
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing biocatalysis as a green tool in antibiotic synthesis and discovery
Guilherme F. S. Fernandes†
 *ab,
Seong-Heun Kim†c and
Daniele Castagnolo*a
*ab,
Seong-Heun Kim†c and
Daniele Castagnolo*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: d.castagnolo@ucl.ac.uk; guilherme.fernandes@ucl.ac.uk
bSchool of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK
cInstitute of Pharmaceutical Science, School of Cancer & Pharmaceutical Science, King's College London, 150 Stamford Street, London SE1 9NH, UK
First published on 24th September 2024
Abstract
Biocatalysis offers a sustainable approach to drug synthesis, leveraging the high selectivity and efficiency of enzymes. This review explores the application of biocatalysis in the early-stage synthesis of antimicrobial compounds, emphasizing its advantages over traditional chemical methods. We discuss various biocatalysts, including enzymes and whole-cell systems, and their role in the selective functionalization and preparation of antimicrobials and antibacterial building blocks. The review underscores the potential of biocatalysis to advance the development of new antibiotics and suggests directions and potential applications of enzymes in drug development.
1. Introduction
The pioneering discovery of penicillin by Sir Alexander Fleming in 1928 revolutionized medicine and marked a significant milestone in scientific history.1,2 Throughout the 20th century, the discovery, commercialization, and widespread use of new antimicrobial compounds allowed to successfully treat life-threatening infections and contributed to the increased average life expectancy in the developed world. However, already in 1945 Fleming predicted that the high public demand of antibiotics would determine an “era of abuse” and that, as a consequence of such abuse, antibiotic-resistant bacterial strains would emerge. This prediction has become a stark reality, and a century after the discovery of penicillin, the world is facing up a global “antibiotic resistance health emergency”.3 In fact, pathogens that were once susceptible to standard antibiotic regimes for decades, have now developed resistance to most antimicrobial therapies, carrying significant economic and health implications.4 The rise and spread of antibiotic resistance present a challenge to science and medicine. Consequently, the scientific community has launched action plans to identify solutions to combat antimicrobial resistance (AMR), including the development of new and alternative therapeutic strategies.5,6 Currently, one of the most significant AMR challenges is the spread of the ‘ESKAPE’ pathogens, (Enterococcus spp., Staphylococcus aureus, Klebsiella spp., Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.), which are becoming resistant to all the currently available antibiotics.7,8According to a recent survey, bacterial AMR contributed to an estimated 4.95 million deaths in 2019. Shockingly, 1.27 million of these deaths were directly caused by drug-resistant infections, surpassing the death toll from both HIV/AIDS (864![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths) and malaria (643000 deaths).9,10 Another report emphasized the criticality of the situation, as it alerts that failing to take immediate action against this crisis could lead to a staggering increase in deaths, with estimates 10 million deaths due to bacterial infections by 2050.11 During the COVID-19 pandemic, bacterial co-infections have complicated patient treatment, with P. aeruginosa being one of the most common bacteria.12,13 With the coming advent of the ‘post-antibiotic era’ and the global threat of AMR, the urgent need for the development of new antibiotics has become increasingly evident.14 Insufficient investments in novel antibiotics is a significant contributor to the problem of AMR. Over the past four decades, no new classes of antibiotics have been discovered, creating a noteworthy innovation gap referred to as the “discovery void.” This term underscores the notable absence of successful introductions of novel antibiotic classes, further exacerbating the issue of AMR.15 Despite challenges, recent endeavours to uncover new antibiotics have shown promise, with several candidates advancing into preclinical and clinical development stages.15–19
000 deaths) and malaria (643000 deaths).9,10 Another report emphasized the criticality of the situation, as it alerts that failing to take immediate action against this crisis could lead to a staggering increase in deaths, with estimates 10 million deaths due to bacterial infections by 2050.11 During the COVID-19 pandemic, bacterial co-infections have complicated patient treatment, with P. aeruginosa being one of the most common bacteria.12,13 With the coming advent of the ‘post-antibiotic era’ and the global threat of AMR, the urgent need for the development of new antibiotics has become increasingly evident.14 Insufficient investments in novel antibiotics is a significant contributor to the problem of AMR. Over the past four decades, no new classes of antibiotics have been discovered, creating a noteworthy innovation gap referred to as the “discovery void.” This term underscores the notable absence of successful introductions of novel antibiotic classes, further exacerbating the issue of AMR.15 Despite challenges, recent endeavours to uncover new antibiotics have shown promise, with several candidates advancing into preclinical and clinical development stages.15–19
The demand for novel and diverse synthetic strategies for the construction and functionalization of antimicrobial compounds in drug discovery and chemical manufacturing is a growing area of research.20–22 New approaches that can enable the synthesis of antimicrobial agents in a more sustainable and efficient way are particularly attractive.23 Biocatalysis is an appealing green technology that can be widely applied throughout different stages of the drug discovery process,24 from early drug discovery to the scale up synthesis of drug candidates for clinical trials.25–30 The successful application of biocatalysis in the synthesis of many drugs has been recently demonstrated showing its potentiality in accelerating and making antibiotic research more cost-efficient and sustainable.31–38 Furthermore, the intrinsic sustainable nature of biocatalysis aligns with the ever-growing drive toward the use of green chemistry in drug and chemical synthesis, through the reduction of environmental impact and the improvement of reaction safety of chemical processes.20,39–41 Typically occurring in aqueous media, at mild temperatures, and atmospheric pressure, biocatalytic reactions utilize biodegradable and recyclable catalysts, namely the enzymes. Consequently, the application of biocatalytic reactions holds substantial potential in advancing environmentally friendly processes for drug discovery and synthesis.
While considerable emphasis is given to optimizing biocatalytic processes in the late-stage development of pharmaceuticals, it is important to acknowledge that biocatalysis also holds significant importance in the earlier stages of the drug development cycle. This review aims to provide an overview of recent research and findings on the implementation of biocatalysis in antibacterial drug discovery. Although various reviews have explored the industrial applications of biocatalysis in drug development,40,42–49 herein we will primarily focus on the utilization of biocatalysis in the discovery and synthesis of novel antibacterial compounds. This review aims to illustrate the potential of biocatalysis as a sustainable and efficient tool to address the challenges associated with traditional synthetic methods and to provide new, greener, and more efficient synthetic routes in the discovery of antimicrobial agents.
To facilitate the organization of the forthcoming section and highlight trends in enzyme sourcing, we have categorised the review according to the Enzyme Commission number (EC number), which is a numerical classification system used to categorize enzymes based on the specific chemical reactions they catalyse. Throughout the text, we will utilize the following abbreviations to denote enzyme sources: EnzymeWT (wild-type enzyme), EnzymeEng (enzyme specifically designed for the synthetic process), or EnzymeComm (commercially available enzyme).
2. Oxidoreductases [EC 1]
Oxidoreductases are a diverse group of enzymes that facilitate the transfer of electrons from an electron donor to an electron acceptor molecule. Typically, these enzymes use nicotinamide adenine dinucleotide phosphate (NADP+) or nicotinamide adenine dinucleotide (NAD+) as cofactors.50 Since many chemical and biochemical reactions involve oxidation/reduction processes, there is a significant interest in drug discovery to exploit oxidoreductases in synthesis.51Glycopeptide antibiotics (GPAs), such as vancomycin, possess complex structures, and their natural biosynthesis involves a complex cyclization cascade mediated by cytochrome P450 (Oxy) enzymes.52 Thus, considerable investigations have been directed towards the use of a chemoenzymatic cascades to synthesize and potentially diversify GPA structures. Forneris and Seyedsayamdost described a chemoenzymatic approach to synthesize the vancomycin aglycone derivative 2.53 The method involves the use of three oxygenases54 (OxyA, OxyB and OxyC) that introduce bisaryl ether linkages and form carbon–carbon crosslinks through a cascade sequence (Fig. 1). While the enzymes OxyB and OxyA had been shown to introduce bisaryl ether linkages,55,56 the final OxyC catalysed crosslink remained elusive. The authors successfully reconstituted the OxyC-catalyzed reaction and applied it in a cascade reaction with OxyA and OxyB. This facilitated the installation of the elusive biaryl bond in a cascade sequence. The OxyC enzyme was cloned from Amycolatopsis orientalis DSM 40040, expressed in E. coli BL21(DE3), and further purified. The chemoenzymatic approach is simple and flexible, and it allowed the authors to obtain a core-modified vancomycin aglycone derivative 2, containing a thioamide bond (Fig. 1). The authors also suggested that the thioamide could be further used as a precursor to produce a vancomycin variant incorporating an amidine moiety. This alteration reinstates crucial hydrogen bonds with the target in the developing peptidoglycan chain of vancomycin-resistant pathogens, thereby maintaining its effectiveness against them.57,58 This enzymatic approach has the potential to facilitate the efficient and selective in vitro synthesis of new vancomycin derivatives, making it an important breakthrough in the development of new antibiotics and the fight against antibiotic resistance,53 as shown by vancomycin derivative synthesized using this approach that exhibited efficacy against vancomycin resistant bacterial strains.59,60
 | ||
| Fig. 1 Chemoenzymatic synthesis of a vancomycin derivative 2 with the oxygenase enzymes. | ||
Following up this work, Tailhades and colleagues reported a thorough analysis to determine the tolerance and the versatility of the Oxy chemoenzymatic cascade towards modifications in the GPA peptide structure.61 The authors evaluated the capacity of the enzymatic cascade to produce GPAs bearing amino acid residues different from those present in the natural compound and they identified peptide modifications that are enzymatically tolerated and can also reveal the mechanistic basis for substrate intolerance where present.61
Olchowik-Grabarek et al. reported the enzymatic synthesis and characterization of antimicrobial aryl iodides derived from phenolic acids, e.g. iodinated gallic (3) and ferulic (4) acids, using laccase enzymes. Laccases are multicopper oxidases found in plants, fungi, and bacteria, and they are able to oxidize a variety of phenolic substrates through one-electron oxidations. The Trametes versicolor laccase was used to convert the gallic acid into the corresponding aryl iodide 3 and ferulic acid into iodine derivative (4) (Fig. 2a). This conversion process was aided by the presence of 2,2′-azino-bis-(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS) as a redox mediator and potassium iodide (KI).62 Iodogallic acid (3) was found to present broad spectrum antibacterial and antiviral activity. Specifically, the minimum inhibitory concentration (MIC) of iodogallic acid against S. aureus was determined to be 118.8 μg mL−1. In comparison, the MIC value for the non-iodinated gallic acid was reported to be 5250 μg mL−1. Thus, the introduction of iodine into the structure of gallic acid resulted in a remarkable 50–100-fold increase in its bacteriostatic properties, highlighting its enhanced potency. On the other side, the iodinated ferulic acid (4) did not result in any improvement in its antibacterial effect.62
 | ||
| Fig. 2 (a) Laccase-assisted iodination of gallic acid; (b) biocatalytic synthesis of dihydroxyequol using monooxygenase enzymes; (c) biotransformation process of p-aminobenzoic acid to p-nitrobenzoic acid. | ||
Nozawa et al. described the biocatalytic synthesis and assessment of 6,3′-dihydroxyequol (8) from equol (5) using two flavin-dependent monooxygenases: HpaBro-3 from Rhodococcus opacus, which hydroxylates the C-3′ position of equol 5 with high regioselectivity, and HpaBpl-1 from Photorhabdus luminescens, which hydroxylates the C-6 position of 5 (Fig. 2b). E. coli cells expressing both HpaBro-3 and HpaBpl-1 were incubated with equol, leading to the formation of 3′-hydroxyequol (6), 6-hydroxyequol (7), and 6,3′-dihydroxyequol (8) (via the intermediates 3′- and 6-hydroxyequols) in a single reaction step. The compound 6-hydroxyequol (7) exhibited inhibition of E. coli growth, with a MIC value of 64 μg mL−1, demonstrating stronger antibacterial activity compared to equol (MIC value of 256 μg mL−1).63
Nóbile et al. identified a N-oxygenase activity in Streptomyces griseus and subsequently exploited such microorganism as whole cell biocatalyst in the biotransformation of p-aminobenzoic acid (9) into p-nitrobenzoic acid (10) (Fig. 2c). Aromatic nitro compounds play a crucial role as building blocks in the synthesis of various nitroaromatic antibiotics, including pretomanid, delamanid, chloramphenicol, azomycin, metronidazole, and benznidazole.64 The synthesis of natural products has greatly benefited from the application of biocatalysis.
Renata's group developed a modular chemoenzymatic approach for the synthesis of GE81112 B1, a tetrapeptides identified in 2006 during a screen for new inhibitors of bacterial protein synthesis (15) (Fig. 3a).65
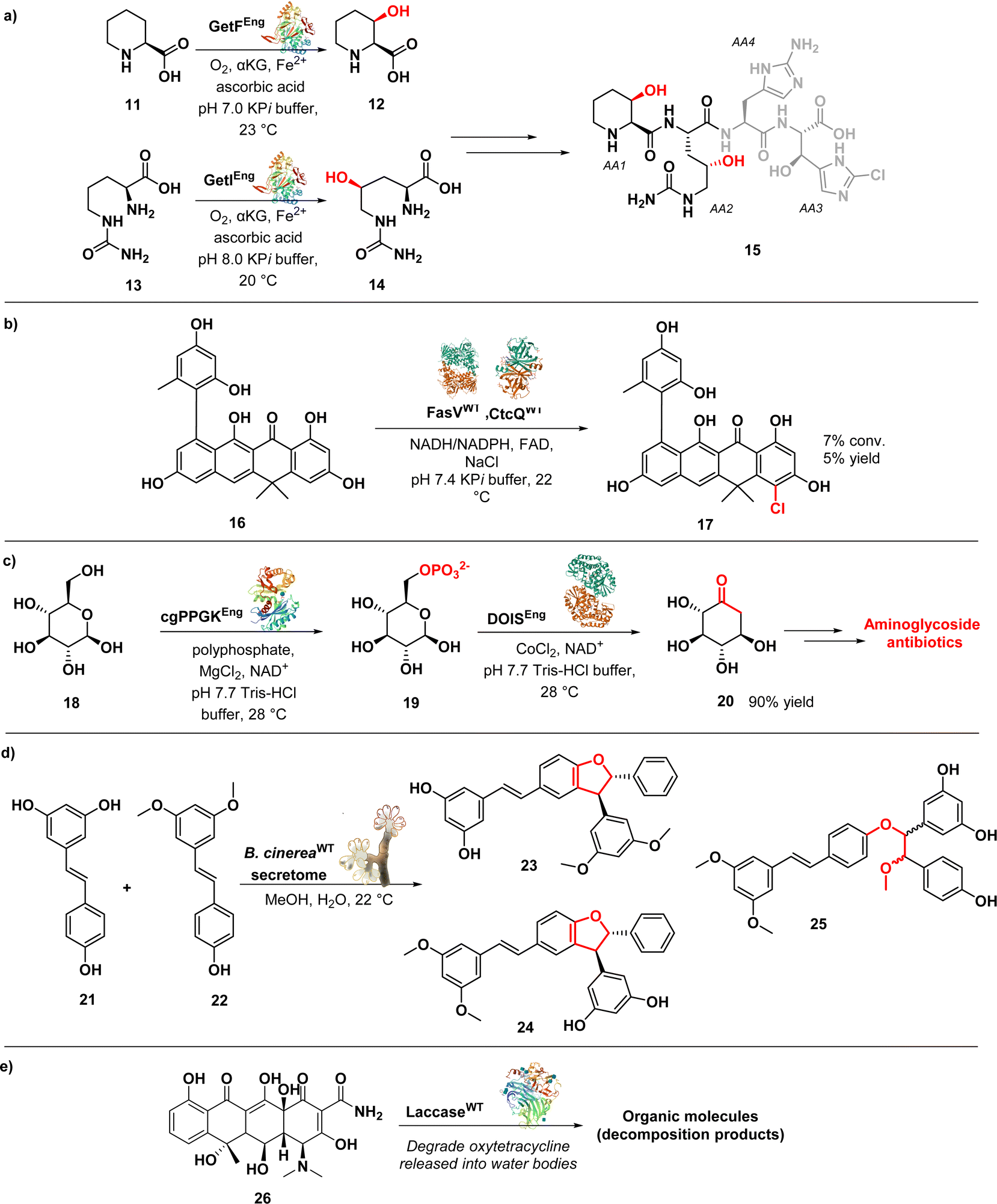 | ||
| Fig. 3 (a) Remote hydroxylation by oxygenase enzymes GetF and Getl; (b) enzymatic chlorination with halogenase FasV; (c) one-pot biocatalytic transformation of D-glucose to 2-deoxy-scyllo-inosose; (d) biotransformation of resveratrol and pterostilbene with the secretome of B. cinerea; (e) laccase-immobilized for enzymatic degradation of oxytetracycline in the environment. | ||
In this approach, the researchers utilized two key enzymes, GetF and GetI (expressed in E. coli strain BL21(DE3)), which are iron and α-ketoglutarate dependent dioxygenases. These enzymes were responsible for the hydroxylation of L-pipecolic acid (11) and L-citrulline (13), generating the necessary intermediates 12 and 14 for constructing monomers AA1 and AA2, respectively. By integrating these enzymatic components with traditional synthetic methods, the authors were able to achieve the synthesis of the natural product in just eleven steps. Building upon this successful approach, the researchers expanded their efforts by synthesizing ten analogues of 15, enabling the identification of key pharmacophores associated with its activity, further enhancing the understanding of its mode of action. Notably, they discovered that the presence of a syn-β-hydroxy amino acid at the AA1 position and a β-OH group on AA4 were critical for the compounds' antibacterial activity. Surprisingly, the scaffold demonstrates a remarkable tolerance for substitutions at the AA2 position. Additionally, they observed that removing the C2–NH2 group in AA3 or the C2–Cl in AA4 only resulted in a modest reduction in activity, indicating the compounds' relative tolerance towards these modifications.65,66 The research group also achieved a significant milestone by successfully conducting a chemoenzymatic synthesis of Fasamycin A (17) (Fig. 3b), a chlorinated naphthacenoid compound known for its potent antibacterial activities against methicillin-resistant Staphylococcus aureus (MRSA) (MIC = 3.1 μg mL−1) and vancomycin-resistant Enterococcus faecalis (VRE) (MIC = 0.8 μg mL−1).67 The potent antibacterial effects of Fasamycin A can be attributed to its inhibition of FabF, a key enzyme involved in the biosynthesis of type II fatty acids.68 The synthesis employed a convergent strategy, merging two fragments via a Sammes annulation reaction to generate a dimethylnaphthacenone system. An enzymatic halogenation step was then utilized to introduce the necessary chlorine substituent. This late-stage enzymatic C–H chlorination reaction was catalysed by FasV, a flavin-dependent halogenase, in combination with CtcQ, an FAD reductase. While the observed conversion achieved by this enzyme pair was the highest recorded (7%), the overall chlorination yield remained modest (5%). Nevertheless, it is noteworthy that the enzymatic chlorination takes place under more gentle reaction conditions in comparison to traditional chemical methods.67
In a study conducted by Kudo et al., an efficient enzymatic method for producing 2-deoxy-scyllo-inosose (2DOI) (20) (Fig. 3c) from D-glucose (18) and polyphosphate was developed. 2DOI is a crucial intermediate in the production of aminoglycoside antibiotics like kanamycin, neomycin, butirosin, and gentamicin. The researchers identified a polyphosphate glucokinase (cgPPGK) derived from Corynebacterium glutamicum, which effectively generated D-glucose 6-phosphate (G6P) (19) from polyphosphate. By combining this enzyme with 2-deoxy-scyllo-inosose synthase (DOIS), they enabled a one-pot enzymatic reaction that yielded 2DOI. Seven different DOISs were assessed to find the most effective one, and ultimately cgPPGK, and BtrC from Bacillus circulans achieved nearly complete conversion of D-glucose to 2DOI. Remarkably, a 90% yield of 2DOI was achieved in a reaction volume of 15 mL.69
Queiroz et al. conducted a study on the generation of stilbene antimicrobials to combat multiresistant strains of Staphylococcus aureus. They employed the enzymatic secretome of Botrytis cinerea to biotransform a mixture of resveratrol (21) and pterostilbene (22) (Fig. 3d), utilizing methanol as a cosolvent. This process resulted in the production of 15 structurally complex stilbenes, including 8 newly isolated compounds. Notably, compounds 23, 24, and 25 exhibited significant antibacterial activity against MRSA, with MIC of 4, 2, and 8 μM, respectively. Furthermore, these compounds displayed remarkable efficacy against vancomycin-resistant strains of S. aureus (VRSA), with MIC values below 4 μM. By leveraging the catalytic promiscuity of enzymes within the bacterial secretome, the study highlights the advantages of using an enriched fraction of enzymes for diverse substrate transformations. This approach allows for simultaneous catalysis of different chemical reactions, surpassing the capabilities of individual purified enzymes.70
Oxidoreductases have found applications beyond drug discovery in addressing antimicrobial resistance. The excessive use of antibiotics has led often to their uncontrolled release into the environment, significantly contributing to the emergence of antimicrobial resistance.71 Pumera et al. introduced a fascinating approach for effectively removing antibiotics from water bodies that involves enzyme-immobilized self-propelled zinc oxide–Au (ZnO–Au) microrobots able to degrade antibiotics. The authors generated a hybrid enzymatic/photocatalytic system, consisting of the enzyme laccase (from Trametes versicolor) physically adsorbed onto the ZnO–Au microrobots. The laccase-immobilized microrobots, driven by light, effectively eliminated oxytetracycline (26) (Fig. 3e), a broad-spectrum tetracycline antibiotic, from water by oxidizing the aromatic rings containing electron-donating groups using molecular oxygen in multicopper-electron redox reactions.72
Finally, in a recent study, a sodium caseinate (SC)-epigallocatechin gallate (EGCG)-carboxymethyl chitosan (CMC) ternary film was developed as an eco-friendly packaging material with antibacterial properties. This film can enhance the storage quality of foods and reduce the risk of foodborne infections caused by drug-resistant bacteria. Tyrosinase was used to synthesize the ternary conjugate, enabling covalent cross-linking between EGCG, SC, and CMC. Notably, EGCG, a polyphenol known for its potent antibacterial properties, played a key role in this process. The films exhibited effective antibacterial activity against S. aureus, a well-known pathogen associated with AMR.73
3. Transferases [EC 2]
Transferases are a class of enzymes that catalyse the transfer of specific functional groups, such as methyl or glycosyl groups, from one molecule (the donor) to another (the acceptor). These enzymes are involved in hundreds of different biochemical pathways and are essential to many important processes in living organisms. While transferases have the potential to be used in industrial processes, their application in biopharmaceutical industry is currently limited. Nevertheless, some glycosyltransferases are utilized for the synthesis of oligosaccharides, indicating the potential for practical applications.74Boger et al. described a chemoenzymatic approach for combating vancomycin resistance by converting vancomycin into maxamycin.75,76 A two-step glycosylation method involving two glycosyltransferases, GtfE and GtfD, was used in this study to introduce a disaccharide onto binding pocket-modified vancomycin analogues aglycons (29) (Fig. 4). The addition of carbohydrates to vancomycin aglycons in a sequential manner with corresponding glucosyl donors, UDP-glucose for GftE and UDP-vancosamine for GtfD, resulted in the production of a set of vancomycin analogues. The subsequent peripheral modification involved introducing a chlorobiphenyl group into these vancomycin analogues through selective reductive amination, resulting in a novel series of derivatives known as maxamycins. This modification significantly improved their binding affinity and efficacy against vancomycin-resistant bacteria.59,77,78
 | ||
| Fig. 4 Enzymatic glycosylation of vancomycin aglycon (27) by glycosyltransferases GtfE and GtfD. | ||
These findings emphasize the potential of a chemoenzymatic strategy to antibiotic redesign in combating resistance and improving therapeutic qualities.79
Bashyal and co-workers described the biocatalytic synthesis of monoterpene O-glycosides using the glycosyltransferase YjiC derived from Bacillus subtilis (Fig. 5a).80 The authors successfully converted seven different monoterpenes into 41 glycoside derivatives through the efficient catalytic action of YjiC, which facilitated the formation of glycosidic bonds. Notably, the broad substrate tolerance of YjiC towards diverse sugar donors was identified. Furthermore, the authors demonstrated robust enzyme activity through a whole-cell biotransformation using recombinant E. coli cells. These approaches have the potential to facilitate both in vitro and in vivo synthesis of monoterpene O-glycosides exhibiting antibacterial and antinematodal activities. This study suggests that the chemoenzymatic approach holds significant promise for industrial applications, owing to its cost-effectiveness, sustainability, robustness, and ease of scalability.
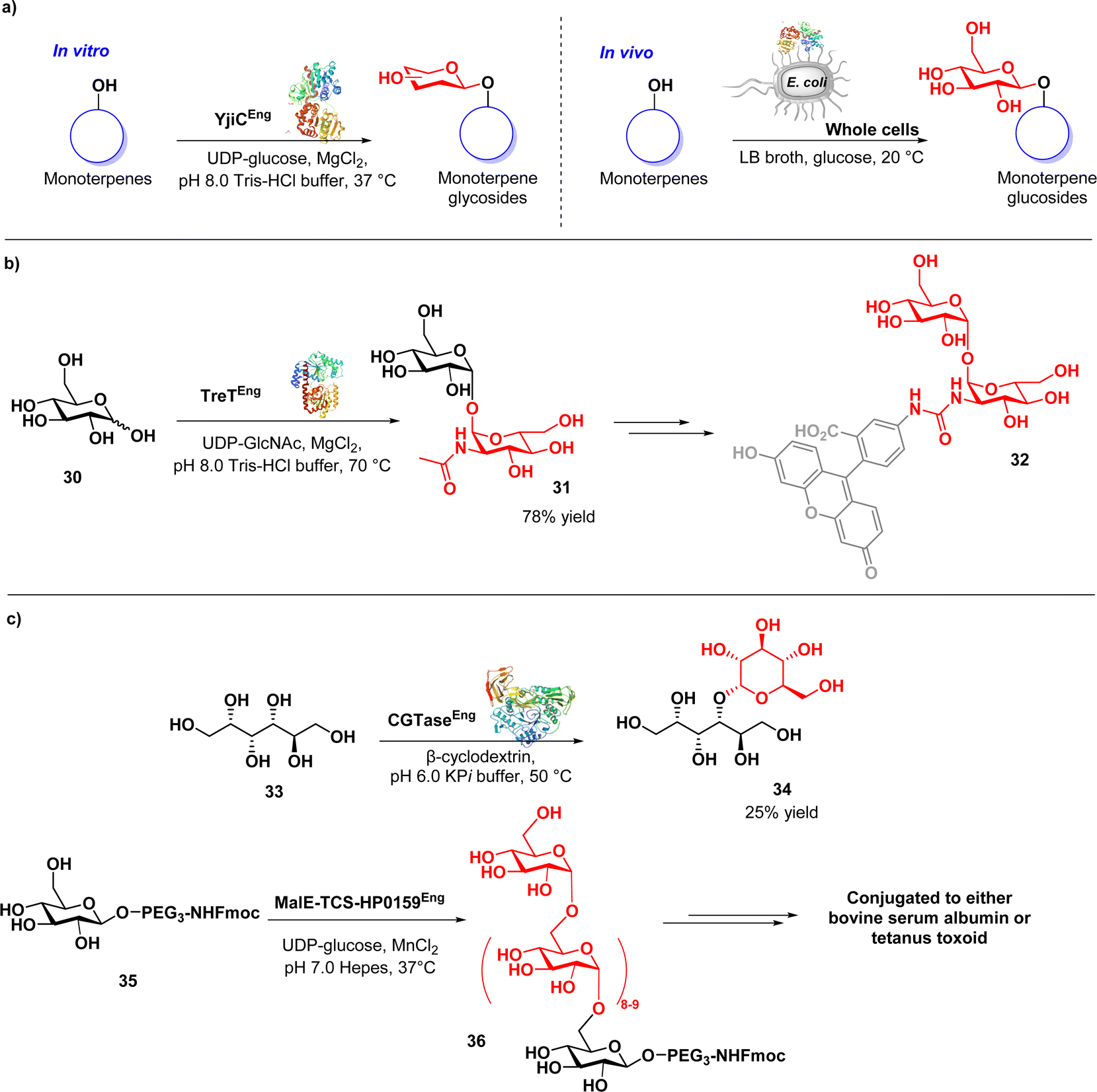 | ||
| Fig. 5 (a) Enzymatic synthesis of monoterpene O-glycosides by glycosyltransferases; (b) chemoenzymatic synthesis of trehalosamine derivatives via trehalose synthase-catalysed glycosylation; (c) chemoenzymatic transglycosylation using glycosyltransferases. | ||
In a study conducted by Groenevelt et al., a chemoenzymatic approach was used to synthesise trehalosamine (31) (Fig. 5b), an aminoglycoside antibiotic known for its activity against M. tuberculosis and M. smegmatis.81 This strategy was implemented to address the challenges associated with the isolation of 31 from natural sources or its chemical synthesis, resulting in improved yield, stereoselectivity, and overall efficiency. The method exploits T. tenax TreT, a trehalose-producing glycosyltransferase, to convert glucose analogues (30) into the corresponding trehalose analogues with complete 1,1-α,α-stereoselectivity (31). Trehalosamine was efficiently synthesized via a highly efficient one-step chemoenzymatic reaction, followed by chemical N-deacetylation, achieving a 50% isolated yield and an overall two-step yield of 39%. This yield significantly surpasses traditional chemical synthesis methods, which are typically characterized by lengthy and low-yield processes involving 8–11 steps, with yields usually below 10%. This method exhibits the ability to efficiently synthesise novel trehalosamine-based antibiotics as well as trehalose-based mycobacteria-specific imaging probes (32) capable of labelling mycobacterial cells.
In a study conducted by Haewpetch et al., a chemoenzymatic reaction utilizing cyclodextrin glucosyltransferase (CGTase) was employed to synthesize maltitol (34) (Fig. 5c) as an anticariogenic agent for dental care.82 The authors initially evaluated the polyol-acceptor specificity of CGTase with β-cyclodextrin, enabling the selection of an appropriate glucosyl acceptor, namely sorbitol (33). Through the chemoenzymatic approach, maltitol was successfully synthesized, achieving an improved yield of 25%. The synthesized maltitol demonstrated growth inhibitory activity against S. mutans, highlighting its potential as an anticariogenic agent. The use of glucosyltransferase was also reported in the synthesis of vaccines. Altman and co-workers conducted a study wherein they successfully prepared an α-1,6-glucan-based conjugate vaccine against H. pylori using a recombinant α-1,6-glucosyltransferase derived from the H. pylori strain (MalE-TCS-HP0159).83 Through a chemoenzymatic synthesis approach, the α-1,6-glucan chain was synthesised and then conjugated to a carrier protein. The resulting synthetic glycoconjugate demonstrated immunogenicity in rabbits and mice, eliciting a robust and specific IgG response. Furthermore, the post-immune sera obtained from rabbits that received the conjugates exhibited cross-reactivity. These findings highlight the versatility and effectiveness of the chemoenzymatic approach in both antibiotics and vaccine development, with the potential to contribute to overcoming antibiotic-resistant bacteria.
Aminoglycosides have a structural complexity that may limit the chemical modifications on the aminoglycoside scaffolds. Ban et al. successfully addressed this limitation by employing a one-pot enzymatic reaction84 to synthesise amikacin derivatives (38 and 39) (Fig. 6a).
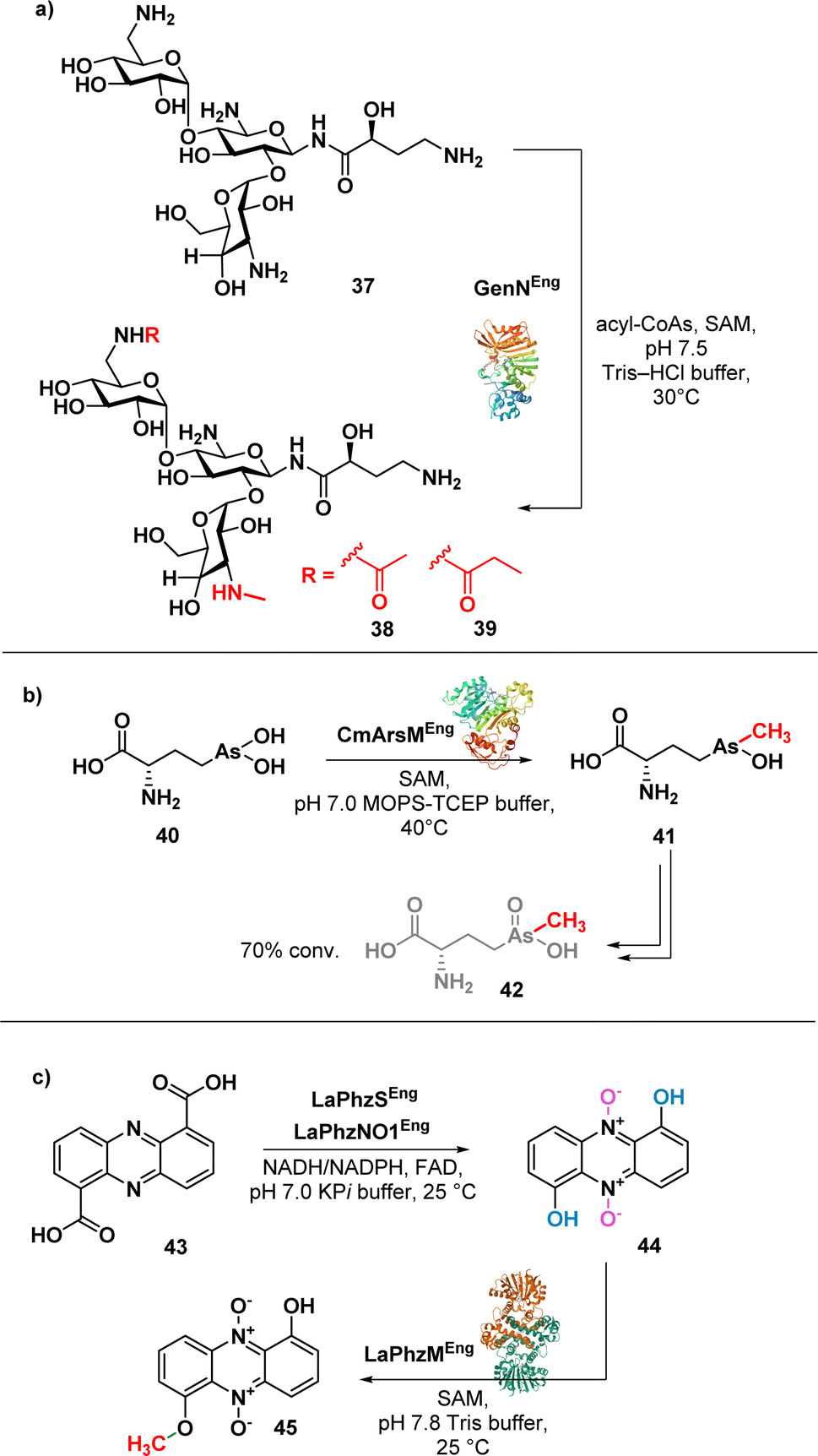 | ||
| Fig. 6 (a) Enzymatic synthesis of amikacin derivatives using transferases GenN; (b) enzymatic synthesis of arsinothricin (42); (c) enzymatic synthesis of myxin (45). | ||
The method involves the use of two transferases, 6′-N-acetyltransferase AAC(6′)-APH(2′′) and 3′′-N-methyltransferase GenN, facilitating the methylation at C3′′-amine position and the acylation at the C6′-amine of amikacin (37), respectively. The chemoenzymatic approach is efficient and selective, allowing the authors to obtain modified amikacin on both 6′ and 3′′-amino groups. Notably, 6′-N-acyl-3′′-N-methylated amikacin analogues exhibited improved antibacterial efficacy against amikacin-resistant pathogens and an improved toxicity profile. The regiospecific enzymatic approach presented in this study not only expands the chemical modification on aminoglycosides but also offers promising candidates for combating antibiotic-resistant pathogens.
Suzol and colleagues described a synthetic strategy for the synthesis of arsinothricin (AST) (42) (Fig. 6b), a potent broad-spectrum antibiotic, starting from 1,2-dichloroethane, with enzymatic methylation as the final step.85 The authors successfully carried out a three-step synthesis of the precursor compound, hydroxyarsinothricin (AST-OH), starting from 1,2-dichloroethane. Subsequently, AST-OH was reduced to its trivalent form, AST-OH(III) (40), in preparation for the subsequent enzymatic reaction. The trivalent 40 was then incubated with CmArsM, an As(III) S-adenosylmethionine methyltransferase derived from Cyanidioschyzon merolae. CmArsM catalysed the transfer of the S-methyl group from S-adenosyl methionine, resulting in the formation of the trivalent form of AST (41). Lastly, spontaneous oxidation of 41 yields the final product 42. This study not only demonstrated the synthetic pathways for AST but also highlighted the potential scalability of both the chemical and enzymatic approaches in AST synthesis for further drug development.
Jiang et al. reported a one-pot biosynthesis approach for the production of the antibiotic myxin (45) (Fig. 6c) from the direct precursor phenazine 1,6-dicarboxylic acid (43).86 This method involves the sequential use of two redox enzymes, LaPhzS and LaPhzNO1, along with the O-methyltransferase LaPhzM. Acid 43 was converted into dihydroxyphenazine through a decarboxylative hydroxylation, facilitated by the enzyme LaPhzS. Subsequently, LaPhzNO1 was added to the reaction mixture to catalyse the phenazine N-oxidation (44).87 Finally, O-methylation of 44 was achieved through LaPhzM, yielding compound 45.
This study demonstrated the broad substrate specificity of LaPhzM as it can use nonoxide, mono-oxide, or dioxide phenazines as substrates, highlighting the versatility of the enzyme in the chemoenzymatic synthesis of phenazine antibiotics. This one-pot biosynthesis approach has the potential to facilitate the efficient synthesis of phenazine antibiotics and the identification of enzymes in the chemoenzymatic synthesis of novel phenazine derivatives.
Daptomycin (46) (Fig. 7), a cyclic lipopeptide antibiotic, is considered a last-resort antibiotic for the treatment of Gram-positive bacterial infections. However, the emergence of daptomycin-resistant strains highlights the need for the development of daptomycin analogues. To address this issue, prenylated DAP were synthesised using the indole prenyltransferase, CdpNPT. Singh et al. performed a comprehensive analysis to assess the flexibility of this chemoenzymatic approach using diverse alkyl pyrophosphates as the prenyl donors.88 The results revealed that CdpNPT could transfer various alkyl groups to DAP with potential alkylation occurring at the N1, C2, C5, and C6 positions of the indole ring (Fig. 7). However, the study identified a limitation in the lack of regiospecificity of the alkyl substitution, which could impact the potency of the compounds. Later, the authors described the regiospecific synthesis of daptomycin derivatives using CdpNPT.89
 | ||
| Fig. 7 Chemoenzymatic synthesis of daptomycin derivatives by prenyltransferase CdpNPT. | ||
They evaluated the capacity of the enzymatic approach to produce regiospecific C6-modified DAP using alkyl and aryl pyrophosphates, including allyl, propargyl, cyclic alkene and aromatics. While successful prenylation of the substrate was achieved in most cases, exceptions were observed with but-2-en1-yl and benzyl groups.
These studies demonstrated the efficient late-stage diversification of a complex scaffold, underscoring the importance of applying biosynthetic strategies. It is noteworthy that Dap derivatives synthesised through this approach exhibited enhanced efficacy against DAP-resistant strains compared to the parent molecules.
Finally, Zeng et al. reported a chemoenzymatic synthesis of sialylated lactulose and their inhibitory effect against S. aureus.90 The authors developed an efficient one-pot two-enzyme sialylation system, using CMP-sialic acid synthetase and a sialyltransferase, PmST1 or Pd26ST for α2,3-linkage or α2,6-linkage, respectively (Fig. 8). The reaction used N-acetylneuraminic acid as the donor and lactulose as the acceptor substrate. The results demonstrated that this chemoenzymatic synthesis approach provides a promising synthetic path in the production of sialylated lactulose with improved antibacterial activity against S. aureus, indicating their potential therapeutic application.
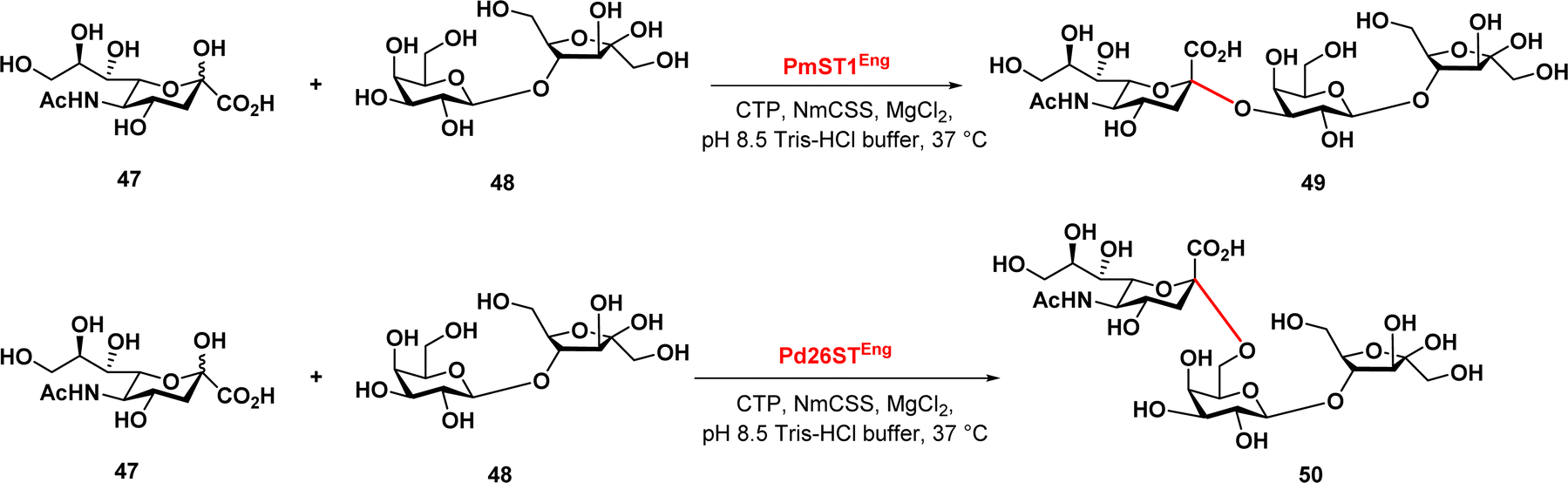 | ||
| Fig. 8 Enzymatic synthesis of sialylated lactuloses via sialyltransferases PmST1 and Pd26ST. | ||
4. Hydrolases [EC 3]
Hydrolases are a wide class of enzymes, which includes lipases, esterases, phosphatases, proteases, glycosylases, and GTPases, that use water to break chemical bonds. They can be considered a special class of transferases, where the donor group is transferred to water. Hydrolases can cleave C–O, C–N, O–P, and C–S bonds, and are commonly used in organic synthesis due to their broad substrate specificity, high stereoselectivity, and ability to hydrolyse a range of functional groups, including esters, amides, and nitriles. Moreover, many of these enzymes are reversible and, depending on the reaction conditions, may operate in the opposite direction, enabling the formation of these same functional groups.91 Unlike some enzymes, hydrolases do not require cofactors and can tolerate water-miscible solvents (e.g., DMSO, DMF). Additionally, hydrolases are often cheap and readily commercially available.92Lipases are widely utilized enzymes in biocatalysis and have emerged as prominent biocatalysts in organic synthesis. They are commonly employed for hydrolysis, esterification, and transesterification reactions.93 Recently, there has been growing interest in exploring lipase promiscuity, leading to unconventional reactions.94,95
Samsonowicz-Górski et al. reported a lipase-catalysed Knoevenagel–phospha-Michael reaction to synthesize β-phosphono malonates with antimicrobial properties. This research introduced a metal-free biocatalytic route for phosphorous–carbon bond formation. Candida cylindracea lipase (CCL) emerged as the most effective catalyst among the enzymes assessed. The process yielded the β-phosphonomalonitriles (51–54) (Fig. 9a) with up to 93% yield at 20 °C. Notably, only minimal amounts of the desired products were formed in the absence of the enzyme. Several selected compounds exhibited MIC values ranging from 0.2 to 1.4 μg mL−1 against a panel of E. coli strains.96
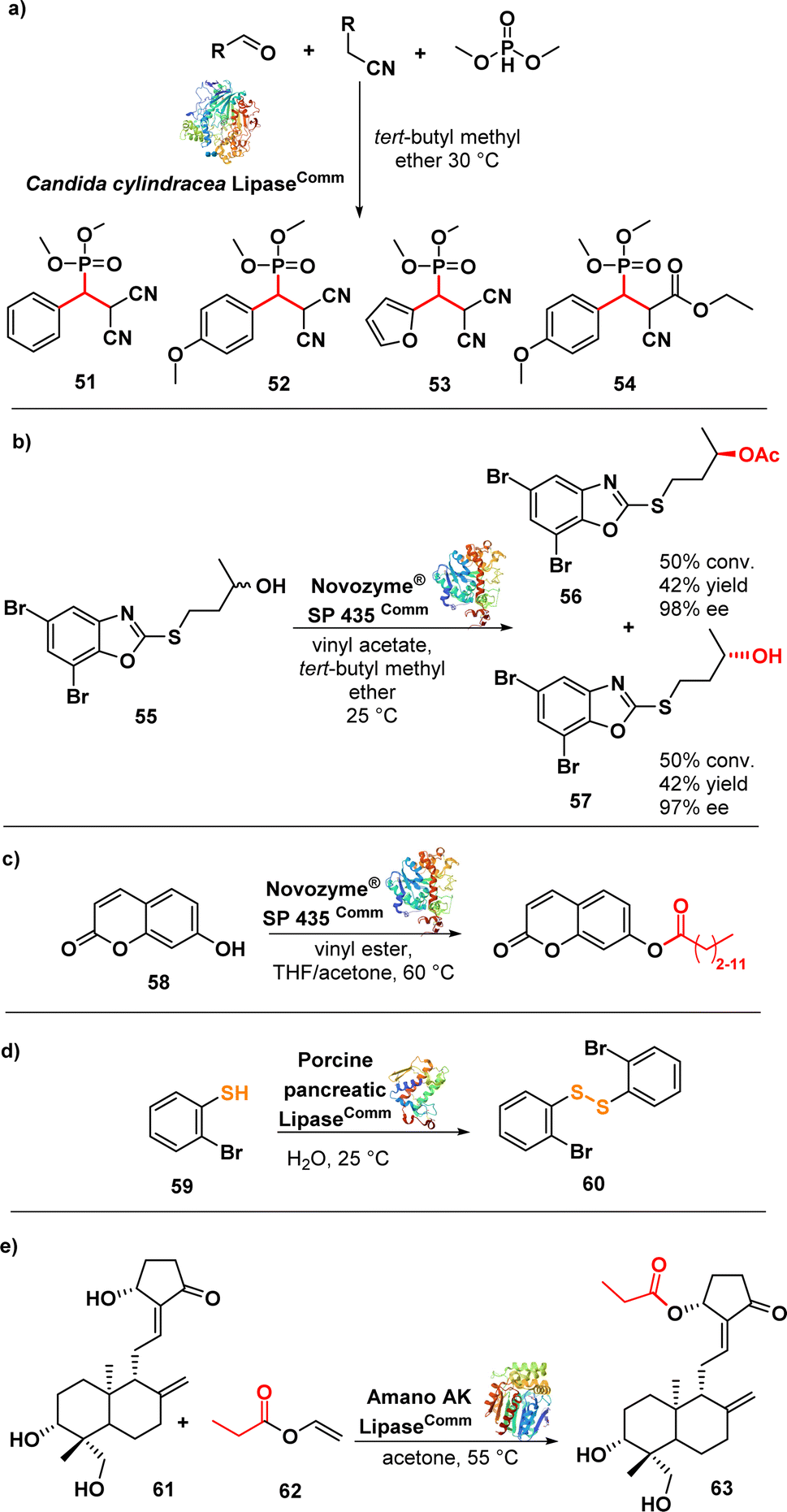 | ||
| Fig. 9 (a) Lipase-catalysed synthesis of β-phosphonomalononitriles; (b) lipase-catalysed transesterification of alcohol and kinetic resolution; (c) transesterification of umbelliferone with lipase; (d) biocatalytic synthesis of disulphide using lipase; (e) lipase-catalysed regioselective acylation of andrographolide. | ||
Łukowska-Chojnacka et al. described a lipase-catalysed kinetic resolution of benzoxazole derivatives endowed with antitubercular activity. They employed a two-step process involving lipase-catalysed transesterification of alcohols and subsequent enzymatic hydrolysis of esters to obtain enantiomerically enriched benzoxazoles. Novozyme® SP 435 (lipase from Candida antarctica-B immobilized on a macroporous acrylic resin) exhibited the highest catalytic performance and maintained the same stereopreference throughout both steps. The synthesized compounds were evaluated against a range of Mycobacterium strains, including drug-resistant ones. Compound 55 (Fig. 9b), in racemic form, displayed good activity against M. avium with a MIC of 12.5 μg mL−1. Interestingly, the isolated enantiomers, (−)-(R) and (+)-(S), did not exhibit any significant difference in activity compared to the racemic mixture. Nevertheless, these compounds did not show promising activity against other strains.97 Despite the limited efficacy of these derivatives, it is noteworthy that chemoenzymatic synthesis holds potential for developing drugs targeting M. tuberculosis and nontuberculous mycobacteria, addressing two significant challenges in the continuing emergence of AMR.98–101
The same lipase, Novozym® 435 (from Candida antarctica, CAL-B), was used in a biocatalytic process to produce umbelliferone ester derivatives (Fig. 9c). These esters were synthesized by acylating 7-hydroxy-2H-chromen-2-one (58) with various long chain vinyl esters, with the lipase serving as the catalyst. Notably, the conversion efficiency was higher for vinyl esters with shorter carbon chains (∼90%) compared to those with longer carbon chains (∼20%). Moreover, the resulting compounds exhibited promising activity against MRSA and several Gram-negative strains with MIC values ranging from 0.25 to 1 μg mL−1.102
Porcine pancreatic lipase (PPL) was investigated as a biocatalyst to synthesize diaryl disulphides, leveraging its capability to catalyse S–S bond formation via oxidative coupling of thiophenol (59) (Fig. 9d). The lipase-catalysed synthesis of the disulfides was carried out in water at room temperature. Interestingly, other lipases such as Candida antarctica (CAL-B) and Candida cylindracea lipase (CCL) did not yield the desired disulphides. Compound 60 exhibited potent inhibitory effects against highly drug-resistant clinical strains of S. aureus, including MRSA and VRSA, with MIC values ranging from 1–2 μg mL−1. Further evaluation demonstrated that compound 60 was non-cytotoxic to Vero cells, exhibited bactericidal effects against S. aureus ATCC 29213, and demonstrated activity comparable to vancomycin against S. aureus biofilm.103 The lipase Amano AK (from Pseudomonas fluorescens) was used in a regioselective and effective enzymatic synthesis of andrographolide (61) (Fig. 9e) derivatives. The lipase demonstrated high efficiency in esterifying the hydroxyl group at the C-14 carbon of andrographolide using acyl donors. Among the synthesized compounds, compound 62 exhibited the strongest activity against S. aureus and E. coli, with MIC values of 4 and 8 μg mL−1, respectively.104
Polymyxin antibiotics, such as polymyxin E (colistin), are natural cyclic lipopeptides that selectively target Gram-negative bacteria by binding to the lipid A moiety of lipopolysaccharides.105 Recent efforts have focused on finding less toxic alternatives to polymyxins, leading to the discovery of a highly effective macrocyclic peptide conjugate (66) (Fig. 10) with remarkable in vivo activity.106 Wood et al. developed a convenient chemoenzymatic synthesis method to create this chimeric bicyclic peptide antibiotic. Using the proteolytic enzyme ficin, they obtained the polymyxin fragment (65) by degrading colistin (64) via hydrolysis of an amide bond. This fragment (65) was then coupled with a second macrocycle synthesized via solid-phase peptide synthesis (SPPS). The resulting conjugates exhibited potent antibacterial activity against a panel of Gram-negative bacteria, including colistin-resistant E. coli strains, with MIC values ranging from 0.1 to 1.6 μg mL−1. As anticipated, the tested compounds did not exhibit activity against MRSA, a representative Gram-positive strain.107
 | ||
| Fig. 10 Chemoenzymatic synthesis of bicyclic chimera 66 by the proteolytic enzyme ficin. | ||
D-p-Hydroxyphenylglycine (D-HPG) (69) (Fig. 11a) serves as a vital precursor for semisynthetic β-lactam antibiotics like cephalexin, cefadroxil, and amoxicillin. Liu et al. described the whole-cell biocatalytic production of D-HPG from DL-hydroxyphenylhydantoin (DL-HPH) (67) in E. coli using two hydrolases, D-hydantoinase and D-carbamoylase. By finely regulating the expression of the D,D-carboxypeptidase dacB gene in E. coli, they successfully disrupted the peptidoglycan synthesis network and the cell wall structure, leading to increased permeability of the inner and outer membranes. This enhanced permeability allowed for greater substrate entry into the cells, resulting in a remarkable yield of 100% using the engineered cells. Importantly, the engineering undertaken aimed to facilitate the cellular penetration of the substrate, enabling its subsequent progression through the biocatalytic pathway involving the natural enzymes.108
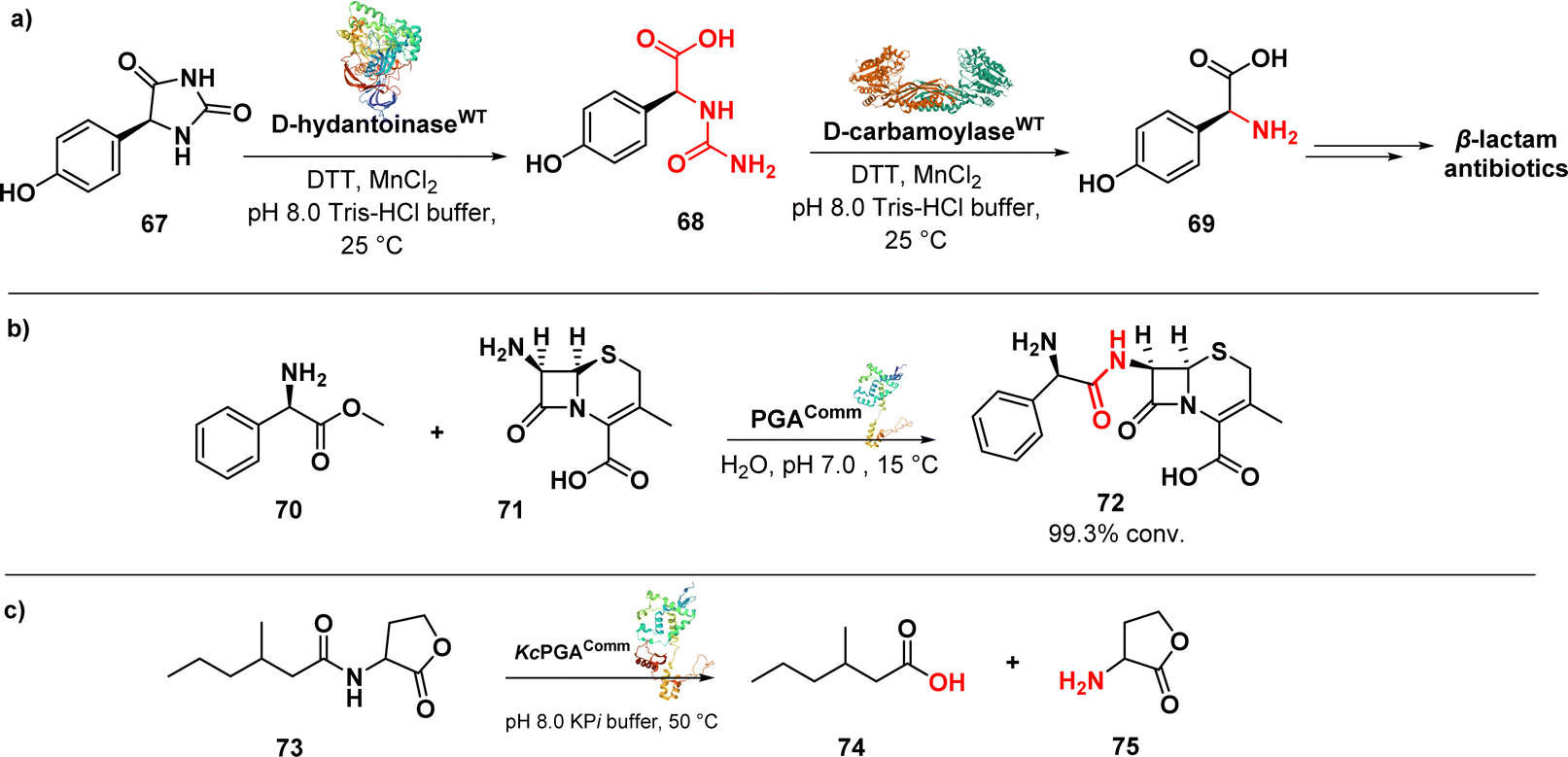 | ||
| Fig. 11 (a) Biocatalytic synthesis of D-p-hydroxyphenylglycine by D-hydantoinase and D-carbamoylase; (b) enzymatic synthesis of cephalexin catalysed by penicillin G acylase; (c) enzymatic reaction catalysed by KcPGA to degrade quorum sensing signal molecules. | ||
An efficient enzymatic synthesis of the β-lactam antibiotic cephalexin (72) (Fig. 11b) was also carried out using a hydrolase enzyme.
In this process, immobilized penicillin G acylase (PGA) served as the catalyst in a suspension aqueous solution system. Cephalexin was synthesized from 7-amino-3-deacetoxycephalosporanic acid (70) and D-phenylglycine methyl ester (71). Optimal conditions were established, resulting in a remarkable 99.3% conversion ratio of 7-amino-3-deacetoxycephalosporanic acid and a productivity of 200 mmol l−1 h−1. These values far exceeded those achieved by the homogeneous aqueous solution systems. Moreover, the acylase demonstrated excellent stability, retaining 95.4% of its initial activity even after 10 cycles of enzymatic synthesis.109
In addition to its known role, penicillin acylases were investigated for a new function, namely the cleavage of bacterial quorum sensing signal molecules acyl homoserine lactones (AHL) (73) (Fig. 11c). Kluyvera citrophila penicillin G acylase (KcPGA) was used to degrade AHL into fatty acids (74) and homoserine lactones (75). Molecular modelling studies supported the enzymatic findings, showing that AHLs fit precisely into the hydrophobic pocket of the enzyme's active site.110
AHLs serve as communication signal molecules among bacteria, and by reducing their accumulation, intercellular communication is disrupted, leading to a state known as quorum quenching. This approach holds promise for the development of sustainable antibacterial therapeutics, as it significantly reduces the likelihood of bacterial resistance.111
5. Ligases [EC 6]
Ligases, also referred to as synthetase enzymes, are responsible for the covalent linkage of two substrate molecules by forming new chemical bond, such as C–O, C–S, or C–N. This process requires energy, which is generally provided by the simultaneous hydrolysis of ATP into either ADP or AMP.112Hamed et al. conducted a study on the biocatalytic production of bicyclic β-lactams with three adjacent chiral centres. They used engineered crotonases to improve the efficiency and selectivity of the production process. The aim of the work was to produce (4,6)-disubstituted-t-carboxymethylproline (CMP) (79) (Fig. 12a) derivatives with the desired (4S,6R)-stereochemistry, similar to clinically used carbapenems, as well as C-4/C-6-trisubstituted products. The study revealed that employing a tandem system of engineered carboxymethylproline synthase and an alkylmalonyl-CoA forming enzyme (specifically, crotonyl-CoA carboxylase reductase) could enhance the formation of certain (4S,6S)-disubstituted-t-CMP derivatives. This approach also expanded the range of accepted substrates. By coupling an appropriately engineered CMPS with a malonyl CoA synthetase, starting from a pyrroline-5-carboxylate derivative (76) and an achiral C2-alkylated malonic acid derivative (77), the stereoselectivity of the CMPS-catalysed process could be enhanced. Carbapenam synthetase (CarA) was able to convert some of these derivatives into bicyclic β-lactams, demonstrating the potential of the Ccr-CMPS-CarA system for producing 1β-methyl-substituted carbapenams. Notably, some of these products showed improved hydrolytic stability compared to unsubstituted 1β-carbapenams. The researchers also found that by making structural substitutions to wild type CMPS, they could tune their bond forming capacity to produce 5-carboxymethylproline derivatives substituted at C-4 and C-6. Moreover, the application of tandem enzyme incubations not only enhanced stereocontrol but also broadened the spectrum of generated products. In summary, the study underscores the utility of tandem enzyme systems featuring engineered crotonases in the synthesis of asymmetric bicyclic β-lactams.113
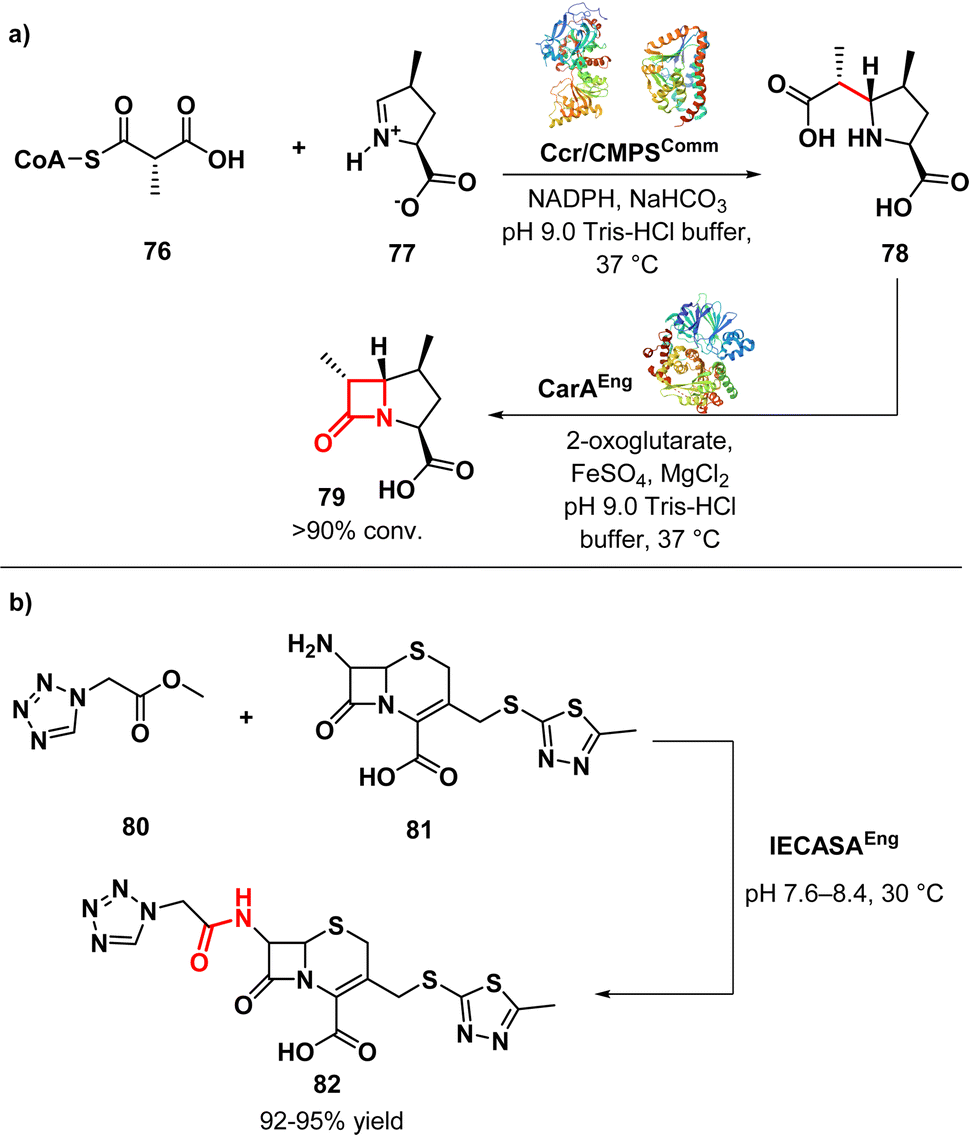 | ||
| Fig. 12 (a) Biocatalytic synthesis of bicyclic β-lactams tandem enzymes Ccr-CMPS-CarA; (b) enzymatic synthesis of cefazolin by cephalosporin-acid synthetase. | ||
Wang et al. developed a method for synthesizing the β-lactam antibiotic cefazolin (82) (Fig. 12b) using enzymatic acylation. They used immobilized cephalosporin-acid synthetase (IECASA) from a recombinant E. coli strain VKPM B-12316. To prevent the precipitation of 7-amino-3-(5-methyl-l,3,4-thiadiazol-2-yl)thiomethyl-3-cephem-4-carboxylic acid during the reaction, a stepwise pH gradient was applied based on investigations on the solubility of the components. This method resulted in a high yield of 82 (92–95%) under optimal conditions. The final reaction mixture contained 65–85 mg mL−1 of 82.114 The approach utilized in this study holds potential for developing other similar processes aimed at synthesizing novel β-lactam derivatives.
6. Conclusions
Against the backdrop of the escalating global threat posed by antimicrobial resistance, spotlighted by the ‘ESKAPE’ pathogens, the need for innovative solutions in antimicrobial drug discovery becomes evident. The substantial death toll and the potential surge in resistant bacterial infections by 2050 underscore the gravity of the situation. In the face of these challenges, biocatalysis emerges as a promising and sustainable pathway to overcome the limitations of conventional synthetic methods in antibacterial drug synthesis and drug discovery. Offering an eco-friendly approach, biocatalysis has demonstrated success in drug synthesis, highlighting potential for cost-efficiency and sustainability. Amidst this urgent need for transformative solutions, all chemical fields find themselves at a crossroads, with sustainability and environmental responsibility at the forefront of global concerns. The utilization of catalysts stands as a key strategy for achieving faster, more efficient chemical reactions, aiming to enhance product yield while minimizing unwanted by-products. While chemocatalysis currently dominates many applications, including pharmaceuticals, biocatalysis presents a sustainable alternative to traditional chemical processes, as it holds the promise of reduced waste, increased energy efficiency, and improved product quality. Despite these advantages, there exists significant reluctance to adopt biocatalysis or chemoenzymatic catalysis within the drug discovery cycle, particularly in the early stages of development. Acknowledging the pivotal role of biocatalysis in these early stages, this review underscores its capacity to carve out new, environmentally conscious, and more efficient synthetic routes, specifically in the exploration and synthesis of novel antibacterial compounds.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
G. F. S. F.: conceptualization, data curation, investigation, review and editing. S. H. K.: conceptualization, data curation, investigation, writing, review and editing. D. C.: conceptualization, formal analysis, writing, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. F. S. F. and S. H. K. acknowledge the University of London for financial support (Maplethorpe Postdoctoral Fellowships). D. C. and G. F. S. F. acknowledge European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101027065 for financial support to G. F. S. F.Notes and references
- S. Y. Tan and Y. Tatsumura, Singap. Med. J., 2015, 56, 366–367 CrossRef PubMed.
- R. Gaynes, Emerg. Infect. Dis., 2017, 23, 849–853 CrossRef.
- M. F. Chellat, L. Raguž and R. Riedl, Angew. Chem., Int. Ed., 2016, 55, 6600–6626 CrossRef PubMed.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- D. G. J. Larsson and C. F. Flach, Nat. Rev. Microbiol., 2022, 20, 257–269 CrossRef.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef PubMed.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1–12 Search PubMed.
- J. N. Pendleton, S. P. Gorman and B. F. Gilmore, Expert Rev. Anti Infect. Ther., 2013, 11, 297–308 CrossRef PubMed.
- Antimicrobial Resistance Collaborators, Lancet, 2022, 399, 629–655 CrossRef PubMed.
- GBD 2019 Diseases and Injuries Collaborators, Lancet, 2020, 396, 17–23 CrossRef.
- The Review on Antimicrobial Resistance, Chaired by Jim O'Neill: Tackling a Crisis for the Health and Wealth of Nations, 2014.
- L. Lansbury, B. Lim, V. Baskaran and W. S. Lim, J. Infect., 2020, 81, 266–275 CrossRef.
- J. A. Bengoechea and C. G. Bamford, EMBO Mol. Med., 2020, 12, e12560 CrossRef.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microbiol., 2019, 10, 539 CrossRef.
- P. Beyer and S. Paulin, ACS Infect. Dis., 2020, 6, 1289–1291 CrossRef.
- M. S. Butler, V. Gigante, H. Sati, S. Paulin, L. Al-Sulaiman, J. H. Rex, P. Fernandes, C. A. Arias, M. Paul, G. E. Thwaites, L. Czaplewski, R. A. Alm, C. Lienhardt, M. Spigelman, L. L. Silver, N. Ohmagari, R. Kozlov, S. Harbarth and P. Beyer, Antimicrob. Agents Chemother., 2022, 66, e0199121 CrossRef PubMed.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed.
- G. H. Talbot, A. Jezek, B. E. Murray, R. N. Jones, R. H. Ebright, G. J. Nau, K. A. Rodvold, J. G. Newland, H. W. Boucher and The Infectious Diseases Society of America, Clin. Infect. Dis., 2019, 69, 1–11 CrossRef.
- M. S. Butler, I. R. Henderson, R. J. Capon and M. A. T. Blaskovich, J. Antibiot., 2023, 76, 431–473 CrossRef PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- F. Zhao, D. Masci, E. Tomarelli and D. Castagnolo, Synthesis, 2020, 52, 2948–2961 Search PubMed.
- J. Schwarz, K. Rosenthal, R. Snajdrova, M. Kittelmann and S. Lütz, Chimia, 2020, 74, 368–377 CrossRef PubMed.
- A. Kinner, P. Nerke, R. Siedentop, T. Steinmetz, T. Classen, K. Rosenthal, M. Nett, J. Pietruszka and S. Lütz, Biomedicines, 2022, 10, 964 CrossRef PubMed.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef PubMed.
- J. I. Ramsden, S. C. Cosgrove and N. J. Turner, Chem. Sci., 2020, 11, 11104–11112 RSC.
- J. Chapman, A. E. Ismail and C. Z. Dinu, Catalysts, 2018, 8, 238 CrossRef.
- M. A. Emmanuel, S. G. Bender, C. Bilodeau, J. M. Carceller, J. S. DeHovitz, H. Fu, Y. Liu, B. T. Nicholls, Y. Ouyang, C. G. Page, T. Qiao, F. C. Raps, D. R. Sorigué, S. Z. Sun, J. Turek-Herman, Y. Ye, A. Rivas-Souchet, J. Cao and T. K. Hyster, Chem. Rev., 2023, 123, 5459–5520 CrossRef.
- H. Yang, H. Yu, I. A. Stolarzewicz and W. Tang, Chem. Rev., 2023, 123, 9397–9446 Search PubMed.
- R. N. Patel, ACS Catal., 2011, 1, 1056–1074 Search PubMed.
- R. N. Patel, Coord. Chem. Rev., 2008, 252, 659–701 CrossRef.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem, 2018, 2, 409–421 CrossRef.
- S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. P. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- P. Intasian, K. Prakinee, A. Phintha, D. Trisrivirat, N. Weeranoppanant, T. Wongnate and P. Chaiyen, Chem. Rev., 2021, 121, 10367–10451 CrossRef CAS PubMed.
- S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715–735 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef PubMed.
- J. A. McIntosh and A. E. Owens, Curr. Opin. Green Sustainable Chem., 2021, 29, 100448 CrossRef.
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2022, 26, 1878–1899 CrossRef.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CrossRef.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef.
- J. B. Pyser, S. Chakrabarty, E. O. Romero and A. R. H. Narayan, ACS Cent. Sci., 2021, 7, 1105–1116 CrossRef.
- E. Romero, B. S. Jones, B. N. Hogg, A. R. Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021, 60, 16824–16855 CrossRef.
- R. J. Young, S. L. Flitsch, M. Grigalunas, P. D. Leeson, R. J. Quinn, N. J. Turner and H. Waldmann, JACS Au, 2022, 2, 2400–2416 CrossRef.
- F. Xu, Ind. Biotechnol., 2005, 1, 38–50 CrossRef.
- A. T. Martínez, F. J. Ruiz-Dueñas, S. Camarero, A. Serrano, D. Linde, H. Lund, J. Vind, M. Tovborg, O. M. Herold-Majumdar, M. Hofrichter, C. Liers, R. Ullrich, K. Scheibner, G. Sannia, A. Piscitelli, C. Pezzella, M. E. Sener, S. Kılıç, W. J. H. van Berkel, V. Guallar, M. F. Lucas, R. Zuhse, R. Ludwig, F. Hollmann, E. Fernández-Fueyo, E. Record, C. B. Faulds, M. Tortajada, I. Winckelmann, J.-A. Rasmussen, M. Gelo-Pujic, A. Gutiérrez, J. C. del Río, J. Rencoret and M. Alcalde, Biotechnol. Adv., 2017, 35, 815–831 CrossRef PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- C. C. Forneris and M. R. Seyedsayamdost, Angew. Chem., Int. Ed., 2018, 57, 8048–8052 CrossRef CAS.
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. Robinson, R. D. Süssmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640–653 CrossRef CAS.
- C. C. Forneris, S. Ozturk, M. I. Gibson, E. J. Sorensen and M. R. Seyedsayamdost, ACS Chem. Biol., 2017, 12, 2248–2253 CrossRef CAS PubMed.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895 CrossRef CAS.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, E5052–E5061 CrossRef CAS PubMed.
- A. Okano, A. Nakayama, K. Wu, E. A. Lindsey, A. W. Schammel, Y. Feng, K. C. Collins and D. L. Boger, J. Am. Chem. Soc., 2015, 137, 3693–3704 CrossRef CAS.
- A. Okano, A. Nakayama, K. Wu, E. A. Lindsey, A. W. Schammel, Y. Feng, K. C. Collins and D. L. Boger, J. Am. Chem. Soc., 2015, 137, 3693–3704 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, E5052–E5061 CrossRef.
- J. Tailhades, Y. Zhao, Y. T. C. Ho, A. Greule, I. Ahmed, M. Schoppet, K. Kulkarni, R. J. A. Goode, R. B. Schittenhelm, J. J. De Voss and M. J. Cryle, Angew. Chem., Int. Ed., 2020, 59, 10899–10903 CrossRef PubMed.
- E. Olchowik-Grabarek, F. Mies, S. Sekowski, A. T. Dubis, P. Laurent, M. Zamaraeva, I. Swiecicka and V. Shlyonsky, Biochim. Biophys. Acta, 2022, 1864, 184011 CrossRef PubMed.
- D. Nozawa, A. Matsuyama and T. Furuya, Bioorg. Med. Chem. Lett., 2022, 73, 128908 CrossRef.
- M. L. Nóbile, A. M. Stricker, A. M. Iribarren and E. S. Lewkowicz, J. Biotechnol., 2021, 327, 36–42 CrossRef.
- C. R. Zwick III, M. B. Sosa and H. Renata, J. Am. Chem. Soc., 2021, 143, 1673–1679 CrossRef.
- L. Brandi, A. Lazzarini, L. Cavaletti, M. Abbondi, E. Corti, I. Ciciliato, L. Gastaldo, A. Marazzi, M. Feroggio, A. Fabbretti, A. Maio, L. Colombo, S. Donadio, F. Marinelli, D. Losi, C. O. Gualerzi and E. Selva, Biochemistry, 2006, 45, 3692–3702 CrossRef PubMed.
- J. Li and H. Renata, J. Org. Chem., 2021, 86, 11206–11211 CrossRef.
- Z. Feng, D. Chakraborty, S. B. Dewell, B. V. B. Reddy and S. F. Brady, J. Am. Chem. Soc., 2012, 134, 2981–2987 CrossRef PubMed.
- F. Kudo, A. Mori, M. Koide, R. Yajima, R. Takeishi, A. Miyanaga and T. Eguchi, Biosci. Biotechnol. Biochem., 2021, 85, 108–114 CrossRef PubMed.
- D. Righi, R. Huber, A. Koval, L. Marcourt, S. Schnee, A. Le Floch, V. Ducret, R. Perozzo, C. C. Ruvo, N. Lecoultre, E. Michellod, S. N. Ebrahimi, E. Rivara-Minten, V. L. Katanaev, K. Perron, J. L. Wolfender, K. Gindro and E. F. Queiroz, J. Nat. Prod., 2020, 83, 2347–2356 CrossRef.
- D. G. J. Larsson and C. F. Flach, Nat. Rev. Microbiol., 2022, 20, 257–269 CrossRef.
- C. M. Oral, M. Ussia and M. Pumera, Small, 2022, 18, 2202600 CrossRef.
- Q. Wang, W. Chen, C. Ma, S. Chen, X. Liu and F. Liu, Int. J. Biol. Macromol., 2022, 222, 509–520 CrossRef PubMed.
- P. E. V. Paul, V. Sangeetha and R. G. Deepika, in Recent Developments in Applied Microbiology and Biochemistry, Academic Press, 2019, pp. 107–125 Search PubMed.
- Z.-C. Wu and D. L. Boger, Acc. Chem. Res., 2020, 53, 2587–2599 CrossRef.
- A. Nakayama, A. Okano, Y. Feng, J. C. Collins, K. C. Collins, C. T. Walsh and D. L. Boger, Org. Lett., 2014, 16, 3572–3575 CrossRef.
- A. Okano, A. Nakayama, A. W. Schammel and D. L. Boger, J. Am. Chem. Soc., 2014, 136, 13522–13525 CrossRef PubMed.
- M. J. Moore, P. Qin, D. J. Keith, Z. C. Wu, S. Jung, S. Chatterjee, C. Tan, S. Qu, Y. Cai, R. L. Stanfield and D. L. Boger, J. Am. Chem. Soc., 2023, 145, 12837–12852 CrossRef.
- R. C. James, J. G. Pierce, A. Okano, J. Xie and D. L. Boger, ACS Chem. Biol., 2012, 7, 797–804 CrossRef PubMed.
- P. Bashyal, R. P. Pandey, S. B. Thapa, M. K. Kang, C. J. Kim and J. K. Sohng, ACS Omega, 2019, 4, 9367–9375 CrossRef.
- J. M. Groenevelt, L. M. Meints, A. I. Stothard, A. W. Poston, T. J. Fiolek, D. H. Finocchietti, V. M. Mulholand, P. J. Woodruff and B. M. Swarts, J. Org. Chem., 2018, 83, 8662–8667 CrossRef.
- P. Haewpetch, P. Rudeekulthamrong and J. Kaulpiboon, Biomolecules, 2022, 12, 167 CrossRef.
- E. Altman, V. Chandan, B. A. Harrison, M. Schur, M. F. Goneau, J. Li and M. Gilbert, Glycobiology, 2022, 32, 691–700 CrossRef CAS.
- Y. H. Ban, M. C. Song, J. H. Jeong, M. S. Kwun, C. R. Kim, H. S. Ryu, E. Kim, J. W. Park, D. G. Lee and Y. J. Yoon, Front. Microbiol., 2021, 12, 725916 CrossRef PubMed.
- S. H. Suzol, A. H. Howlader, A. E. Galván, M. Radhakrishnan, S. F. Wnuk, B. P. Rosen and M. Yoshinaga, J. Nat. Prod., 2020, 83, 2809–2813 CrossRef CAS.
- J. Jiang, D. G. Beltran, A. Schacht, S. Wright, L. Zhang and L. Du, ACS Chem. Biol., 2018, 13, 1003–1012 CrossRef CAS.
- Y. Zhao, G. Qian, Y. Ye, S. Wright, H. Chen, Y. Shen, F. Liu and L. Du, Org. Lett., 2016, 18, 2495–2498 CrossRef CAS PubMed.
- E. M. Scull, C. Bandari, B. P. Johnson, E. D. Gardner, M. Tonelli, J. You, R. H. Cichewicz and S. Singh, Appl. Microbiol. Biotechnol., 2020, 104, 7853–7865 CrossRef CAS.
- N. Mupparapu, Y. H. C. Lin, T. H. Kim and S. I. Elshahawi, Chem.–Eur. J., 2021, 27, 4176–4182 CrossRef PubMed.
- J. Zeng, Y. Hu, T. Jia, R. Zhang, T. Su, J. Sun, H. Gao, G. Li, M. Cao and M. Song, PLoS One, 2018, 13, e0199334 CrossRef PubMed.
- P. Coelho, F. Arnold and J. Lewis, in Comprehensive Organic Synthesis, 2014, 2nd edn, pp. 390–420 Search PubMed.
- A. R. Alcántara, M.-J. Hernaiz and J. V. Sinisterra, in Comprehensive Biotechnology, Academic Press, 2011, 2nd edn, pp. 309–331 Search PubMed.
- P. Chandra, Enespa, R. Singh and P. K. Arora, Microb. Cell Factories, 2020, 19, 169 CrossRef PubMed.
- B. P. Dwivedee, S. Soni, M. Sharma, J. Bhaumik, J. K. Laha and U. C. Banerjee, ChemistrySelect, 2018, 3, 2441–2466 CrossRef.
- A. Patti and C. Sanfilippo, Int. J. Mol. Sci., 2022, 23, 2675 CrossRef PubMed.
- J. Samsonowicz-Górski, D. Koszelewski, P. Kowalczyk, P. Śmigielski, A. Hrunyk, K. Kramkowski, A. Wypych, M. Szymczak, R. Lizut and R. Ostaszewski, Int. J. Mol. Sci., 2022, 23, 8819 CrossRef.
- E. Łukowska-Chojnacka, A. Kowalkowska and A. Napiórkowska, Chirality, 2018, 30, 457–468 CrossRef PubMed.
- M. D. Johansen, J. L. Herrmann and L. Kremer, Nat. Rev. Microbiol., 2020, 18, 392–407 CrossRef CAS.
- G. F. S. Fernandes, A. M. Thompson, D. Castagnolo, W. A. Denny and J. L. Dos Santos, J. Med. Chem., 2022, 65, 7489–7531 CrossRef CAS.
- D. Castagnolo, M. Radi, F. Dessì, F. Manetti, M. Saddi, R. Meleddu, A. De Logu and M. Botta, Bioorg. Med. Chem. Lett., 2009, 19, 2203–2205 CrossRef CAS.
- D. Castagnolo, F. Dessì, M. Radi and M. Botta, Tetrahedron: Asymmetry, 2007, 18, 1345–1350 CrossRef CAS.
- V. Soares, M. B. Marini, L. A. De Paula, P. S. Gabry, A. C. F. Amaral, C. A. Malafaia and I. C. R. Leal, Biotechnol. Lett., 2021, 43, 469–477 CrossRef CAS.
- I. S. Saima, A. G. Lavekar, M. Shukla, D. Equbal, A. K. Sinha and S. Chopra, Drug Dev. Res., 2019, 80, 171–178 CrossRef PubMed.
- H. S. Patil, D. D. Jadhav, A. Paul, F. A. Mulani, S. J. Karegaonkar and H. V. Thulasiram, Bioorg. Med. Chem. Lett., 2018, 28, 1132–1137 CrossRef CAS PubMed.
- V. Stojanoski, B. Sankaran, B. V. V. Prasad, L. Poirel, P. Nordmann and T. Palzkill, BMC Biol., 2016, 14, 81 CrossRef.
- A. Luther, M. Urfer, M. Zahn, M. Müller, S.-Y. Wang, M. Mondal, A. Vitale, J.-B. Hartmann, T. Sharpe, F. Lo Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M.-A. Westwood, N. Desjonquères, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithié, G. Upert, A. Lederer, P. Zbinden, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452–458 CrossRef CAS PubMed.
- T. M. Wood, C. J. Slingerland and N. I. Martin, J. Med. Chem., 2021, 64, 10890–10899 CrossRef CAS PubMed.
- Y. Liu, L. Zhu, W. Qi and B. Yu, Appl. Microbiol. Biotechnol., 2019, 103, 8839–8851 CrossRef CAS.
- Y. Fan, Y. Li and Q. Liu, Biotechnol. Appl. Biochem., 2021, 68, 136–147 CrossRef CAS PubMed.
- R. Mukherji, N. K. Varshney, P. Panigrahi, C. G. Suresh and A. Prabhune, Enzyme Microb. Technol., 2014, 56, 1–7 CrossRef CAS PubMed.
- C. Fuqua, M. R. Parsek and E. P. Greenberg, Annu. Rev. Genet., 2001, 35, 439–468 CrossRef CAS.
- G. L. Holliday, S. A. Rahman, N. Furnham and J. M. Thornton, J. Mol. Biol., 2014, 426, 2098–2111 CrossRef CAS PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, S. Warhaut, T. D. W. Claridge and C. J. Schofield, Commun. Chem., 2019, 2, 7 CrossRef PubMed.
- L. Wang, A. V Sklyarenko, D. Li, A. I. Sidorenko, C. Zhao, J. Li and S. V Yarotsky, Bioprocess Biosyst. Eng., 2018, 41, 1851–1867 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |