
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,5-Hydrogen atom transfer of α-iminyl radical cations: a new platform for relay annulation for pyridine derivatives and axially chiral heterobiaryls†
Yu-Qiang
Zhou
,
Kui-Cheng
He
,
Wei-Hao
Zheng
,
Jing-Fang
Lv
,
Shi-Mei
He
,
Ning
Yu
,
Yun-Bo
Yang
,
Lv-Yan
Liu
,
Kun
Jiang
* and
Ye
Wei
 *
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: weiye712@swu.edu.cn; kjiang@swu.edu.cn
First published on 23rd April 2024
Abstract
The exploitation of new reactive species and novel transformation modes for their synthetic applications have significantly promoted the development of synthetic organic methodology, drug discovery, and advanced functional materials. α-Iminyl radical cations, a class of distonic ions, exhibit great synthetic potential for the synthesis of valuable molecules. For their generation, radical conjugate addition to α,β-unsaturated iminium ions represents a concise yet highly challenging route, because the in situ generated species are short-lived and highly reactive and they have a high tendency to cause radical elimination (β-scission) to regenerate the more stable iminium ions. Herein, we report a new transformation mode of the α-iminyl radical cation, that is to say, 1,5-hydrogen atom transfer (1,5-HAT). Such a strategy can generate a species bearing multiple reactive sites, which serves as a platform to realize (asymmetric) relay annulations. The present iron/secondary amine synergistic catalysis causes a modular assembly of a broad spectrum of new structurally fused pyridines including axially chiral heterobiaryls, and exhibits good functional group tolerance. A series of mechanistic experiments support the α-iminyl radical cation-induced 1,5-HAT, and the formation of several radical species in the relay annulations. Various synthetic transformations of the reaction products demonstrate the usefulness of this relay annulation protocol for the synthesis of significant molecules.
Introduction
Reactive species, such as carbocations, carbanions, and radicals play a considerable role in the synthesis of small molecules and macromolecules. Therefore, much effort has been devoted to the exploration of new types of species and their synthetic applications to create valuable compounds. In this context, distonic ions,1,2 a class of species with separated charge and radical sites, have been extensively investigated in the past a few years.3,4 For example, the classic McLafferty rearrangement involves the fragmentation of a carbonyl-containing distonic radical cation, which is applied in the structural elucidation using mass spectroscopy.5–7 Moreover, the distonic ions have been recognized as important active intermediates for several organic reactions, which can be exemplified by the well-established Hofmann–Löffler–Freytag reaction which related to the formation of a nitrogen-containing distonic radical cation,8,9 and the cyclization reactions involving alkene radical cations.10–13 Despite these great achievements, the methods for the generation of the distonic ions, the type of these species, and the related synthetic transformations remain limited. Therefore, it is highly desirable to explore new approaches for structurally accessing new distonic ions, and then to apply them into organic transformations to produce valuable compounds.The α-iminyl radical cations represent an important class of distonic ions, and their synthetic uses have attracted much attention from the synthetic community in the past few years. Two approaches have been explored to access these species. One is one-electron oxidation of a transient enamine via singly occupied molecular orbital (SOMO) activation, coined by MacMillan.14 The resultant radical cation can participate in several enantioselective α-functionalization reactions.15–22 The other is the conjugate addition of nucleophilic radicals to α,β-unsaturated iminium ions formed by condensation of α,β-unsaturated carbonyls with amines (Scheme 1a).23,24 Compared to the SOMO activation of the enamines, the radical conjugate addition mode is highly challenging, and related examples remain scarce.25–30 One obstacle to these reactions would be the formation of short-lived and highly reactive α-iminyl radical cations that have a high tendency to create radical elimination (β-scission) to regenerate the more stable iminium ions.25,31 To address this problem, an “electron transfer strategy” has been utilized to convert the α-iminyl radical cation into a relatively stable enamine by acquiring an electron from the electron-rich species (Scheme 1b). In this context, Melchiorre and co-workers recently made a breakthrough, and designed a novel chiral primary amine bearing a carbazole redox-active moiety. The in situ generated α-iminyl radical cation could be reduced by the intramolecular carbazole via single electron transfer (SET).25,26 In addition, the α-iminyl radical cation may gain an electron from the reduced state of the photocatalyst (PC), which were demonstrated by the groups of Melchiorre and Yu in the photocatalytic synthesis of chiral aldehydes.27–30 In addition to the “electron transfer strategy”, it was be necessary to exploit more new transformation modes to convert the α-iminyl radical cations into other types of reactive species which could induce new transformations, such as cascade reactions, to synthesize value-added molecules. As such, the α-iminyl radical cation based synthetic methods will find extensive applications in organic, drug, and material synthesis.
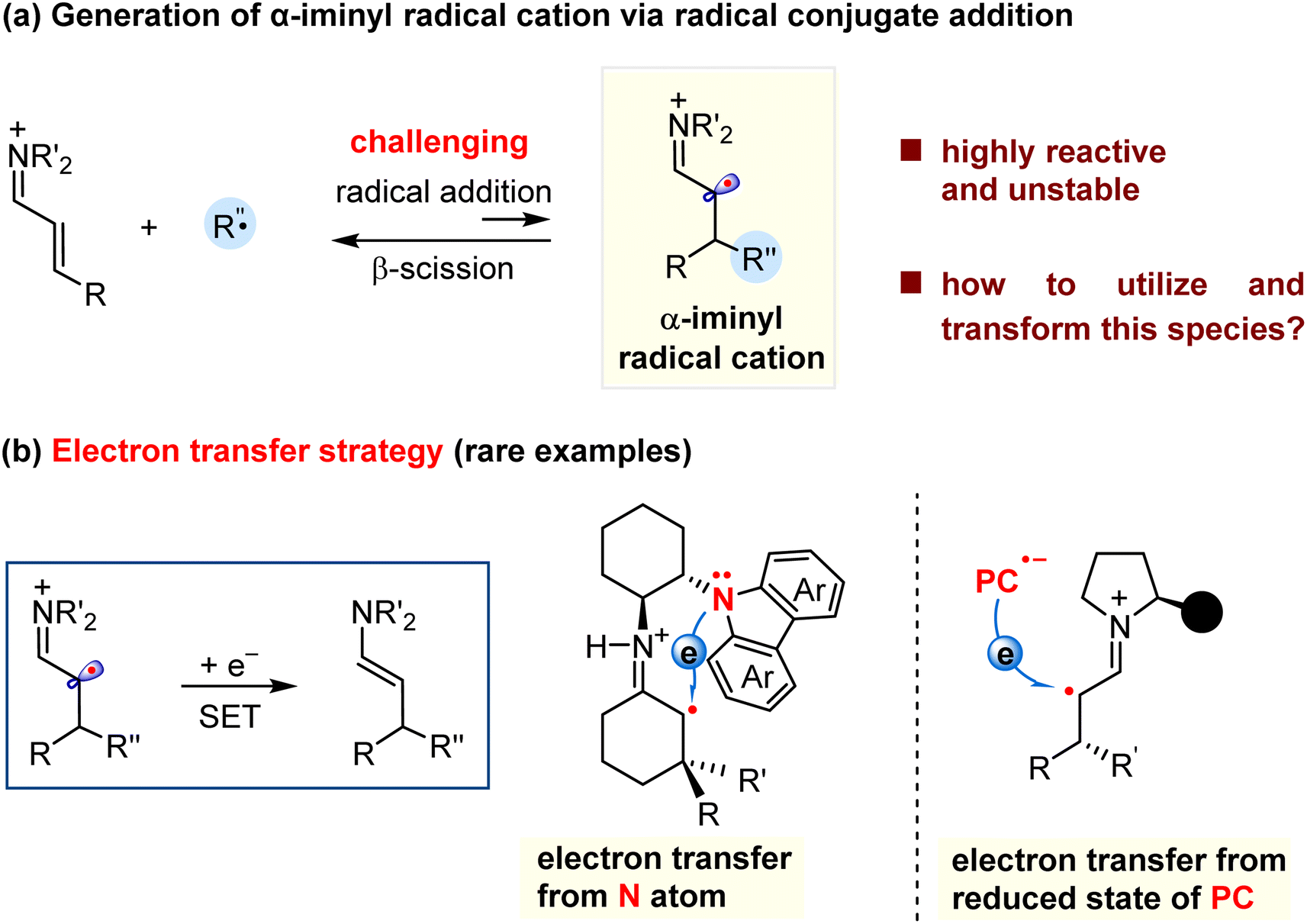 | ||
| Scheme 1 Generation of the α-iminyl radical cation via radical conjugate addition and related examples. | ||
To exploit new transformations based on the α-iminyl radical cations, we turned our attention to the radical-induced 1,5-hydrogen atom transfer (1,5-HAT) strategy.32–41 We recognized that the α-iminyl radical cation-triggered 1,5-HAT would be feasible, because such a process could generate a relatively stable radical cation species, which could be a driving force for the reaction. It is worth noting that the newly formed species is remarkably interesting, because of its multiple reactive sites, including a carbon radical, an electrophilic imine carbon atom, and a nucleophilic enamine carbon atom, as well as a functional group (FG) derived from the radical precursor. As such, the α-iminyl radical cation-triggered 1,5-HAT would provide a new platform for the realization of cascade reactions (Scheme 2a). On the other hand, the iminyl radical has exhibited great usefulness in the synthesis of numerous compounds.42–45 A remarkably significant type of transformation of the iminyl radical would be hydrogen atom transfer reactions,38,46 including 1,5-HAT and formal 1,3-HAT. Generally, 1,5-HAT dominates formal 1,3-HAT when α-H and γ-H are concurrent in the substrate, possibly because the former proceeds via a kinetically favored six-membered cyclic transition state. Nevertheless, we questioned whether the formal 1,3-HAT may occur prior to the 1,5-HAT. In our previous studies, we found that 1,3-HAT could selectively occur when a position for 1,5-HAT exists simultaneously, which was partially supported by the DFT calculations.47 This work inspired us to combine the selective 1,3-HAT of the iminyl radical with the attractive transformation of the α-iminyl radical cation, which may involve an intriguing process consisting of an α-carbon radical conjugate addition to an α,β-unsaturated iminium ion, and an α-iminyl radical cation-triggered 1,5-HAT, as shown in Scheme 2b. To the best of our knowledge, no examples regarding such a radical relay protocol have so far been reported. The success of this proposition requires the capability to tackle several challenges, such as: (1) the control of the order of HAT, the chemo-, regio-, and stereo-selectivities, (2) the inhibition of the β-C and β-H scissions, and (3) the realization of the compatibility of the amines and transition metals used to generate the iminyl radical. Herein, we describe a novel relay annulation strategy that showcases an unprecedented α-iminyl radical cation-induced 1,5-HAT (Scheme 2c). This iron/secondary amine synergistic catalysis constructs three new bonds and two rings in one step, which delivers a broad spectrum of fused pyridines, and exhibits good functional group tolerance. More importantly, this relay annulation approach can be upgraded to an asymmetric version, affording a series of axially chiral pyridine-based biaryls. These results further emphasize the great synthetic potential of the α-iminyl radical cation-induced HAT in the synthesis of enantiopure molecules.
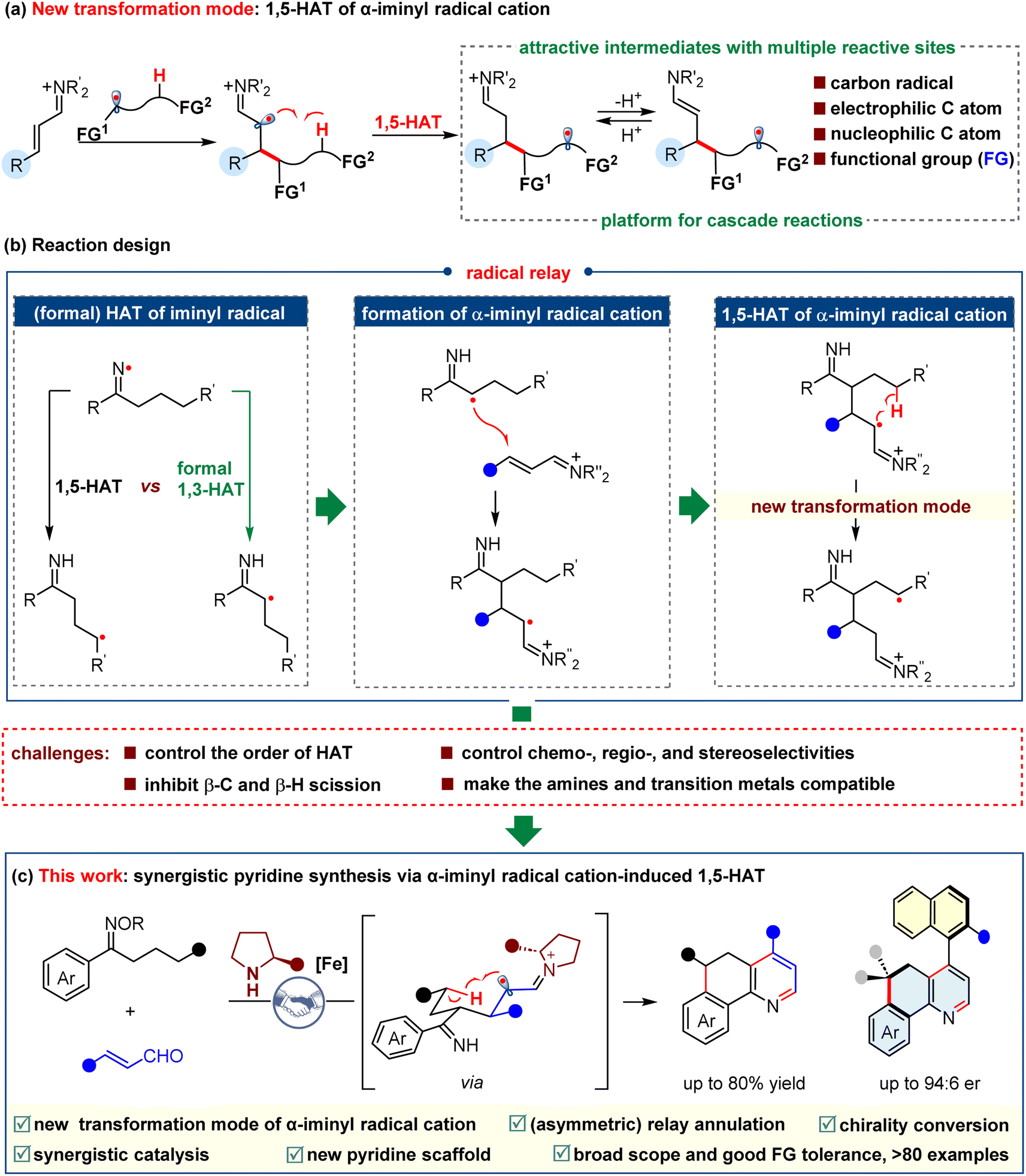 | ||
| Scheme 2 Design plan based on α-iminyl radical cation-induced 1,5-HAT. | ||
Results and discussion
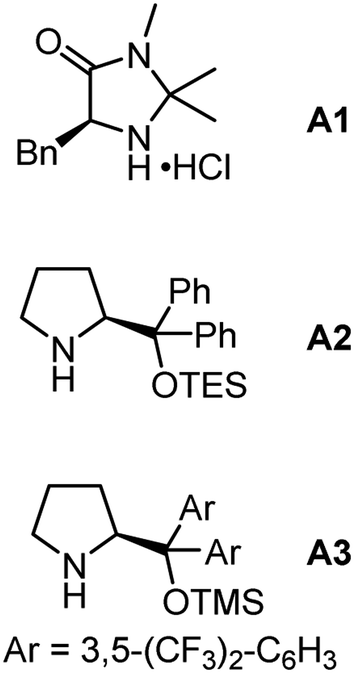
To probe the feasibility of our relay strategy, we chose the reaction of oxime ester 1 derived from 4-methyl-1-phenylpentan-1-one with cinnamaldehyde 2 for the reaction condition optimization (Table 1; please see Table S1 in the ESI† for detailed results). With Fe(acac)2 as a catalyst, several secondary amines could catalyze the reactions to produce a fused pyridine 3 plus a non-fused pyridine 3′ (entries 1–4). The formation of the former might involve the α-iminyl radical cation-triggered 1,5-HAT, and the latter would be generated by a [3 + 3]-type reaction.48 Among these amines, pyrrolidine·HClO4 showed the best reactivity, affording 3 with a 59% yield (entry 1). The structure of 3 was unambiguously confirmed by single crystal X-ray diffraction.49 In addition, the MacMillan catalyst A1 was unreactive in the transformation (entry 5), and the Jørgensen–Hayashi catalysts A2 and A3 showed lower reactivities (entries 6 and 7). Decreasing the reaction temperature from 100 to 60 °C resulted in a lower yield (entry 8). No product was detected in the absence of Fe(acac)2 or a secondary amine, demonstrating the need for both components (entries 9 and 10). Gratifyingly, the reaction delivered the target product with a 70% isolated yield when increasing the reaction temperature to 120 °C (entry 11). It should be noted that, in many reactions, 4-methyl-1-phenylpentan-1-one oxime, and 4,4-dimethyl-3,4-dihydronaphthalen-1(2H)-one50 as well as other unidentified byproducts were observed, which would result from the use of an excess amount of 1.|
|
||||
|---|---|---|---|---|
| Entry | Secondary amine | Yieldb (%) | ||
| 3 | 3′ | |||
| a Reaction conditions: 1 (0.3 mmol), 2 (0.1 mmol), Fe(acac)2 (10 mol%), amine (20 mol%), 1,4-dioxane (1 mL), 100 °C, 12 h, under N2. b Determined by GC using n-tridecane as an internal standard. c Reaction was run at 60 °C. d In the absence of Fe(acac)2. e Reaction was run at 120 °C. f Isolated yield. Abbreviations: Bz = benzoyl, TES = triethylsilyl, TMS = trimethylsilyl. | ||||
| 1 | Pyrrolidine·HClO4 | 59 | 13 |
|
| 2 | Piperidine·HClO4 | 51 | 15 | |
| 3 | Morpholine·HClO4 | 55 | 12 | |
| 4 | iPr2NH·HClO4 | 34 | 7 | |
| 5 | A1 | 0 | 0 | |
| 6 | A2 | 18 | 18 | |
| 7 | A3 | 17 | 7 | |
| 8c | Pyrrolidine·HClO4 | 51 | 10 | |
| 9c | None | 0 | 0 | |
| 10c,d | Pyrrolidine·HClO4 | 0 | 0 | |
| 11e | Pyrrolidine·HClO4 | 72 (70)f | 12 | |
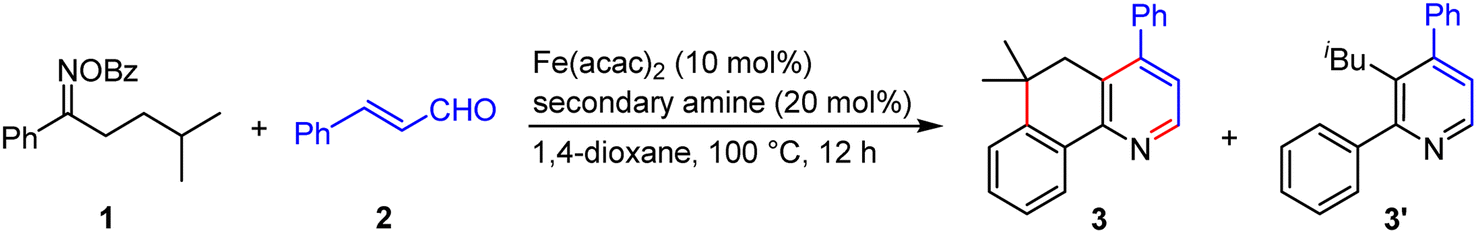
With the viable reaction conditions in hand, we then demonstrated the generality of this relay annulation approach by evaluating a variety of oxime esters. As shown in Scheme 3, diverse functionalities can easily be installed into the aromatic rings, including methyl (4 and 12), methoxy (5 and 16), methylthio (6), chloro (7 and 13), iodo (8), trifluoromethyl (9), cyano (10), ester (11), cyclopropylmethoxy (15), and difluoromethoxy (15) groups. For the oxime esters with a meta-substitution on the aryl ring, the Csp3–Csp2 formation occurred at the less hindered site (12 and 13). The steric hindrance significantly influenced the reaction because the presence of an ortho methyl group resulted in none of the desired products being formed. Interestingly, various heterocycle-containing oxime esters, including benzodioxole (17), thiophene (18), furan (19), and pyridine (20), reacted with cinnamaldehyde to give rise to the target products in synthetically useful or moderate yields. It should be noted that the pyridine-derived oxime ester possesses two potential sites, C2 and C4 of the pyridine ring, for the Csp3–Csp2 formation, whereas only the C2 functionalized product was detected. This result was consistent with the usual regioselectivity observed in the classic Minisci reaction, which would partly support the reaction mechanism hypothesis regarding the α-iminyl radical cation-induced 1,5-HAT process. We further probed the functionalities of the aliphatic chain of the oxime esters, besides having the dimethyl groups at the γ position, it can be a cyclic moiety. As such, the fused pyridine with a quaternary spiro carbon center was readily prepared (21). The substituents can also be ethyl/ethyl (22) and n-butyl/methyl (23) groups, and the reactions produced the corresponding products with moderate yields. The reactions of the oxime esters bearing a mono-substituent at the γ position with cinnamaldehyde occurred sluggishly, which afforded the desired products with low yields and other byproducts, including the parent ketones of the oxime esters (24–27). These results suggested that the γ-carbon radical stability would significantly affect the transformation. More interestingly, two oxime esters derived from pharmaceuticals, probenecid and adapalene, can also participate in the relay annulation to generate the target products with 79% and 40% yields, respectively (28 and 29). In some cases, competitive intramolecular C–C formation side products (benzocyclohexanone derivatives) were observed, which accounts for the moderate yields of the fused pyridines in some reactions.
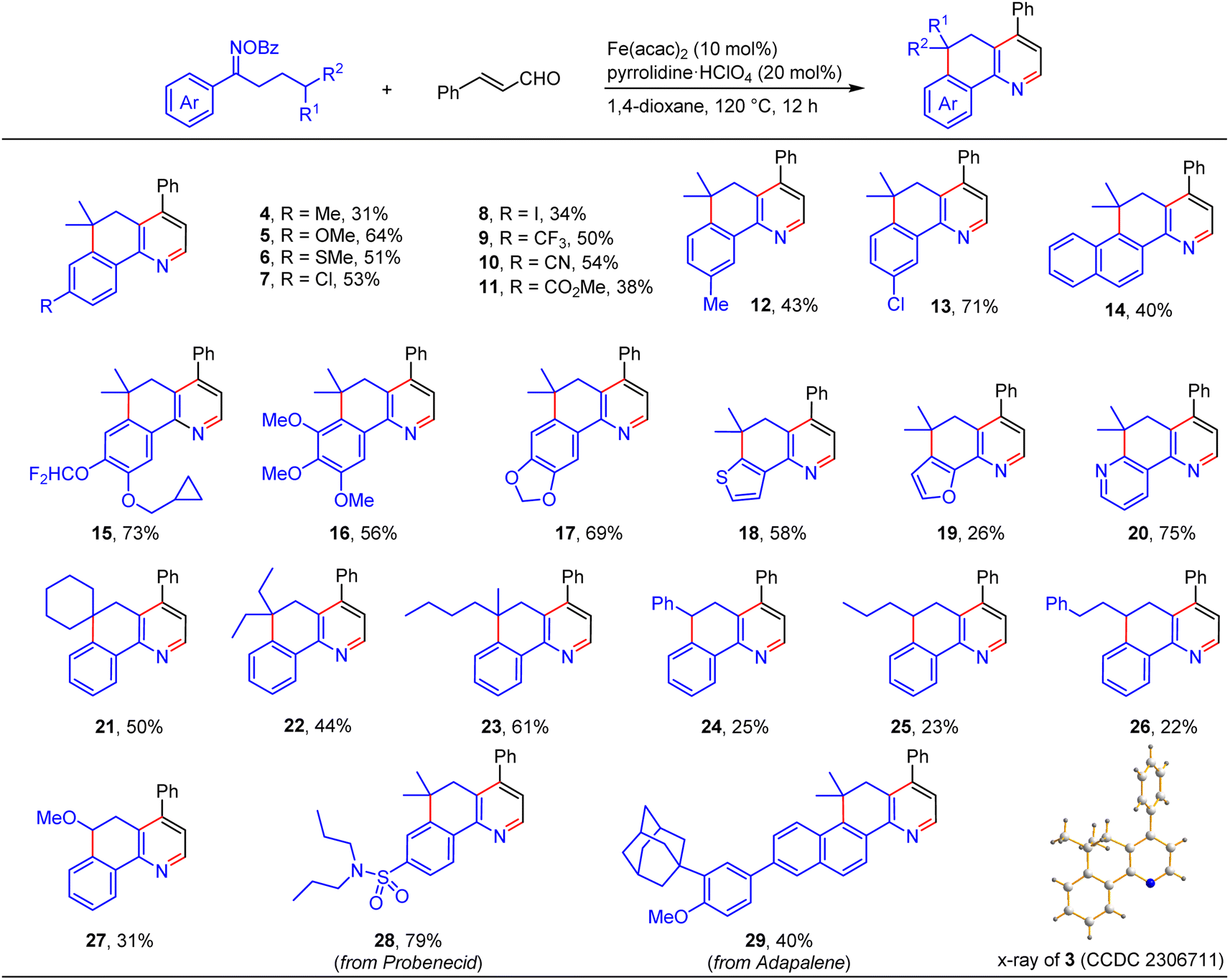 | ||
| Scheme 3 Substrate scope of the oxime esters. Reaction conditions: oxime ester (0.6 mmol), enal (0.2 mmol), Fe(acac)2 (10 mol%), pyrrolidine·HClO4 (20 mol%), 1,4-dioxane (2 mL), 120 °C, 12 h, under N2. | ||
We further showed the generality of this relay annulation approach by surveying the scope of the enals (Scheme 4). We found that substituted cinnamaldehydes were generally good components for the transformation, which delivered the target fused pyridines with moderate to good yields and a variety of functional groups were tolerated, such as methoxy (31 and 45), methylthio (33), halogen (34–37, and 44), ester (38), trifluoromethyl (39), cyano (40), trifluoromethoxy (43), boron (41), alkynyl (42), and cyclopropylmethoxy (46) groups. These diverse functionalities will provide synthetic opportunities to prepare more valuable molecules. In addition, we examined the effect of heteroaromatics on the reactions. Notably, enals containing furan (48), benzofuran (49), pyrrole (50), thiophene (51), pyridine (52), and quinoline (53) moieties can be employed to provide the desired products in moderate to good yields. Moreover, enals with aliphatic (54), alkynyl (55), and ester (56) groups were also amenable to the transformations. Enals bearing an α-substituent are generally challenging substrates in the previous radical conjugate additions, yet our method could give rise to the desired products with synthetically useful yields (57 and 58). It should be noted that four-fold annulations can afford the target product 59 with a 36% yield. This result particularly underlines the power of the present relay annulation, as the conventional methods would not allow its synthesis with such great ease. More interestingly, enals containing sterically bulky naphthyl groups can take part in the reactions to give the corresponding atropisomeric biaryls with moderate yields (62–64), which provided a platform for the atroposelective construction of axially chiral heterobiaryls via an asymmetric relay annulation strategy.
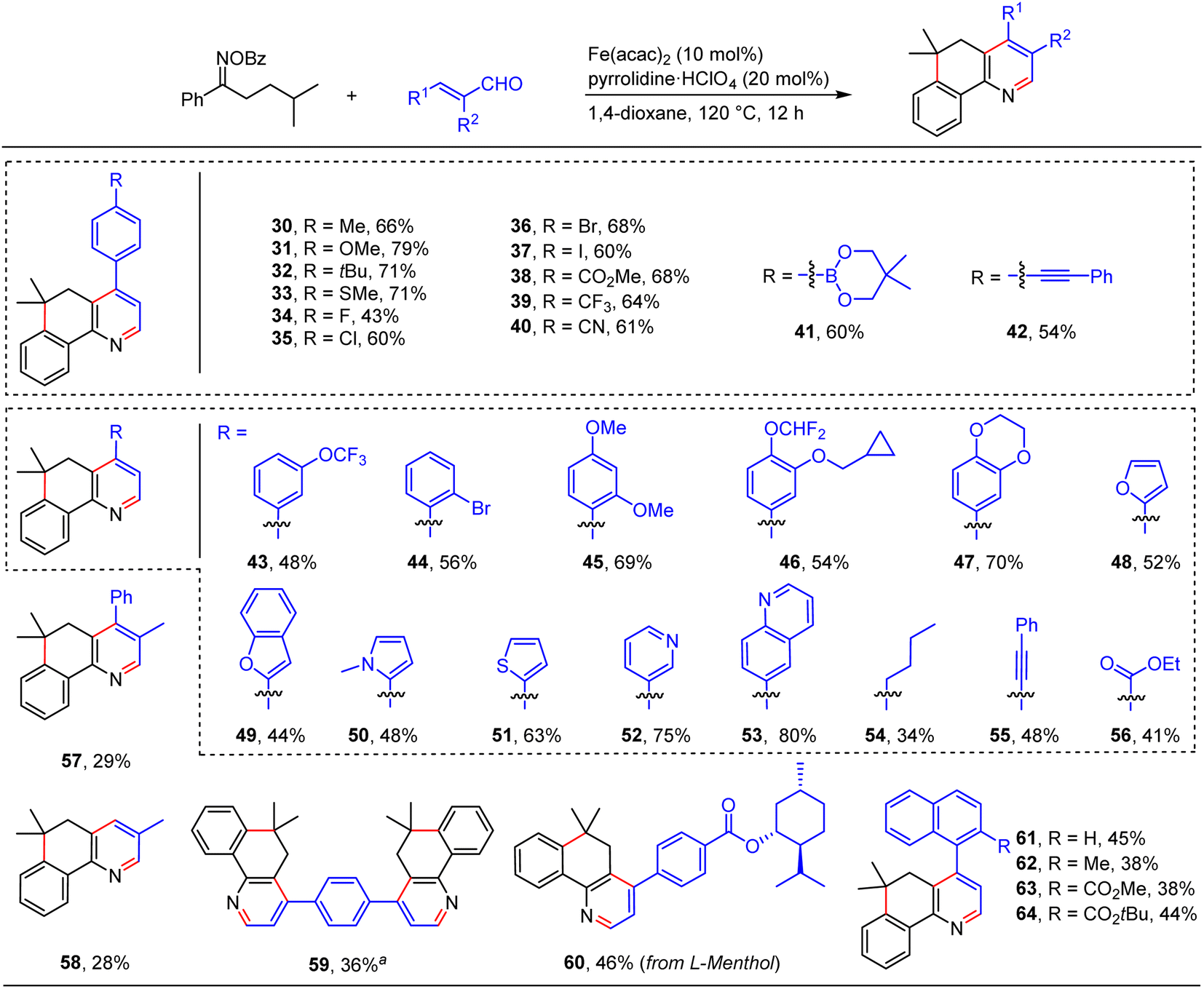 | ||
| Scheme 4 Substrate scope of enals. Reaction conditions: oxime ester (0.6 mmol), enal (0.2 mmol), Fe(acac)2 (10 mol%), pyrrolidine·HClO4 (20 mol%), 1,4-dioxane (2 mL), 120 °C, 12 h, under N2. aOxime ester (1.2 mmol), cinnamaldehyde (0.2 mmol), Fe(acac)2 (20 mol%), pyrrolidine·HClO4 (40 mol%), 1,4-dioxane (4 mL), 120 °C, 12 h, under N2. | ||
The obtained fused pyridines exhibited diverse types of transformations (Scheme 5). The methylene group in compound 3 can be easily oxidized with CrO3 to a ketone group with a 67% yield (Scheme 5a). The pyridine moiety is a commonly used directing group to facilitate C–H activation. As such, aryl C–H bonds smoothly underwent benzoylation and bromination under Pd catalysis to produce the target products (Schemes 5b and c). In addition, the pyridine ring can be efficiently converted into a pyridine N-oxide with 3-chloroperbenzoic acid (m-CPBA) as an oxidant (Scheme 5d). This pyridine ring can be easily converted into an aromatic ring with a 71% yield by Studer's skeletal editing approach (Scheme 5e).51 The existence of a bromo group provided a platform to realize an amination reaction by means of a classic Pd-catalyzed Buchwald–Hartwig amination reaction (Scheme 5f).52 In addition, compound 24 created oxidative dehydrogenation efficiently to give rise to a fused quinoline 71 with a good yield (Scheme 5g).
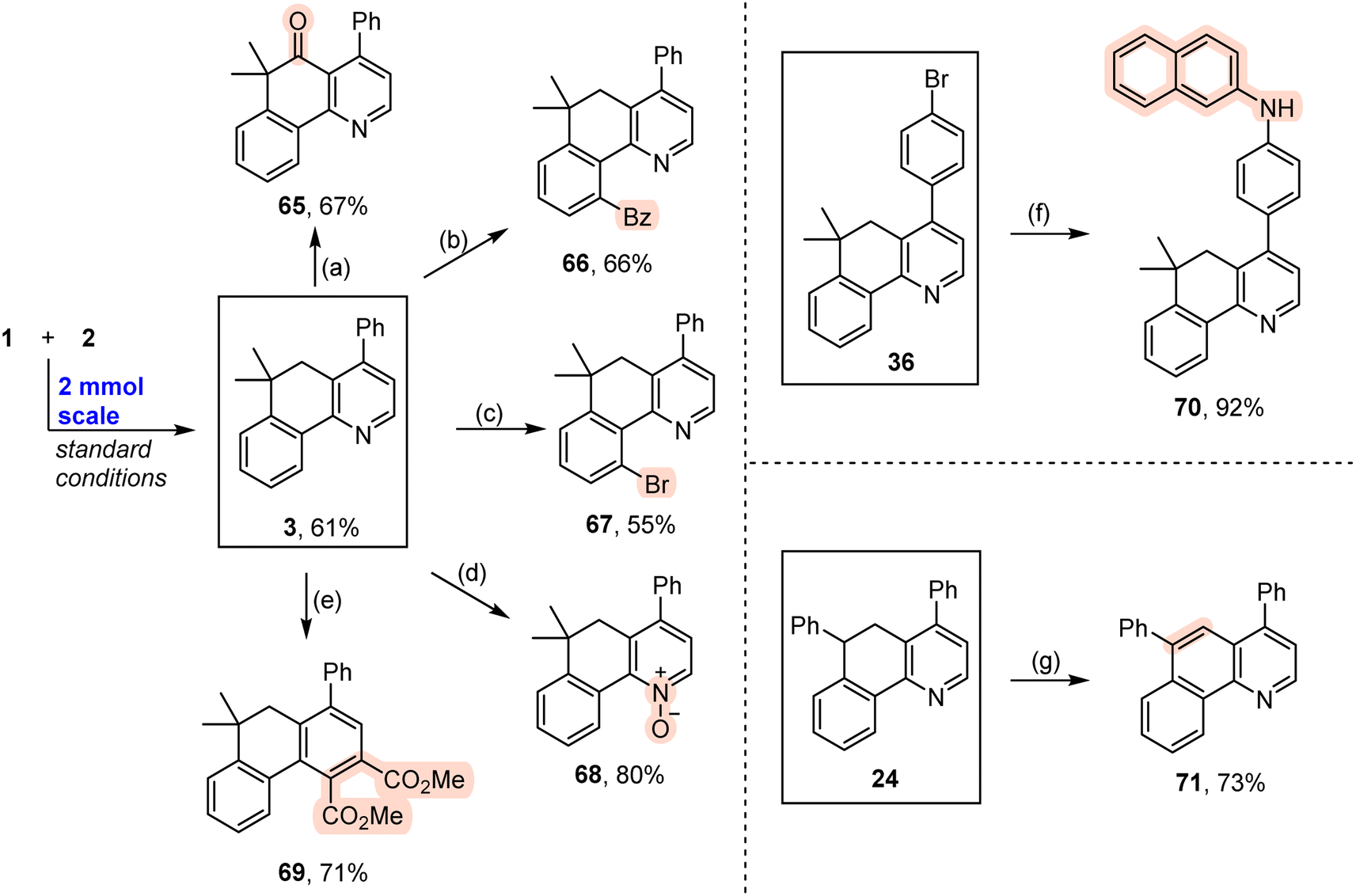 | ||
| Scheme 5 Synthetic applications of the fused pyridines. Reaction conditions: (a) 3 (0.4 mmol), CrO3 (2 equiv.), Ac2O (3 mL), 25 °C, 3 h. (b) 3 (0.1 mmol), benzil (1.5 equiv.), Pd(OAc)2 (5 mol%), TBHP (3 equiv.), THF (1 mL), 100 °C, 20 h. (c) 3 (0.1 mmol), NBS (1.2 equiv.), Pd(OAc)2 (5 mol%), MeCN (1.5 mL), 100 °C, 48 h. (d) 3 (0.1 mmol), m-CPBA (4 equiv.), DCM, 0 → 25 °C, 12 h. (e) 3 (0.15 mmol), dimethyl acetylenedicarboxylate (6 equiv.), methyl pyruvate (6 equiv.), MeCN (1 mL), rt for 48 h, then 80 °C for 48 h. (f) 36 (0.1 mmol), 2-naphthylamine (1.2 equiv.), Pd(OAc)2 (5 mol%), rac-BINAP (10 mol%), NaOtBu (1.5 equiv.), toluene (1 mL), 100 °C, 12 h. (g) 24 (0.1 mmol), DDQ (2 equiv.), toluene (1 mL), 80 °C, 48 h. | ||
To gain insights into the relay annulation mechanism, we performed a series of mechanistic investigations (Scheme 6). Firstly, an iminium salt 72 derived from cinnamaldehyde and pyrrolidine was reacted with 1 in the presence of 10 mol% Fe(acac)2 to afford 3 with a 74% yield (Scheme 6a). In addition, the iminium salt 72 also served as a cocatalyst for the reaction of 1 and 2, producing 3 with a 70% yield (Scheme 6b). These results supported the intermediacy of an iminium ion in the present reaction. Secondly, a radical clock experiment with an oxime ester 73 with 2 afforded a fused pyridine 74, which might involve a ring-opening process of the cyclopropyl group (75 → 76, Scheme 6b), thus, an α-carbon radical would be formed in the reaction. Thirdly, the model reaction of 1 with 2 in the presence of 2,6-di-tert-butyl-4-methylphenol (BHT) offered a polysubstituted pyridine 77 with a 23% yield plus a trace amount of 3 (Scheme 6c). The formation of 77 would involve the trapping of the α-iminyl radical cation by the BHT (78 → 79), which inhibited the 1,5-HAT of the α-iminyl radical cation. Moreover, it was observed that the reaction between 1 and an enal 80 in the presence of BHT cannot produce the desired product 83, while generating a substituted pyridine 81 and a substituted phenol 82 with 34% and 18% yields, respectively (Scheme 6c). We speculated that an α-iminyl radical cation 84 underwent β-C scission to yield an iminium ion 85 and a radical 86. The former created cyclization to produce compound 81, and the latter was trapped by BHT to form compound 82. These two experiments suggested the existence of an α-iminyl radical cation in the relay annulation. In order to elucidate that the α-iminyl radical cation triggered 1,5-HAT, we carried out an experiment with oxime ester 87 and 4-methoxycinnamaldehyde 88 as the substrates, which furnished two substituted pyridines 89 and 90 (Scheme 6d). The former would involve an α-iminyl radical cation 91 triggered 1,5-HAT (91 → 92) and ring-opening processes (92 → 93), the latter might be generated via a cyclization reaction of 91. It should be noted that a reaction between 4,4-dimethyl-3,4-dihydronaphthalen-1(2H)-one 94, 2, and ammonium acetate in the presence of Fe(acac)2, pyrrolidinium perchlorate, and PhCO2H in 1,4-dioxane at 120 °C under air, only produced a trace amount of 3, indicating that a process involving a condensation of a ketimine with an enal would be ruled out (Scheme 6e).53,54
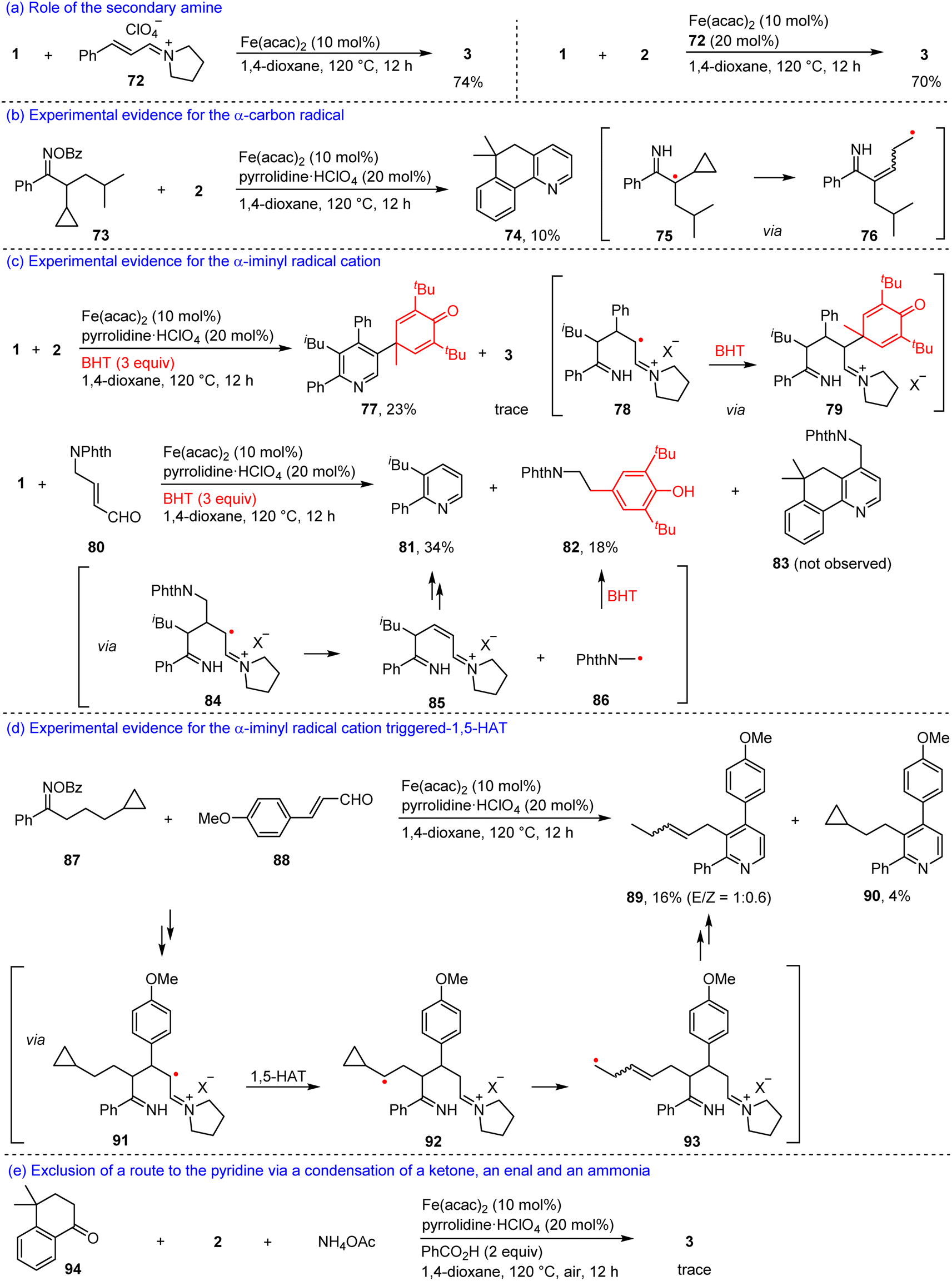 | ||
| Scheme 6 Mechanistic studies (X = ClO4, OBz, or acac). | ||
Based on these results and the work related to the iminyl radical transformations,42–45 we postulated a possible synergistic catalysis for the present reaction (Scheme 7). Upon single electron transfer from a Fe(II) complex to 1, an iminyl radical A and a Fe(III) complex are formed. The former may quickly isomerize to an α-carbon radical B. Such a species then undergoes the radical conjugate addition to an iminium ion C derived from a condensation between pyrrolidine and cinnamaldehyde to produce an α-iminyl radical cation D. This reactive intermediate would cause 1,5-HAT to generate a tertiary dimethyl γ-carbon radical E, and then homolytic aromatic substitution occurs to form a benzo six-membered ring radical F. Subsequently, an oxidation reaction of F with the Fe(III) complex furnishes a compound G that would undergo intramolecular cyclization to afford a 1,4-dihydropyridine I as well as the pyrrolidine catalyst, thus closing the iminium catalytic cycle. Finally, I would be oxidized by the Fe(III) complex into the target product 3, with an accompanying regeneration of the Fe(II) catalyst.
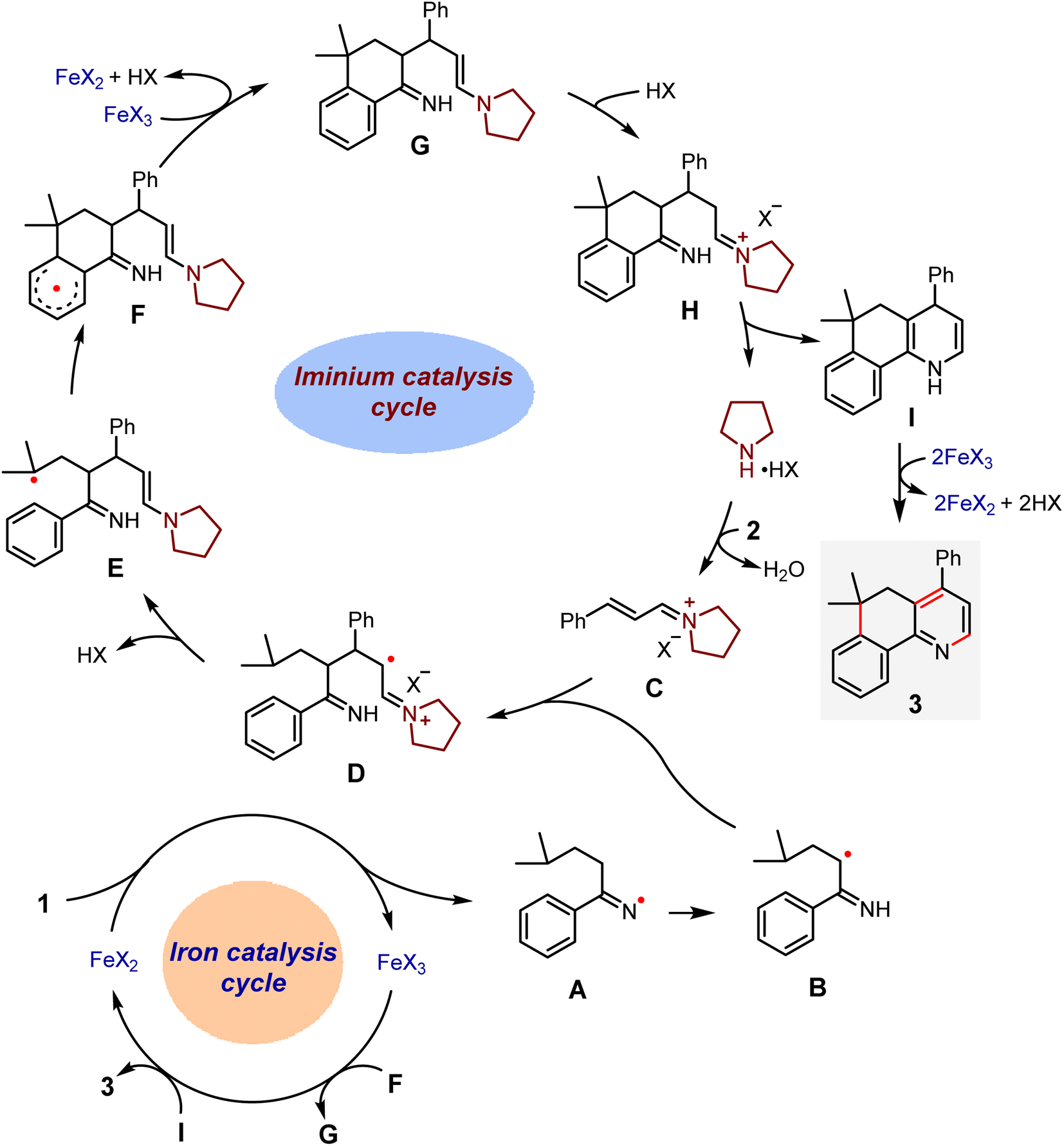 | ||
| Scheme 7 Proposed mechanism for the relay annulation via synergistic iron and iminium catalysis (X = ClO4, OBz, or acac). | ||
Axially chiral biaryls exist in numerous natural products, bioactive compounds, privileged chiral ligands/catalysts, and optically pure materials.55–58 Over the past few decades, much effort has been devoted to constructing the axially chiral biaryls, and several synthetic tactics have been explored, such as asymmetric aryl–aryl coupling and de novo aromatic ring formation.59–68 Nevertheless, this field remains in its infancy, and efficient synthetic methods still need to be identified. Encouraged by the success in the synthesis of fused pyridines via relay annulation, we then planned to develop an asymmetric relay annulation strategy to construct the axially chiral biaryls. As shown in Scheme 7, the 1,4-dihydropyridine I would be formed in the present transformation. If a central chiral 1,4-dihydropyridine could be generated through an asymmetric relay annulation, an atropisomeric aza-biaryl may be produced by means of central-to-axial chirality conversion (Scheme 8a).69–71 With this in mind, we perceived that the use of a chiral secondary amine as the iminium activation catalyst would enable the atroposelective relay annulation, because its chiral skeleton could enantioselectively control several steps, such as carbon radical conjugate addition and cyclization. In addition, a highly electron-withdrawing group, pentafluorobenzoyl (FBz), was installed on the oxygen atom of the oxime, which would cause the N–O bond cleavage under a mild reaction temperature. After considerable investigation of various reaction parameters, we found the reaction of oxime ester 95 with enal 96 afforded the desired product 97 with a 53% yield and a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 enantiomeric ratio (er) with A4 as the amine catalyst (Scheme 8b; please see Table S2 in the ESI† for detailed results of the optimization of the reaction conditions). Please see Scheme S5† for the proposed model for the central-to-axial chirality conversion.
:7 enantiomeric ratio (er) with A4 as the amine catalyst (Scheme 8b; please see Table S2 in the ESI† for detailed results of the optimization of the reaction conditions). Please see Scheme S5† for the proposed model for the central-to-axial chirality conversion.
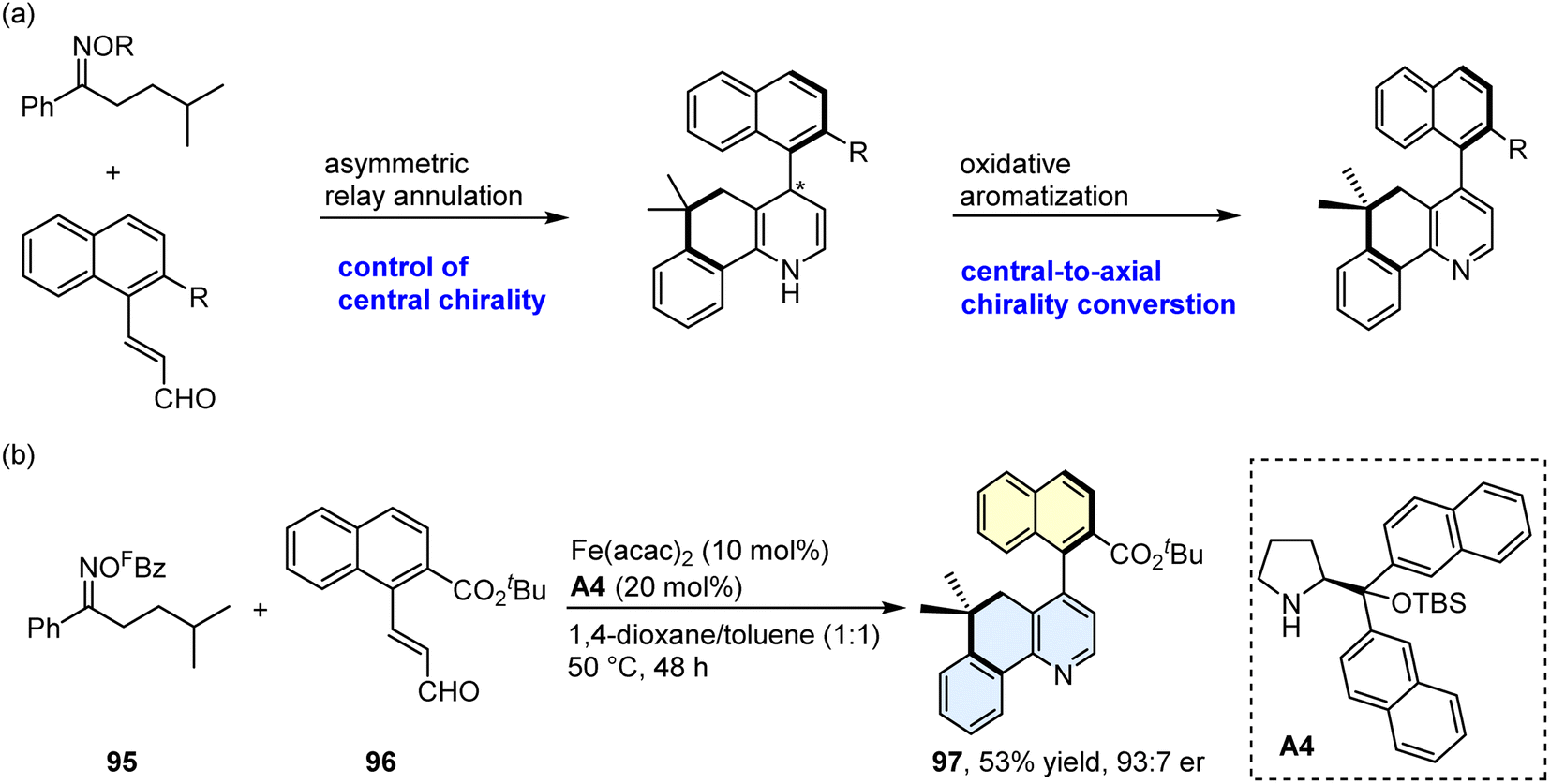 | ||
| Scheme 8 Asymmetric relay annulation. (a) Axially chiral aza-biaryl formation via central-to-axial chirality conversion. (b) Construction of an axially chiral biaryl by the newly developed method. Abbreviation: FBz = pentafluorobenzoyl. TBS = tert-butyl(dimethyl)silyl. | ||
We subsequently expanded the generality of this asymmetric relay annulation with a set of oxime esters and enals (Scheme 9). First, the effect of the electronic properties of the substituents on the oxime aryl ring was examined. As shown in the synthesis of 98–104, the introduction of methyl, methoxy, halogen, ester, and trifluoromethyl substituents can be tolerated, affording the corresponding axially chiral biaryls in moderate yields with up to 94:6 er. The pyridine-derived oxime ester and an oxime ester bearing a cyclohexyl group at the β position both underwent the relay annulations to furnish the expected products in synthetic yields with 93:7 er (105 and 106). Encouraged by these results, we next evaluated the substituents on the naphthyl ring, and found that different ester groups slightly affected the enantioselectivities (107–111). However, the installation of OTs, OTf, and OtBu groups at the β position of the naphthyl ring reduced the stereoselectivities (112–114). Furthermore, enals containing moieties derived from gemfibrozil and oxaprozin participated in the reactions to provide the target products with moderate yields with good er values (115 and 116). It should be noted that a 2 mmol scale reaction of 95 and 96 was performed, and a comparable result was obtained (51% yield, 93:7 er). The absolute configuration of 97 was determined as (aS) by X-ray diffraction analysis,72 and those of the other products were assigned by analogy. It is of great significance that the ester group in the product provides a platform for further functional group elaboration to give several other axially chiral aza-biaryls (117–120).
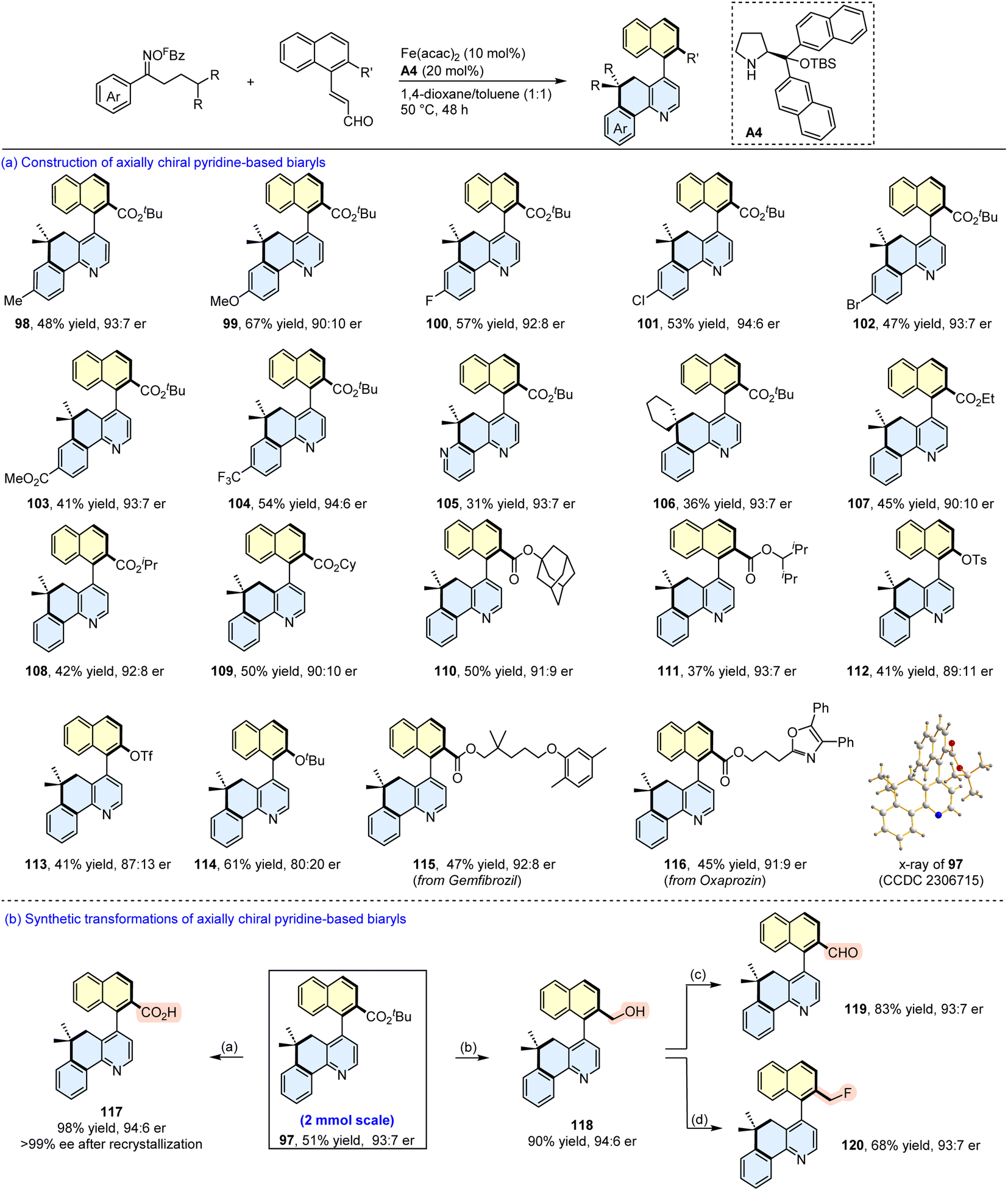 | ||
| Scheme 9 (a) Construction of axially chiral pyridine-based biaryls. Reaction conditions: oxime ester (0.6 mmol), enal (0.2 mmol), Fe(acac)2 (10 mol%), A4 (20 mol%), 1,4-dioxane/toluene (2 mL, v/v = 1/1), 50 °C, 48 h, under N2. (b) Synthetic transformations of 97. Reaction conditions: (a) NaOH (2 M), 95% EtOH, 100 °C, 24 h. (b) DIBAL-H (2.5 equiv.), DCM, −78 °C, 2 h. (c) IBX (1.2 equiv.), HOAc (1.2 equiv.), MeCN, rt, 6 h. (d) DAST (4 equiv.), DCM, 0 °C to rt, 12 h. | ||
Conclusion
In summary, we have explored a new transformation mode of the highly reactive α-iminyl radical cation, that is 1,5-HAT, which provides a platform for the realization of relay annulations. The present strategy can modularly assemble a wide range of structurally novel pyridine derivatives and axially chiral aza-biaryls that would be difficult to access by conventional methods. A series of mechanistic experiments have supported the α-iminyl radical cation-induced 1,5-HAT, and the formation of several radical species in the relay annulations. We believe that the successful exploration of the α-iminyl radical cation-induced 1,5-HAT would open a new avenue for discovering new asymmetric relay annulations for the synthesis of chiral molecules. This work also enriches the knowledge of iron-catalyzed asymmetric radical reactions.73–77 Several projects based on the α-iminyl radical cations are ongoing in our group.Data availability
All experimental data, and detailed experimental procedures are available in the ESI.†Author contributions
Y. W. and K. J. conceived the project. Y.-Q. Z., K.-C. H., W.-H. Z., J.- F. L., S.-M. H., N. Y., Y.-B. Y., and L.-Y. L. performed the experiments and analyzed the data. Y. W. and Y.-Q. Z. prepared the manuscript and the ESI.† All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 22271237), Natural Science Foundation of Chongqing (No. CSTB2023NSCQ-MSX0281), Southwest University (No. SWU120046), Key Project of Innovation Research 2035 Pilot Plan of Southwest University (No. SWU-XDZD22007), and Chongqing Municipal Education Commission (Nos. KJZD-K202200201, KJQN202200222) is greatly appreciated.References
- W. J. Bouma, J. K. MacLeod and L. Radom, Experimental Evidence for the Existence of a Stable Isomer of CH3OH+·: The Methylenoxonium Radical Cation, CH2OH2+·, J. Am. Chem. Soc., 1982, 104, 2930–2931 CrossRef CAS.
- B. F. Yates, W. J. Bouma and L. Radom, Detection of the Prototype Phosphonium (CH2PH3), Sulfonium (CH2SH2) and Chloronium (CH2ClH) Ylides by Neutralization-Reionization Mass Spectrometry: A Theoretical Prediction, J. Am. Chem. Soc., 1984, 106, 5805–5808 CrossRef CAS.
- S. Hammerum, Distonic Radical Cations in Gaseous and Condensed Phase, Mass Spectrom. Rev., 1988, 7, 123–202 CrossRef CAS.
- K. M. Stirk, L. M. Kiminkinen and H. I. Kenttamaa, Ion-Molecule Reactions of Distonic Radical Cations, Chem. Rev., 1992, 92, 1649–1665 CrossRef CAS.
- F. W. McLafferty, Mass Spectrometric Analysis Broad Applicability to Chemical Research, Anal. Chem., 1956, 28, 306–316 CrossRef CAS.
- J. T. Bursey, M. M. Bursey and D. G. I. Kingston, Intramolecular Hydrogen Transfer in Mass Spectra. I. Rearrangements in Aliphatic Hydrocarbons and Aromatic Compounds, Chem. Rev., 1974, 74, 215–242 CrossRef.
- C. S. Sevov and O. Wiest, Selectivity in Radical Cation Cycloadditions, in Carbon-centered Free Radicals and Radical Cations: Structure, Reactivity, and Dynamics, ed. M. D. Forbes, Wiley, 2010, pp. 61–82 Search PubMed.
- A. Hofmann, Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef.
- K. Löffler and C. Freytag, Über eine neue Bildungsweise von N-alkylierten pyrrolidinen, Ber. Dtsch. Chem. Ges., 1909, 42, 3427–3431 CrossRef.
- N. L. Bauld, D. J. Bellville, B. Harirchian, K. T. Lorenz, R. A. Pabon Jr, D. W. Reynolds, D. D. Wirth, H. S. Chiou and B. K. Marsh, Cation Radical Pericyclic Reactions, Acc. Chem. Res., 1987, 20, 371–378 CrossRef CAS.
- K. A. Margrey and D. A. Nicewicz, A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions Via Acridinium Photoredox Catalysis, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed.
- R. Feng, J. A. Smith and K. D. Moeller, Anodic Cyclization Reactions and the Mechanistic Strategies That Enable Optimization, Acc. Chem. Res., 2017, 50, 2346–2352 CrossRef CAS PubMed.
- M.-J. Luo, Q. Xiao and J.-H. Li, Electro-/Photocatalytic Alkene-Derived Radical Cation Chemistry: Recent Advances in Synthetic Applications, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Enantioselective Organocatalysis Using SOMO Activation, Science, 2007, 316, 582–585 CrossRef CAS.
- H.-Y. Jang, J.-B. Hong and D. W. C. MacMillan, Enantioselective Organocatalytic Singly Occupied Molecular Orbital Activation: The Enantioselective α-Enolation of Aldehydes, J. Am. Chem. Soc., 2007, 129, 7004–7005 CrossRef CAS PubMed.
- J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, Enantioselective α-Arylation of Aldehydes Via Organo-SOMO Catalysis. An Ortho-Selective Arylation Reaction Based on an Open-Shell Pathway, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRef CAS PubMed.
- T. Kano, H. Mii and K. Maruoka, Metal-Free Direct Asymmetric Aminoxylation of Aldehydes Catalyzed by a Binaphthyl-Based Chiral Amine, Angew. Chem., Int. Ed., 2010, 49, 6638–6641 CrossRef CAS PubMed.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Photochemical Activity of a Key Donor–Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Direct, Enantioselective α-Alkylation of Aldehydes Using Simple Olefins, Nat. Chem., 2017, 9, 1073–1077 CrossRef CAS PubMed.
- L. Ye, Q.-S. Gu, Y. Tian, X. Meng, G.-C. Chen and X.-Y. Liu, Radical Asymmetric Intramolecular α-Cyclopropanation of Aldehydes Towards Bicyclo [3.1.0] Hexanes Containing Vicinal All-Carbon Quaternary Stereocenters, Nat. Commun., 2018, 9, 227 CrossRef PubMed.
- N. T. Jui, J. A. O. Garber, F. G. Finelli and D. W. C. MacMillan, Enantioselective Organo-SOMO Cycloadditions: A Catalytic Approach to Complex Pyrrolidines from Olefins and Aldehydes, J. Am. Chem. Soc., 2012, 134, 11400–11403 CrossRef CAS PubMed.
- R. J. Comito, F. G. Finelli and D. W. C. MacMillan, Enantioselective Intramolecular Aldehyde α-Alkylation with Simple Olefins: Direct Access to Homo-Ene Products, J. Am. Chem. Soc., 2013, 135, 9358–9361 CrossRef CAS PubMed.
- G. Lelais and D. W. C. MacMillan, Modern Strategies in Organic Catalysis: The Advent and Development of Iminium Activation, Aldrichimica Acta, 2006, 39, 79–87 CAS.
- A. Erkkilä, I. Majander and P. M. Pihko, Iminium Catalysis, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Asymmetric Catalytic Formation of Quaternary Carbons by Iminium Ion Trapping of Radicals, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- A. Bahamonde, J. J. Murphy, M. Savarese, É. Brémond, A. Cavalli and P. Melchiorre, Studies on the Enantioselective Iminium Ion Trapping of Radicals Triggered by an Electron-Relay Mechanism, J. Am. Chem. Soc., 2017, 139, 4559–4567 CrossRef CAS PubMed.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Enantioselective Radical Conjugate Additions Driven by a Photoactive Intramolecular Iminium-Ion-Based EDA Complex, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- J.-J. Zhao, H.-H. Zhang, X. Shen and S. Yu, Enantioselective Radical Hydroacylation of Enals with α-Ketoacids Enabled by Photoredox/Amine Cocatalysis, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed.
- E. Le Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, A General Organocatalytic System for Enantioselective Radical Conjugate Additions to Enals, Angew. Chem., Int. Ed., 2021, 60, 5357–5362 CrossRef CAS PubMed.
- T. H.-F. Wong, D. Ma, R. Di Sanza and P. Melchiorre, Photoredox Organocatalysis for the Enantioselective Synthesis of 1, 7-Dicarbonyl Compounds, Org. Lett., 2022, 24, 1695–1699 CrossRef CAS PubMed.
- H. Jakobsen, S.-O. Lawesson, J. Marshall, G. Schroll and D. Williams, Studies in Mass Spectrometry. Part XII. Mass Spectra of Enamines, J. Chem. Soc. B, 1966, 940–946 RSC.
- J. Robertson, J. Pillai and R. K. Lush, Radical Translocation Reactions in Synthesis, Chem. Soc. Rev., 2001, 30, 94–103 RSC.
- Ž. Čeković, Reactions of δ-Carbon Radicals Generated by 1, 5-Hydrogen Transfer to Alkoxyl Radicals, Tetrahedron, 2003, 59, 8073–8090 CrossRef.
- S. Chiba and H. Chen, sp3 C–H Oxidation by Remote H-Radical Shift with Oxygen-and Nitrogen-Radicals: A Recent Update, Org. Biomol. Chem., 2014, 12, 4051–4060 RSC.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- X. Q. Hu, J. R. Chen and W. J. Xiao, Controllable Remote C−H Bond Functionalization by Visible-Light Photocatalysis, Angew. Chem., Int. Ed., 2017, 56, 1960–1962 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Recent Advances in Radical C–H Activation/Radical Cross-Coupling, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Remote C–H Functionalization via Selective Hydrogen Atom Transfer, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed.
- X. Wu and C. Zhu, Recent Advances in Alkoxy Radical-Promoted C–C and C–H Bond Functionalization Starting from Free Alcohols, Chem. Commun., 2019, 55, 9747–9756 RSC.
- D. L. Golden, S.-E. Suh and S. S. Stahl, Radical C(sp3)–H Functionalization and Cross-Coupling Reactions, Nat. Rev. Chem, 2022, 6, 405–427 CrossRef CAS PubMed.
- Z. Zhang, P. Chen and G. Liu, Copper-Catalyzed Radical Relay in C(sp3)–H Functionalization, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- J. C. Walton, The Oxime Portmanteau Motif: Released Heteroradicals Undergo Incisive EPR Interrogation and Deliver Diverse Heterocycles, Acc. Chem. Res., 2014, 47, 1406–1416 CrossRef CAS PubMed.
- H. Huang, X. Ji, W. Wu and H. Jiang, Transition Metal-Catalyzed C–H Functionalization of N-Oxyenamine Internal Oxidants, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC.
- D. S. Bolotin, N. A. Bokach, M. Y. Demakova and V. Y. Kukushkin, Metal-Involving Synthesis and Reactions of Oximes, Chem. Rev., 2017, 117, 13039–13122 CrossRef CAS PubMed.
- R. Wang and C. Wang, Asymmetric Imino-Acylation of Alkenes Enabled by HAT-Photo/Nickel Cocatalysis, Chem. Sci., 2023, 14, 6449–6456 RSC.
- G. Kumar, S. Pradhan and I. Chatterjee, N-Centered Radical Directed Remote C−H Bond Functionalization via Hydrogen Atom Transfer, Chem.–Asian J., 2020, 15, 651–672 CrossRef CAS PubMed.
- Q.-P. Liu, X. Chen, N. Yu, Y.-L. Li, K.-C. He, W.-H. Zheng, Y.-Q. Zhou, K. Jiang, L. Yang and Y. Wei, Relay C–H Functionalization Enables De Novo Synthesis of Pyridines and Pyridones, ACS Catal., 2023, 13, 5795–5807 CrossRef CAS.
- Y. Wei and N. Yoshikai, Modular Pyridine Synthesis from Oximes and Enals through Synergistic Copper/Iminium Catalysis, J. Am. Chem. Soc., 2013, 135, 3756–3759 CrossRef CAS PubMed.
- Deposition number 2306711 (for 3) contains the supporting crystallographic data for this paper.
- Y. Zhang, Z. Yin and X. F. Wu, Iron-Catalyzed Synthesis of Dihydronaphthalenones from Aromatic Oxime Esters, Adv. Synth. Catal., 2019, 361, 3223–3227 CrossRef CAS.
- Q. Cheng, D. Bhattacharya, M. Haring, H. Cao, C. Mück-Lichtenfeld and A. Studer, Skeletal Editing of Pyridines through Atom-Pair Swap from CN to CC, Nat. Chem., 2024, 1–8 CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, The Buchwald–Hartwig Amination after 25 Years, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- F. Lieby-Muller, C. Allais, T. Constantieux and J. Rodriguez, Metal-Free Michael Addition Initiated Multicomponent Oxidative Cyclodehydration Route to Polysubstituted Pyridines from 1, 3-Dicarbonyls, Chem. Commun., 2008, 4207–4209 RSC.
- C. Allais, T. Constantieux and J. Rodriguez, Use of β, γ-Unsaturated α-Ketocarbonyls for a Totally Regioselective Oxidative Multicomponent Synthesis of Polyfunctionalized Pyridines, Chem.–Eur. J., 2009, 15, 12945–12948 CrossRef CAS PubMed.
- J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, The Challenge of Atropisomerism in Drug Discovery, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed.
- G. Bringmann, T. Gulder, T. A. Gulder and M. Breuning, Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. Miller and P. J. Edwards, Assessing Atropisomer Axial Chirality in Drug Discovery and Development, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS PubMed.
- W. Fu and W. Tang, Chiral Monophosphorus Ligands for Asymmetric Catalytic Reactions, ACS Catal., 2016, 6, 4814–4858 CrossRef CAS.
- E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- J. Wencel-Delord, A. Panossian, F. Leroux and F. Colobert, Recent Advances and New Concepts for the Synthesis of Axially Stereoenriched Biaryls, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- Y.-B. Wang and B. Tan, Construction of Axially Chiral Compounds Via Asymmetric Organocatalysis, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Recent Advances in the Synthesis of Axially Chiral Biaryls Via Transition Metal-Catalysed Asymmetric C–H Functionalization, Chem. Commun., 2019, 55, 8514–8523 RSC.
- T. Z. Li, S. J. Liu, W. Tan and F. Shi, Catalytic Asymmetric Construction of Axially Chiral Indole-Based Frameworks: An Emerging Area, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed.
- J. A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Atroposelective Transformation of Axially Chiral (Hetero) Biaryls. From Desymmetrization to Modern Resolution Strategies, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Recent Advances in Catalytic Asymmetric Construction of Atropisomers, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- W. Qin, Y. Liu and H. Yan, Enantioselective Synthesis of Atropisomers Via Vinylidene Ortho-Quinone Methides (VQMs), Acc. Chem. Res., 2022, 55, 2780–2795 CrossRef CAS PubMed.
- H. H. Zhang, T. Z. Li, S. J. Liu and F. Shi, Catalytic Asymmetric Synthesis of Atropisomers Bearing Multiple Chiral Elements: An Emerging Field, Angew. Chem., 2024, 136, e202311053 CrossRef.
- O. Quinonero, M. Jean, N. Vanthuyne, C. Roussel, D. Bonne, T. Constantieux, C. Bressy, X. Bugaut and J. Rodriguez, Combining Organocatalysis with Central-to-Axial Chirality Conversion: Atroposelective Hantzsch-Type Synthesis of 4-Arylpyridines, Angew. Chem., Int. Ed., 2016, 55, 1401–1405 CrossRef CAS PubMed.
- V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, Enantioselective Syntheses of Furan Atropisomers by an Oxidative Central-to-Axial Chirality Conversion Strategy, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS PubMed.
- S. Yan, W. Xia, S. Li, Q. Song, S.-H. Xiang and B. Tan, Michael Reaction Inspired Atroposelective Construction of Axially Chiral Biaryls, J. Am. Chem. Soc., 2020, 142, 7322–7327 CrossRef CAS PubMed.
- Deposition number 2306715 (for 97) contains the supporting crystallographic data for this paper.
- L. Ge, H. Zhou, M.-F. Chiou, H. Jiang, W. Jian, C. Ye, X. Li, X. Zhu, H. Xiong, Y. Li, L. Song, X. Zhang and H. Bao, Iron-Catalysed Asymmetric Carboazidation of Styrenes, Nat. Catal., 2021, 4, 28–35 CrossRef CAS.
- R. R. Surgenor, X. Liu, M. J. H. Keenlyside, W. Myers and M. D. Smith, Enantioselective Synthesis of Atropisomeric Indoles via Iron-Catalysed Oxidative Cross-Coupling, Nat. Chem., 2023, 15, 357–365 CrossRef CAS PubMed.
- T. A. Trinh and J. M. Schomaker, Unnatural α-Amino Acid Synthesis, Nat. Synth., 2023, 2, 600–601 CrossRef.
- W.-C. C. Lee, D.-S. Wang, Y. Zhu and X. P. Zhang, Iron(III)-Based Metalloradical Catalysis for Asymmetric Cyclopropanation via A Stepwise Radical Mechanism, Nat. Chem., 2023, 15, 1569–1580 CrossRef CAS PubMed.
- C.-X. Ye, D. R. Dansby, S. Chen and E. Meggers, Expedited Synthesis of α-Amino Acids by Single-Step Enantioselective α-Amination of Carboxylic Acids, Nat. Synth., 2023, 2, 645–652 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2306711 and 2306715. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01858c |
| This journal is © The Royal Society of Chemistry 2024 |