resulted in reversible luminescence on–off photoswitching behavior through alternating UV and visible light irradiation (Fig. 22).116 Ring-opening DTEs exhibited no absorption in the wavelength range of 400–800 nm and Ln-MOFs showed the characteristic color and brightness of Eu3+ ions. After irradiating with 300 nm UV light, DTEs adopted their ring-closure form and a new absorption band in the range of 500–700 nm appeared, which had good overlap with the emission spectra of Ln-MOFs, resulting in luminescence quenching due to FRET from Eu3+ to DTEs. When irradiated with visible light, DTE molecules switched from ring-closure form to ring-opening form, and the luminescence of LnMOFs was restored.
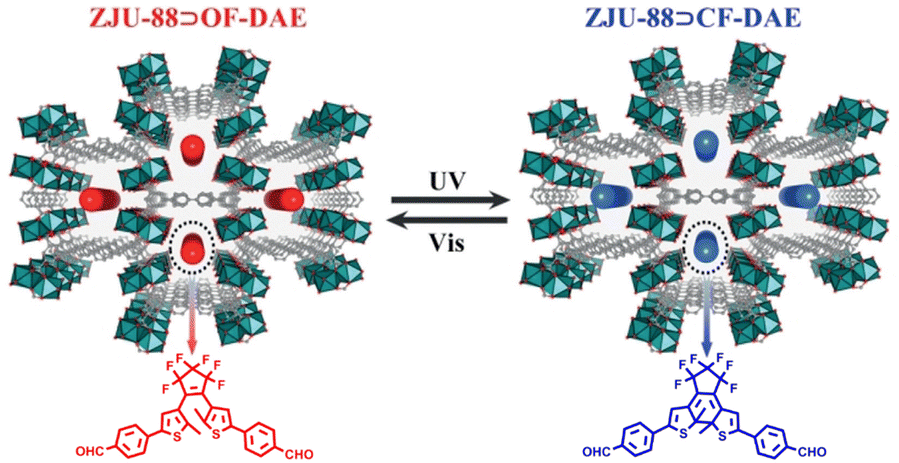 | ||
| Fig. 22 Structure of LnMOFs and the molecular structures of photoresponsive DTEs. Reproduced with permission from ref. 116. Copyright: 2019, Wiley-VCH. | ||
4.3. Intelligent anticounterfeiting
With the rapid development of social economy and technology, counterfeit and inferior goods are emerging. The counterfeiting of valuable and confidential documents, such as currency, passports, certificates, and movies, poses a significant economic and security threat to both consumers and governments.33,117,118 Photoluminescent printing is a common means of anti-counterfeiting. Many countries use anticounterfeiting labels on banknotes due to their high efficiency, facile design, and tunable optical properties. However, the use of static luminescence signals can be easily imitated, and their encrypted information can be easily decrypted.119–121 Therefore, the design and development of high-security anti-counterfeiting materials with different luminescent colors that respond to external stimuli and change their optical outputs is of great significance in eliminating counterfeit products.Based on Ln3+ ions and the photoresponse of DTEs, we constructed a series of photoresponsive lanthanide luminescent materials to be used in intelligent anticounterfeiting. The luminescence on–off switching behavior was realized by controlling the FRET from Eu3+ ions to DTEs by alternating UV and visible light irradiation, and potential applications in the intelligent anti-counterfeiting field were further explored. In 2019, photoresponsive lanthanide luminescent hydrogels with high transparency, excellent tensile properties, and good luminescence were prepared using an in situ copolymerization strategy of the functionalized lanthanide complex, DTEs, and the acrylamide monomer (Fig. 23A–C).122 Hydrogel blocks with various luminescent colors, including red, green, yellow, and colorless were arranged to form hydrogel matrix codes, allowing for the encryption and decryption of information on demand. Considering that inkjet printing is currently the most widely used printing method and the incompatibility between the printers and existing security inks due to their high viscosity or large particle content, a photoresponsive supramolecular polyelectrolyte was constructed through hierarchical self-assembly of Ln3+ ions, bis-ligand and DTEs driven by coordination and ionic interaction (Fig. 23D and E).123 The supramolecular polyelectrolyte was directly utilized as an anti-counterfeiting ink in commercial printers to produce high-resolution QR codes on blue PET film, allowing for light-controlled remote multiple reversible information erasure and display. To meet different needs, multi-stimuli-responsive anisotropic hydrogels were prepared by copolymerizing magnetic alumina platelets, lanthanide complexes, DTEs, and N-isopropylacrylamide in a directed magnetic field.124 The hydrogel blocks were assembled into a QR code, and the information could be hidden/recovered by UV/visible light irradiation or heating/cooling. The promising applicability in the field of multi-dimensional intelligent anti-counterfeiting could provide new research horizons for expanding the applications of functional lanthanide luminescent hydrogels.
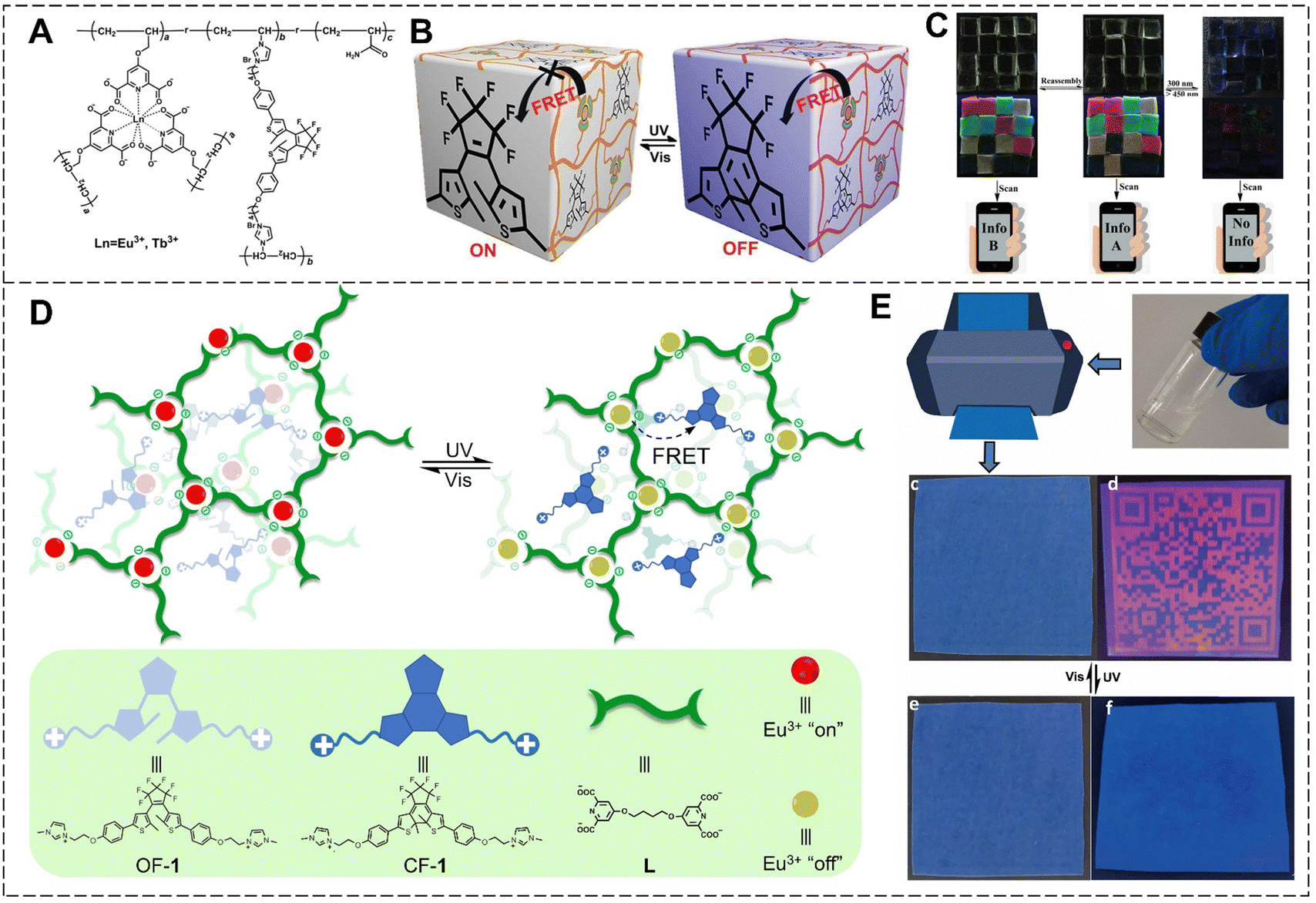 | ||
| Fig. 23 (A) Chemical structure of the photoresponsive lanthanide luminescent hydrogel. (B) Schematic illustration of the luminescence on–off switching behavior by alternating UV and visible light irradiation. (C) Photos of the hydrogel matrix codes constructed by the photoresponsive hydrogel under daylight and UV light. Reproduced with permission from ref. 122. Copyright: 2019, Wiley-VCH. (D) Schematic illustration of supramolecular polyelectrolyte and chemical structures of DTEs as well as bis-ligand. (E) Schematic illustration of the printing process and photos of the QR code on blue PET film under daylight (left) and UV light (right). Reproduced with permission from ref. 123. Copyright: 2021, Nature Publishing Group. | ||
Yang et al. loaded SP into LnMOFs to prepare a luminescent host–guest system for dynamic anti-counterfeiting (Fig. 24).125 Changes in emission intensity and luminescence color can be achieved by simple UV/visible light irradiation for different times, eliminating external interfering factors. Under UV light irradiation, SP was gradually transformed into MC, which exhibited a strong absorption band at 550 nm. Energy was transferred from Ln3+ ions to MC through FRET, so the color of luminescence turned continuously green to red. Under visible light irradiation, MC converted back into SP, which did not overlap spectrally with Ln3+ ions, resulting in the cessation of FRET. Therefore, this system presented dynamic luminescent colors with prolonged irradiation time, facilitating the creation of complicated anti-counterfeiting patterns. This strategy provides novel materials to enhance the reliability of anti-counterfeiting, and practical guidance for further application in the field of dynamic anti-counterfeiting and information storage.
 | ||
| Fig. 24 (A) Synthetic process of loading SP into Tb-MOFs and (B) advanced anti-counterfeiting based on the SP@Tb-MOF/PDMS (PDMS = polydimethylsiloxane) film with a dual optical response. Reproduced with permission from ref. 125 Copyright: 2022, American Chemical Society. | ||
4.4. Theranostics
Fluorescence imaging with high sensitivity is a non-invasive method for studying biomolecular mechanisms and pathways in living cells. However, it faces challenges when applied to real samples due to the optical interference caused by the inhomogeneity of biological samples and their own fluorescence. Compared to irreversible changes in luminescence signal, luminescence that can respond reversibly provides an effective way to avoid optical interference. Therefore, designing novel luminescent probes capable of modulating the signal output is crucial for research.Cheng et al. designed terpyridine-modified DTEs (DTE@Tpy), and coordinated them with Eu(tta)3·3H2O to prepare a new luminescent lanthanide molecular switch, DTEs@TpyEu(tta)3. Reversible luminescence on–off switching behaviors in both solution and poly(methyl methacrylate) films were realized under external UV and visible light irradiation (Fig. 25).126 To be noted, it can serve as a highly sensitive luminescent probe to enter living cells for imaging without optical interference, and luminescence on–off switching was also observed in living cells, showing potential for application in more sophisticated imaging.
 | ||
| Fig. 25 (A) Schematic illustration of the photoresponsive luminescence on–off switching. (B) Confocal images of NIH 3T3 cells after incubation with CF-DTE@TpyEu(tta)3. Reproduced with permission from ref. 126. Copyright: 2016, American Chemical Society. | ||
In addition to being used in cell imaging, photoresponsive lanthanide luminescent materials can also be used in drug delivery platforms.127,128 Chemotherapy remains the primary treatment for cancer. However, it can damage normal biological tissues and has a short circulation time in the body, which affects its safety and therapeutic efficacy. Light-triggered drug delivery platforms can remotely control the release of drugs at the desired location and time to realize on-demand therapy. This can significantly reduce side effects and avoid any effects toward healthy biological tissue. While most photoresponsive systems are sensitive to UV and visible light, their limited tissue penetration and photodamage restrict their further use in living systems.129,130 Introducing NIR light-responsive Ln-UCNPs into the system can address these issues. Ln-UCNPs have the ability to absorb NIR light and convert it into UV or visible light, which can then drive the photoresponsive system to release the drug. The main mechanisms for the drug release of NIR-responsive Ln-UCNP materials are photolysis and photoisomerization.
For the photolysis strategy, photolabile molecule-coupled anticancer drugs are loaded into Ln-UCNPs. The UV/visible light emitted by Ln-UCNPs drive photolabile molecule cracking and release of anticancer drugs. Li et al. designed hydrophobic Ln-UCNPs with a core–shell structure (NaYF4:Yb, Tm@NaLuF4-mSiO2) by photolysis (Fig. 26A).131 They then loaded coumarin-conjugated anticancer drug chloramphenicol into the nanoparticles through physisorption. Upon 980 nm laser irradiation, the Ln-UCNPs emitted UV light, which photolytically cleaved the coumarin, releasing chloramphenicol and inhibiting tumor growth.
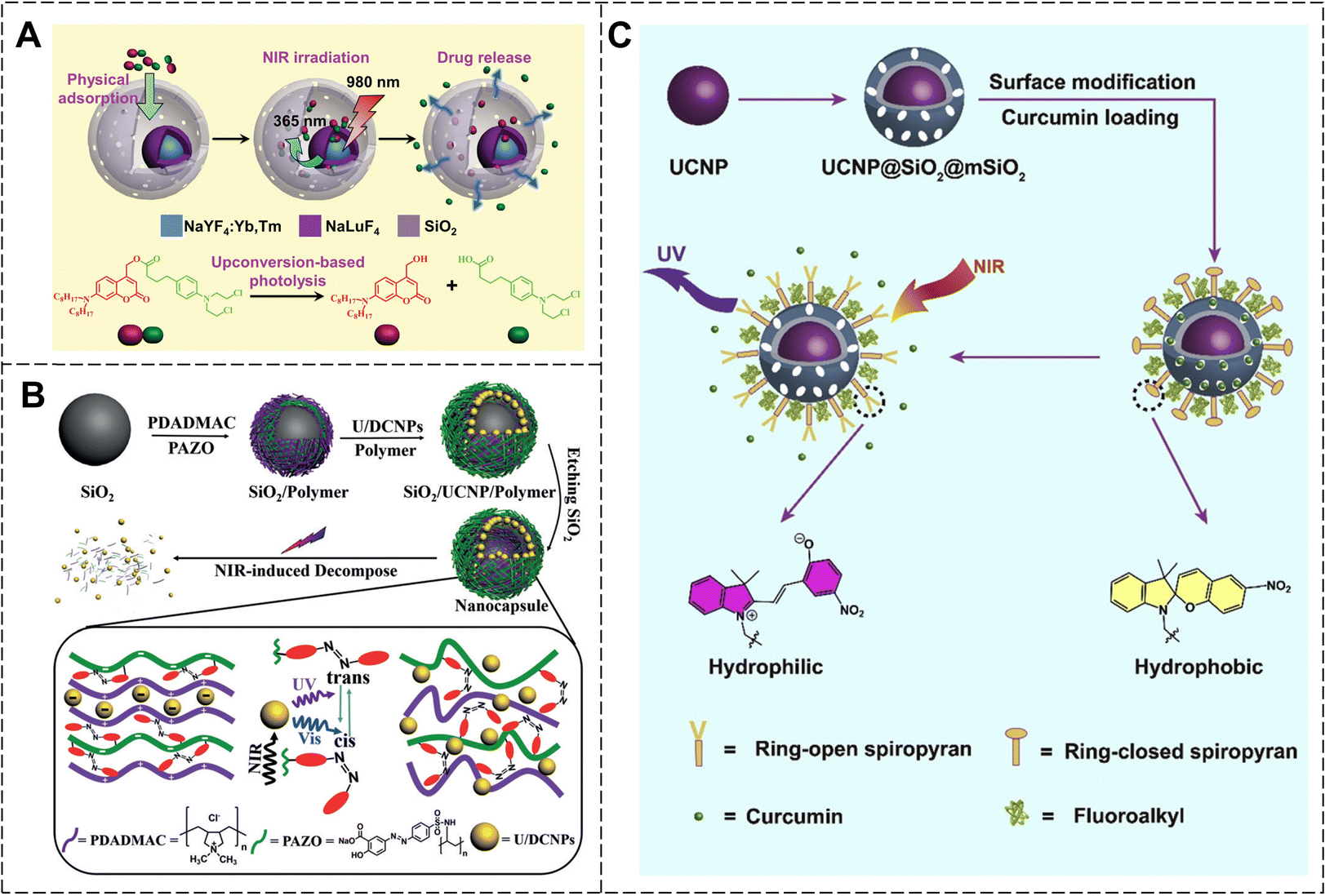 | ||
| Fig. 26 (A) Schematic illustration of the NIR-responsive drug delivery and chemical structures of prodrug photolysis. Reproduced with permission from ref. 131. Copyright: 2014, Wiley-VCH. (B) The synthetic process of nanocapsules and NIR-triggered photoisomerization of azobenzene. Reproduced with permission from ref. 132. Copyright: 2018, Wiley-VCH. (C) Schematic illustration of the NIR-triggered system and photoisomerization of spiropyran. Reproduced with permission from ref. 133. Copyright: 2017, Elsevier. | ||
As another photoresponsive drug delivery platform, the photoisomerization strategy can be realized by introducing photoisomeric molecules into Ln-UCNP systems. NIR light was upconverted into UV/visible light by Ln-UCNPs. The UV/visible light triggered the photoisomerization of the photoresponsive molecules, thereby controlling the release of the drug loaded in the system. Zhao et al. modified spherical silica with positively charged polymers, and then assembled it with azobenzene-containing polymers and negatively charged dual-mode up/down-converted nanoparticles (Ln-U/DCNPs) via electrostatic interactions to prepare nanocapsules (Fig. 26B).132 Under NIR light irradiation, Ln-U/DCNPs could convert NIR light into UV light. This, in turn, caused cis/trans isomerization of the azobenzene, leading to the decomposition of the nanocapsules and the release of the anticancer drug. Spiropyran is another photoisomeric molecule. Ring-opening SP after irradiation with visible light is hydrophobic, whereas ring-closure MC after irradiation with UV light is hydrophilic. Qu et al. prepared mesoporous silica-coated Ln-UCNPs, and then modified SP on the surface of silica. The hydrophobic anticancer drug curcumin was loaded into mesoporous silica (Fig. 26C).133 Prior to NIR illumination, SP formed a dense hydrophobic layer on the surface of the silica, which prevented the premature release of curcumin. Upon NIR light irradiation, Ln-UCNPs efficiently converted NIR light into UV light, causing MC to isomerize into SP while also becoming hydrophilic, thereby enabling drug release. Additionally, the UV/visible light produced by Ln-UCNPs could activate curcumin to its triplet state and reacted with oxygen to generate reactive oxygen species, enhancing the antitumor impact.
5. Conclusions and outlook
In conclusion, this review summarizes the response mechanism, preparation method, and applications of photoresponsive lanthanide luminescent materials in luminescent actuators, photoswitches, intelligent anticounterfeiting, and theranostics. Compared to other stimuli-responsive materials, photoresponsive lanthanide luminescent materials exhibit a number of inherent advantages, including accurate control of location and wavelength, low cost-effectiveness, and environmentally friendly nature.This research field is rapidly evolving, with increasing emphasis on gaining a deeper understanding of the mechanism of action of photoresponsive lanthanide materials and finding simpler and more effective preparation strategies to expand their potential applications in various fields. However, challenges still exist. First, it is challenging for lanthanide luminescent materials to maintain their luminescence in aqueous environments, limiting their aqueous application, in particular in biological fields. This difficulty arises mainly due to the coupling of the high-frequency stretching vibrations of –OH in water molecules with lanthanide ions, leading to nonradiative transitions of the central lanthanide ions and luminescence quenching. Maintaining the aqueous phase luminescence of lanthanide materials often requires the elaborate design of organic ligands to increase the coordination number thus protect the lanthanide ions from water molecules. In terms of the chemical structure of ligands, it is possible to design polydentate chelating ligands, which can enhance the luminescence in aqueous solution by using synergistic coordination. Furthermore, the coordination number and structure of lanthanide complexes should also be investigated in-depth. Alternatively, some amphiphilic ligands can be utilized to prepare complexes that can self-assemble into micelles in aqueous solution to protect the lanthanide luminescence center. Secondly, the majority of current photoresponsive lanthanide luminescent materials are driven by UV/visible light. This makes it difficult to penetrate biological tissues, can cause damage to the tissues, and they can be interfered with by the self-fluorescence of the biological tissues. The assembly of lanthanide-doped upconversion nanoparticles with photoresponsive molecules to prepare composites that utilize the energy of lanthanide-doped upconversion nanoparticles to drive photoresponsive molecules is a growing area of research. Lanthanide-doped upconversion nanoparticles are capable of converting NIR light into UV and visible light, thereby enabling the delivery and controlled release of guest molecules. Thirdly, although lanthanide luminescent materials have demonstrated effective intelligent anticounterfeiting properties, their long response time (seconds or even minutes) is still far from the requirements of actual applications. Shorter times are necessary to meet future equipment needs. Efforts are being made to investigate the potential of new photoresponsive molecules and mechanisms, or to modify existing photoresponsive molecules. In parallel, efforts can be made to enhance the understanding of the relationship between suitable structures and photoresponse by integrating theoretical calculations into the structural design and synthesis of materials. This approach could facilitate more effective and precise control over the resulting materials.
Author contributions
All authors have given approval to the final version of the manuscript. Lingna Su: data curation and writing – original draft; Xiao Liu: conceptualization (table of contents) and writing – original draft; Qingyu Niu: data curation (sensing mechanisms) and writing – original draft; and Zhiqiang Li: funding acquisition, data validation, and writing – reviewing and editing.Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22171069), the Natural Science Foundation of Hebei Province (B2021202026), and the Educational Committee of Hebei Province (JZX2024012).Notes and references
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374–6458 CrossRef CAS PubMed.
- X. Qin, X. Liu, W. Huang, M. Bettinelli and X. Liu, Chem. Rev., 2017, 117, 4488–4527 CrossRef CAS PubMed.
- Y. Liu, D. Tu, H. Zhu and X. Chen, Chem. Soc. Rev., 2013, 42, 6924–6958 RSC.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- D. Chen, Y. Wang and M. Hong, Nano Energy, 2012, 1, 73–90 CrossRef CAS.
- G. Wang, Q. Peng and Y. Li, Acc. Chem. Res., 2011, 44, 322–332 CrossRef CAS PubMed.
- P. Singh, S. Kachhap, P. Singh and S. K. Singh, Coord. Chem. Rev., 2022, 472, 214795 CrossRef CAS.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- E. R. Trivedi, S. V. Eliseeva, J. Jankolovits, M. M. Olmstead, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2014, 136, 1526–1534 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC.
- S.-J. Li, F. Li, N. Kong, J.-R. Liu and X. Zhu, Adv. Healthcare Mater., 2023, 12, 2302276 CrossRef CAS PubMed.
- Z. Wang, M. Zhang, S. Chi, M. Zhu, C. Wang and Z. Liu, Adv. Healthcare Mater., 2022, 11, 2200521 CrossRef CAS PubMed.
- J. Xu, A. Gulzar, P. Yang, H. Bi, D. Yang, S. Gai, F. He, J. Lin, B. Xing and D. Jin, Coord. Chem. Rev., 2019, 381, 104–134 CrossRef CAS.
- P. Du, R. An, Y. Liang, P. Lei and H. Zhang, Coord. Chem. Rev., 2022, 471, 214745 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- A. A. Ansari, M. R. Muthumareeswaran and R. Lv, Coord. Chem. Rev., 2022, 466, 214584 CrossRef.
- S. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2009, 253, 1099–1201 CrossRef CAS.
- K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413–6416 CrossRef CAS PubMed.
- Z. Li, Z. Hou, H. Fan and H. Li, Adv. Funct. Mater., 2017, 27, 1604379 CrossRef.
- Q. Zhu, L. Zhang, K. Van Vliet, A. Miserez and N. Holten-Andersen, ACS Appl. Mater. Interfaces, 2018, 10, 10409–10418 CrossRef CAS PubMed.
- Z. Gao, B. Xu, T. Zhang, Z. Liu, W. Zhang, X. Sun, Y. Liu, X. Wang, Z. Wang and Y. Yan, Angew. Chem., Int. Ed., 2020, 59, 19060–19064 CrossRef CAS PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- P. Li and H. Li, Coord. Chem. Rev., 2021, 441, 213988 CrossRef CAS.
- H.-Q. Yin, X.-Y. Wang and X.-B. Yin, J. Am. Chem. Soc., 2019, 141, 15166–15173 CrossRef CAS PubMed.
- K. Wu, X.-Y. Liu, Y.-L. Huang, M. Xie, X. Xiong, J. Zheng, W. Lu and D. Li, Inorg. Chem. Front., 2022, 9, 1714–1721 RSC.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- M.-K. Tsang, G. Bai and J. Hao, Chem. Soc. Rev., 2015, 44, 1585–1607 RSC.
- X. He, J. Wang, X. Liu, Q. Niu, Z. Li, B. Chen and Q. Xiong, Adv. Healthcare Mater., 2024, 2400747 CrossRef PubMed.
- R. Gao, X. Fang and D. Yan, J. Mater. Chem. C, 2019, 7, 3399–3412 RSC.
- M. Radisic, H. Park, H. Shing, T. Consi, F. J. Schoen, R. Langer, L. E. Freed and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18129–18134 CrossRef CAS PubMed.
- M.-C. Wong, L. Chen, G. Bai, L.-B. Huang and J. Hao, Adv. Mater., 2017, 29, 1701945 CrossRef PubMed.
- F. Yang, Y. Yuan, R. P. Sijbesma and Y. Chen, Macromolecules, 2020, 53, 905–912 CrossRef CAS.
- Y. Gao, P. Jing, N. Yan, M. Hilbers, H. Zhang, G. Rothenberg and S. Tanase, Chem. Commun., 2017, 53, 4465–4468 RSC.
- T. Feng, Y. Ye, X. Liu, H. Cui, Z. Li, Y. Zhang, B. Liang, H. Li and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 21752–21757 CrossRef CAS PubMed.
- X. Liu, B. Li, Y. Xu, Z. Li, Y. Zhang, Z.-J. Ding, H. Cui, J. Wang, H.-B. Hou and H. Li, Dalton Trans., 2019, 48, 7714–7719 RSC.
- X. Zhang, K. Jiang, H. He, D. Yue, D. Zhao, Y. Cui, Y. Yang and G. Qian, Sens. Actuators, B, 2018, 254, 1069–1077 CrossRef CAS.
- Z. Dai, L. Tian, B. Song, Z. Ye, X. Liu and J. Yuan, Anal. Chem., 2014, 86, 11883–11889 CrossRef CAS PubMed.
- L. Lu, C. Chen, D. Zhao, J. Sun and X. Yang, Anal. Chem., 2016, 88, 1238–1245 CrossRef CAS PubMed.
- P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed.
- X. Li, Y. Xie, B. Song, H.-L. Zhang, H. Chen, H. Cai, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 2689–2693 CrossRef CAS PubMed.
- Y. Liu, Z. Wei, X. Liao and J. Zhou, Acc. Mater. Res., 2020, 1, 225–235 CrossRef CAS.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- B. Yan, Acc. Chem. Res., 2017, 50, 2789–2798 CrossRef CAS PubMed.
- D. Parker, J. D. Fradgley and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 8193–8213 RSC.
- H. Liu, S. Wei, H. Qiu, M. Si, G. Lin, Z. Lei, W. Lu, L. Zhou and T. Chen, Adv. Funct. Mater., 2022, 32, 2108830 CrossRef CAS.
- X. Feng, C. Wang, S. Shang, H. Liu, X. Huang, J. Jiang and H. Zhang, Chem. Eng. J., 2022, 450, 138356 CrossRef CAS.
- Y. Xie, G. Sun, J. Li and L. Sun, Adv. Funct. Mater., 2023, 33, 2303663 CrossRef CAS.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS PubMed.
- Q. Xiao, X. Yin, L. Lv, X. Dong, N. Zhou, K. Liu and X. Luo, J. Rare Earths, 2023, 41, 981–988 CrossRef CAS.
- M. M. Upadhyay and K. Kumar, J. Rare Earths, 2023, 41, 1295–1301 CrossRef CAS.
- X. Liu, Y. Ye, X. He, Q. Niu, B. Chen and Z. Li, Angew. Chem., Int. Ed., 2024, 63, e202400195 CrossRef CAS PubMed.
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed.
- B. Li, Z. Song, K. Zhu, Q. Niu, Z. Li and H. Li, ACS Appl. Mater. Interfaces, 2021, 13, 20633–20640 CrossRef CAS PubMed.
- W.-M. Liu, P. H. J. Keizers, M. A. S. Hass, A. Blok, M. Timmer, A. J. C. Sarris, M. Overhand and M. Ubbink, J. Am. Chem. Soc., 2012, 134, 17306–17313 CrossRef CAS PubMed.
- D. Mouchel Dit Leguerrier, R. Barré, J. K. Molloy and F. Thomas, Coord. Chem. Rev., 2021, 446, 214133 CrossRef CAS.
- W.-L. Chan, C. Xie, W.-S. Lo, J.-C. G. Bünzli, W.-K. Wong and K.-L. Wong, Chem. Soc. Rev., 2021, 50, 12189–12257 RSC.
- A. R. Lippert, T. Gschneidtner and C. J. Chang, Chem. Commun., 2010, 46, 7510–7512 RSC.
- B. Li, Z.-J. Ding, Z. Li and H. Li, J. Mater. Chem. C, 2018, 6, 6869–6874 RSC.
- X. Liu, B. Li, W. Wang, Z. Li and Q. Xiong, Inorg. Chem. Front., 2022, 9, 4194–4200 RSC.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- G. Weng, S. Thanneeru and J. He, Adv. Mater., 2018, 30, 1706526 CrossRef PubMed.
- L. K. Truman, S. Comby and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 9624–9627 CrossRef CAS PubMed.
- S.-L. Hou, J. Dong, M.-H. Tang, X.-L. Jiang, Z.-H. Jiao and B. Zhao, Anal. Chem., 2019, 91, 5455–5460 CrossRef CAS PubMed.
- D. Zhao, J. Yang, X. Tian, J. Wei, Q. Li and Y. Wang, Chem. Eng. J., 2022, 434, 134806 CrossRef CAS.
- K. Luan, R. Meng, C. Shan, J. Cao, J. Jia, W. Liu and Y. Tang, Anal. Chem., 2018, 90, 3600–3607 CrossRef CAS PubMed.
- Y. Zhou, H. Ye, Y. Chen, R. Zhu and L. Yin, Biomacromolecules, 2018, 19, 1840–1857 CrossRef CAS PubMed.
- Y. Sun, Y. Sha, G. Cui, F. Meng and Z. Zhong, Adv. Drug Delivery Rev., 2023, 192, 114624 CrossRef CAS PubMed.
- X. Sun, J. Sun, B. Dong, G. Huang, L. Zhang, W. Zhou, J. Lv, X. Zhang, M. Liu, L. Xu, X. Bai, W. Xu, Y. Yang, X. Song and H. Song, Biomaterials, 2019, 201, 42–52 CrossRef CAS PubMed.
- R. M. Supkowski, J. P. Bolender, W. D. Smith, L. E. L. Reynolds and W. D. Horrocks Jr, Coord. Chem. Rev., 1999, 185–186, 307–319 CrossRef CAS.
- X. Liu, Z. Tang, B. Song, H. Ma and J. Yuan, J. Mater. Chem. B, 2017, 5, 2849–2855 RSC.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- J. Zhang and H. Tian, Adv. Opt. Mater., 2018, 6, 1701278 CrossRef.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- S. Fredrich, R. Göstl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- B. Slepetz and M. Kertesz, J. Am. Chem. Soc., 2013, 135, 13720–13727 CrossRef CAS PubMed.
- J. Fregoni, G. Granucci, E. Coccia, M. Persico and S. Corni, Nat. Commun., 2018, 9, 4688 CrossRef CAS PubMed.
- D.-Y. Kim, W.-J. Yoon, Y.-J. Choi, S.-I. Lim, J. Koo and K.-U. Jeong, J. Mater. Chem. C, 2018, 6, 12314–12320 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- T. Fukaminato, T. Sasaki, T. Kawai, N. Tamai and M. Irie, J. Am. Chem. Soc., 2004, 126, 14843–14849 CrossRef CAS PubMed.
- H. Miyasaka, M. Murakami, A. Itaya, D. Guillaumont, S. Nakamura and M. Irie, J. Am. Chem. Soc., 2001, 123, 753–754 CrossRef CAS PubMed.
- H.-Q. Zheng, Y. Yang, Z. Wang, D. Yang, G. Qian and Y. Cui, Adv. Mater., 2023, 35, 2300177 CrossRef CAS PubMed.
- Z. He, Y. Li, H. Wu, Y. Yang, Y. Chen, J. Zhu, Q. Li and G. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 48133–48142 CrossRef CAS PubMed.
- Z. Zhang, X. Kang, X. Zhao, X. Dai, X. Su, B. Yang, Y. Luo, C. Xiong, H. Chang and X. Li, J. Mater. Chem. C, 2024, 12, 5191–5202 RSC.
- J. H. Kim, S. V. Lindeman and J. K. Kochi, J. Am. Chem. Soc., 2001, 123, 4951–4959 CrossRef CAS PubMed.
- M. Li, A. D. Schlüter and J. Sakamoto, J. Am. Chem. Soc., 2012, 134, 11721–11725 CrossRef CAS PubMed.
- H. Wang, L. Fang, Z. Zhang, J. Epaarachchi, L. Li, X. Hu, C. Lu and Z. Xu, Composites, Part A, 2019, 125, 105525 CrossRef CAS.
- L. Fang, T. Fang, X. Liu, Y. Ni, C. Lu and Z. Xu, Compos. Sci. Technol., 2017, 152, 190–197 CrossRef CAS.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- T. M. Khang, P. Q. Nhien, T. T. K. Cuc, C.-C. Weng, C.-H. Wu, J. I. Wu, B. T. B. Hue, Y.-K. Li and H.-C. Lin, Small, 2023, 19, 2205597 CrossRef CAS PubMed.
- S. Lin, K. G. Gutierrez-Cuevas, X. Zhang, J. Guo and Q. Li, Adv. Funct. Mater., 2021, 31, 2007957 CrossRef CAS.
- H.-B. Cheng, H.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2013, 135, 10190–10193 CrossRef CAS PubMed.
- J. Lai, Y. Zhang, N. Pasquale and K.-B. Lee, Angew. Chem., Int. Ed., 2014, 53, 14419–14423 CrossRef CAS PubMed.
- S.-L. Huang, T. A. Hor and G.-X. Jin, Coord. Chem. Rev., 2017, 346, 112–122 CrossRef CAS.
- X.-D. Huang, Y. Xu, K. Fan, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Angew. Chem., Int. Ed., 2018, 57, 8577–8581 CrossRef CAS PubMed.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- M. Behl and A. Lendlein, Mater. Today, 2007, 10, 20–28 CrossRef CAS.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed.
- H. Yuk, S. Lin, C. Ma, M. Takaffoli, N. X. Fang and X. Zhao, Nat. Commun., 2017, 8, 14230 CrossRef CAS PubMed.
- C. Ma, X. Le, X. Tang, J. He, P. Xiao, J. Zheng, H. Xiao, W. Lu, J. Zhang, Y. Huang and T. Chen, Adv. Funct. Mater., 2016, 26, 8670–8676 CrossRef CAS.
- C.-H. Zhu, Y. Lu, J. Peng, J.-F. Chen and S.-H. Yu, Adv. Funct. Mater., 2012, 22, 4017–4022 CrossRef CAS.
- S. Wei, W. Lu, X. Le, C. Ma, H. Lin, B. Wu, J. Zhang, P. Theato and T. Chen, Angew. Chem., Int. Ed., 2019, 58, 16243–16251 CrossRef CAS PubMed.
- G. A. Mandl, P. A. Rojas-Gutierrez and J. A. Capobianco, Chem. Commun., 2018, 54, 5847–5850 RSC.
- Z. Li, G. Wang, Y. Wang and H. Li, Angew. Chem., Int. Ed., 2018, 57, 2194–2198 CrossRef CAS PubMed.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- J. Volarić, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 RSC.
- H.-J. Yu, H. Wang, F.-F. Shen, F.-Q. Li, Y.-M. Zhang, X. Xu and Y. Liu, Small, 2022, 18, 2201737 CrossRef CAS PubMed.
- W.-L. Zhou, X.-Y. Dai, W. Lin, Y. Chen and Y. Liu, Chem. Sci., 2023, 14, 6457–6466 RSC.
- Z. Li, G. Wang, Y. Ye, B. Li, H. Li and B. Chen, Angew. Chem., Int. Ed., 2019, 58, 18025–18031 CrossRef CAS PubMed.
- S. Shikha, T. Salafi, J. Cheng and Y. Zhang, Chem. Soc. Rev., 2017, 46, 7054–7093 RSC.
- P. Kumar, S. Singh and B. K. Gupta, Nanoscale, 2016, 8, 14297–14340 RSC.
- F. Li, X. Wang, Z. Xia, C. Pan and Q. Liu, Adv. Funct. Mater., 2017, 27, 1700051 CrossRef.
- X. Ji, R.-T. Wu, L. Long, X.-S. Ke, C. Guo, Y.-J. Ghang, V. M. Lynch, F. Huang and J. L. Sessler, Adv. Mater., 2018, 30, 1705480 CrossRef PubMed.
- X. Chen, Q. Jin, L. Wu, C. Tung and X. Tang, Angew. Chem., Int. Ed., 2014, 53, 12542–12547 CrossRef CAS PubMed.
- Z. Li, H. Chen, B. Li, Y. Xie, X. Gong, X. Liu, H. Li and Y. Zhao, Adv. Sci., 2019, 6, 1901529 CrossRef CAS PubMed.
- Z. Li, X. Liu, G. Wang, B. Li, H. Chen, H. Li and Y. Zhao, Nat. Commun., 2021, 12, 1363 CrossRef CAS PubMed.
- X. Liu, B. Li, W. Wang, Y. Zhang, H. Li and Z. Li, Chem. Eng. J., 2022, 449, 137718 CrossRef CAS.
- Y. Yang, Y. Li, Y. Chen, Z. Wang, Z. He, J. He and H. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 21330–21339 CrossRef CAS PubMed.
- H.-B. Cheng, G.-F. Hu, Z.-H. Zhang, L. Gao, X. Gao and H.-C. Wu, Inorg. Chem., 2016, 55, 7962–7968 CrossRef CAS PubMed.
- W. Zhao, Y. Zhao, Q. Wang, T. Liu, J. Sun and R. Zhang, Small, 2019, 15, 1903060 CrossRef CAS PubMed.
- T. Zhao, L. Chen, Q. Li and X. Li, J. Mater. Chem. B, 2018, 6, 7112–7121 RSC.
- Y.-P. Gu, R. Cui, Z.-L. Zhang, Z.-X. Xie and D.-W. Pang, J. Am. Chem. Soc., 2012, 134, 79–82 CrossRef CAS PubMed.
- J. Yan, B. Li, P. Yang, J. Lin and Y. Dai, Adv. Funct. Mater., 2021, 31, 2104325 CrossRef CAS.
- L. Zhao, J. Peng, Q. Huang, C. Li, M. Chen, Y. Sun, Q. Lin, L. Zhu and F. Li, Adv. Funct. Mater., 2014, 24, 363–371 CrossRef CAS.
- T. Zhao, P. Wang, Q. Li, A. A. Al-Khalaf, W. N. Hozzein, F. Zhang, X. Li and D. Zhao, Angew. Chem., Int. Ed., 2018, 57, 2611–2615 CrossRef CAS PubMed.
- C. Liu, Y. Zhang, M. Liu, Z. Chen, Y. Lin, W. Li, F. Cao, Z. Liu, J. Ren and X. Qu, Biomaterials, 2017, 139, 151–162 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |