
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-dependent CO2 thermolysis on barium titanate nanocatalysts†
Smita
Takawane
,
Masatoshi
Miyamoto
,
Takumi
Watanabe
and
Tomonori
Ohba
 *
*
Graduate School of Science, Chiba University, 1-33 Yayoi, Inage, Chiba 263-8522, Japan. E-mail: ohba@chiba-u.jp
First published on 1st July 2024
Abstract
Rising CO2 levels pose a significant threat to global warming, extreme weather events, and ecosystem disruption. Mitigating these effects requires a reduction in CO2 concentration using innovative technologies for CO2 capture, storage, and utilization. Perovskite-type barium titanate nanocatalysts have the potential for high CO2 conversion into valuable solid carbon products at low temperatures. In this study, we investigated the pressure-dependent CO2 conversion activity of barium titanate nanocatalysts at 700 K. A key focus of this study is the impact of pressure on the interaction between CO2 molecules and barium titanate nanocatalysts to evaluate the CO2 conversion mechanism. The primary structures of the nanocatalysts remained unchanged after CO2 thermolysis, whereas carbon was deposited on the nanocatalysts above 0.05 MPa. The reactant carbons after CO2 conversion at various pressures between 0.01 and 1.0 MPa at 700 K were evaluated by temperature-programmed desorption in an O2 atmosphere. The desorption peaks observed at approximately 500 K, 800–900 K, and 900–1300 K were the results of desorption of chemisorbed CO2, less- and high-crystalline graphitic carbons. Chemisorbed CO2 and less-crystalline graphitic carbon were observed at 0.05 MPa. Highly crystalline graphitic carbons were observed on the nanocatalysts after CO2 thermolysis at 0.1–1.0 MPa as well as chemisorbed CO2, although the amount of carbon at 1.0 MPa was smaller than the others. Therefore, the approach of CO2 thermolysis at a low temperature of 700 K and 0.1–0.5 MPa is promising for producing valuable solid carbon products and mitigating the environmental impact of CO2 emissions.
Sustainability spotlightRising CO2 levels significantly influence global warming, extreme weather events, and disruptions to ecosystems. Mitigating these effects requires innovative CO2 capture, storage, and utilization technologies. Nanocatalysts have the potential for high CO2 conversion into valuable solid carbon products at low temperatures. In this study, we investigated the pressure-dependent CO2 conversion activity of perovskite-type BaTiO3 nanocatalysts at 700 K. In particular, we investigate the impact of pressure on the interaction between CO2 molecules and BaTiO3 nanocatalysts and evaluate the key reaction mechanism and pressure dependence of CO2 thermolysis. This study is essential for optimizing reaction conditions and maximizing CO2 conversion efficiency using more sustainable technology. |
Introduction
CO2 is a prominent element in the progression of climate alterations, such as extreme weather phenomena encompassing wildfires, droughts, hurricanes, heatwaves, and rising sea levels due to a rise in worldwide temperature.1 CO2 concentration in the atmosphere rose to 417.2 ppm in 2022, with a 2.5 ppm increase every year, despite the concentration of approximately 280 ppm during the 1800 s.2 This upward trend has been a concern for humanity. The surge in CO2 concentration is primarily attributable to heightened energy consumption, deforestation, and combustion of fossil fuels. The objective of the Paris Agreement is to limit the global average temperature rise to significantly less than 2 °C compared with that in the preindustrial era. To realize this goal, CO2 emissions must be cut by 50% by 2030, and a net-zero status should be achieved by 2050.3 Carbon capture, utilization, and storage technology is the key to combating global warming, in which CO2 emissions are mitigated by capturing and storing CO2 and converting it into highly valuable products.4Adsorption is the most mature technology for capturing CO2. Various novel nanomaterials have been employed to capture CO2 from flue gas using zeolites, metal–organic frameworks (MOFs), carbon materials, mesoporous silica, and metal oxides. These materials possess high specific surface areas, exceptional selectivity, superior thermal and chemical stability, and various other advantageous properties. For instance, the CO2 adsorption capacities of zeolites ZSM-5, LEZ-Zeolite Na(X), and Na(Y) are 5.2, 0.7, 5.14, and 7.06 mmol g−1, respectively, at room temperature and ambient pressure.5,6 MOFs MOF-177, MIL-100(Fe), and MiL-101(Cr) exhibit CO2 adsorption capacities of 33.5, 0.67, and 1.05 mmol g−1, respectively.7,8 Chen and co-workers reported CO2 adsorption capacities of 2.7–5 and 0.2–3 mmol g−1 on activated carbons and carbon nanotubes (CNTs) at 273 K below 0.1 MPa.9 Zafanelli and co-workers reported activated carbons adsorbed up to 3.17 mmol g−1 CO2 at 586 K and 0.12 MPa.10 Osler and co-workers reported that multiwalled CNT/chitosan composite had an adsorption capacity of 0.07 mmol g−1.11 Amine-based adsorbents exhibit an adsorption capacity of 0.03 mmol g−1 at 348 K and ambient pressure.12 Metal oxides, such as Ce and Mn–CaO, demonstrate high CO2 adsorption capacities, reaching 14 mmol g−1 at a high temperature of 1046 K and 0.1 MPa.13
Converting CO2 through chemical processes is a challenging endeavor, because of the remarkable thermodynamic stability of CO2 molecules.14 The breaking of the chemical bonds in CO2 requires substantial energy. Researchers have devised innovative approaches to overcome this challenge using catalysts. An example involves the use of nanomaterial-based catalysts, along with operating under high pressure and temperature conditions.15 Although these techniques effectively satisfy the energy demands for CO2 conversion, rendering the process more attainable, high temperature and pressure reactions can be energetically costly. Therefore, approaches for catalytic CO2 conversion that are less energetic are required. Electrocatalytic, photocatalytic, and thermocatalytic methods are crucial techniques for converting CO2 into valuable chemicals and fuels. In addition, CO2 can be harnessed for mineral carbonation by leveraging metals such as calcium or magnesium to form carbonates.16 Moreover, microalgal cultivation is a promising method for capturing CO2 from waste streams and converting it into biofuels. However, it requires huge land areas, and process control is complex.17 Photocatalytic CO2 reduction exhibits remarkable selectivity, enabling the conversion of CO2 into various carbon products, such as carbon monoxide (2003 μmol g−1 h−1), formic acid (46 μmol g−1 h−1), formaldehyde (1 μmol g−1 h−1), methanol (186 μmol g−1 h−1), methane (4200 μmol g−1 h−1), acetic acid (39 μmol g−1 h−1), acetaldehyde (572 μmol g−1 h−1), and ethanol (52 μmol g−1 h−1), using ruthenium-based catalysts, gold nanoparticles supported on TiO2 sheets, carbon nitride-CdS quantum dots, alumina silicates, and titanium-based catalysts.18–25
Thermocatalytic CO2 conversion is another potential method for achieving efficient CO2 conversion. Thermocatalytic methanation involves the conversion of CO2 into methane using a Ca-inserted NiTiO3 perovskite catalyst with H2 as a reduction gas at 623 K, achieving an impressive 87.32% conversion rate,26 which is called the Sabatier reaction. In the chemical scheme, CO2 and H2 combine in the presence of a suitable catalyst, often a metal catalyst such as nickel, to yield methane and water, typically within the temperature range of 473–823 K.27 The reverse water–gas shift reaction is predominant to generate CO from CO2 and H2 above 873 K. CO can serve as a precursor for a wide array of chemical applications, including its role in the Fischer–Tropsch synthesis to produce liquid fuels.28 The hydrogenation of CO2 can also result in the formation of methanol when hydrogen gas is present along with a catalyst-based metal.29 CO2 is directly converted into solid carbon materials, including CNTs or graphene, by increasing the temperature and pressure. The direct conversion of CO2 into solid carbon is the subject of ongoing research for carbon capture and utilization. Carbon materials are widely used in various applications, including energy storage, batteries, supercapacitors, drug delivery, water purification, electrodes, and catalyst support.30–32 Diamond can be generated from CO2 through the LiCl–Li2O molten salt method; CO2 conversion into diamond involves subsequent conversion into Li2CO3 crystals.33 Graphite is synthesized at 1048 K using a CO2 and SO2 gas mixture in molten salts of Li2CO3–Na2CO3–K2CO3–Li2SO4.34 The electrochemical method for carbon material production from CO2 requires high operating temperatures exceeding 1023 K, because of the use of molten salts. Kim and co-workers produced CNTs at 773–873 K and 0.1 MPa using NaBH4 and metallic Ni.35 Despite this achievement, the development of CO2 conversion technique at lower temperatures is required. We succeeded in converting CO2 to graphitic carbon at 700 K and ambient pressure using perovskite-type titanium nanocatalysts with high reaction rates ranging from 1600 to 3300 μmol g−1 h−1.36 Nano-barium titanate is an effective adsorbent material, because of its substantial surface area and numerous active sites. In this study, we demonstrate pressure-dependent thermocatalytic CO2 conversion to solid carbons from 0.01 to 1 MPa at 700 K using barium titanate nanocatalysts and evaluate the key mechanism with an elementary reaction process.
Experimental
Preparation of barium titanate nanocatalyst
Barium titanate nanocatalysts were synthesized using the solvothermal method in an N2-filled glove box. Each barium ethoxide and titanium tetraisopropoxide (>99%; Kojundo Chemical Laboratory Co., Saitama, Japan) were dissolved in a 10 mL methanol and methoxyethanol mixed solution, with a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (>99%; Fujifilm Wako Pure Chemical Co., Saitama, Japan) to prepare a 200 mM solution and then vigorously agitated for 3 h. Following the successful blending of the precursors, water was added to the precursor solution to maintain a molar ratio of Ba:Ti:H2O = 1:1:5. The mixture was heated in an autoclave at 400 K for 24 h. Finally, barium titanate nanocatalysts were obtained after drying at 333 K for one day in ambient air.
:2 (>99%; Fujifilm Wako Pure Chemical Co., Saitama, Japan) to prepare a 200 mM solution and then vigorously agitated for 3 h. Following the successful blending of the precursors, water was added to the precursor solution to maintain a molar ratio of Ba:Ti:H2O = 1:1:5. The mixture was heated in an autoclave at 400 K for 24 h. Finally, barium titanate nanocatalysts were obtained after drying at 333 K for one day in ambient air.
Characterization of barium titanate nanocatalyst
The crystallinity of barium titanate nanocatalysts was evaluated by X-ray diffraction (XRD) (SmartLab, Rigaku Co., Tokyo, Japan) with Cu Kα radiation (wavelength λ = 0.1541 nm) at an X-ray generator voltage of 40 kV and current of 40 mA, within the 2θ range of 10–90°, employing an angular step size of 0.01°. The crystallite sizes of the nanocatalysts were determined using the Scherrer equation,37 which relies on the full peak widths at half maximum. A Scherrer constant of 0.89 was used for calculations.Catalytic activity test
Barium titanate nanocatalysts were first heated at 700 K in an O2 atmosphere for 24 h to eliminate surface contaminants before the catalytic activity test. The reaction stainless-steel cell with a quartz tube was also preheated at 700 K. A 200 mg barium titanate nanocatalyst was placed in a quartz tube supported by quartz wool. CO2 adsorption and reactions on the nanocatalysts were initiated at 700 K after heating at a rate of 10 K min−1. CO2 gas at 0.01, 0.05, 0.1, 0.5, and 1.0 MPa flowed in the reaction cell for 24 h. The catalyst weights before and after CO2 adsorption/reaction were evaluated to determine the reaction amount of CO2. Thermogravimetric analysis (TGA; Thermo Plus EVO2, Rigaku Co., Tokyo, Japan) was also conducted to evaluate the amount of carbon reduced from CO2 using the nanocatalysts after CO2 adsorption/reaction. Reduced carbon was evaluated by Raman scattering spectroscopy with a Nd:YAG laser (532 nm) at a power of 0.1 mW (NRS-3000; JASCO Co., Tokyo, Japan) with an accumulation time of 300 s and 10–15 point average, TGA and transmission electron microscopy at 120 keV (TEM; JEM-2100F, JEOL Co., Tokyo, Japan). The D- and G-bands in the Raman spectra indicate the presence of amorphous and crystalline graphitic carbon in the barium titanate nanocatalysts, respectively. The weight changes of the nanocatalysts with reduced carbon were examined by TGA in an O2 atmosphere (>99.7%) at a flow rate of 100 mL min−1 at 300–1373 K. The rate of temperature increase was 10 K min−1 and the air was replaced with Ar gas (>99.99%) at 300 K for 5 h before TGA measurements. We simultaneously employed carbon detection using a custom mass spectrometry system that includes BELLMASS II (MicrotracBEL Co., Osaka, Japan) for temperature-programmed desorption (TPD) analyses in O2 atmosphere. TPD measurements were performed using a TGA device. The evolved gases during the oxidation of nanocatalysts after CO2 thermolysis are systematically analyzed in thermal reactions with O2 gas (100 mL min−1) involving Ar gas (100 mL min−1) as a flowing gas in the temperature range of 300–1373 K. The exhaust gases were evaluated using mass spectroscopy.Results and discussion
Barium titanate nanocatalysts have a high potential for CO2 thermolysis, as reported elsewhere.36 CO2 was adsorbed/reduced onto the nanocatalyst surface, which influenced the nanocatalyst weights. CO2 gas at 0.01–1.0 MPa was first introduced in a stainless cell with barium titanate nanocatalysts, and CO2 thermolysis was performed on the nanocatalysts at 700 K. The amount of reduced carbon attached to the nanocatalysts was evaluated from the weight-decreasing curves of the nanocatalysts after CO2 thermolysis in an O2 atmosphere (Fig. S1†). That is, the weights of nanocatalyst with chemisorbed/reduced CO2 were decreased by the removal of chemisorbed CO2 and/or the oxidation of reduced carbon by heating in an O2 atmosphere. The first decreasing step was a result of the removal of chemisorbed CO2 at 300–600 K, and the reduced carbons were then oxidized above 600 K. The slight gradual increase in the amount at high temperatures above 1200 K was due to additional oxidation of nanocatalyst itself in the O2 atmosphere. Fig. 1a shows the amount of chemisorbed/reduced CO2 at 0.01–1.0 MPa, as evaluated from the TGA in Fig. S1.† The CO2 chemisorbed/reduced amounts considerably increased up to 670 μmol g−1 in the low-pressure region between 0 and 0.05 MPa and gradually increased to 800 μmol g−1 below 1 MPa. Barium titanate nanocatalysts had a 10.2 nm crystallite size without any impurities (Fig. 1b). The crystal structures were hardly changed by the CO2 reduction reaction at 700 K and 0.01–1.0 MPa, although tiny amounts of impurities attributed to barium carbonate appeared by BaCO3-like structure formation on the nanocatalyst surface via CO2 chemisorption. In our preceding report, we indicated that surface-chemisorbed CO2 and reduced carbon would be released by preheating in the O2 atmosphere and the catalytic activity would be recovered.36,38 In addition, the crystal structure of nanocatalyst was maintained during heating. Meanwhile, a peak attributed to amorphous/graphitic carbons was hardly observed, whereas the phase periodicity of carbon is necessary for apparent diffraction to generate distinct XRD peaks.39 On the other hand, sp2 hybridized carbons could be identified by observing the D- and G-bands at 1300–1340 and 1600 cm−1 in Raman spectra, respectively.40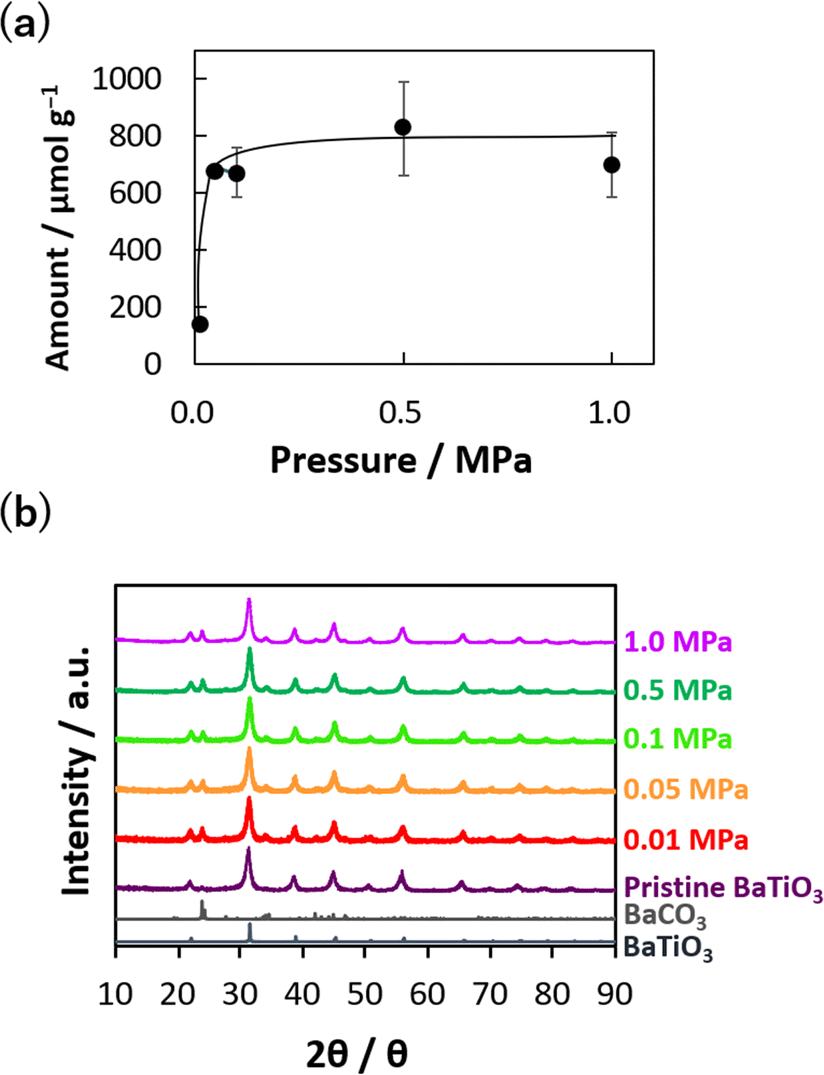 | ||
| Fig. 1 (a) Weight increase of nanocatalysts after CO2 thermolysis at 0.01–0.5 MPa. (b) XRD patterns of barium titanate nanocatalysts after CO2 thermolysis. XRD patterns of barium carbonate and crystalline barium titanate are also shown as references. | ||
Fig. 2 shows the Raman scattering spectra of the nanocatalysts after CO2 thermolysis and the pristine nanocatalyst (see also in Fig. S2†). The barium titanate nanocatalysts originally had a peak at 1500 cm−1; thus, the peak intensities were normalized using the peak at 1500 cm−1. Additional peaks on the D- and G-bands were increasingly observed with higher-pressure treatment in CO2 thermolysis, accompanied by peak broadening. The Raman scattering spectra of typical carbons, such as acetylene black and Madagascar graphite, are also shown in Fig. 2. Acetylene black has distinctive D- and G-bands at 1336 and 1592 cm−1, respectively, as expected from a lack of well-defined structural order,41 whereas Madagascar graphite has only G-band at 1580 cm−1. Ferrari and Robertson revealed that the G-band peak aligns with the in-plane bond stretching of sp2 C atom pairs, while the D-band peak is associated with the breathing modes of rings.42 Defects such as vacancies and dislocations yield D-band peaks. Notably, the Raman scattering spectra of the nanocatalysts resembled that of less graphitized carbon, and the CO2-reduced carbon amounts increased with reaction pressure. In addition, a broad peak that emerged in the range of 1410–1460 cm−1 was observed for high-pressure CO2 thermolysis, associated with a defective D-band caused by structural anomalies.43,44 The Raman scattering spectra at 1400–1470 cm−1 were an indicator of structural disorder and defects in carbon materials.42,43 We summarize the formation of carbon by CO2 thermolysis at different pressures. By CO2 thermolysis at 0.01 and 0.05 MPa, carbon structures were slightly observed, indicating slight/partial CO2 reduction. Graphitic carbons with disordered (amorphous) structures were observed at 0.1 and 0.5 MPa.43 The disordered structures were significant after CO2 thermolysis at 1.0 MPa. This result indicated that high-pressure CO2 gas was reduced even on less active sites in a chain reaction.
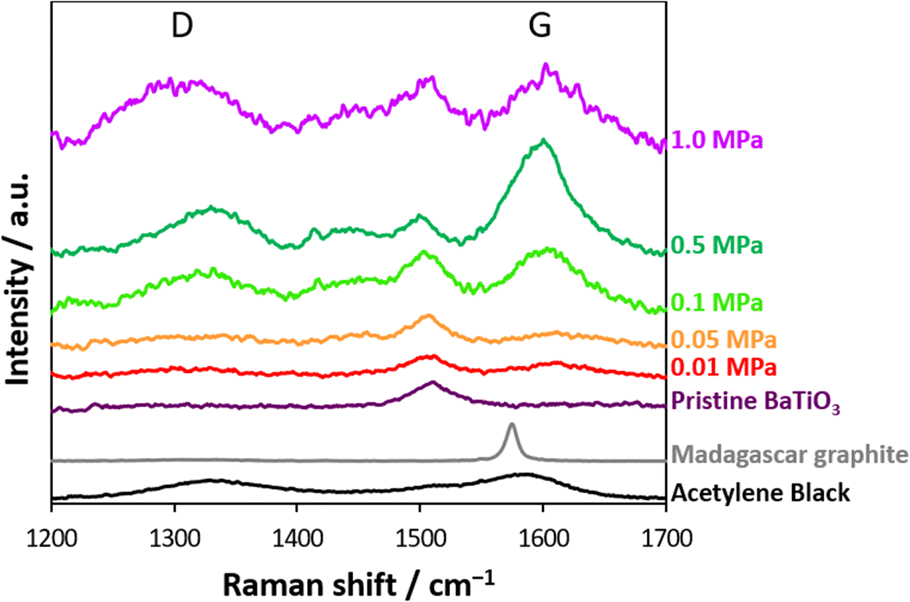 | ||
| Fig. 2 Raman scattering spectra of the nanocatalysts before and after CO2 thermolysis. Raman scattering spectra of acetylene black and Madagascar graphite are shown for reference. | ||
The TEM images in Fig. 3 show carbon coating on particles of barium titanate nanocatalysts after CO2 thermolysis at 700 K under pressures of 0.01, 0.05, 0.1, 0.5, and 1.0 MPa for 24 h. Carbon products resulting from CO2 reduction considerably increased with CO2 pressure, which was also expected from the optical color change from white to grayish-black. CO2 thermolysis at 0.01 MPa hardly produced carbons, whereas the TEM images above 0.05 MPa indicated catalytic reduction of CO2, corresponding to the isothermal weight change analysis in Fig. 1a. The weight change observed in the low-pressure CO2 reactions was primarily attributed to chemisorbed CO2, while chemisorbed CO2 was hardly observed in the TEM images in Fig. 3b. Reduced carbon was observed from 0.05 MPa in Fig. 3c. A large amount of carbon products was observed on the nanocatalysts after CO2 thermolysis above 0.1 MPa (Fig. 3d–f), reaching a detectable level of carbon deposited on nanocatalysts in Raman spectroscopy (Fig. 2).
 | ||
| Fig. 3 TEM images of (a) pristine nanocatalyst and nanocatalyst after CO2 thermolysis at (b) 0.01, (c) 0.05, (d) 0.1, (e) 0.5, and (f) 1.0 MPa. The insets show the optical images of the nanocatalysts. | ||
The dependence of CO2 thermolysis duration was also examined for CO2 thermolysis at 0.5 MPa, which was the best pressure condition for producing graphitic carbon (Fig. 2). The reduced carbon attached to the nanocatalysts after CO2 thermolysis was quantitively analyzed by the TGA; the amount after thermolysis at 0.5 MPa for 1, 5, and 10 days were 830 ± 160, 1060, and 1450 μmol g−1, respectively. Fig. 4a shows the XRD patterns of the nanocatalysts after CO2 thermolysis. The nanocatalyst structures hardly changed, while BaCO3 peaks appeared increasingly, probably due to surface carbonate formation on the Ba sites. Crystalline and amorphous graphitic structures were also observed in the Raman scattering spectra (Fig. 4b); D-, defective D-, and G-bands increased with heating duration in CO2 thermolysis. Graphitic carbons mostly increased, because of an increase in the G-band intensity.
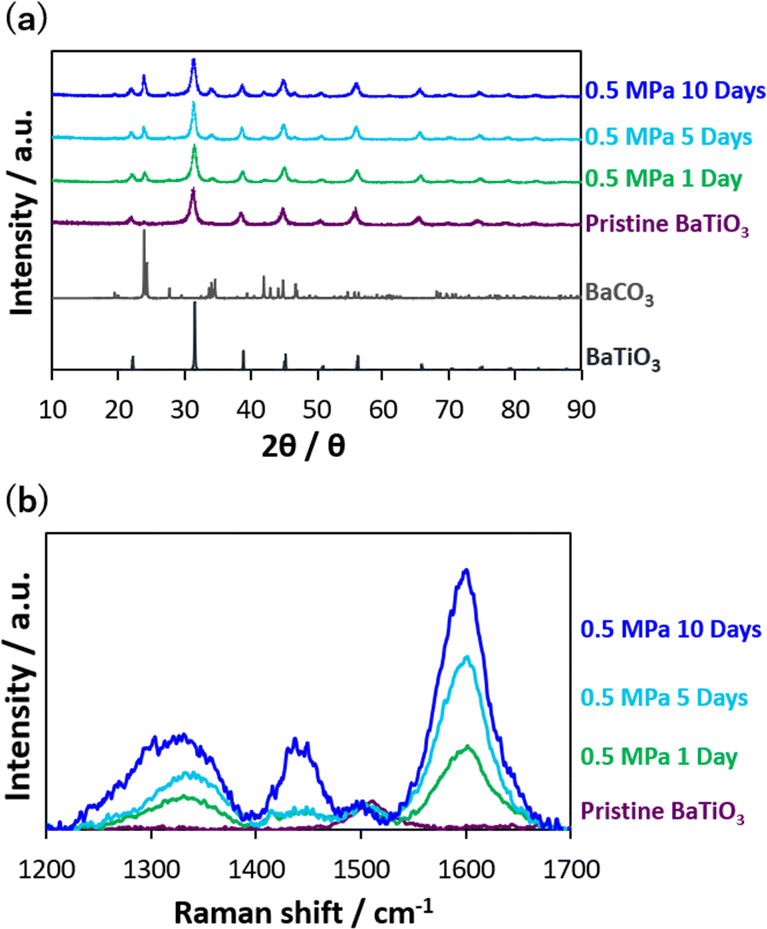 | ||
| Fig. 4 (a) XRD of nanocatalysts at 700 K and 0.5 MPa for 1–10 days. (b) Raman spectra of pristine barium titanate and nanocatalysts after CO2 thermolysis. | ||
Fig. 5 shows carbon removal from nanocatalysts after CO2 thermolysis by CO2-TPD-mass spectra in an O2 atmosphere in which CO2 was produced on nanocatalysts with carbons by oxidation. Carbon structures were thus assessed from distinctive temperatures during TPD measurements.45 The CO2-TPD-mass spectra of acetylene black and Madagascar graphite are shown in Fig. S3.† Broad CO2 peaks on carbon materials appeared at 400 K, probably due to the removal of surface oxygen groups, which were also observed elsewhere.46–49 The oxidation temperature of carbon materials associated with the strong, sharp peaks indicated that less- and high-crystalline carbons were oxidized at 1000 and 1270 K, respectively. The nanocatalysts after CO2 thermolysis at 0.01 MPa, as well as the pristine nanocatalyst, had no CO2 peak, indicating no detectable level of CO2 reduction. Meanwhile, CO2 mass peaks were observed for nanocatalysts above 0.05 MPa. The CO2 peak between 300–500 K was due to the desorption of chemisorbed CO2 on the nanocatalyst surface. Carbon dioxide evolved at 800–900 K and slightly at 900–1100 K for the nanocatalysts at 0.05 MPa, in which amorphous and graphitic carbons on the nanocatalysts were oxidized, respectively. Here, oxidation of typical carbon materials is observed in the range of 600–700 K for activated carbons, 900–1100 K for graphitized carbon, and 500–1100 K for high-surface-area graphite, polycrystalline graphite, nonporous carbon, and diamond powders.46,47,50–54 Chemisorbed CO2 peaks were also observed for the nanocatalysts above 0.1 MPa, while CO2 peaks by the oxidation of amorphous carbons between 800 and 900 K were weakened, and alternatively, the peaks by the oxidation of graphitic carbons above 900 K were distinct. This was expected based on the Raman spectra shown in Fig. 2. Therefore, various carbon structures were formed on the nanocatalysts after CO2 thermolysis. Previous studies also indicated graphitic carbons on Pt/TiO2 and Ru/TiO2 catalysts,55 filamentous carbons on Pt/MgO–Al2O3 catalysts, Ru support on modified SiO2 catalysts, and Rh/γ-Al2O3–La2O3 catalysts.56–58 Ghelamallah and Grangers identified four types of carbon on Rh/La2O3; the CO2 peaks below 523 K, within 635–661 K, within 693–798 K, and above 973 K demonstrated weakly bound carbon deposited directly on the rhodium metal, carbons at the boundary between the metal and lanthanum oxide support, carbons on the lanthanum oxide, and graphitic carbon, respectively.59 This indicated that the nanocatalysts had strong CO2 reduction activity, assumed from graphitic carbon production, which was evaluated by the high-temperature oxidation of carbons above 900 K. The heating duration during CO2 thermolysis also influenced the carbon structure of the nanocatalysts, as evaluated from the CO2-TPD-mass spectra in Fig. 5. The oxidation peak for the 0.5 MPa-based CO2 thermolysis at 1050 K was intense in five days and was split into two peaks at 1000 and 1150 K in ten days. Graphitic carbon changed to less- and higher-crystalline graphitic carbons during long-term CO2 thermolysis by aging associated with crysal growth of carbons and remained amorphous region.
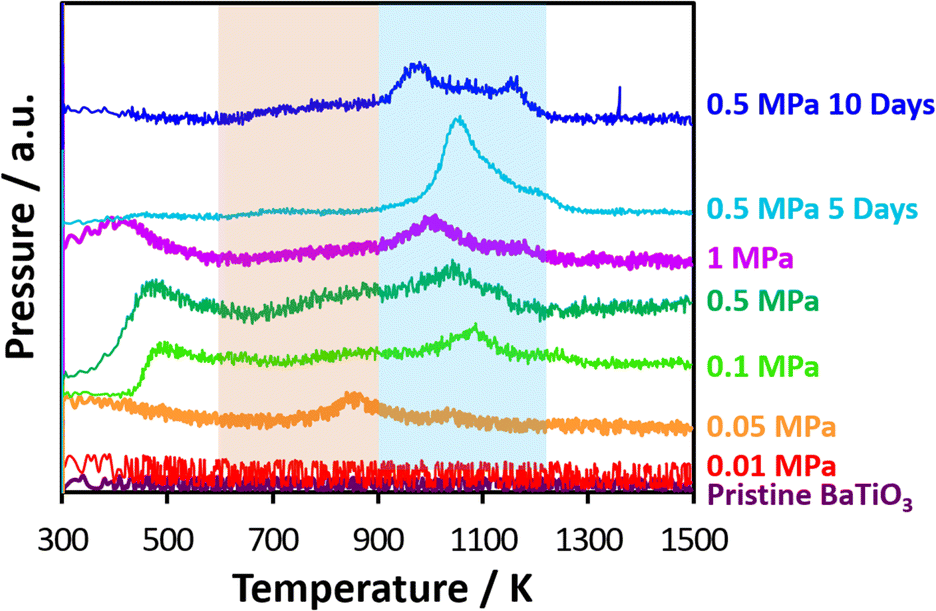 | ||
| Fig. 5 TPD spectra of CO2-reduced carbons on nanocatalysts in O2 atmosphere after CO2 thermolysis at 0.01, 0.05, 0.1, 0.5, and 1.0 MPa for one day and 0.5 MPa for five and ten days. | ||
The possible reaction mechanism for CO2 reduction on the nanocatalysts may originate from CO2 chemisorption onto an active metal surface to the dissociation of carbon dioxide to carbon monoxide (2CO2 → CO + ½O2(g)) and then further reaction to solid carbon with reduction and a disproportionation reaction (2CO → C(s) + ½O2(g) and 2CO → C(s) + CO2(g)), although O2 production was not detected in the TPD. A high temperature is normally required for the exothermic reduction reactions, but the above reduction reactions of CO2 and CO on the nanocatalysts are promoted at higher pressure by Le Chatelier's principle. On the other hand, the disproportionation reaction promotes carbon solidification (2CO → C(s) + CO2(g)) at temperatures lower than 900 K. Therefore, superior CO2 reduction activity on barium titanate nanocatalysts at 700 K facilitated further reduction to solid carbons.
Conclusions
In this study, CO2 conversion into solid carbons using barium titanate nanocatalysts at 700 K and various pressures was demonstrated to reveal the unique reaction mechanism and pressure dependence of CO2 thermolysis. The nanocatalysts facilitated CO2 reduction to amorphous and graphitic carbons at 700 K and high pressure, especially at 0.1–0.5 MPa, where a greater amount of reduced carbon was observed from TEM and optical images, Raman scattering spectroscopies, and TPD-Mass analyses of the nanocatalysts after CO2 thermolysis reaction. The findings of this study indicated that CO2 reduction to CO on the nanocatalysts facilitated further reduction to graphitic carbon at high pressures, even at 700 K. This knowledge is essential for optimizing reaction conditions and maximizing CO2 conversion efficiency using more sustainable technology. However, further investigations are required to evaluate the CO2 reduction mechanism associated with theoretical analyses in order to develop highly active CO2 reduction catalysts.Data availability
The data supporting this article have been included as part of the ESI.† The data that support the findings of this study are available from the corresponding author, T. O., upon reasonable request.Author contributions
ST: methodology, investigation, analyzing the data, writing the original draft of the manuscript; MM: analyzing the data; TW: methodology; TO; supervision, conceptualization, writing, reviewing, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TEM measurements were conducted at the Center for Analytical Instrumentation in Chiba University. This research was supported by JSPS KAKENHI Grant Number 23H01999 and Suzuki Foundation.References
- M. M. Ramirez-Corredores, M. R. Goldwasser and E. Falabella de Sousa Aguiar, in Decarbonization as a Route Towards Sustainable Circularity, ed. M. M. Ramirez-Corredores, M. R. Goldwasser and E. Falabella de Sousa Aguiar, Springer International Publishing, Cham, 2023, pp. 1–14 Search PubMed.
- P. Friedlingstein, M. O'Sullivan, M. W. Jones, R. M. Andrew, L. Gregor, J. Hauck, C. Le Quéré, I. T. Luijkx, A. Olsen, G. P. Peters, W. Peters, J. Pongratz, C. Schwingshackl, S. Sitch, J. G. Canadell, P. Ciais, R. B. Jackson, S. R. Alin, R. Alkama, A. Arneth, V. K. Arora, N. R. Bates, M. Becker, N. Bellouin, H. C. Bittig, L. Bopp, F. Chevallier, L. P. Chini, M. Cronin, W. Evans, S. Falk, R. A. Feely, T. Gasser, M. Gehlen, T. Gkritzalis, L. Gloege, G. Grassi, N. Gruber, Ö. Gürses, I. Harris, M. Hefner, R. A. Houghton, G. C. Hurtt, Y. Iida, T. Ilyina, A. K. Jain, A. Jersild, K. Kadono, E. Kato, D. Kennedy, K. Klein Goldewijk, J. Knauer, J. I. Korsbakken, P. Landschützer, N. Lefèvre, K. Lindsay, J. Liu, Z. Liu, G. Marland, N. Mayot, M. J. McGrath, N. Metzl, N. M. Monacci, D. R. Munro, S. I. Nakaoka, Y. Niwa, K. O'Brien, T. Ono, P. I. Palmer, N. Pan, D. Pierrot, K. Pocock, B. Poulter, L. Resplandy, E. Robertson, C. Rödenbeck, C. Rodriguez, T. M. Rosan, J. Schwinger, R. Séférian, J. D. Shutler, I. Skjelvan, T. Steinhoff, Q. Sun, A. J. Sutton, C. Sweeney, S. Takao, T. Tanhua, P. P. Tans, X. Tian, H. Tian, B. Tilbrook, H. Tsujino, F. Tubiello, G. R. van der Werf, A. P. Walker, R. Wanninkhof, C. Whitehead, A. Willstrand Wranne, R. Wright, W. Yuan, C. Yue, X. Yue, S. Zaehle, J. Zeng and B. Zheng, Earth Syst. Sci. Data, 2022, 14, 4811–4900 CrossRef.
- R. Falkner, Int. Aff., 2016, 92, 1107–1125 CrossRef.
- T. M. Gür, Prog. Energy Combust. Sci., 2022, 89, 100965 CrossRef.
- Y. Cho, J.-Y. Lee, A. D. Bokare, S.-B. Kwon, D.-S. Park, W.-S. Jung, J.-S. Choi, Y.-M. Yang, J.-Y. Lee and W. Choi, J. Ind. Eng. Chem., 2015, 22, 350–356 CrossRef CAS.
- L. Yu, J. i. Gong, C. Zeng and L. Zhang, Sep. Purif. Technol., 2013, 118, 188–195 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- S. Xian, J. Peng, Z. Zhang, Q. Xia, H. Wang and Z. Li, Chem. Eng. J., 2015, 270, 385–392 CrossRef CAS.
- L. Chen, T. Watanabe, H. Kanoh, K. Hata and T. Ohba, Adsorpt. Sci. Technol., 2017, 36, 625–639 CrossRef.
- L. F. A. S. Zafanelli, A. Henrique, H. Steldinger, J. L. Diaz de Tuesta, J. Gläsel, A. E. Rodrigues, H. T. Gomes, B. J. M. Etzold and J. A. C. Silva, Microporous Mesoporous Mater., 2022, 335, 111818 CrossRef CAS.
- K. Osler, N. Twala, O. O. Oluwasina and M. O. Daramola, Energy Proc., 2017, 114, 2330–2335 CrossRef CAS.
- T. Chitsiga, M. O. Daramola, N. Wagner and J. Ngoy, Energy Proc., 2016, 86, 90–105 CrossRef CAS.
- H. Guo, X. Kou, Y. Zhao, S. Wang, Q. Sun and X. Ma, Chem. Eng. J., 2017, 334, 237–246 CrossRef.
- A. Ateka, P. Rodriguez-Vega, J. Ereña, A. T. Aguayo and J. Bilbao, Fuel Process. Technol., 2022, 233, 107310 CrossRef CAS.
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Chem. Eng. J., 2021, 409, 128138 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- L. N. Nguyen, M. T. Vu, H. P. Vu, M. A. H. Johir, L. Labeeuw, P. J. Ralph, T. M. I. Mahlia, A. Pandey, R. Sirohi and L. D. Nghiem, Crit. Rev. Environ. Sci. Technol., 2023, 53, 216–238 CrossRef.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS.
- K. Maeda, R. Kuriki, M. Zhang, X. Wang and O. Ishitani, J. Mater. Chem. A, 2014, 2, 15146–15151 RSC.
- W. Hou, W. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- A. Li, T. Wang, C. Li, Z. Huang, Z. Luo and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 3804–3808 CrossRef CAS.
- J. Albero, H. Garcia and A. Corma, Top. Catal., 2016, 59, 787–791 CrossRef CAS.
- F. Yu, X. Jing, Y. Wang, M. Sun and C. Duan, Angew. Chem., Int. Ed., 2021, 60, 24849–24853 CrossRef CAS PubMed.
- X. Qian, W. Yang, S. Gao, J. Xiao, S. Basu, A. Yoshimura, Y. Shi, V. Meunier and Q. Li, ACS Appl. Mater. Interfaces, 2020, 12, 55982–55993 CrossRef CAS.
- R. Das, K. Das, B. Ray, C. P. Vinod and S. C. Peter, Energy Environ. Sci., 2022, 15, 1967–1976 RSC.
- J. Y. Do, N.-K. Park, M. W. Seo, D. Lee, H.-J. Ryu and M. Kang, Fuel, 2020, 271, 117624 CrossRef CAS.
- B. Alrafei, I. Polaert, A. Ledoux and F. Azzolina-Jury, Catal. Today, 2020, 346, 23–33 CrossRef CAS.
- Y. Qi, Y.-A. Zhu and D. Chen, Green Chem. Eng., 2020, 1, 131–139 CrossRef.
- G. Lombardelli, M. Mureddu, S. Lai, F. Ferrara, A. Pettinau, L. Atzori, A. Conversano and M. Gatti, J. CO2 Util., 2022, 65, 102240 CrossRef CAS.
- K. Turcheniuk and V. N. Mochalin, Nanotechnology, 2017, 28, 252001 CrossRef CAS PubMed.
- X. Zhang, B. Gao, A. Creamer, C. Cao and Y. Li, J. Hazard. Mater., 2017, 338, 102–123 CrossRef CAS.
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed.
- A. R. Kamali, Carbon, 2017, 123, 205–215 CrossRef CAS.
- Z. Chen, Y. Gu, L. Hu, W. Xiao, X. Mao, H. Zhu and D. Wang, J. Mater. Chem. A, 2017, 5, 20603–20607 RSC.
- G. M. Kim, W.-G. Lim, D. Kang, J. H. Park, H. Lee, J. Lee and J. W. Lee, Nanoscale, 2020, 12, 7822–7833 RSC.
- T. Watanabe and T. Ohba, ACS Sustain. Chem. Eng., 2021, 9, 3860–3873 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- T. Watanabe and T. Ohba, Nanoscale, 2022, 14, 8318–8325 RSC.
- K. Judai, N. Iguchi and Y. Hatakeyama, J. Chem., 2016, 2016, 7840687 Search PubMed.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2477–2512 CrossRef CAS PubMed.
- D. R. Tallant, T. A. Friedmann, N. A. Missert, M. P. Siegal and J. P. Sullivan, MRS Online Proc. Libr., 1997, 498, 37–48 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- T. Kyotani, J.-i. Ozaki and T. Ishii, Carbon Rep., 2022, 1, 188–205 CrossRef.
- B. Marchon, J. Carrazza, H. Heinemann and G. A. Somorjai, Carbon, 1988, 26, 507–514 CrossRef CAS.
- B. Marchon, W. T. Tysoe, J. Carrazza, H. Heinemann and G. A. Somorjai, J. Phys. Chem., 1988, 92, 5744–5749 CrossRef CAS.
- P. Novák, J. Ufheil, H. Buqa, F. Krumeich, M. E. Spahr, D. Goers, H. Wilhelm, J. Dentzer, R. Gadiou and C. Vix-Guterl, J. Power Sources, 2007, 174, 1082–1085 CrossRef.
- T. Ishii, Y. Kaburagi, A. Yoshida, Y. Hishiyama, H. Oka, N. Setoyama, J.-i. Ozaki and T. Kyotani, Carbon, 2017, 125, 146–155 CrossRef CAS.
- A. Dandekar, R. T. K. Baker and M. A. Vannice, Carbon, 1998, 36, 1821–1831 CrossRef CAS.
- S. Haydar, C. Moreno-Castilla, M. A. Ferro-García, F. Carrasco-Marín, J. Rivera-Utrilla, A. Perrard and J. P. Joly, Carbon, 2000, 38, 1297–1308 CrossRef CAS.
- M. R. Cuervo, E. Asedegbega-Nieto, E. Díaz, S. Ordóñez, A. Vega, A. B. Dongil and I. Rodríguez-Ramos, Carbon, 2008, 46, 2096–2106 CrossRef CAS.
- H. F. Gorgulho, J. P. Mesquita, F. Gonçalves, M. F. R. Pereira and J. L. Figueiredo, Carbon, 2008, 46, 1544–1555 CrossRef CAS.
- G. Hotová and V. Slovák, Anal. Chem., 2017, 89, 1710–1715 CrossRef PubMed.
- S. A. Singh and G. Madras, Appl. Catal., A, 2016, 518, 102–114 CrossRef CAS.
- D. C. Carvalho, H. S. A. de Souza, J. M. Filho, A. C. Oliveira, A. Campos, É. R. C. Milet, F. F. de Sousa, E. Padron-Hernandez and A. C. Oliveira, Appl. Catal., A, 2014, 473, 132–145 CrossRef CAS.
- S. Das, M. Shah, R. K. Gupta and A. Bordoloi, J. CO2 Util., 2019, 29, 240–253 CrossRef CAS.
- M. Ghelamallah and P. Granger, Fuel, 2012, 97, 269–276 CrossRef CAS.
- M. Ghelamallah and P. Granger, Appl. Catal., A, 2014, 485, 172–180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00253a |
| This journal is © The Royal Society of Chemistry 2024 |