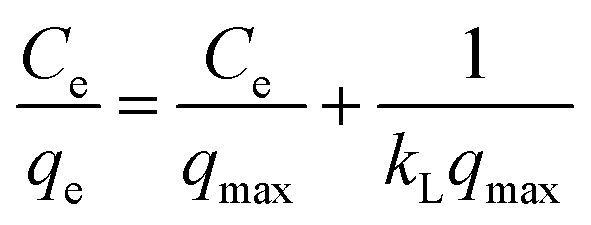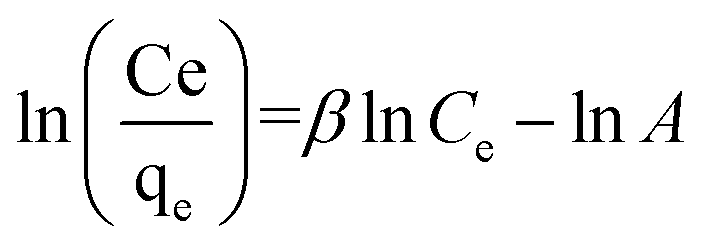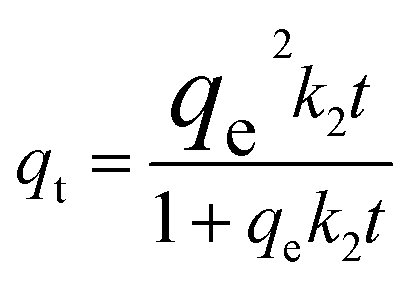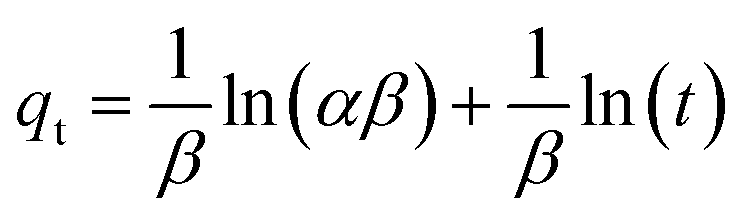DOI:
10.1039/D4RA04365K
(Review Article)
RSC Adv., 2024,
14, 26091-26102
Investigation of pantoprazole loading and release from a magnetic-coated chitosan-modified zirconium-based metal–organic framework (MOF) as a nanocarrier in targeted drug delivery systems
Received
14th June 2024
, Accepted 7th August 2024
First published on 19th August 2024
Abstract
This study reports a novel magnetic and porous nanocomposite, Fe3O4@CS@UIO-66-NH2(Zr), developed by growing a zirconium-based metal–organic framework on magnetite–chitosan. It is designed for targeted and delayed pantoprazole delivery, the nanocomposite exhibits pH-sensitive behavior and functions as an efficient nanocarrier. The synthesis process involved coating magnetite nanoparticles with chitosan, followed by the growth of UIO-66-NH2(Zr) on the coated nanoparticles. The nanocomposite demonstrated high drug loading efficiency (DLE) in acetate buffer (pH 5.0) and deionized water, with loading percentages of 79% and 75%, respectively, within 48 hours. The corresponding drug loading content (DLC) was approximately 14% and 10%. The Freundlich and Langmuir models accurately described the multilayer adsorption behavior of pantoprazole on the nanocomposite's active sites. BET and EDX-map analyses confirmed that the drug was loaded into the nanocomposite's pores and uniformly adsorbed on its surface. The drug release kinetics were best described by the pseudo-second-order model. Due to its porosity, magnetic properties, and favorable drug loading characteristics, the Fe3O4@CS@UIO-66-NH2(Zr) nanocomposite shows potential as an efficient targeted drug delivery system for in vivo applications.
1 Introduction
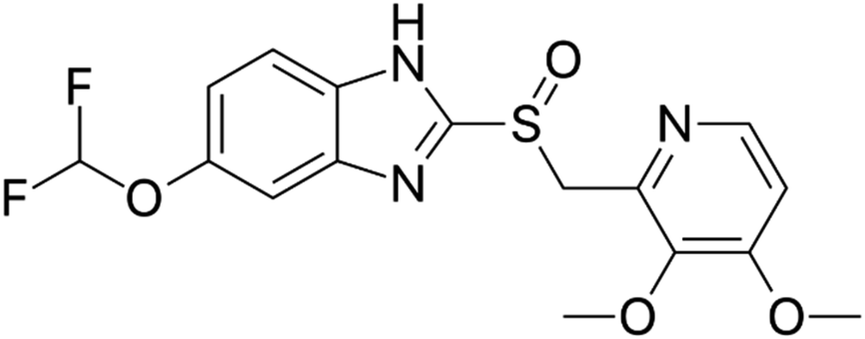
Proton pump inhibitors (PPIs) potently inhibit the H+/K+-ATPase enzyme in the secretory canaliculi of stimulated parietal cells, blocking the final common pathway for hydrochloric acid (HCl) secretion by irreversibly binding to free –SH groups on the ATPase enzyme. Like other PPIs, pantoprazole, 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl) methyl] sulfinyl]-benzimidazole sodium (Scheme 1), is an acid-activated prodrug that accumulates in the secretory canaliculi when parietal cells are secreting acid.1,2 Pantoprazole is rapidly absorbed, with peak plasma concentrations occurring approximately 2.5 hours after oral administration of 40 mg, experiencing minimum first-pass metabolism, and highly bioavailable, with a mean absolute bioavailability of 77%.3 PPIs have exhibited a range of other properties that may have therapeutic benefits, such as antioxidant activity, dual protection against gastric ulcers, that can reduce oxidative stress in gastrointestinal ulcers, providing both anti-acid and anti-inflammatory effects.4 Sevki Taskiran et al.'s study revealed that pantoprazole possesses antiepileptic properties by attenuating PTZ-induced seizures in rats, protecting neuroblastoma cells against PTZ-induced toxicity, and ameliorating memory impairment caused by epileptic seizures. These effects appear to be mediated by its antioxidant action and induction of BDNF release.5 Zhang et al.'s findings showed that pantoprazole through accumulation of polyubiquitinated proteins impaired proteasome function and subsequently activated the autophagy pathway as a compensatory mechanism to degrade protein aggregates is involved in antitumor activity.6 Furthermore, it has been suggested that pantoprazole and other PPIs reduce interferon-induced neurotoxicity by inhibiting the signal transducer and activator of the transcription protein 3 (STAT3) pathway.7 Despite all the above mentioned uses and benefits, it has been found that PPIs negatively affect bone metabolism, leading to changes in bone density and an increased risk of fractures. The exact mechanism of these adverse effects in vivo remains unknown. An osteopetrosis-like effect, suggested by Hyun et al., could explain the increased fracture risk but requires further confirmation.8–10 In this regard a bilayer tablet was introduced with the aim of formulating a novel gastro-retentive tablet to deliver a combination of a fixed dose of two drugs, Clarithromycin and Pantoprazole, reducing side effects and antibiotic resistance to eliminate Helicobacter pylori (H. pylori) in the gastrointestinal tract.11 Additionally, targeted drug delivery systems can help reduce the side effects of drugs by lowering the dosage to only the amount needed for the target tissue. Comoglu et al. prepared pantoprazole loaded microspheres by emulsion-solvent evaporation using two different types of enteric-coating polymers: Eudragit S 100 and hydroxypropyl methylcellulose phthalate for the purpose of delayed release formulation.12 Kishore Babu et al. developed gastro-resistant double-walled microspheres. The primary wall consists of mucoadhesive polymer HPMC and release-controlling polymer sodium alginate. The secondary wall, made of Eudragit RS 100, ensures sustained pantoprazole release for up to 14 hours at pH 7.4.13
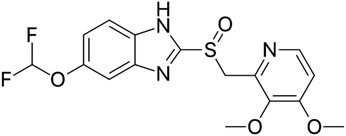 |
| | Scheme 1 Pantoprazole structure. | |
Biocompatible metal–organic frameworks (MOFs) synthesized from transition metal ions (e.g., Zr, Zn, Fe, Cu) exhibit high drug loading capacity, stable chemical structure, and ease of modification. These properties make them promising candidates for drug delivery systems (DDSs). Their potential is further enhanced by tunable pore shapes and sizes, which can be tailored by selecting various organic binders and metals.14–19 University of Oslo (UIO) series of metal organic frameworks consist of an interconnected network of zirconium ions and ligands, such as terephthalic acid. UIOs exhibit both water stability and biocompatibility, making them excellent candidates for drug delivery applications.20 Functionalized terephthalic acid linkers enable the creation of various MOF derivatives based on the UiO-66 structure, such as UiO-66-NO2, UiO-66-Br, UiO-66-NH2, and UiO-66-SO3H.21 Among these, UiO-66-NH2 stands out due to its large specific surface area and numerous active amino groups, making it suitable for drug delivery via dermal, oral, and intravenous routes.22 Core–shell compounds consist of diverse organic or inorganic materials that can encapsulate organic cores with mineral shells, or vice versa. The physical and chemical properties of these compounds are primarily influenced by the materials and structure of both the core and shell.23 Magnetite nanoparticles (Fe3O4) have been extensively studied for their potential in targeted drug delivery systems due to their magnetic responsiveness.24 However, their tendency to aggregate and high surface energy have hindered their clinical application.25,26 To address these issues, a hybrid system combining Fe3O4 nanoparticles with metal–organic frameworks (MOFs), specifically UiO-NH2, has been proposed. This approach facilitates the easy separation of magnetic nanoparticles using external magnetic fields,27–30 enabling targeted drug delivery.31 Given the hydrophilic nature of UiO-NH2, introducing an interlayer of biocompatible polymers like chitosan can bridge the compatibility gap between MOFs and magnetite. Chitosan, a natural biopolymer with hydroxyl groups, not only enhances the interaction between MOFs and magnetite32,33 but also imparts pH-sensitive and mucoadhesive properties, improving gastro-retentive functionality and overall drug delivery efficiency.11 This innovative combination of MOFs, magnetic nanoparticles, and biopolymers holds great promise for revolutionizing targeted drug delivery systems. Nasrabadi et al. synthesized the nanoporous metal–organic framework UiO-66 using a thermal solvent method to study drug delivery and release of pantoprazole. Evaluations in phosphate-buffered (pH 7.4) and acetate-buffered (pH 5.0) solutions showed 80% and 87% drug release, respectively, within 3 days.34 Shahin et al. synthesized a Fe3O4@UiO-66-NH2@PEI magnetic nanocomposite using a Schiff base reaction with glutaraldehyde. They studied the drug delivery and release of imatinib in buffer solutions at pH 3, 5.5, 7.2, and 9 at 37 °C over 48 hours, finding maximum release at pH 3 and 5.5.35 To our knowledge, there has been no research on using UiO-66-NH2(Zr) supported on magnetite–chitosan for targeted drug delivery. So far, various porous materials like super porous hydrogels, nanofibers, and polymeric nanoparticles have been used for loading and releasing pantoprazole.36–38
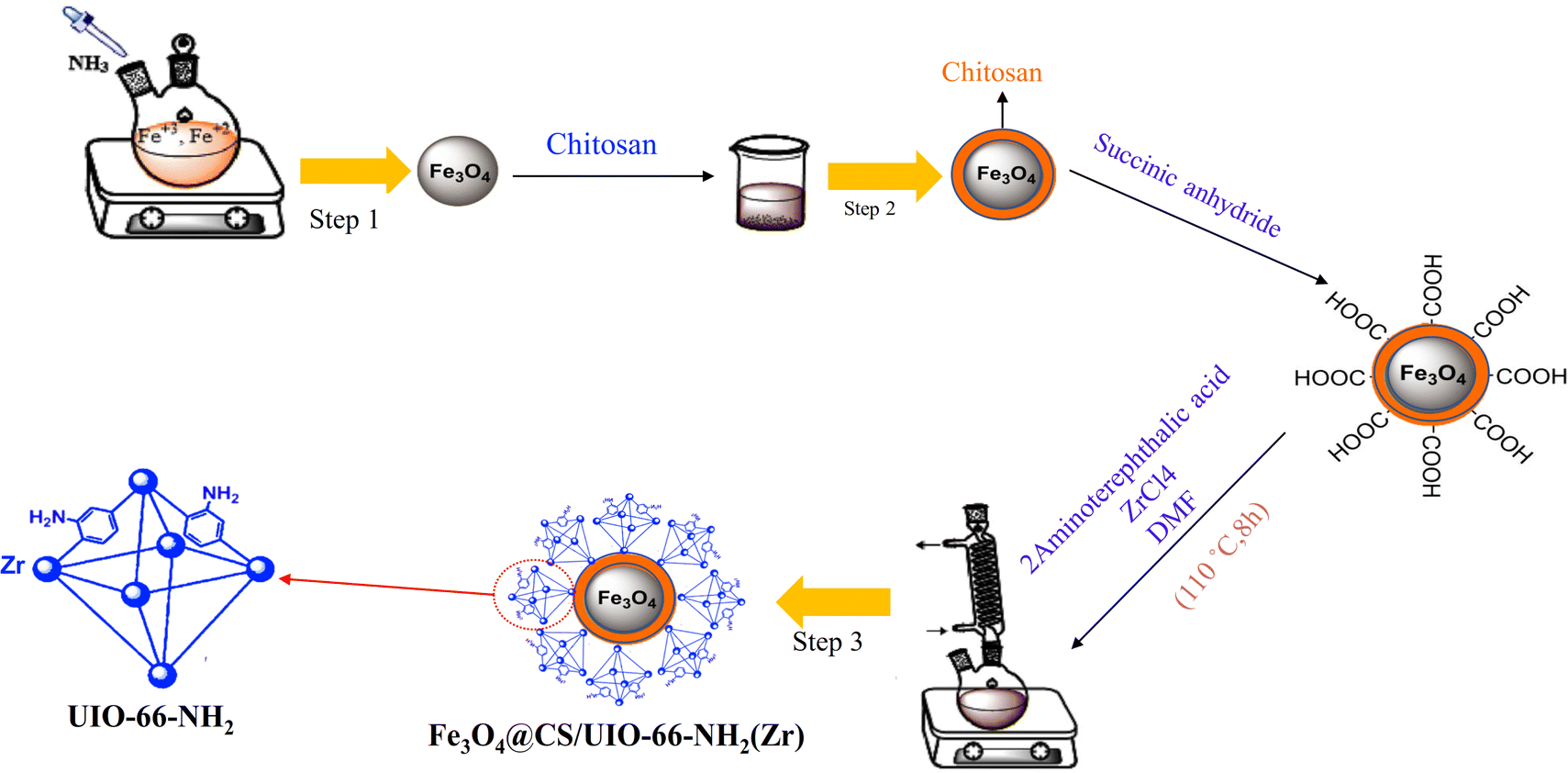
In targeted drug delivery, biocompatible nanocomposites that can direct the drug to the target tissue and reduce the drug dosage to minimize side effects can be very beneficial. According to this, in this study, a magnetic and porous nanocarrier (Fe3O4@CS/UiO-66-NH2(Zr)) was prepared by synthesizing UiO-66-NH2(Zr) onto magnetite–chitosan nanoparticles (Fe3O4@CS). To ensure proper MOF growth on the magnetic core, surface functionalization with –COOH groups is necessary. Some previous studies have functionalized magnetite structures using SiO2 followed by APTES, a two-step process involving SiO2 coating and subsequent APTES functionalization to introduce –NH2 groups, which are then modified with –COOH groups.35 However, in this study, chitosan was used to coat magnetite, eliminating the need for two synthesis steps and functionalizing the magnetite surface with –NH2 and –OH groups. This approach is advantageous for drug delivery applications, as SiO2 coating is not biocompatible, whereas chitosan provides a fully biocompatible layer. The synthesized nanocomposite was successfully employed for delivering pantoprazole (PNT). The magnetic nanocomposite was synthesized using a three-step procedure outlined in Scheme 2. The loading and release behavior of pantoprazole (PNT) were studied in PBS (phosphate buffer saline, pH 7.4) and AB (acetate buffer, pH 5.0) solutions.
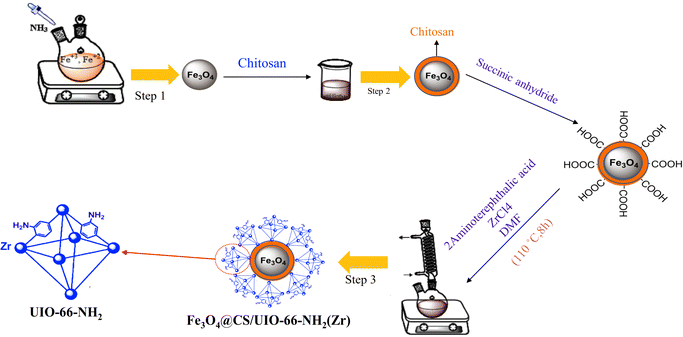 |
| | Scheme 2 Synthetic pathway of Fe3O4@CS/UIO-66-NH2(Zr). | |
2 Experimental
2.1 Chemicals and instrumentations
All chemicals included ferric chloride hexahydrate, ferrous chloride tetrahydrate, ammonia, succinic anhydride, glacial acetic acid, ethanol, DMF, 2-aminoterephthalic acid, sodium hydroxide, pantoprazole and zirconium chloride were purchased from Merck Co. (Germany) and Sigma-Aldrich (USA) and used without further purification. pH meter (Starious, Germany) used for pH adjustment. X-ray powder diffraction (XRD) patterns of samples were obtained with a Philips Xpert X-ray powder diffractometer (Cu Kα radiation, λ = 0.154056 nm). Fourier transform infrared spectra (FT-IR) were recorded as KBr pellets using a PerkinElmer 781 spectrophotometer. UV-visible spectra were recorded in the range 200–800 nm on Photonix Ar 2015 UV-visible spectrophotometer. Magnetization curves were recorded by Meghnatis Daghigh Kavir Co Vibrating sample magnetometer (VSM) at room temperature. The structure and surface morphology of the prepared samples were investigated by field emission scanning electron microscope (FE-SEM ZEISS-Sigma-300). TGA-DTA curves were recorded by a Rheometric Scientific Inc. 1998 thermal analysis apparatus under a N2 atmosphere. Nitrogen adsorption–desorption isotherms were measured at 196 °C using a Belsorp mini II (Microtrac Bel Crop Japan) automatic adsorption instrument after degassing the samples at 150 °C for 5 h.
2.2 Synthesis of Fe3O4 nano-particles (MNPs)
Magnetic nanoparticles (MNPs) were synthesized using a co-precipitation method.39,40 Initially, FeCl2·4H2O (0.5 g, 2.56 mmol) and FeCl3·6H2O (1 g, 3.7 mmol) were separately added to 12.5 mL and 10 mL of ionized water, respectively. The mixture was allowed to blend for 10 minutes at 50 °C. Subsequently, the temperature was gradually raised to 80 °C. Next, a 25% ammonia solution was meticulously added drop by drop to the vigorously stirred mixture, resulting in the formation of a black solid product. This product consists of magnetite nanoparticles (MNPs), which were subsequently separated using an external magnet. The MNPs were then washed three times with deionized water and ethanol before being dried under vacuum conditions at 60 °C for 12 hours.
2.3 Synthesis of Fe3O4@CS core–shell
Fe3O4@CS nano-spheres were synthesized by modifying the previous method.41 Initially, magnetite particles (0.5 g) and chitosan (0.5 g) were sonicated in a mixture containing 5 mL of deionized water and 50 mL of ethanol for 25 minutes. The resulting mixture was stirred for 45 minutes at 35 °C. The product was subsequently separated using an external magnet, washed three times with deionized water and ethanol, and then dried at room temperature for 24 hours.
2.4 Synthesis of Fe3O4@CS@UIO-66-NH2(Zr)
First, Fe3O4@CS (0.04 g) and succinic anhydride (0.5 g) were sonicated separately in 15 mL of deionized water for 25 minutes. The mixture was then refluxed at 80 °C for 4 hours under continuous stirring. The resulting product was washed multiple times with ethanol and deionized water and finally dried under vacuum conditions at 60 °C for 4 hours. Finally, 0.02 g of the obtained product was mixed with ZrCl4 (0.09 g) and 2-aminoterephthalic acid (0.07 g) in a solution containing 60 μL of acetic acid and 30 mL of DMF. The mixture was sonicated for 25 minutes and then refluxed at 110 °C for 8 hours to complete the reaction. The resulting sediment was washed several times with DMF and deionized water before being dried under vacuum conditions at 70 °C for 4 hours.35
2.5 PNT encapsulation in Fe3O4@CS@UIO-66-NH2(Zr)
To prepare a drug delivery system, 50 mg of Fe3O4@CS@UIO-66-NH2(Zr) was dispersed in 5 mL of acetate buffer at pH 5.0. Subsequently, 10 mg of pantoprazole was added to the suspension. The sealed mixture was stirred for 24 hours at room temperature. The resulting solid was washed with deionized water to remove any adsorbed PNT on the external surfaces and then dried at 60 °C. The drug loading efficiency (DLE), drug loading content (DLC), drug loading capacity (qe), and encapsulation efficiency (EE%) were calculated using the following formulas:42
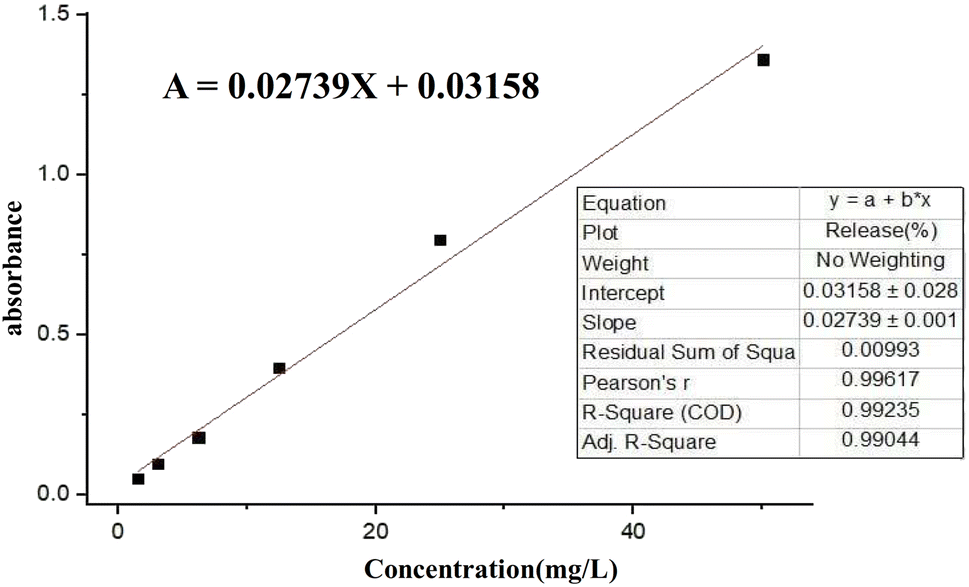
where; Co (mg L−1) is the initial drug (PNT) concentration, Ce (mg L−1) is the PNT concentration in supernatant, V(L) is the volume of the drug solution, and m(g) is the mass of the Fe3O4@CS/UIO-66-NH2(Zr). The calibration curve was used to obtain the adsorption concentration data (Fig. 1).
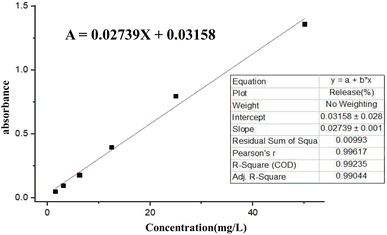 |
| | Fig. 1 Calibration curve of pantoprazole in deionized water (DIW). | |
2.6 Drug release from loaded-Fe3O4@CS@UIO-66-NH2(Zr)
The release of pantoprazole was conducted in buffer solutions at pH 7.4 and 5.0. The loaded Fe3O4@CS@UIO-66-NH2(Zr) (0.03 g) was suspended in a buffer medium (200 mL) under slow stirring. At specific intervals, 5 mL of the supernatant was collected, and an equal amount of fresh buffer solution was added to the suspension. All collected samples were separated by centrifugation at 5000 rpm for 5 minutes. The amount of released drug from the drug-loaded nanocarrier was measured by UV-visible spectrophotometry using the calibration curve (Fig. 1). The percentage of drug released (PR%) at consecutive time points (tn, hours) was calculated using the following eqn:
3 Results and discussion
3.1 Materials characterization
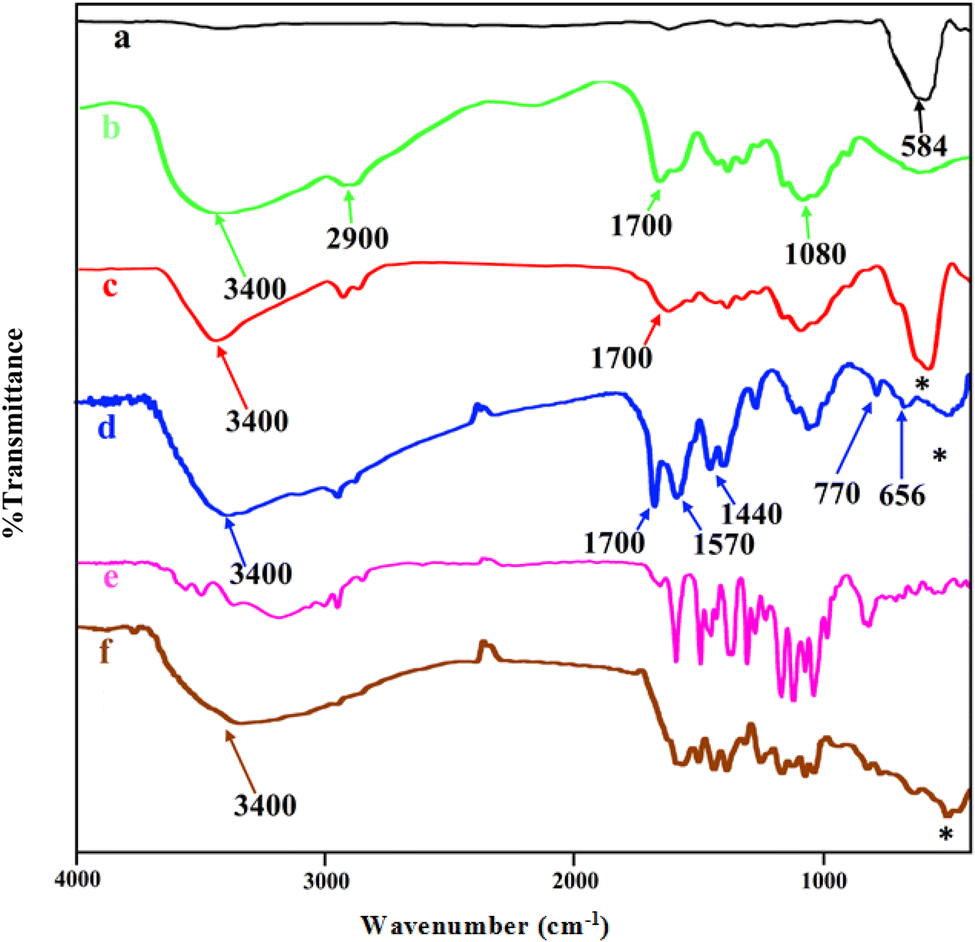
The FT-IR spectra of MNPs (Fe3O4), chitosan, core–shell magnetite–chitosan (Fe3O4@CS), Fe3O4@CS@UIO-66-NH2(Zr), pantoprazole and Fe3O4@CS@UIO-66-NH2(Zr)-pantoprazole are shown in Fig. 2a–f. In Fig. 1a, the absorption band around 584 cm−1 is assigned to the Fe–O stretching vibration43–45 denoted by the (*) symbol. This absorption band remains present in the magnetic-core nanocomposites (Fig. 2a, c, d and f) with a minor shift and decreased intensity. Fig. 2b displays the spectrum of chitosan. The absorption band around 2900 cm−1 corresponds to the C–H stretching of the backbone polymer, while the bands at 1400, 1033 and 1080 cm−1 are respectively assigned to the C–O stretching of the primary alcoholic group and amide I. Additionally, the absorption bands at 1700 and 3400 cm−1 correspond to N–H and amide II of the primary amine.41,46 The formation of Fe3O4@CS core–shell is confirmed by the absorption bands at approximately 1080, 1033, 1400, 1700, 2900, 3400, and 467 cm−1. Fig. 2d presents the spectrum of Fe3O4@CS@UIO-66-NH2(Zr). In this spectrum, the Fe–O stretching band is observed around 584 cm−1. Peaks located at approximately 656 and 770 cm−1 are assigned to the Zr–O bonds within the structure of the UIO-66-NH2 framework, indicating the successful growth of the MOF on the Fe3O4@CS magnetic core.47 Bands in the range of 1400−1700 cm−1 are related to asymmetric and symmetric stretching vibrations of carboxyl functional groups (–COO–).48–50 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic ring vibrational bands are detectable at 1570 cm−1, and the absorption band at 3400 cm−1 corresponds to the –NH2 stretching vibration of primary amines in the 2-aminoterephthalic acid. The band appearing around 2850−3050 cm−1 is attributed to the C–H stretching vibration of saturated and unsaturated carbons (Fig. 2d).50–52 Fig. 2e shows the spectrum of PNT and Fig. 2f shows the FT-IR spectrum of Fe3O4@CS@UIO-66-NH2(Zr)-PNT. (Fig. 2f) demonstrates the drug confinement inside the pores.
C aromatic ring vibrational bands are detectable at 1570 cm−1, and the absorption band at 3400 cm−1 corresponds to the –NH2 stretching vibration of primary amines in the 2-aminoterephthalic acid. The band appearing around 2850−3050 cm−1 is attributed to the C–H stretching vibration of saturated and unsaturated carbons (Fig. 2d).50–52 Fig. 2e shows the spectrum of PNT and Fig. 2f shows the FT-IR spectrum of Fe3O4@CS@UIO-66-NH2(Zr)-PNT. (Fig. 2f) demonstrates the drug confinement inside the pores.
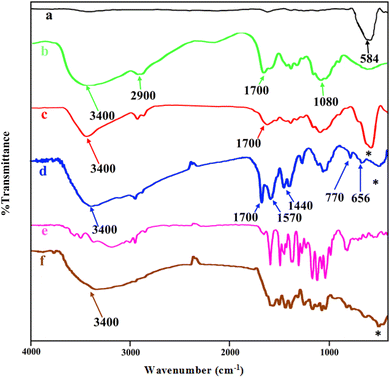 |
| | Fig. 2 FTIR spectra of (a) Fe3O4, (b) chitosan, (c) Fe3O4@CS, (d) Fe3O4@CS/UIO-66-NH2(Zr), (e) pantoprazole, (f) PNT-Fe3O4@CS/UIO-66-NH2(Zr). | |
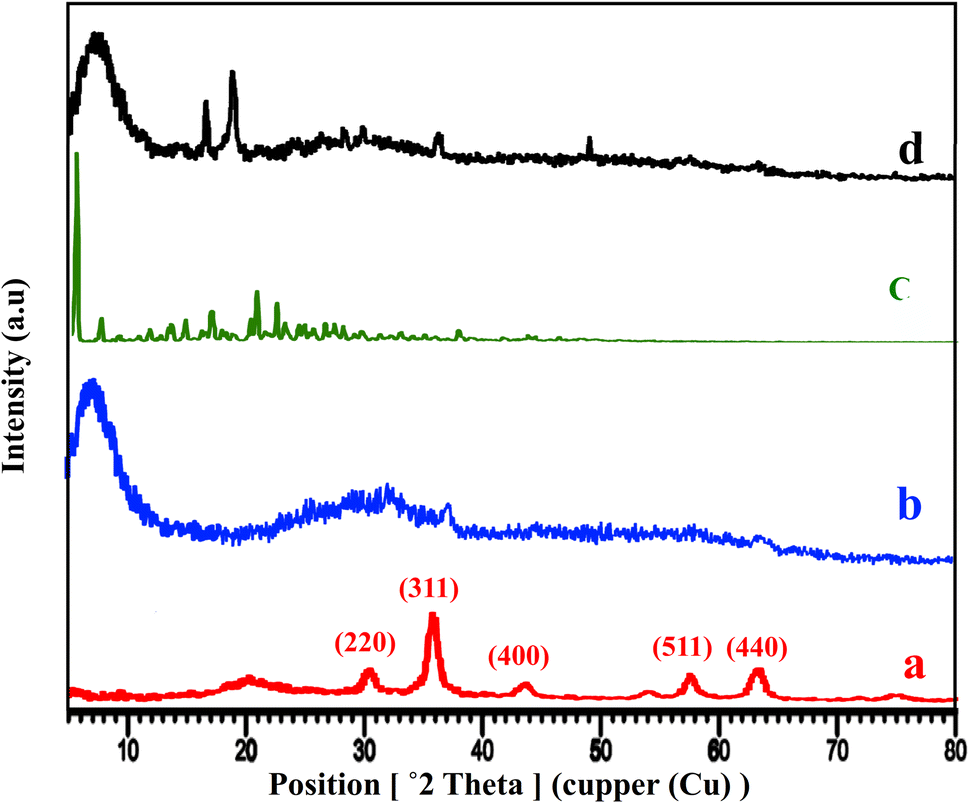
The XRD patterns of Fe3O4@CS, free-PNT, Fe3O4@CS/UIO-66-NH2(Zr), and PNT-Fe3O4@CS/UIO-66-NH2(Zr) are shown in Fig. 3. As displayed in Fig. 3a, the peaks at 2θ = 30.45°, 36.10°, 43.67°, 50.14°, 57.68°, and 64.32° correspond, respectively, to the (220), (311), (400), (422), (511), and (440) reflections of the crystalline structure of the Fe3O4 sample and chitosan broad peak centered around 20.29°.45,53,54 Additionally, a broad peak at 2θ = 5.16–28.68° is attributed to the UIO-66-NH2 (Fig. 3b).55 These results are consistent with previous findings.35,47 The crystalline nature of Fe3O4@CS/UIO-66-NH2(Zr) is preserved after the encapsulation of pantoprazole in the magnetically modified MOF. However, a slight shift and change in the intensity of Fe3O4@CS/UIO-66-NH2(Zr) and magnetite peaks are observed (Fig. 3d). Interestingly, the sharp shifted peaks of pantoprazole (Fig. 3c) around 20 indicate the filling of the pores with the drug.
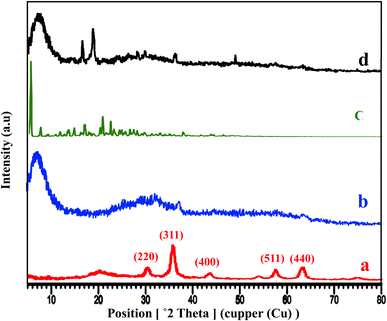 |
| | Fig. 3 XRD patterns of (a) Fe3O4@Cs, (b) Fe3O4@CS/UIO-66-NH2(Zr), (c) pantoprazole, (d) PNT-Fe3O4@CS/UIO-66-NH2(Zr). | |
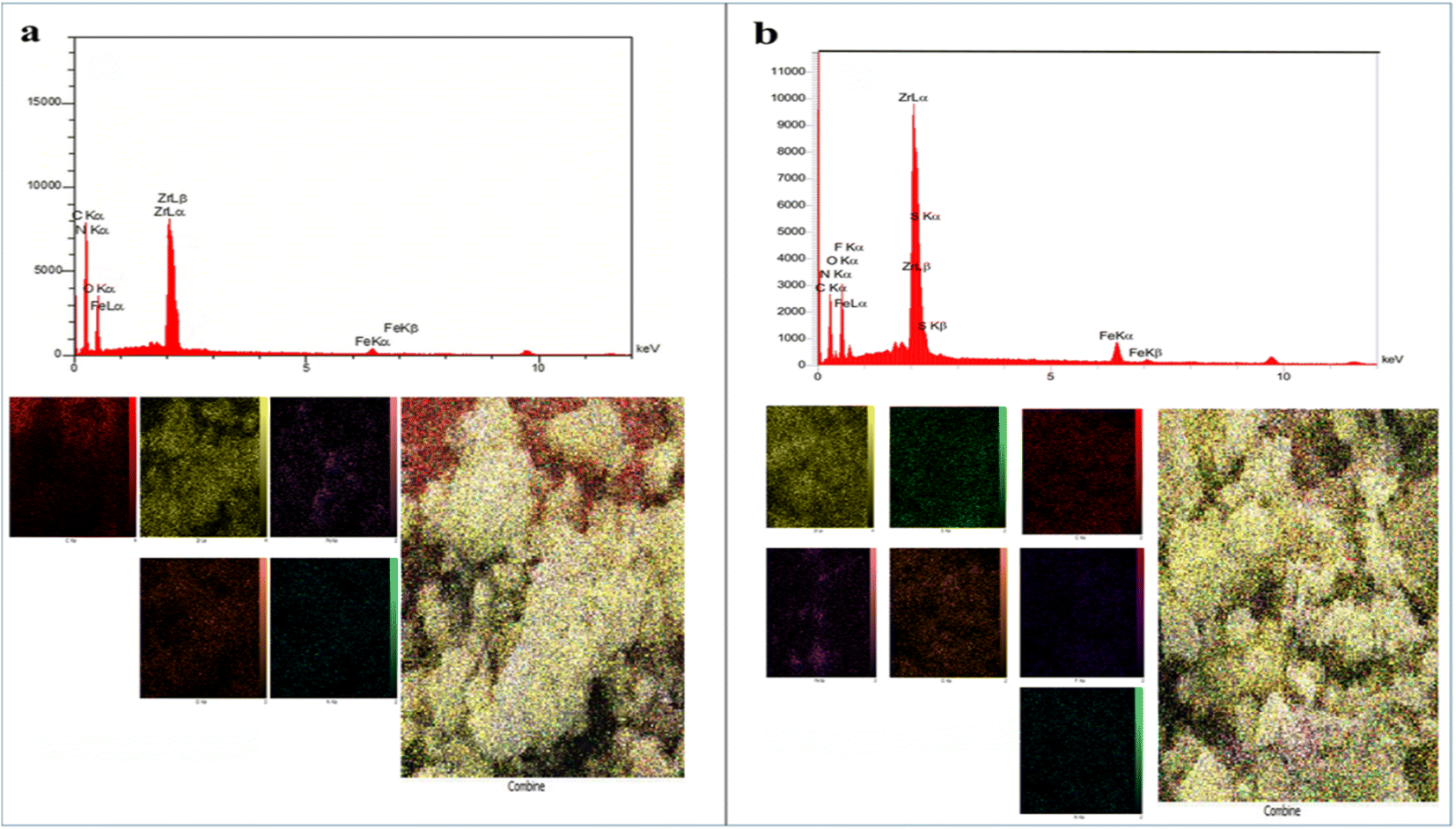
Field emission scanning electron microscopy (FE-SEM) images of Fe3O4@CS/UIO-66-NH2(Zr) (Fig. 4a and b) and PNT-Fe3O4@CS/UIO-66-NH2(Zr) (Fig. 4c and d) were obtained to determine the morphology and particle size. The nanoparticles exhibit non-uniformity, and to some extent, they are interconnected. In Fig. 5a, EDX (energy-dispersive X-ray spectroscopy) and EDX-mapping of Fe3O4@CS/UIO-66-NH2(Zr) reveal the expected elements in the sample: Fe (0–1, 6–7 keV), C (0–1 keV), O (0–1 keV), N (0–1 keV), Si (1–2 keV), and Zr (2–3 keV). The EDX-mapping demonstrates uniform distribution of these elements throughout the sample. Fig. 5b displays EDX and EDX-mapping of PNT-Fe3O4@CS/UIO-66-NH2(Zr). The presence of F and S elements in the EDX spectrum indicates the successful loading of the drug on Fe3O4@CS/UIO-66-NH2(Zr). Furthermore, the nearly uniform distribution of these elements, evident in the EDX-map, confirms that the drug is uniformly distributed on the nanocarrier.
 |
| | Fig. 4 FESEM images of (a and b) Fe3O4@CS/UIO-66-NH2(Zr) and (c and d) PNT-Fe3O4@CS/UIO-66-NH2(Zr). | |
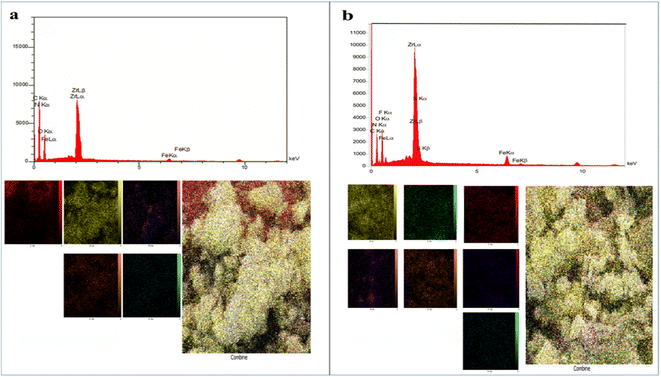 |
| | Fig. 5 EDX analysis and EDX elemental mapping of (a) Fe3O4@CS/UIO-66-NH2(Zr) and (b) PNT-Fe3O4@CS/UIO-66-NH2(Zr). | |
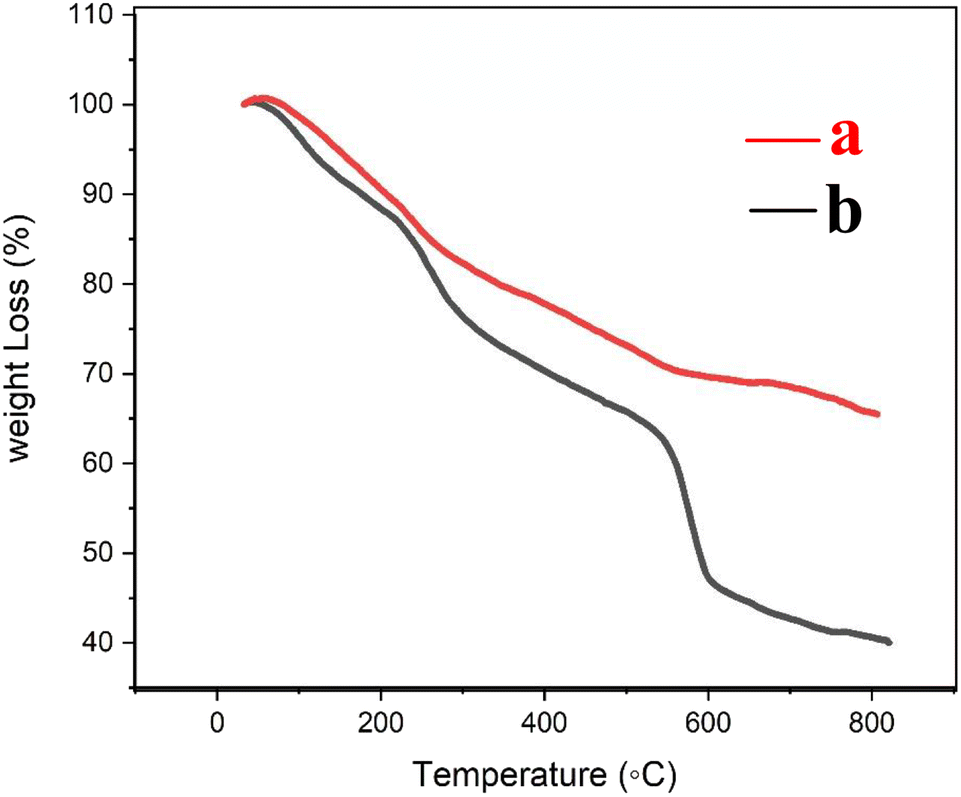
The thermogravimetric (TG) curves of Fe3O4@CS/UIO-66-NH2(Zr) and PNT-Fe3O4@CS/UIO-66-NH2(Zr) are shown in Fig. 6. As the temperature increases from room temperature to approximately 700 °C, the mass of magnetite remains relatively constant, while the MOF structure grown on it decomposes. In Fig. 6a, weight loss between 25 and 800 °C is observed in the TGA curve of Fe3O4@CS/UIO-66-NH2(Zr). The initial weight loss (up to 200 °C) is likely due to the removal of water from the sample pores. Between 250–500 °C, the weight loss may be attributed to the dehydroxylation of metal oxo-clusters and decomposition of the bridging carboxylates of the framework linker.35,56 Fig. 6b shows that the drug experiences weight loss beyond 200 to 300 °C after its melting point.57 The total percentage of weight loss is approximately 35%. Interestingly, PNT-Fe3O4@CS/UIO-66-NH2(Zr) exhibits approximately 25% more weight loss.
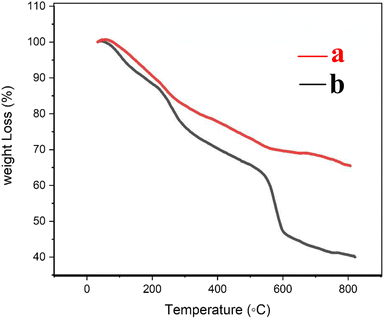 |
| | Fig. 6 TG curves (a) Fe3O4@CS/UIO-66-NH2(Zr) and (b) PNT-Fe3O4@CS/UIO-66-NH2(Zr). | |
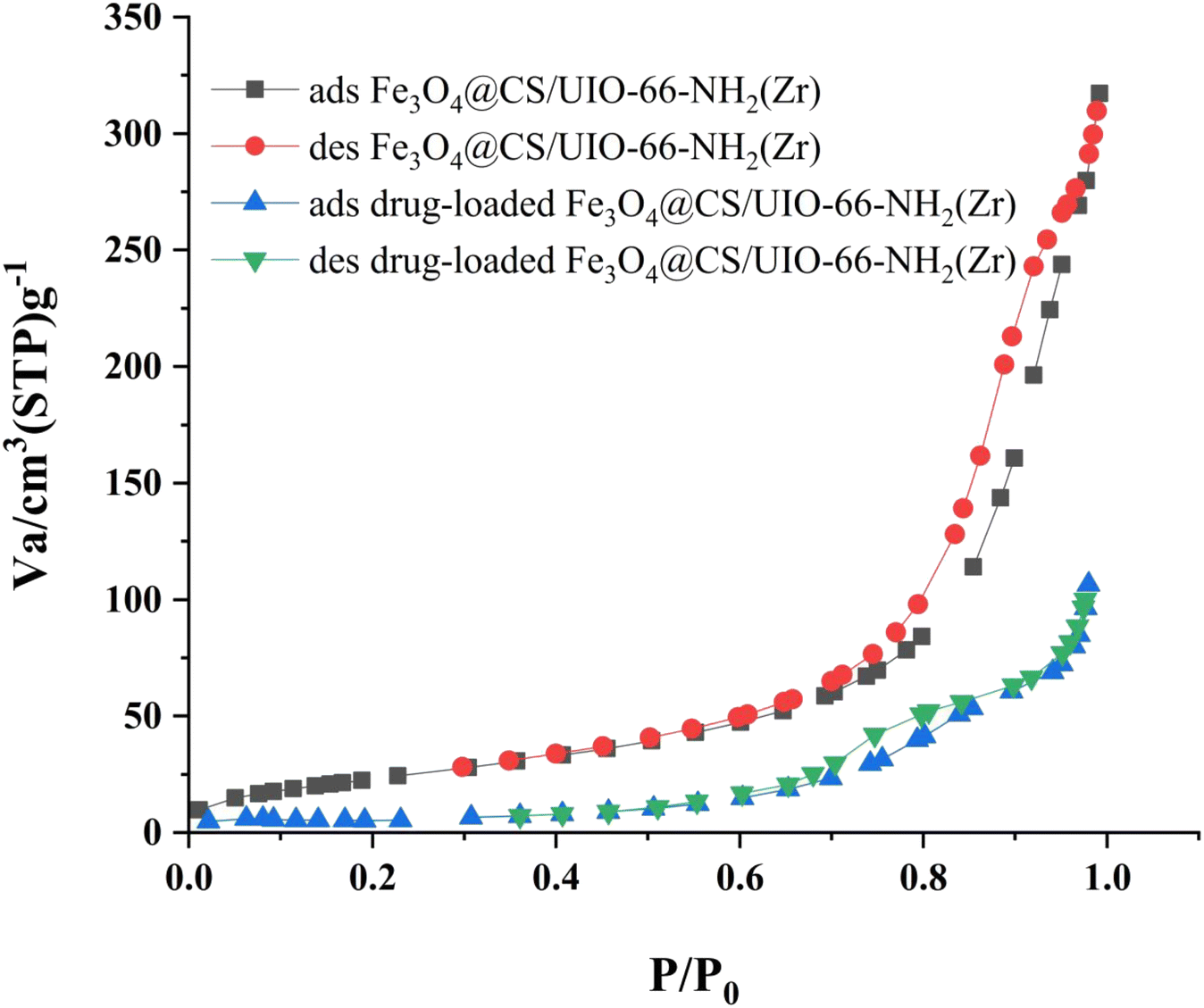
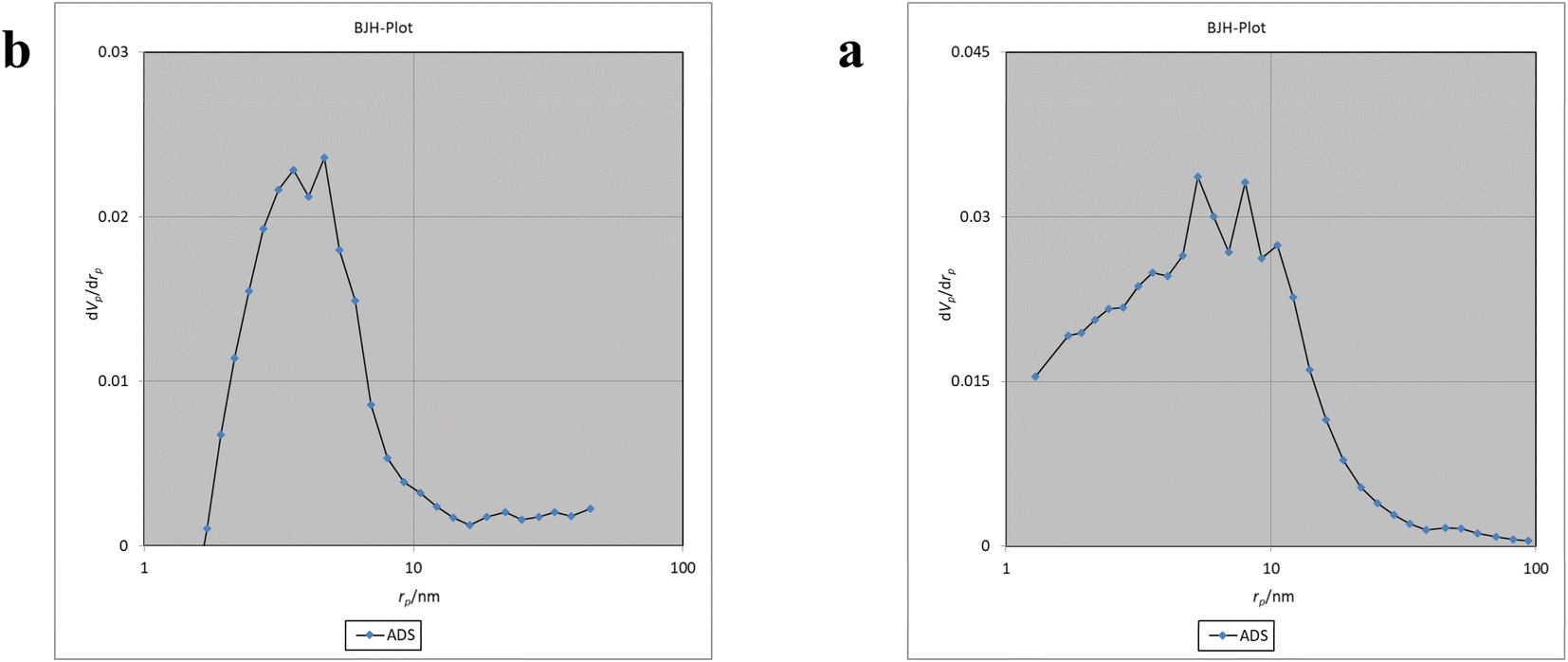
The Brunauer–Emmett–Teller (BET) analysis is commonly used to investigate specific surface area, pore volume, and study the porous structure of nanomaterials. In Fig. 7, the adsorption–desorption isotherms for Fe3O4@CS/UIO-66-NH2(Zr) and PNT-Fe3O4@CS/UIO-66-NH2(Zr) are shown. As depicted in this figure, the isotherms for Fe3O4@CS/UIO-66-NH2(Zr) and PNT-Fe3O4@CS/UIO-66-NH2(Zr) exhibit type IV behavior, indicating mesoporous structures. The size of the pores falls between 2 and 50 nm, as illustrated in the size of the pores falls between 2 and 50 nm, as illustrated in Fig. 8. From the comparison of the curves, it becomes clear that the pores are occupied by the drug. Table 1 presents the surface area, pore volume, and average pore diameter obtained from the results of the BJH analysis before and after loading pantoprazole. The BET specific surface area and pore volume of Fe3O4@CS/UIO-66-NH2(Zr) are 91.182 m2 g−1 and 0.4817 cm3 g−1 respectively. For PNT-Fe3O4@CS/UIO-66-NH2(Zr) these values are 27.018 m2 g−1 and 0.1646 cm3 g−1 respectively. The BJH results reveal that pantoprazole was loaded into a large portion of the pores within the metal–organic framework.
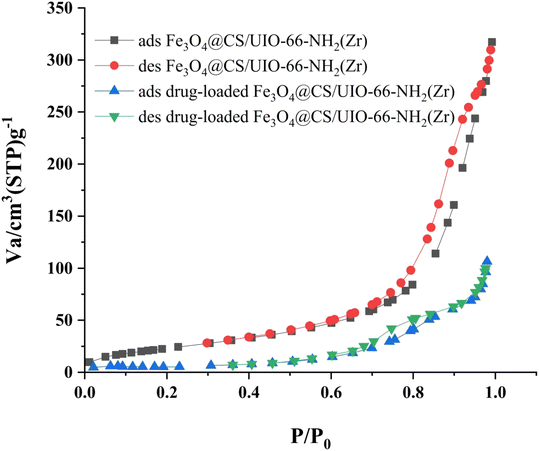 |
| | Fig. 7 N2 adsorption–desorption Fe3O4@CS/UIO-66-NH2(Zr) and drug-load Fe3O4@CS/UIO-66-NH2(Zr). | |
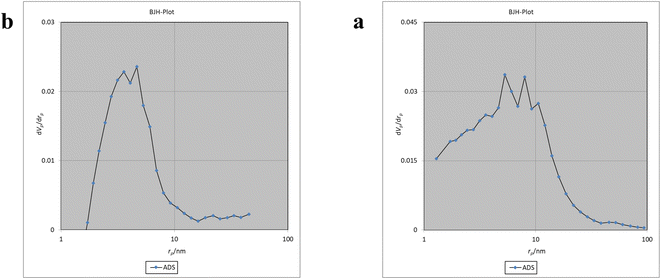 |
| | Fig. 8 BJH of a: Fe3O4@CS/UIO-66-NH2(Zr) and b: drug-load Fe3O4@CS/UIO-66-NH2(Zr). | |
Table 1 The parameters of BET for Fe3O4@CS/UIO-66-NH2(Zr) and PNT-Fe3O4@CS/UIO-66-NH2(Zr)
| Parameters |
FE3O4@CS/UIO-66-NH2(ZR) |
PNT-FE3O4@CS/UIO-66-NH2(ZR) |
| Vm [cm3 (STP) g−1] |
20.59 |
6.2076 |
| SBET (m2 g−1) |
91.182 |
27.018 |
| Mean pore diameter (nm) |
21.117 |
24.365 |
| Total pore volume (Vp) (cm3 g−1) |
0.4814 |
0.1646 |
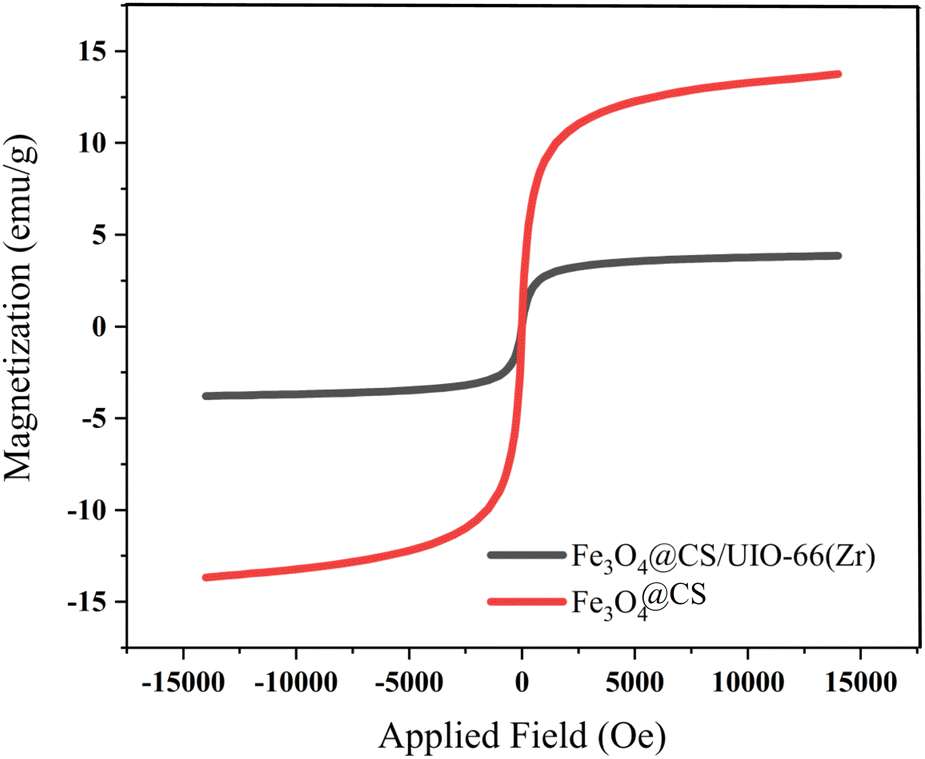
The magnetic properties of the samples were investigated using Vibrating Sample Magnetometry (VSM) at room temperature. The VSM curves for Fe3O4@CS and Fe3O4@CS/UIO-66-NH2(Zr) are shown in Fig. 9. As depicted in this figure, the saturation magnetization (Ms) values for the Fe3O4@CS and Fe3O4@CS/UIO-66-NH2(Zr) particles are approximately 13.7 and 3.8 emu g−1, respectively. The VSM test was also performed on the drug-loaded sample, but no significant difference was found in the saturation magnetization between the drug-loaded and unloaded nanocomposites. The samples exhibit zero coercivity (HC) at room temperature, and no hysteresis loop is clearly observed, indicating that both are superparamagnetic.58 Interestingly, after the growth of the 3D UIO-66-NH2(Zr) structure on Fe3O4@CS, the Ms value decreases significantly. This phenomenon is attributed to the decrease in the Fe3O4@CS mass ratio following the growth of the dielectric UIO-66-NH2(Zr) onto Fe3O4@CS. The Ms value obtained in this work for Fe3O4@CS/UIO-66-NH2(Zr) is lower than in similar studies.35
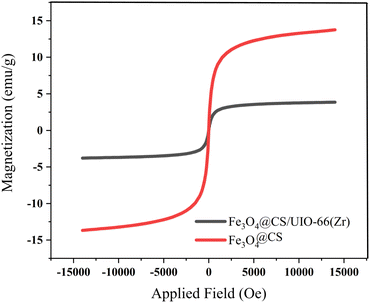 |
| | Fig. 9 VSM magnetization of Fe3O4@CS and Fe3O4@CS/UIO-66-NH2(Zr). | |
3.2 Adsorption of pantoprazole
The pH value plays a crucial role in drug loading onto nanocarriers. To investigate the optimal loading capacity of Fe3O4@CS/UIO-66-NH2(Zr), we utilized deionized water (pH 7) and an acetate buffer (pH 5) as impregnation media. The drug loading percentages were 75% for deionized water (pH 7) and 79% for the acetate buffer at pH 5. Consequently, the highest drug loading occurred at pH 5. It appears that drug loading is higher in an acidic environment compared to a neutral one. However, the difference is not substantial, and the acidic environment does not significantly enhance drug loading.
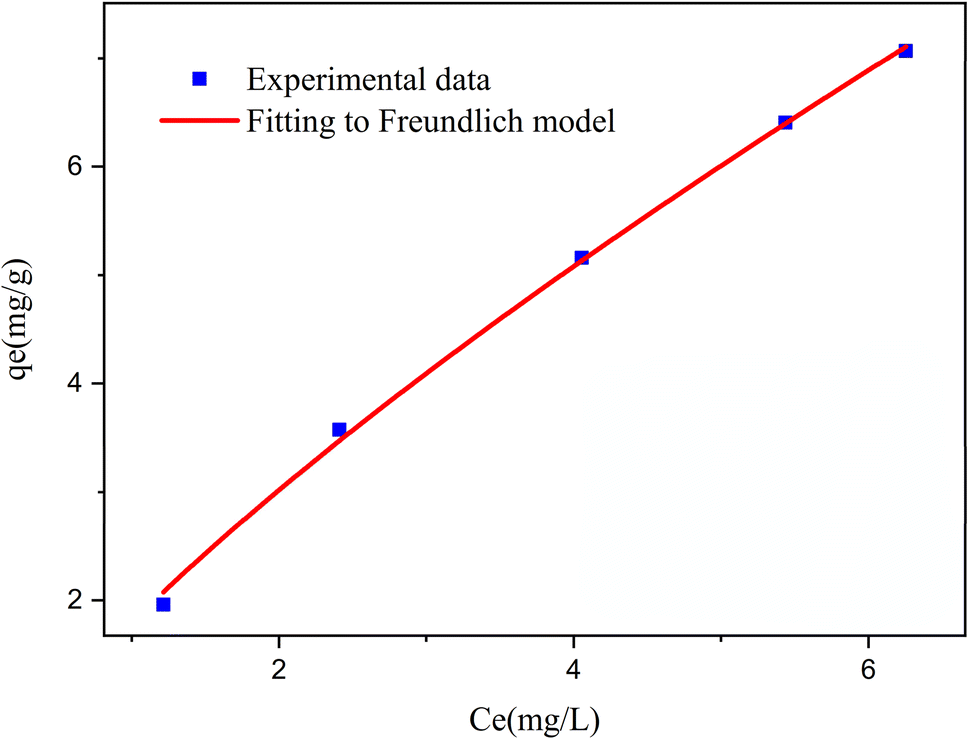
3.2.1 Modeling of adsorption isotherms. The adsorption isotherm represents a mathematical relationship between the amount of adsorbate at the initial time and the equilibrium time during the adsorption process at a constant temperature and pH. In our study, we conducted adsorption isotherms by adding 0.01 g of MOF to 50 mL of deionized water (pH 7) containing initial PNT concentrations of 20, 50, 40, 100, and 200 ppm. After stirring for 24 hours and centrifuging the supernatant, we calculated the amount of adsorbed drug. We evaluated several models—Langmuir, Freundlich, Dubinin–Radushkevich (D–R), Temkin, and Redlich–Peterson—to describe the adsorption isotherm. The resulting equations and parameters are summarized in Table 2. Specifically. qe (mg g−1) and qmax (mg g−1) represent the capacity of PNT adsorbed per gram onto MOF at equilibrium and the maximum PNT sorption capacity. Ce (mg L−1) corresponds to the equilibrium drug concentration in the buffer solution. KL (L mg−1), KF (mg g−1) (L mg−1)1/n, KDR (mol2 kJ−2), and KT (L g−1) are equilibrium constants for Langmuir, Freundlich, D–R, and Temkin models. n denotes the surface heterogeneity factor. B = RT/b is a constant associated with the heat of adsorption. ε represents the Polanyi potential (J2 (mol2)−1), which is calculated as ε = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(1 + 1/Ce). Our analysis reveals that the Freundlich and Redlich–Peterson models exhibit the best fit for the adsorption of the drug onto the adsorbent, with an R2 value of 0.99. Notably, the Freundlich adsorption isotherm model characterizes both multilayer sorption and sorption on heterogeneous surfaces. Furthermore, the numerical value of 1/n < 1 indicates physical adsorption, suggesting that the adsorption capacity is only slightly suppressed at lower equilibrium concentrations and becomes favorable at higher concentrations. The adsorption isotherm represents a mathematical relationship between the initial amount of adsorbate and the equilibrium time during an adsorption process at constant temperature and pH. In our study, we conducted adsorption isotherms by adding 0.01 g of MOF to 50 mL of deionized water (with a pH of 7) containing initial PNT concentrations of 20, 50, 40, 100, and 200 ppm. After stirring for 24 hours and centrifuging the supernatant, we calculated the amount of adsorbed drug. In this study, Langmuir, Freundlich, Dubinin–Radushkevich (D–R), Temkin and Redlich–Peterson models were evaluated to describe the adsorption isotherm. The equations and parameters obtained are summarized in Table 2. Here Ce (mg L−1) was the equilibrium drug concentration in the buffer solution. qe (mg g−1) and qmax (mg g−1) were the capacity of PNT adsorbed per gram onto MOF at equilibrium and the maximum PNT sorption capacity. KF (mg g− 1) (L mg− 1)1/n, KL (L mg− 1), and KDR (mol2 kJ− 2), KT (L g−1) are equilibrium constants for Freundlich, Langmuir, D–R and Temkin. ε is Polanyi potential (J2 (mol2)−1) which is equal to ε = RTln(1 + 1/Ce). n is the surface heterogeneity factor, B = RT/b is a constant associated with the heat of adsorption. In the table presented (Table 2), both the Freundlich and Langmuir models exhibit the best fitting for the adsorption of a drug onto an adsorbent, with an R2 value of 0.99. The Freundlich adsorption isotherm model is applied for characterizing both multilayer sorption and sorption on heterogeneous surfaces. The numerical value of 1/n < 1 indicates physical adsorption and shows that adsorption capacity is only slightly suppressed at lower equilibrium concentrations and adsorption is favorable only in high concentrations.59 The Freundlich adsorption isotherm of pantoprazole is given in Fig. 10. In the Dubinin–Kaganer–Radushkevich (DKR) isotherm β (mol2 J−2) is the activity coefficient related to mean sorption energy
ln(1 + 1/Ce). Our analysis reveals that the Freundlich and Redlich–Peterson models exhibit the best fit for the adsorption of the drug onto the adsorbent, with an R2 value of 0.99. Notably, the Freundlich adsorption isotherm model characterizes both multilayer sorption and sorption on heterogeneous surfaces. Furthermore, the numerical value of 1/n < 1 indicates physical adsorption, suggesting that the adsorption capacity is only slightly suppressed at lower equilibrium concentrations and becomes favorable at higher concentrations. The adsorption isotherm represents a mathematical relationship between the initial amount of adsorbate and the equilibrium time during an adsorption process at constant temperature and pH. In our study, we conducted adsorption isotherms by adding 0.01 g of MOF to 50 mL of deionized water (with a pH of 7) containing initial PNT concentrations of 20, 50, 40, 100, and 200 ppm. After stirring for 24 hours and centrifuging the supernatant, we calculated the amount of adsorbed drug. In this study, Langmuir, Freundlich, Dubinin–Radushkevich (D–R), Temkin and Redlich–Peterson models were evaluated to describe the adsorption isotherm. The equations and parameters obtained are summarized in Table 2. Here Ce (mg L−1) was the equilibrium drug concentration in the buffer solution. qe (mg g−1) and qmax (mg g−1) were the capacity of PNT adsorbed per gram onto MOF at equilibrium and the maximum PNT sorption capacity. KF (mg g− 1) (L mg− 1)1/n, KL (L mg− 1), and KDR (mol2 kJ− 2), KT (L g−1) are equilibrium constants for Freundlich, Langmuir, D–R and Temkin. ε is Polanyi potential (J2 (mol2)−1) which is equal to ε = RTln(1 + 1/Ce). n is the surface heterogeneity factor, B = RT/b is a constant associated with the heat of adsorption. In the table presented (Table 2), both the Freundlich and Langmuir models exhibit the best fitting for the adsorption of a drug onto an adsorbent, with an R2 value of 0.99. The Freundlich adsorption isotherm model is applied for characterizing both multilayer sorption and sorption on heterogeneous surfaces. The numerical value of 1/n < 1 indicates physical adsorption and shows that adsorption capacity is only slightly suppressed at lower equilibrium concentrations and adsorption is favorable only in high concentrations.59 The Freundlich adsorption isotherm of pantoprazole is given in Fig. 10. In the Dubinin–Kaganer–Radushkevich (DKR) isotherm β (mol2 J−2) is the activity coefficient related to mean sorption energy 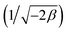 , this value is below 8 kJ mol−1 and the adsorption type can be explained by physical adsorption.60
, this value is below 8 kJ mol−1 and the adsorption type can be explained by physical adsorption.60
Table 2 Parameters and regression coefficients for isotherm adsorbsion models
| Isotherm |
Eqn |
Parameters |
| Langmuir |
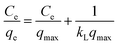 |
qmax = 1.8497, KL = 1.023, R2 = 0.99 |
| Freundlich |
qe = KF(Ce)1/n |
n = 1.3315, KF = 1.7936, R2 = 0.99 |
| Temkin |
qe = Bln(KT) + Bln(Ce) |
B = 3.0869, KT = 1.4406, R2 = 0.98 |
| Dubinin–Radushkevich |
ln(qe) = ln(Xm) − βε2 |
Xm = 14.4833, E = 0.6198 (kJ mol−1), R2 = 0.95 |
| Redlich–Peterson |
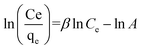 |
β = 0.2227, A = 1.7278, R2 = 0.96 |
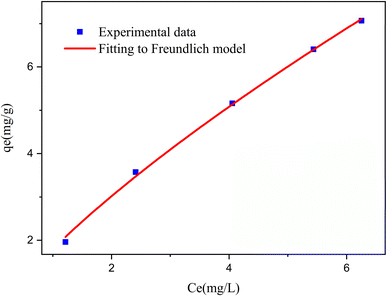 |
| | Fig. 10 The loading curve for pantoprazole and the corresponding Freundlich model. | |
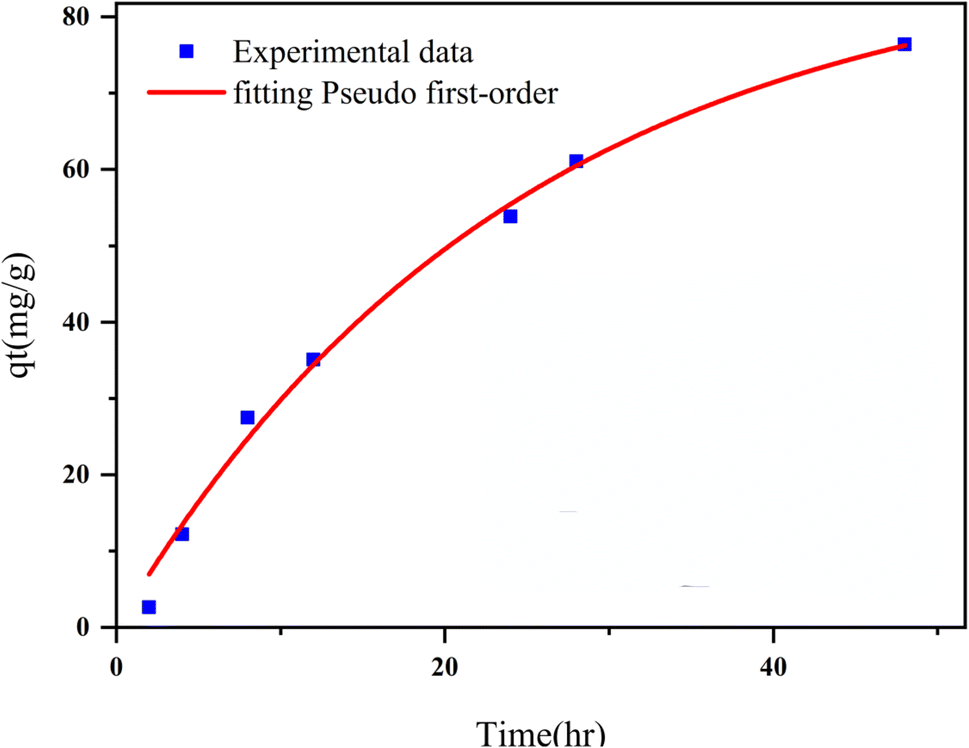
3.2.2 Adsorption kinetics. The adsorption kinetics play a crucial role in determining the time required to reach equilibrium between a drug and a nanocarrier. In our study, we investigated the rate of drug uptake using deionized water (pH 7), and the results are summarized in Table 3. We explored several kinetic models to express the adsorption processes, including zero-order, pseudo-first order, pseudo-second order, and Elovich. In these equations: K represents the adsorption rate constant for each eqn. α (mg g−1 min−1) and β (g mg−1) denote the initial rate constants for adsorption and desorption. As shown in Fig. 11, the pseudo-first-order model provided a better fit for the kinetic adsorption of pantoprazole on the nanocarrier compared to the other models.
Table 3 Adsorption kinetic equations of Fe3O4@CS/UIO-66-NH2(Zr)
| Kinetic model |
Eqn |
Parameters |
| Zero-order |
qt = q0 + k0t |
q0 = 10.37, k0 = 1.56, R2 = 0.92 |
| Pseudo-first-order |
qt = qe (1 − exp(−k1t)) |
qe = 86.65, k1 = 0.04, R2 = 0.99 |
| Pseudo-second-order |
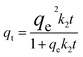 |
qe = 33.33, k2 = 0.0012, R2 = 0.91 |
| Elovich |
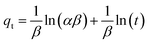 |
α = 51.39, β = 0.043, R2 = 0.98 |
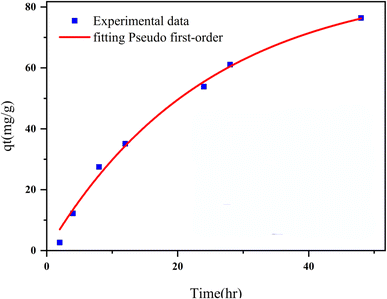 |
| | Fig. 11 Adsorption kinetics curve fit Pseudo 1st order model for Fe3O4@CS/UIO-66-NH2(Zr). | |
3.3 Pantoprazole release
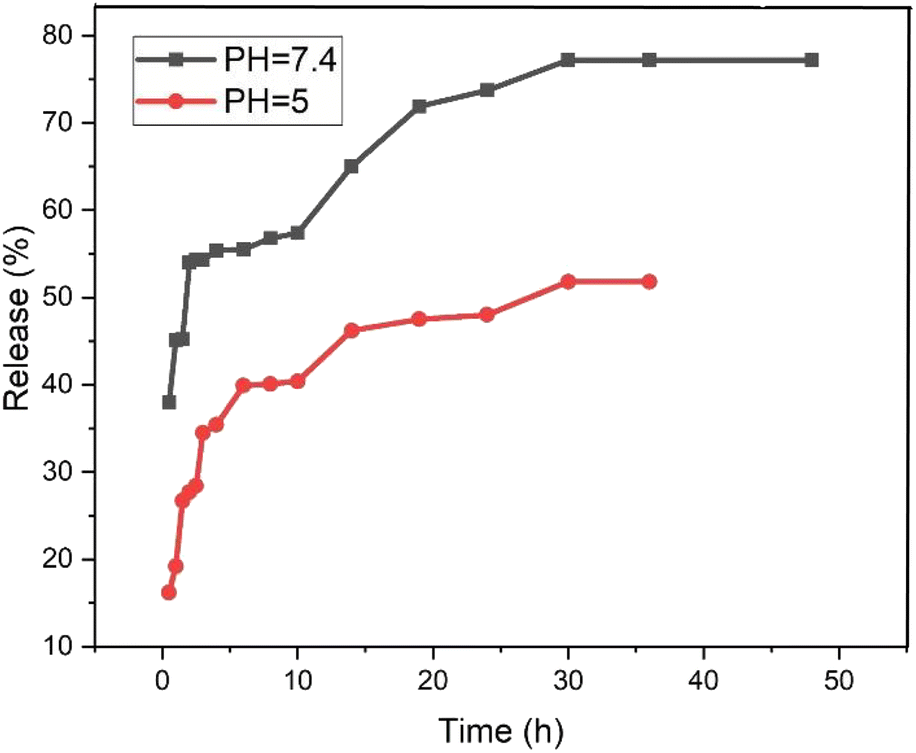
The release behavior of pantoprazole was assessed in both neutral and acidic environments. According to Fig. 12, at pH 7.4 and pH 5, PNT-Fe3O4@CS/UIO-66-NH2(Zr) exhibited approximately 54% and 35% release, respectively, within the first 2 hours. Consequently, under physiological conditions, the “burst effect” was observed, while controlled release occurred in an acidic medium. Furthermore, the release pattern in phosphate buffer can be divided into three segments: the primary burst release of the drug within the initial 2 hours (approximately 54%) is attributed to the diffusion of pantoprazole from the nanocarrier interfaces. A secondary slow release (around 3% over the next 8 hours) is related to the diffusion of drug molecules from the voids into the surrounding environment. The remaining 20% release is governed by drug molecules close to the cavity walls, exhibiting strong host–guest interactions such as cation–anion electrostatic interactions, hydrogen bonding, and interactions between the aromatic rings of the ligand and pantoprazole. Finally, the complete drug release from the PNT-Fe3O4@CS/UIO-66-NH2(Zr) composite was achieved after 36 hours, reaching a maximum value of approximately 77%. In contrast, at acidic pH, the evaluated release amount was approximately 48% during the same 36 hours period. The interaction between the drug and the surface of the MOF is stronger at acidic pH, leading to the observed controlled release at this pH.59,61
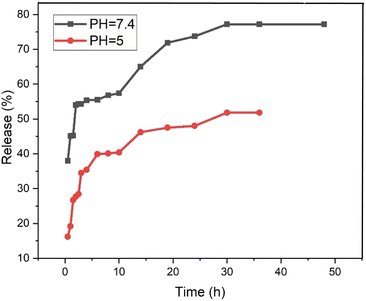 |
| | Fig. 12 The release of pantoprazole at acidic and neutral pH. | |
Table 4 provides an overview of the release of pantoprazole in other systems and the same system with another drug for comparison with the present work.
Table 4 Comparison of the release of the pantoprazole with other reported nanocarriers
| Carrier |
Drug |
Drug loading |
Drug release(%) |
Time |
Ref. |
| Hydroxypropyl methylcellulose phthalate, Eudragit S 100 |
Pantoprazole (microsphere) |
Encapsulation efficiency: from 60–78% |
From 55–98% |
120 min |
12 |
| PAA-g-Chi, PAA-g-Chi-cly |
Pantoprazole |
— |
pH 2.0:100% |
250 min |
62 |
| pH 7.4:35% |
| LRS microballoons |
Pantoprazole |
Encapsulation efficiency: LRS-1(10%), LRS-2(71%), LRS-3(26%), LRS-4(77%) |
pH 6.8: LRS-1(75%), LRS-2(99%), LRS-3(95%), LRS-4(71%) |
12 hours |
63 |
| HPMC phthalate HP55, Eudragit S100 |
Pantoprazole |
Encapsulation efficiency: from 25–80% |
From 84–100% |
12 hours |
1 |
| Fe3O4@UIO-66-NH2@PEI |
Imatinib |
Encapsulation efficiency: (94%) |
pH 9:38% |
48 hours |
35 |
| pH 3.0:98% |
| Fe3O4@CS@UIO-66-NH2(Zr) |
Pantoprazole |
Drug-loading efficiency: pH 5(79%), pH 7.4(75%) |
pH 7.4:78% |
32 hours |
This work |
| pH 5.0:48% |
Table 4 presents a comparison between the nanocarrier prepared in this study and other carriers for pantoprazole delivery. Additionally, the table reveals that the amount of drug loading is comparable to that of other carriers. However, in terms of drug delivery, the carrier developed in this work demonstrates enhanced control over drug release compared to other carriers.
4 Conclusion
In this study, a novel magnetic nanocomposite (Fe3O4@CS@UIO-66 NH2(Zr)) was synthesized by growing a biocompatible and stable MOF in water on the Fe3O4@Cs structure. The SiO2-APTES coating on the magnetite core requires two synthesis steps. Additionally, SiO2 is not a biocompatible coating, whereas chitosan is a fully biocompatible coating and requires only one synthesis step. The nanocomposite was characterized using various techniques (FT-IR, FE-SEM, EDX, EDX-map, XRD, BET, TGA, and VSM) for its potential as a nanocarrier for pantoprazole adsorption and release. EDX-map results confirmed the uniform adsorption of pantoprazole on the nanocomposite surface, while BET and BJH analyses demonstrated substantial drug loading within the metal–organic framework's pores, with smaller pores preferentially occupied. Isotherm and kinetic models were employed to examine the drug adsorption and release behavior, with drug loading efficiency reaching 79% at pH 5 and 75% at pH 7.4. In contrast, drug release was observed at 48% at pH 5 and 77% at pH 7.4 after 36 hours. The differences in drug release were attributed to stronger hydrogen interactions between the drug and nanocarrier under acidic conditions. The release profile in pH 5 during the first hours, lowest of amount, and the burst release in physiological conditions demonstrated the potential of this system as a gastro-retentive and enteric transfer mechanism for oral administration, as well as a targeted system with controlled release for intravenous administration.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Conflicts of interest
There are no conflicts to declare.
References
- A. M. Nasef, A. R. Gardouh and M. M. Ghorab, Futur J. Pharm. Sci., 2017, 3, 103–117 Search PubMed.
- G. Sachs, Pharmacotherapy, 1997, 17, 22–37 CrossRef CAS.
- P. Poole, Am. J. Health Syst. Pharm., 2001, 58, 999–1008 CrossRef CAS.
- M. N. Abed, F. A. Alassaf, M. H. M. Jasim, M. Alfahad and M. E. Qazzaz, Pharmacology, 2020, 105, 645–651 CrossRef CAS PubMed.
- A. S. Taskiran, M. Ergul, H. Gunes, A. Ozturk, B. Sahin and E. Ozdemir, Cell. Mol. Neurobiol., 2021, 41, 173–183 CrossRef CAS PubMed.
- Y. Cao, M. Chen, D. Tang, H. Yan, X. Ding, F. Zhou, M. Zhang, G. Xu, W. Zhang and S. Zhang, Cell Death Dis., 2018, 9, 604–622 CrossRef PubMed.
- S. Hashioka, A. Klegeris and P. L. Mcgeer, Glia, 2011, 59, 833–840 CrossRef.
- K. Mizunashi, Y. Furukawa, K. Katano and K. Abe, Calcif. Tissue Int., 1993, 53, 21–25 CrossRef CAS.
- J. J. Hyun, H. J. Chun, B. Keum, Y. S. Seo, Y. S. Kim, Y. T. Jeen, H. S. Lee, S. H. Um, C. D. Kim and H. S. Ryu, Int. J. Mol. Med., 2010, 26, 877–883 CAS.
- M. Prause, C. Seeliger, M. Unger, M. V. Griensven and A. T. Haug, Injury, 2014, 45, 1156–1164 CrossRef.
- G. Ullah, A. Nawaz, M. S. Latif, K. U. Shah, S. Ahmad, F. Javed, M. Alfatama, S. A. Abd Ghafar and V. Lim, Gels, 2023, 9, 43–60 CrossRef CAS PubMed.
- T. Comoglu, N. Gonul, A. Dogan and N. Basci, Drug Deliv., 2008, 15, 295–302 CrossRef CAS.
- A. K. Babu, N. B. Teja, B. Ramakrishna, B. B. Kumar and G. V. Reddy, Methods, 2011, 15, 28–37 Search PubMed.
- K. Liang, R. Wang, M. Boutter, C. M. Doherty, X. Mulet and J. J. Richardson, Chem. Commun., 2017, 53, 1249–1252 RSC.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS.
- A. Kohan Hoosh Nejad, H. Ahmad Panahi, E. Keshmirizadeh and N. Torabi Fard, Int. J. Polym. Mater., 2023, 72, 1083–1092 CrossRef CAS.
- A. Datt, D. Fields and S. C. Larsen, J. Phys. Chem., 2012, 116, 21382–21390 CAS.
- Y. Cheng, R. A. Morshed, B. Auffinger, A. L. Tobias and M. S. Lesniak, Adv. Drug Delivery Rev., 2014, 66, 42–57 CrossRef CAS.
- J. Ren, N. M. Musyoka, H. W. Langmi, M. Mathe and S. Liao, Int. J. Hydrog. Energy, 2017, 42, 289–311 CrossRef CAS.
- L. Li, Y. Xu, D. Zhong and N. Zhong, Colloids Surf., A, 2020, 586, 124255–124289 CrossRef CAS.
- M. N. Timofeeva, V. N. Panchenko, J. W. Jun, Z. Hasan, M. M. Matrosova and S. H. Jhung, Appl. Catal., A, 2014, 471, 91–97 CrossRef CAS.
- C. Y. Li, J. M. Liu, Z. H. Wang, S. W. Lv, N. Zhao and S. Wang, J. Hazard. Mater., 2020, 384, 121348–121356 CrossRef CAS PubMed.
- M. Ebadi, K. Buskaran, S. Bullo, M. Z. Hussein, S. Fakurazi and G. Pastorin, Alexandria Eng. J., 2021, 60, 733–747 CrossRef.
- V. K. Varadan, L. F. Chen and J. Xie, Nanomedicine: design and applications of magnetic nanomaterials, nanosensors and nanosystems, John Wiley & Sons, 2008 Search PubMed.
- S. Ahmadi, M. Fazilati, H. Nazem and S. M. Mousavi, BioMed Res. Int., 2021, 2021, 1–14 Search PubMed.
- N. Matinise, K. Kaviyarasu, N. Mongwaketsi, S. Khamlich, L. Kotsedi, N. Mayedwa and M. Maaza, Appl. Surf. Sci., 2018, 446, 66–73 CrossRef CAS.
- L. Huang, M. He, B. Chen and B. Hu, Chemosphere, 2018, 199, 435–444 CrossRef CAS.
- S. Ahmadipouya, M. H. Haris, F. Ahmadijokani, A. Jarahiyan, H. Molavi and F. M. Moghaddam, J. Mol. Liq., 2021, 322, 114910–114950 CrossRef CAS.
- C. M. Navarathna, N. B. Dewage, A. G. Karunanayake, E. L. Farmer, F. Perez, E. B. Hassan and T. E. Mlsna, J. Inorg. Organomet. Polym. Mater., 2020, 30, 214–229 CrossRef CAS.
- F. Ahmadijokani, S. Tajahmadi, M. H. Haris, A. Bahi, M. Rezakazemi, H. Molavi, F. Ko and M. Arjmand, Chemosphere, 2021, 275, 130087–130097 CrossRef CAS.
- M. Meng, S. Wang, J. Dai, Y. Li, Y. Feng, Y. Zhu, H. Zhang and Y. Liu, Colloids Surf., A, 2022, 644, 128830–128839 CrossRef CAS.
- Y. Jiang, W. Cai, W. Tu and M. Zhu, J. Chem. Eng. Data, 2018, 64, 226–233 CrossRef.
- A. Lajevardi, M. H. Sadr, A. Badiei and M. Armaghan, J. Mol. Liq., 2020, 307, 112996–113005 CrossRef CAS.
- M. Nasrabadi, M. A. Ghasemzadeh and M. R. Zand Monfared, New J. Chem., 2019, 43, 16033–16044 RSC.
- R. Shahin, M. Yousefi, H. Ziyadi, M. Bikhof and M. Hekmati, Inorg. Chem. Commun., 2023, 147, 110186–110195 CrossRef CAS.
- S. M. ACheer, A. Prakash and D. Faulds, Drugs, 2003, 63, 101–133 CrossRef PubMed.
- N. V. Gupta and H. G. Shivakumar, Curr. Drug Deliv., 2009, 6, 505–510 CrossRef CAS.
- A. M. Nasef, A. R. Gardouh and M. M. Ghorab, Futur J. Pharm. Sci., 2017, 3, 1–15 Search PubMed.
- F. Nemati, M. M. Heravi and R. S. Rad, Chin. J. Catal., 2012, 33, 1825–1831 CrossRef CAS.
- M. Setoodehkhah and S. Momeni, J. Inorg. Organomet. Polym. Mater., 2018, 28, 1098–1106 CrossRef CAS.
- D. N. Cuong, T. T. Hoa, D. Q. Khieub, T. D. Lamd, N. D. Hoac and N. V. Hieu, J. Alloys Compd., 2012, 523, 120 CrossRef.
- M. Pooresmaeil and H. Namazi, Int. J. Biol. Macromol., 2020, 162, 501–511 CrossRef CAS.
- R. Baharfar and S. Mohajer, Catal. Lett., 2016, 146, 1729–1742 CrossRef CAS.
- W. Xie and M. Huang, Catalysts, 2019, 9, 850 CrossRef CAS.
- F. Parsa, M. Setoodehkhah and S. M. Atyabi, Inorg. Chem. Commun., 2023, 155, 111056 CrossRef CAS.
- M. Mohammadikish and S. H. Hashemi, J. Mater. Sci., 2019, 54, 6164–6173 CrossRef CAS.
- J. B. Huo, L. Xu, X. Chen, Y. Zhang, J. C. E. Yang, B. Yuan and M. L. Fu, Micropor. Mesopor. Mat., 2019, 276, 68–75 CrossRef CAS.
- H. Molavi, A. Hakimian, A. Shojaei and M. Raeiszadeh, Appl. Surf. Sci., 2018, 445, 424–436 CrossRef CAS.
- Z. H. Rada, H. R. Abid, J. Shang, H. Sun, Y. He, P. Webley and S. Liu, Ind. Eng. Chem. Res., 2016, 55, 7924–7946 CrossRef CAS.
- M. Aghajanzadeh, M. Zamani, H. Molavi, H. K. Manjili, H. Danafar and A. Shojaei, J. Inorg. Organomet. Polym. Mater., 2018, 28, 177–186 CrossRef CAS.
- H. Molavi, A. Eskandari, A. Shojaei and S. A. Mousavi, Microporous Mesoporous Mater., 2018, 256, 193–201 CrossRef.
- H. Molavi, M. Zamani, M. Aghajanzadeh, H. K. Manjili, H. Danafar and A. Shojaei, Appl. Organomet. Chem., 2018, 32, 4221–4240 CrossRef.
- X. Y. Xu, C. Chu, H. Fu, X. D. Du, P. Wang, W. Zheng and C. C. Wang, Chem. Eng. J., 2018, 15, 436–444 CrossRef.
- W. Cai, Z. Li, J. Wei and Y. Liu, Chem. Eng. Res. Des., 2018, 140, 23–32 CrossRef CAS.
- N. A. Bee, R. Massart and S. Neveu, J. Magn. Magn. Mater., 1995, 149, 6–9 CrossRef.
- L. Huang, M. He, B. Chen and B. Hu, J. Mater. Chem., 2016, 4, 5159–5166 RSC.
- A. K. Awasthi, L. Kumar, P. Tripathi, M. Golla, M. A. Aga, C. S. Reddy and P. Kumar, ACS Omega, 2017, 2, 5460–5469 CrossRef CAS.
- C. A. Coles and R. N. Yong, Eng. Geol., 2006, 85, 19–25 CrossRef.
- J. Nawrocki, D. Prochowicz, A. Wiśniewski, I. Justyniak, P. Goś and J. Lewiński, Eur. J. Inorg. Chem., 2020, 10, 796–800 CrossRef.
- C. C. Wang, L. C. Juang, C. K. Lee, T. C. Hsu, J. F. Lee and H. P. Chao, J. Colloid Interface Sci., 2004, 280, 27–35 CrossRef CAS PubMed.
- Z. Nadizadeh, M. R. Naimi-Jamal and L. Panahi, J. Solid State Chem., 2018, 259, 35–42 CrossRef CAS.
- V. Teofilović, B. Agan, J. Pavličević, D. Lacin and A. Z. Aroguz, React. Kinet. Mech. Catal., 2022, 135, 1423–1437 CrossRef.
- P. Gupta, M. Kumar and D. Kaushik, Adv. Pharm. Bull., 2017, 7, 461–467 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Fatemeh Parsa
* and
Fatemeh Parsa
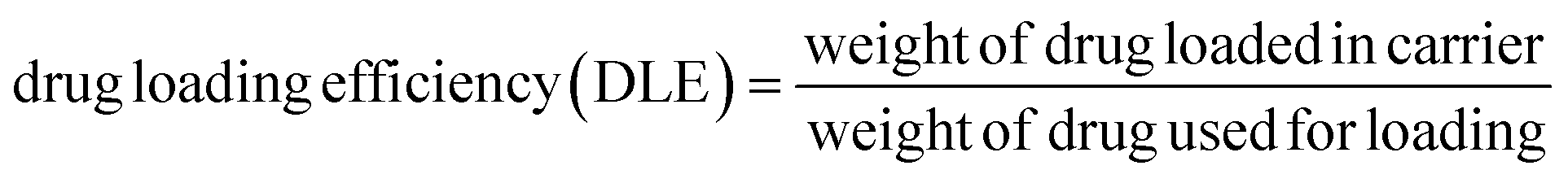
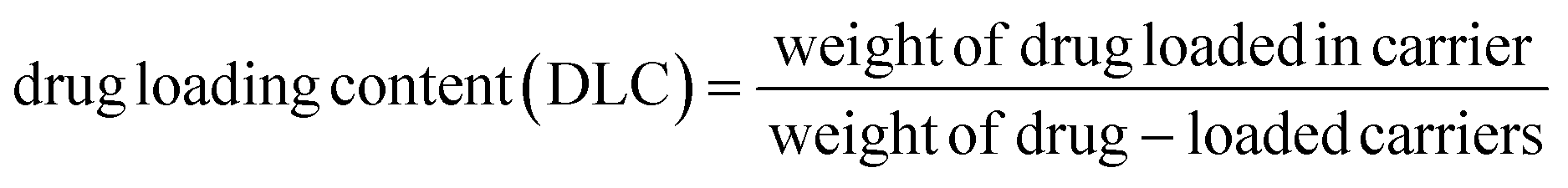
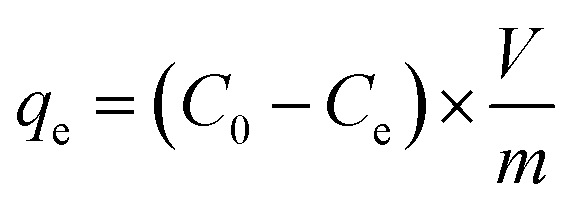
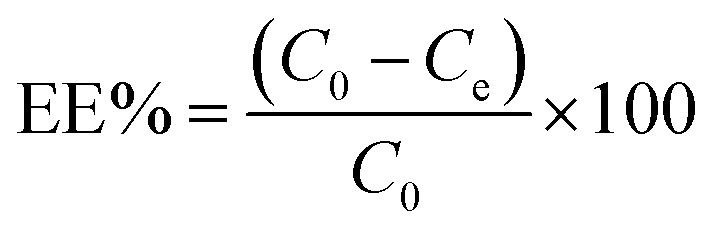
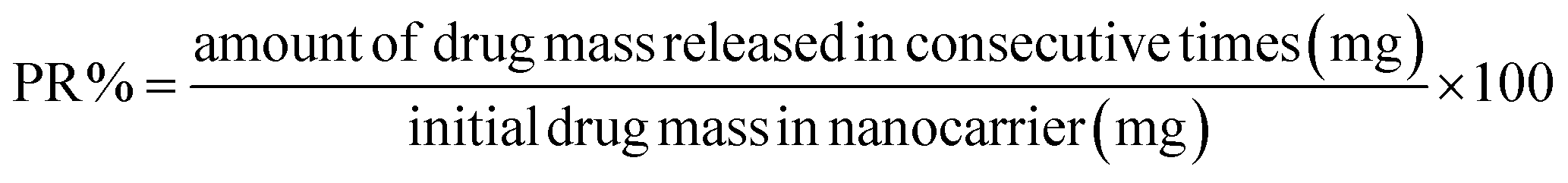

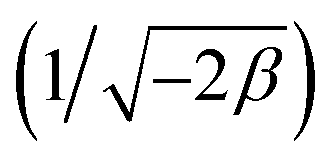 , this value is below 8 kJ mol−1 and the adsorption type can be explained by physical adsorption.60
, this value is below 8 kJ mol−1 and the adsorption type can be explained by physical adsorption.60