
Recent advances in upgrading CO2 to C3+ products via electrochemical and complementary engineering†
Xian
Zhong
ab,
Hong-Jie
Peng
a,
Chuan
Xia
 *bd and
Xinyan
Liu
*ac
*bd and
Xinyan
Liu
*ac
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731, P. R. China. E-mail: xinyanl@uestc.edu.cn
bSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 611731, P. R. China. E-mail: chuan.xia@uestc.edu.cn
cKey Laboratory of Quantum Physics and Photonic Quantum Information, Ministry of Education, University of Electronic Science and Technology of China, Chengdu 611731, P. R. China
dYangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou 313001, P. R. China
First published on 24th June 2024
Abstract
Upgrading CO2 to various carbon-containing products through renewable electrochemical routes offers a promising solution to achieve a “Net Zero” and circular economy. Multicarbon C3+ products are especially energy-rich and economically valuable. However, due to the diverse possibilities of C–C coupling and the complexities of reaction pathways, the efficient and selective electrochemical reduction of CO2 to C3+ products remains a tremendous challenge. Summarizing the latest advances in generating C3+ products from CO2, this review focuses on both key material development and process design in electrochemical and complementary engineering approaches. For the methodologies involving only electrochemical reactions, we categorize them based on the catalysts adopted, summarizing the specific design strategies and mechanistic understandings of copper and non-copper catalysts, respectively. To further improve the efficiency of C3+ synthesis, the concept of “electrochemical + X” is introduced. “X” herein refers to a complementary sector to direct CO2 electrolysis, encompassing the homogeneous non-electrocatalytic reactions in a one-pot electrochemical process and the sequential thermochemical or biological processes after electrochemical CO2 conversion. Lastly, we discuss the challenges of pure electrochemical as well as “electrochemical + X” approaches and outline promising future directions. We believe that this review contains a comprehensive summary of the means to optimize for C3+ compounds, and can motivate researchers to develop innovative strategies to further enhance C3+ production efficiency, paving the way towards the ultimate renewable-driven chemical industries.
 Xian Zhong | Xian Zhong graduated from Chang'an University with a bachelor’s degree in 2021. Now she is pursuing a master’s degree under the joint supervision of Dr Xinyan Liu and Prof. Chuan Xia at the University of Electronic Science and Technology of China (UESTC). Her research interests mainly focus on first-principles calculations for applications such as electrocatalytic CO2 reduction, water splitting, and high-energy secondary batteries. |
 Hong-Jie Peng | Dr Hong-Jie Peng is a Professor at the Institute of Fundamental and Frontier Sciences (IFFS), University of Electronic Science and Technology of China (UESTC). He obtained B.S. and PhD degrees at the Department of Chemical Engineering, Tsinghua University, in 2013 and 2018, respectively. He was a postdoctoral fellow at SUNCAT Center, Stanford University, during 2018–2020. He was selected as a Highly Cited Researcher from 2019 to 2023 by Clarivate Analytics. His current research interests are advanced energy chemistry and energy materials, including high-energy rechargeable batteries, renewable electrocatalysis, theoretical tools, and data-driven methods. |
 Chuan Xia | Dr Chuan Xia is a Professor of Materials and Energy at the University of Electronic Science and Technology of China (UESTC). His group focuses on developing novel catalysts and device architectures that can be applied in electrocatalysis. He has won the J. Evans Attwell-Welch postdoctoral fellowship (2019), Best Applied Paper Award of AIChE (2020), and Falling Walls Science Breakthroughs of the Year Award (2022), Top 10 News Stories of Scientific and Technological Progress in China (2022), Sichuan Youth May Fourth Medal (2023), and Chinese Chemical Society Young Chemist Award (2023). More information about his research can be found here: https://www.chuan-lab.com. |
 Xinyan Liu | Dr Xinyan Liu is a Research Professor at the Institute of Fundamental and Frontier Sciences (IFFS), University of Electronic Science and Technology of China (UESTC). Having obtained her bachelor’s and PhD degree from Tsinghua University and Stanford University in 2013 and 2018 respectively, she has also worked at Meta Inc. for two years as a Research Data Scientist before she joined full time at UESTC. Dr Liu's research interest has been mainly focused on energy chemistry-related interdisciplinary study combining theoretical simulations and artificial intelligence, such as electrocatalysis, catalyst high-throughput screening, and battery prognosis. |
1. Introduction
The increase in carbon dioxide (CO2) emissions inevitably induces global warming and detrimental threats to the environment.1,2 Based on this aspect, upgrading CO2 to value-added chemical feedstocks through renewable approaches remains extremely valuable, as it presents a sustainable approach to alleviate environmental stress and complete the anthropogenic carbon cycle.3 Various technologies of CO2 reutilization have been developed in recent years, including chemical,4 thermochemical,5,6 biochemical,7,8 photochemical,9–11 and electrochemical methods.12–15 Among these technologies, the electrochemical CO2 reduction reaction (eCO2RR) powered by electricity from sustainable sources emerges as a promising candidate, due to its mild reaction conditions (e.g. room temperature and ambient pressure) and the substantially lower cost of renewable electricity. As the eCO2RR consumes renewable energies while producing multiple base chemicals,16 it is anticipated to supplement or even replace the current fossil-resource-based chemical manufacture, manifesting great potential to achieve “Net Zero” industrial CO2 emissions.Remarkable progress has been reported for the eCO2RR to C1 and C2 hydrocarbons and oxygenates (e.g., carbon monoxide (CO),17–19 methanol (CH3OH),20–22 ethanol (CH3CH2OH)23–25 and ethylene (C2H4)26–28), where outstanding reaction activities and selectivities can be achieved. Compared to their C1 or C2 counterparts, C3+ products usually exhibit higher energy densities and are more economically valuable. For instance, 1-propanol (CH3CH2CH2OH) possesses high volumetric energy density (27.0 kJ mL−1) and research octane number (118). The most typical C4 monohydric alcohol, 1-butanol (CH3CH2CH2CH2OH), exhibits even higher volumetric energy density (29.2 kJ mL−1).29 Both C3+ alcohols are promising substitutes for petroleum-derived gasoline. Along with other C3 and C4 products such as allyl alcohol, acetone, and propionaldehyde,30 they serve as important raw chemicals for fine chemical industries. Long-chain aliphatic and aromatic hydrocarbons, though challenging to produce from CO2, are desirable synthetic fuels due to their high heat value and lower volatility compared to light hydrocarbons/oxygenates, and remain more compatible with the existing fuel storage and transportation infrastructures.29,31,32 If the scope of C3+ products is further expanded to nutrients such as saccharides, amino acids, and lipids, electrochemical CO2 upgrading will further provide a sustainable avenue towards synthetic foods to cope with the growth of the global population.33,34 However, electrochemically converting CO2 to C3+ products with practically relevant efficiency still remains a formidable challenge owing to the complexity in reaction pathways and the insufficiency in key materials and new process design.
In this work, we focus on recent advances in upgrading CO2 to C3+ products through direct electrochemical and “electrochemical + X” approaches, where “X” refers to complementary engineering with homogeneous reactions, thermochemical conversions, and biological processes (Fig. 1). We begin with a brief introduction on the principles of the eCO2RR, followed by C3+ formation mechanisms with a primary focus on the most widely adopted copper (Cu)-based catalysts. Then, we summarize the design and optimization principles of both Cu-based and non-Cu catalysts to enhance their catalytic activities towards C3+ chemicals in direct electrochemical CO2 conversion. The latest research progress in coupling the eCO2RR with other complementary engineering approaches, as well as the significance of such a combinatorial “electrochemical + X” strategy in achieving high C3+ production efficiency, is then presented. Lastly, we conclude this work by providing a perspective on challenges and opportunities for both approaches, hoping to shed light on future research studies.
 | ||
| Fig. 1 Schematic illustration of direct electrochemical approaches (eCO2RR and eCORR) and “electrochemical + X” approaches for upgrading CO2 to C3+ products. | ||
2. Direct electrochemical approaches
2.1. Principles of the eCO2RR
For CO2 upgrading to C3 and C4, it is viable to adopt direct electrochemical approaches through the eCO2RR in an electrolysis system (Fig. 1). The eCO2RR at the cathode is paired with oxidative reactions, normally the oxygen evolution reaction, at the anode. The electrolysis system can be designed in a one-pot or sequential fashion. The latter one in most cases consists of two electrolyzers in series;35 where CO2 reduces to CO in the first and the subsequent electrochemical CO reduction reaction (eCORR) take place in the second. The advantages of sequential design primarily lie in possibilities to optimize catalytic materials and reaction environments separately to maximize the efficiency of CO2-to-CO conversion and the eCORR. For instance, the bulk and local electrolyte pH have been experimentally observed and theoretically validated to largely influence the product selectivities of the eCO2RR/eCORR. Alkaline conditions have been demonstrated to promote the eCORR towards C2+ multicarbon products with exceptional selectivity.36–42 Nevertheless, direct eCO2RR in alkaline solutions suffers from severe reactions between CO2 and hydroxide, as well as the resultant decrease in local pH and decrease in carbon efficiency. The sequential electrolysis system design allows for addressing such a dilemma.For the catalysts at the cathode side, metallic Cu and Cu-based materials have been intensively investigated for the eCO2RR/eCORR in a direct electrocatalytic process and remain as the most well-known catalysts to drive the formation of C2+ products.16,43 Despite the great progress in improving the activity and selectivity of the eCO2RR/eCORR towards C2 products, strategies for enhancing carbon-chain growth to C3+ products remain limited. In an early study, Hori et al. reported n-PrOH as an eCO2RR product on Cu with a faradaic efficiency (FE) of 4.2% at a potential of −1.4 V vs. the normal hydrogen electrode (NHE) in KClO4,44 which establishes a C3 production baseline on Cu. As CO* (* refers to a surface adsorption site) has been probed as a key surface intermediate for C–C coupling,45–47 challenges of efficient C3+ production include both insufficient surface coverage of CO*48,49 and the large number of competing reaction pathways against C3+ formation.50 For instance, the inadequate stabilization of 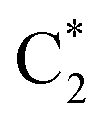 intermediates on pristine Cu surfaces leads to species desorption rather than further intermolecular coupling with CO*. In addition, the coupling of certain intermediates always suffers from competitions with corresponding hydrogenation/protonation reactions under reductive conditions. Therefore, it remains essential to acquire a deeper understanding of the reaction mechanisms and develop catalysts capable of stabilizing key
intermediates on pristine Cu surfaces leads to species desorption rather than further intermolecular coupling with CO*. In addition, the coupling of certain intermediates always suffers from competitions with corresponding hydrogenation/protonation reactions under reductive conditions. Therefore, it remains essential to acquire a deeper understanding of the reaction mechanisms and develop catalysts capable of stabilizing key  intermediates and favoring the specific coupling pathway towards C3+.
intermediates and favoring the specific coupling pathway towards C3+.
2.2. C3 formation mechanisms through the eCO2RR
As Cu and oxide-derived Cu (OD-Cu) catalysts have been investigated the most for the eCO2RR to C2+ products, the majority of mechanistic studies leveraging theoretical computations or spectroscopic characterization also focus on these catalysts. Compared to C1 and C2 products, the C3 formation mechanism remains more elusive due to the complexity in reaction pathways and the large number of possible steps (>103) involved in the entire reaction network. Enlightened by the widely recognized CO* dimerization mechanism to yield OCCO* as a common precursor for C2 formation in the eCO2RR, some density functional theory (DFT)-based attempts have been used to investigate the sequential (first, 2CO* → OCCO* + *, and then, OCCO* + CO* → OCCOCO* + *) or concerted (3CO* → OCCOCO* + 2*) CO* trimerization mechanism,51–53 where the OCCOCO* trimer was proposed as the initial C3 backbone for further reduction. However, recent studies by Abild-Pedersen et al.54 and López et al.55 both argued that the high activation barrier (ΔGa of at least 1 eV at −0.9 V vs. the reversible hydrogen electrode (RHE, pH 7), relative to 3CO*) for this process on Cu(100) or OD-Cu makes it less favorable than other C2–C1 coupling steps, and that the direct trimerization is kinetically inaccessible as the protonation of the negatively charged OCCO* dimer dominates over the OCCO* + CO* coupling under reducing conditions. To validate the direct trimerization mechanism, spectroscopic evidence for the OCCOCO* trimer or its mono-hydrogenated derivatives can be valuable and is still currently lacking.Alternatively, statistical analyses of experiments and DFT computations were combined to balance the efficiency and accuracy of mechanism exploration, unveiling new mechanistic insights. Abild-Pedersen et al.54 first correlated the C2 and C3 formation rates based on the pioneering work of the eCO2RR on Cu single-crystal substrates by Hori et al.,56 revealing the higher possibilities of late C2 intermediates (e.g. hydrocarbons C2Hx, where x = 1–3, or mono-oxygenates such as acetaldehyde CH3CHO) to participate in C3 formation than late C1 intermediates (e.g. CHx). In addition, the correlations of 1-propanol with C2H4 or ethanol formation exhibit great differences regarding the correlation coefficient as well as the Cu facets, implying the existence of at least two possible parallel reaction pathways towards C3 (Fig. 2a). Through the analysis of explicitly calculated barriers for all coupling steps between CO* and the likely C2Hx intermediates, they identified the coupling of HCCH* and CO* (HCCH* + CO* → CHCHCO* + *) as the most kinetically feasible step to compete with the corresponding hydrogenation on both flat Cu(100) and stepped Cu(511) facets featuring four-fold square sites. Specifically, facets combining (100)-like square sites and step sites, such as Cu(511), were found to exhibit notably lower HCCH*–CO* coupling barriers (ΔGa = 0.51 eV at −0.9 V vs. RHE (pH 7)) than flat Cu(100) (ΔGa = 0.80 eV at −0.9 V vs. RHE). This finding well explains the experimentally observed higher C3 selectivity on stepped Cu(100) facets.56 In addition, they further elucidated the feasibility of CO*–CH3CHO* coupling  under alkaline conditions. Thanks to the substantial stabilization of an interfacial electric field on its transition state, this step exhibits a low barrier of ΔGa = 0.61 eV at −0.6 V vs. RHE (pH 13) on Cu(100), which opens up a supplementary pathway towards C3 on non-stepped Cu surfaces. As the (100)-like square sites are identified to be crucial for C–C coupling, Tsai et al. investigated the strain effect on Cu(100) model surfaces and found that biaxial strains, namely compression along one axis and elongation along the other, significantly lowered C2–CO coupling barriers although two different C2 intermediates of CCH* and CCOH* were considered.57 The symmetry distortion from standard square sites to a rectangle or parallelogram site was shown to enable two scales for stabilizing the
under alkaline conditions. Thanks to the substantial stabilization of an interfacial electric field on its transition state, this step exhibits a low barrier of ΔGa = 0.61 eV at −0.6 V vs. RHE (pH 13) on Cu(100), which opens up a supplementary pathway towards C3 on non-stepped Cu surfaces. As the (100)-like square sites are identified to be crucial for C–C coupling, Tsai et al. investigated the strain effect on Cu(100) model surfaces and found that biaxial strains, namely compression along one axis and elongation along the other, significantly lowered C2–CO coupling barriers although two different C2 intermediates of CCH* and CCOH* were considered.57 The symmetry distortion from standard square sites to a rectangle or parallelogram site was shown to enable two scales for stabilizing the  and CO* adsorbates, respectively.
and CO* adsorbates, respectively.
 | ||
| Fig. 2 C3 formation mechanisms through the eCO2RR on Cu and OD-Cu. (a) CO coupling mechanisms with late C2 species such as HCCH* and CH3CHO* to yield C3 species (red), as well as the corresponding competing protonation steps of HCCH* (blue) and CH3CHO* (green) that result in C2 species such as ethylene and ethanol. DFT-calculated barrier ΔGa is presented at two typical potentials vs. RHE (URHE). The presented data were reproduced from ref. 54 for the Cu(100) surface. (b) Typical CxHyOz backbones, and reaction and activation electronic energies (ΔE and Ea) for CHxCHy–CHzO coupling steps on an OD-Cu surface model. The most likely steps are indicated in bold. Adapted from ref. 55 with permission from The Royal Society of Chemistry. | ||
Since OD-Cu has been experimentally probed as an excellent catalyst promoting C3 formation, theoretical efforts have been devoted to this specific system as well.55,58 By devising a divide-and-conquer strategy that combines reaction network graphs, DFT calculations equipped with an implicit solvation model and a voltage polarization correction, and model co-reduction experiments, López et al. thoroughly examined the reaction thermodynamics and kinetics of 586 C1–C2 coupling steps and identified the coupling between C2 hydrocarbons (e.g. CH2CH*) and C1 mono-oxygenates (e.g. CO* and CHO*) as the most likely step (Fig. 2b).55 Although the proposed intermediates to derive C3 backbones are slightly different from those identified by Abild-Pedersen et al.,54 this distinction could be attributed to the OD-Cu model considered for DFT calculations, where the Cu sites are polarized and thus different from the metallic sites in pure Cu models. Nevertheless, both studies identified a late  hydrocarbon intermediate as the key precursor for C3 products in the eCO2RR. While the previous work focused only on the C3 backbone,54 López et al. further explained the absence of propylene (CH3CH
hydrocarbon intermediate as the key precursor for C3 products in the eCO2RR. While the previous work focused only on the C3 backbone,54 López et al. further explained the absence of propylene (CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) in eCO2RR products on routine Cu-based catalysts, which was ascribed to the inaccessible allyl alkoxy (CH2CHCH2O*) intermediate as a kinetic trap.
CH2) in eCO2RR products on routine Cu-based catalysts, which was ascribed to the inaccessible allyl alkoxy (CH2CHCH2O*) intermediate as a kinetic trap.
In addition to DFT calculations, in situ spectroscopic observations provide experimental evidence to elucidate the C3 formation mechanism. Employing isotopic labeling and in situ surface enhanced infrared absorption spectroscopy, Xu et al. investigated alkaline eCORR behaviors on OD-Cu with an addition of CH3CHO in the electrolyte, revealing that CO attacks the carbonyl carbon of CH3CHO during the coupling and the carbon from CO ends up in the hydroxymethyl (–CH2OH) group of the 1-propanol product.59 Interestingly, only a small fraction of C3 was observed to come from CH3CHO + CO coupling, while the majority remained as the result of self-coupling of CO. This suggested that there is an intermediate that forms before CH3CHO, and can be derived from both CH3CHO and CO. This intermediate was then proposed to be methylcarbonyl (CH3CO*), and the possibility of it acting as a precursor for C3 formation is also supported by López et al.'s calculation results, as the coupling barrier between CH3CO* and CHO* could be as low as 0.32 eV.55 So far, the in situ spectroscopic evidence is mostly obtained for the coupling step involving a C2 oxygenate intermediate. The in situ characterization of the C2 hydrocarbon-dominated cross-coupling mechanism, however, still remains limited.
The aforementioned mechanistic studies shed light on the C3 formation mechanism during the eCO2RR or eCORR on Cu-based catalysts, greatly promoting the rational design of catalytic materials and reactors to optimize C3 yields. Yet it is still arguable which pathway is the most dominant and what the most crucial C2 intermediate is. As C2 formation could proceed via multiple pathways, the dominance of which is dependent on the potential, microenvironment, and catalytic surface, the governing C3 formation pathways are anticipated to be dynamic as well. More in-depth theoretical and in situ/operando spectroscopic investigations would be necessary. Moreover, while Cu is focused on in the current mechanistic investigations, other catalysts may exhibit distinctive reaction mechanisms. In the next sections, we will discuss the strategies to further improve Cu-based catalysts and the efforts in seeking for alternative non-Cu catalysts.
2.3. Cu-based catalysts
As Cu-based materials are recognized as the most widely adopted catalysts for direct eCO2RR or eCORR to C3+ chemicals, a variety of strategies have been proposed to regulate the nature and number of Cu active sites. Furthermore, catalyst engineering is often combined with reaction microenvironment modulation, synergistically shifting the concentration and/or coverage of key reaction intermediates (e.g. CO) to the optimal range for selective C3+ formation. Overall, these strategies can be categorized as micro-/nanostructure engineering, defect engineering, oxidation state regulation, and foreign element regulation. In addition, the rare case of selective eCO2RR to C3 by single-site Cu catalysts is also discussed.CHCH2OH)) at a constant current density of 5 mA cm−2 in an electrolyte of 0.1 M KHCO3.56 The single-crystal Cu electrode follows the microfacet notation proposed by Somorjai et al.,61 and Cu(S)-[n(100) × (111)] refers to the Cu(100) facet modified by close-packed (111) steps. The fact that the best C3 FE of 12% can be achieved on Cu(711) with a moderate density of step sites points out the significance of both (100)-like terrace and (111) step sites. While the former type of site is essential for selective eCO2RR to C2 hydrocarbon intermediates, the coupling of these C2 hydrocarbon fragments with CO was theoretically predicted to be more facile on under-coordinated sites.54 The adoption of single-crystal substrates is, however, not practical for industrial-scale catalytic processes, due to the limited scalability of single-crystal material fabrication. Although the possibility of scalable synthesis of single-crystal metal foil has been recently showcased,62,63 a great number of pieces of single-crystal Cu foil exhibited strong tendency to reconstruct under routine eCO2RR conditions.64 Therefore, alternative approaches to tailor active site types and distributions on Cu catalysts are required.
Since the general understanding on nanocatalysts has revealed that a decrease in nanoparticle size gives rise to an increase in step site densities,65 it becomes straightforward to adopt Cu nanoparticles (NPs) and other nanostructures to enrich under-coordinated sites in the eCO2RR. Moreover, coating nano-particulate Cu catalysts onto the gas diffusion layer (GDL) or integrating them into a membrane electrode assembly (MEA) is also rather convenient, endowing these approaches with desirable applicability in practical electrolyzers. For instance, Cuellar et al. investigated the particle size effect of four commercial Cu powders loaded on the GDL and demonstrated the advantage of CO as a reactant over CO2 in improving the selectivity towards both C2 and C3.35 Specifically, the partial current density for C3 formation through the eCORR was roughly 4 times higher than that of the eCO2RR. The highest C3 selectivity (FE of ∼28%) was achieved using Cu NPs of a size < 100 nm for the eCORR, which presents around a 6-fold increase compared to Cu powders of 5 μm. Further experiments in a flow cell for the eCORR at a constant current density of 300 mA cm−2 still present a high 1-propanol FE of 18%. These results demonstrate the new possibility to enhance C3 selectivity through the combination of Cu NPs and the eCORR.
Despite their great catalytic potential, CuNPs may suffer from inevitable structural reconstruction, which can alter the initial morphology and active sites. Either precise control over catalyst reconstruction or suppression of such reconstruction through rational nanostructure design is therefore highly desirable. For instance, the Yang group has proposed an ensemble catalyst derived from well-assembled Cu NPs for selective eCO2RR to C2–C3 products at low overpotentials66 (Fig. 3a) and unraveled a dynamic “electrochemical scrambling” mechanism that drove the reconstruction of Cu NPs to disordered Cu nanocrystals through systematic ex situ, passivated ex situ and in situ characterization.67 The electrochemically scrambled Cu nanocrystal was probed as the critical component leading to enhanced selectivity towards C2–C3 products. Note that although this type of catalyst facilitated overall C–C coupling, the enhancement in C3 selectivity was not as profound as that of C2. More precise manipulation of the reconstruction process therefore remains valuable to further optimize the yields of higher-carbon products beyond those of C2. Alternatively, the reconstruction can be suppressed by the design of robust nanostructured Cu catalysts. For instance, Zhao et al. developed a facile surfactant-free synthesis method to obtain Cu2O nanocrystals with various architectures, which can be further transformed into Cu catalysts under eCORR conditions without much change in the morphology (Fig. 3b).68 Among all the directed architectures, the catalyst with a morphology of a branching cubic framework (BCF-Cu2O) exhibited fivefold higher partial current density of 1-propanol at −0.45 V vs. RHE than the surfactant-coated nanocube catalyst. According to DFT calculations, the clean surface and the exposure of Cu(100) and Cu(110) were found to facilitate 1-propanol production, which corroborated with the high catalytic activity of BCF-Cu2O exhibiting the highest exposure of Cu(100) and Cu(110) facets. In addition to the sophisticated engineering of synthesis procedures, the adoption of a support to anchor the nanoparticles presents another possibility to stabilize the Cu nanostructure. According to Lu et al., dispersing Cu/Cu2O nanoparticles on nitrogen-doped graphene enabled nearly a one-fold increase in the 1-propanol FE during the eCO2RR.69 In brief, all above studies highlight the importance of morphology control for Cu catalysts in enhancing C3 formation.
 | ||
| Fig. 3 Micro-/nanostructure engineering of Cu-based catalysts. (a) Schematic illustration of the transformation process of Cu NP ensembles into an active catalyst for C3 product formation. Adapted from ref. 66 with permission from National Academy of Sciences. (b) Partial current density of 1-propanol through the eCORR on a variety of catalysts derived from surfactant-free Cu2O nanocrystals at −0.45 V vs. RHE. Adapted from ref. 68 with permission from American Chemical Society. (c) Schematic illustration of Cu2O HoMSs with different numbers of layers. Adapted from ref. 70 with permission from Wiley-VCH GmbH. | ||
In addition to regulating the characteristics and densities of active sites, micro-/nanostructure engineering on Cu catalysts can also tune the concentration and coverage of key reaction intermediates such as C2 and CO. For instance, studies on porous or hollow-structured catalysts have demonstrated the effectiveness of exploiting the confinement effect to steer the selectivity from C1 to C2+.71,72 This idea can be further leveraged to increase C3 yields. Sargent et al. developed a methodology to synthesize open Cu nanocavities and adjust their geometry by varying the acid-etching time of Cu2O pre-catalysts.73 The finite-element method (FEM) was adopted to verify the restricted diffusion of locally produced C2 species in the cavity, a higher concentration of which was found to be responsible for increasing its probability to couple with CO, leading to a higher C3 production rate. With an optimized Cu nanocavity, the FE of 1-propanol from the eCORR can reach a maximum of 21 ± 1% at a conversion rate of 7.8 ± 0.5 mA cm−2. Alternatively, Zeng et al. leveraged a different synthetic method based on Ostwald ripening and fabricated a series of Cu2O hollow multi-shell structures (HoMSs) with tunable shell numbers (Fig. 3c). They demonstrated that the 3-shell HoMSs with a stronger nanoconfinement effect exhibited a sharp increase of 1-propanol FE from negligible (1-shell HoMSs) and <2% (2-shell HoMSs) to >15% (at −0.65 V vs. RHE).70 Similar effects of multi-shell structures on promoting C3 formation were also demonstrated with the eCORR.74 These studies showcase rational micro-/nanostructuring as a promising physical route, in addition to the conventional active site regulation approach, to steer the product selectivity towards C3 products during the eCO2RR and eCORR.
 | ||
| Fig. 4 Defect engineering of Cu-based catalysts. (a) Schematic illustration of HF-Cu with enriched (100)/(111) grain boundaries. (b) The Cu(111):(100) interface per area, measured from the high-resolution transmission electron microscopy images, plotted against the 1-propanol selectivity performance. Adapted from ref. 51 with permission from Springer Nature. (c) Simulated initial models and final configurations after oxygen removal (from CuO to CuOD-Cu and from Cu2O to Cu2OD-Cu) using neural network potential-based molecular dynamics simulation. (d) Coordination number dependent surface Cu density on CuOD-Cu and Cu2OD-Cu. (e) FE of products on CuOD-Cu (left) and Cu2OD-Cu (right) during the eCO2RR in a H-cell. Adapted from ref. 83 with permission from American Association for the Advancement of Science. (f) Schematic illustration of the 1-propanol formation mechanism on adjacent CuSx-DSV. Adapted from ref. 75 with permission from Springer Nature. | ||
Besides grain boundaries, other types of defects in OD-Cu or other Cu catalysts derived from compound pre-catalysts can also efficiently promote C3 formation. Shown as under-coordinated protuberances on ordinary crystal planes, Cu adparticles were proved by DFT calculations to be excellent site motifs to enhance the chemisorption of CO as well as its coupling with C2 surface species such as OCCOH* and  76 Further manipulating the reconstruction of an OD-Cu catalyst in a CO-enriched environment allowed for the creation of an adparticle structure, which achieved an exceptional FE of 23% for 1-propanol, surpassing that of OD-Cu only rich in grain boundaries with no adparticles. The density of under-coordinated sites can be regulated by not only a gas atmosphere but also OD-Cu precursors. For instance, Gong et al. systematically compared different precursors (i.e. Cu2O, CuO, and Cu(OH)2) of OD-Cu and found that OD-Cu from Cu(OH)2 possessed a relatively high density of stepped Cu(110) and Cu(100), which allowed for higher CO coverage and more facile CO–CO coupling, respectively. The highest FE of ∼11% for 1-propanol (at −0.98 V vs. RHE) can be consequently achieved among the three types of OD-Cu.77 Leveraging neural network potential based molecular dynamics and in situ X-ray absorption spectroscopy, Tang et al. also demonstrated that OD-Cu from CuO possessed an intrinsically higher population of undercoordinated Cu sites than the counterpart from Cu2O owing to the vigorous oxygen-removal-induced structural collapse during CuO electroreduction (Fig. 4c and d).83 The high-density under-coordinated defect sites were suggested to increase the population of CO* and *HOCCOH intermediates by both in situ spectroscopy and computational simulations, leading to a promising FE of 17.9% for 1-propanol at −0.94 V vs. RHE (Fig. 4e). The reconstruction process can be further regulated by decorating CuO with Au NPs, which aided the formation of more disordered Cu structures and resulted in a record high 1-propanol FE of 46.6% obtained in a flow cell.78
76 Further manipulating the reconstruction of an OD-Cu catalyst in a CO-enriched environment allowed for the creation of an adparticle structure, which achieved an exceptional FE of 23% for 1-propanol, surpassing that of OD-Cu only rich in grain boundaries with no adparticles. The density of under-coordinated sites can be regulated by not only a gas atmosphere but also OD-Cu precursors. For instance, Gong et al. systematically compared different precursors (i.e. Cu2O, CuO, and Cu(OH)2) of OD-Cu and found that OD-Cu from Cu(OH)2 possessed a relatively high density of stepped Cu(110) and Cu(100), which allowed for higher CO coverage and more facile CO–CO coupling, respectively. The highest FE of ∼11% for 1-propanol (at −0.98 V vs. RHE) can be consequently achieved among the three types of OD-Cu.77 Leveraging neural network potential based molecular dynamics and in situ X-ray absorption spectroscopy, Tang et al. also demonstrated that OD-Cu from CuO possessed an intrinsically higher population of undercoordinated Cu sites than the counterpart from Cu2O owing to the vigorous oxygen-removal-induced structural collapse during CuO electroreduction (Fig. 4c and d).83 The high-density under-coordinated defect sites were suggested to increase the population of CO* and *HOCCOH intermediates by both in situ spectroscopy and computational simulations, leading to a promising FE of 17.9% for 1-propanol at −0.94 V vs. RHE (Fig. 4e). The reconstruction process can be further regulated by decorating CuO with Au NPs, which aided the formation of more disordered Cu structures and resulted in a record high 1-propanol FE of 46.6% obtained in a flow cell.78
In contrast to the “protuberant” under-coordinated defect sites out of a standard crystal facet, vacancies can be deemed as a “dented” counterpart by removing either cationic or anionic atoms and leaving holes on the surfaces. Sargent et al. reported that the modification of the Cu2S core with Cu surface vacancies led to an impressive FE of 8 ± 0.7% for 1-propanol production, but it was also notable that the Cu vacancies were not only selective to 1-propanol but also ethanol.79 An alternative strategy to selectively enhance 1-propanol production over other alcohols is to precisely generate double sulfur vacancies (DSVs) on CuS using a controllable lithium electrochemical tuning method.75 As demonstrated by Zheng et al., DSV-rich CuSx catalysts, obtained after 10 charge/discharge cycles in a lithium electrochemical cell, exhibited the highest FE of 15.4% for 1-propanol; whereas neither decreasing nor increasing the cycle numbers gave rise to better yields of 1-propanol, highlighting the pivotal role of DSV that possessed an optimized Cu–Cu distance in facilitating CO trimerization (Fig. 4f). Similar to the observations on grain boundaries, defect sites such as vacancies, albeit enabling an increase in C3 selectivity in some cases, require delicate structural control to realize the C3-specific eCO2RR. The structure–property relationships are so far case-specific, and somewhat lack generality. The synthetic methods to manipulate the types and population of, if C3-specific, certain active defect sites also diverge.
Manipulating the phase segregation of initial alloys could be a useful strategy to isolate CO formation and C–C coupling on different domains. For instance, Broekmann et al. prepared a binary CuPd alloy with a nominal composition of Cu91Pd9 and further adopted a sequential treatment of air annealing and in situ reduction to induce phase segregation, which resulted in Pd-rich and Cu-rich domains, respectively (Fig. 5a).101 While the Pd-rich domains promoted CO formation during the eCO2RR, CO was then transported to the Cu-rich ones for subsequent C–C coupling. The increased CO availability induced by Pd-rich domains led to higher propensity of C–C coupling. Interestingly, the seminal work by Kenis et al. unraveled the key role of phase segregation in generating more C2 than C1 chemicals during the eCO2RR.97 The distinctive product distributions of phase-separated CuPd alloy systems in the above two studies can be rationalized through the different ratios of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu. While the equal-molar system (Cu:Pd = 1:1) primarily facilitates C2 products such as ethylene (FE > 40% at −0.65 V vs. RHE) and ethanol (FE > 14% at −0.65 V vs. RHE), as shown by Kenis et al.,97 the work by Broekmann et al.101 showed that decreasing Pd content favored C3 (FE = 13.7 ± 0.8% at −0.65 V vs. RHE) over C2 products (e.g. FEethanol = 7.1 ± 0.3% at −0.65 V vs. RHE).A sSimilar effect by dilute alloying was also demonstrated by Ye et al., who introduced a family of facet-defined dilute CuAu alloy nanorods (NRs) with precisely controlled Au atomic content from ca. 1% to 16%.102 All these NR alloy catalysts possessed uniform crystal shapes with preferentially exposed (100) surfaces, among which the NR2 catalyst with a nominal composition of Cu98Au2 exhibited the highest FE of 18.2 ± 0.3% for 1-propanol at −0.41 V vs. RHE (Fig. 5b). Note that NR5 and NR6 with more than 12 at% Au exhibited phase separation, yielding nanocrystalline Au. However, phase separation in this case favored C1 formation rather than C3. Therefore, it is pivotal to precisely control both the mixing patterns and content of the foreign element in the Cu-based alloys for selective eCO2RR towards C3 products.
:Cu. While the equal-molar system (Cu:Pd = 1:1) primarily facilitates C2 products such as ethylene (FE > 40% at −0.65 V vs. RHE) and ethanol (FE > 14% at −0.65 V vs. RHE), as shown by Kenis et al.,97 the work by Broekmann et al.101 showed that decreasing Pd content favored C3 (FE = 13.7 ± 0.8% at −0.65 V vs. RHE) over C2 products (e.g. FEethanol = 7.1 ± 0.3% at −0.65 V vs. RHE).A sSimilar effect by dilute alloying was also demonstrated by Ye et al., who introduced a family of facet-defined dilute CuAu alloy nanorods (NRs) with precisely controlled Au atomic content from ca. 1% to 16%.102 All these NR alloy catalysts possessed uniform crystal shapes with preferentially exposed (100) surfaces, among which the NR2 catalyst with a nominal composition of Cu98Au2 exhibited the highest FE of 18.2 ± 0.3% for 1-propanol at −0.41 V vs. RHE (Fig. 5b). Note that NR5 and NR6 with more than 12 at% Au exhibited phase separation, yielding nanocrystalline Au. However, phase separation in this case favored C1 formation rather than C3. Therefore, it is pivotal to precisely control both the mixing patterns and content of the foreign element in the Cu-based alloys for selective eCO2RR towards C3 products.
 | ||
| Fig. 5 Foreign element regulation of Cu-based catalysts. (a) Phase segregation from the as-prepared CuPd alloy (ap-Pd9Cu91) to the activated sample (od-Pd9Cu91), as revealed by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and STEM energy-dispersive X-ray spectroscopy (EDS) images. Adapted from ref. 101 with permission from The Royal Society of Chemistry. (b) (Upper) schematic illustration of Au/Cu NR synthesis, (bottom left) HAADF-STEM image and STEM-EDS elemental maps of a single NR with 2.1 ± 0.3 at%Au, and (bottom right) FEs of C1, C2, and C3 products vs. the Au contents of NRs. Adapted from ref. 102 with permission from American Chemical Society. (c) DFT-calculated activation energy (Ea) for C1–C1 and C1–C2 coupling on screened Ag–X–Cu systems (X = Au, Pd, Pt, Ni, Fe and Ru). Adapted from ref. 103 with permission from Springer Nature. (d) Schematic illustration of a supersaturated CO2-stabilized CuAg alloy with highly dispersed Ag atoms. Adapted from ref. 104 with permission from Springer Nature. | ||
Compared to the eCO2RR, the eCORR could take advantage of alkaline electrolysis and direct gas feeding. The former enables higher C2 yields than acidic or neutral electrolysis, and the latter leads to better mass transport management. In addition, the absence of a CO2-to-CO pathway in the eCORR allows for better mechanistic understanding of the effect induced by foreign elements on C2/C3 selectivity tuning. Combining Cu-based dilute alloys with the eCORR has been demonstrated to yield significant improvement in C3 selectivity. For example, Sargent et al. theoretically designed an Ag-doped Cu alloy surface with asymmetric C–C coupling active sites, which exhibited the lowest activation barriers for both C1–C1 and C1–C2 coupling to form C3 products among various single metal (i.e. Ag, Au, Ru, Rh, and Pd) doped Cu surfaces.105 Such a design was experimentally realized using a dilute Cu96Ag4 alloy for the eCORR in a flow cell reactor with a GDE, which exhibited a promising FE of 33 ± 1% for 1-propanol at −0.46 V vs. RHE (in 1 M KOH solution). The FE towards 1-propanol can be further enhanced to 37 ± 3% at a high current density of 300 mA cm−2 in a MEA-based reactor, by introducing only 1 at% of Ru to the dilute Ag-in-Cu system.103 The theoretical calculations further revealed that the additional isolated Ru dopant can induce higher coverage of CO* and lower activation barriers for both C1–C1 and C1–C2 coupling (Fig. 5c). Alternatively, the specific CuAg system for alkaline eCORR can be engineered by altering the precursor from pure Cu to Cu compounds. Fontecave et al. investigated a series of catalysts derived from metal-doped Cu nitrides (denoted as CuMx%Nt h with x giving the mol% of the doping metal M and t the time for nitridation), among which CuAg5%N20 h showed an optimized FE of 45% for 1-propanol at 150 mA cm−2.106 The higher C3 selectivity of CuAg5%N20 h compared to CuAg5% was attributed to increased roughness and porosity, while the introduction of Ag, consistent with other reports, was reported to lower C1–C2 coupling barriers.
Interestingly, the product distributions within the dilute bimetallic CuAg systems that deliver the most promising C3 selectivity are still highly tunable by regulating the reaction conditions and local environments. For example, in the above work by Fontecave et al., systematic investigations were performed to unravel the effect of the CO feeding rate and electrolyte properties.106 Key findings include that (1) a moderate CO feeding rate enables a delicate balance between surface-adsorbed CO* and C2 intermediates inclined to coupling, which corroborates well with the competitive co-adsorption study of CO and acetaldehyde by Koper et al.;107 (2) the formation of 1-propanol exhibits an exceptional interfacial electric field effect, which becomes profound on increasing the concentration of cations or changing the cation from K+ to Cs+; (3) a too-high alkaline concentration is detrimental to 1-propanol production due to the reaction pathway bifurcation to acetate,42 which offsets the advantage of the cation-induced field effect at high concentrations. The regulation of reaction environments can even alter the identity of major C3 products. Adopting CO2-supersaturated conditions, Voiry et al. reported the electrosynthesis of a rarely observed C3 product, 2-propanol, from the eCO2RR on a Cu94Ag6 alloy catalyst with a high FE of 56.7% at −0.70 V vs. RHE (in 1 M CsHCO3 solution with ∼3 M CO2).104 The CO2-supersaturated condition was found to provide combinatorial effects by suppressing the phase segregation and modulating CO* adsorption strength/coverage (Fig. 5d). The resultant highly dispersed Ag atoms in Cu weakened the surface–O binding and thus protected the secondary C–O bond from cleavage, leading to favorable formation of 2-propanol over 1-propanol. This mechanism is possibly dominated by the ligand effect of Ag, as evidenced by the charge transfer between Ag and Cu.
In addition to the ligand effect, the doping atom could induce a strain effect through atomic size misfitting. Zhang et al. reported that doping Pb (∼2.9 at%), an extremely heavy element with a large atomic radius, in Cu resulted in numerous atomic Pb-concentrated grain boundaries and induced a large number of under-coordinated Cu sites after geometric structural distortion.108 The FE of CO-to-1-propanol can consequently reach 47 ± 3% at −0.68 V vs. RHE. Note that Pb is classified as a formate-producing rather than CO-producing element in the eCO2RR. This work clearly showcased an approach of regulating Cu with an “inactive” foreign element, distinctive from designing Cu-based alloys with CO-active elements.
Despite the great potential of dilute Cu-based alloys for selective eCO2RR/eCORR to C3, it should be noted that due to the complexity in reaction pathways and sensitivity of key steps to reaction conditions/environments, the operational window for attaining desirable selectivity is rather narrow. Thus, combinatorial investigation is advocated in future research when focusing on complex products like C3.
2.4. Non-Cu catalysts
Cu-based catalysts, though yielding impressive FEs towards C3+ products, still require rather large overpotentials to achieve satisfactory production rates. And the vast possibilities in coupling routes also make the selectivity control of Cu-based catalysts challenging. The suitable operational window for Cu-based materials to selectively catalyze C3 formation largely hinges on the type of reactors, the carbon source (e.g. CO2, CO, or their mixtures), and the electrolyte. One reason for such sensitivity could be attributed to the highly polar nature of key intermediates such as and its protonated forms, the CO dimer, as well as other carbonyl compounds. These polar intermediates are prone to complex interfacial effects from the solvent, ion, and electric field. Thus, the journey to search for non-Cu-based catalysts with lower overpotentials or alternative C–C coupling mechanisms never ceases.
and its protonated forms, the CO dimer, as well as other carbonyl compounds. These polar intermediates are prone to complex interfacial effects from the solvent, ion, and electric field. Thus, the journey to search for non-Cu-based catalysts with lower overpotentials or alternative C–C coupling mechanisms never ceases.
Promising substitutes for Cu include various metal compounds such as molybdenum (Mo)-based and nickel (Ni)-based materials. The original pure metallic Mo and Ni exhibit much stronger CO* adsorption than Cu and present negligible eCO2RR activity due to the severe CO poisoning and undesirable reaction energetics of CO protonation or CO–CO coupling. Their compounds with nonmetal elements, however, have generally weaker binding due to the more filled metal d-states, thereby exhibiting potential catalytic capability of interest. For instance, Lewis et al. observed 1-propanol to be the major eCO2RR product on single-crystal or thin-film MoS2 electrodes, although the maximum FE (∼3.5%) was far below that of the competing hydrogen evolution reaction (HER, FE of ∼50–60%).112 They also revealed the active sites for the eCO2RR to 1-propanol to be MoS2 terraces rather than its edges. Despite the substantial HER activity and self-decomposition, MoS2 indeed demonstrates unique eCO2RR properties compared with Cu. Understanding the origin of such unique eCO2RR activity would be valuable for guiding the design of non-Cu catalysts.
Other than MoS2, Ni-based catalysts present another group of promising candidates to yield higher-order products from the eCO2RR. In the seminal work by Paris and Bocarsly, Ni3Al alloy films on glassy carbon electrodes have been demonstrated to produce 1-propanol (FE = 1.9 ± 0.3%) in addition to a substantial FE (∼30%) of forming CO.113 Their subsequent work further pointed out the necessity of the intermetallic character at the atomic scale to provide distinct yet complementary sites for the eCO2RR to 1-propanol.114 The investigation of the H/D isotope effect also suggested a reaction mechanism different from that on Cu. Combining theory and the experiment, Yeo et al. achieved success in stabilizing the polarized Niδ+ active sites, which exhibited moderate CO binding and generated a variety of mixed long-chain C3–C6 hydrocarbons with a total FE of up to 6.5% in the electrolyte of 0.1 M KHCO3.115 In contrast, metallic Ni remained inactive. In addition, the authors proposed a novel reaction mechanism reminiscent of the Fischer–Tropsch synthesis: COOH + CHx coupling followed by successive CHx insertions, which remains rather distinctive from CO–C2 coupling or even CO trimerization on other catalysts (Fig. 6a). This mechanism was further validated through the invariant C3+ yield with the addition of CO or formaldehyde (CH2O) feedstock.
 | ||
| Fig. 6 Non-Cu catalysts. (a) Initial steps in the C–C coupling towards long-chain hydrocarbons on polarized Ni sites. The insertion mechanism, schematically depicted at the bottom of the figure, is expected to sustain the production of C3+ products. Adapted from ref. 115 with permission from Springer Nature. (b) Standard Gibbs free energy changes of possible C–C bond forming reactions (298 K and pH 7), supporting the formaldehyde condensation mechanism on NixPy catalysts at near-thermoneutral potentials. Adapted from ref. 116 with permission from The Royal Society of Chemistry. | ||
Similarly, new reaction mechanisms have also been proposed on nickel phosphides (NixPy). For instance, Dismukes et al. verified the capability of P-rich NixPy (Ni12P5, Ni2P, Ni5P4, and NiP2) to convert CO2 to C3 (methylglyoxal) and C4 (2,3-furandiol) products at a very low overpotential (<50 mV) with high selectivity (maximum FE = 84% for C3 on NiP2 at −0.1 V vs. RHE; maximum FE = 71% for C4 on Ni2P at 0 V vs. RHE), successfully simulating the catalytic performance of Ni-based enzymes.116 In addition, the authors suggested a reaction mechanism radically different from that of Cu; formation of formate as the oxygenated precursor happens first, followed by the formation of CH2O, and self-condensation of CH2O (Fig. 6b). Rappe et al. further adopted DFT calculations to investigate such a mechanism on Ni2P, revealing that the rate-determining step was surface hydride transfer to physisorbed CO2, a non-electrochemical step.117 Therefore, simultaneous engineering of both thermal and electrochemical steps remains a valuable strategy to further optimize NixPy-based catalysts.
According to the above studies, it is evident that the reaction steps on non-Cu catalysts are significantly different from the ones reported on Cu-based systems. The precursors for C–C coupling vary from CO (for the majority of Cu-based materials) to hydrocarbons and formate (for non-Cu catalysts). Consequently, more comprehensive understanding of the specific reaction mechanism remains extremely vital to ensure rational design of future catalysts with enhanced catalytic activity and selectivity towards C3+ products. Another challenge and opportunity lies in the suppression of the HER side reaction. A Mo- and Ni-based compound catalyst, albeit with decent C3+ selectivity in eCO2RR products, normally suffer from severe HER competition. The most promising C3+ activity and selectivity are mainly observed at low overpotential and low current density. In fact, at higher overpotential where industrially desired current density is achievable, most metal sulfides and phosphides were found to be predominantly active towards the HER rather than the eCO2RR.118 Seeking for compatible HER-suppression strategies might offer opportunities to fully exert the catalytic properties of non-Cu catalysts. In this sense, Asadi et al. reported imidazolium-functionalized Mo3P NPs with an ionomer coating for selective eCO2RR to propane.119 Both the imidazolium functionalization and ionomer coating created a relatively hydrophobic environment and increased the CO2/water ratio near the catalytic surface, resulting in suppressed HER. The positively charged imidazolium further modified the interfacial electric field and adsorption energies of carbon-based intermediates on Mo atoms, directing to a propane-formation pathway in which the trimerization was initiated from co-adsorption of CO* + CH* + CO*. Thanks to the synergistic effects between Mo3P, imidazolium, and ionomer coating, a promising FE of 91% for propane was obtained at −0.8 V vs. RHE, demonstrating the great potential of non-Cu catalysts for C3 electrosynthesis from CO2/CO with desirable selectivity/activity after rational catalyst-reaction microenvironment co-design.
3. “Electrochemical + X” approaches
Direct electrochemical approaches for converting CO2 to C3+ chemicals have seen significant progress in novel catalyst development and industrially applicable reactor design. C3 molecules such as 1-propanol have been reported as the primary product from direct electrocatalysis.66,68 While the success of generating species beyond C3 has been established in a few cases mostly with non-Cu catalysts having distinctive reaction mechanisms,115,116 the production of C4 molecules such as 1-butanol (CH3CH2CH2CH2OH) has rarely been reported to occur with measurable quantities through a single electrochemical process. Although studies have suggested that minor and yet-to-be-quantified amounts of 1-butanol can be produced through the eCO2RR120 or co-electroreduction of acetaldehyde and CO on OD-Cu electrodes,59 the extremely low selectivity makes these processes impractical for direct C4 production. More importantly, a greater gap exists between the products from direct eCO2RR and desirable C5+ target chemicals, such as synthetic gasoline (C4–C12), sustainable aviation fuels (C8–C16), aromatics (C6+), and life-related organic chemicals including saccharides, amino acids, and lipids. Electrochemical upgrading of CO2 towards more complex chemicals therefore requires complementary processes to leverage eCO2RR-attainable chemicals for advanced synthesis in tandem.3.1. “Electrochemical + homogeneous reaction” approaches
Before the discussion on coupling different processes, it is prerequisite to reveal the C4 formation mechanism on the most investigated Cu-based catalysts. In this regard, Yeo et al. conducted systematic eCO2RR experiments on OD-Cu in a GDE-based flow cell.121 By improving the detection and quantification of liquid products with low FEs through a flow cell and headspace gas chromatography, they did observe the formation of 1-butanol in alkaline electrolyte, but the FE was as low as 0.056% and the partial current density was only 0.080 mA cm−2. Unlike the formation of C3 species that usually involves one individual C1 adsorbate (e.g. *CO), the aldol condensation of two acetaldehyde molecules was found to account for C–C bond formation, yielding an initial formation of 3-hydroxybutanal (CH3CH(OH)CH2CHO) (Fig. 7a). The subsequent dehydration then produces crotonaldehyde (CH3CHCHCHO) as a critical C4 intermediate, the further 2e−-reduced products of which could either be crotyl alcohol (CH2CHCHCH2OH) or butanal (CH3CH2CH2CHO). Parallel electrolysis experiments of acetaldehyde, crotonaldehyde, crotyl alcohol, and butanal confirmed that the synthetic route towards 1-butanol followed the sequence of acetaldehyde → crotonaldehyde → butanal → 1-butanol while crotyl alcohol remained electrochemically inert. This mechanism was supported by DFT calculations, as well as the promotion of aldol condensation by hydroxide ions (OH−) and dehydration of 3-hydroxybutanal on Cu surfaces. Collectively, such a mechanism can well explain the low selectivity of C4 products on Cu during the eCO2RR or eCORR: (1) the low selectivity towards acetaldehyde on Cu; (2) the competition between acetaldehyde electroreduction to ethanol and aldol condensation; and (3) the hydration of crotonaldehyde to an unreactive form in alkaline solution. Overcoming the above limitations could be realized through a cascade reaction engineering approach, where CO2-to-acetaldehyde conversion, aldol condensation, and crotonaldehyde electroreduction are optimized separately. Such a design might be worthy of future investigations.
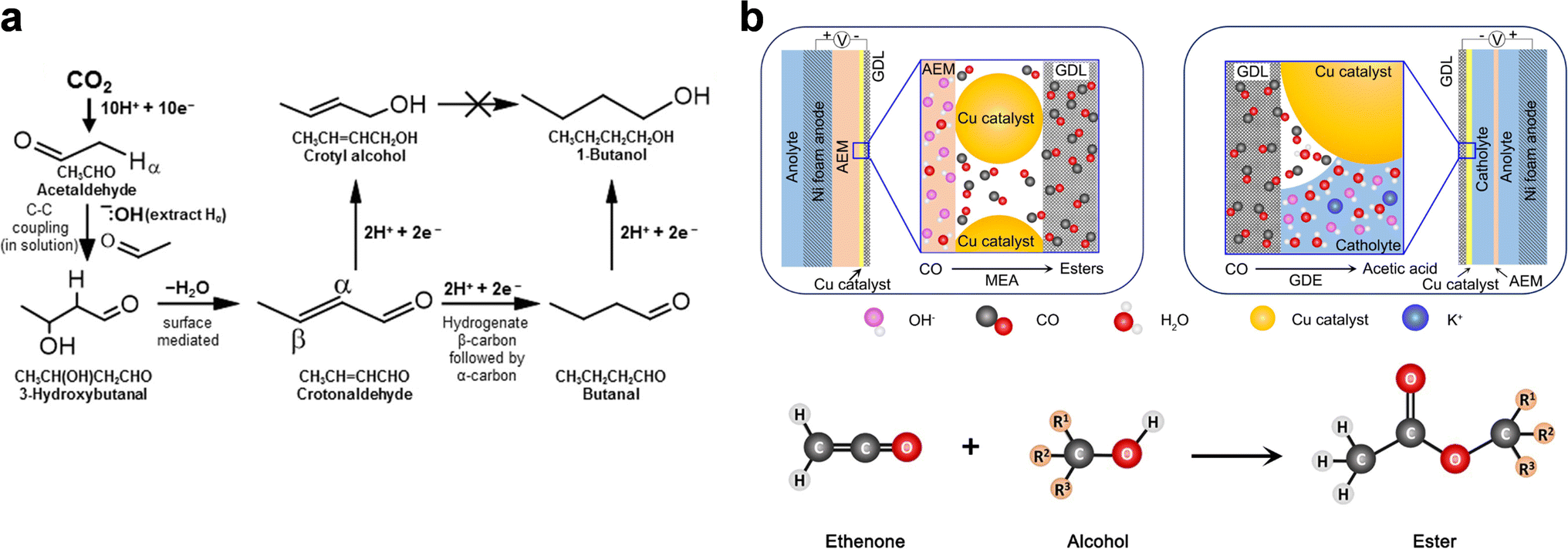 | ||
| Fig. 7 “Electrochemical + homogeneous reaction” approaches. (a) Simplified domino reaction mechanism for the eCO2RR to 1-butanol. Adapted from ref. 121 with permission from Wiley-VCH GmbH. (b) Schematic illustration of (upper) the eCORR process in the cathodic compartment of a MEA cell and a GDE embedded in a flow cell, and (bottom) ester formation from the reaction between ethenone and alcohol. Adapted from ref. 122 with permission from Wiley-VCH GmbH. | ||
In line with the above mechanistic study, it is fundamentally feasible to couple direct electrolysis with controllable homogeneous reactions to steer the product selectivity. Interestingly, Yeo et al. demonstrated the production of C3–C6 acetate esters through the eCORR in an MEA cell (Fig. 7b).122 The absence of esters was attributed to the “water-rich” interfacial conditions in conventional H-cells or GDE-based flow cells, which favor the nucleophilic reaction between OH− and the key intermediate ethenone (CH2CO) that leads to acetate production. In the MEA-based eCORR, in contrast, ethenone preferentially reacts with alcohols to yield esters, resulting in an unprecedented total FE of 22% for esters (∼20% for ethyl acetate) at 250 mA cm−2. It should be noted that as the major alcohol product during the eCORR was ethanol, the dominant ester product was ethyl acetate (C4). C5 and C6 ester production thus critically relies on the yields of C3 and C4 alcohols.
Since the rates of homogeneous reactions remain independent of electrochemical potential, on-site homogeneous reactions can be rate-limiting in the entire integrated process. While introducing catalysts for homogeneous reactions may facilitate the post-eCO2RR transformation, the alignment of different reaction conditions and the separation of homogeneous catalysts from the original eCO2RR systems can be challenging. In addition, the number of homogeneous reactions that can operate under mild ambient conditions is limited. Therefore, we will then move on to discussing the coupling of an electrochemical approach with another type of complementary process, heterogeneous thermocatalytic reactions, in a tandem or cascade manner. Compared to homogeneous reactions, heterogeneous thermocatalytic reactions are more common in industrial manufacture of bulk chemicals and thus offer greater opportunities for process coupling.
3.2. “Electrochemical + thermochemical” approaches
The dependence on fossil-fuel-derived feedstocks (e.g. H2 production via extremely energy-consuming steam reforming) in a conventional thermochemical approach for chemical manufacture usually results in positive net CO2 emissions.123 Switching from fossil fuels to renewable feedstocks, such as CO, hydrocarbons, and oxygenates derived from direct electrochemical CO2 reduction, could then potentially offer an alternative to alleviate CO2 emissions in the conventional chemical industry. The coupling of the eCO2RR with downstream thermochemical synthesis, namely the “electrochemical + thermochemical” approach, seems to be simple and straightforward. However, the separation of required feedstocks from a mixture of products derived from the eCO2RR can be energetically demanding. Integrating a separation unit between the electrochemical and thermochemical modules may offset the advantage of reducing CO2 emissions but remains necessary in some cases to optimize the reaction conditions of thermochemical reactors. For instance, Wang et al. demonstrated that simply inserting a CO2-absorption procedure between an eCO2RR reactor and a cascade C2H4 oxidation reactor can double the production rate of ethylene glycol.124To make the entire process more economically favorable, it is of course more desirable to leverage the mixed products as feedstocks directly for thermochemical conversion without further purification. Reactions requiring mixed reactants therefore naturally emerge as suitable candidates. Due to the ubiquity of CO as the most common product of the eCO2RR, various efforts have been reported to integrate carbonylation reactions in the tandem process. For instance, Chen et al. proposed a two-step tandem electrochemical-thermochemical approach to synthesize value-added C3 oxygenate molecules from CO2 (Fig. 8a),125 in which CO2 was first electrochemically reduced to mixed gas products of C2H4, CO, and H2 on Cu with a MEA reactor. Due to the low solubility of these gas products in aqueous solution, it is convenient to separate them from other liquid products. A thermochemical hydroformylation reaction was then employed to produce propanal (C2H4 + CO + H2 → CH3CH2CHO) and 1-propanol (CH3CH2CHO + H2 → CH3CH2CH2OH) from a mixed C2H4/CO/H2 feedstock (Fig. 8). It was shown that both the Cu-catalyst-dependent gas inlet composition and the reaction temperature can influence C3 yields and selectivity. The optimized C3 oxygenate selectivity on a reduced CO2-basis was up to 18%, corresponding to a 4-fold improvement compared to direct electrochemical CO2 conversion in a flow cell. Meanwhile, dilute C3 oxygenate products could be easily isolated from the gaseous outlet stream of the second hydroformylation reactor. A similar concept was also leveraged by Li et al.,126 who adopted an optimized Rh-complex catalyst for the hydroformylation reaction, increasing the C3 oxygenate selectivity to 44%. Other carbonylation reactions such as hydrocarboxylation, alkoxycarbonylation, and aminocarbonylation are also possible, as suggested by some very recent reports.127,128
 | ||
| Fig. 8 “Electrochemical + thermochemical” approaches. (a) Schematic illustration of the eCO2RR + hydroformylation tandem process for the synthesis of C3 oxygenate products. Adapted from ref. 125 with permission from American Chemical Society. (b) (Upper) schematic illustration of the eCO2RR + C2H4 dimerization cascade process for the synthesis of C4 hydrocarbons, and (bottom) DFT-based reaction energy diagram of C2H4 dimerization towards C4H10 (solid lines) and C4H8 (short dashed lines) under 0 ML (beige) and 1/2 ML (blue) CO* coverage, respectively. Adapted from ref. 129 with permission from Springer Nature. | ||
In addition to the major reactants, the eCO2RR or eCORR can also supply activity- or selectivity-regulating species that do not directly participate in the reaction to thermochemical reactors. For instance, Sargent et al. showcased a different electrochemical and thermochemical route for converting CO2 to butane (C4H10) via a C1 (CO/CO2)–C2 (C2H4)–C4 (C4H10) cascade (Fig. 8b).129 CO in the humidified outlet stream of the direct CO2 or CO electrolyzer was found to promote C2H4 dimerization to C4H10 with a selectivity of up to 95% in a secondary thermochemical reactor under ambient conditions. The best overall CO-to-C4H10 cascade selectivity was as high as 43%. Mechanistic insights into the role of CO were obtained through DFT calculations, demonstrating that an increase in CO coverage can result in lowered dimerization and hydrogenation barriers, both of which promote the conversion of C2H4 to C4H10 according to the Cossee–Arlman olefin polymerization mechanism. More interestingly, a recent work by Chen et al. presents a proof-of-the-concept of leveraging eCO2RR-derived compositionally tunable syngas (i.e. CO + H2) for thermocatalytic synthesis of carbon nanofibers on the FeCo alloy under relatively mild conditions (370–450 °C, 1 atm).130 The co-feeding of both CO and H2 was necessary for high solid carbon yields; while with CO feed only, the FeCo alloy catalyst deactivated rapidly.
The above success, as well as the vast number of well-established thermochemical reactions, has demonstrated the great potential of the “electrochemical + thermochemical” approach to extend the reach of electrochemical CO2 upgrading to the synthesis of C3+ products or even advanced solid carbon materials. Other promising reactions include Fischer–Tropsch synthesis and syngas–methanol–aromatic conversion.131,132 While the gas product stream of the eCO2RR has been primarily focused on so far, the liquid product stream, such as alcohols and carboxylic acids, remains less considered for further thermochemical conversion, probably due to their low concentration in the blended electrolyte solution. A number of opportunities therefore still remain for engineering tandem or cascade “electrochemical + thermochemical” CO2 conversion processes, which call for further exploration by future researchers.
3.3. “Electrochemical + biological” approaches
Similar to thermochemical reactions, there are many well-established biocatalytic reactions or processes capable of producing C3+ chemicals with high selectivity and product specificity. More interestingly, biological processes can produce long-chain natural products that are absent in the product spectra of conventional electrolysis or thermocatalysis. For instance, glucose and fatty acids are key elements for human nutrition and important raw materials for fine chemical engineering and the food industry.133,134 Producing them by mimicking the natural process of CO2 fixation therefore presents great potential to revolutionize agriculture, reshape the bioeconomy, maintain biodiversity, and minimize the carbon footprint.135–137 In light of this, coupling biological processes with electrocatalysis provides a potential route to this goal, where renewable electricity is adopted to transform CO2 into reduced products, and various organisms or extracted enzymes are employed to produce value-added long-chain compounds from the reduced products.138 Note that in a broader context of bioelectrocatalysis, the electrochemical and biochemical processes can be integrated directly or indirectly. In direct bioelectrocatalysis, organisms compatible with the complex electrochemical environments are attached to catalytic electrodes directly, the number of which however remains limited. Thus, we primarily focus on the indirect coupling strategy, namely the spatially separated electro-biosystem, in this review.Along this line, Schmid et al. described a scalable hybrid system by placing a fermentation module after a GDE-based CO2 electrolyzer yielding syngas for subsequent anaerobic fermentation (Fig. 9a).139 The CO2 electrolyzer was carefully engineered with a commercial Ag-based GDE, exhibiting high energy efficiency of converting CO2 to syngas at hundreds of mA cm−2. Such a high current density is generally unattainable in the direct bioelectrolysis scheme. Two kinds of anaerobic fermenters were then adopted for the relay biochemical conversion; C. autoethanogenum was first responsible for converting syngas to acetate and ethanol, C. kluyveri was then used to convert acetate and ethanol to butyrate and hexanoate, and finally C. autoethanogenum was adopted again to produce 1-butanol and 1-hexanol from their corresponding carboxylates. The total carbon selectivity to C4 and C6 alcohols was found to be significantly higher than that of the pure electrochemical approach.
 | ||
| Fig. 9 “Electrochemical + biological” approaches. (a) Schematic illustration of the CO2 electrolyzer coupled to a fermentation module for the synthesis of 1-butanol and 1-hexanol. Adapted from ref. 139 with permission from Springer Nature. (b) Schematic illustration of the in vitro artificial sugar synthesis system based on the two-step eCO2RR–eCORR and yeast fermentation. Adapted from ref. 33 with permission from Springer Nature. | ||
Nevertheless, the low solubility of the gas feed could limit the productivity, volumetric efficiency, and economic viability of the “electrochemical + biological” system. In this regard, liquid carbon sources remain more desirable. While formic acid and methanol have high biotoxicity, acetic acid, or acetate as its salt form, is more readily metabolizable by a broad range of organisms. Separate studies have demonstrated the feasibility of leveraging eCO2RR-derived acetic acid/acetate for the biocatalytic production of long-chain organic compounds. As proposed by Xia et al., acetate can be employed to organically link the two-step eCO2RR-eCORR and yeast fermentation, generating glucose with high yields (Fig. 9b).33 In the two-step eCO2RR-eCORR process, a Ni–N–C single atom catalyst and a defect-rich Cu catalyst were employed as the CO producer and acetate producer, achieving a FE of nearly 100% for CO from the eCO2RR and a FE of 52% for acetate from the eCORR, respectively. Moreover, the adoption of a porous solid electrolyte in the second CO electrolyzer allowed for continuous generation of pure and concentrated acetic acid without pollution from electrolyte salts. The authors then genetically engineered S. cerevisiae to realize glucose production from the as-electrogenerated pure acetic acid in vitro with a high glucose yield of 8.90 μmol per gram yeast per hour. In contrast, acetate with salt impurities resulted in no glucose production. The wide applicability of this work has attracted much attention, as it can be readily extended to the production of other chemicals as well (e.g. free fatty acids). Nearly at the same time, Jinkerson et al. reported a similar two-step electrochemical CO2-to-acetate conversion route and used acetates for the heterotrophic cultivation of food-producing organisms such as yeast, fungi, algae, and crop plants, presenting another valid example of the promising perspectives of a decoupled electro-biosystem.34 The available products of the “electrochemical + biological” scheme have been further extended to polymers. Dai et al. presented a systematic multi-tier chem-bio design to seamlessly integrate the eCO2RR with microbial metabolism.140 By taking soluble C2 intermediates as the feedstock, microorganisms showed better metabolic kinetics than C1 intermediates and H2 to produce medium-chain-length polyhydroxyalkanoates for applications such as biodegradable plastics.
In general, the technical feasibility and huge potential of “electrochemical + biological” approaches have been demonstrated by a handful of studies. The ability to produce long-chain compounds that are rarely observed in electrochemical and thermochemical CO2 conversion by the “electrochemical + biological” approach makes it particularly attractive. In addition, biological processes usually require only mild reaction conditions, in contrast to the majority of thermochemical processes demanding elevated temperature or pressure, which can be valuable for process decentralization.
4. Summary and outlook
Due to the higher energy density and economic value of C3+ fuels and chemicals than their C1 and C2 counterparts, upgrading CO2 to C3+ products with renewable electricity is undoubtedly a promising direction to realize “Net Zero” emission and lay the foundation for a sustainable chemical industry. In the past few decades, many efforts have been devoted to the development and optimization of direct eCO2RR technologies, showing a great leap in C3 selectivity and activity. Aiming to produce C4+ species with higher molecular weights, coupling the eCO2RR with a complementary process has emerged in recent years to allow for tandem or cascade conversion leveraging reduced products in the outlet stream of the eCO2RR. Despite the continuous research progress and constantly renewed records of C3+ selectivity/activity, challenges towards industrialization persist. Here we discuss some perspectives on these challenges and opportunities for addressing them.4.1. Direct electrochemical approach
The direct electrochemical approach presents the simplest way of converting CO2 to C3+ products as the least number of processes are involved. The overall process can often run at near room temperature and ambient pressure. Along with the scalability of electrolyzers, this approach is extremely suitable for decentralized applications. To reveal the general trend of the eCO2RR/eCORR to C3+ products, the optimal FE and partial current density (jC3+) of C3+ products are summarized in Table S1† and Fig. 10. It can be generally concluded that after decades of research, the conversion efficiency of CO2 to a specific C3+ product is still the major bottleneck of direct CO2 electrolysis. | ||
| Fig. 10 A summary on catalytic performance of direct electrochemical CO2/CO-to-C3+ conversion using different types of catalysts (including single-crystal Cu, nanostructured Cu or OD-Cu, doped or alloyed Cu with foreign element regulation, Cu SAC, and Mo- or Ni-based non-Cu catalysts). The first three types of catalysts are further divided into two groups to reveal the influence of electrolyte pH; cyan for the alkaline conditions where KOH electrolytes are adopted, and red for the neutral or acidic conditions where bicarbonates, halides, or other oxysalts are adopted. The size of markers indicates the magnitude of partial current density of C3+ formation. Data for plotting are tabulated in Table S1.† | ||
In addition to the above general conclusion, several key aspects can be extracted from Fig. 10: (1) the structural sensitivity of Cu-based catalysts. With Cu-based materials still being the only widely justified catalytic systems for the eCO2RR to C3+ chemicals, most of the reports have demonstrated optimal FEs of 5–15%, which is quite close to the results obtained on single-crystal Cu(S)-[n(100) × (111)] electrodes according to the 20 year-old report by Hori et al.56 This finding accentuates the necessity of combining different Cu sites with distinct coordination numbers, local geometries and electronic structures, and binding affinities to critical surface species (e.g. CO and C2 precursors). Therefore, a focus shift from active site to active site ensemble may facilitate future Cu-based catalyst design. By comparing the nanostructure Cu/OD-Cu (shown as circles in Fig. 10) systems with doped/alloyed Cu (shown as squares in Fig. 10) systems, it is found that dilute alloying presents the most promising route for selective C3 production; nevertheless, we also note that these bimetallic or multimetallic materials still benefit from desirable micro-/nanostructure and defect-rich surfaces. Untangling the intertwined structural and electronic effects is still of great interest from a fundamental perspective.
(2) The conditional sensitivity of direct electrolysis. CO2/CO electrolysis under alkaline conditions (shown in cyan in Fig. 10) generally demonstrates higher catalytic efficiency than electrolysis under neutral or acidic conditions (shown in red in Fig. 10). This is in accordance with the well-understood pH effect that high pH favors C2 formation, giving rise to higher coverage of C2 for further coupling reactions.36–42 Also, the HER is suppressed at high alkalinity. However, it must be pointed out that the inevitable reaction between CO2 and hydroxide is detrimental to the local alkalinity maintenance and overall carbon efficiency. The conversion from CO2 to CO is suggested to be spatially decoupled from the subsequent CO-to-C3+ process; otherwise, improving the catalytic efficiency in acidic media would be another promising direction.
(3) The narrow optimal operational window. Through combinatorial efforts in engineering catalysts, CO2/CO feedings, reaction microenvironments, and reactors, the highest C3 FEs were reported in the range of approximately 45–55% (highlighted as the region in yellow, Fig. 10). However, such an optimal performance is only attainable in a quite narrow potential (or current-density) window,104,106 severely limiting the compatibility of pure electrochemical systems with renewable power generations showing huge variations in output current. Therefore, it is necessary to further widen the high-C3-selectivity window for practical decentralized applications.
(4) The inherent mechanistic limitations of the carbonyl-dominated C–C coupling mechanism. It is rare for Cu-based catalytic systems to yield products beyond C3 through direct eCO2RR. Although recent studies have validated the possibility to produce C4+ through domino reactions coupling homogeneous non-electrocatalytic and heterogeneous electrocatalytic steps in one-pot electrolysis, their FEs are at the level of ∼20%, far from satisfactory for industrialization.121,122 The lack or scarcity of C4+ products in the Cu-catalyzed eCO2RR suggests the inherent limitation of the carbonyl-dominated C–C coupling mechanism. On one hand, moderately binding catalytic surfaces like Cu are essential for suppressing the HER. On the other hand, high CO* coverage is essential for the initial C–C bond formation. Nonetheless, it is difficult to obtain sufficient coverage of higher-order C2+ as well under such a prerequisite. Balancing the surface distribution of various key C1–C3 intermediates would require a variety of active site ensembles possessing different propensities to bind specific intermediates. This could explain the high structural sensitivity of Cu-based catalysts. However, Cu surfaces are often extremely sensitive to the environment and quite dynamic under specific reaction conditions,66–68 which is in stark contradiction to the above requirements to stabilize C3-specific active site ensembles and balance the propensities of key surface intermediates. The origin of the above limitation still remains a missing piece in the current mechanistic understanding of the eCO2RR. Further investigations combining advanced characterization and simulation tools to reveal corresponding mechanistic understandings therefore remain nontrivial.
(5) The promise and problem of non-Cu catalysts. Exploring non-Cu catalysts having different mechanisms (e.g. hydrocarbon oligomerization and oxygenate condensation) can effectively circumvent the above fundamental limitations that conventional Cu-based catalysts may pose. As a consequence, some non-Cu catalysts such as Mo and Ni phosphides have shown very promising FEs (>90%) and overpotentials (<300 mV) for C3 production (Fig. 10).116,119 These Mo- and Ni-based non-Cu catalysts generally feature intermediate chemisorption strengths between Cu and strong-binding surfaces known for being HER-active only (e.g. Ni metal), allowing for favorable stabilization of certain C2+ surface species. However, the enhancement of the HER can emerge as an undesirable side effect.118 Therefore, the increased carbon selectivity towards C3+ usually comes at the price of lowered electron selectivity, which is also the main reason why C3 FE decreases drastically with increasing overpotential (Fig. 10). Seeking appropriate HER-suppression strategies without sacrificing the unique C3+ productivity is a promising future direction. As the exploration of novel non-Cu catalysts is less reported than that of Cu-based catalysts, more justification on the validity of non-Cu catalysts for both carbon- and electron-selective eCO2RR would be necessary.
4.2. “Electrochemical + X” approaches
Compared to pure electrochemical approaches, “electrochemical + X” approaches present several unique advantages (Table 1). The first exceptional advantage lies in the high production efficiency (combining both selectivity and activity) of C3+ products from CO2. This is attributed to the discrete nature of process units, which allows for individual design and separate optimization of each unit without interference from other processes, as opposed to reactions in one pot. Moreover, complementing a spatially decoupled “X” process can strictly isolate the needs for CO2 activation and C–C coupling into different sectors. And the vast possibilities in complementary processes significantly expand the product scope from single-electrolysis-accessible C3/C4 alcohols and hydrocarbons to inaccessible C5+ chemicals. In addition, we propose that the spatially separated yet still in-place tandem process is more advantageous than deploying multiple individual plants in different locations, due to the elimination of safety risks associated with long-range transportation and long-term storage of hazardous chemicals (e.g. CO and H2).| “Electrochemical + X” | Direct electrochemical | |
|---|---|---|
| Advantages | • High conversion efficiency | • Simple processes |
| • Decoupled process optimization | • Mild conditions | |
| • Expanded scope of available products | • Suitable for decentralized applications | |
| • Eliminate risky chemical transportation and storage | ||
| Challenges | Overall | Cu-based catalytic systems |
| • Complex processes | • Low conversion efficiency | |
| • Increased capitalized costs | • Limited availability of products | |
| • Interface or connection between modules (e.g. stream purification) | • Narrow operational window | |
| “X” – homogeneous reactions | Non-Cu catalytic systems | |
| • Reaction rate alignment | • HER competition | |
| • Mass-transport management of reactive intermediates | • Wider and more rigorous demonstration | |
| “X” – thermochemical | ||
| • Additional energy consumption and CO2 emission | ||
| • Unsuitable for decentralized applications | ||
| “X” – biological | ||
| • Extra energy input for non-autotrophic organisms, and CO2 emission | ||
| • Product separation from organisms |
However, “electrochemical + X” approaches also face a series of challenges for practical implementation: (1) the system complexity is higher than that of the direct electrochemical approaches as there are more unit processes, which inevitably results in increased costs for construction, operation, and maintenance, offsetting the benefits of more valuable C4+ chemicals as well as their enhanced yields in a tandem process. More importantly, some of the “X” processes, especially thermochemical reactions demanding high temperature and pressure, are obviously energy-intense and carbon-emission-positive, while biological processes often rely on non-autotrophic organisms, the life maintenance of which also needs extra energy inputs and emits CO2. Both aspects mitigate the ability of such hybrid approaches to reduce CO2 emission compared to their purely renewable-powered electrochemical counterparts. Therefore, critical techno-economic and carbon footprint analyses are of utmost urgency and importance in future research, especially for unexplored products (e.g. aromatics).
(2) Another key aspect is the interaction between electrochemical and complementary modules. The product outlet streams of the eCO2RR are typically mixed gas and liquid products, as currently it is only possible to control the product selectivity to near-unity for 2e− products (CO and formate) via the eCO2RR. Although some studies have showcased the possibility of leveraging a mixture feedstock without purification,125,129 the influence of impurities is usually nontrivial and deserves in-depth investigations.33,34 Implementation of extra separation units can further introduce economic burden. In this case, adoption of solid-electrolyte-based electrolyzers may alleviate the issue of separating liquid products from electrolyte salts.33 Seeking complementary approaches with high tolerance to impurities or capabilities to utilize multiple feedstocks presents another promising future direction. Apart from the inlet stream purification, the separation of products and organisms also presents a technical hurdle and deserves further techno-economic analyses and investigations.
(3) The actual efficiencies of “electrochemical + X” approaches require more rigorous and comprehensive evaluation. There are a number of factors leading to deteriorated efficiency. For “electrochemical + homogeneous reaction” approaches, homogeneous reactions (e.g. aldol condensation or esterification) under ambient conditions often possess lower rates than the prior electrochemical reactions, limiting the conversion of C1–3 intermediates to C4+ products.121,122 The mass transport of reactive intermediates also needs to be well managed towards full utilization. When “X” stands for thermochemical or biological processes, the overall energy efficiency might not be as promising as the conversion efficiency (based on either carbon or electron utilization). The extra energy required for driving thermochemical reactions or sustaining organism lifespan is nontrivial, and developing better practices to maintain the superiority of “electrochemical + X” remains indispensable.
In sum, the electrochemical upgrading of CO2 to high-value C3+ products has remained and will stay a challenging yet attractive research area for decades to come. Collaborative research studies from multidisciplinary and interdisciplinary perspectives are highly desirable to spur innovations on advanced materials and processes. And we believe that the successful industrialization of electrochemical and sustainable CO2 upgrading will eventually present a viable solution to the environmental and energy crises faced by human society.
Data availability
No primary research results have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22109020, 22109082, 22379021 and 22102018) and Sichuan Science and Technology Program (2023NSFSC0115). C. X. acknowledges the Huzhou Science and Technology Bureau (2022GZ45).References
- J. D. Shakun, P. U. Clark, F. He, S. A. Marcott, A. C. Mix, Z. Liu, B. Otto-Bliesner, A. Schmittner and E. Bard, Nature, 2012, 484, 49–54 CrossRef CAS.
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 184–187 CrossRef CAS PubMed.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- S. Mehla, A. E. Kandjani, R. Babarao, A. F. Lee, S. Periasamy, K. Wilson, S. Ramakrishna and S. K. Bhargava, Energy Environ. Sci., 2021, 14, 320–352 RSC.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- K. Schuchmann and V. Müller, Nat. Rev. Microbiol., 2014, 12, 809–821 CrossRef CAS.
- Y. Song, J. S. Lee, J. Shin, G. M. Lee, S. Jin, S. Kang, J.-K. Lee, D. R. Kim, E. Y. Lee, S. C. Kim, S. Cho, D. Kim and B.-K. Cho, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7516–7523 CrossRef CAS.
- G. Yang, P. Qiu, J. Xiong, X. Zhu and G. Cheng, Chin. Chem. Lett., 2022, 33, 3709–3712 CrossRef CAS.
- S. Bai, H. Qiu, M. Song, G. He, F. Wang, Y. Liu and L. Guo, eScience, 2022, 2, 428–437 CrossRef.
- S. Ng, J. J. Foo and W. Ong, InfoMat, 2022, 4, e12279 CrossRef CAS.
- T. Jia, L. Wang, Z. Zhu, B. Zhu, Y. Zhou, G. Zhu, M. Zhu and H. Tao, Chin. Chem. Lett., 2024, 35, 108692 CrossRef CAS.
- Q. Zhu, J. Ma, X. Kang, X. Sun, J. Hu, G. Yang and B. Han, Sci. China: Chem., 2016, 59, 551–556 CrossRef CAS.
- Y. Luo, K. Zhang, Y. Li and Y. Wang, InfoMat, 2021, 3, 1313–1332 CrossRef CAS.
- D. Song, Y. Lian, M. Wang, Y. Su, F. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS.
- K. Xu, S. Zheng, Y. Li, H. Chu, Q. Xiong, Z. Mei, Q. Zhao, L. Yang, S. Li and F. Pan, Chin. Chem. Lett., 2022, 33, 424–427 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- B. Yang, L. Li, Z. Jia, X. Liu, C. Zhang and L. Guo, Chin. Chem. Lett., 2020, 31, 2627–2633 CrossRef CAS.
- J. Du, S. Li, S. Liu, Y. Xin, B. Chen, H. Liu and B. Han, Chem. Sci., 2020, 11, 5098–5104 RSC.
- Y. Yang, J. Fu, Y. Ouyang, T. Tang, Y. Zhang, L.-R. Zheng, Q.-H. Zhang, X.-Z. Liu, J. Wang and J.-S. Hu, Natl. Sci. Rev., 2023, 10, nwac248 CrossRef CAS PubMed.
- H. H. Wong, M. Sun, T. Wu, C. H. Chan, L. Lu, Q. Lu, B. Chen and B. Huang, eScience, 2024, 4, 100140 CrossRef.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS.
- T. Qin, Y. Qian, F. Zhang and B.-L. Lin, Chin. Chem. Lett., 2019, 30, 314–318 CrossRef CAS.
- L. Tao, E. C. D. Tan, R. McCormick, M. Zhang, A. Aden, X. He and B. T. Zigler, Biofuels, Bioprod. Biorefin., 2014, 8, 30–48 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- P. Dürre, Biotechnol. J., 2007, 2, 1525–1534 CrossRef PubMed.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS.
- T. Zheng, M. Zhang, L. Wu, S. Guo, X. Liu, J. Zhao, W. Xue, J. Li, C. Liu, X. Li, Q. Jiang, J. Bao, J. Zeng, T. Yu and C. Xia, Nat. Catal., 2022, 5, 388–396 CrossRef CAS.
- E. C. Hann, S. Overa, M. Harland-Dunaway, A. F. Narvaez, D. N. Le, M. L. Orozco-Cárdenas, F. Jiao and R. E. Jinkerson, Nat. Food, 2022, 3, 461–471 CrossRef CAS.
- N. S. Romero Cuellar, K. Wiesner-Fleischer, M. Fleischer, A. Rucki and O. Hinrichsen, Electrochim. Acta, 2019, 307, 164–175 CrossRef CAS.
- K. G. Schulz, U. Riebesell, B. Rost, S. Thoms and R. E. Zeebe, Mar. Chem., 2006, 100, 53–65 CrossRef CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- J. C. Bui, C. Kim, A. Z. Weber and A. T. Bell, ACS Energy Lett., 2021, 6, 1181–1188 CrossRef CAS.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- H.-J. Peng, M. T. Tang, J. Halldin Stenlid, X. Liu and F. Abild-Pedersen, Nat. Commun., 2022, 13, 1399 CrossRef CAS.
- J. Lee, Y. Kwon, R. L. Machunda and H. J. Lee, Chem.–Asian J., 2009, 4, 1516–1523 CrossRef CAS PubMed.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Chem. Soc. Chem. Commun., 1988, 17 RSC.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS.
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. I. Goddard, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- L. Chen, C. Tang, Y. Zheng, E. Skúlason and Y. Jiao, J. Mater. Chem. A, 2022, 10, 5998–6006 RSC.
- M. T. Tang, H.-J. Peng, J. H. Stenlid and F. Abild-Pedersen, J. Phys. Chem. C, 2021, 125, 26437–26447 CrossRef CAS.
- S. Pablo-García, F. L. P. Veenstra, L. R. L. Ting, R. García-Muelas, F. Dattila, A. J. Martín, B. S. Yeo, J. Pérez-Ramírez and N. López, Catal. Sci. Technol., 2022, 12, 409–417 RSC.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- Y.-T. Chan, I.-S. Huang and M.-K. Tsai, Phys. Chem. Chem. Phys., 2019, 21, 22704–22710 RSC.
- C.-C. Chang, M.-S. Ku, W.-H. Lien and S.-F. Hung, J. Phys. Chem. C, 2022, 126, 5502–5512 CrossRef CAS.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- M. A. Van Hove and G. A. Somorjai, Surf. Sci., 1980, 92, 489–518 CrossRef CAS.
- M. Wu, Z. Zhang, X. Xu, Z. Zhang, Y. Duan, J. Dong, R. Qiao, S. You, L. Wang, J. Qi, D. Zou, N. Shang, Y. Yang, H. Li, L. Zhu, J. Sun, H. Yu, P. Gao, X. Bai, Y. Jiang, Z.-J. Wang, F. Ding, D. Yu, E. Wang and K. Liu, Nature, 2020, 581, 406–410 CrossRef CAS PubMed.
- S. Jin, M. Huang, Y. Kwon, L. Zhang, B.-W. Li, S. Oh, J. Dong, D. Luo, M. Biswal, B. V. Cunning, P. V. Bakharev, I. Moon, W. J. Yoo, D. C. Camacho-Mojica, Y.-J. Kim, S. H. Lee, B. Wang, W. K. Seong, M. Saxena, F. Ding, H.-J. Shin and R. S. Ruoff, Science, 2018, 362, 1021–1025 CrossRef CAS.
- C. Zhu, Z. Zhang, L. Zhong, C.-S. Hsu, X. Xu, Y. Li, S. Zhao, S. Chen, J. Yu, S. Chen, M. Wu, P. Gao, S. Li, H. M. Chen, K. Liu and L. Zhang, Chem, 2021, 7, 406–420 CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- Y. Li, D. Kim, S. Louisia, C. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194–9201 CrossRef CAS.
- J. Liu, F. You, B. He, Y. Wu, D. Wang, W. Zhou, C. Qian, G. Yang, G. Liu, H. Wang, Y. Guo, L. Gu, L. Feng, S. Li and Y. Zhao, J. Am. Chem. Soc., 2022, 144, 12410–12420 CrossRef CAS PubMed.
- W.-Y. Zhi, Y.-T. Liu, S.-L. Shan, C.-J. Jiang, H. Wang and J.-X. Lu, J. CO2 Util., 2021, 50, 101594 CrossRef CAS.
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202113498 CrossRef CAS PubMed.
- P.-P. Yang, X.-L. Zhang, F.-Y. Gao, Y.-R. Zheng, Z.-Z. Niu, X. Yu, R. Liu, Z.-Z. Wu, S. Qin, L.-P. Chi, Y. Duan, T. Ma, X.-S. Zheng, J.-F. Zhu, H.-J. Wang, M.-R. Gao and S.-H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- H. Du, L.-X. Liu, P. Li, Q. Min, S. Guo and W. Zhu, ACS Nano, 2023, 17, 8663–8670 CrossRef CAS.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- D. Zhong, Z.-J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 CrossRef CAS PubMed.
- C. Long, K. Wan, Y. Chen, L. Li, Y. Jiang, C. Yang, Q. Wu, G. Wu, P. Xu, J. Li, X. Shi, Z. Tang and C. Cui, J. Am. Chem. Soc., 2024, 146, 4632–4641 CrossRef CAS.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 11642–11645 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS.
- C. Long, X. Liu, K. Wan, Y. Jiang, P. An, C. Yang, G. Wu, W. Wang, J. Guo, L. Li, K. Pang, Q. Li, C. Cui, S. Liu, T. Tan and Z. Tang, Sci. Adv., 2023, 9, eadi6119 CrossRef CAS.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- F. Dattila, R. García-Muelas and N. López, ACS Energy Lett., 2020, 5, 3176–3184 CrossRef CAS.
- H.-Y. Wang, M. Soldemo, D. Degerman, P. Lömker, C. Schlueter, A. Nilsson and P. Amann, Angew. Chem., Int. Ed., 2022, 61, e202111021 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824–830 RSC.
- Z. Lian, F. Dattila and N. López, Nat. Catal., 2024, 7, 401–411 CrossRef CAS.
- T. He, G. Kour, X. Mao and A. Du, J. Catal., 2020, 382, 49–56 CrossRef CAS.
- D. Liu, Y. Hu, E. Shoko, H. Yu, T. T. Isimjan and X. Yang, Electrochim. Acta, 2021, 365, 137343 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS.
- J. Li, H. Xiong, X. Liu, D. Wu, D. Su, B. Xu and Q. Lu, Nat. Commun., 2023, 14, 698 CrossRef CAS PubMed.
- M. Rahaman, K. Kiran, I. Z. Montiel, V. Grozovski, A. Dutta and P. Broekmann, Green Chem., 2020, 22, 6497–6509 RSC.
- S. Jeong, C. Huang, Z. Levell, R. X. Skalla, W. Hong, N. J. Escorcia, Y. Losovyj, B. Zhu, A. N. Butrum-Griffith, Y. Liu, C. W. Li, D. Reifsnyder Hickey, Y. Liu and X. Ye, J. Am. Chem. Soc., 2024, 146, 4508–4520 CrossRef CAS.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O'Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- H. Phong Duong, J. G. Rivera de la Cruz, N.-H. Tran, J. Louis, S. Zanna, D. Portehault, A. Zitolo, M. Walls, D. V. Peron, M. W. Schreiber, N. Menguy and M. Fontecave, Angew. Chem., Int. Ed., 2023, 62, e202310788 CrossRef CAS PubMed.
- A. H. M. da Silva, Q. Lenne, R. E. Vos and M. T. M. Koper, ACS Catal., 2023, 13, 4339–4347 CrossRef CAS PubMed.
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- S. A. Francis, J. M. Velazquez, I. M. Ferrer, D. A. Torelli, D. Guevarra, M. T. McDowell, K. Sun, X. Zhou, F. H. Saadi, J. John, M. H. Richter, F. P. Hyler, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Chem. Mater., 2018, 30, 4902–4908 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, ACS Catal., 2017, 7, 6815–6820 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, Faraday Discuss., 2019, 215, 192–204 RSC.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- S. Banerjee, A. Kakekhani, R. B. Wexler and A. M. Rappe, ACS Catal., 2021, 11, 11706–11715 CrossRef CAS.
- A. T. Landers, M. Fields, D. A. Torelli, J. Xiao, T. R. Hellstern, S. A. Francis, C. Tsai, J. Kibsgaard, N. S. Lewis, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 1450–1457 CrossRef CAS.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 127, 14914–14918 CrossRef.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Angew. Chem., Int. Ed., 2020, 59, 21072–21079 CrossRef CAS PubMed.
- Y. Zhou, R. Ganganahalli, S. Verma, H. R. Tan and B. S. Yeo, Angew. Chem., Int. Ed., 2022, 61, e202202859 CrossRef CAS PubMed.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- L. Fan, Y. Zhao, L. Chen, J. Chen, J. Chen, H. Yang, Y. Xiao, T. Zhang, J. Chen and L. Wang, Nat. Catal., 2023, 6, 585–595 CrossRef CAS.
- A. N. Biswas, Z. Xie, R. Xia, S. Overa, F. Jiao and J. G. Chen, ACS Energy Lett., 2022, 2904–2910 CrossRef CAS.
- J. Zhang, X. Kang, Y. Yan, X. Ding, L. He and Y. Li, Angew. Chem., Int. Ed., 2024, 63, e202315777 CrossRef CAS PubMed.
- G. Mei, Y. Lu, X. Yang, S. Chen, X. Yang, L.-M. Yang, C. Tang, Y. Sun, B. Y. Xia and B. You, Angew. Chem., Int. Ed., 2024, 63, e202314708 CrossRef CAS.
- Q. Li, D.-D. Ma, S. Zhou, W.-B. Wei, S.-G. Han and Q.-L. Zhu, Adv. Funct. Mater., 2024, 34, 2316187 CrossRef CAS.
- M. G. Lee, X.-Y. Li, A. Ozden, J. Wicks, P. Ou, Y. Li, R. Dorakhan, J. Lee, H. K. Park, J. W. Yang, B. Chen, J. Abed, R. dos Reis, G. Lee, J. E. Huang, T. Peng, Y.-H. (Cathy) Chin, D. Sinton and E. H. Sargent, Nat. Catal., 2023, 6, 310–318 CrossRef CAS.
- Z. Xie, E. Huang, S. Garg, S. Hwang, P. Liu and J. G. Chen, Nat. Catal., 2024, 7, 98–109 CrossRef CAS.
- G. Tian, X. Liu, C. Zhang, X. Fan, H. Xiong, X. Chen, Z. Li, B. Yan, L. Zhang, N. Wang, H.-J. Peng and F. Wei, Nat. Commun., 2022, 13, 5567 CrossRef CAS PubMed.
- G. Tian, Z. Li, C. Zhang, X. Liu, X. Fan, K. Shen, H. Meng, N. Wang, H. Xiong, M. Zhao, X. Liang, L. Luo, L. Zhang, B. Yan, X. Chen, H.-J. Peng and F. Wei, Nat. Commun., 2024, 15, 3037 CrossRef CAS PubMed.
- Y. Zhu, Z. Huang, Q. Chen, Q. Wu, X. Huang, P.-K. So, L. Shao, Z. Yao, Y. Jia, Z. Li, W. Yu, Y. Yang, A. Jian, S. Sang, W. Zhang and X. Zhang, Nat. Commun., 2019, 10, 4049 CrossRef.
- C. de Carvalho and M. Caramujo, Molecules, 2018, 23, 2583 CrossRef.
- L. Chen, C. Tang, Y. Zheng, E. Skúlason and Y. Jiao, J. Mater. Chem. A, 2022, 10, 5998–6006 RSC.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- S. A. Bonke, M. Wiechen, D. R. MacFarlane and L. Spiccia, Energy Environ. Sci., 2015, 8, 2791–2796 RSC.
- Y. Liu, P. Cruz-Morales, A. Zargar, M. S. Belcher, B. Pang, E. Englund, Q. Dan, K. Yin and J. D. Keasling, Cell, 2021, 184, 1636–1647 CrossRef CAS.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- P. Zhang, K. Chen, B. Xu, J. Li, C. Hu, J. S. Yuan and S. Y. Dai, Chem, 2022, 8, 3363–3381 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03088e |
| This journal is © The Royal Society of Chemistry 2024 |