
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali and alkaline earth metals in liquid salts for supercapatteries
Qiang
Guo
 abc,
Peiying
Fan
a,
Yuhan
Zhang
a,
Li
Guan
a,
Han
Wang
a,
Anna
Croft
*ad and
George Zheng
Chen
*a
abc,
Peiying
Fan
a,
Yuhan
Zhang
a,
Li
Guan
a,
Han
Wang
a,
Anna
Croft
*ad and
George Zheng
Chen
*a
aDepartment of Chemical and Environmental Engineering, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: george.chen@nottingham.ac.uk
bNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, P. R. China
cDepartment of Chemical and Environmental Engineering, Faculty of Science and Engineering, University of Nottingham Ningbo China, Ningbo 315100, P. R. China
dDepartment of Chemical Engineering, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK. E-mail: a.k.croft@lboro.ac.uk
First published on 29th November 2023
Abstract
The full oxidation of lithium metal (4Li + O2 ⇌ 2Li2O) offers a mass normalised Gibbs energy change greater than that for the combustion of carbon (C + O2 ⇌ CO2) or any hydrocarbon fuel (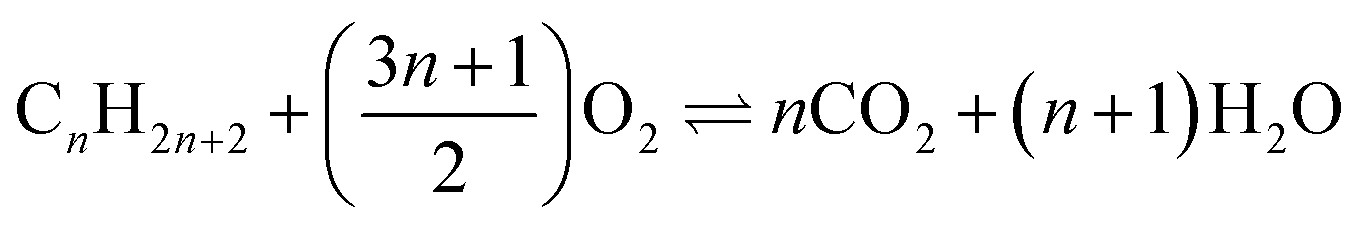 ). This thermodynamic comparison promises a lithium–oxygen (air) battery with a petrol comparable energy density. Similar analyses apply to other abundant alkali and alkaline earth metals (AAEMs) which all feature very high specific charge capacity and the most negative electrode potentials. The success of lithium ion batteries (LIBs) in both research and commercial development confirms these thermodynamic predictions. However, the experimentally demonstrated energy capacities of all AAEM-based batteries are only small fractions of the thermodynamic values. A main cause is that a satisfactory oxygen positive electrode (positrode) is still to be developed, whilst the very few options of AAEM storage positrodes still do not match AAEM negative electrodes (negatrodes) in charge capacity. Another challenge results from the complicated interactions between AAEMs and the currently used organic carbonate electrolytes that not only reduce the negatrode capacity but also exert restrictions on both electron and ion transfers. The flammability of currently used organic electrolytes is another major concern with respect to the safety of AAEM batteries. Herein, we introduce the concept and potential, and review the relevant practices of a promising ionic liquid supercapattery that couples an AAEM negatrode with a supercapacitor positrode to bypass the thermodynamic and kinetic difficulties of an oxygen or AAEM storage positrode. The further discussion aims at the selection of ionic liquid-based electrolytes that can enable the reversible anodic dissolution of AAEMs and a wide potential window for the supercapacitor positrode. The use of molten salt-based electrolytes is also postulated and analysed, not only because of their high ionic conductivity, low cost and unique applications, but also their high temperatures that eliminate dendritic growth on the liquid AAEM negatrode and heat buildup in the cell.
). This thermodynamic comparison promises a lithium–oxygen (air) battery with a petrol comparable energy density. Similar analyses apply to other abundant alkali and alkaline earth metals (AAEMs) which all feature very high specific charge capacity and the most negative electrode potentials. The success of lithium ion batteries (LIBs) in both research and commercial development confirms these thermodynamic predictions. However, the experimentally demonstrated energy capacities of all AAEM-based batteries are only small fractions of the thermodynamic values. A main cause is that a satisfactory oxygen positive electrode (positrode) is still to be developed, whilst the very few options of AAEM storage positrodes still do not match AAEM negative electrodes (negatrodes) in charge capacity. Another challenge results from the complicated interactions between AAEMs and the currently used organic carbonate electrolytes that not only reduce the negatrode capacity but also exert restrictions on both electron and ion transfers. The flammability of currently used organic electrolytes is another major concern with respect to the safety of AAEM batteries. Herein, we introduce the concept and potential, and review the relevant practices of a promising ionic liquid supercapattery that couples an AAEM negatrode with a supercapacitor positrode to bypass the thermodynamic and kinetic difficulties of an oxygen or AAEM storage positrode. The further discussion aims at the selection of ionic liquid-based electrolytes that can enable the reversible anodic dissolution of AAEMs and a wide potential window for the supercapacitor positrode. The use of molten salt-based electrolytes is also postulated and analysed, not only because of their high ionic conductivity, low cost and unique applications, but also their high temperatures that eliminate dendritic growth on the liquid AAEM negatrode and heat buildup in the cell.
Sustainability spotlightAlkali and alkaline earth metals have non-exhaustible resources on the earth and are particularly attractive for making the negative electrode of supercapatteries. With the aid of liquid salts, including the low cost and resource-rich molten salts, and supercapacitor positive electrodes of high anodic stability, supercapatteries promise clean and sustainable energy storage solutions to the capture and conversion of various forms of renewable energy and the affordable electric passenger aircrafts. |
1 Introduction
Driven by the rapid market expansion of portable electronics and electric transportation, the demand for cost-effective and high-efficiency energy storage has become increasingly important in the last two decades.1 Electrochemical energy storage devices (EESDs), such as rechargeable batteries and supercapacitors, are ideal candidates due to their modular nature, commercial attractiveness, and potential fossil-comparable energy capacity.Commercial lithium (Li) ion batteries usually show satisfactorily high specific energy. Nevertheless, they demonstrate low power density due to the sluggish diffusion of Li ions in intercalation-type electrode materials.1 Especially, the fast intercalation of Li ions is not kinetically supported by the Li storage positive electrode (positrode) (LiCoO2 or LiNi1−x−yCoxMnyO2).2 Moreover, because of their relatively low earth crust abundances, Co, Ni and Mn are or become very expensive, whilst their extraction from the respective minerals and decommission after service life exert a huge environmental impact and sustainability concern with respect to the supply chains. Although, as a successful commercial product, the LiFePO4 positrode shows improved thermal and chemical stability, cell safety, and longer cycle life, its moderate potential (<3.5 V vs. Li+/Li) and high self-discharge rate only support a cell with relatively low exploitable energy density.3 On the other hand, traditional supercapacitors typically offer high power capability (e.g., 10 kW kg−1) and long cycle life but only low to moderate specific energy (<50 W h kg−1) due to the limited capacity for electrostatic adsorption and desorption of ions at the electrolyte|electrode interface.1
Neither batteries nor supercapacitors alone can satisfy all the current and future commercial requirements. Thus, to achieve large energy capacity and high power capability in one EESD, hybrid devices combining capacitive and Nernstian charge storage mechanisms in the positrode and negative electrode (negatrode) without invoking Li intercalation have been proposed. A supercapattery (= supercapacitor + battery) is an innovative hybrid EESD, aiming to combine the advantages of rechargeable batteries and supercapacitors. Supercapatteries can provide sufficient electron transfer reactions and fast ion diffusion in the negatrode and positrode, thus showing higher power capability and longer cycle life than batteries, and larger energy capacity than supercapacitors (Fig. 1).4 Accordingly, supercapatteries can be fabricated, for example, by pairing a supercapacitor positrode with a battery negatrode. The battery negatrode usually stores charge through the reversible transfer of localised valence electrons according to the Nernst equation, i.e., the Nernstian storage mechanism. In the supercapacitor positrode, the charge is stored via EDL capacitance, pseudocapacitance, or even a combination of both mechanisms. The pseudocapacitance results from a capacitive Faradaic process according to the transfer of zone-delocalised valence electrons.4 The best performance of a supercapattery depends mainly on the optimal coupling of electrode materials and electrolytes to utilise effectively the potential windows and charge capacities of both electrodes without compromising the respective electron transfer kinetics and ion transport dynamics. While this coupling principle is further exemplified and discussed in the following sections, it is worth mentioning here that simply combining a Nernst electrode with a capacitive electrode does not necessarily lead to a better performing supercapattery. A key engineering strategy is to follow the two golden rules: the amount of charge, Q, and the amplitude of the current, I, passing through the two electrodes in a supercapattery (and any other EESD) must be equal.4
| Qn = mnQsp,n = mcCsp,cΔEc = Qc | (1) |
| In = U/(Rn + Rc + Rel) = Ic | (2) |
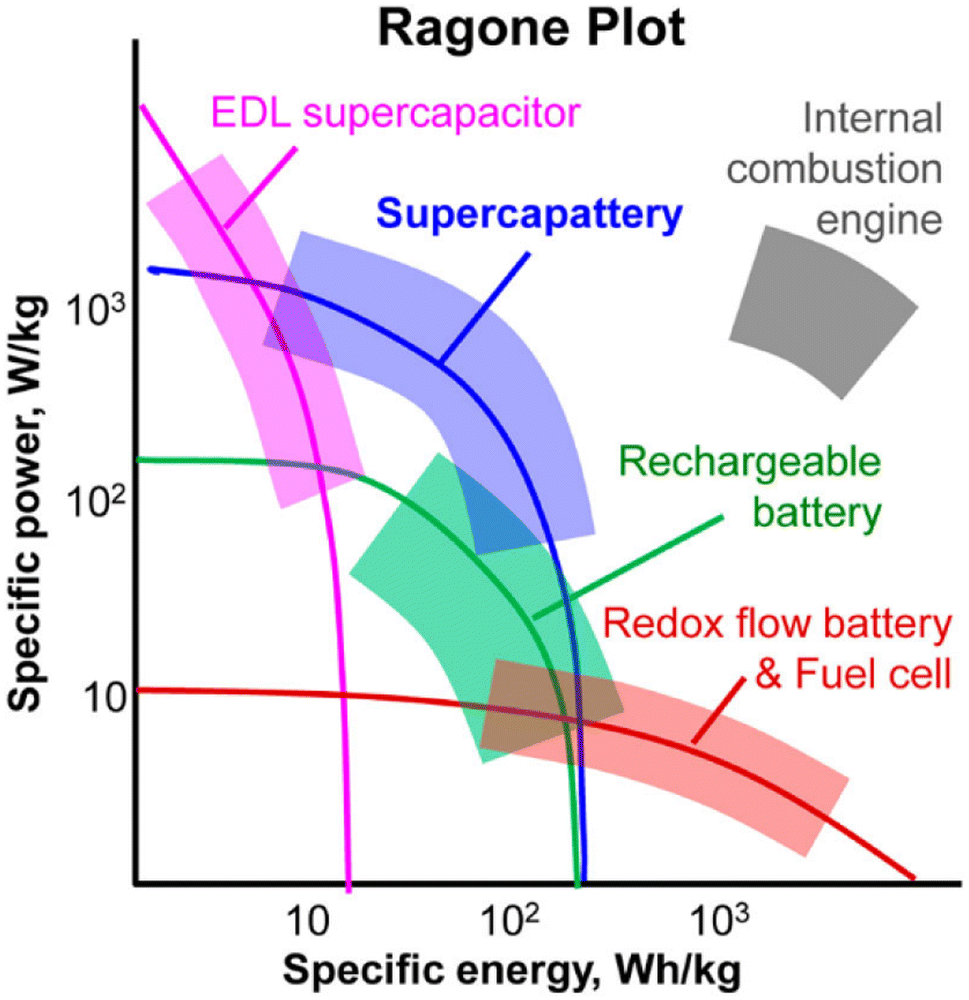 | ||
| Fig. 1 A schematic Ragone plot demonstrating the position of the supercapattery in comparison with other energy devices.4 | ||
It should be noted that although Rel is the only term for electrolyte appearing in these two equations, the electrolyte also influences ΔEc and U and both Qn and Qc. Further, for a given Nernstian electrode, Qn is a constant, but Qc is proportional to ΔEc, which means U may be extended with using a smaller mc as long as ΔEc ≤ CPR, the capacitive potential range beyond which the electrode experiences Nernstian or irreversible reactions. On the other hand, Nernstian electrodes have usually high values of Qsp,n, particularly those based on alkali or alkaline earth metals (AAEMs), which means that the ratio mn/mc (= Csp,cΔEc/Qsp,n) may be much smaller than 1. Therefore, if the positrode and negatrode masses of a supercapattery are made the same or similar as is the common practice in battery or supercapacitor manufacturing, the supercapattery highly likely underperform relative to its battery or supercapacitor counterpart.
In principle, the energy capacity of any EESD can be calculated according to eqn (3) in which Umax is the maximum cell voltage that is determined by the maximum potential difference between the positrode and negatrode.
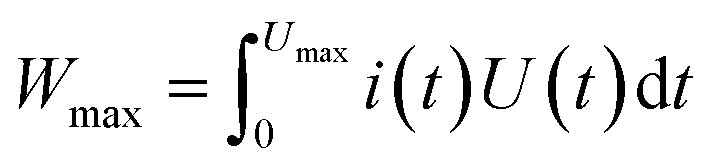 | (3) |
In rechargeable batteries, the positrode and negatrode potential windows are each relatively narrow and should be as wide apart from each other as possible to maximise the cell voltage. Unfortunately, this strategy also limits the minimum cell voltage to avoid over-discharging. However, in supercapatteries, the capacitive electrode can have a very wide potential window that can overlap with that of the Nernstian electrode. As a result, a supercapattery can be discharged to a practically meaningful low cell voltage, if not zero. Further, because of their narrow potential windows, the positrode and negatrode in a battery have to be matched carefully in mass loading to equalise the charge capacity. In a supercapattery, because the charge capacity of the capacitive electrode is a function of both mass loading and working potential range, there is a much larger space to manipulate the electrode mass loading for an optimal cell performance.
As a key component in EESDs, electrolytes maintain electronic insulation but ionic conduction between the positrode and negatrode, and also assist charge transfer processes on the electrodes.5 In eqn (3), Umax can be the same as but not wider than the electrochemical stability window (ESW) of the electrolyte. Thus, electrolyte selection is a key design element and strongly determines the working or cell voltage of any EESD. It is particularly important for designing a supercapacitor whose linear voltage–charge relationship simplifies eqn (3) to eqn (4) below,
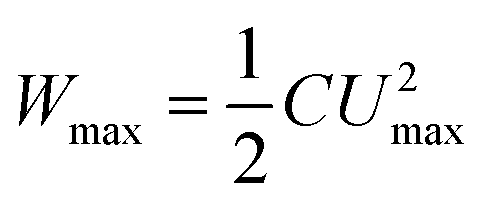 | (4) |
In comparison with aqueous and organic electrolytes, ILs are pure liquid salts and widely recognized for their widest ESWs, more ionized environments, negligible volatility, low flammability, and high thermal, chemical, and electrochemical stabilities.7 However, ILs usually show ionic conductivity lower than 15 mS cm−1 at room temperature when they are viscous, which could lead to reduced achievable energy capacity and power capability in EESDs. Fortunately, many performance parameters of EESDs such as specific energy and power, or operation temperature can be further improved by employing mixed solvents of different ILs or ILs with organic solvent, or redox additives in IL electrolytes.8
Another key component in EESDs is electrode materials. In addition to the capacitance or charge capacity, the cell voltage of EESDs derived from the potential gap between negatrode and positrode materials also greatly impacts the specific energy of EESDs. Thus, cell voltage should be given more attention. For example, it may be feasible to obtain a pseudocapacitive material with a specific capacitance value of 800 F g−1, but it would be difficult to make a symmetrical supercapacitor from such a material to approach a cell voltage of 3.0 V or higher. Thus, a unique strategy for improving the performance is to combine a supercapacitor positrode (either EDL capacity or pseudocapacity) with an AAEM negatrode of high theoretical specific capacity and very negative potential in a supercapattery with optimized IL electrolytes. For instance, Li metal, as the ultimate battery-type negatrode for high specific energy EESDs, is arousing wide attention owing to its very negative potential (−3.04 V vs. standard hydrogen electrode) and high theoretical specific charge capacity (3860 mA h g−1) and charge density (2061 mA h cm−3) as well as low mass density (0.53 g cm−3).9–11 The cell voltages of supercapatteries can be effectively broadened, which is beneficial to enhancing the energy storage capacity. A hypothetical supercapattery composed of a Li metal negatrode and a supercapacitor positrode of assumed 300 F g−1 is evaluated here. Considering that the specific charge capacity of Li metal (3860 mA h g−1) is much larger than that of the supercapacitor electrode, the mass of the Li metal incorporated into the supercapattery can be negligible. The theoretical specific energy would be ∼656 Wh kg−1 for a cell voltage varying from 4.0 V to 0.5 V. This theoretical value is even higher than that for conventional Li ion batteries (LIBs).5
Following the success of Li-based batteries, other AAEMs such as Na12 or Mg13 have also been increasingly studied as the negatrodes for batteries, mainly because of the concerns regarding the limited Li resources. A bottleneck in non-Li AAEM ion batteries exists due to the lack of suitable positrode host materials,8 but it may be easily bypassed by using a supercapacitor positrode to match the AAEM negatrode. However, metal negatrodes also suffer from a series of drawbacks, such as dendrite growth and unstable solid electrolyte interphase (SEI) formation, resulting in capacity fading, volume expansion, or poor power capability. Impressively, as electrolytes in AAEM supercapatteries, IL engineering technology also shows promising advantages to alleviate the problems mentioned above. For example, key additives in IL are critical to helping metal-based cells achieve a stable reversible deposition. It was shown that novel non-flammable IL electrolytes composed of 1-ethyl-3-methylimidazolium and high-concentration bis(fluorosulfonyl)imide (FSI) with sodium bis(trifluoromethanesulfonyl)imide (NaTFSI) as a key additive could improve the Li metal deposition/dissolution behaviour. The formation of hybrid passivation interphases was found to contribute to dendrite-free Li deposition owing to the introduction of Na ions.14 Therefore, it seems that the ingenious design of IL paring with an AAEM negatrode is key to enhancing the electrochemical performance of supercapatteries.
In this article, we first review the recent relevant research progress of IL-based electrolytes for typical AAEM supercapatteries, focusing on lithium (Li), sodium (Na), magnesium (Mg), potassium (K), and calcium (Ca). In the course of the discussion, we make necessary comparisons with AAEM ion supercapacitors and batteries. It is worth noting that many reported AAEM ion supercapacitors (e.g., Li-ion or Na-ion capacitors) are composed of an AAEM ion host electrode and an EDL capacitive or pseudocapacitive electrode. Because the ion host electrode works according to ion intercalation enabled redox chemistry, it is the same as a Nernstian or battery electrode. Thus, these ion capacitors are basically supercapatteries as well, although they are still different from those supercapatteries with a metal negatrode.
We then discuss the challenges and prospects of developing high-performance IL-based AAEM supercapatteries. The focus is firstly on the unfavourable aspects of ILs, such as high viscosity caused low ionic conductivity, and corrosivity to metals. The AAEM negatrodes also have intrinsic but problematic performances, mainly dendritic deposition and unwanted reactive interactions with ILs. Potential mitigations of such challenging issues are then sought from the literature and discussed.
In particular relevance to overcoming the low conductivity of ILs and dendritic deposition of AAEMs, we review the literature on batteries and supercapacitors with molten salts as the electrolyte, and then offer some preliminary considerations on the prospect of molten salt supercapatteries with an AAEM negatrode. Molten salts, including molten hydroxides and oxides, are the high temperature counterpart of ionic liquids and they both are liquid salts in nature. (Another type of liquid salt is the so-called deep eutectic solvents15,16 whose application in EESDs deserves a separate coverage.) They offer some unique advantages over ionic liquids. For example, LiCl (m.p. = 605 °C, b.p. = 1383 °C) remains stable in a very wide working temperature window from 650 °C to 1000 °C, which means a negatrode of Li metal (m.p. = 180.5 °C) is in the liquid state, avoiding all problems associated with dendritic deposition. Further, at temperatures slightly higher than its melting point (e.g., by 50 °C), an inorganic molten salt becomes water-like in viscosity and hence offers high ionic conductivity, which is beneficial to high power capability. Last but not least, salt mixtures of inorganic and/or organic nature often show eutectic melting behaviour, which lowers the working temperature. For example, the so-called solar salt is used for heat transfer and storage in various concentrated solar power (CSP) plants. It is the mixture of NaNO3 and KNO3 with an equimolar eutectic temperature at about 222 °C, although the actual NaNO3 content is higher on balancing the benefits from reduced cost, enhanced heat capacity and increased liquidus temperature.17–19
2 Ionic liquid-based AAEM supercapatteries
When the two electrode materials of an EESD have been determined, the selection of electrolytes that are more suitable for each electrode material can greatly improve the cell stability and enable greater energy capacity and power capability.20 Since its first report in 1994, the aqueous electrolyte of 5 M LiNO3 for rechargeable lithium batteries has attracted growing interest due to its low price, environmental friendliness, good conductivity, and easy preparation.21 The development of new aqueous electrolytes has been ongoing.22 In 2020, Adelowo et al.23 developed a high-energy aqueous Li-ion on-chip capacitor based on interdigitated 3D carbon microelectrode arrays, which can achieve 5.03 μW h cm−2 areal energy, which is five times higher than that of other aqueous electrolytes. Organic electrolytes are often compared to aqueous electrolytes. The theoretical decomposition voltage of water is 1.23 V. Considering the overpotential of hydrogen or oxygen, the highest cell voltage of a traditional aqueous electrolyte is only about 2.0 V (such as that in lead-acid batteries). In LIBs, the operating cell voltage can be usually a high value between 3.0 and 4.0 V. Compared with aqueous electrolytes, organic electrolytes do not have advantages in price, availability, and conductivity, but can provide a wider and more stable ESW and wider temperature range (e.g., −30 to 70 °C).24 In recent years, the emerging sulfolane, ether and nitrile electrolytes have shown the potential to meet the requirements for high performance batteries.25Compared with aqueous and organic electrolytes, IL electrolytes have obvious advantages. As pure liquid salts at room temperature, they are highly ionized, negligibly volatile, non-explosive and non-flammable, and offer great safety. Further, their ESWs can usually reach beyond 5.0 V.26 For example, 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14TFSI) can remain stable up to 5.9 V as quoted by Susantyoko et al.27 Such wide ESWs are perhaps the most important reason for using ILs in supercapacitors against eqn (4). However, unlike electrolytes with water and organic solvents of small molecules, ILs are mostly prepared from organic synthesis and present wide spectra of structures, compositions and properties. Thus, the understanding of charge (energy) storage mechanisms and performances derived mostly from aqueous and organic electrolytes in supercapacitors and consequently the manufacturing strategy may likely change when such new ILs become available. It has to be acknowledged that research on ionic liquids for supercapacitors has been growing fast and well covered in several recent reviews.6,28–30 Considering space and topic, the following discussion will be based on selected literature on ILs relevant to supercapatteries.
It is worth mentioning that ILs can be used not only directly as liquid electrolytes but also dissolve in traditional organic solvents or electrolytes to form new electrolytes with the merits of both. Such mixture electrolytes could achieve higher safety than organic electrolytes only, and better electrochemical performance than pure ILs.31 Fleischmann et al.32 developed high-pressure supercapatteries using 1-methyl-1-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr13TFSI) as the electrolyte. This cell achieved 100 W h kg−1 specific energy, 2 kW kg−1 specific power, and over 1500 stable cycles, and could work satisfactorily at 80 °C.32
In addition, adding suitable redox agents to the electrolyte can also greatly improve the performance of electrochemical devices. Redox electrolytes of quinones dissolved in ILs enabled high-performance supercapatteries with energy densities three times higher than when ILs alone were used as electrolytes.33 The current problem with IL electrolytes is that they may become cathodically unstable at potentials more negative than 1.0 V (vs. Li/Li+), which could limit the voltage across the device to about 4.3 V. The ESWs of IL electrolytes are mainly affected by the nature of the solvent, conductive salts (i.e., cations and anions), and trace amounts of water and impurities. The cell voltage can be controlled by rationally adjusting these parameters.34,35 Furthermore, the high viscosity and low ionic conductivity of IL electrolytes are detrimental to the cycling stability and rate capability of hard carbons.
In the following sub-sections, centring on IL-based AAEM supercapatteries, we analyse typical examples of IL-based AAEM-ion supercapacitors, AAEM batteries and AAEM-ion supercapatteries, and the impact of the cell configuration and electrolyte composition of these IL–AAEM based EESDs on the development of supercapatteries. The relevant electrode materials and electrolyte compositions and the respective electrochemical performances are summarised in Table 1. The order of introduction of these EESDs is lithium, sodium, magnesium, potassium, and calcium according to their atomic numbers in the periodic table.
| Positrode | Negatrode | Ionic liquid | Salt | Configuration | Cycle | CE (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Electrode materials: MESO, mesoporous carbon; AC, activated carbon; PBA, Prussian blue analogue. Cations: Pyr13, 1-methyl-1-propylpyrrolidinium; EMI, 1-ethyl-3-methylimidazolium; Pyr14, 1-butyl-1-methylpyrrolidinium. Anions: FSI, bis(fluorosulfonyl)imide; TFSI, bis(trifluoromethanesulfonyl)imide; FAP, tri(pentafluoroethyl)trifluorophosphate. Salts: EtAlCl2: ethylaluminium dichloride. | |||||||
| N/S co-doped MESO | Na metal | Pyr13-FSI | NaFSI | 1 | 3000 | 100 | 46 |
| LiMn2O4 | MESO | EMI-TFSI | LiTFSI | 4 | 1000 | 100 | 47 |
| LiNixMn2−xO4 | MESO | EMI-TFSI | LiTFSI | 4 | 1500 | 85 | 48 |
| AC | Li4Ti5O12 | Pyr13-TFSI | LiTFSI | 4 | 1500 | 70 | 32 |
| LiNi1/3Co1/3Mn1/3O | Li metal | Pyr14-TFSI | LiTFSI | 3 | 100 | 99 | 49 |
| AC | Hard carbon | EMI-FSI | LiFSI | 2 | 3000 | 95.33 | 50 |
| AC | Hard carbon | Pyr13-FSI | LiFSI | 2 | 3000 | 88.44 | 50 |
| PBA | K metal | EMI-Cl | EtAlCl2 and KFSI | 3 | 820 | 99.9 | 51 |
| Graphite | AC | Pyr14-TFSI | Mg(TFSI)2 | 4 | 50 | 98 | 52 |
| AC | Li metal | Pyr14-FAP | LiClO4 | 1 | — | — | 5 |
2.1 ILs in lithium-based supercapatteries
One of the first supercapatteries is the Li-ion-based battery–supercapacitor hybrid devices, invented by Amatucci and co-workers.36 The negatrode of the Li ion supercapattery was nanostructured Li4Ti5O12, and the positrode was made of activated carbon (AC). This supercapattery could use either aqueous or organic electrolytes. It could also be assembled using Li ion host materials as the positrode, such as Ti-based oxides,37 Fe3O4,38 Nb2O5,39 and MnO.40 However, due to the inherent disadvantages of the intercalation-type positrode, the reported Li ion supercapatteries still had the problem of slow energy storage kinetics, and the specific power and energy were limited within 900 W kg−1 and 40 W h kg−1, respectively.41Li metal is considered the ultimate choice for the negatrode of Li batteries due to its high theoretical capacity and extremely negative potential,42 which is necessary for high energy EESDs.43 More recently, designs that use Li metal as the negatrode of supercapatteries have emerged. In 2021, Zhong et al.44 first reported flexible Li metal capacitors with an in situ prepared PETEA-based polymer gel electrolyte.44 In the same year, Liu et al.45 coupled a Li metal foil negatrode with a three-dimensional scaffold activated carbon (3D-SAC) positrode into a cell with an organic carbonate electrolyte. The positrode showed a specific capacitance up to 280 F g−1 in a potential range from 1.5 to 4.3 V vs. Li/Li+. The galvanostatic charging and discharging (GCD) curve of the Li-carbon cell was very triangular in shape, similar to that of a capacitor thanks to the very flat GCD profile of the Li foil. A remarkable specific energy of 633 W h kg−1 was derived from the GCD curve of the cell. It is noted that only the positrode mass was used to calculate the cell specific energy, which is still creditable because the mass change of the Li negatrode should be insignificant compared to the total mass of the carbon positrode.
However, the Li dendrites formed during multiple charge and discharge cycles would pierce the separator and cause an internal short circuit of the EESDs. To solve this problem, several measures have been proposed, such as designing a highly stable artificial SEI between the metal electrode materials and the electrolyte to help Li metal deposit and dissolve more uniformly and prevent Li dendrites from piercing the separator.53 Selecting appropriate electrolyte and electrolyte additives (e.g., NaTFSI)14 and developing new electrolytes are necessary to better prevent the generation of Li dendrites from the source.54
The specific energy and power of supercapatteries with IL electrolytes have been demonstrated to be comparable to those using aqueous and traditional organic electrolytes, while maintaining excellent cycling stability.47 To design the Li ion supercapattery, some studies have used mesoporous carbon as a capacitive electrode and 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI) as the electrolyte. The positrode was LiMn2O4 (relevant electrochemical performance and electrode morphology are shown in Fig. 2).47 The same research group also investigated the performance of a Ni-doped LiMn2O4 positrode in supercapatteries with a lithium salt/imidazolium IL electrolyte.48
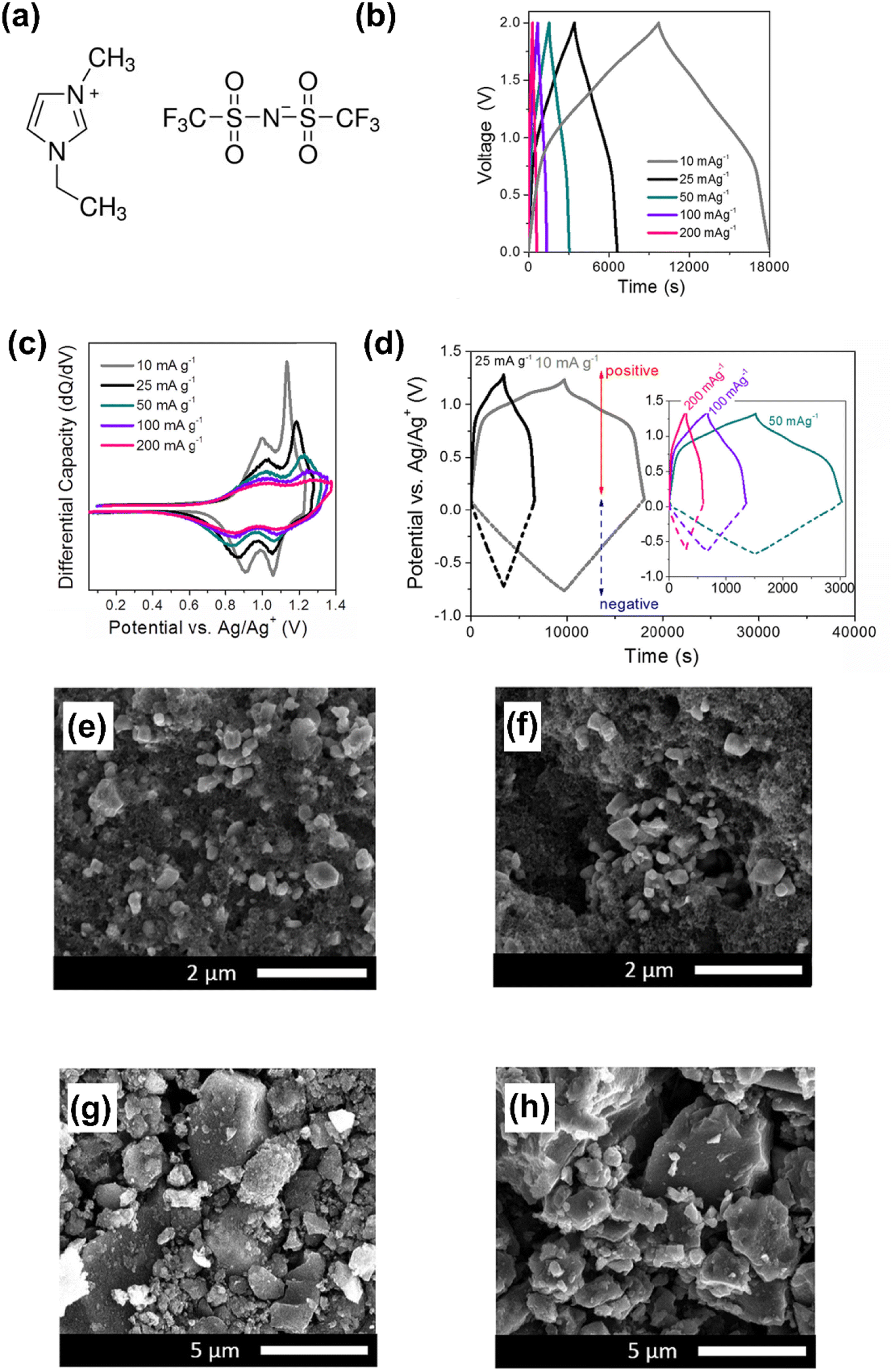 | ||
| Fig. 2 (a) Molecular structures of the cation and anion in the ionic liquid of 1-ethyl-3-methylimidazolium-bis(trifluoromethanesulfonyl)imide (EMITFSI). (b) Galvanostatic charge–discharge curves, (c) differential capacity vs. potential (dQ/dV) curves for the positrode, (d) evolution of the potentials of the electrodes at different current densities (10–200 mA g−1) for the LiMn2O4‖MESO cell in 1 M LiTFSI in EMITFSI. SEM images of the positrode: (e) before and (f) after cycling; SEM images of the negatrode: (g) before and (h) after cycling.47 (Reprinted from ref. 47 with permission from Rightslink). | ||
Fleischmann et al.32 used Li4Ti5O12 as the negatrode, activated carbon as the positrode, and an IL containing Li salt as the electrolyte to form a supercapattery with a wide electrochemical window and a maximum voltage of 4.0 V. The reported EESD could achieve a specific energy of 100 W h kg−1 and a specific power of 2 kW kg−1 (Fig. 3).
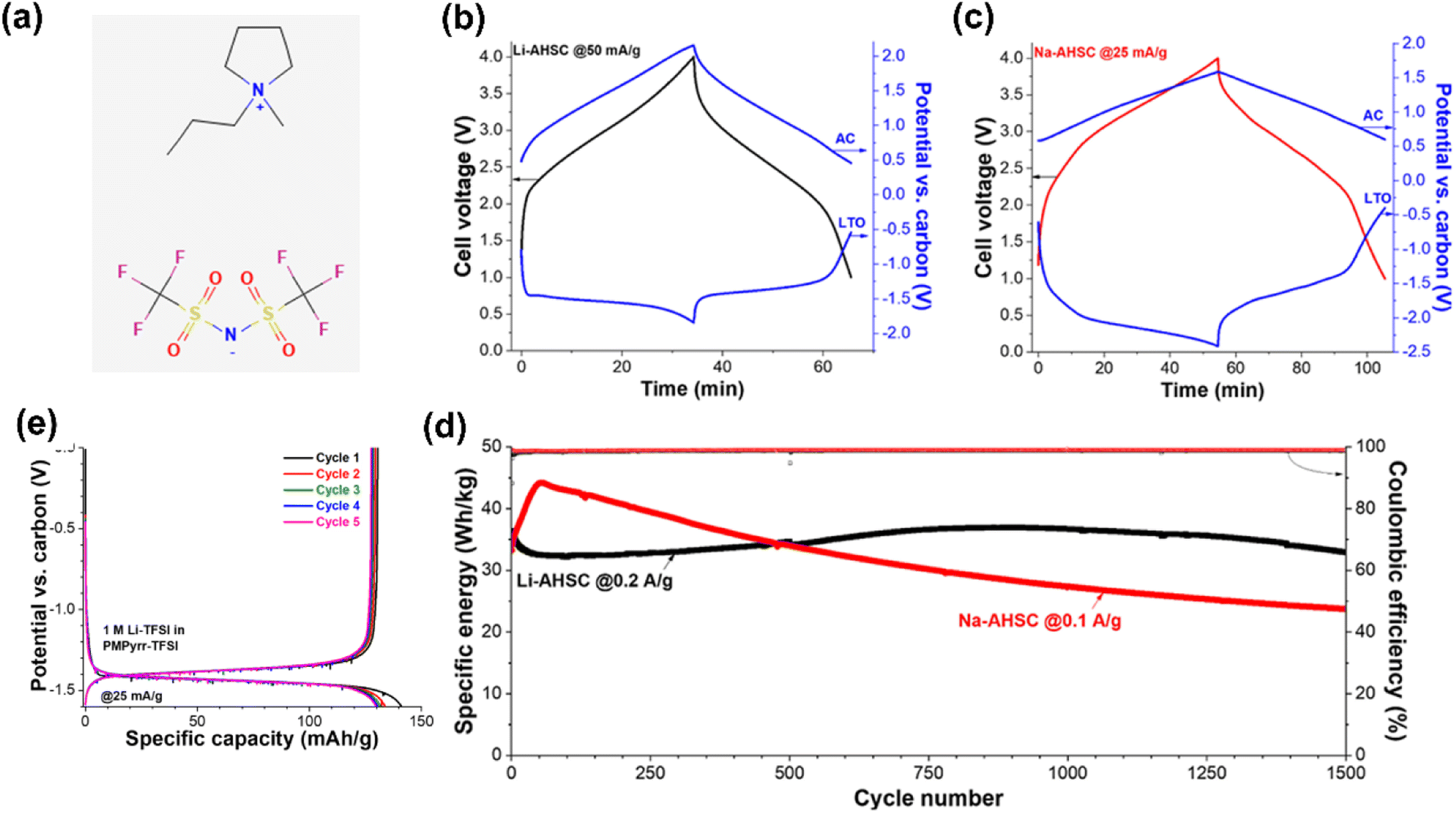 | ||
| Fig. 3 (a) 1-Methyl-1-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr13TFSI) ionic liquid molecular structure. Voltage profiles of (b) Li-based supercapatteries cycled at 50 mA g−1 and (c) Na-based supercapatteries cycled at 25 mA g−1, including potential development at the negatrode (Li4Ti5O12) and positrode (activated carbon). (d) The cycling stability of Li-based supercapatteries cycled at 0.2 A g−1, and Na-based supercapatteries cycled at 0.1 A g−1 over 1500 cycles between 1 V and 4 V cell voltage. (e) Comparison of the first 5 galvanostatic cycles in 1 M LiTFSI in Pyr13TFSI electrolyte at 25 mA g−1.32 (Reprinted from ref. 32 with permission from Rightslink). | ||
The first IL-based supercapattery with a Li metal negatrode and an activated carbon positrode was reported in 2016 by our group.5 The electrolyte was a mixture of 1-butyl-1-methylpyrrolidinium tri(pentafluoroethyl)trifluorophosphate (Pyr14FAP) and gamma-butyrolactone (γ-GBL) (v/v-1/1) containing 0.5 mol L−1 LiClO4. The GCD curves of this IL cell were measured within a voltage range from 4.3 to 1.7 V and exhibited highly capacitive features, leading to a high specific energy of 232 W h kg−1 (Fig. 4). In the future, with the exploration of more positrode materials for IL-based supercapacitors and the development of Li metal as a negatrode technology for EESDs, the application of Li metal in supercapatteries with IL electrolytes is likely to achieve higher energy storage and more stable cycling performances.
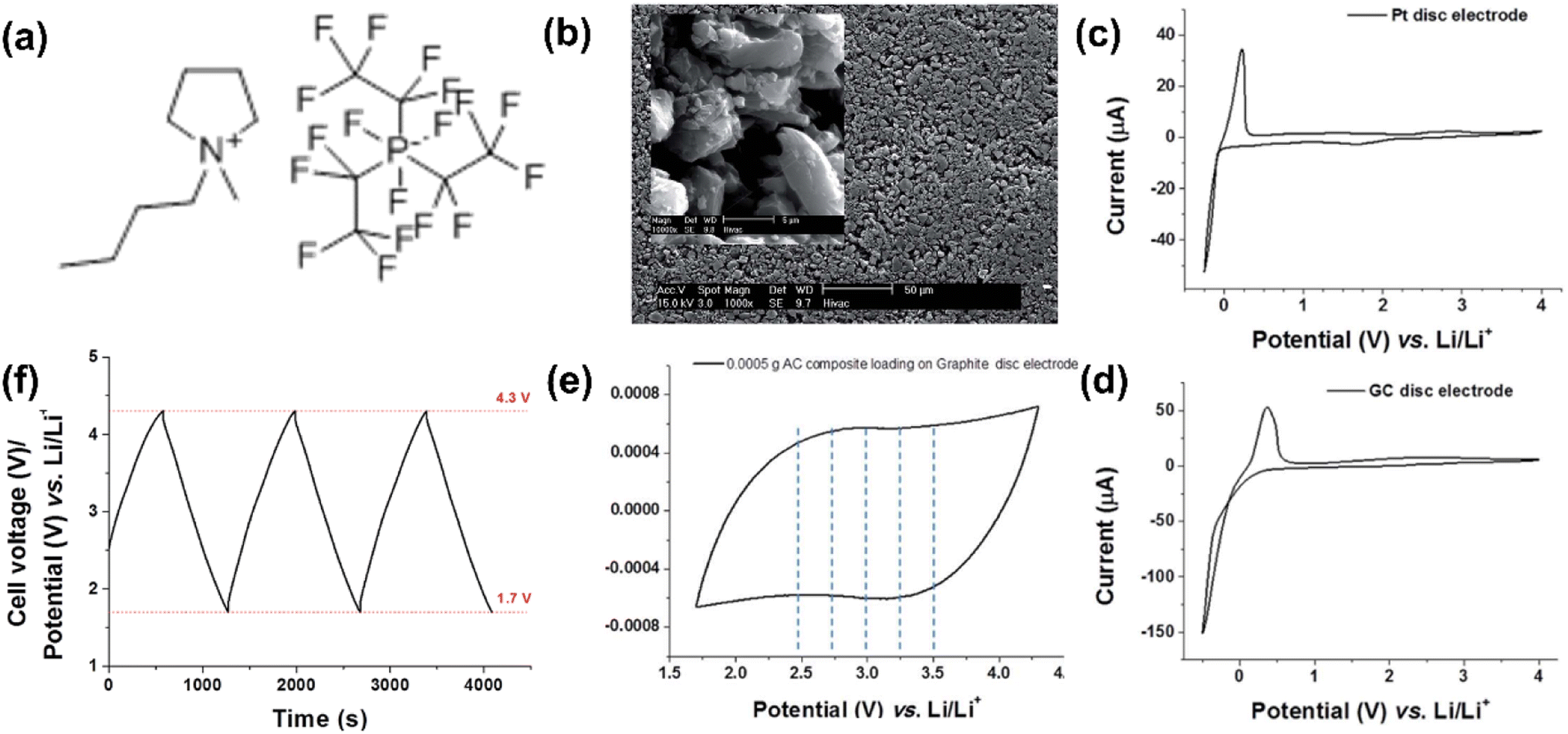 | ||
Fig. 4 (a) Molecular structure of the ionic liquid of 1-butyl-1-methylpyrrolidinium tri(pentafluoroethyl)trifluorophosphate (Pyr14FAP), (b) SEM image of the activated carbon pellet. The inset of (b) is a high-magnification SEM image of an AC pellet. Cyclic voltammograms using (c) a Pt disc electrode of 2 mm diameter and (d) a glass carbon disc electrode of 3 mm diameter, in a mixture of Pyr14FAP and γ-GBL, containing 0.01 mol L−1 LiClO4 at a scan rate of 10 mV s−1. (e) Cyclic voltammogram of 0.5 mg AC composite on a graphite disc electrode of 5 mm diameter in a mixture of Pyr14FAP and γ-GBL, containing 0.5 mol L−1 LiClO4 at a scan rate of 10 mV s−1. (f) Galvanostatic charge–discharge curves of a pellet of 0.5 mg AC composite on a graphite disc electrode of 5 mm diameter in a mixture of Pyr14FAP and γ-GBL, containing 0.5 mol L−1 LiClO4. Current density: 1 mA cm−2, the volume ratio of Pyr14FAP to γ-GBL in the mixture was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 (Reprinted from ref. 5 with permission from Rightslink). :1.5 (Reprinted from ref. 5 with permission from Rightslink). | ||
2.2 ILs in sodium-based supercapatteries
Li-based electrochemical devices occupy a leading position in the global EESDs market, but Li resources are not very abundant. In contrast, sodium (Na) resources are far greater than those of Li. Because the physical and chemical properties of Li and Na are remarkably similar, Na-based energy storage materials have become one of the candidates to replace or complement Li-based ones.55 TiS2 was the earliest reported reversible Na intercalation material.56 Initially, considering the excellent performance of graphite electrodes in LIBs, scientists had tried but failed to use graphite as an intercalation material for Na-ion batteries.57,58 In 2000, Stevens et al.59 reversibly inserted Na+ ions into hard carbon at room temperature and achieved a reversible Na capacity of 300 mA h g−1. This is a big development in the field of carbon-based Na intercalation materials, even if this value is lower than the Li capacity of the same hard carbon.59 In another study, an aqueous Na-ion capacitor with carbon microspheres as the negatrode and cobalt hexacyanoferrate (CoHCF, which is a good AAEM ion host material) as the positrode achieved 54.4 W h kg−1 specific energy.60In order to obtain higher energy density, some scientists have proposed Na metal batteries because Na has a theoretical capacity of about 1166 mA h g−1.61 In 2019, a hybrid device composed of a Na metal negatrode and a capacitive material positrode was reported.62 The negatrode of this supercapattery was designed using a catalytic carbon nanotemplate (C-CNTP), whose ordered graphitic structure enabled reversible Na metal deposition/stripping. The positrode employed nanoporous pyroproteins (N-PPts) to store Na ions. The voltage window of such Na metal supercapatteries could reach 4.0 V which helped the delivery of high specific energy and power of 238 W h kg−1 and 462 W kg−1, respectively. In the following year, the same group developed another Na metal supercapattery based on a nanoembossing pyropolymer catalytic layer (NE-P-CL) coupled with a nanopore-engineered pyropolymer (NE-PP). In the new devices, NE-P-CL was designed for reversible Na deposition whilst NE-PP was fabricated as a capacitive positrode. The cell exhibited high specific energy and power of 348 W h kg−1 and 85.3 kW kg−1, respectively.63
At present, the difficulty encountered in the development of Na metal batteries is the high reactivity between metallic Na and electrolytes. In 2015, Iermakova and co-workers64 reported the cyclic GCD profiles of Li/Li and Na/Na symmetrical cells in conventional alkyl carbonate electrolytes. Their observations revealed very large and electrolyte dependent resistance in the Na/Na cell (about 6 times larger than that of the Li/Li cell), indicating the formation of an unfavourable and resistive SEI from reactions between Na and the carbonate electrolytes. Further, Na metal is fragile and difficult to process, whilst its low mechanical flexibility and poor electrolyte wettability compromise the interfacial stability. Thus, it is not easy to form a stable SEI at the “Na negatrode|carbonate electrolyte” interface,65 which then leads to the growth of Na dendrites, the entry of free metallic Na into the electrolyte, and the hindrance of ion flux.66
In contrast, some IL electrolytes have been shown to be capable of offering much higher cycling stability than conventional electrolytes for Na metal deposition and dissolution. For example, using Na metal as the negatrode, N and S co-doped mesoporous carbon as the positrode, and the IL 1-methyl-1-propylpyrrolidinium bis(fluorosulfonyl)imide (Pyr13FSI) as the electrolyte, the supercapattery could stably cycle 3000 times at 100% coulombic efficiency (CE), and the capacity remains almost unchanged (Fig. 5).46
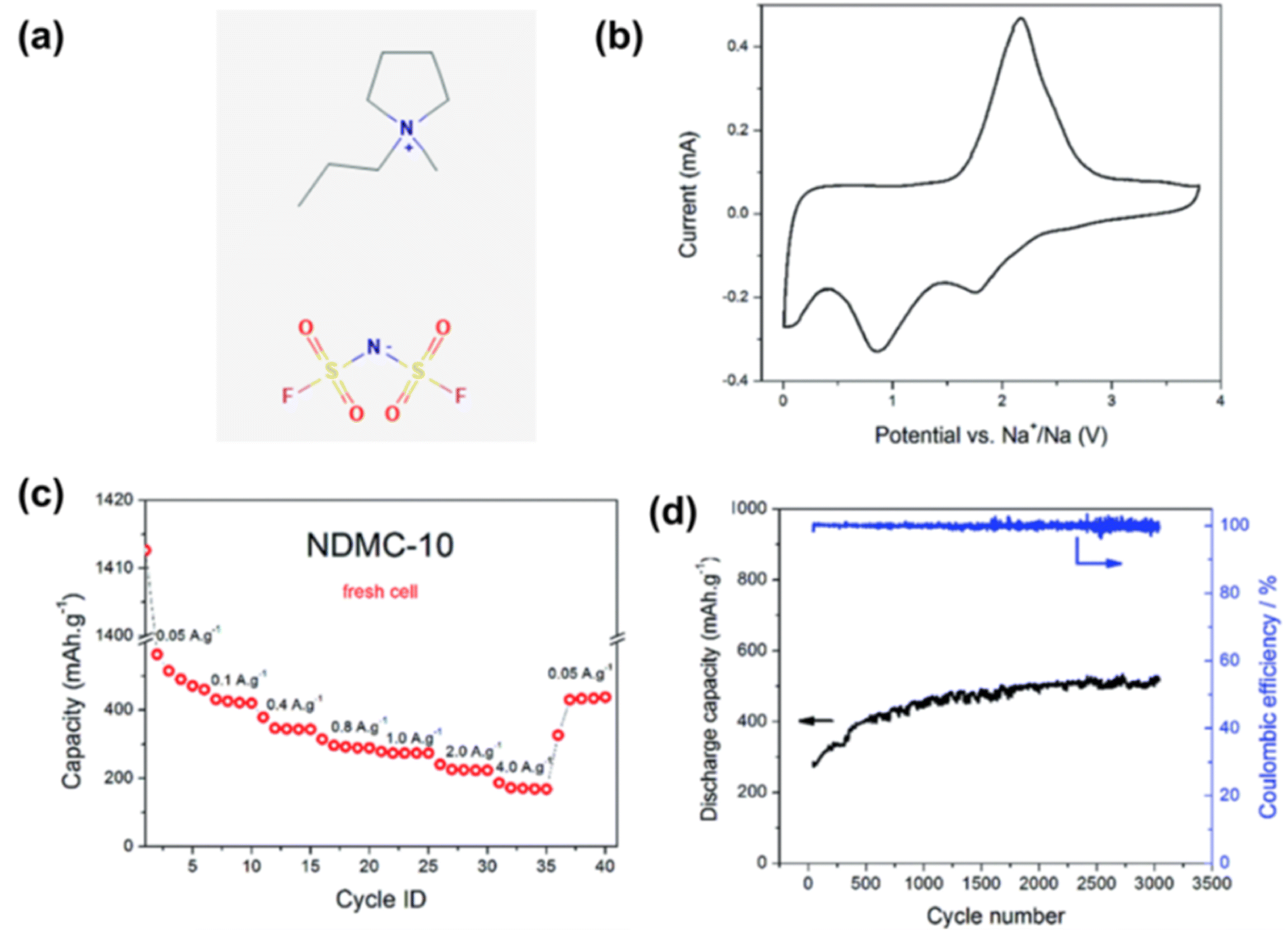 | ||
| Fig. 5 (a) 1-Methyl-1-propylpyrrolidinium bis(fluorosulfonyl)imide (Pyr13FSI) ionic liquid molecular structure. (b) Cyclic voltammogram at 1 mV s−1, (c) rate capability at different current densities and (d) long-term cycling (3000 cycles) at 1.0 A g−1 of the IL-based Na metal supercapatteries.46 (Reprinted from ref. 46 with permission from Rightslink). | ||
2.3 ILs in magnesium-based supercapatteries
Unlike Li- and Na-based supercapatteries, magnesium (Mg) metal has unique advantages for use as the negatrode. Mg metal is an excellent negatrode material because it is neither as easy to form dendrites as Li metal nor as easy to react with an electrolyte as Na metal. In Mg supercapatteries, Mg metal and its alloys can be directly used as negatrodes without an additional Mg metal pre-doping process. In 2014, Yoo et al.67 developed, for the first time, a prototype Mg supercapattery as a successful conceptual EESD. The negatrode of this supercapattery was a Mg foil and the positrode was made from a cloth of activated carbon. The problem of premature saturation of voids in AC, before the electrode potential reaches the limit, was solved by adding suitable electrolyte additives.67However, it is known that the Mg2+ cannot penetrate the passivation film formed on the Mg metal negatrode, whilst reversible Mg deposition requires special electrolytes of Mg organo-haloaluminate complexes in ether solvents.68 In such cases, the compatibility of every component of the electrolyte with the positrode material needs to be carefully considered. Complex electrolyte additives often make it difficult to match suitable positrode materials for existing Mg-based EESDs. The Mg organo-haloaluminate electrolytes are often nucleophilic and can therefore chemically react with the electrophilic oxide positrode. Furthermore, the large ionic sizes of Mg ion complexes tend to limit the use of a porous carbon positrode in organo-Mg haloaluminate electrolytes.67 Apparently, these non-ideal electrolytes are unfavourable for the development of Mg supercapatteries.
With the successive breakthroughs in high-power Mg metal batteries in the past two years, new ideas have emerged for solving problems in metal Mg supercapatteries. Meister et al.52 reported a novel dual-ion capacitor containing an IL electrolyte of magnesium bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2) dissolved in 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14TFSI). When charging this device, the TFSI− ion was stored in the graphite positrode through intercalation which is Faradaic (Nernstian) in nature, whilst Mg2+ and Pyr14+ ions were physically adsorbed/desorbed at the porous carbon negatrode in accordance with the EDL capacitive mechanism (Fig. 6). Obviously, this cell configuration falls within the scope of supercapatteries.
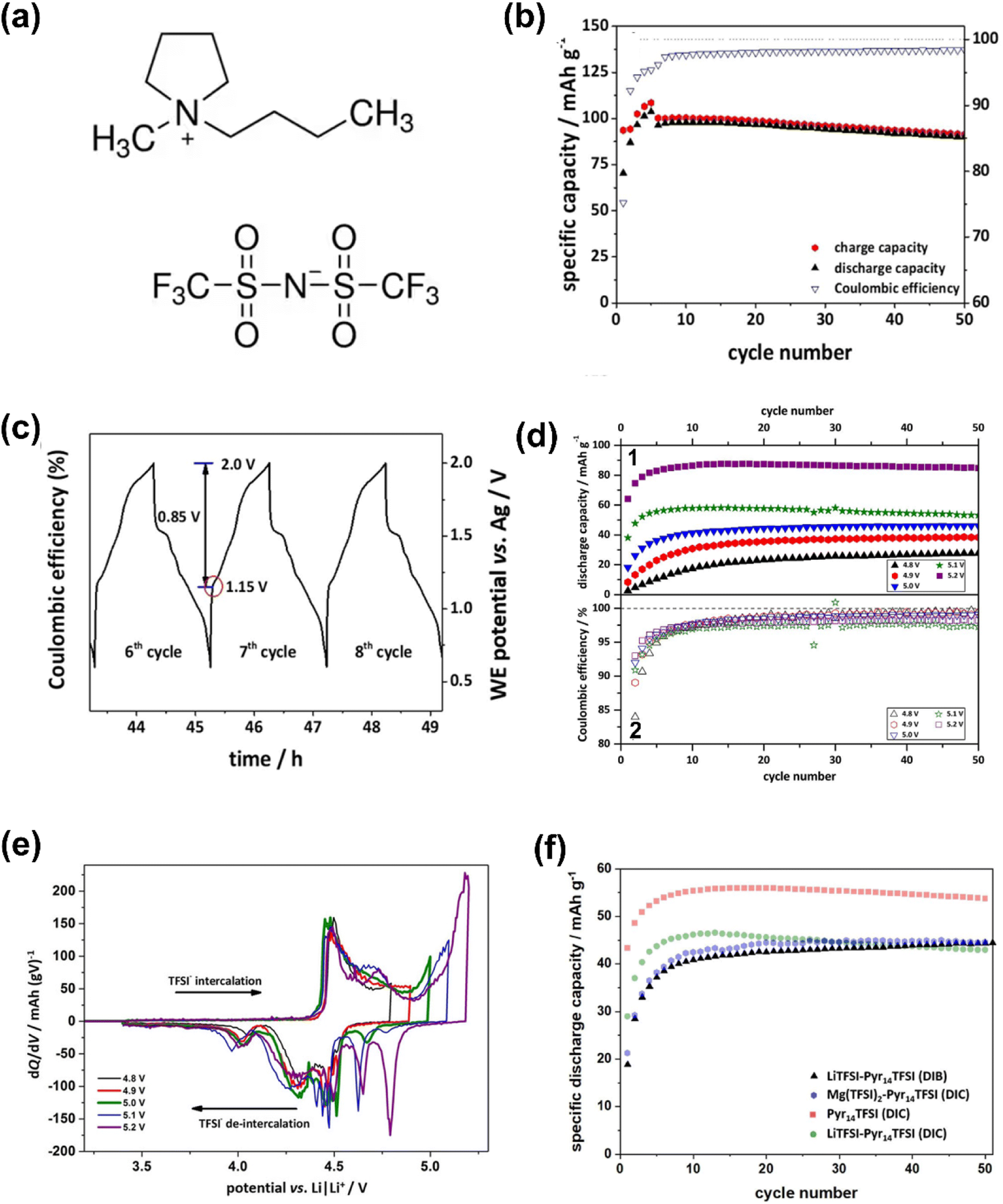 | ||
| Fig. 6 (a) 1-Butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14TFSI) ionic liquid molecular structure. (b) Cycling performances and (c) the related working electrode potential vs. time profiles of the graphite‖AC supercapattery during galvanostatic charge–discharge cycling at 100 mA g−1. Electrolyte: 0.3 M Mg(TFSI)2 in Pyr14TFSI. (d) (1) Specific discharge capacities and (2) Coulombic efficiencies vs. cycle number and (e) comparison of specific differential capacity profiles (dQ/dV) of the graphite‖AC supercapattery at 50 mA g−1. Potential range: 3.4 V to 4.8–5.2 V vs. Li/Li+; electrolyte: 0.3 M Mg(TFSI)2 in Pyr14TFS. (f) Cycling performances of the graphite‖AC supercapattery (DIC) at 50 mA g−1 using either Pyr14TFSI (red), 0.3 M LiTFSI in Pyr14TFSI (green) or 0.3 M Mg(TFSI)2 in Pyr14TFSI (blue) as electrolyte. In addition, the graphite‖Li metal (DIB) cell (0.3 M LiTFSI in Pyr14TFSI; black) at 50 mA g−1 is illustrated. Potential range: 3.4 V to 5.0 V vs. Li/Li+.52 (Reprinted from ref. 52 with permission from Rightslink). | ||
In 2020, Yan and co-workers reported a high-power Mg battery based on a heterogeneous redox enolization mechanism and a weakly coordinated electrolyte. This work uncovered a positrode reaction mechanism for rapid storage of Mg2+ ions, and at the same time created a Mg electrolyte based on ether-mixed solvents and weakly coordinating anions (CB11H12−), enabling Mg metal deposition with no dendrite formation at relatively high current densities, e.g., 20 mA cm−2. The battery achieved 30.4 kW kg−1 specific power, which is almost two orders of magnitude higher than the highest power output of a previously reported Mg battery.69
Furthermore, this work highlighted the discovery and application of a series of methoxyethylamine chelating agents that facilitate interfacial charge transfer kinetics in rechargeable bivalent metal batteries. The solvent shell recombination process can also suppress the side reactions occurring on the layered oxide positrode and metal negatrode, leading to a rechargeable Mg metal battery with a specific energy of 412 W h kg−1.70 Designing an artificial interface protective layer on the metal negatrode not only effectively inhibited the harmful decomposition reaction of the common Mg electrolyte on the Mg metal negatrode, but also promoted uniform Mg deposition, avoiding the occurrence of dendrites which cause a short circuit in the battery.71 The development of electrode materials and electrolytes in batteries, capacitors, and supercapatteries is different but interlinked. Therefore, the development of Mg metal batteries is bound to bring new development opportunities for Mg metal supercapatteries.
2.4 ILs in potassium-based supercapatteries
In addition to Li, Na, and Mg, the other two relatively abundant AAEMs are calcium (Ca) and potassium (K) which are also gradually emerging in research for using as the negatrode in EESDs. Relatively mature Li- and Na-based negatrode materials and the matching electrolytes can form the base for the development of K- and Ca-based EESDs.72 In comparison with Li+, Na+ and Mg2+ ions, both K+ and Ca2+ are much larger in ionic radius, causing an obvious volume change during charging and discharging, parasitic reactions, and dendritic growth. Meanwhile, there are fewer studies reported on using Ca as the negatrode.73 K is clearly more popular in research, although the electrochemical devices are still far from practical applications.In 2020, Hundekar et al.74 achieved in situ healing of dendrites in K metal batteries by rationally controlling the self-heating behaviour of K electrodes. This opens the door to K supercapatteries with high energy density.74 Fabrication of metal K-containing negatrodes by infiltrating aligned carbon nanotube membranes with molten K was attempted to provide sufficient electrode/electrolyte contact for charge transfer. Such a K metal negatrode showed stable plating/stripping profiles and low polarisation during charge and discharge. In addition, this design could also effectively suppress the growth of dendrites. It was paired with a Prussian blue positrode when assembled into a full cell whose very good performance confirmed a high compatibility between these two electrode materials.75 This approach was also considered as an effective solution for the design of K metal negatrodes.
In recent years, the use of IL electrolytes for high-performance K-based batteries has gradually increased. In 2019, Yoshii et al.76 developed the Pyr13TFSI-based stable and safe IL electrolytes with potassium bis(trifluoromethanesulfonyl)imide (KTFSI) to work with new high voltage layered positrode materials for high-voltage K-ion batteries. In 2020, Sun et al.51 reported a battery using an IL, 1-ethyl-3-methylimidazolium chloride, with two important additives, i.e., potassium bis(fluorosulfonyl)imide (KFSI) and EtAlCl2, as the electrolyte and metal K as the negatrode (Fig. 7). The IL electrolyte specified was found to be able to provide a robust K-containing passivating interphase in batteries to achieve excellent cycling performance.
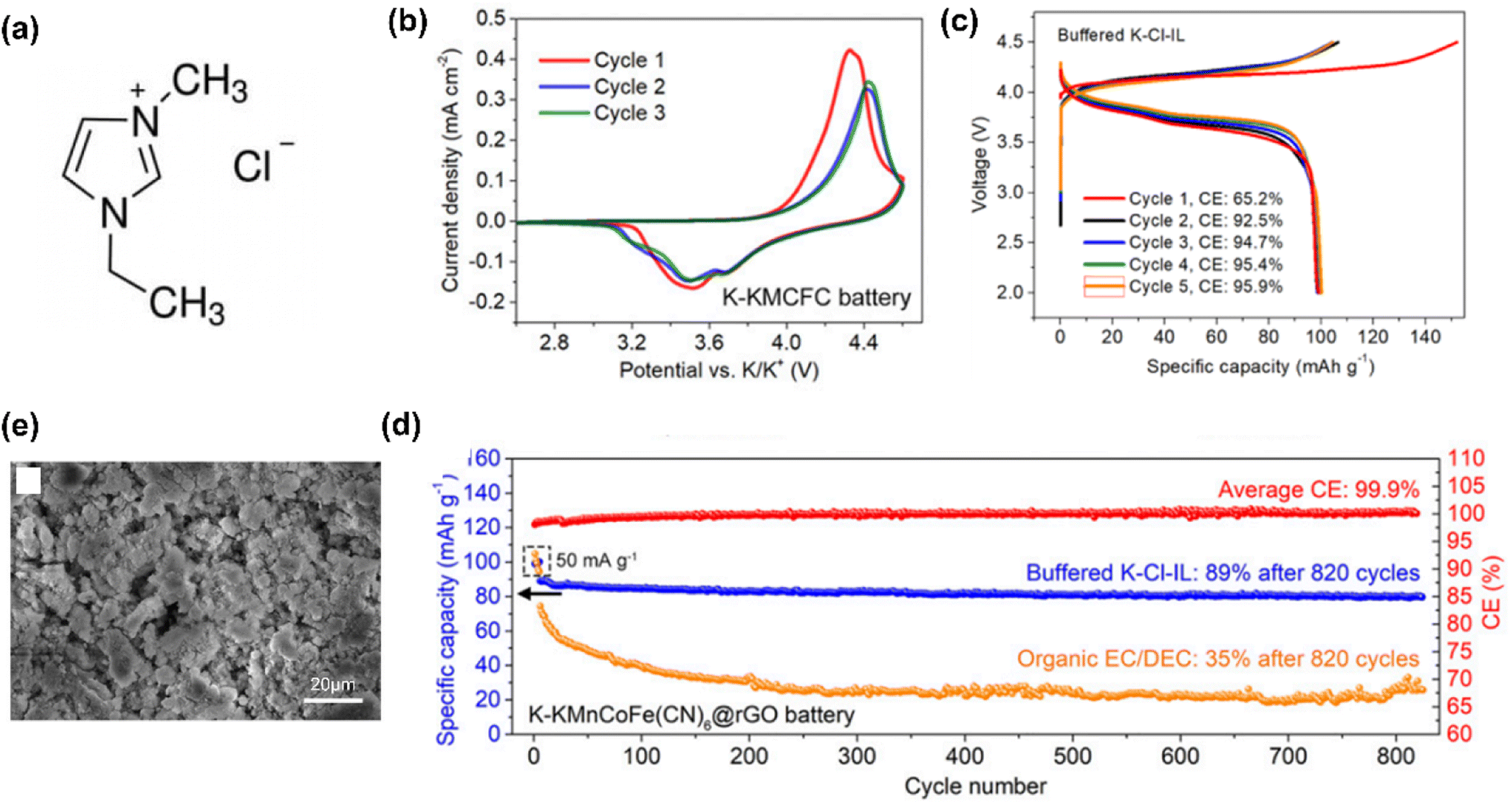 | ||
| Fig. 7 (a) Molecular structure of 1-ethyl-3-methylimidazolium chloride (EMICl) ionic liquid. (b) Cyclic voltammograms of a K metal‖KMCFC battery using buffered K–Cl-IL electrolyte at a scan rate of 0.1 mV s−1. (c) The first five galvanostatic charge–discharge curves of K metal‖KMCFC@rGO batteries at 50 mA g−1 using buffered K–Cl-IL electrolyte. (d) Cycling performances of K metal‖KMCFC@rGO batteries using buffered K–Cl-IL and organic electrolytes at 100 mA g−1. (e) SEM image of a K negatrode cycled in a K metal‖KMCFC@rGO cell. EtAlCl2 and KFSI were added in a mixture of AlCl3 and EMICl at a molar ratio of 1.2, to obtain the final KCl-buffered chloroaluminate IL (buffered K–Cl-IL) electrolyte. KMCFC represents K1.90Mn0.92Co0.08[Fe(CN)6]0.96. Reduced graphene oxide was introduced to KMCFC (referred to as KMCFC@rGO).51 (Reprinted from ref. 51 with permission from Copyright (2020), National Academy of Sciences). | ||
In the same year, a unique potassium monocation ionic liquid (K-SCIL) was developed in Japan for use in K-ion batteries. The IL electrolyte worked well with the graphite negatrode and supported high current densities.77 Even though no supercapattery that combines K metal or K ions with IL electrolytes has been reported yet, with the research on high-quality positrode materials and functional ILs, there will be more and more K-based supercapatteries based on ILs in future research.
2.5 ILs in calcium-based supercapatteries
Calcium (Ca) is considered as a promising metal for rechargeable batteries due to its theoretically very negative working potential. However, the passivation layer formed on the Ca metal surface in the proton electrolyte cannot effectively conduct Ca2+, making the deposition process difficult to achieve. In addition, it is difficult to find electrode materials for Ca EESDs that can efficiently store and release Ca reversibly.78 Recently, to address this issue, Park et al.79 applied density functional theory (DFT)-based high-throughput quantum mechanical calculations to predict the battery-related properties of various layered materials incorporating Ca and transition metal oxides. CaCo2O4 was most recently found to have an optimal balance of properties including thermodynamic stability, average voltage, energy density, and synthesisable properties.The direct use of metallic Ca as the negatrode is far from meeting the performance standards of commercial batteries because the coulombic efficiency of the Ca metal negatrode was found to be unsatisfactory at low power.
Optimization of the SEI structure via tuning IL is an effective way to enhance the coulombic efficiency of Ca-based batteries. Recently, Passerini et al.80 achieved an extraordinary initial discharge capacity of 332 mA h g−1 and reversible capacity of 244 mA h g−1 using the optimized IL-based electrolyte ([Ca(BH4)2]0.05[N07TFSI]0.95) in V2O5‖Ca cells. It was revealed by quantitative analysis that the polyether chains could effectively replace TFSI− from the Ca2+ coordination sphere, fostering the reversible Ca deposition/dissolution process. Meanwhile, an organic-rich, but inorganic-poor SEI layer was formed, enabling Ca2+ diffusion rather than passivating the Ca metal. Among the Ca-based EESDs that have been reported so far, the best performer is a supercapattery in which tin which is capable of alloying with Ca was used as the negatrode and activated carbon served as the positrode.81 It exhibited a fairly large reversible capacity of 92 mA h g−1, unrivalled rate capability (full recovery of the discharge capacity upon rate variation), and high capacity retention of 84% after 1000 cycles at room temperature. Among the IL electrolytes for Ca-based EESDs, Pyr14TFSI and PyrH4TFSI based IL electrolytes showed good transport performance and electrochemical stability in EDL capacitors. However, the compatibility of such electrolytes with the TiS2 positrode is not optimistic, which makes the application of IL electrolytes in Ca-based energy storage devices still a big challenge.82
3 Challenges and prospects
In this part, the current main challenges in terms of ILs and AAEM negatrodes for developing the respective supercapatteries are discussed. The prospects of developing high-performance IL-based AAEM supercapatteries are also analysed and speculated on.3.1 Challenges
However, low ionic conductivity, possible risk of leakage to the environment, high selectivity to material structure/composition, strong corrosivity, and unsatisfactory functionality are currently the main challenges for their use in AAEM supercapatteries.
The design via synthesis and selection of appropriate additives in the IL electrolyte are important ways to modify and improve any undesirable physical and chemical properties of IL, e.g., low ionic conductivity.84 First of all, low ionic conductivity at room temperature, owing to high viscosity, sometimes results in reduced energy capacity and power capability in AAEM supercapatteries. Modification of the cation structure, e.g., replacing the linear alkyl substituents of the ammonium cation with curled ether groups, was shown to significantly lower the viscosity of the ILs with the same anion by a factor of 0.2 to 0.1.85
The addition of some molecular solvents in IL electrolytes can also effectively improve their ionic conductivity.86 Lalia et al.87 developed a new binary mixture of non-flammable additives composed of triethylphosphate (TEP) and ethylene carbonate (EC) to improve the performance of IL electrolytes. After adding TEP and EC in the 0.4 M LiTFSI in N-methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl)imide (PP13TFSI), the ionic conductivity at room temperature was enhanced from 8.2 × 10−4 S cm−1 to 3.5 × 10−3 S cm−1. No change in the ESW of PP13TFSI was observed. Novel anion and cation designs are an effective alternative strategy to modify the low ionic conductivity of ILs. Chen et al.88 fabricated a novel IL of 1-trimethylsilylmethyl-3-butylimidazole bis(trifluoromethanesulfonyl)imide ([SiM-BIM]TFSI) for Li metal rechargeable batteries. The imidazolium cation with a silicon-containing substituent could reduce the viscosity and improve the ionic conductivity of IL electrolytes. Further, the heteroatom Si substituent was found to make the C-2 position of the imidazolium cation less active, which stabilised the cation against cathodic polarisation. This effect in turn helped achieve uniform Li deposition/dissolution and contributed to increasing the CE and stability of the cell.
Many ILs themselves are unfriendly or even harmful to the environment and must be fully confined within the EESDs during their whole service life. The liquid nature of ILs means high mass mobility which makes it challenging to achieve full utilisation efficiency during long-term dis-/charging cycling. At any level, the loss of IL will not only increase the cost but also lower the energy capacity of the EESDs, or even bring about the risk of leakage beyond the legal limits. Compared with traditional organic electrolytes, polymer electrolytes have the advantages of no flow and no leakage. Thus, by combining ILs and polymer materials, the leakage and utilisation issues are in principle resolved, whilst the conductivity, stability and safety of polymer electrolytes are also improved. Rupp et al.89 combined polyethylene oxide (PEO), 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14TFSI), and LiTFSI into PEO/IL/LiTFSI ternary composites. Not only was the conductivity increased, but also the safety issues associated with LIBs were improved. Lavall et al.90 obtained a new electrolyte based on thermoplastic polyimide esters with different proportions of LiTFSI, propylene carbonate (PC) and N-ethyl(methylether)-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (RYRA1201TFSI). These polymer electrolytes showed good thermal stability, wide ESWs and a maximum ionic conductivity of 10−4 S cm−1.
Sometimes, large IL ions effectively block the pores in different ways, including size mismatch and strong/permanent adsorption that hinder further ion movement in and out of the pores. Thus, ILs may show a high selectivity towards the structure of positrode/negatrode materials (e.g., surface area, pore size or porosity). Shiraishi et al.91 discussed the electrochemical capacitance of activated carbon fibres in 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIBF4) versus a propylene carbonate solution of 0.5 M (C2H5)4NBF4 (TEABF4/PC). The activated carbon fibres showed a very stable cycling performance but decreased capacitance due to the mismatch between the micropore sizes of the activated carbon fibres and the TEA+ cation in TEABF4/PC. Activated carbon fibres showed a higher capacitance in EMIBF4, but their cycle performance was relatively poor because of a mismatch between the narrow micropores and the size of the EMI+ ion. The analysis showed that the EMI+ cation could strongly adsorb on a flat carbon surface. Therefore, in the slit-shaped micropores of the activated carbon fibres, EMI+ ions could irreversibly adsorb on the walls of the micropores. As a result, the discharge capacity decayed fast. Complementary to the pore size, the positrode must have a sufficiently large number of pores to maintain an adequate number of redox species and products (from the charging reaction) inside the pores for improving the Nernstian charge storage.92
Some halides in ILs show strong corrosion to the current collector, which can affect the practical application of IL in EESDs. IL-based electrolytes have been studied for secondary Al batteries as early as 1933 (ref. 83) and have recently gained increasing attention in Al metal batteries. A special issue with Al batteries is that the SEI layers which can form during dis-/charging often passivate the Al surface, both ionically and electrically. Meanwhile, surface passivation of Al can be alleviated using ILs containing AlCl3 according to the work of Jayaprakash et al.93 Thus, Al ion conductivity in the SEI can be improved using this reported IL. However, the Cl in the Al salts (Al2Cl7−) is quite corrosive, even to stainless-steel current collectors.94 In addition, the Cl in Mg organohaloaluminate salts (among other variants) is very corrosive to the current collectors.95 Therefore, when selecting the anions of ILs, their corrosiveness to battery components should be carefully considered, especially in practical applications. In addition, necessary anti-corrosion measures should be taken. For example, an anti-corrosion coating may be sprayed on the surface of battery components e.g., current collectors to effectively reduce the corrosive effects of ILs.
The thermochemical and electrochemical stability of ILs is one of the basic reasons why they are regarded as excellent green solvents and electrolytes. However, high stability also means poor functionality for some specific tasks, but it can be altered or improved by the addition of other additives, salts and/or solvents. For instance, redox additives or mediators can be dissolved in the IL electrolyte to improve the energy capacity of EESDs, particularly supercapacitors. Such additives undergo reversible electron transfer reactions inside a porous electrode and can contribute to extra charge storage capacity.92,96 Further, the ionic conductivity of IL electrolytes can be enhanced to a certain degree with the addition of ionic redox additives, which improves the specific power of EESDs.97,98
Navalpotro et al.33 dissolved 0.4 M para-benzoquinone (p-BQ) in 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr14TFSI). This redox IL was used in asymmetrical hybrid supercapacitors (which were in principle the same as supercapatteries). One pair of very broad redox current peaks appeared on the CV (Fig. 8a) in a symmetrical two-electrode cell with Vulcan carbon and commercial activated carbon electrodes, indicating the presence of a Faradaic contribution from the redox processes of p-BQ.
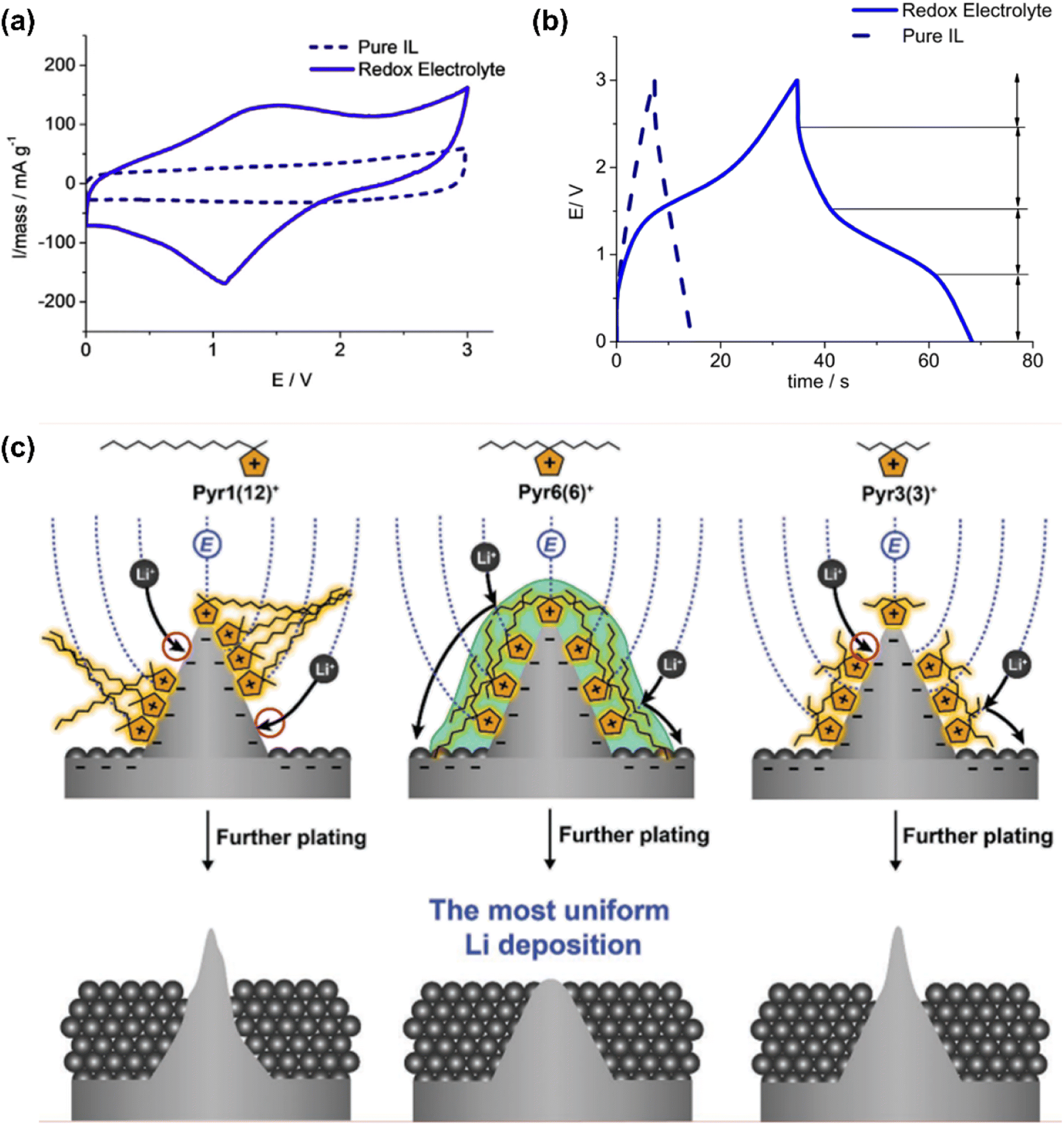 | ||
| Fig. 8 (a) Cyclic voltammograms and (b) galvanostatic charging and discharging profiles up to 3.0 V of a symmetrical cell of Vulcan carbon. Electrolytes: redox IL electrolyte of 0.4 M p-BQ in PYR14TFSI (solid blue lines) and pure PYR14TFSI (dashed black lines). Temperature: 60 °C. Current density: 10 mA cm−2.33 (c) The impact of three different IL cations, Pyr1(12)+ (left), Pyr6(6)+ (middle), and Pyr3(3)+ (right), on inducing uniform Li metal deposition owing to the formation of lithiophobic protective layers on Li protuberances.99 (Reprinted from ref. 33 and 99 with permission from Rightslink). | ||
The GCD profiles as shown in Fig. 8b illustrated approximately four different regions, including the plateau region typical of the Faradaic contribution between 1.5 and 0.8 V during discharging. These CV and GCD features show that charge storage in the Vulcan carbon-based supercapacitors is composed of the EDL capacitance and Faradaic reactions (Nernstian process). The specific energy of the cell with the redox IL operating at different working voltages (2.0, 3.0, and 3.5 V) was obviously greater than that with pure PYR14TFSI. Interestingly, the specific energy of Vulcan carbon (240 m2 g−1 specific area) was more significant than pica carbon (2400 m2 g−1) with or without using the same redox IL. This feature also confirms that the effectiveness of applying redox ILs is strongly influenced by electrode materials and structures.
Some special physical and chemical properties of ILs can significantly improve the electrochemical performance of AAEMs, especially for metal deposition on the negatrodes without dendritic growth.100,101 ILs with an appropriate Li salt concentration can effectively improve the stability of the electrode interface. A solution of 2 M LiTFSI in 1-methyl-1-propylpyrrolidinium bis(trifluoromethanesulfonyl)amide (Pyr13TFSI) with added ether solvent to form a hybrid IL electrolyte was reported by Li et al.102 Li dendrite growth and the corrosion of Li metal in the batteries was effectively alleviated by surface passivation in this hybrid IL electrolyte. They found that there was a synergistic effect of Pyr13TFSI IL and Li salt, remarkably enhancing the reversibility of Li plating. The stability of SEI layers, including passivation substances (Li3N and LiF) on Li metal, can be dramatically improved by including Pyr13TFSI in the electrolyte.
Targeted designs for anions and cations are also an important strategy to improve battery performance. For instance, cations with lithiophobic symmetric alkyl chains were introduced to 1,1-dihexylpyrrolidium bis(fluorosulfonyl)imide ([Pyr6(6)FSI]) to shield this moiety from the Li negatrode (Li tips) and mitigate the continuous growth of Li dendrites by Jang et al.99 This effect could have resulted from firstly pyrrolidium cations (Pyr+) being preferentially attracted to the protuberances on the Li surface by the electric field and secondly the reduction potential of Pyr+ was more negative than that of the Li ions (−3.04 V vs. standard hydrogen electrode, SHE). In other words, the Pyr+ cations with symmetric alkyl chains led to the most densely assembled shielding layer compared with the conventional cations with asymmetric alkyl chains (Fig. 8c). Thus, alkyl chains in this cation enabled the cation to be assembled on protuberant tips, forming a protective layer against further Li deposition. Uniform Li deposition and higher CE become more realistic because of this protective layer on any protuberances on the surface of the Li deposit.99
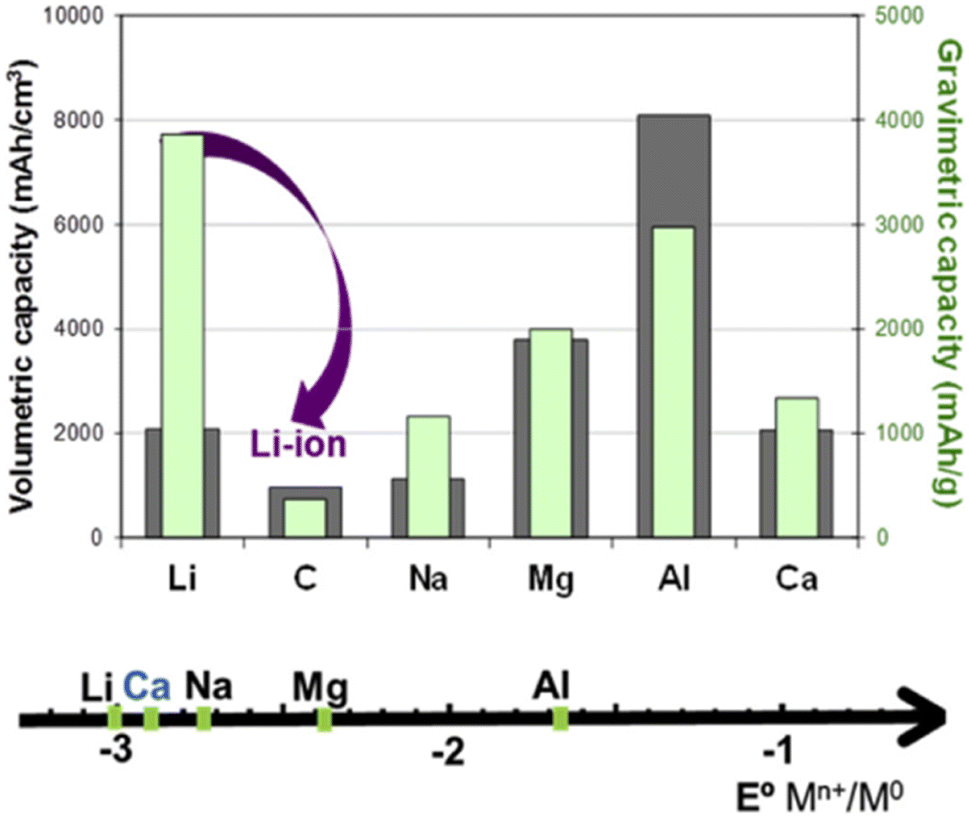 | ||
| Fig. 9 Comparison of the standard reduction potential versus the charge density (left, volumetric capacity) or specific charge capacity (right, gravimetric capacity) for different metals and graphite in the LIB.108 (Reprinted from ref. 108 with permission from Rightslink). | ||
The demand for high energy storage capacity in LIBs has revitalised research on AAEM negatrodes. Unfortunately, two electrochemical challenges strongly limit the practical application of a pure AAEM negatrode in EESDs. One is the plating properties of the AAEM negatrode material. The other is the nature of the electrolyte decomposition on the AAEM negatrode.109
Dendrite formations are relatively common on many metal negatrodes. For example, Li metal batteries were plagued with well-known dendrite formation that can lead to thermal runaway or explosion.110 Compared with Li metal, Na or K metal batteries will likely result in even more dangerous thermal-runaway accidents due to the combination of higher reactivity and lower melting point of Na and K metals.111 In addition to the safety issues related to dendrite formation, continuous exposure to fresh metals can lead to electrolyte decomposition and then capacity loss, resulting in electrolyte depletion and low CE. The severity of these issues is strongly related to the operating conditions (e.g., current density, areal capacity, and electrolyte composition) and most vitally, the nature of the AAEM negatrode.112
3.1.2.1 Solid electrolyte interphase (electrolyte decomposition). The SEI as a passivation film on the metal negatrode can isolate the continuous direct contact between the electrolyte and the metal and provide channels for the transport of desolvated ions. Thus, it must be carefully designed.113 Although stable SEI formation with the same level of performance as that in LIBs is difficult to obtain on Li, other AAEM negatrodes seem to be more problematic than the Li negatrode. For instance, a stable SEI cannot be formed on the Na metal owing to the bulky and porous nature of the SEI formed in carbonate-based electrolytes, which will enhance the direct contact between the Na negatrode and the electrolyte.114 Furthermore, the SEI on the Na metal is more soluble in carbonate electrolytes due to the higher number of inorganic species and weaker ionic bonds (larger ionic radius) in the SEI compared with that on the Li metal.115 Diglyme-based electrolytes can facilitate the formation of higher inorganic material and minimal polymeric film, inducing improved electrochemical performance of Na metal batteries owing to the decreased SEI solubility in the ether-based electrolyte.116
The dication of Mg117 or Ca118 suffers from great difficulty in diffusing through the SEI layer. For the SEI on Mg and Ca, the metal is not only electrically passivated but also ionically insulating, which greatly restricts the transfer of both cations and electrons.109 For this reason, Mg Grignard reagents were developed as an electrolyte that does not decompose over Mg in the Mg metal battery. Thus SEI-free Mg can be formed during cycling.119 Later, Mg organohaloaluminate salt-based electrolytes were developed by Aurbach et al.,120 which can render the electrolyte sufficiently stable toward Mg metal to enable pairing with a positrode and facilitate stable Mg ion conductive SEI formation.
The highly tuneable properties of ILs have given rise to very specific electrolyte compositions. Specifically, ILs in Mg batteries need to be functionalized with ether to facilitate stable stripping and plating. Without the ether solvent, the Mg2+ dication is coordinated with the bis(trifluoromethanesulfonyl)imide (TFSI) anion. The chance of TFSI reduction and Mg passivation film formation is significantly increased during plating Mg because of the close proximity between the TFSI anion and the Mg atom.121 Interestingly, recent work demonstrated that pre-fabricated artificial SEI coatings composed of cyclised polyacrylonitrile and Mg(CF3SO3)2 over Mg could improve the Mg2+ ion conductivity to 1.19 × 10−6 S cm−1, while remaining electronically insulating.122 This work can further allow the use of a wide range of electrolyte solvents.
The commercial availability of Ca metal batteries is still unknown, possibly due to the highly reactive nature of Ca and relatively low specific energy estimation, which has attracted limited research on such EESDs.123 However, according to a recent breakthrough, reversible plating of Ca with Ca(BH4)2 in a tetrahydrofuran (THF) electrolyte was achieved for ∼50 cycles,124 where a CaH2 layer formed on the surface of Ca served as a poor-quality SEI. More impressively, a recent work on the Ca metal negatrode showed that the SEI formed on Ca could conduct Ca2+ ions at mild temperatures ranging from 75 to 100 °C, enabling an extended range of applications.125
3.1.2.2 Polymorphous metal deposit (plating properties). The formation of polymorphous metal deposits, e.g., dendrites, is a common phenomenon in many electrochemical metal plating processes. Generally, there are four different modes of electrodeposition: root growth (whiskers), tip growth (dendrite), a combination of root and tip growth, and higher-dimensional plating (non-dendritic growth).126 Many efforts have been devoted to suppressing the formation of dendrites, such as through electrolyte engineering,127 forming an artificial SEI or coating layer,9,128 and development of 3D current collectors.129,130 Fundamentally, metal electrodeposition consists of five sequential processes. First, the metal ions transfer from the bulk electrolyte. Secondly, desolvation of the metal ion from the electrolyte occurs. The process of surface adsorption is followed by charge transfer and ultimately surface diffusion to the deposition site. The final morphology and quality of the metal deposit are affected by each of these steps.131
There are three modes describing polymorphous metal growth, as shown in Fig. 10a. For example, for Li, these are whisker-like (Li whisker) growth, mossy-like (Li moss) growth, and tree-like (Li dendrite) growth. These each follow different growth patterns. Li whiskers follow a root growth pattern because there is no compressive stress whilst electronic/ionic conductivities are higher at the root. Li moss belongs to the surface growth pattern because of a higher deposition rate than SEI formation. Li dendrites follow a tip growth pattern due to the higher electric field at the tips.132–134 Mossy metal usually forms before Li whiskers or serious Li dendrite growth and originates from the difference in the SEI layer's electronic conductivity.
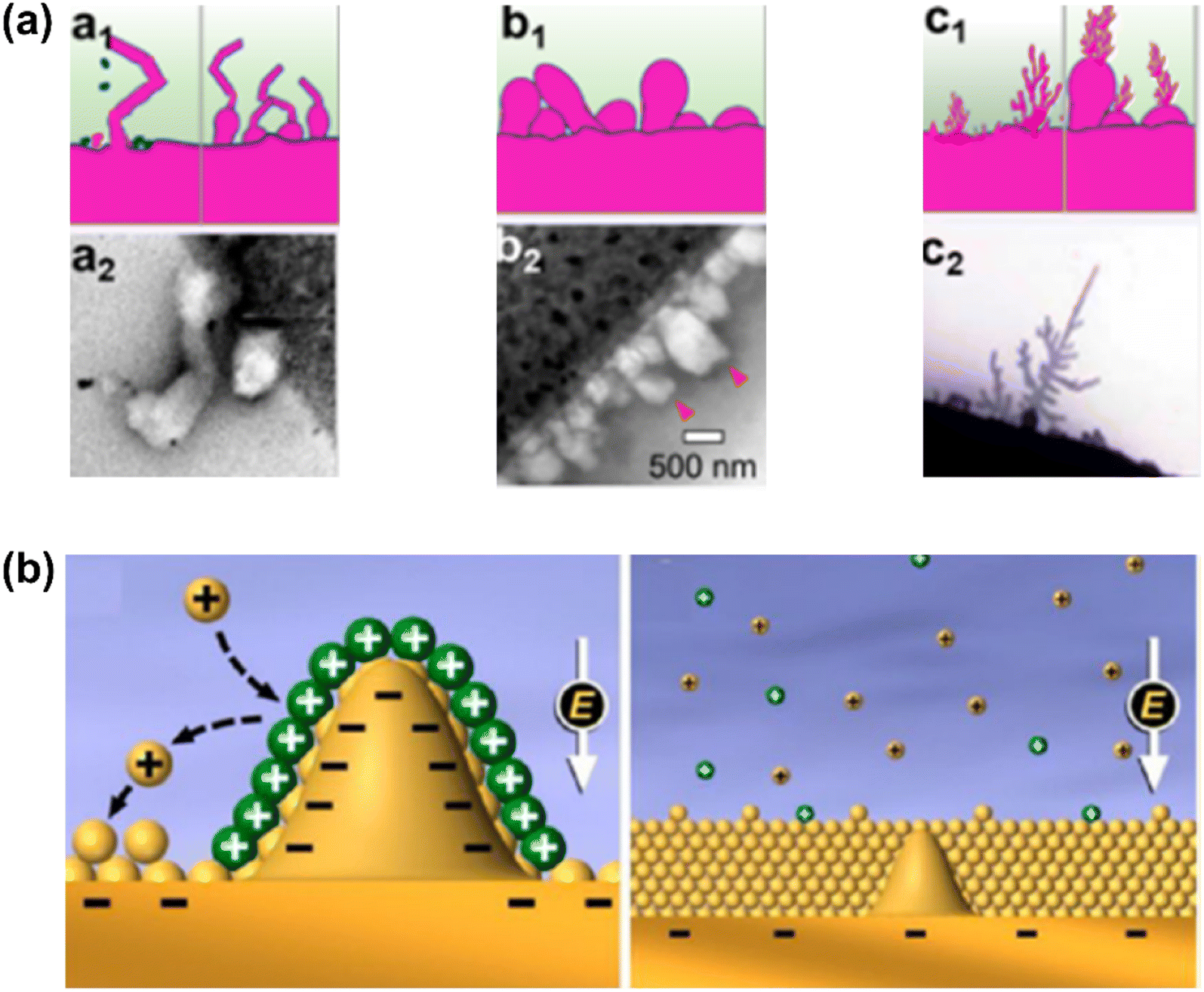 | ||
| Fig. 10 (a) Three modes describing polymorphous Li metal growth: (a1, b1, and c1) schematic morphologies, (a2, b2, and c2) real microscopic images for whisker-like, mossy-like, and tree-like (dendrite) deposits.132 (b) Diagram of Li deposition based on the self-healing electrostatic shield mechanism.135 (Reprinted with permission from132 Copyright 2021, reprinted with permission from ref. 135. Copyright 2013, American Chemical Society). | ||
Interestingly, almost no mossy dendrite growth on multivalent AAEM negatrodes has been found, possibly because of the lack of special SEI that can induce mossy growth.109 The most noteworthy property of Mg negatrodes is their dendrite-free deposition. A possible reason may be the surface energy of Mg being larger than that of alkali metals (Li > Na > K).136 This explanation is in agreement with DFT calculations and experimental results which indicate that Mg does not plate dendritically (large surface area) due to the higher surface energy relative to Li.137,138 However, it was reported that the absence of dendrites in Mg might not be an inherent property arising from surface energy, but rather a result of surface diffusion of Mg atoms.139 The surface diffusion of Mg atoms on an Mg bulk surface was calculated to be relatively faster than that of Na and Li.140 Thus, slower surface diffusion can lead to tip growth. To alleviate this dendrite growth, electrolyte/metal ion pairings with faster surface diffusion could be implemented.
Metal ions in the electrolyte are not fast enough to match the electron flux at currents beyond a certain limiting level and deposition time, causing severely polarised potential in cells.141 The metal plating switches from high-dimensional or mossy plating to tip dendritic growth. At Sand's time (dendrite formation time), the concentration of the cation for plating is zero at the metal surface and pure dendritic growth begins.142 The electrolyte cannot supply sufficient cations for plating, and sharp dendrites are rapidly formed to maintain a constant current density. The presence of a 3D current collector is reported to effectively decrease the current density and prolong Sand's time, alleviating dendrite formation.129 Enhancing salt concentration can decrease the local spatial charge variation and mitigate dendrite formation owing to the increased plating cation's transference number (Li,143 Na,144 and K145). However, high viscosity and related slow charge and mass transfer at a higher current are the main problems, especially in IL electrolytes. Another alternative method is to add small amounts of cations such as Cs+ with a more negative effective reduction potential relative to Li, to reduce variations in spatial charge (Fig. 10b).135
3.2 Prospects
IL electrolytes can play an important role in the construction of high performance AAEM supercapatteries. Creating economical, functional, and stable IL electrolytes is not a trivial task, but it is worthy of more effort and attempts, since the physical and chemical properties of ILs are largely tuneable by a judicious combination of cations and anions. Notably, appropriate additives can be rationally selected or designed to improve the drawbacks of ILs (e.g., low ionic conductivity). Meanwhile, film-forming properties such as SEI on AAEMs, corrosion toward current collector/AAEMs, and structure matching with the positrode should also be carefully considered.
The SEI with electron passivation and Li+ ion conduction is a fortunate compromise between highly reactive AAEM negatrodes and relatively unstable electrolytes in LIBs. However, such SEIs from LIBs are hard to mimic in other EESDs. For example, the SEI is often absent on Mg negatrodes. Theoretically, nearly 100% CE renders exceptional electrochemical properties for Mg without the SEI. Furthermore, many tricky problems that are related to SEI formation such as the electrolyte drying out, SEI-induced deviation in spatial charge, and metal negatrode consumption may not be present in other multivalent metal batteries. Last but not least, faster dis-/charging processes may be easily achieved owing to the absence of a barrier in the absence of an SEI. For example, an aluminium (Al)–chalcogen battery may achieve the absence of an SEI on Al metal using molten-salt electrolyte composed of NaCl–KCl–AlCl3 and show excellent capacity retention at an ultrahigh current rate (200 C).148
From current viewpoints, the key technological point for AAEM metal and ion EESDs lies in whether or not the electrolyte decomposition can form a uniform SEI layer that conducts the respective AAEM ions. However, if there is the possibility of developing an SEI-free metal negatrode from an IL electrolyte with high positrode stability, it will be much more attractive for achieving highly stable and efficient electrochemical performances (e.g., higher CE). This strong anti-reduction nature of the cation and anion from ILs and AAEMs is expected. Meanwhile, other challenges of ILs e.g., poor negatrode stability and high cost or corrosion toward the current collector, are not expected to be evident. Alternatively, developing AAEM ion conducting interphases could be the most viable choice to facilitate similar high cycle durability of commercial LIBs for AAEM supercapatteries.
However, to the best of our knowledge, there is not yet any purposely reported study on molten salt supercapatteries, although a noticeable portion of the literature is on high temperature rechargeable batteries of which AAEMs were used to make the negatrode. In more recent years, molten salt supercapacitors are emerging. Therefore, it is not unreasonable to postulate molten salt supercapatteries as a concept and explore their prospects and challenges.
The effort in the past to utilise molten salts for EESDs was focussed on batteries in which the dominant charge storage follows the Nernstian mechanism. Molten salt batteries have regained significant attention in recent years due to their high specific energy, long cycle life, and ability to operate at high temperatures. It is interesting to note that in conventional high power batteries with organic electrolytes such as LIBs, heat build-up inside the batteries during dis-/charging is a technical and safety challenge and must be dissipated effectively and quickly. Consequently, sophisticated heat management is needed in terms of design, manufacture and material selection, further increasing the cost. However, for molten salt EESDs which are especially suitable for high power applications, there is no need for heat management because the Joule heat resulting from current passing through the electrode and electrolyte is needed to maintain the working temperature.
One nontrivial component of molten salt batteries is the negatrode material. Compared to other materials, the metal negatrode reveals numerous advantages as discussed before, including high theoretical specific capacity, very negative electrode potentials, high electronic conductivities and considerable compatibility with molten salt electrolyte and metal-free positrodes (e.g., O2).150 As in room temperature electrolytes, metallic negatrodes, such as Li, Na, Mg, Al and Fe, have been investigated extensively in molten salt batteries.151–162 For instance, Giordani et al.152 reported a Li–O2 battery with 95% energy efficiency achieved in a molten LiNO3–KNO3 eutectic at 150 °C. However, the cycling stability was poor (<50 cycles, 2.6 mA h cm−2 at 0.6 mA cm−2), due to the oxidation of carbon and consequent formation of Li2CO3 at the positrode.
Xia et al.153 also studied the Li–O2 battery in the molten eutectic mixture of LiNO3–KNO3 with a nanostructured Ni/LiNO3–KNO3 composite positrode in which a thin layer of LixNiO2 was formed in situ on individual Ni nanoparticles and functioned as the catalyst for O–O bond cleavage and formation. A coulombic efficiency of ∼100% was achieved, also at 150 °C, with promising stability (150 cycles, 0.5 mA h cm−2 at 0.2 mA cm−2). It should be noted that this work was focused on the O2 positrode improvement, mentioning little about the performance of the Li negatrode. Since the testing temperature was 150 °C, the Li negatrode, a disk, was in a solid state and prone to dendritic deposition. Although this was not mentioned, the Li negatrode was separated from the molten salt by a solid electrolyte, Li1.5Al0.5Ge1.5(PO4)3 (LAGP), which obviously helped to mitigate the impact of dendritic deposition. Further, the LAGP functioned as a barrier preventing any product from the positrode to reach the Li negatrode, enhancing the coulombic efficiency of the cell.
Yin et al.154 investigated the liquid metal battery using a liquid Li–Pb alloy negatrode and a liquid Pb positrode in molten LiCl–KCl containing PbCl2. With a porous TiN membrane, the cell achieved a coulombic efficiency of 92% with a round-trip energy efficiency of 71% at 150 mA cm−2 at 410 °C. The very unusual and novel feature of this work is the use of an electron conducting TiN membrane. In conventional EESDs, a porous and/or ion conducting membrane is used to physically separate and electronically insulate the positrode from the negatrode. In terms of function, this membrane forms a part of the electrolyte. In contrast, in the work of Yin et al., the TiN membrane was actually used to hold the liquid Li–Pb negatrode above the liquid Pb positrode. It is therefore a separator for physical prevention of the direct contact and mixing between the two liquid metal electrodes, but the electronic insulation between the positrode and negatrode was achieved by a layer of molten salt. Therefore, the TiN membrane formed a part of the negatrode, instead of the electrolyte. In principle, this (−) Pb–Li‖Pb (+) cell should involve only the reversible conversion between Pb(0) and Pb(II) alternately on the positrode and negatrode. In the liquid Pb–Li negatrode, Li functioned as a solvent to lower the activity of Pb(0). However, because of the presence of Li+ in the molten salt electrolyte, the occurrence of the conversion between Li(0) and Li(I) may not be excluded completely.
Nevertheless, the relatively high cost of lithium because of the limited earth crust abundance could be a major problem inhibiting its wide usage. A viable alternative is a sodium negatrode which costs less and Na is far more abundant on the earth than Li. One attractive example for both transportation and stationary applications is the sodium–metal halide (Na–MH or ZEBRA) battery using a solid membrane of Na+ ion conductor, i.e. Na–β–alumina as the primary electrolyte and molten sodium tetrachloroaluminate (NaAlCl4) as the secondary electrolyte, which has been produced commercially by the FzSoNick Group. Relatively high specific energy (120 W h kg−1) can be achieved at 300 °C with energy efficiency varying from 90 to 95%.163 Besides, Shamim et al.156 reported a 90% energy efficiency at 265 °C for a ZEBRA battery module (48TL200) from FzSoNick with promising stability (a degradation rate of 0.0046%/cycle over 150 cycles). In 2022, Zhu et al.157 reported, as shown in Fig. 11a, a β-Al2O3 membrane enabled Na–O2 battery in a molten NaNO3–KNO3–CsNO3 eutectic at 270 °C with considerable areal energy and power (33 mW h cm−2 and 19 mW cm−2, respectively). The work also revealed dynamic and kinetic complications around the oxygen positrode and the β-Al2O3 membrane, restricting the battery from deep discharging and high power operation. Nonetheless, no issue was mentioned on the liquid Na negatrode.
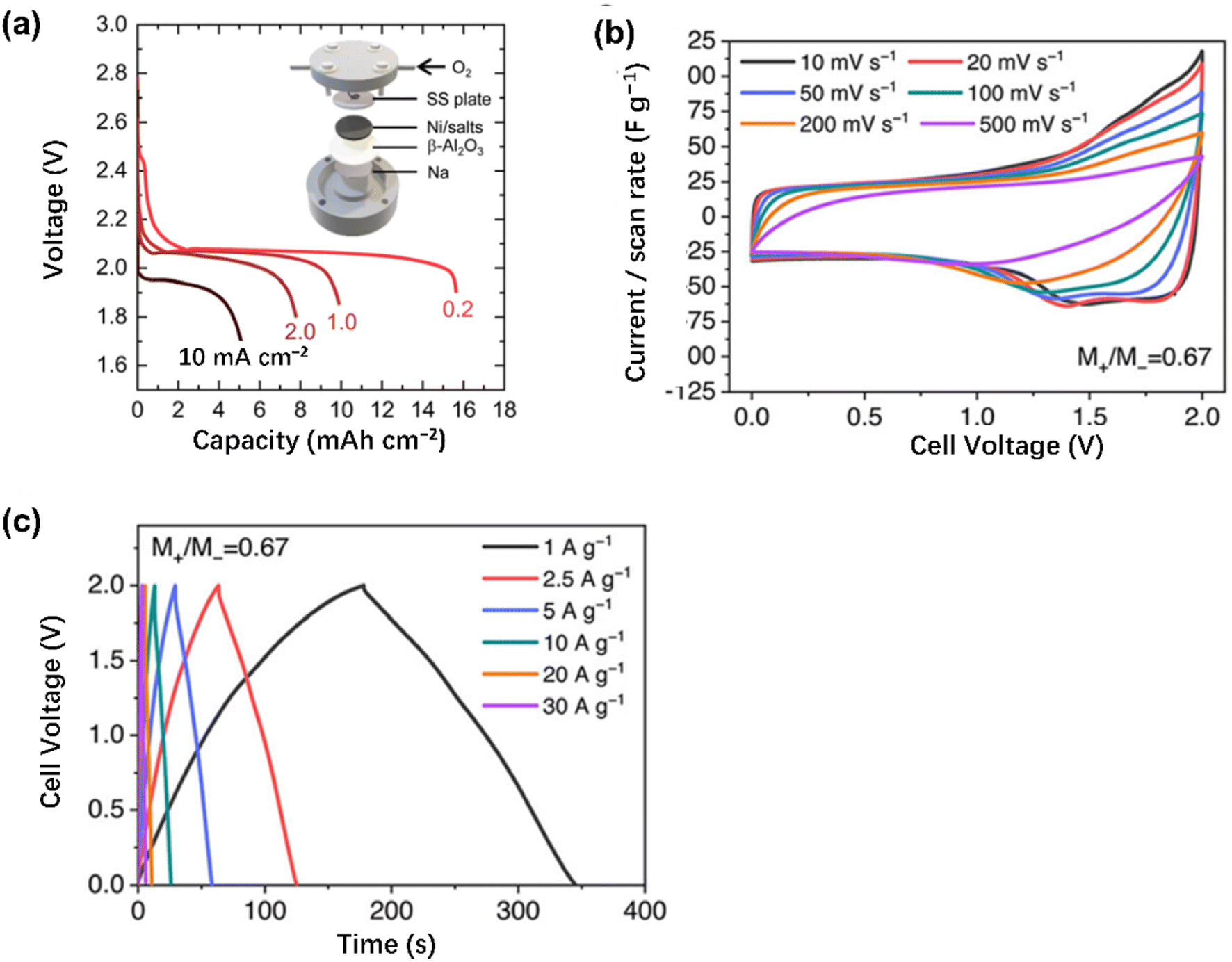 | ||
| Fig. 11 (a) Discharge curves at indicated current densities of a laboratory molten-salt Na–O2 battery as schematically illustrated with a liquid Na negatrode, a two-phase electrolyte of β-Al2O3 membrane plus a molten mixture of NaNO3–KNO3–CsNO3, and a sintered Ni powder positrode attached to a stainless steel (SS) mesh, current collector, through which the inlet and outlet of O2 gas occur. (b) CVs and (c) GCDs at indicated potential scan rates and specific currents, respectively, of an asymmetrical cell of a carbon positrode and negatrode at the mass ratio of M+/M− = 0.67 in molten AlCl3–LiCl–KCl (molar ratio = 0.6:0.2:0.2) at 125 °C. (Adapted with permission from ref. 157 and 164, Copyright 2022, Royal Society of Chemistry). | ||
Molten salt batteries utilising other earth abundant metals as negatrodes (e.g., Mg, Ca, Al and Fe) have also been reported in MgCl2–KCl–NaCl,158 LiCl–NaCl–CaCl2,159 AlCl3–NaCl–LiCl–KCl160 and Fe3O4–Na2CO3–K2CO3 (ref. 162) with remarkable efficiency and life span. It is worth noting that a strong competitor to AAEMs is Al which has also been researched due to the large theoretical specific charge capacity/charge density caused by three-electron transfer in one redox couple (Al3+/Al). Song et al.160 developed an Al-ion battery in molten NaAlCl4 with a coulombic efficiency higher than 99% at 120 °C. An extremely long cycle life (up to 9000 cycles) was also achieved at a 4000 mA g−1. Furthermore, AlCl3–NaCl–LiCl–KCl was utilised by Tu et al. for lowering the working temperature of an Al-ion battery.161 A coulombic efficiency of 91.3% was obtained with a specific capacity of 114.9 mA h g−1 at 90 °C over 1500 cycles.
These few selected studies on molten salt batteries are by no means exhaustive of the literature, but they have already provided valuable insights into the electrochemical performance of these molten salts to accommodate reversible and stable dis-/charging of AAEM and other metal negatrodes. It is particularly worth mentioning that these studies have never encountered problems from dendrite and/or SEI formation, pronouncing a clear advantage of molten salts over both organic and IL electrolytes. However, the positrode design and material selection for Nernstian storage remain a case by case challenge to molten salt batteries, leaving an opportunity for the development of capacitive positrodes and molten salt supercapattery.
In more recent efforts to develop molten salt EESDs, apart from rechargeable batteries, supercapacitors have emerged, owing to their unmatched power capability and cycle life proven in aqueous and organic electrolytes at room temperature. In comparison with their aqueous or organic counterparts, inorganic molten salt electrolytes offer a complementary choice with wide electrochemical stability windows (ESWs) comparable with those of organic electrolytes, high ionic conductivity and low material cost, matching those of aqueous electrolytes.165 Whilst it is possible to use molten salt EESDs in an ambient environment, their high temperature applications are unmatched by their counterparts with aqueous and organic electrolytes.
The first supercapacitor with an inorganic molten salt electrolyte was perhaps reported in 2013.166 The eutectic mixture of LiNO3, NaNO3 and CsNO3 was used as the electrolyte in a symmetrical supercapacitor of activated carbon (1700 m2 g−1 specific surface area). Capacitive storage was investigated by CV at 140 °C, exhibiting a satisfactory rectangular feature without any current peaks. The specific cell capacitance was measured to reach 31.5 F g−1 which is comparable to that of an aqueous supercapacitor. The work revealed mismatching wetting between the molten salt and activated carbon and offered a simple solution by pre-soaking the activated carbon in the aqueous solution of 0.1 M NaNO3 followed by drying before use in the molten salt. The cell worked to a maximum cell voltage of 1.6 V, giving rise to a specific energy of 22.8 W h kg−1.
A significant improvement was reported by Wang et al.,164 employing activated carbon electrodes in molten AlCl3–NaCl–LiCl to form a supercapacitor which exhibited a fairly high specific energy (50.4 W h kg−1) at 125 °C and a specific power of 1.1 kW kg−1. The achieved high cycling stability (99.8% capacitance retention after 10000 cycles) revealed the feasibility of molten salt supercapacitors. Specifically, the enhanced storage capacity was attributed mainly to the intercalation of the AlCl4− anion into the carbon via reaction (5) below.
| Cn + AlCl4− = Cn[AlCl4−] + e | (5) |
The electrode potential of reaction (5) is highly anodic, which means that at high cell voltages, the forward process of reaction (5) should occur on the carbon positrode, but the reverse on the carbon negatrode. This feature is reflected by CVs at higher cell voltages showing increased discharging and charging currents with small peaks, and by GCDs exhibiting smaller gradients (dV/dt) as shown in Fig. 11b and c. Because reaction (5) and its CV and GCD features are indicative of the presence of the Faradaic or Nernstian storage mechanism, the cell was actually a supercapattery.
In the same study,164 other types of molten salt consisting of redox active chloro-aluminate ions, bromine ions, and iodine ions were also examined with a promising specific capacitance (268 to 379 F g−1). These values are comparable to those of non-aqueous supercapacitors, promising the employment of molten salt in supercapacitors for various high-temperature applications.
Unfortunately, the familiar respective drawbacks of supercapacitors (small energy capacity) and rechargeable batteries (low power capability) shown at room temperature also appear at high temperatures. It is therefore natural to consider the prospects of molten salt supercapatteries, although there is a paucity of research in this direction, except for the unnoticed finding in the work of Wang et al.164 Experiences and skills can be learnt from research on batteries and supercapacitors with ionic liquids or molten salts as the electrolyte. For example, based on the past studies in ref. 155–157 on the liquid Na negatrode and ref. 166 and 164 on the activated carbon positrode, it can be anticipated that the first molten salt supercapattery can be made from coupling these two electrodes in a variety of inorganic molten salts with or even without using a Na+ ion conducting ceramic membrane. Introducing redox additives in the molten salts should also be considered to enhance the charge capacity of the capacitive positrode.156
It is worth mentioning that because molten salts freeze at room temperature, a fully charged molten salt EESD can be kept on a shelf for a long period until the next designated time of discharge without suffering from any self-discharge. This is because the solidified salt is an insulator to both electrons and ions. This storage advantage is unmatchable by other electrolyte based EESDs in which self-discharge is inevitable, particularly supercapacitors. A storage life over a year or longer is needed in many remote areas such as in the south and north poles and on the moon or Mars. Of course, to bring the solidified salts back to the working temperature would require pre-heating. This can be achieved by, for example, using the so-called thermite (thermate) that is a composite of metal fuel and oxidant and can, upon ignition, undergo highly exothermic but non-explosive redox reaction for fast heating in confined areas.167
Further, molten salt supercapatteries are suitable for both low and high temperature applications, but it is their high temperature uses that make them unique EESDs for unusual applications in, for example, concentrated solar power (CSP) plants in which molten salts are used for thermal energy transfer and storage. Apparently, with molten salt EESDs, the CSP plant can engage in direct electricity storage, taking advantage of the sunlight heated high temperature molten salts. Particularly, with their expected relatively low cost, capability of high energy and power density storage, and durable services, molten salt supercapatteries could be an ideal choice to help the CPS plant to achieve storage of both heat and electricity.
Like other emerging technologies, molten salt AAEM supercapatteries also have technical challenges. The most common one is the supporting materials for making the molten salt container. In the authors' experience, molten salts themselves are non-corrosive to metals, but can become aggressive in the presence of moisture and/or oxygen. Therefore, drying the salt before melting and sealing the EESD completely from air are crucial to maintaining stability and durability. On the other hand, very little is known about the transferability of various existing room temperature supercapacitor positrodes for molten salts. Last but not least, cell design and material selection are very important to accommodate the high temperature liquid salt without any internal and external leak so that it is easy and safe to position the EESD in any orientation.
4 Conclusion
In summary, supercapatteries combining a supercapacitor positrode with an AAEM negatrode of high theoretical charge capacity and the most negative potentials with optimised IL electrolytes are a promising strategy to approach the next generation of high performance EESDs. Although various studies on IL-based electrolytes for supercapacitors and AAEM-based batteries are widely reported, research related to IL-AAEM supercapatteries is still climbing a long and steep hill. There are challenges from unfavourable IL properties such as low ionic conductivity, possible leakage risk, high selectivity to material structure/composition, strong corrosivity, and unknown functionality. These drawbacks of ILs are twinned with challenges from using AAEM negatrodes such as SEI formation, electrolyte depletion, and polymorphous metal deposition. Also, the relevant mechanism of combining capacitive and Nernstian charge storage still needs further detailed study. Particularly, innovative synthesis of low cost, functional, and stable IL electrolytes to solve both the issues of IL electrolytes and AAEM negatrodes is worth putting the greatest effort in. As the saying goes, “there are always more ways than difficulties” and this is also true for supercapattery development to serve our future energy needs. Along these lines, we propose to utilise molten salts, the high temperature counterpart of ILs, in EESDs, particularly AAEM supercapatteries, in search for solutions to almost all kinetic, dynamic and mechanistic difficulties encountered in IL and organic electrolytes, in combination with careful and innovative designs in materials selection and processing and cell manufacture.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to express our appreciation for the current and past financial supports from the CSC (File No. 202108330377, Q. G.), DTP Programme of the UNNC and NIMTE, CAS (2019 -2023, Q. G.), student families (P. Y. F. and Y. H. Z.), Innovate UK (Smart Grants, 10017140), Daphne Jackson Fellowship (2020–2022, L. G.), Propulsion Futures Beacon of Excellence of the UoN (2021–2022), Municipal Bureau of Science and Technology (3315 Plan and 2014A35001-1), E.ON AG (Energy Storage Award 2007), Royal Society (Braine Mercer Feasibility Award, 2006), Leverhulme Trust (A. K. C.) and EPSRC (EP/J000582/1, GR/R68078).References
- F. Yu, C. Zhang, F. Wang, Y. Gu, P. Zhang, E. R. Waclawik, A. Du, K. Ostrikov and H. Wang, Mater. Horiz., 2020, 7, 495–503 RSC.
- W. Liu, P. Oh, X. Liu, M. J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew Chem. Int. Ed. Engl., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- J. Hu, W. Huang, L. Yang and F. Pan, Nanoscale, 2020, 12, 15036–15044 RSC.
- G. Z. Chen, Int. Mater. Rev., 2016, 62, 173–202 CrossRef.
- L. Yu and G. Z. Chen, Faraday Discuss., 2016, 190, 231–240 RSC.
- L. Yu and G. Z. Chen, Front. Chem., 2019, 7, 272 CrossRef CAS PubMed.
- H. Sakaebe and H. Matsumoto, Electrochem. Commun., 2003, 5, 594–598 CrossRef CAS.
- L. Guan, G. Z. Chen, A. K. Croft and D. M. Grant, J. Electrochem. Soc., 2022, 169, 030529 CrossRef CAS.
- Q. Guo, W. Deng, S. Xia, Z. Zhang, F. Zhao, B. Hu, S. Zhang, X. Zhou, G. Z. Chen and Z. Liu, Nano Res., 2021, 14, 3585–3597 CrossRef CAS.
- Q. Guo, Y. Yu, S. Xia, C. Shen, D. Hu, W. Deng, D. Dong, X. Zhou, G. Z. Chen and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 46043–46055 CrossRef CAS PubMed.
- Q. Guo, S. Wang, Y. Li, J. Wang, Y. Wu, Y. Yu, S. Xia, D. Hu, B. Hu, Z. Ye, X. Zhou, G. Z. Chen and Z. Liu, J. Power Sources, 2023, 580, 233401 CrossRef CAS.
- S. A. Ferdousi, L. A. O'Dell, J. Sun, Y. Hora, M. Forsyth and P. C. Howlett, ACS Appl. Mater. Interfaces, 2022, 14, 15784–15798 CrossRef CAS PubMed.
- H. Wang, X. Feng, Y. Chen, Y.-S. Liu, K. S. Han, M. Zhou, M. H. Engelhard, V. Murugesan, R. S. Assary, T. L. Liu, W. Henderson, Z. Nie, M. Gu, J. Xiao, C. Wang, K. Persson, D. Mei, J.-G. Zhang, K. T. Mueller, J. Guo, K. Zavadil, Y. Shao and J. Liu, ACS Energy Lett., 2019, 5, 200–206 CrossRef.
- H. Sun, G. Zhu, Y. Zhu, M. C. Lin, H. Chen, Y. Y. Li, W. H. Hung, B. Zhou, X. Wang, Y. Bai, M. Gu, C. L. Huang, H. C. Tai, X. Xu, M. Angell, J. J. Shyue and H. Dai, Adv. Mater., 2020, 32, e2001741 CrossRef PubMed.
- S. Azmi, M. F. Koudahi and E. Frackowiak, Energy Environ. Sci., 2022, 15, 1156–1171 RSC.
- J. Wu, Q. Liang, X. Yu, Q.-F. Lü, L. Ma, X. Qin, G. Chen and B. Li, Adv. Funct. Mater., 2021, 31, 2011102 CrossRef CAS.
- M. Durth, C. Prieto, A. Rodríguez-Sánchez, D. Patiño-Rodríguez and L. F. Cabeza, Sol. Energy, 2019, 182, 57–63 CrossRef CAS.
- R. Benages-Vilau, T. Calvet, M. A. Cuevas-Diarte and H. A. J. Oonk, Phase Transitions, 2016, 89, 1–20 CrossRef CAS.
- B. D'Aguanno, M. Karthik, A. N. Grace and A. Floris, Sci. Rep., 2018, 8, 10485 CrossRef PubMed.
- T. van Ree, Curr. Opin. Electrochem., 2020, 21, 22–30 CrossRef CAS.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- E. Adelowo, A. R. Baboukani, O. Okpowe, I. Khakpour, M. Safa, C. Chen and C. Wang, J. Power Sources, 2020, 455, 227987 CrossRef CAS.
- S. Kaipannan and S. Marappan, Sci. Rep., 2019, 9, 1104 CrossRef PubMed.
- Q. Gao, J. Energy Chem., 2019, 38, 219–224 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- K. C. Lethesh, A. Bahaa, M. Abdullah, M. O. Bamgbopa and R. A. Susantyoko, Front. Chem., 2022, 10, 859304 CrossRef CAS PubMed.
- L. Miao, Z. Song, D. Zhu, L. Li, L. Gan and M. Liu, Energy Fuels, 2021, 35, 8443–8455 CrossRef CAS.
- J. Feng, Y. Wang, Y. Xu, Y. Sun, Y. Tang and X. Yan, Energy Environ. Sci., 2021, 14, 2859–2882 RSC.
- S. Pan, M. Yao, J. Zhang, B. Li, C. Xing, X. Song, P. Su and H. Zhang, Front. Chem., 2020, 8, 261 CrossRef CAS PubMed.
- A. Guerfi, M. Dontigny, P. Charest, M. Petitclerc, M. Lagacé, A. Vijh and K. Zaghib, J. Power Sources, 2010, 195, 845–852 CrossRef CAS.
- S. Fleischmann, M. Widmaier, A. Schreiber, H. Shim, F. M. Stiemke, T. J. Schubert and V. Presser, Energy Storage Mater., 2019, 16, 391–399 CrossRef.
- P. Navalpotro, J. Palma, M. Anderson and R. Marcilla, J. Power Sources, 2016, 306, 711–717 CrossRef CAS.
- I. Plitz, A. DuPasquier, F. Badway, J. Gural, N. Pereira, A. Gmitter and G. Amatucci, Appl. Phys. A: Mater. Sci. Process., 2006, 82, 615–626 CrossRef CAS.
- H. A. Andreas, J. Electrochem. Soc., 2015, 162, A5047 CrossRef CAS.
- G. G. Amatucci, F. Badway, A. Du Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930 CrossRef CAS.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 RSC.
- E. Lim, H. Kim, C. Jo, J. Chun, K. Ku, S. Kim, H. I. Lee, I.-S. Nam, S. Yoon and K. Kang, ACS Nano, 2014, 8, 8968–8978 CrossRef CAS PubMed.
- H. Wang, Z. Xu, Z. Li, K. Cui, J. Ding, A. Kohandehghan, X. Tan, B. Zahiri, B. C. Olsen and C. M. Holt, Nano Lett., 2014, 14, 1987–1994 CrossRef CAS PubMed.
- Y.-G. Wang, J.-Y. Luo, C.-X. Wang and Y.-Y. Xia, J. Electrochem. Soc., 2006, 153, A1425 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS.
- Q. Zhong, B. Liu, B. Yang, Y. Li, J. Li and X. Yan, Chin. Chem. Lett., 2021, 32, 3496–3500 CrossRef CAS.
- B. Liu, J. Chen, B. Yang, Z. Lin, C. J. Zhang, Z. Zeng, M. Jiao, L. Liu, Y. Sun and R. Hou, Energy Storage Mater., 2021, 42, 154–163 CrossRef.
- T. C. Mendes, F. Zhou, A. J. Barlow, M. Forsyth, P. C. Howlett and D. R. MacFarlane, Sustainable Energy Fuels, 2018, 2, 763–771 RSC.
- G. A. dos Santos Junior, V. D. Fortunato, G. G. Silva, P. F. Ortega and R. L. Lavall, Electrochim. Acta, 2019, 325, 134900 CrossRef.
- G. A. dos Santos Junior, V. D. Fortunato, G. A. Bastos, G. G. Silva, P. F. Ortega and R. L. Lavall, ACS Appl. Energy Mater., 2020, 3, 9028–9039 CrossRef CAS.
- Y. S. Yun, J. H. Kim, S.-Y. Lee, E.-G. Shim and D.-W. Kim, J. Power Sources, 2011, 196, 6750–6755 CrossRef CAS.
- N. Hirota, K. Okuno, M. Majima, A. Hosoe, S. Uchida and M. Ishikawa, Electrochim. Acta, 2018, 276, 125–133 CrossRef CAS.
- H. Sun, P. Liang, G. Zhu, W. H. Hung, Y.-Y. Li, H.-C. Tai, C.-L. Huang, J. Li, Y. Meng, M. Angell, C.-A. Wang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27847–27853 CrossRef CAS PubMed.
- P. Meister, V. Küpers, M. Kolek, J. Kasnatscheew, S. Pohlmann, M. Winter and T. Placke, Batteries Supercaps, 2021, 4, 504–512 CrossRef CAS.
- S. Chen, C. Niu, H. Lee, Q. Li, L. Yu, W. Xu, J.-G. Zhang, E. J. Dufek, M. S. Whittingham, S. Meng, J. Xiao and J. Liu, Joule, 2019, 3, 1094–1105 CrossRef CAS.
- M. S. Park, S. B. Ma, D. J. Lee, D. Im, S.-G. Doo and O. Yamamoto, Sci. Rep., 2014, 4, 3815 CrossRef PubMed.
- S. W. Kim, D. H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- G. H. Newman and L. P. Klemann, J. Electrochem. Soc., 1980, 127, 2097 CrossRef CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Skaarup, J. Power Sources, 1989, 26, 341–345 CrossRef CAS.
- J. M. Tarascon and G. W. Hull, Solid State Ionics, 1986, 22, 85–96 CrossRef CAS.
- D. Stevens and J. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- K. Lu, B. Song, X. Gao, H. Dai, J. Zhang and H. Ma, J. Power Sources, 2016, 303, 347–353 CrossRef CAS.
- C. Zhao, L. Liu, X. Qi, Y. Lu, F. Wu, J. Zhao, Y. Yu, Y.-S. Hu and L. Chen, Adv. Energy Mater., 2018, 8, 1703012 CrossRef.
- H. W. Kwak, M. E. Lee, H.-J. Jin and Y. S. Yun, J. Power Sources, 2019, 418, 218–224 CrossRef CAS.
- S. Park, J. C. Hyun, J. H. Kwak, M. E. Lee, H.-J. Jin and Y. S. Yun, Appl. Surf. Sci., 2020, 513, 145848 CrossRef CAS.
- D. Iermakova, R. Dugas, M. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060 CrossRef CAS.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS PubMed.
- R. Wibowo, L. Aldous, E. I. Rogers, S. E. Ward Jones and R. G. Compton, J. Phys. Chem. C, 2010, 114, 3618–3626 CrossRef CAS.
- H. D. Yoo, I. Shterenberg, Y. Gofer, R. E. Doe, C. C. Fischer, G. Ceder and D. Aurbach, J. Electrochem. Soc., 2014, 161, A410 CrossRef CAS.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203–217 CrossRef CAS.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P.-f. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- H. Chu, Z. Zhang, Z. Song, A. Du, S. Dong, G. Li and G. Cui, Chem. Commun., 2021, 57, 9430–9433 RSC.
- W. Zuo, R. Li, C. Zhou, Y. Li, J. Xia and J. Liu, Adv. Sci., 2017, 4, 1600539 CrossRef PubMed.
- X. Deng, L. Li, G. Zhang, X. Zhao, J. Hao, C. Han and B. Li, Energy Storage Mater., 2022, 53, 467–481 CrossRef.
- P. Hundekar, S. Basu, X. Fan, L. Li, A. Yoshimura, T. Gupta, V. Sarbada, A. Lakhnot, R. Jain and S. Narayanan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5588–5594 CrossRef CAS PubMed.
- L. Qin, Y. Lei, H. Wang, J. Dong, Y. Wu, D. Zhai, F. Kang, Y. Tao and Q. H. Yang, Adv. Energy Mater., 2019, 9, 1901427 CrossRef.
- K. Yoshii, T. Masese, M. Kato, K. Kubota, H. Senoh and M. Shikano, ChemElectroChem, 2019, 6, 3901–3910 CrossRef CAS.
- H. Yamamoto, C.-Y. Chen, K. Kubota, K. Matsumoto and R. Hagiwara, J. Phys. Chem. B, 2020, 124, 6341–6347 CrossRef CAS PubMed.
- Y. Wuhai, Z. Jingwen, W. Cunguo and C. Guanglei, Energy Storage Sci. Technol., 2019, 8, 26 Search PubMed.
- H. Park, C. J. Bartel, G. Ceder and P. Zapol, Adv. Energy Mater., 2021, 11, 2101698 CrossRef CAS.
- X. Gao, X. Liu, A. Mariani, G. A. Elia, M. Lechner, C. Streb and S. Passerini, Energy Environ. Sci., 2020, 13, 2559–2569 RSC.
- N. Wu, W. Yao, X. Song, G. Zhang, B. Chen, J. Yang and Y. Tang, Adv. Energy Mater., 2019, 9, 1803865 CrossRef.
- T. Stettner, R. Dugas, A. Ponrouch and A. Balducci, J. Electrochem. Soc., 2020, 167, 100544 CrossRef CAS.
- G. A. Giffin, J. Mater. Chem. A, 2016, 4, 13378–13389 RSC.
- F. Philippi and T. Welton, Phys. Chem. Chem. Phys., 2021, 23, 6993–7021 RSC.
- D. Rauber, F. Philippi, B. Kuttich, J. Becker, T. Kraus, P. Hunt, T. Welton, R. Hempelmann and C. W. M. Kay, Phys. Chem. Chem. Phys., 2021, 23, 21042–21064 RSC.
- S. Tang, G. A. Baker and H. Zhao, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC.
- B. S. Lalia, N. Yoshimoto, M. Egashira and M. Morita, J. Power Sources, 2010, 195, 7426–7431 CrossRef CAS.
- N. Chen, Y. Guan, J. Shen, C. Guo, W. Qu, Y. Li, F. Wu and R. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 12154–12160 CrossRef CAS PubMed.
- B. Rupp, M. Schmuck, A. Balducci, M. Winter and W. Kern, Eur. Polym. J., 2008, 44, 2986–2990 CrossRef CAS.
- R. L. Lavall, S. Ferrari, C. Tomasi, M. Marzantowicz, E. Quartarone, A. Magistris, P. Mustarelli, S. Lazzaroni and M. Fagnoni, J. Power Sources, 2010, 195, 5761–5767 CrossRef CAS.
- S. Shiraishi, N. Nishina, A. Oya and R. Hagiwara, Electrochemistry, 2005, 73, 593–596 CrossRef CAS.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054–A5059 CrossRef CAS.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610–12612 RSC.
- L. D. Reed and E. Menke, J. Electrochem. Soc., 2013, 160, A915 CrossRef CAS.
- J. Muldoon, C. B. Bucur, A. G. Oliver, J. Zajicek, G. D. Allred and W. C. Boggess, Energy Environ. Sci., 2013, 6, 482–487 RSC.
- L. Zhang, S. Yang, J. Chang, D. Zhao, J. Wang, C. Yang and B. Cao, Front. Chem., 2020, 8, 413 CrossRef CAS PubMed.
- H. Yu, J. Wu, L. Fan, K. Xu, X. Zhong, Y. Lin and J. Lin, Electrochim. Acta, 2011, 56, 6881–6886 CrossRef CAS.
- H. Yu, J. Wu, L. Fan, Y. Lin, K. Xu, Z. Tang, C. Cheng, S. Tang, J. Lin, M. Huang and Z. Lan, J. Power Sources, 2012, 198, 402–407 CrossRef CAS.
- J. Jang, J. S. Shin, S. Ko, H. Park, W. J. Song, C. B. Park and J. Kang, Adv. Energy Mater., 2022, 12, 2103955 CrossRef CAS.
- M. Y. Yang, S. V. Zybin, T. Das, B. V. Merinov, W. A. Goddard, E. K. Mok, H. J. Hah, H. E. Han, Y. C. Choi and S. H. Kim, Adv. Energy Mater., 2022, 13, 2202949 CrossRef.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- N. W. Li, Y. X. Yin, J. Y. Li, C. H. Zhang and Y. G. Guo, Adv. Sci., 2017, 4, 1600400 CrossRef PubMed.
- G. N. Lewis and F. G. Keyes, J. Am. Chem. Soc., 1913, 35, 340–344 CrossRef CAS.
- J. B. Goodenough and H. Gao, Sci. China: Chem., 2019, 62, 1555–1556 CrossRef CAS.
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed.
- J. Ding, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457–6498 CrossRef CAS PubMed.
- X. Gao, H. Wu, C. Su, C. Lu, Y. Dai, S. Zhao, X. Hu, F. Zhao, W. Zhang, I. P. Parkin, C. J. Carmalt and G. He, Energy Environ. Sci., 2023, 16, 1364–1383 RSC.
- A. Ponrouch, J. Bitenc, R. Dominko, N. Lindahl, P. Johansson and M. R. Palacin, Energy Storage Mater., 2019, 20, 253–262 CrossRef.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 CrossRef CAS.
- D. Fouchard and J. B. Taylor, J. Power Sources, 1987, 21, 195–205 CrossRef CAS.
- N. S. Gingrich and L. Heaton, J. Chem. Phys., 1961, 34, 873–878 CrossRef CAS.
- S. Jiao, J. Zheng, Q. Li, X. Li, M. H. Engelhard, R. Cao, J.-G. Zhang and W. Xu, Joule, 2018, 2, 110–124 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- W. Luo, C. F. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2016, 7, 1601526 CrossRef.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- D. Aurbach, Y. Gofer, A. Schechter, O. Chusid, H. Gizbar, Y. Cohen, M. Moshkovich and R. Turgeman, J. Power Sources, 2001, 97–98, 269–273 CrossRef CAS.
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536 CrossRef CAS.
- C. Liebenow, J. Appl. Electrochem., 1997, 27, 221–225 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- T. Watkins, A. Kumar and D. A. Buttry, J. Am. Chem. Soc., 2016, 138, 641–650 CrossRef CAS PubMed.
- S. B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS PubMed.
- K. A. See, J. A. Gerbec, Y.-S. Jun, F. Wudl, G. D. Stucky and R. Seshadri, Adv. Energy Mater., 2013, 3, 1056–1061 CrossRef CAS.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 CrossRef CAS PubMed.
- A. Ponrouch, C. Frontera, F. Barde and M. R. Palacin, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed.
- S. Li, M. Jiang, Y. Xie, H. Xu, J. Jia and J. Li, Adv. Mater., 2018, 30, e1706375 CrossRef PubMed.
- L. P. Hou, X. Q. Zhang, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 15109–15113 CrossRef CAS PubMed.
- G. He, Q. Li, Y. Shen and Y. Ding, Angew. Chem., Int. Ed., 2019, 58, 18466–18470 CrossRef CAS PubMed.
- S. Xia, Q. Guo, Y. Yu, Y. Li, S. Wang, D. Dong, Z. Liu, H. Zhou, X. Zhou and Z. Liu, Carbon, 2023, 203, 743–752 CrossRef CAS.
- S. Zhang, W. Deng, X. Zhou, B. He, J. Liang, F. Zhao, Q. Guo and Z. Liu, Mater. Today Energy, 2021, 21, 100770 CrossRef CAS.
- A. Aryanfar, D. J. Brooks, A. J. Colussi, B. V. Merinov, W. A. Goddard 3rd and M. R. Hoffmann, Phys. Chem. Chem. Phys., 2015, 17, 8000–8005 RSC.
- P. Zou, Y. Sui, H. Zhan, C. Wang, H. L. Xin, H. M. Cheng, F. Kang and C. Yang, Chem. Rev., 2021, 121, 5986–6056 CrossRef CAS PubMed.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790–801 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- H. L. Skriver and N. M. Rosengaard, Phys. Rev. B, 1992, 46, 7157–7168 CrossRef CAS PubMed.
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2018, 4, 375–376 CrossRef.
- M. Jackle and A. Gross, J. Chem. Phys., 2014, 141, 174710 CrossRef PubMed.
- J. H. Han, E. Khoo, P. Bai and M. Z. Bazant, Sci. Rep., 2014, 4, 7056 CrossRef CAS PubMed.
- H. J. S. Sand, Lond. Edinb. Dublin philos. mag. j. sci., 1901, 1, 45–79 CrossRef CAS.
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- L. Schafzahl, I. Hanzu, M. Wilkening and S. A. Freunberger, ChemSusChem, 2017, 10, 401–408 CrossRef CAS PubMed.
- N. Xiao, W. D. McCulloch and Y. Wu, J. Am. Chem. Soc., 2017, 139, 9475–9478 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS.
- Q. Pang, J. Meng, S. Gupta, X. Hong, C. Y. Kwok, J. Zhao, Y. Jin, L. Xu, O. Karahan, Z. Wang, S. Toll, L. Mai, L. F. Nazar, M. Balasubramanian, B. Narayanan and D. R. Sadoway, Nature, 2022, 608, 704–711 CrossRef CAS PubMed.
- W. Brockner, K. Toerklep and H. A. Oeye, J. Chem. Eng. Data, 1981, 26, 250–253 CrossRef CAS.
- C. Wei, L. Tan, Y. Zhang, Z. Wang, B. Xi, S. Xiong, J. Feng and Y. Qian, Energy Storage Mater., 2022, 50, 473–494 CrossRef.
- H. Kim, D. A. Boysen, J. M. Newhouse, B. L. Spatocco, B. Chung, P. J. Burke, D. J. Bradwell, K. Jiang, A. A. Tomaszowska, K. Wang, W. Wei, L. A. Ortiz, S. A. Barriga, S. M. Poizeau and D. R. Sadoway, Chem. Rev., 2013, 113, 2075–2099 CrossRef CAS PubMed.
- V. Giordani, D. Tozier, H. Tan, C. M. Burke, B. M. Gallant, J. Uddin, J. R. Greer, B. D. McCloskey, G. V. Chase and D. Addison, J. Am. Chem. Soc., 2016, 138, 2656–2663 CrossRef CAS PubMed.
- C. Xia, C. Y. Kwok and L. F. Nazar, Science, 2018, 361, 777–781 CrossRef CAS PubMed.
- H. Yin, B. Chung, F. Chen, T. Ouchi, J. Zhao, N. Tanaka and D. R. Sadoway, Nat. Energy, 2018, 3, 127–131 CrossRef CAS.
- J. Prakash, L. Redey and D. R. Vissers, J. Power Sources, 1999, 84, 63–69 CrossRef CAS.
- N. Shamim, E. C. Thomsen, V. V. Viswanathan, D. M. Reed, V. L. Sprenkle and G. Li, Materials, 2021, 14, 2280 CrossRef CAS PubMed.
- Y. G. Zhu, G. Leverick, A. Accogli, K. Gordiz, Y. Zhang and Y. Shao-Horn, Energy Environ. Sci., 2022, 15, 4636–4646 RSC.
- D. J. Bradwell, H. Kim, A. H. C. Sirk and D. R. Sadoway, J. Am. Chem. Soc., 2012, 134, 1895–1897 CrossRef CAS PubMed.
- H. Kim, D. A. Boysen, T. Ouchi and D. R. Sadoway, J. Power Sources, 2013, 241, 239–248 CrossRef CAS.
- Y. Song, S. Jiao, J. Tu, J. Wang, Y. Liu, H. Jiao, X. Mao, Z. Guo and D. J. Fray, J. Mater. Chem. A, 2017, 5, 1282–1291 RSC.
- J. Tu, J. Wang, H. Zhu and S. Jiao, J. Alloys Compd., 2020, 821, 153285 CrossRef CAS.
- S. Zhang, Y. Yang, L. Cheng, J. Sun, X. Wang, P. Nan, C. Xie, H. Yu, Y. Xia, B. Ge, J. Lin, L. Zhang, C. Guan, G. Xiao, C. Peng, G. Z. Chen and J.-Q. Wang, Energy Storage Mater., 2021, 35, 142–147 CrossRef.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734–749 RSC.
- K. Wang, Z. Chen, K. Liu, C. Yang, H. Zhang, Y. Wu, Y. Long, H. Liu, Y. Jin, M. Li and H. Wu, Energy Environ. Sci., 2022, 15, 5229–5239 RSC.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- D. W. Kirk and J. W. Graydon, ECS Trans., 2013, 53, 27 CrossRef.
- W. Zhao, H. Ren, T. Yan, Y. Ou, Q. Jiao, H. Wang, D. J. Kline and M. R. Zachariah, Chem. Eng. J., 2020, 396, 124559 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |