
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New sources of genipin-rich substances for crosslinking future manufactured bio-based materials†
Liliana B.
Hurtado Colmenares
 ab,
Maryam
Nejati
c,
Yuan
Fang
d,
Boyang
Guo
e,
Amparo
Jiménez-Quero
c,
Antonio J.
Capezza
*b and
Marcos A.
Sabino
*a
ab,
Maryam
Nejati
c,
Yuan
Fang
d,
Boyang
Guo
e,
Amparo
Jiménez-Quero
c,
Antonio J.
Capezza
*b and
Marcos A.
Sabino
*a
aDepartment of Chemistry, B5IDA Research Group, Simon Bolivar University, Caracas 89000, Venezuela. E-mail: msabino@usb.ve
bDepartment of Fibre and Polymer Technology, Polymeric Materials Division, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm 10044, Sweden. E-mail: ajcv@kth.se
cDivision of Glycoscience, Department of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, AlbaNova University Centre, SE-106 91, Stockholm, Sweden
dDepartment of Materials Science and Engineering, Uppsala University, Box 35, 751 03, Uppsala, Sweden
eDepartment of Chemistry, Ångström Laboratory, Molecular Biomimetics, Microbial Chemistry, Uppsala University, Box 523, 751 20 Uppsala, Sweden
First published on 9th November 2023
Abstract
Genipap (Genipa americana L.), also known as caruto, is a fruit native to Central and South America and presents a novel source of a crosslinking substance containing genipin for biopolymers in various applications. In this study, the fruit's core was used to extract the genipin-rich genipap oil, and a complete characterization of the oil as an inexpensive replacement for commercial genipin powder is included. The extracted genipap oil shows a high phenolic content and remarkable non-hemolytic, antioxidant, and antimicrobial activity. The potential of genipap oil is further demonstrated by its advantage over commercial genipin powder, which did not show antioxidant activity. The crosslinking capacity of the genipap oil was tested with chitosan films and hot-pressed sheets of protein blends from agro-industrial biomass, including zein, wheat gluten, and potato protein. The results indicated that incorporating genipap oil in these blends allowed for manufacturing homogenous structures and improved their mechanical performance compared to the non-crosslinked blends. The use of the oil represents an advantage from a material engineering perspective as it allows for better distribution of genipin during the thermal processing of the materials compared with the commercial genipin. Further, commercial genipin requires solvents and extensive purification processes, which hinders its upscalability. These results support the use of the extracted fruit oil as a green, inexpensive, efficient crosslinking agent, opening new avenues for several applications.
Sustainability spotlightManufacturing biopolymers with competing properties to synthetic plastics typically requires crosslinkers (which are often cytotoxic). We are interested in finding a non-cytotoxic crosslinker that can also be effectively processed with the biopolymer. We envisioned a novel strategy of using extracted oil from the genipap fruit containing genipin as a crosslinker and providing an alternative to the expensive purified genipin, which currently requires many solvents for its processing. We have characterized the extracted oil and evaluated its reactivity towards different biopolymers. Using this source of genipin is beneficial in terms of processing and costs, increasing the possibility of material upscaling. Our work conforms to the UN's Sustainable Development Goals: industry, innovation, and infrastructure (SDG 9); sustainable cities and communities (SDG 11); and responsible consumption and production (SDG 12). |
Introduction
Genipap (Genipa Americana L.) is a tropical fruit native to Central and South America, also known as caruto (Venezuela) or genipapo (Brazil).1–3 Genipap belongs to the Rubiaceae family1 and has been used by indigenous people for the production of body coloring, juice, liqueurs, and jams.3 Studies have shown that the fruit of Genipa Americana contains several bioactive compounds, including flavonoids, tannins, and iridoids, such as geniposide and genipin.3–5 Genipin has received considerable attention owing to its unique chemical structure and crosslinking properties of natural polymers.6–12 In addition, genipin is considered the bioactive compound responsible for the remarkable properties of the unripe Genipap fruit, and it is a powerful colorant, antioxidant, antimicrobial, and anticancer agent.1,12,13 It is also one of the few crosslinking agents of biomacromolecules that is not cytotoxic like glutaraldehyde. Thus, it is an important reagent to produce future bio-based materials approved for human contact.14–19Genipin is an iridoid, a secondary plant metabolite that is part of the monoterpenoids and has hydroxyl groups in its structure that give it a polar character. Genipin can also form covalent bonds with amino groups, forming polysaccharide or protein crosslinking networks.1,12,15 This process is interesting because it can improve physical and mechanical properties of bio-based materials, making them more stable and resistant to degradation.9,19,20 Incorporating genipin into biomaterial provides opportunities to utilize polysaccharides and proteins as raw materials, especially those obtained from aquaculture and agricultural by-products/residues that, in the absence of crosslinkers, lack competitive mechanical properties compared to fossil-based plastics. An important example of these by-products from the aquaculture industry is fishing activities, generating 4.1–2.2 million ton of exoskeletal debris from crustaceans, such as shrimp, which are rich in chitin.21,22 On the other hand, agroindustry generates about 1.3 billion tonnes of co-products, according to the Food and Agriculture Organization of the United Nations FAO.23,24 Most of these co-products are protein-rich powders, such as zein (corn gluten meal), wheat gluten, and potato protein (protein-rich powders from starch extraction).23
Unfortunately, the high cost of pure genipin and the high CO2 impact from the extensive chemical procedures needed for its purification limits its use in large-scale production. This is because genipin is commercially obtained by enzymatic hydrolysis of geniposide in Gardenia Jasminoide fruit.25 Alternative works have reported using unripe genipap fruit to obtain fatty acids and a genipin-rich extract by combining supercritical fluid and pressure solvent extractions. The extraction costs were even reduced 8 times when integrating recovery processes, resulting in a purified genipin powder.13 Further, recent works show the need to find new sources of genipin to increase the availability of genipin powder.26 Here, genipin-rich powders are considered a bottleneck for manufacturing bio-based materials using continuous polymer processing techniques with temperatures as low as 100 °C,27,28 well below the melting temperature of genipin particles. Thus, it becomes important to look for alternatives to obtain genipin in a more functional and less expensive form, allowing for an advance in the sustainability of the current extraction conditions and promoting more agro-industrial co-products as raw materials, thus enabling a circular economy.17,23,29
In this article, the extraction of an oil-containing genipin from the fruit of the genipap is reported as an alternative to commercial genipin powder for its use in materials manufacturing. We report a full characterization of the extracted genipap oil, including chemical composition and physicochemical properties required for its use in materials in contact with humans. Further, we evaluated the potential of the genipap oil to cross-link the most common natural polymers used to manufacture bioplastics and can also be obtained as co-products from the agro-food industry, i.e., chitin, zein, wheat gluten, and potato protein. The study demonstrated the possibility of making materials with a more efficient crosslinking process when using the extracted genipap oil containing genipin, which could be used as films in various biotechnological applications or to produce low-cost, environmentally friendly porous absorbents for single-use personal hygiene products.
Experimental
Materials
Unripe fruits of Genipa Americana L. were collected between December and January from a Venezuelan National Park, Parque del Este “Generalísimo Francisco de Miranda,” located in Caracas, Venezuela. The fruits were cooled and kept at −4 °C until they were used. The extraction solvent was chloroform (CHCl3), purchased from Fisher Scientific. All chemicals and reagents used for assays of protein content, lipid content, ash content, monosaccharide composition, total phenolic content, antioxidant activity, and antimicrobial activity were purchased from Sigma-Aldrich (Sweden) unless otherwise stated.Chitosan (CS) (molecular weight 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–375000 Da, >75% deacetylated chitin) and poly(D-glucosamine) were purchased from Sigma-Aldrich. Wheat gluten concentrate (WG) was obtained from Lantmännen Reppe AB (Sweden). The protein content present in the WG was 86.3 ± 0.3% (N × 6.25). Potato protein concentrate (PP) was supplied by Lyckeby Starch AB (Sweden). In this case, the protein content in PP was 82 ± 2%. The WG and PP are agro-food industry co-products. Zein (Z) ≥98% (product number Z3625) was purchased from Sigma-Aldrich. Glycerol (ACS ≥99.5% reagent) and sodium bicarbonate (SBC, NaHCO3, ACS ≥98%) were purchased from Sigma-Aldrich. Commercial genipin ≥98% was purchased from Zhixin Biotechnology (China).
000–375000 Da, >75% deacetylated chitin) and poly(D-glucosamine) were purchased from Sigma-Aldrich. Wheat gluten concentrate (WG) was obtained from Lantmännen Reppe AB (Sweden). The protein content present in the WG was 86.3 ± 0.3% (N × 6.25). Potato protein concentrate (PP) was supplied by Lyckeby Starch AB (Sweden). In this case, the protein content in PP was 82 ± 2%. The WG and PP are agro-food industry co-products. Zein (Z) ≥98% (product number Z3625) was purchased from Sigma-Aldrich. Glycerol (ACS ≥99.5% reagent) and sodium bicarbonate (SBC, NaHCO3, ACS ≥98%) were purchased from Sigma-Aldrich. Commercial genipin ≥98% was purchased from Zhixin Biotechnology (China).
Extraction of genipap oil
Unripe genipap was used in its natural state, i.e., without prior drying, to avoid possible modification that could affect the extraction process. The unripe fruit's peel (exocarp or epicarp) was removed to extract genipap oil (GO). The core (consisting of mesocarp, endocarp, and seeds) was grated with a domestic grater. 200 mL chloroform was used as a solvent for every hundred grams of grated fruit. The maceration was carried out for 3 h with mechanical agitation. The suspension was filtered, and the solid residue was wrapped in filter paper and placed in a Soxhlet apparatus connected to a solvent flask containing the previously used CHCl3. After that, the system was heated to boiling, and the reflux continued for 6 h to completely remove the solvent with a roto-evaporator at 61–62 °C (Fig. 1). The resulting extract was an oil with a viscous appearance resembling honey and dark color. The genipap oil was stored in a desiccator under vacuum. Thin-layer chromatography of the genipap oil was performed as a preliminary characterization test compared with commercial genipin. To obtain pure genipin from the genipap oil, 4 g of the oil was diluted in ethyl acetate, and the precipitation of genipin was induced by adding cyclohexane. Solid genipin was washed with n-hexane and recrystallized from a 7:1 ethyl acetate/cyclohexane mixture.
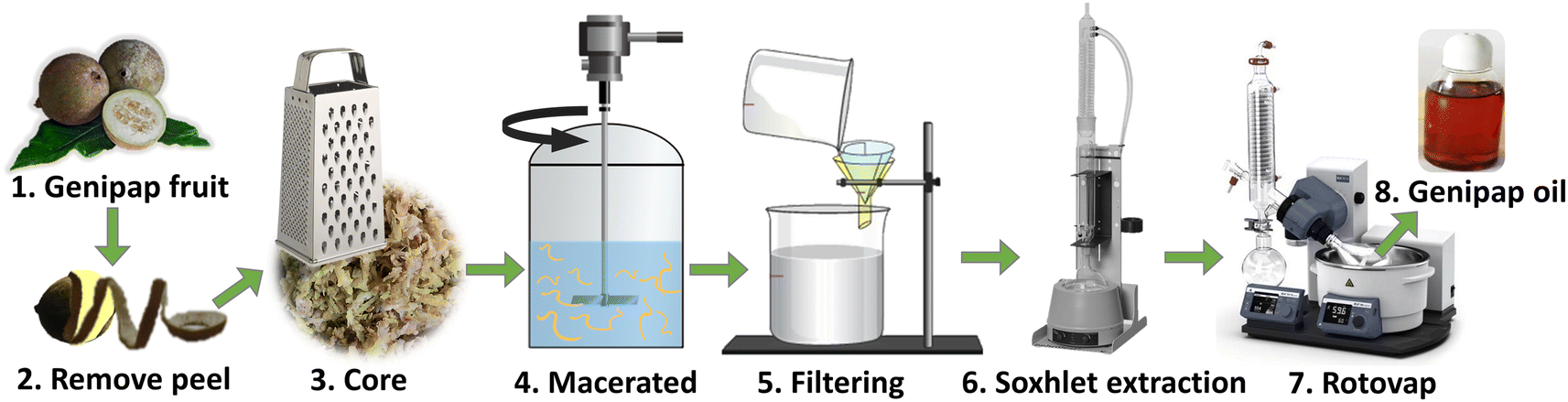 | ||
| Fig. 1 A scheme of the genipap oil extraction process. | ||
Compositional analysis of the genipap fruit and the genipap oil
The water content in the genipap oil was determined by the difference between the initial and the final weight after being lyophilized for 3 days. The result was reported as the percentage of water, calculated as 100 × (initial weight-dried weight)/initial weight.The ash content was determined by pulverizing the genipap peel, core, and genipap oil and weighed in dried crucibles in triplicate. The samples were then charred at 200 °C and transferred to an oven. After that, they were kept at 550 °C for 24 h and allowed to cool down. The produced ash was weighed, and the samples were kept in a desiccator between steps. The results were reported as the percentage of ash.30
The lipid content was analyzed according to the method by Bligh and Dyer (1959)31 using the proportion of chloroform:methanol:water = 1:2:0.8. The samples were first vortexed to solubilize the lipids. Additional chloroform and water were then added to reach a ratio of chloroform:methanol:water = 2:2:1.8 and the two phases were separated.32 The results are the average of triplicates, and the standard deviation is reported.
The protein content of the soluble extracts of the peel, core, and genipap oil was determined using the Bio-Rad protein assay kit according to the Bradford assay.33 Bovine serum albumin (BSA) in six concentrations (0.05–0.5 mg mL−1) was used as a standard.
The monosaccharide composition of the fruit parts (peel and core) and the genipap oil was determined by the hydrolysis of trifluoroacetic acid (TFA).34 1–2 mg of the sample was mixed with 1 mL TFA and kept at 120 °C for 3 h. The hydrolyzed solutions were then filtered through Chromacol 0.2 μm nylon filters (Thermo Scientific, USA). 100 μL of each sample was air-dried and reconstituted in Milli-Q water. The final solution was analyzed by high-performance anion exchange chromatography-pulse amperometric detection (HPAEC-PAD) using a Dionex CarboPac PA20 column. Elution was performed at a constant flow of 0.4 mL min−1. The eluents used were (A) water as the main eluent, (B) 200 mM NaOH (aqueous), and (C) 100 mM NaOH with 100 mM sodium acetate (aqueous). The elution started with 1.2% B at time zero, followed by 50% B at 20 min, 100% C at 30 min, 100% B at 46 min, and back to 1.2% B at 50 min. The elution was carried out at 30 °C for a total time of 60 min. The utilized monosaccharide standards used were glucose, galactose, mannose, xylose, rhamnose, arabinose, fucose, galacturonic acid, and glucuronic acid at concentrations ranging from 0.001 to 0.1 mg mL−1. All these measurements were performed in triplicate.
The phenolic content of the samples (10 mg) was measured after saponification with 2 M NaOH (500 μL). The samples were flushed with nitrogen and incubated at 60 °C overnight. Next, the samples were neutralized with 12 M HCL, and the phenolic compounds were extracted by partitioning the solution by the same volume of ethyl acetate (×4). The ethyl acetate phase was collected and air-dried at room temperature.36 The dried compounds were reconstituted in methanol. Subsequently, 100 μL of these methanolic solutions were mixed with 2% acetic acid (1:1 v/v) and analyzed by an HPLC-UV/Vis system (Agilent Technologies, Santa Clara, CA), with SB-C18 column (Zorbax, 4.6 × 250 mm, 5 μm particle size, Agilent, USA). The eluents were (A) 2% acetic acid in water and (B) absolute methanol. The flow rate was set at 1 mL min−1, and the signals were recorded at 325 nm. The elution started with 100% A and was followed by ramping up from 25% to 65% B, beginning from 11 min to 35 min. The flow was then changed to 100% A at 40 min, followed by a 5 min column wash at 55 to 60 min.36 Finally, the amount of caffeic acid, coumaric acid, ferulic acid, sinapic acid, and cinnamic acid present was calculated by comparison with the standard curves of the respective compounds.
The genipap oil extracted from the fruit of Genipa Americana L. was also characterized and compared with commercial genipin by Nuclear Magnetic Resonance (NMR) using a Bruker Avance III 500 MHz spectrometer equipped with a DIFF30 probe. The spectra were acquired with a 90° pulse, with a delay time of 10 s between scans. A total of eight scans were acquired for each measurement. CDCl3 was used as a solvent, and the spectra were obtained at room temperature.
FTIR spectra were obtained using a Thermo Fisher Nicolet iS5 FTIR-ATR Spectrophotometer with a deuterated triglycine sulfate detector. All scans were obtained using a step and resolution of 1.0 and 4.0 cm−1, respectively, with 32 consecutive scans per sample.
Structural and biological analysis of the genipap fruit and the genipap oil
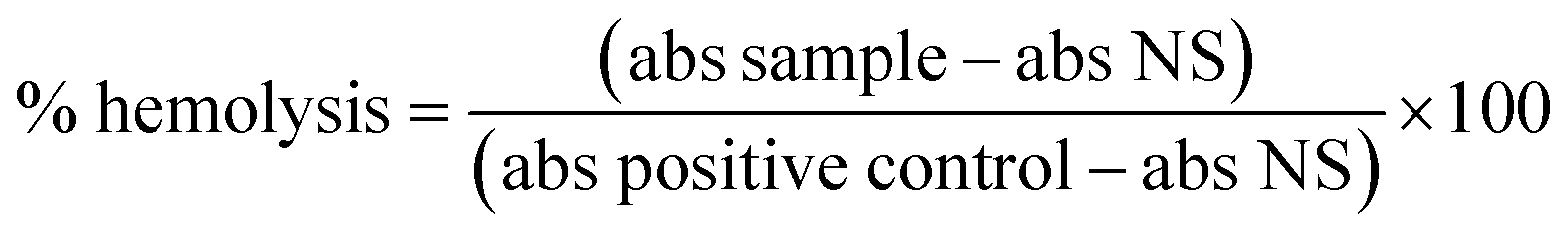
Sheep blood from Håtunalab AB was provided in compliance with the relevant ethical approval laws to protect animals (1988:534) and institutional guidelines (Approval for Laboratory Animal Facility and Business License to use Laboratory Animals) approved by The Swedish Board of Agriculture.
Evaluation of the crosslinking capacity of genipap oil
| Sample | Protein | GO x g/10 g proteina |
|---|---|---|
| a When genipap oil was used for the sample preparation, the nomenclature is as shown in the table, whereas when commercial genipin was used, the name is changed from GO to GEN. All samples contained 5 g glycerol/10 g protein and 0.5 g SBC/10 g protein. | ||
| Z | Zein | 0 |
| Z/GO0.125 | 0.125 | |
| Z/GO0.250 | 0.250 | |
| Z/GO0.500 | 0.500 | |
| WG | Wheat gluten | 0 |
| WG/GO0.125 | 0.125 | |
| WG/GO0.250 | 0.250 | |
| WG/GO0.500 | 0.500 | |
| PP | Potato protein | 0 |
| PP/GO0.125 | 0.125 | |
| PP/GO0.250 | 0.250 | |
| PP/GO0.500 | 0.500 | |
The best-performing samples from the initial assessment were used to prepare porous sheets resembling absorbent pads. The sheets were produced by blending the proteins and processed by hot-pressing. Previous works reported that the proteins used in this study could be processed by hot-pressing, and the sheets can be used to produce packaging and other biodegradable absorbent products.29,42,43 The composition of these samples is shown in Table 2. All the components of the formulations were mixed by mechanical stirring. The mixtures were placed between preheated aluminum plates with antiadhesion paper. The mold used was a (10 × 4) cm rectangular shape and a thickness of 1 mm. A molding force of 150 kN and a temperature of 110 °C were applied for both plates.
| Samplea | Zein (%) | Wheat gluten (%) | Potato protein (%) | Glycerol 5 g/10 g protein | SBC 0.5 g/10 g protein | GO 0.250 g/10 g protein |
|---|---|---|---|---|---|---|
| a The samples prepared using commercial genipin had the same mass composition as the genipap oil samples. In the name, GO was replaced by GEN. | ||||||
| 75Z/25WG | 75 | 25 | 0 | + | + | − |
| 75Z/25WGO | 75 | 25 | 0 | + | + | + |
| 60Z/25WG/15PP | 60 | 25 | 15 | + | + | − |
| 60Z/25WG/15PPGO | 60 | 25 | 15 | + | + | + |
| 25Z/25WG/50PP | 25 | 25 | 50 | + | + | − |
| 25Z/25WG/50PPGO | 25 | 25 | 50 | + | + | + |
The processing temperature was decided from the thermal stability data of the extracted genipap oil. The total processing time was 10 min. The mold was kept under pressure for the first 5 min and the last 5 min without pressure (always at 110 °C). The force was removed to allow the material to degas, expand, and increase the porosity. The pad sheets were cooled before removing them from the mold and kept in desiccators.
The morphology of these bio-based pad sheets was observed by scanning electron microscopy (SEM) using a JEOL JSM6390 (30 kV voltage). The tensile test was performed using a universal testing machine, Instron 5944, with a 500 N load cell. The crosshead speed was 10 mm min−1. All samples were pre-conditioned in 50 ± 2.5% relative humidity (RH) for 72 h before the test, according to ASTM D1623-03.
Results and discussion
Composition
The composition of the genipap peel and core and the extracted genipap oil are summarized in Table 3. The analysis showed the presence of genipin in a detectable amount only in the genipap oil, corresponding to previous reports showing that unripe genipap contains genipin after the extraction procedure.3 Additionally, the oil's carbohydrate content was lower than the peel and core, and the non-detectable soluble protein suggests that the extraction process is highly selective for genipin and phenolic compounds. This result was favored by the use of chloroform (polarity index of 4.4)44 instead of ethanol (polarity index of 5.2),44 which, being more polar, could extract a greater amount of bioactive and carbohydrates and compromise the crosslinking capacity of the genipap oil product. The core showed a higher lipid content than the peel, 14.4 ± 0.5 and 12.1 ± 0.4, respectively, which are higher than those reported in the literature.1 Generally, lipid content in genipap is low, for the whole fruit from 0.34 to 3%,1,45 peel 3.69%, mesocarp 4%, endocarp 5.2%, endocarp + seeds 5.6%, and seeds 7.6%.1 Even though the fruit layers were not analyzed separately, the tendency for the peel to have the lowest lipid content was demonstrated (see Table 3).| Sample | Water content (%) | Carbohydrate content (%) | Lipid content (%) | Ash content (%) | Soluble protein (%) | Total phenols (mg GAE per g dry sample) | Genipin (% dry mass) |
|---|---|---|---|---|---|---|---|
| a NM: not measured. The peel and core compositional analyses were performed after freeze-drying these and the genipap oil compositional analysis corresponds to the product from the reported extraction method. b ND: not detectable. The results are acquired from the water content, monosaccharide analysis by acid hydrolysis, lipid content, ash content, Bradford protein assay for soluble proteins, total phenolics measurement by Folin–Ciocalteu assay, and genipin content quantitation by HPLC. | |||||||
| Peel | NMa | 28.6 ± 1.3 | 12.1 ± 0.4 | 5.90 ± 0.30 | 0.095 ± 0.008 | 5.32 ± 0.43 | NDb |
| Core | NMa | 33.9 ± 3.2 | 14.4 ± 0.5 | 7.44 ± 0.04 | 0.867 ± 0.071 | 6.98 ± 0.81 | NDb |
| Genipap oil | 11.9 ± 0.0 | 0.86 ± 0.52 | 77.4 ± 1.8 | 0.76 ± 0.21 | NDb | 26.09 ± 2.43 | 55.4 ± 2.4 |
Of the peel and core analyzed, the lowest carbohydrate content was obtained for the peel. The large difference between the core and the peel can be because the endocarp is in the core, the place of nutrient reserve deposition for the embryo.3 This also correlates with the protein content shown by the core being 9 times higher than in the peel. A previous work reported a protein content in the whole fruit in the range of 0.21–6.6%,46 and Náthia-Neves et al.1 reported 4.4% for the peel and 10.2% for the endocarp + seeds; these values were higher than those found in this research. However, it must be taken into account that the chemical and physical characteristics of the fruit may vary depending on various factors such as maturity, time of harvest, climatic and soil conditions, and post-harvest handling.45 On the other hand, the compositional analysis obtained for genipap oil has not previously been reported, highlighting the importance of this work, especially if this product is to be used in materials as an alternative to current genipin forms (Table 3).
Quantitative analysis of compounds in the genipap oil
Fig. 2a and Table S1† show the most prevalent monosaccharides in the samples, which were glucose, xylose, arabinose, and glucuronic acid. Glucose is the main sugar in the genipap fruit, together with fructose (another isomeric form). The amount of glucose is three times higher in the core as the main nutritional part of the fruit. The xylose amount was four times higher in the peel as part of hemicellulose fractions in those protective and more recalcitrant parts of the fruit.47 The genipap oil did not contain a high amount of carbohydrates compared to the peel and core (see Table 3). However, the detected carbohydrates in the genipap oil were represented mostly by glucuronic acid and glucose (i.e., the major part of the monosaccharide composition).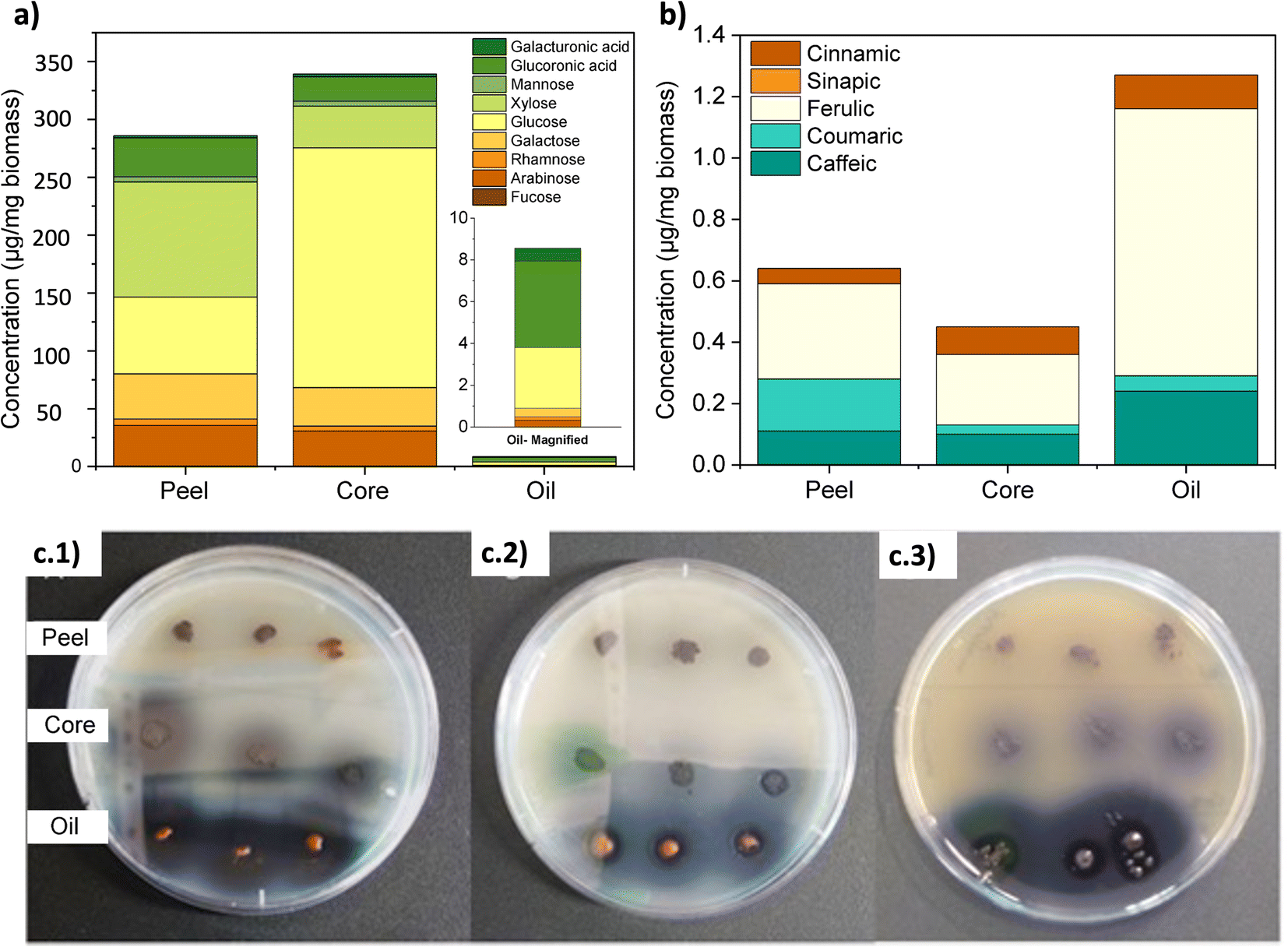 | ||
| Fig. 2 (a) The monosaccharide composition of the genipap peel, core, and genipap oil. The compositional analysis was done through TFA hydrolysis, followed by HPEAC-PAD detection of the monosaccharide. (b) Quantitative measurements of phenolic compounds through the saponification of the samples and identification through HPLC-UV-vis. (c) The antimicrobial activity of genipap fruit parts (peel and core) and genipap oil against (c.1) E. coli, (c.2) B. cereus, and (c.3) L. innocua. | ||
Fig. 2b and Table S2† show the presence of diverse phenolic acids in the samples by HPLC analysis. The higher concentration of ferulic acids in the genipap oil was due to the extraction process for genipin that was preserved other antioxidant compounds as the ones identified here, ferulic acid being the most abundant compound (Fig. 2b). The presence of these compounds can influence final products in having favorable biological activities such as antioxidant activity that can be beneficial in cases such as the production of active food packaging.48
Biological activity
The effective antimicrobial activity of the genipap oil is correlated with the presence of genipin. It has been reported that genipin possesses various biological activities, including antimicrobial properties against bacteria, fungi, and viruses. A study published by Koudouna et al. found that genipin had significant antimicrobial activity against strains of S. aureus, P. aeruginosa, and C. albicans.8 The results obtained in the genipap oil could open the possibility for future use in biomedical applications.
The absence of antioxidant activity of genipin has also been reported in the literature. Koo et al.50 reported that genipin did not show any scavenging effect on the free radicals of DDPH. However, genipin was shown to have a significant antilipoperoxidative capacity. This suggests that genipin can effectively scavenge hydroxyl radicals formed in a Fe2+/ascorbate system.50 Additionally, Hughes et al. suggest that the antioxidant activity of genipin may depend on concentration, source, and solvent. This is because their results indicated that 50 μM genipin has a free radical scavenging capacity comparable to ascorbic acid.51
Several studies have investigated the antioxidant potential of genipap fruit and found that it contains high levels of phenolic compounds, flavonoids, and carotenoids, all known to have antioxidant activity.52 Additionally, there are reports that the aqueous extract of genipap fruit has potent antioxidant activity, as demonstrated by its ability to inhibit the oxidation of low-density lipoprotein (LDL), a type of cholesterol that can contribute to the development of heart disease.53,54
Genipin has many potential biomedical applications, including as a crosslinking agent for tissue engineering, drug delivery, and natural food preservative.14 However, there have been concerns about the hemotoxicity of genipin, which refers to its potential to damage red blood cells (erythrocytes) and other blood components.10 In a study published by Gholami et al.,56 it was found that genipin induced hemolysis dose-dependently, with higher concentrations of genipin inducing hemolysis.56 It is important to note that the concentrations used in these studies were often higher than those typically used in biomedical applications.
Moreover, many studies have also shown that genipin can be used safely in certain applications, such as crosslinking tissue engineering scaffolds.57 In this case, where the hemo-compatibility of the genipap oil as a new genipin-derived component in future biomaterials is evaluated, and given the potential application of these biobased products in feminine hygiene products,17 the genipin content in the oil is low. Therefore, none of its components have adverse effects on blood cells.
Chemical characterization of the oil
Fig. 3a shows the thin layer chromatogram demonstrating the presence of genipin in the extracted genipap oil. The Rf values obtained for the oil (0.63 ± 0.01) were comparable with those of commercial genipin (0.61 ± 0.01). However, the genipap oil also showed a spot between the red dotted lines, indicating that it may contain a mixture of other iridoids found in genipap fruits.58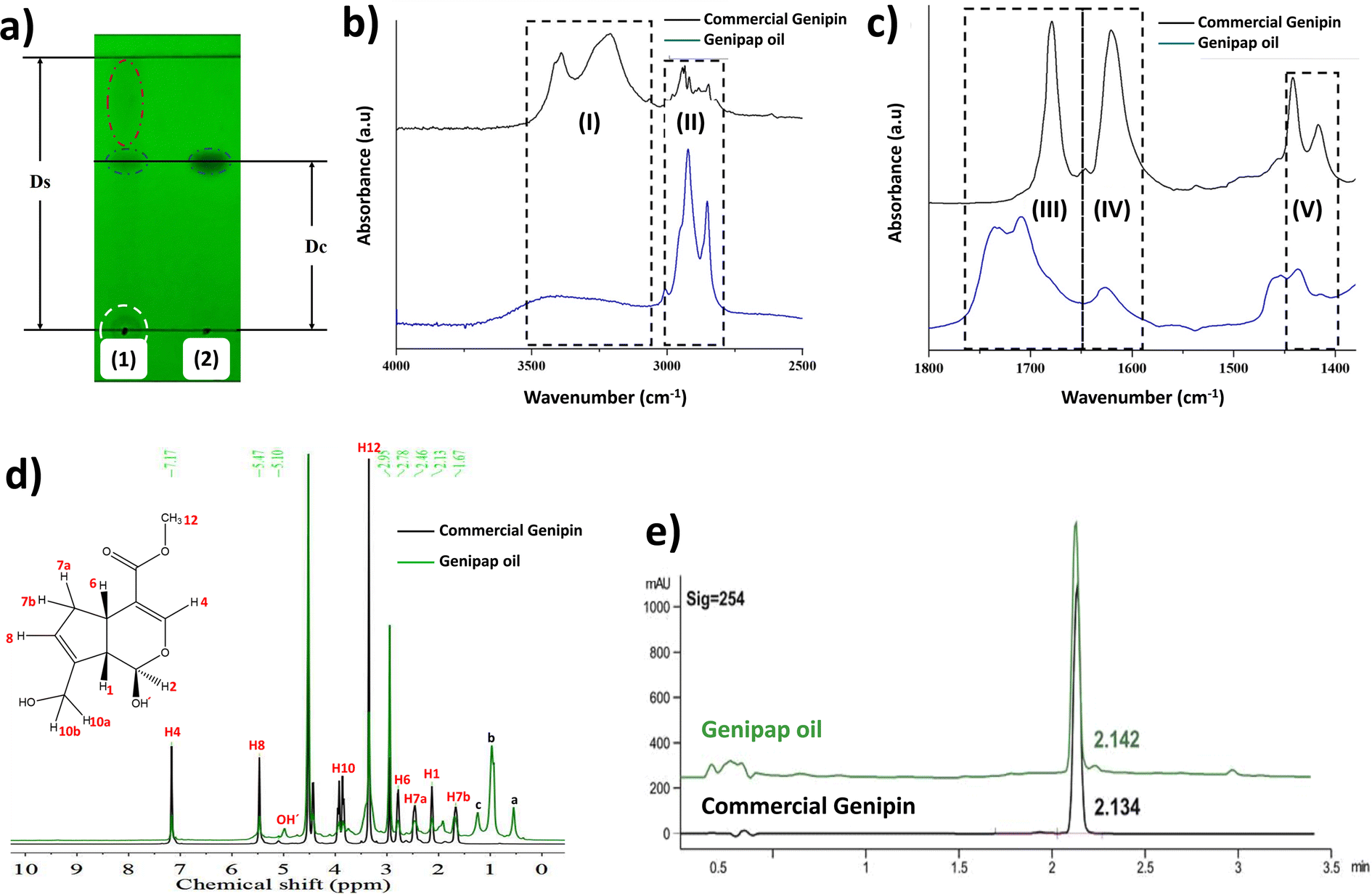 | ||
| Fig. 3 The characterization of the genipap oil. (a) The thin layer chromatogram (TLC) of the genipap oil (1) and commercial genipin (2) observed under UV light. FTIR-ATR spectrum of the oil extracted from genipap and commercial genipin (b) in the range 4000–2500 cm−1 and (c) 1800–1400 cm−1. (d) The 1H-NMR spectrum of the genipap oil compared to commercial genipin, (e) HPLC analysis of the genipap oil vs. commercial genipin. | ||
The FTIR spectrum of the genipap oil was similar to that of commercial genipin, with the main difference being the intensities in some peaks (Fig. S1, ESI†). The broad band in the range of 3600–3300 cm−1 in the genipap oil corresponds to the stretching of the –OH (Region (I), Fig. 3b). Genipin showed a double band in this region, one corresponding to the axial deformation of free OH and the other to the deformation of the OH bound to intermolecular hydrogen.59 The difference may result from the genipap oil presenting a more heterogenous composition than the commercial genipin, preventing the distinction between the two types of OH bonds. In the 3000–2800 cm−1 Region (II), signals corresponding to the stretching of the C–H bonds and asymmetric stretching of the H–C–H bonds were observed.60 Region (III) (Fig. 3c) corresponds to the stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl groups, shifting to a higher wavenumber in the genipap oil than commercial Genipin. The shifting in the carbonyl peak is possibly due to the stretching of CO groups of the acidic compounds present in the oil, such as geniposide acid,61 whose bands occur in the range of 1700–1760 cm−1,62 unlike the CO bonds of commercial genipin that appear below 1700 cm−1.60 The Region (IV) of Fig. 3c shows the same band for commercial genipin and genipap oil, with different intensities and assigned to the stretching of the CC bond.62 The higher peak intensity in commercial genipin results from its purity, allowing the distinguishing of the vibrations of each group. Lastly, in Region (IV), genipin and genipap oil share bands with different intensities, with commercial genipin being more intense than the oil. However, the observed bands are characteristic of the stretching of the CC bond of the cyclic structure.62
O carbonyl groups, shifting to a higher wavenumber in the genipap oil than commercial Genipin. The shifting in the carbonyl peak is possibly due to the stretching of CO groups of the acidic compounds present in the oil, such as geniposide acid,61 whose bands occur in the range of 1700–1760 cm−1,62 unlike the CO bonds of commercial genipin that appear below 1700 cm−1.60 The Region (IV) of Fig. 3c shows the same band for commercial genipin and genipap oil, with different intensities and assigned to the stretching of the CC bond.62 The higher peak intensity in commercial genipin results from its purity, allowing the distinguishing of the vibrations of each group. Lastly, in Region (IV), genipin and genipap oil share bands with different intensities, with commercial genipin being more intense than the oil. However, the observed bands are characteristic of the stretching of the CC bond of the cyclic structure.62
The 1H NMR spectrum of the genipap oil and commercial genipin is shown in Fig. 3d. The signals in the lower field were those observed at 7.17 and 5.47 ppm, corresponding to protons attached to sp2 carbons (H4 and H8). In addition, a broad band centered at 5.10 ppm, characteristic of the least shielded hydroxyl group (OH′), was also observed. The signals corresponding to the protons attached to sp3 carbons of methoxy were observed as a very intense singlet at 3.42 ppm (see Fig. 3d). At a higher field, the H6 proton was observed at 2.78 ppm, H7a at 2.46 ppm, H1 at 2.13 ppm, and the H7b proton at 1.67 ppm. Additionally, it can be observed that the genipap oil has three signals between 0.5 and 1.5 ppm that are not present in commercial genipin, resulting from the other molecules present in the genipap oil. These additional signals in the genipap oil can be attributed to compounds or fractions of the aliphatic type of lipid components present in the genipap oil.
Fig. 3e shows that the HPLC peak of the genipap oil corresponds well with that of commercial genipin (2.142 vs. 2.134 min elution time, respectively), which further confirms the presence of genipin in the genipap oil.
Similar studies reporting the characterization of genipap oil have not been found in the literature. The literature on the genipap fruit mostly focuses on extracting its active components and purifying genipin (not in the oil as an intermediate genipin-derived substance). Thus, the signals between 0.5 and 1.5 ppm in genipap oil may correspond to CH3 and CH2 from saturated acyl groups, which could be attributed to oleic and linoleic acid.63 Ávila et al. carried out the fatty acid profile of the G. Americana seed oil and reported that linoleic acid was the main acidic component in the biomass, with a percentage of 61.5%.64
The results in this study indicate that the extracted and dried genipap oil contains 55.4 ± 2.4 wt% genipin. Moreover, 1H-NMR analysis allowed us to accurately estimate the amount of genipin contained in the genipap oil (Fig. 3d). The concentration (mg mL−1) ratio of the commercial genipin and the genipap oil peak corresponding to genipin is 100:41.8793. Thus, the amount of genipin in the genipap oil is 14%. Pure genipin was also extracted from the genipap oil, using the extraction method described in Fig. 1, followed by solvent extraction, yielding 3.8 mg genipin per g of the genipap core. Various methods have been developed to extract and purify genipin using different organic solvents, reporting yields as high as 4.4 mg per g of fruit.37 Another work by Ramos de la Peña et al. reports the enzymatic hydrolysis of geniposide with β-glycosidase with a yield of 7.9 mg g−1 of fruit.2 The greater genipin content obtained in the genipap oil compared to previous results1,2 can be ascribed to the herein combination of maceration, followed by Soxhlet extraction (Fig. 1). The interaction of the solvent, at its boiling temperature, with the solids from the peeled fruit could increase the solubility of the extracted compounds. In addition, using chloroform as an extraction solvent facilitates a genipap product with high genipin content. Here, we tested ethanol, while other works report the use of water, which resulted in a genipap oil without genipin13 or solid extracts.1 Here, despite the use of chloroform, the extraction process remains sustainable because the chloroform used was recycled at least 5 times, showing no differences in the genipap oil.
Oil thermal stability
Thermogravimetric analysis assessed the genipap oil thermal stability for suggesting processing windows for its utilization as an additive in processed bioplastics. Fig. 4 shows the TGA profile of the extracted genipap oil and its first derivative. The first mass loss was at a temperature of 120 °C with a percentage loss of 4–5% and the second at T = 204 °C with almost 20%. According to Table 3, the water content of the genipap oil is 11–12%, which correlates with the first mass loss at ca. 120 °C. The mass loss is also associated with the different compounds in the genipap oil (Table 3 and Fig. 4). The weight loss over 200 °C can be related to the decomposition of genipin, which agrees with previous literature reporting the thermal degradation temperature at ≈200 °C with at least 40%.6 The monosaccharide and phenolic compounds reported here can also be thermally degraded at these temperatures. Therefore, it is suggested to use genipap oil at temperatures below 120 °C to guarantee maximum thermal stability.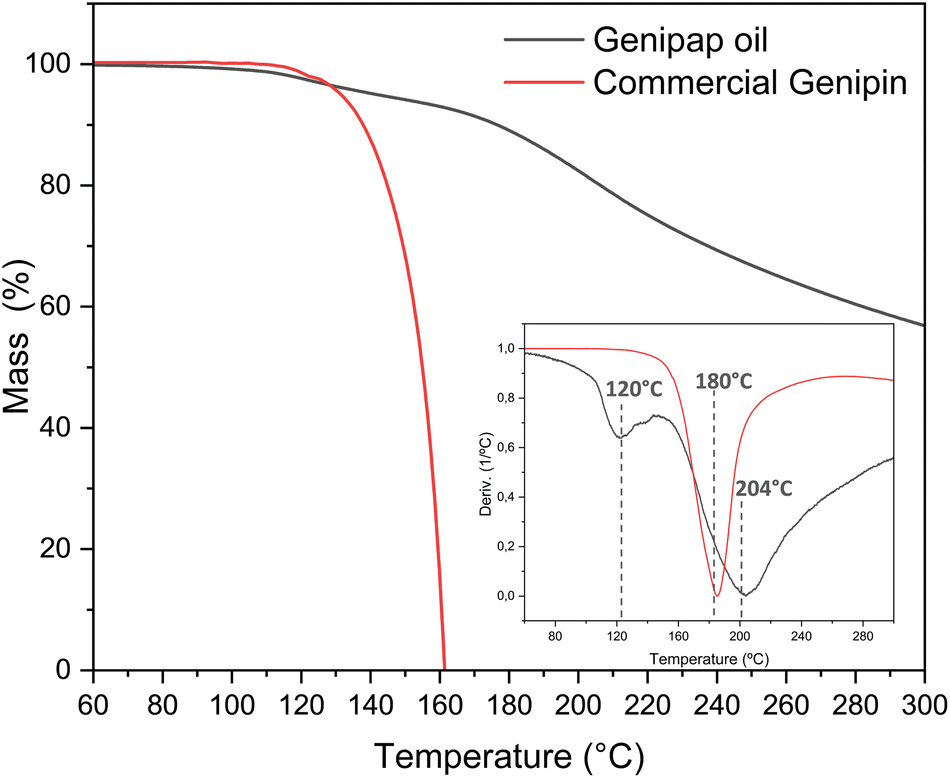 | ||
| Fig. 4 The TGA and first derivate of the genipap oil and the commercial genipin. | ||
Despite the low concentration of genipin in genipap oil, having genipin in a suspension form represents an advantage from a processing perspective. Fig. S2† shows the DSC of commercial genipin with a melting peak at 121 °C. These high temperatures can be problematic during the thermal manufacturing of biomaterials because temperatures below 120 °C are often used to avoid excessive endogenous crosslinking reactions or thermal degradation of the natural polymer.27,28,65 As the next step, it was assessed if the genipap oil (with 14% content of genipin in the as-extracted stage, Fig. 3d) has a crosslinking capacity with the most reactive and used natural polymers for producing genipin crosslinked bio-based products, i.e., chitosan and proteins.
Efficiency of genipap oil as a crosslinking reagent
The crosslinking capacity of the oil was preliminarily studied using chitosan due to its well-known high reactivity with genipin as a common crosslinking agent. These preliminary results are shown in Fig. 5a, showing the appearance of the chitosan films and the protein mixtures with/without genipap oil. The chitosan films without genipap oil (CS) were semi-transparent, brittle, and did not retain the shape of the mold. However, the films with genipap oil rapidly changed color to dark blue, and the increase in the genipap oil content from 0.05 to 0.10 wt% made the films harder and more mechanically stable (see Fig. 5a). Du et al.16 fabricated similar chitosan films with improved permselectivity using genipin as an alternative “green” crosslinking agent to replace glutaraldehyde.16 In general, crosslinked chitosan with genipin has been extensively used as biomaterial to produce films, hydrogels, and scaffolds for tissue engineering.7,16,57,66 The reason for the extensive use of chitosan with genipin is the presence of primary amino groups in the chitosan structure, making it more susceptible to forming chemical crosslinks with genipin.7,16 Further, chitosan is readily obtained from the deacetylation of chitin, a linear polysaccharide with poor solubility but highly available as a by-product of the marine industry.16,66 | ||
| Fig. 5 The appearance of (a) chitosan films obtained by solvent casting and solvent evaporation, (b) protein foams prepared at 110 °C for 30 min in the oven. The physical appearance of the structures obtained with genipap oil at T = 110 °C over the curing time: (c) color change observed from the top of the reaction beakers and (d) reaction beakers containing the mixtures. | ||
No significant difference in the color intensity was observed with increasing genipap oil concentration in the chitosan films higher than 0.500 g genipap oil/10 g chitosan. Still, the higher concentration of genipap oil does not allow the formation of homogeneous films (see Fig. S3, ESI†). The result suggests that the sample CS0.500 contained sufficient genipin to reach a maximum crosslinking degree, possibly due to the depletion of free amino groups available in the chitosan. Additionally, it is also possible that genipin cannot bind to all free amino groups even at high concentrations due to its complex crosslinking mechanism.19 With these preliminary results on chitosan, we demonstrate the effectiveness of the crosslinking action of genipap oil, allowing us to move on to assessing the capacity of the genipap oil to crosslink porous protein blend systems. Future work should focus on the effect of genipap oil on the intrinsic properties of chitosan films.
Fig. 5b shows that the selected proteins also changed color from yellow (without genipap oil) to dark blue (with genipap oil). These color changes have been reported both for chitosan films and protein sponges/foams crosslinked with commercial genipin.9,16,17 Genipin crosslinks with the free primary amino groups of chitosan, amino acids, peptides, or proteins to produce a dark blue color.18,67 This color change is related to a possible polymerization induced by oxygen radicals of genipin molecules already linked to amino groups.3 The exact mechanism of blue pigment formation is unclear, but it is believed that the formation of genipin copolymers with high CC conjugation is responsible for the characteristic dark blue color.3,11,15,68
Fig. 5c shows that the increase from 0.125 to 0.250 g genipap oil/10 g in the protein resulted in a more intense dark blue color (samples observed from the top of the reaction beaker). However, the protein mixtures' color changes when increasing the genipap oil concentration to 0.500 g/10 g does not seem significant, possibly indicating that the maximum crosslinking has been reached. Moreover, all proteins rapidly changed color, showing a darkening at 10 min in the oven (see Fig. 5c). It is worth noticing that the PP samples with genipap oil remained powdery for all the study conditions, even though the characteristic color change of genipin.9,18,19 PP is the protein with the highest content of lysine residues (amine groups) among the proteins tested with at least 40% of a highly soluble protein fraction (patatin).23,69 Thus, crosslinking with genipin should form stable 3D structures.69 However, the PP used is obtained as a by-product from starch extraction, and the industrial recovery process denatures the protein to a large extent, making it insoluble and non-food grade. Thus, this material can be readily obtained at an industrial scale and at a price as low as 1.4–1.5 € per kg, making it attractive as an alternative source of biomaterials.69
Fig. 5d shows the mixtures in their reaction beaker, revealing that the initially powdered WG sample with 0.500 genipap oil (WG/GO0.500 and 30 min in the oven) compacted, forming a rigid spherical structure. The result can be related to the formation of a highly crosslinked gluten network by the genipap oil, which could favor the polymer chains to undergo a shrinking process. WG is an abundant co-product of the starch industry,17,70 with a reported production of at least 1.1 million tons in 2020.71 WG is one of the most studied plant proteins and the most suitable candidate for producing stable 3D biopolymer networks to replace plastic in adsorbent applications.20,70
On the other hand, Z and PP adopted the mold's shape and could be easily removed from the mold (see Fig. 5d). Interestingly, the Z changed color at 10 min with the lowest genipap oil content, despite the Z reporting no detectable lysine residues72 (Fig. 5d). All the protein systems herein reported changed color to dark blue when crosslinked with the genipap oil up to 2 times faster than when crosslinked with commercial genipin, despite the genipin content being 86% lower in the genipap oil (Fig. S4†). The result suggests that the reactivity of the genipap oil favors the crosslinking reaction even for low lysine-containing proteins. Here, Z is an interesting protein candidate found in corn,29,73 and its non-toxicity and processing versatility make it a strong candidate to replace synthetic plastics.23,74 Thus, the results suggest that the most appropriate genipap oil concentration for producing films and foams is 0.500 g/10 g for chitosan and 0.250 g/10 g for protein. It should be noted that according to this study, the actual genipin concentration of the genipap oil is ca. 14%, which reveals the high reactivity of this oil extract even at such a low crosslinking concentration.
The protein-based materials were blended and thermo-formed into thin 0.4 mm sheets to demonstrate the possibilities of producing bio-based materials with common plastic processing techniques (i.e., hot-pressing). The samples were produced as porous structures by adding SBC and blending the proteins to compare with our previous works, where lyophilized porous crosslinked structures were made with commercial genipin powder as an alternative to absorbent plastics in single-use sanitary materials.17,75 The different crosslinking processing methods used to evaluate the crosslinking capacity of the genipap oil are summarized in Table S3.†
Fig. 6 shows the appearance of the samples obtained by hot pressing the selected protein mixtures. All blends formed a stable structure that could be bent up to 45° (see Fig. 6a and b). A brown-black coloration was observed in the formulations containing genipin or genipap oil, in agreement with the results above when the individual proteins were used (see Fig. 5). The microstructure of the sheets revealed homogenous porous structures with no visual evidence of phase separation, suggesting good compatibility between the proteins, glycerol, SBC, and the crosslinking agent used (Fig. 6c–k).
 | ||
| Fig. 6 Samples obtained by hot press bent up to 45°: (a) control (60Z/25WG/15PP) and (b) genipap oil (60Z/25WG/15PPGO). The hot-pressed samples and their respective SEM of surface and cross-section: (c) 75Z/25WG, (d) 75Z/25WGGEN, (e) 75Z/25WGGO, (f) 60Z/25WG/15PP, (g) 60Z/25WG/15PPGEN, (h) 60Z/25WG/15PPGO, (i) 25Z/25WG/50PP, (j) 25Z/25WG/50PPGEN, and (k) 25Z/25WG/50PPGO. | ||
The sheet fabricated from blending 25:25:50 Z:WG:PP (75Z/25WG/50PP) had the most rough and discontinuous surface among the samples (see Fig. 6i). The result is ascribed to the high amount of PP in the sample, which formed powdery materials when processed only with PP, as shown in Fig. 5b. Incorporating genipap oil in the above blend (75Z/25WG/50PPGO) decreased the roughness and allowed the structure to be smoother and more continuous, despite the high PP content. The SEM cross-section of the sheets revealed a porous and homogeneous structure with large voids of ca. 200 μm in diameter. Fig. 6c–k shows that a more porous network is favored with a low amount of WG and PP, while the increase in PP hindered the formation of pores. A slight decrease in pore size was also observed by adding genipin or genipap oil to the blends (compare Fig. 6c–e with Fig. 6f–h). The reduction or absence of pores can be related to increased crosslinking due to a more elastic network collapsing the pores once the pressure by the foaming agent is reduced.15 Nonetheless, no major changes in the microstructure were observed between the control and the samples having the crosslinking added.
Fig. 7a shows the maximum stress (σmax), strain at break (εmax), and Young's modulus of the different protein blends processed by hot pressing (see the samples in Fig. 6). The results show that incorporating the genipap oil on the samples doubled the σmax and εmax compared to the non-crosslinked sheets, especially those containing potato protein (i.e., 60Z/25WG/15PPGO and 25Z/25WG/50PPGO). Further, the mechanical properties of these samples were enhanced compared to the sample containing commercial genipin. Fig. 7a demonstrated that the samples with commercial genipin increased in σmax compared to those without the crosslinking agent, but the εmax decreased considerably. Garavand et al.43 indicated that the bond produced by the crosslinking reaction results in a denser and more rigid protein network and less mobility between protein chains (i.e., higher tensile strength and lower elongation at break). The results are consistent with the increase in σmax and εmax reported when genipin is used but differ when genipap oil is used as a crosslinking agent. The observed effect in the mechanical properties suggests that the additional components of the genipap oil could act as a plasticizer of the formulation and/or allow the formation of longer crosslink bridges and should be explored in future studies.
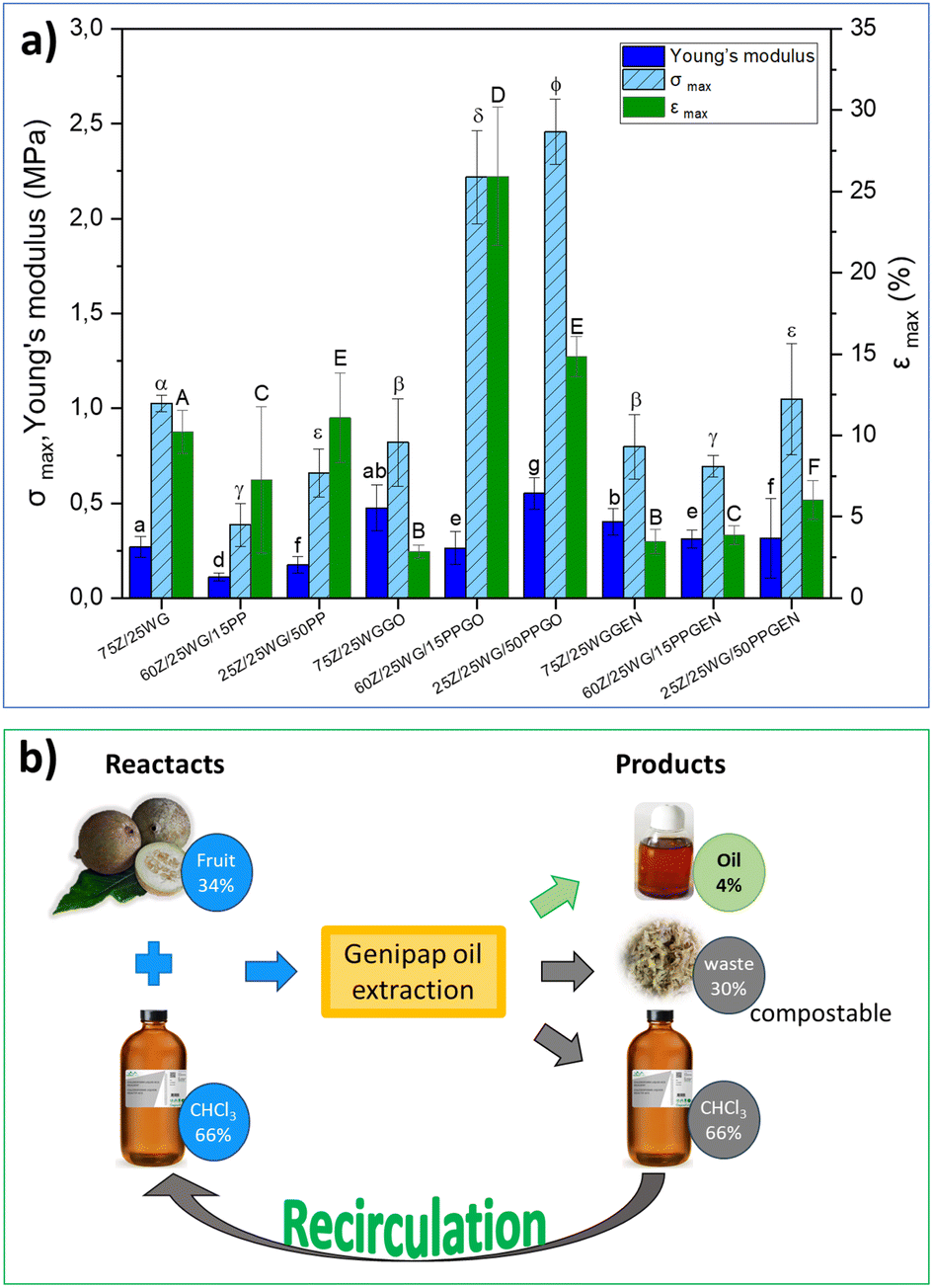 | ||
| Fig. 7 (a) Maximum stress (σmax), strain at break (εmax), and Young's modulus obtained in tensile tests of the different mix of agro-food proteins and (b) the mass balance of the reactants and product/sub-products. Different letters indicate significant differences (p < 0.05). | ||
All in all, it has been demonstrated that genipap oil allows for the formation of stable chitosan films and well-defined porous structures when combined with protein blends and polymer processing techniques. The physical and mechanical properties of the fabricated materials are similar to that of commercial genipin despite the use of ca. 10 times less genipin when adding the genipap oil, which shows the potential of the genipap oil as a genipin-containing suspension for the fabrication of future bio-based materials.
A life-cycle costing (LCC) based on the cost of raw material, solvent, and energy consumed was performed to assess the preliminary potential of the genipap oil as a future genipin-containing substance for bio-based materials crosslinking. The initial mass for the genipap oil extraction requires 34% of genipap fruits and 66% chloroform, from which 4% of genipap oil is extracted (see Fig. 7b). Therefore, our estimations show that the genipap oil will cost $9.5 per g, which can be reduced by 66% if the chloroform is recirculated for new extractions. The extraction process of the genipap oil resulted in ca. 30% biomass waste, which could be composted to increase the circularity of the overall process.
The purification of genipap oil to genipin yielded approximately 0.125 g genipin per g genipap oil and required ethyl acetate, cyclohexane, and n-hexane (Fig. S5†). Such purification steps can further increase the production cost and the amount of waste generated. Furthermore, such solvents used for the genipin purification from the oil fall into the hazardous/problematic reagents category and are not recirculated (see Table S4†). The results here show that genipap oil, without any purification, can be used directly as a potential candidate for crosslinking chitosan and agro-industrial proteins. This means fewer solvents are needed, and the material's properties using genipap oil can be kept compared to pure genipin while reducing 93% of the material production costs. However, even considering the purification steps, the price of genipin extracted from the genipap oil is $19 per 125 mg, approximately 36 times lower than commercial genipin ($140 per 25 mg).
Conclusions
Genipap oil from the Genipa American fruit was extracted using a circular approach to produce a new source of genipin as a crosslinker of natural polymers. It was shown that the oil could crosslink chitosan and proteins obtained as co-products from the agroindustry using traditional polymer processing techniques. The genipap oil containing genipin impacted the physical and mechanical properties of the bio-based materials produced, resulting in improved mechanical properties compared to the control samples with no crosslinking agent. The immediate effect of using the oil as a crosslinking substance was shown by the properties of the materials resembling those crosslinked with commercial genipin while using 10 times less genipin content when the oil is used. In addition to crosslinking, the oil also resulted in remarkable antioxidant capacity with no cytotoxicity, which is of high relevance for applications in the healthcare area. Also, the oil has an advantage over solid genipin pure particles because the traditional low temperatures used to process biopolymers do not allow the melting of genipin crystals, thus decreasing the efficiency of the crosslinking process. From a sustainability perspective, the advantages of using the oil-containing genipin for fabricating the bio-based materials relied on a cost reduction of up to 93% while saving solvents. Thus, this opens up bioplastics manufacturing with improved mechanical properties using genipin, which was a bottleneck before for upscaling such bioplastics recipes.Author contributions
Liliana Hurtado: conceptualization, formal analysis, investigation, methodology, visualization, and writing – original draft. Maryam Nejati: investigation, data curation, writing – original draft. Yuan Fang: validation, and writing – review & editing. Boyang Guo: validation, and writing – review & editing Amparo Jiménez: validation, supervision, writing – review & editing. Antonio J. Capezza: conceptualization, methodology, supervision, writing – review & editing and funding acquisition. Marcos Sabino: conceptualization, methodology, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the BoRydins Foundation (Grant F30/19) for the financial support to the project and the Universitets och högskolerådet (Linnaeus-Palme Grant 3.3.1.34.15281-2021) for the funding of academic activities between Venezuela (USB) and Sweden (KTH). The work of M. Nejati and A. Jiménez-Quero in this scientific publication was supported by funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 101037796. Also, MA. Sabino would like to thank the USB Academic ViceRectorate for his sabbatical leave that allowed him to establish this collaboration with KTH and FAPESP (SP, Brazil) (2021/13949-5) for the financial support for his sabbatical stay at the CTI - Renato Archer.References
- G. Náthia-Neves, A. G. Tarone, M. M. Tosi, M. R. Marostica Junior and M. A. A. Meireles, Food Res. Int., 2017, 102, 595–604 CrossRef.
- A. M. Ramos-de-la-Pena, C. M. G. C. Renard, L. Wicker, J. C. Montanez, L. A. Garcia-Cerda and J. C. Contreras-Esquivel, Ultrason. Sonochem., 2014, 21, 43–49 CrossRef CAS PubMed.
- S. Bentes Ade, H. A. de Souza, J. Amaya-Farfan, A. S. Lopes and L. J. de Faria, J. Food Sci. Technol., 2015, 52, 3919–3924 Search PubMed.
- A. D. Bentes and A. Z. Mercadante, J. Agric. Food Chem., 2014, 62, 10800–10808 CrossRef CAS PubMed.
- L. M. P. Silva, J. S. F. Alves, E. M. D. Siqueira, M. A. D. Neto, L. S. Abreu, J. F. Tavares, D. L. Porto, L. D. Ferreira, D. P. Demarque, N. P. Lopes, C. F. S. Aragao and S. M. Zucolotto, Molecules, 2018, 23, 2521 CrossRef PubMed.
- R. Meena, K. Prasad and A. K. Siddhanta, J. Appl. Polym. Sci., 2007, 104, 290–296 CrossRef CAS.
- N. Kildeeva, A. Chalykh, M. Belokon, T. Petrova, V. Matveev, E. Svidchenko, N. Surin and N. Sazhnev, Polymers, 2020, 12, 1086 CrossRef CAS.
- E. Koudouna, M. Huertas-Bello, C. N. Rodriguez, S. C. Henao, M. L. Navarrete and M. Y. Avila, Transl. Vis. Sci. Technol., 2021, 10, 31 CrossRef PubMed.
- V. J. Alessio Bucciarelli, Y. Yang, G. Fredi, A. Pegoretti, A. Motta and D. Maniglio, Cell Rep. Phys. Sci., 2021, 2, 100605 CrossRef.
- W. Liu, X. Yang, N. Li, G. H. Xi, M. S. Wang, B. Liang, Y. K. Feng, H. Chen, C. C. Shi and W. Z. Li, Polym. Adv. Technol., 2018, 29, 2632–2642 CrossRef CAS.
- S. Ilkar Erdagi, F. Asabuwa Ngwabebhoh and U. Yildiz, Int. J. Biol. Macromol., 2020, 149, 651–663 CrossRef CAS.
- A. S. Belle, C. R. Hackenhaar, L. S. Spolidoro, E. Rodrigues, M. P. Klein and P. F. Hertz, Food Chem., 2018, 246, 266–274 CrossRef CAS PubMed.
- G. V. Náthia-Neves, R. Renata, T. Hatami and M. A. Meireles, J. Supercrit. Fluids, 2020, 164, 104897 CrossRef.
- D. J. Lim, Int. J. Mol. Sci., 2022, 23, 5444 CrossRef CAS PubMed.
- A. C. Alavarse, E. C. G. Frachini, R. da Silva, V. H. Lima, A. Shavandi and D. F. S. Petri, Int. J. Biol. Macromol., 2022, 202, 558–596 CrossRef CAS PubMed.
- J. R. H. Du, L. H. Hsu, E. S. Xiao, X. Guo, Y. F. Zhang and X. S. Feng, Sep. Purif. Technol., 2020, 244, 116843 CrossRef CAS.
- A. J. Capezza, Q. Wu, W. R. Newson, R. T. Olsson, E. Espuche, E. Johansson and M. S. Hedenqvist, Acs Omega, 2019, 4, 18257–18267 CrossRef CAS PubMed.
- M. I. Neves, E. K. Silva, M. A. A. Meireles, I. Elena and A. Cifuentes, Food Res. Int., 2022, 157, 111240 CrossRef CAS.
- A. Bigi, G. Cojazzi, S. Panzavolta, N. Roveri and K. Rubini, Biomaterials, 2002, 23, 4827–4832 CrossRef CAS PubMed.
- A. J. Capezza, Y. X. Cui, K. Numata, M. Lundman, W. R. Newson, R. T. Olsson, E. Johansson and M. S. Hedenqvist, Adv. Sustainable Syst., 2020, 4, 2000110 CrossRef CAS.
- J. S. A. Reboucas, F. P. S. Oliveira, A. C. D. Araujo, H. L. Gouveia, J. M. Latorres, V. G. Martins, C. P. Hernandez and M. B. Tesser, Crit. Rev. Biotechnol., 2023, 43, 50–66 CrossRef CAS PubMed.
- N. Suryawanshi, S. E. Jujjavarapu and S. Ayothiraman, Int. J. Environ. Sci. Technol., 2019, 16, 3877–3898 CrossRef CAS.
- E. Alvarez-Castillo, M. Felix, C. Bengoechea and A. Guerrero, Foods, 2021, 10, 981 CrossRef CAS PubMed.
- Y. F. Tsang, V. Kumar, P. Samadar, Y. Yang, J. Lee, Y. S. Ok, H. Song, K. H. Kim, E. E. Kwon and Y. J. Jeon, Environ. Int., 2019, 127, 625–644 CrossRef CAS PubMed.
- V. G. Tacias-Pascacio, E. Garcia-Parra, G. Vela-Gutierrez, J. J. Virgen-Ortiz, A. Berenguer-Murcia, A. R. Alcantara and R. Fernandez-Lafuente, Catalysts, 2019, 9, 1035 CrossRef CAS.
- R. Rosado-Ramos, G. M. Poças and D. Marques, et al. , Nat. Commun., 2023, 14, 1918 CrossRef CAS PubMed.
- A. J. Capezza, E. Robert, M. Lundman, W. R. Newson, E. Johansson, M. S. Hedenqvist and R. T. Olsson, Polymers, 2020, 12, 459 CrossRef CAS PubMed.
- M. A. Bettelli, A. J. Capezza, F. Nilsson, E. Johansson, R. T. Olsson and M. S. Hedenqvist, Biomacromolecules, 2022, 23, 5116–5126 CrossRef CAS PubMed.
- W. Shi and M. J. Dumont, J. Mater. Sci., 2014, 49, 1915–1930 CrossRef CAS.
- K. Helrich and Association of Official Analytical Chemists, Official Methods of Analysis of the Association of Official Analytical Chemists, Association of Official Analytical Chemists, Arlington, VA, 15th edn, 1990 Search PubMed.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS.
- K. Helrich, Official methods of analysis of the Association of Official Analytical Chemists, Association of Official Analytical Chemists, USA, 1990 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- D. Zhang, R. C. Rudjito, S. Pietiainen, S. C. Chang, A. Idstrom, L. Evenas, F. Vilaplana and A. Jimenez-Quero, Food Chem., 2023, 413, 135660 CrossRef CAS PubMed.
- V. L. Singleton, R. Orthofer and R. M. Lamuela-Raventos, Methods Enzymol., 1999, 299, 152–178 CAS.
- S. Yilmaz-Turan, P. Lopez-Sanchez, A. Jimenez-Quero, T. S. Plivelic and F. Vilaplana, Food Hydrocolloids, 2022, 128, 107575 CrossRef CAS.
- G. Nathia-Neves, G. C. Nogueira, R. Vardanega and M. A. D. Meireles, Food Sci. Technol., 2018, 38, 116–122 CrossRef.
- E. Rincon, E. Espinosa, M. T. Garcia-Dominguez, A. M. Balu, F. Vilaplana, L. Serrano and A. Jimenez-Quero, Carbohydr. Polym., 2021, 272, 118477 CrossRef CAS PubMed.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT–Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- S. Yilmaz-Turan, A. Jimenez-Quero, C. Menzel, D. M. de Carvalho, M. E. Lindstrom, O. Sevastyanova, R. Moriana and F. Vilaplana, Carbohydr. Polym., 2020, 250, 116916 CrossRef CAS PubMed.
- ISO, Biological Evaluation of Medical Devices — Part 5: Tests for In Vitro Cytotoxicity, 2009, 10993-5:2009(E), p. 7 Search PubMed.
- F. Muneer, E. Johansson, M. S. Hedenqvist, T. S. Plivelic and R. Kuktaite, Int. J. Mol. Sci., 2019, 20, 58 CrossRef PubMed.
- F. Garavand, M. Rouhi, S. H. Razavi, I. Cacciotti and R. Mohammadi, Int. J. Biol. Macromol., 2017, 104, 687–707 CrossRef CAS PubMed.
- P. Mukherjee, in Quality Control and Evaluation of Herbal Drugs, Evaluating Natural Products and Traditional Medicine, 2019, pp. 195–236 Search PubMed.
- V. R. de Souza, P. A. P. Pereira, F. Queiroz, S. V. Borges and J. D. S. Carneiro, Food Chem., 2012, 134, 381–386 CrossRef CAS.
- G. M. R. Carmo, A. Brito, C. Soraya and S. da Silva, Food Biosci., 2023, 53, 102514 CrossRef.
- D. Liu, W. Tang, J. Y. Yin, S. P. Nie and M. Y. Xie, Food Hydrocolloids, 2021, 116, 106641 CrossRef CAS.
- M. Soto-Hernandez, P.-T. Mariana and M. Garcías-Mateos, Phenolic Compounds: Biological Activity, 2017 Search PubMed.
- F. B. dos Santos, M. I. L. Ramos and L. Miyagusku, Ciência Rural., 2017, 47, 8 Search PubMed.
- H. J. Koo, Y. S. Song, H. J. Kim, Y. H. Lee, S. M. Hong, S. J. Kim, B. C. Kim, C. Jin, C. J. Lim and E. H. Park, Eur. J. Pharmacol., 2004, 495, 201–208 CrossRef CAS PubMed.
- R. H. Hughes, V. A. Silva, I. Ahmed, D. I. Shreiber and B. Morrison, Brain Res., 2014, 1543, 308–314 CrossRef CAS.
- J. B. H. Ribeiro, E. Macedo, S. Gualberto, A. Silva, C. Souza, M. Zanuto and M. Silva, J. Culin. Sci. Technol., 2021, 21, 215–237 CrossRef.
- E. F. L. C. Bailao, I. A. Devilla, E. C. da Conceicao and L. L. Borges, Int. J. Mol. Sci., 2015, 16, 23760–23783 CrossRef CAS PubMed.
- C. M. B. Omena, I. B. Valentim, G. D. Guedes, L. A. Rabelo, C. M. Mano, E. J. H. Bechara, A. C. H. F. Sawaya, M. T. S. Trevisan, J. G. da Costa, R. C. S. Ferreira, A. E. G. Sant'Ana and M. O. F. Goulart, Food Res. Int., 2012, 49, 334–344 CrossRef CAS.
- M. F. Elahi, G. P. Guan, L. Wang and M. W. King, J. Appl. Polym. Sci., 2014, 131, 2517–2526 CrossRef.
- M. Gholami, M. Tajabadi, A. Khavandi and N. Azarpira, Front. Bioeng. Biotechnol., 2023, 10, 1075166 CrossRef PubMed.
- M. A. G. Sabino and R. A. Garcia, Int. J. Adv. Med. Biotechnol., 2020, 3, 2–15 Search PubMed.
- M. Ono, N. Ishimatsu, C. Masuoka, H. Yoshimitsu, R. Tsuchihashi, M. Okawa, J. Kinjo, T. Ikeda and T. Nohara, Chem. Pharm. Bull., 2007, 55, 632–634 CrossRef CAS PubMed.
- G. Magalhães, Master, Universidade Federal Do Ceará, 2007 Search PubMed.
- N.-A. Rangel-Vázquez, N.-A. Rangel-Vazquez, I. Xplore and P. River, Structural Analysis Using Computational Chemistry, River Publishers, Denmark, Netherlands, 1st edn, 2016 Search PubMed.
- M. Ono, M. Ueno, C. Masuoka, T. Ikeda and T. Nohara, Chem. Pharm. Bull., 2005, 53, 1342–1344 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley and Sons, Danvers, MA, 7th edn, 2005 Search PubMed.
- M. H. A. Rania, I. M. Almoselhy, M. H. El-Kalyoubi and A. A. El-Sharkawy, Ann. Agric. Sci., 2014, 59, 201–206 CrossRef.
- O.V. Ávila, I. Fernández, H. N. Rocha, A. A. Filho, R. Carvalho and P. Ribeiro, J. Agric. Sci., 2018, 10, 244 Search PubMed.
- M. Jimenez-Rosado, J. E. Maigret, D. Lourdin, A. Guerrero and A. Romero, J. Appl. Polym. Sci., 2022, 139, 51630 CrossRef CAS.
- P. Guerrero, A. Muxika, I. Zarandona and K. de la Caba, Carbohydrate Polym., 2019, 206, 820–826 CrossRef CAS PubMed.
- M. I. L. Neves, A. Valdes, E. K. Silva, M. A. A. Meireles, E. Ibanez and A. Cifuentes, Food Res. Int., 2022, 157, 111240 CrossRef CAS.
- Z. Wang, H. Liu, W. Wenbin, T. Cai, Z. Li, Y. Liu, W. Gao, Q. Wan, X. Wang, J. Wang and X. Yang, J. Tissue Eng., 2020, 11 DOI:10.1177/2041731420974861.
- W. R. Newson, F. Rasheed, R. Kuktaite, M. S. Hedenqvist, M. Gallstedt, T. S. Plivelic and E. Johansson, RSC Adv., 2015, 5, 32217–32226 RSC.
- B. Lagrain, B. Goderis, K. Brijs and J. A. Delcour, Biomacromolecules, 2010, 11, 533–541 CrossRef CAS PubMed.
- IndexBox, World – Wheat Gluten – Market Analysis, Forecast, Size, Trends and Insights Update, COVID-19 Impact, 2022 Search PubMed.
- A. J. Capezza, Novel Superabsorbent Materials Obtained from Plant Proteins, Sveriges Lantbruksuniversitet, Alnarp, 2017, pp. 1–54 Search PubMed.
- Y. F. Gao, H. M. Zheng, J. J. Wang, J. Y. Wu, X. Y. Li and G. Liu, Polym. Bull., 2022, 79, 4647–4665 CrossRef CAS.
- A. C. Jaski, F. Schmitz, R. P. Horta, L. Cadorin, B. J. G. da Silva, J. Andreaus, M. C. D. Paes, I. C. Riegel-Vidotti and L. M. Zimmermann, Ind. Crop. Prod., 2022, 186, 115250 CrossRef CAS.
- Q. W. C. E. Federico, R. T. Olsson and A. J. Capezza, Polym. Test., 2022, 116, 107753 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00303e |
| This journal is © The Royal Society of Chemistry 2024 |