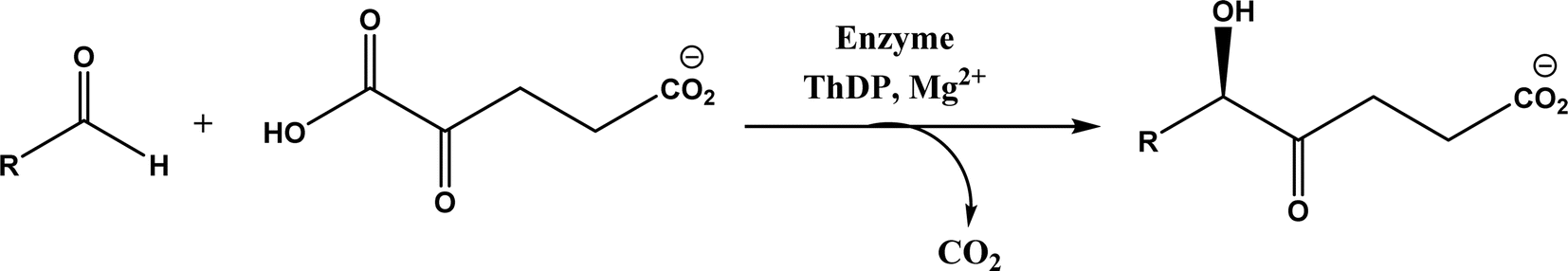DOI:
10.1039/D4RA03885A
(Review Article)
RSC Adv., 2024,
14, 25932-25974
Recent advances in enzymatic carbon–carbon bond formation
Received
27th May 2024
, Accepted 6th August 2024
First published on 19th August 2024
Abstract
Enzymatic carbon–carbon (C–C) bond formation reactions have become an effective and invaluable tool for designing new biological and medicinal molecules, often with asymmetric features. This review provides a systematic overview of key C–C bond formation reactions and enzymes, with the focus of reaction mechanisms and recent advances. These reactions include the aldol reaction, Henry reaction, Knoevenagel condensation, Michael addition, Friedel–Crafts alkylation and acylation, Mannich reaction, Morita–Baylis–Hillman (MBH) reaction, Diels–Alder reaction, acyloin condensations via Thiamine Diphosphate (ThDP)-dependent enzymes, oxidative and reductive C–C bond formation, C–C bond formation through C1 resource utilization, radical enzymes for C–C bond formation, and other C–C bond formation reactions.
1. Introduction
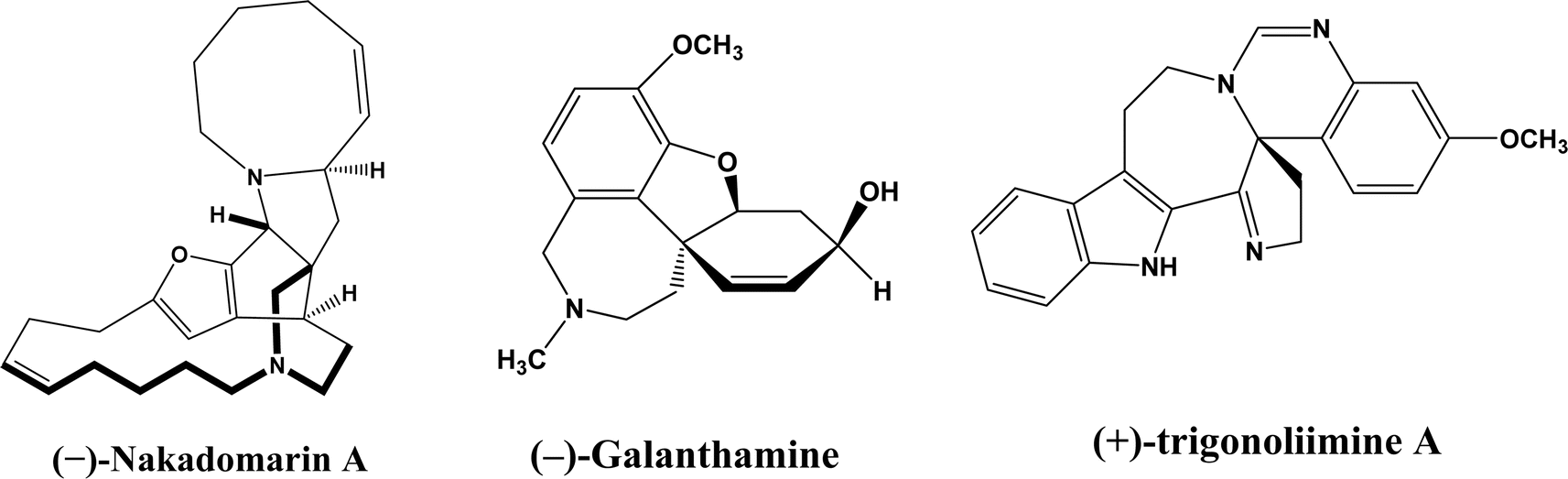
Enzymatic carbon–carbon (C–C) bond formation reactions (such as the Michael addition, Friedel–Crafts alkylation, and the aldol, Mannich, Morita–Baylis–Hillman, Henry, and Diels–Alder reactions) often lead to asymmetric molecules that are essential to the synthesis of many pharmaceutical ingredients such as monoterpene indole [MIAs] and benzylisoquinoline alkaloids.1–6 As an example, asymmetric Michael reaction is a key step for the preparation of pharmaceutical ingredients (Fig. 1) such as marine alkaloid (–)-nakadomarin A (an anticancer, antifungal and antibacterial compound),7 hydrodibenzofuran alkaloids such as (–)-galanthamine (treating Alzheimer's disease),8 and (+)- and (–)-trigonoliimine A (anti-HIV and anti-cancer activities).9 Michael reactions often require complex and expensive chiral organocatalysts to achieve high enantioselectivities, which can be easily accomplished by judicious selection and design of enzymes. It is very important to point out that in addition to their natural catalytic activities, some enzymes could catalyze completely different types of reactions, which is known as catalytic promiscuity.
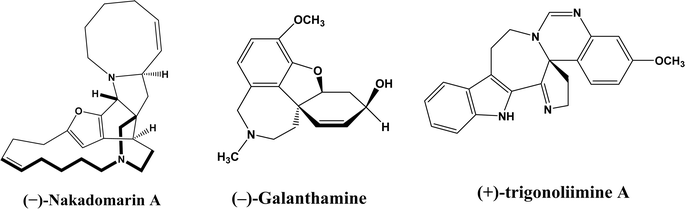 |
| | Fig. 1 Structures of several pharmaceutical ingredients. | |
Over the past decade, there have been several excellent general reviews on related topics focusing on the formation of tetrasubstituted carbon stereocenters catalyzed by aldolases (including those accepting fluoropyruvates as nucleophiles10), hydroxynitrile lyases, and thiamine diphosphate (ThDP)-dependent enzymes,11 and promiscuous enzyme activities of hydrolases (e.g., lipases, proteases, and trypsin), transglutaminase, hydroxynitrile lyases, 4-oxalocrotonate tautomerase, transketolases, ThDP-dependent enzymes, as well as those acylases-catalyzed aldol condensation, Michael addition, Knoevenagel condensation, Mannich reaction, and Henry reactions.12–14 This review intends to provide a more systematic overview of key C–C bond formation reactions and enzymes with more recent examples, and focus on catalytic mechanisms. However, it is not the main goal of this review to discuss C–C bond formations through biosynthesis15 such as DNA methylation,16 polyketide C-methylation,17 biosynthesis of L-sorbose and L-psicose using biocatalytic aldol addition in the Corynebacterium glutamicum strain,18 biosynthetic pathway of the phosphonate phosphonothrixin,19 and cytochrome P450 enzymes-catalyzed biosynthesis of mycocyclosin and guatyromycine,20 etc. To provide a high-level glance of this comprehensive topic, Table 1 presents key reaction types and enzymes with examples of recent advances in the field.
Table 1 Summary of enzymatic carbon–carbon (C–C) bond formation reactions
| Type of reaction |
Enzyme |
Examples of recent advances |
| Aldol addition |
Aldolases |
• Threonine aldolase from Pseudomonas sp. was mutated to improve or invert its stereoselectivity towards aromatic aldehydes32 |
| Based on mechanisms (Fig. 2): |
| (a) Type I aldolases (known as lysine-dependent) |
• Protein engineering and computational de novo enzyme design to develop more robust and more substrate-tolerant aldolases21–24 |
| (b) Types II aldolases (known as metal-dependent) |
| Based on their donor specificity: |
• Ketones were used as acceptors in aldol addition.27–30 |
| (a) Pyruvate, phosphoenolpyruvate, oxaloacetate, or 2-oxobutyrate |
| (b) Dihydroxyacetone phosphate (DHAP) |
• DHAP-dependent aldolase mechanism was illustrated through electronic structure calculations via the DFT method31 |
| (c) Dihydroxyacetone (DHA) and other unphosphorylated analogues (e.g., D-fructose-6-phosphate aldolase) |
| (d) Pyridoxal 5′-phosphate (PLP) (also known as threonine aldolases or glycine/alanine-dependent) |
|
| (e) Acetaldehyde [i.e. 2-deoxy-D-ribose 5-phosphate aldolase (DERA)] |
| Non-aldolases: lipases and proteases |
• Lipases catalyzed the aldol reaction between benzaldehyde derivatives with acetone33 |
| • Alcalase (protease from Bacillus licheniformis) catalyzed the aldol addition of 4-nitrobenzaldehyde and acetone34 |
| • Porcine pancreas lipase (PPL) favored the aldol product (vs. olefin products) especially in more hydrophobic deep eutectic solvent (DES)35 |
| Henry reaction (nitroaldol addition): hydroxy nitrile lyases, transglutaminase, lipases, and D-aminoacylase |
• Alcalase was able to catalyze the Henry reaction between 4-nitrobenzaldehyde and nitromethane34 |
| • Enzymatic Henry reaction in in TX-100/H2O/[BMIM][PF6] microemulsions was examined36 |
| • Gelatin and collagen proteins showed great potential as catalysts for Henry reactions37 |
| Knoevenagel condensation |
Lipases, α-amylase, protease, papain, D-aminoacylase, Baker's yeast, ene-reductase (NerA), and bovine serum albumin (BSA) |
• Immobilized lipase B from Candida antarctica (CALB) catalyzed decarboxylative aldol reactions of aromatic aldehydes and β-ketoesters38 |
| • But no promiscuous catalytic activity for the decarboxylative aldol addition and Knoevenagel reaction between 4-nitrobenzaldehyde and ethyl acetoacetate catalyzed by CALB39 |
| • PPL displayed higher reaction rates and yields for Knoevenagel condensation in water-mimicking ionic liquids (ILs) than tert-butanol, glymes, and [BMIM][Tf2N]. But tertiary amide solvents allowed 8.2–11.1 folds of increases in the initial reaction rate than dual-functionalized ILs40 |
| • Baker's yeast as the whole cell biocatalyst catalyzed the Knoevenagel condensations between aryl aldehydes and malononitrile (or ethyl cyanoacetate, or 2,4-thiazolidinedione)41 |
| Michael addition (1,4-addition) |
Lipases, proteases, D-aminoacylase, duplex DNA, G-quadruplex DNA, and DNA/RNA-derived hybrid catalysts |
• CALB mutant exhibited much faster Michael addition rates than the wild type42 |
| • Acetamide acted as co-catalyst of CALB to promote Michael additions of aromatic nitroolefins and less-activated ketones43 |
| • In contrast to other studies, one study44 reported no stereoselectivity for lipase-catalyzed Michael additions |
| • Hydroxy-functionalized ionic liquids (ILs) led to higher Michael addition yields than longer alkyl chain-substituted ILs45 |
| Friedel–Crafts alkylation and acylation |
Peptides, methyltransferases, dimethylallyl-tryptophan synthases, biosynthetic enzyme CylK, squalene hopene cyclases (SHCs), artificial metalloenzyme, and acyltransferase (ATase) |
• Several methyltransferases originally found in bacteria catalyzed Friedel–Crafts alkylations of coumarins, naphthalenediols, and aromatic amino acids46–49 |
| • The artificial LmrR metalloenzyme promoted the enantioselective Friedel–Crafts alkylation50 |
| • A mutant of ATase (known as PpATaseCH) showed five-time higher activities than the wild type51 |
| Mannich reaction |
Acylase, lipases, trypsin, α-amylase, and Alcalase |
• Neat organic solvents resulted in the Schiff base product (>90%) instead of the Mannich product while the addition of water favored the Mannich reaction when catalyzed by lipases52 |
| • Trypsin from hog pancreas was found a more effective catalyst than lipases and α-amylase for Mannich reactions53 |
| Morita–Baylis–Hillman (MBH) reaction |
Lipases, esterases, and Alcalase, |
• The MBH reaction catalyzed by Alcalase was non-specific protein catalysis because the denatured protease produced similar yields under the same conditions34 |
| • A primitive computationally designed protein acted as an efficient and enantioselective MBHase to promote the MBH reaction between activated alkenes and aldehydes54 |
| Diels–Alder reaction |
Diels–Alderases such as macrophomate synthase (MPS) and AbyU, solanapyrone synthase, and ribozymes |
• For MPS-catalyzed Diels–Alder reactions, the C–C bond forming step was previously debated whether it is Michael-aldol process or Diels–Alder reaction.55 Later, this step was suggested to be a stepwise Michael-aldol reaction instead of a Diels–Alder reaction56 |
| • A de novo computational method was used to design the active site that is suitable for catalyzing a model Diels–Alder reaction57 |
| Acyloin condensations via thiamine diphosphate (ThDP)-dependent enzymes |
Acetohydroxy acid synthase (AHAS, EC 2.2.1.6), benzoylformate decarboxylase (BFD, EC 4.1.1.7), benzaldehyde lyase (BAL, EC 4.1.2.38), pyruvate decarboxylase (PDC, EC 4.1.1.1), phenylpyruvate decarboxylase (PhPDC, EC 4.1.1.43), keto acid decarboxylase (EC 4.1.1.72), transketolase (TK, EC 2.2.1.1), 1-deoxy-D-xylulose 5-phosphate synthase (DXPS, EC 2.2.1.7), flavoenzyme cyclohexane-1,2-dione hydrolase (CDH, EC 3.7.1.11), flavoenzyme YerE, Bacillus stearothermophilus acetylacetoin synthase, and ThDP-dependent PigD and MenD |
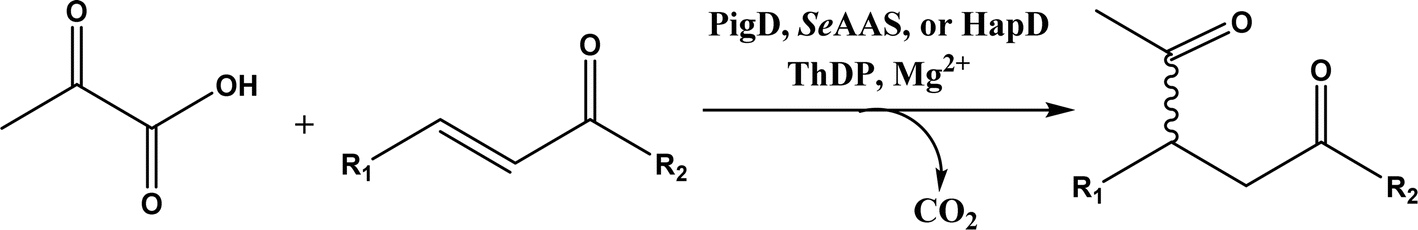
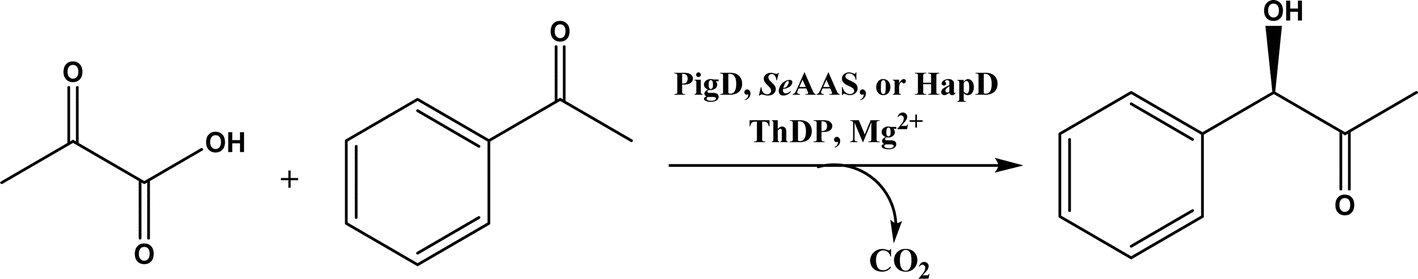
• Two new ThDP-dependent enzymes, SeAAS from Saccharopolyspora erythraea and HapD from Hahella chejuensis were identified to catalyze intermolecular Stetter reactions and benzoin condensation with high enantioselectivity58 |
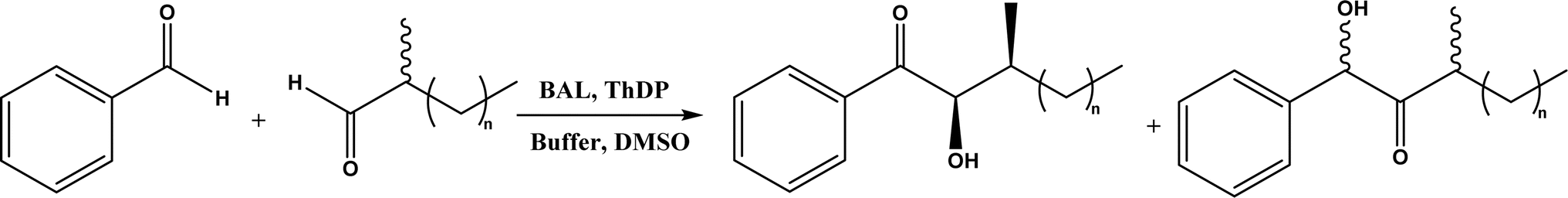
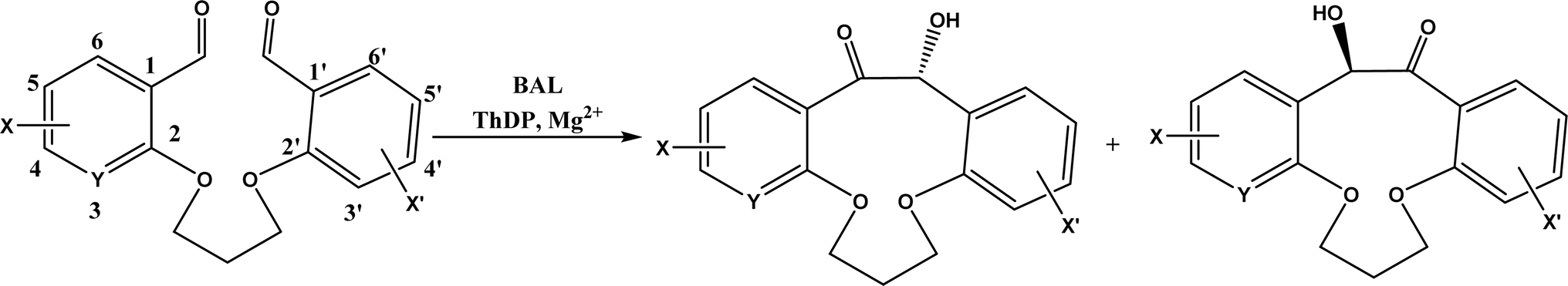
| • Benzaldehyde lyase (BAL) in mixtures of deep eutectic solvents (DES) and water exhibited high activities and good enantioselectivities (27–99% ee) for carboligation reactions of aldehydes59 |
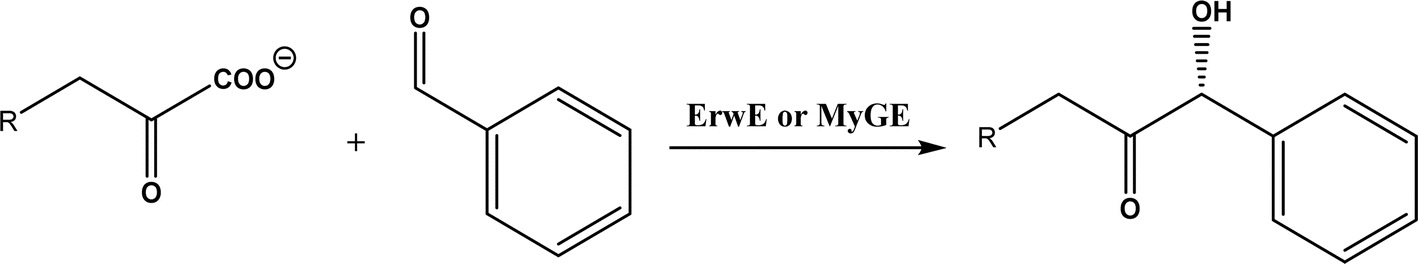
| • A subclass of (myco)bacterial ThDP-dependent enzymes (e.g., ErwE and MyGE) could extend the donor substrate range from achiral α-keto acids and simple aldehydes to customized chiral α-keto acids60 |
| Oxidative and reductive C–C bond formation |
Cytochrome P450 enzymes, redG, nonheme iron mono- and dioxygenases, flavoproteins (such as berberine bridge enzyme), radical S-adenosylmethionine enzymes, laccase, and peroxidases |
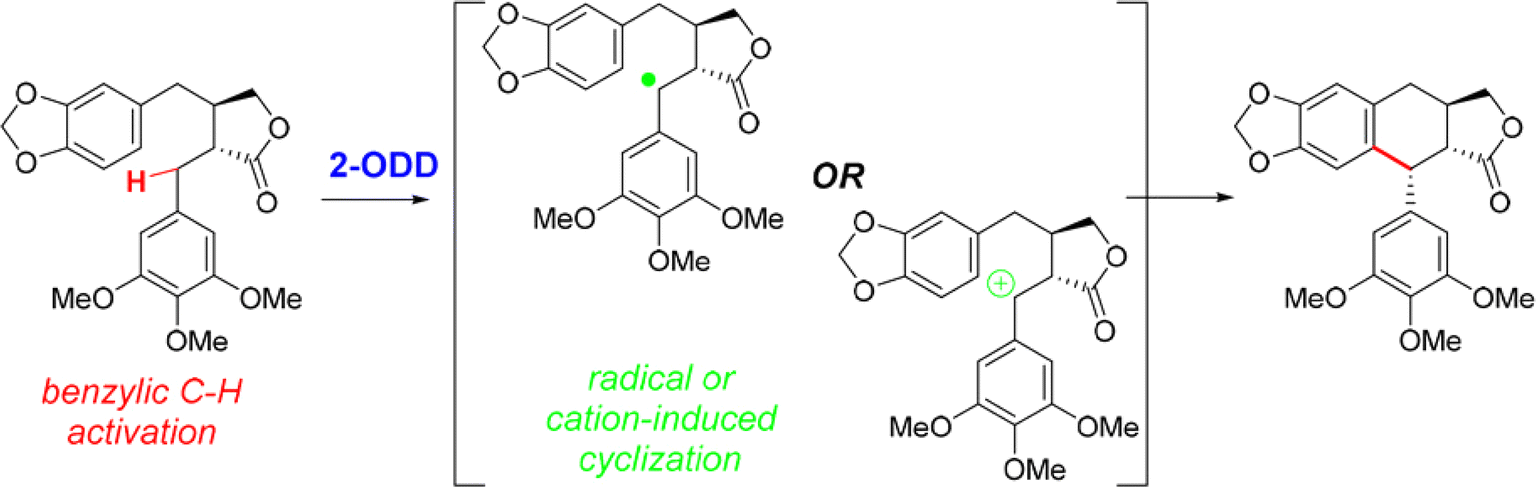
• A nonheme iron enzyme, 2-oxoglutarate/Fe(II)-dependent dioxygenase (2-ODD), promoted the oxidative cyclization in the etoposide biosynthetic pathway61 |
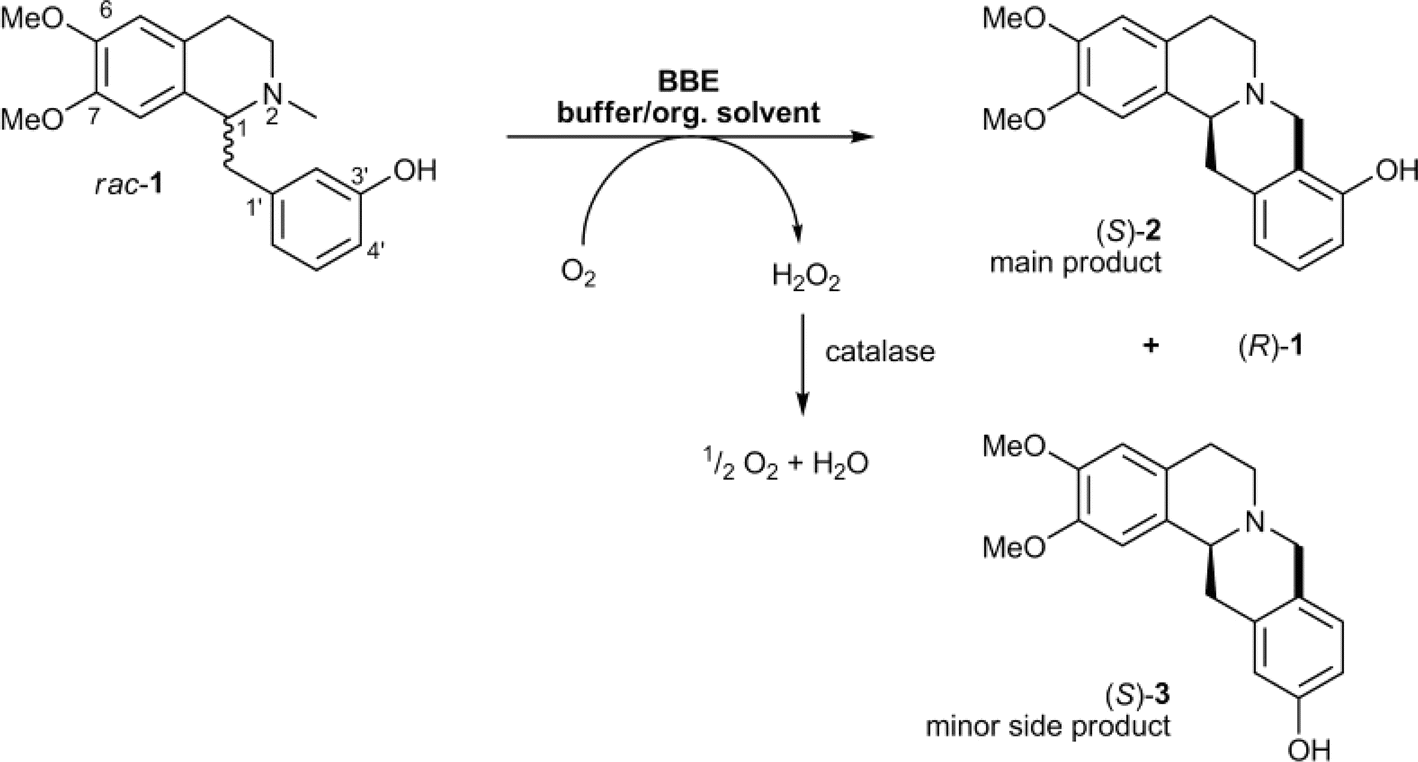
| Flavin-dependent ‘ene’-reductases (EREDs), the ‘ene’-reductase from Caulobacter segnis (CsER), and wild-type ene-reductases from the Old Yellow Enzyme (OYE) |
• The wild-type ene-reductases from the Old Yellow Enzyme (OYE) family favored the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond reduction instead of carbocyclization; however, single-site replacement of the critical proton donor Tyr residue (e.g., Tyr190 in OPR3, Tyr169 in YqjM) with a non-protic Phe or Trp led to more cyclization products62 C double bond reduction instead of carbocyclization; however, single-site replacement of the critical proton donor Tyr residue (e.g., Tyr190 in OPR3, Tyr169 in YqjM) with a non-protic Phe or Trp led to more cyclization products62 |
| C–C bond formation through C1 resource utilization |
Formaldehyde to valuable chiral molecules by using aldolases and ThDP-dependent enzymes, CO2 conversions using carboxylases, formaldehyde transformations using C–C ligases, CO and formate conversions via C–C ligases, CO2 and succinyl coenzyme A (SCoA) conversion to 2-oxoglutarate and CoA |
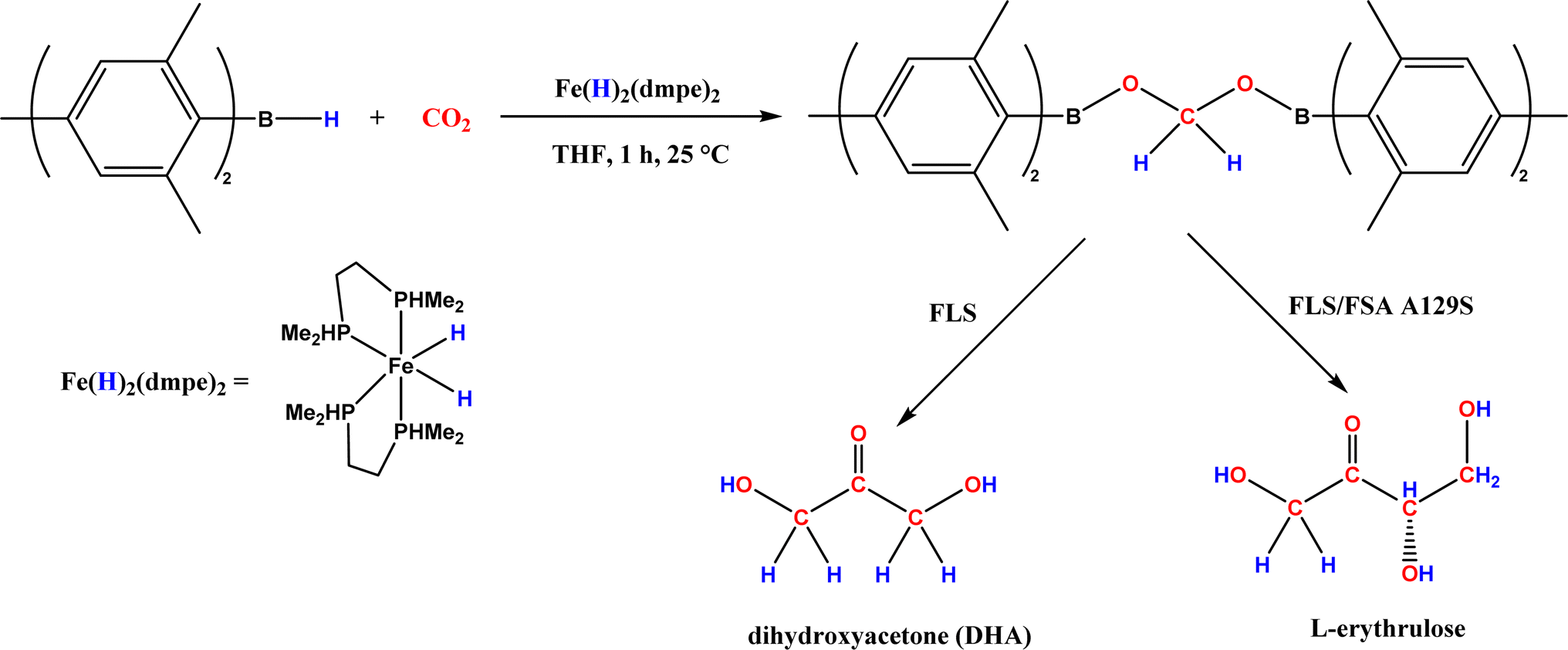
• Formaldehyde was converted to glycolaldehyde by formolase or its variants, and glycolaldehyde was further converted to erythrulose (C4 sugar) by another formolase variant63 |
| • CO2 was converted to a bis(boryl)acetal compound first, followed by selective enzymatic reactions to afford C3 (dihydroxyacetone, DHA) by using a formolase (FLS), or optically pure C4 (L-erythrulose) through a cascade reaction using FLS and D-fructose-6-phosphate aldolase (FSA) A129S variant64 |
| Radical enzymes for C–C bond formation |
Radical S-adenosylmethionine (SAM) enzymes such as pyruvate-formate lyase (PFL), spore photoproduct lyase (SPL), and benzylsuccinate synthase (BSS), O2-sensitive and hydrocarbon activating glycyl radical enzymes (GREs) including a subset known as x-succinate synthases [e.g., benzylsuccinate synthase (BSS), 4-isopropylbenzylsuccinate synthase (IBSS), hydroxybenzylsuccinate synthase (HBSS), naphthyl-2-methylsuccinate synthase (NMSS), and 1-methylalkylsuccinate synthase (MASS)], cytochrome P450 |
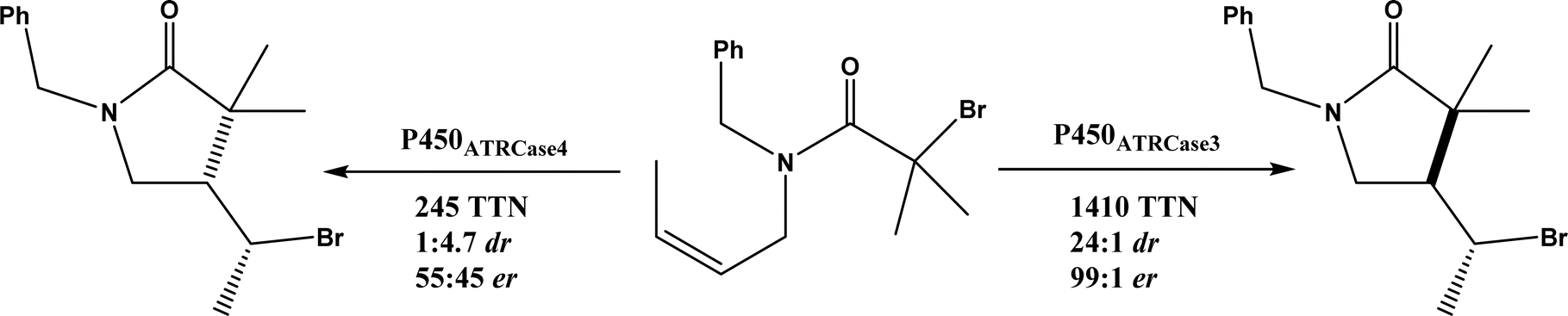
• Cytochrome P450 could be engineered to have a fine control of the radical addition step and the halogen rebound step during stereoselective atom-transfer radical cyclization (ATRC)65 |
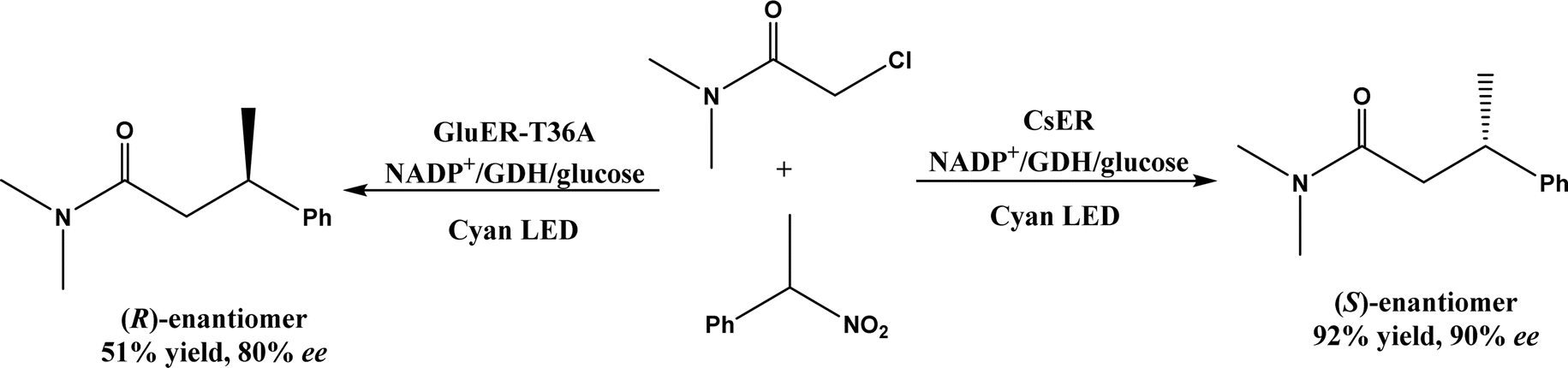
| • Recent examples include SAM for enzymatic redox reactions in C–C bond formation,66 the benzylic radical/carbocation intermediate initiating the C–C bond formation for a nonheme iron enzyme called 2-oxoglutarate/Fe(II)-dependent dioxygenase (2-ODD),61 and the formation of nitro radical anion during ‘ene’-reductase CsER-catalyzed cross-electrophile couplings (XECs) between alkyl halides and nitroalkanes67 |
| Other C–C bond formation mechanisms |
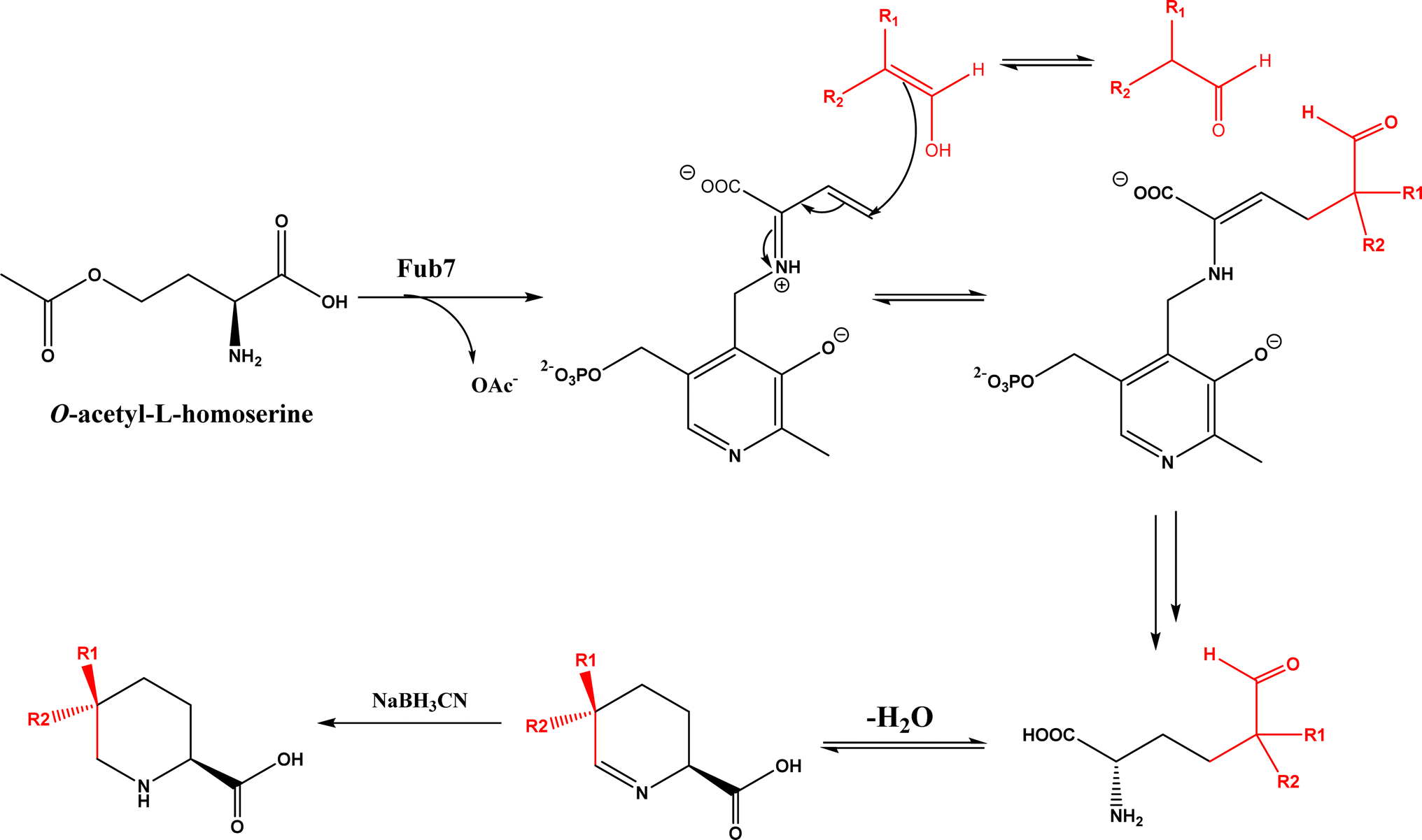
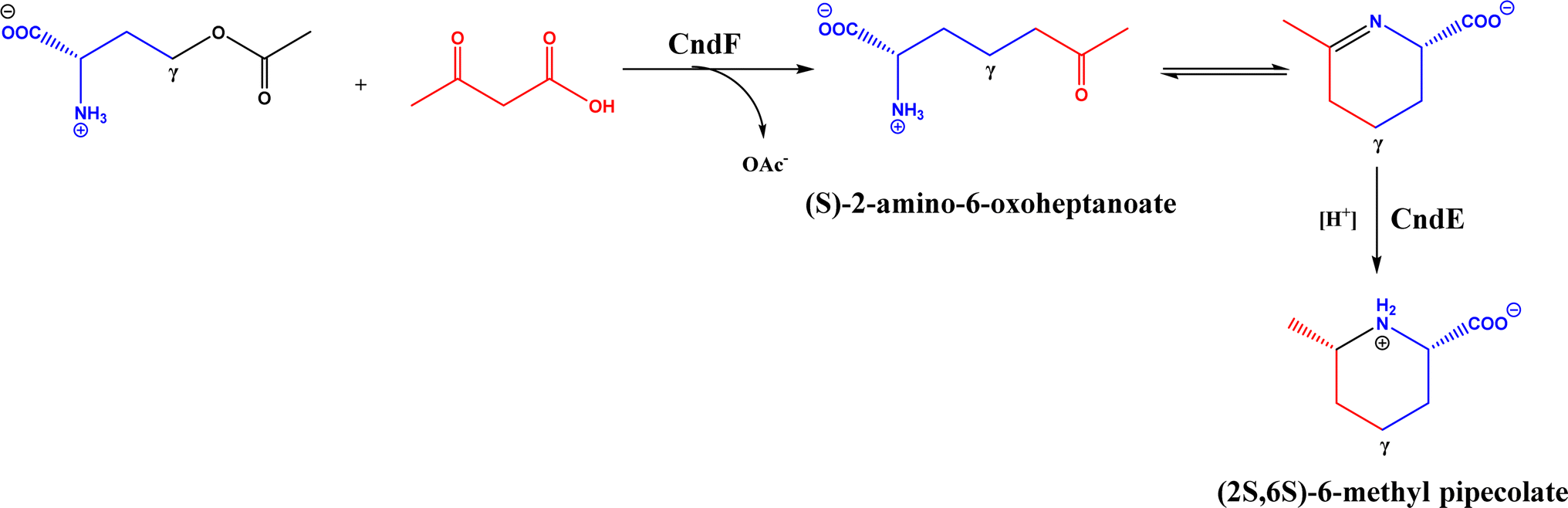
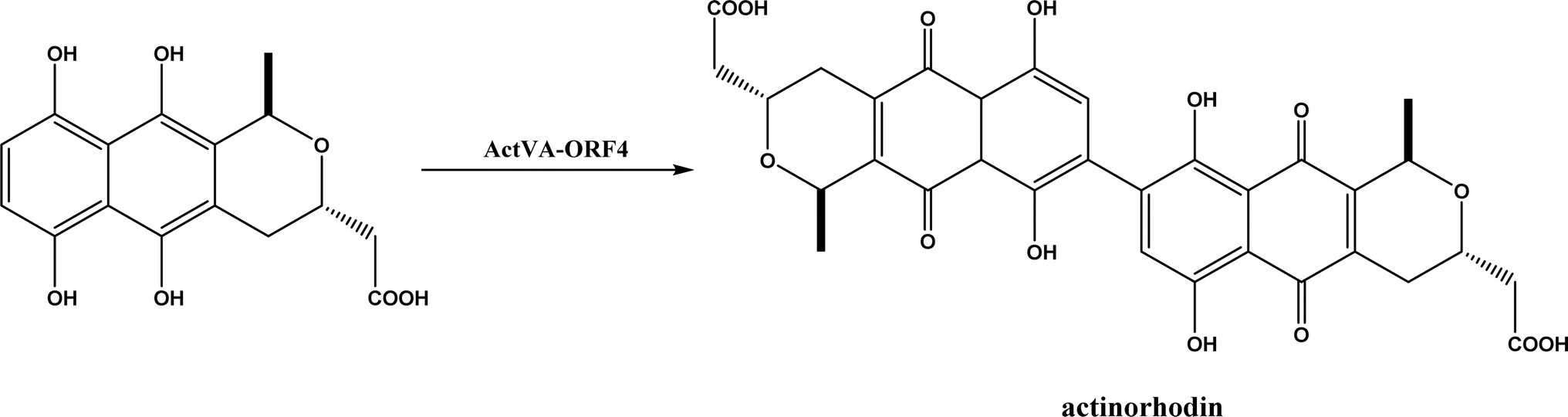
PLP-dependent enzymes such as CndF and Fub7, hydroxynitrile lyases (HNLs) or oxynitrilases, NAD(P)H-dependent ActVA-ORF4, cytochrome P411, ketosynthase, deoxypodophyllotoxin synthase, cis-isoprenyl diphosphate synthase, carboxymethylproline synthase, engineered SAM-dependent sterol methyltransferase |
• CndF catalyzed the C–C coupling of O-acetyl-L-homoserine with 3-oxobutanoic acid to form (S)-2-amino-6-oxoheptanoate, which equilibrates with a cyclic Schiff base; a further reduction by a stereoselective imine reductase CndE gave (2S, 6S)-6-methyl pipecolate68 |
| • Engineered SAM-dependent sterol methyltransferase for C-methylation of unactivated alkenes in mono-, sesqui- and diterpenoids to yield C11, C16 and C21 derivatives with high chemo- and regioselectivity69 |
2. Aldol reaction
2.1. Aldolases
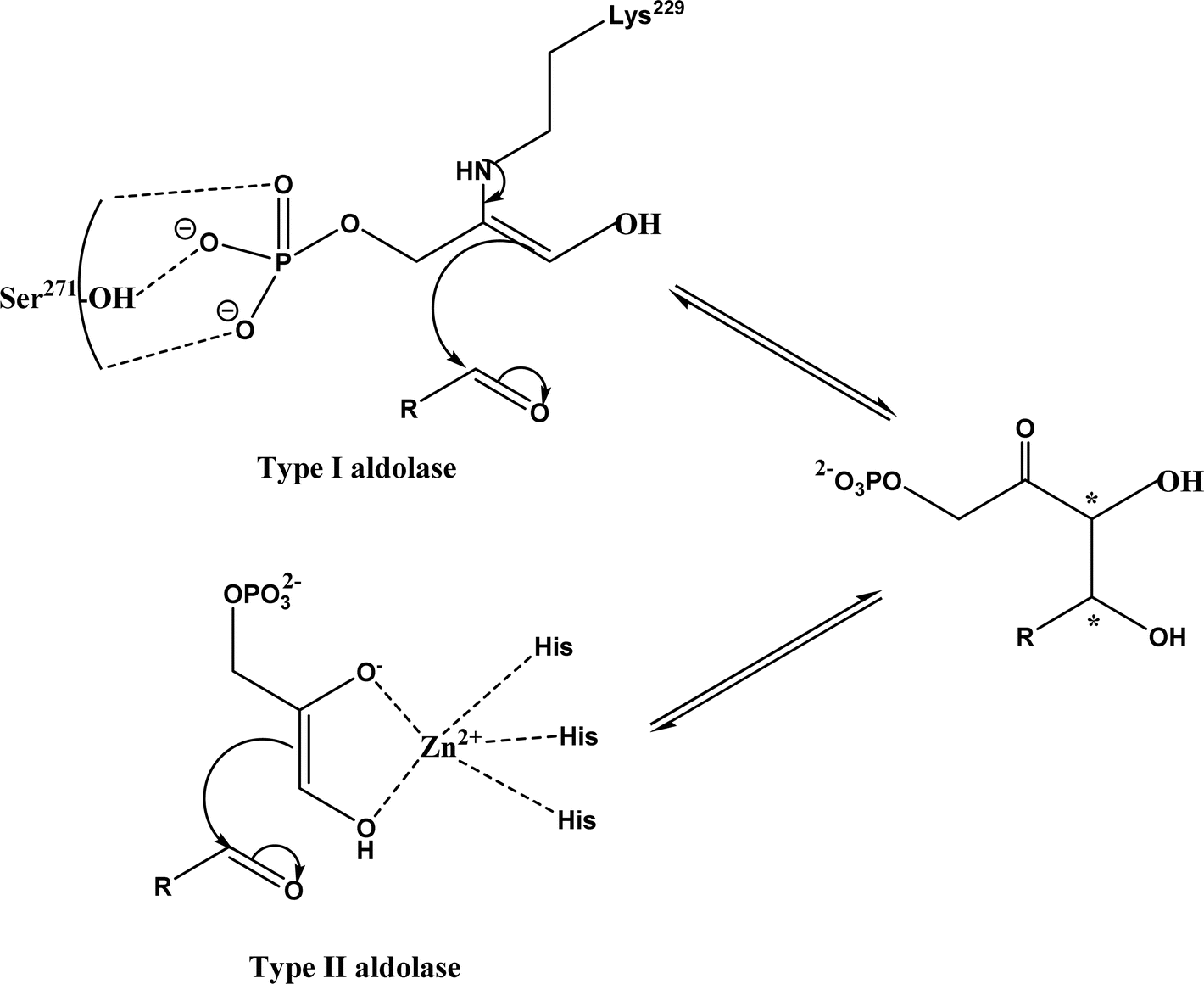
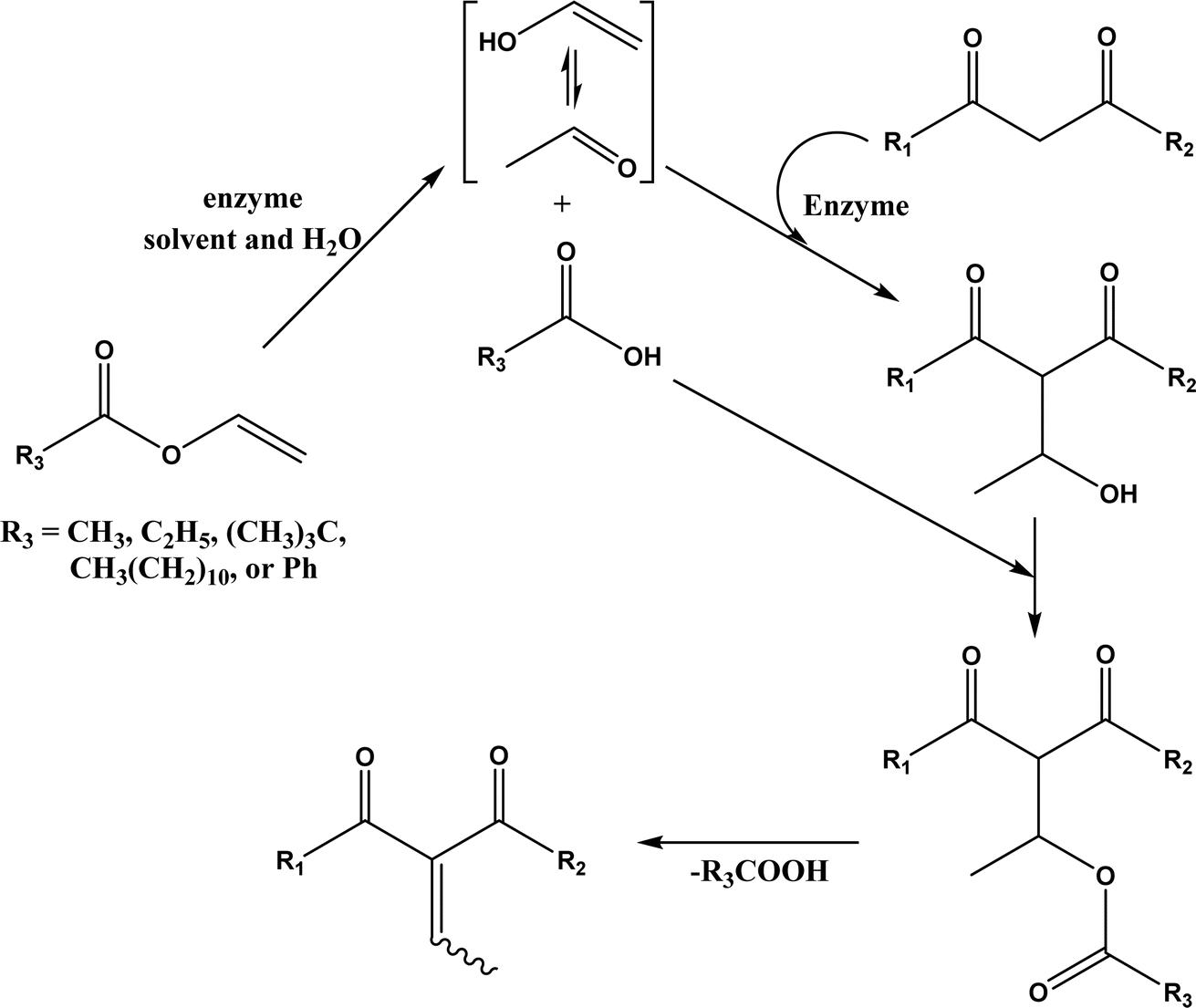
Aldol addition catalyzed by different aldolases is a power tool to facilitate C–C bond ligations and form up to two asymmetric centers as depicted by earlier reviews.13,14,21,23,24,70–78 In particular, formaldehyde as an emerging C1 source can be converted to valuable β- and γ-hydroxycarbonyl compounds (especially carbohydrates) by aldolases and thiamine diphosphate (ThDP)-dependent enzymes.79 Aldolases belong to a subset of lyases (EC 4), and promote the addition of a ketone donor (nucleophile) to an aldehyde acceptor stereoselectively. Aldolases abstract α-proton of the carbonyl group to produce a carbon nucleophile bound at the active site, which attacks the acceptor component (i.e., electrophile) such as aldehyde's carbonyl carbon. Based on the reaction mechanism (Fig. 2), there are two types of aldolases, where Type I (known as lysine-dependent; found in animals and plants) promotes the enamine formation from an imine (a Schiff base) between carbonyl group and lysine residue of the enzyme, and Types II (known as metal-dependent; found in bacteria and fungi) forms an enolate via chelation to a Lewis-acidic transition metal cation (usually Zn2+).21,23,70,72 Conversely, based on their donor specificity, aldolases can be categorized into five types based on different donor substrates:23,24,71,72,74,80,81 (a) pyruvate, phosphoenolpyruvate, oxaloacetate, or 2-oxobutyrate, (b) dihydroxyacetone phosphate (DHAP), (c) dihydroxyacetone (DHA) and other unphosphorylated analogues (e.g., D-fructose-6-phosphate aldolase), (d) pyridoxal 5′-phosphate (PLP) (also known as threonine aldolases or glycine/alanine-dependent; threonine aldolases and serine hydroxymethyltransferase catalyze the addition of glycine/alanine to aldehydes),24 and (e) acetaldehyde [i.e. 2-deoxy-D-ribose 5-phosphate aldolase (DERA)]. It is interesting to note that 4-fluorothreonine transaldolase from Streptomyces sp. MA37 (FTaseMA) possesses both serine hydroxymethyltransferase and aldolase catalytic domains to catalyze transaldol reactions, and the aldolase domain is Zn2+-dependent; basically, this is the PLP-dependent enzyme fused with a metal-binding domain.82 Since the forementioned review articles have discussed various types of aldolases and their applications, this paper intends not to duplicate the effort but rather to focus on recent advances in several areas.
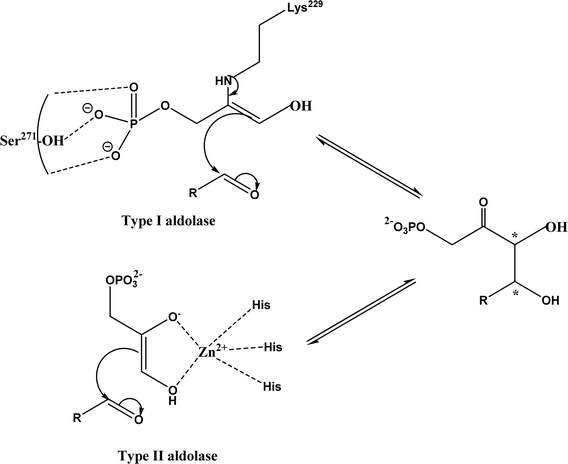 |
| | Fig. 2 Aldol addition mechanisms by Type I and II aldolases (dihydroxyacetone phosphate (DHAP)-dependent enzyme as an example).23,72,83 | |
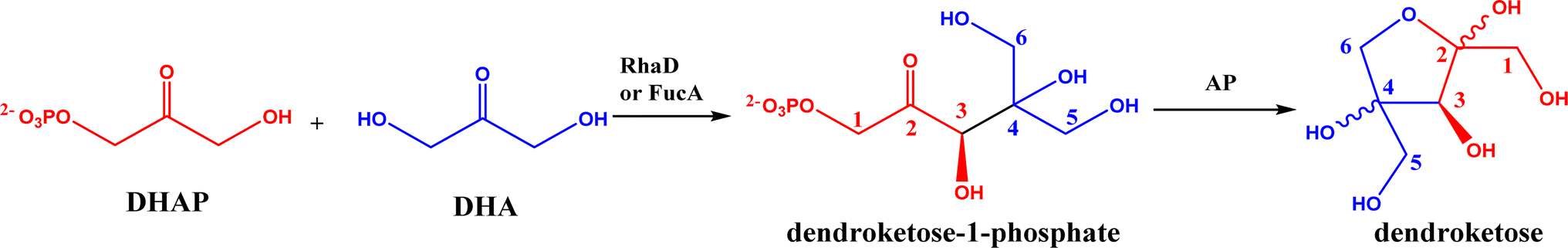
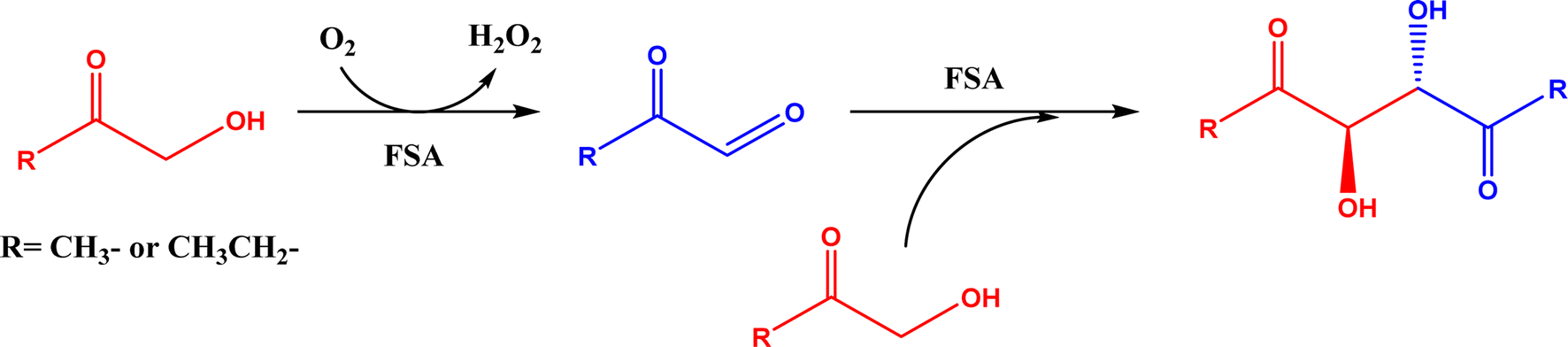
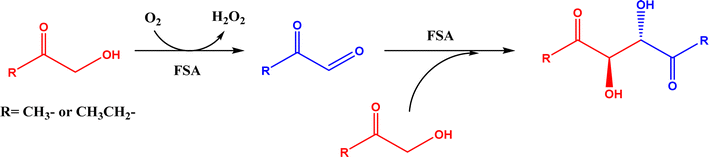
2.1.1 Aldolase donors and acceptors. Aldolases have high substrate specificity for donor structures, but are more tolerant to various aldol acceptor structures.72 For this reason, one bottleneck of aldolase-catalyzed C–C bond formation is the limited choice of donors.23 One solution is to rely on direction evolution, protein engineering, and computational de novo enzyme design to develop more robust and more substrate-tolerant aldolases [e.g., the transaldolase family84,85 and the discovery of fructose-6-phosphate aldolase (FSA) by serendipity86,87].21–24 Meanwhile, several aldolases have been identified to take ketones as acceptors in enzymatic aldol addition. Wang and co-workers27 reported that 4-hydroxy-4-methyl-2-oxoglutarate/4-carboxy-4-hydroxy-2-oxoadipate (HMG/CHA) aldolase from Pseudomonas putida F1 in the presence of Mg2+ or Mn2+ could catalyze the homo-aldol addition of pyruvate, or the addition of pyruvate to 4-hydroxy-2-keto acids including oxaloacetate (Fig. 3). In another study,28 DHAP-dependent L-rhamnulose-1-phosphate aldolase (RhaD) from Bacteroides thetaiotaomicron in the presence of Co2+ is capable of stereoselectively catalyzing the aldol reaction between DHAP and several α-hydroxylated ketones (e.g., hydroxyacetone, 1-hydroxybutanone, hydroxypyruvate, and L-erythrulose) affording optically pure tertiary alcohols with 76–95% yields, although no reaction was observed for non-activated ketones such as acetone, butanone, cyclopentanone, and 4-hydroxybutan-2-one. Yang et al.29 examined the catalytic behavior of L-rhamnulose-1-phosphate aldolase (RhaD) and L-fuculose-1-phosphate aldolase (FucA) from Escherichia coli in the aldol reaction of DHAP and DHA, and the subsequent catalysis by acid phosphatase (AP) to remove phosphate group and also form dendroketose (Fig. 4). A more recent study30 indicated that D-fructose-6-phosphate aldolase (FSA) catalyzed the oxidation and then aldol addition of hydroxyacetone or 1-hydroxy-2-butanone to form diketones, and suggested the likely mechanism being that hydroxy groups in hydroxyketones are oxidized to aldehydes (2-oxoaldehydes), which act as acceptors to react with hydroxyketones to form aldol products (Fig. 5).
 |
| | Fig. 3 HMG/CHA aldolase-catalyzed aldol addition. | |
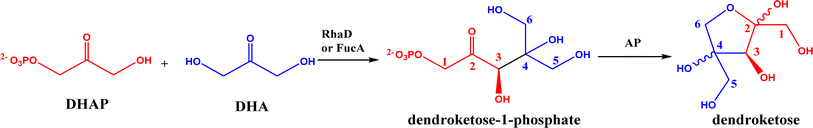 |
| | Fig. 4 Aldol addition of DHAP with DHA to form dendroketose. | |
 |
| | Fig. 5 Aldol reaction of hydroxyketones catalyzed by D-fructose-6-phosphate aldolase (FSA). | |
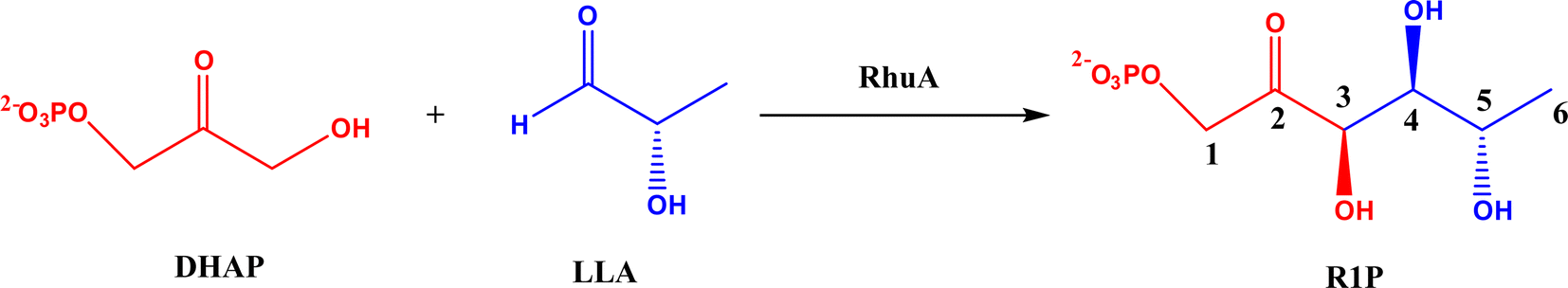
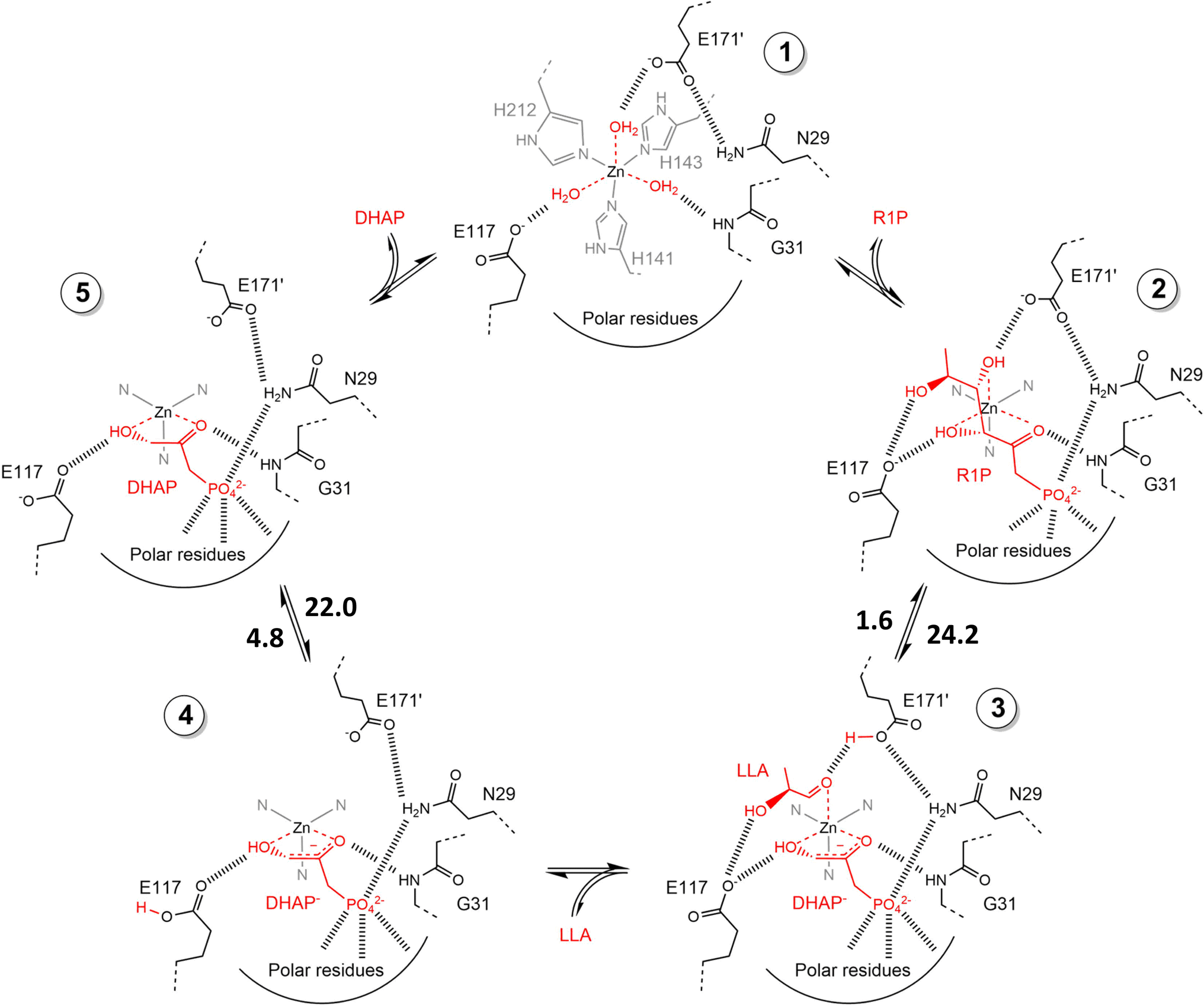
2.1.2 DHAP-dependent aldolase mechanism. To elucidate the catalytic mechanism of DHAP-dependent rhamnulose-1-phosphate aldolase (RhuA)-catalyzed aldol reaction in Fig. 6, electronic structure calculations via the DFT method were completed by considering the substrate molecules, Zn2+, and 13 neighboring residues.31 The calculations led to a five-step mechanism for the aldol cleavage as illustrated in Fig. 7: (1) the substrate R1P binds with Zn2+ through points of Zn–O interactions, and is stabilized by H-bonds and polar attraction with amino acid residues; (2) there is a proton transfer from –OH to E171′ causing the cleavage of C3–C4 bond, where the activation energy is estimated to be 24.2 kcal mol−1; (3) the release of LLA and proton transfer from E171′ to a residue E117; (4) the protonation of DHAP moiety at C-3 by E117, which requires a low activation energy of 4.8 kcal mol−1; and (5) the release of DHAP. Among these five steps, the C–C bond cleavage (Ea = 24.2 kcal mol−1) and the DHAP deprotonation (Ea = 22.0 kcal mol−1) are rate-controlling steps for retro- and aldolic reactions, respectively. Several amino acid residues (i.e., E117, E171′, G31, and N29) and the Zn2+ co-factor are key players in the mechanism; in particular, E117 and E171′ act as two acid/base catalytic residues, and E171′ is directly involved in the C–C bond formation.
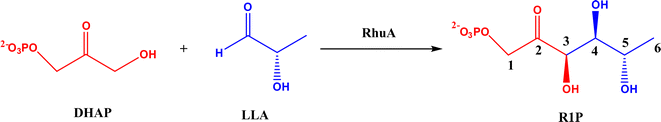 |
| | Fig. 6 Aldol addition of dihydroxyacetone phosphate (DHAP) and LLA = (S)-lactaldehyde to form L-rhamnulose-1-phosphate (R1P) catalyzed by dihydroxyacetone phosphate (DHAP)-dependent rhamnulose-1-phosphate aldolase (RhuA). | |
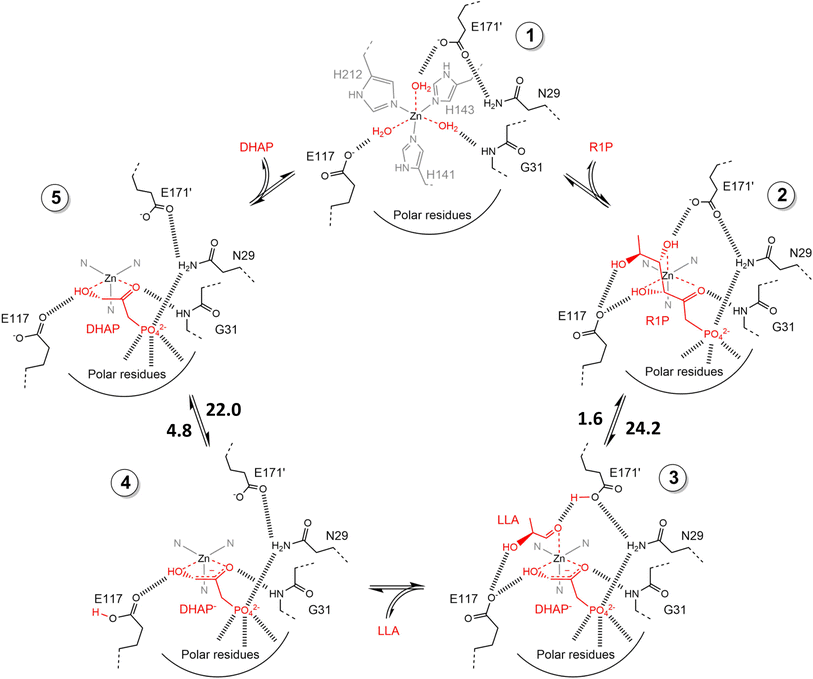 |
| | Fig. 7 Schematic view of catalytic mechanism of rhamnulose-1-phosphate aldolase (RhuA)-catalyzed retro- and aldolic reaction. These structures are geometrically optimized at the DFT level (B3LYP/LANL2DZ). The estimated activation energies are given in kcal mol−1. DHAP = dihydroxyacetone phosphate, R1P = L-rhamnulose-1-phosphate, and LLA = (S)-lactaldehyde [Reprinted/adapted with permission from reference (Fig. S11 in its ESI data).31 Copyright 2020 Elsevier]. | |
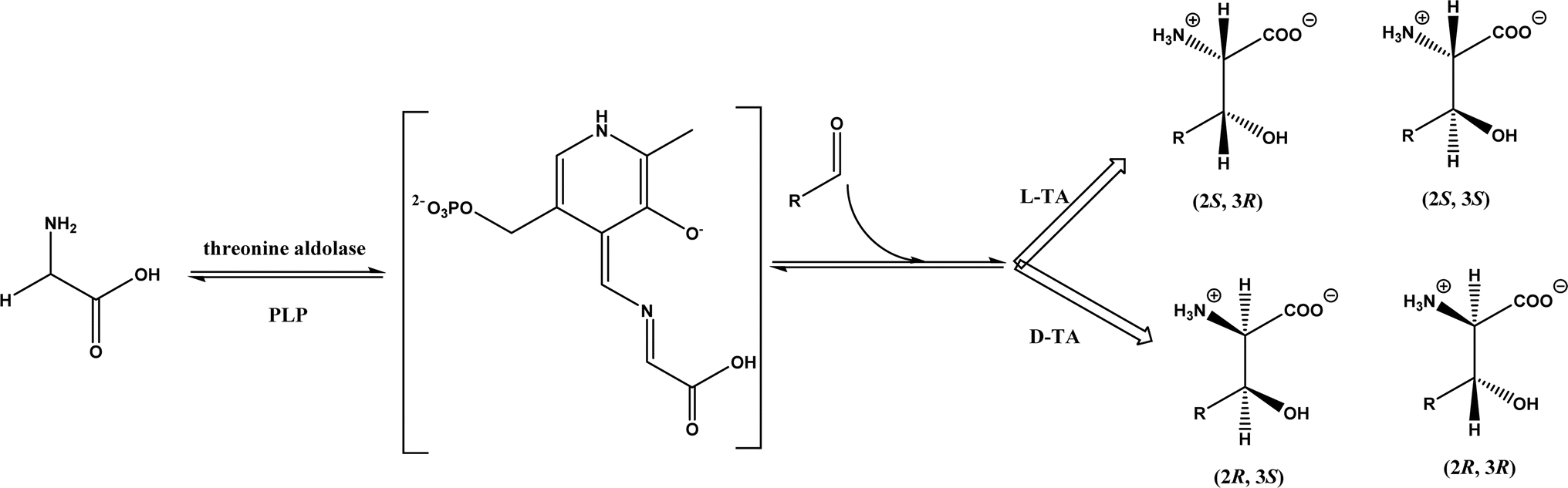
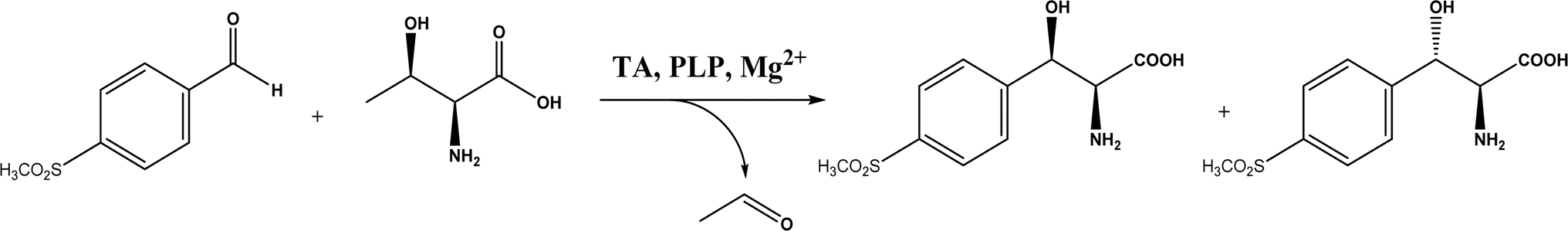
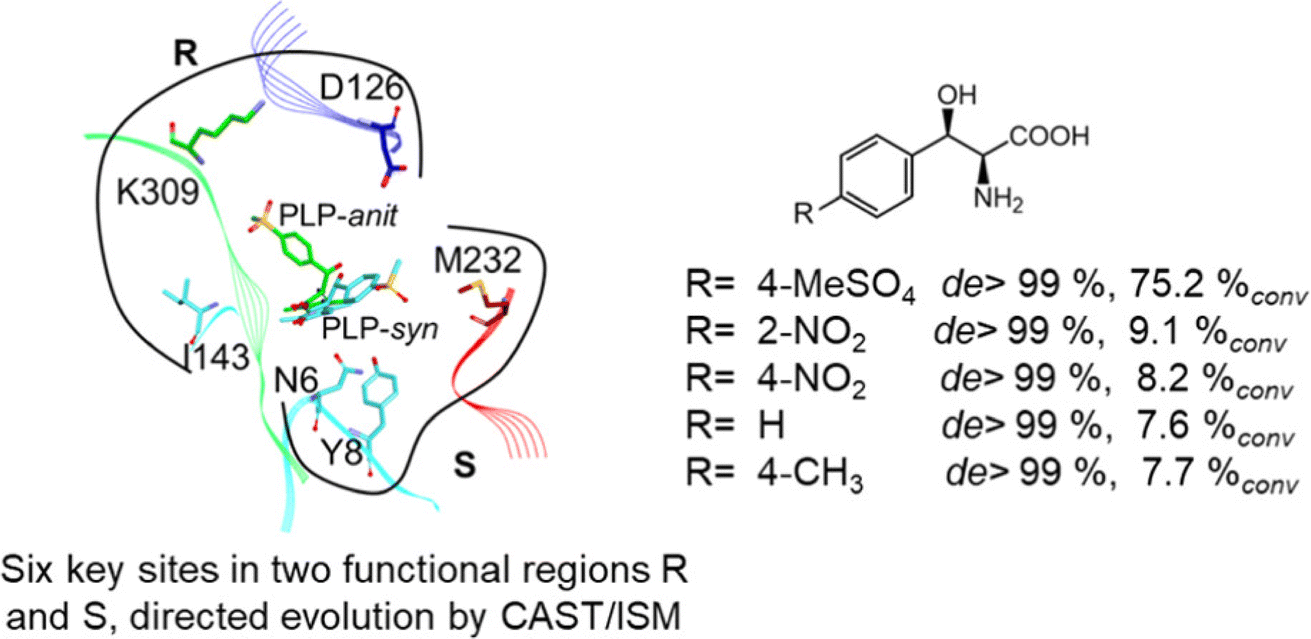
2.1.3 Threonine aldolases. As PLP-dependent enzymes, threonine aldolases (TAs) catalyze C–C coupling with various aldehydes through C–H bond activation (Fig. 8) although wild-type threonine aldolases accommodate few D-amino acids as donors. Both wild-type L-threonine aldolase from Aeromonas jandaei and D-threonine aldolase from Pseudomonas sp. were evaluated in aldol addition reactions of D- or DL-alanine with various of aliphatic and aromatic aldehydes, producing a large pool of β-hydroxy-α,α-dialkyl-α-amino acids with conversions up to >80%; in general, D-threonine aldolase showed higher diastereoselectivities than L-threonine aldolase.88 Three L-threonine aldolases (i.e., Aeromonas jandaei L-allothreonine aldolase, Escherichia coli L-threonine aldolase, and Thermotoga maritima L-allo-threonine aldolase) were evaluated for the addition of glycine to various aldehyde acceptors; it was identified that A. jandaei L-allo-TA gave the best conversion and diastereomeric excess, and preparative-scale reactions (2.0 mmol of aldehyde and 10 mmol glycine) led to 16–50% isolated yields.89 The Lin group90 studied L-threonine transaldolase from Pseudomonas sp. in Escherichia coli whole cells for catalyzing p-methylsulfonyl benzaldehyde and L-threonine to form L-p-methylsulfonylphenylserine in the presence of Mg2+ (Fig. 9), observing 67.1% conversion and 94.5% diastereomeric excess (de) under optimized conditions. In general, when catalyzing the aldol formation of β-hydroxy-α-amino acids, threonine aldolase (LTA) has a high selectivity for the Cα position but a varied selectivity for Cβ, resulting in a moderate diastereoselectivity. To further improve or invert its stereoselectivity towards aromatic aldehydes, threonine aldolase from Pseudomonas sp. was mutated for its amino acid residues that interact with amino and hydroxyl groups of the substrate; the change in the Cβ-stereoselectivity was explained by molecular docking that the distances were modified between hydroxyl group of the substrate and imidazole groups of H133 and H89.32 A combinatorial active-site saturation test/iterative saturation mutagenesis (CAST/ISM) was used to categorize 27 amino acid residues residing in the substrate pocket into two groups based on their functional region prior to the combinatorial mutation of L-threonine aldolase. One of the variants, known as RS1 (mutations Y8H, Y31H, I143R, and N305R), enabled an improved synthesis of L-syn-3-[4-(methylsulfonyl)phenylserine] in a 20 L reactor with 99.5% diastereomeric excess (de) and 73.2% yield; this variant also improved the diastereoselectivity for other aromatic aldehydes (Fig. 10).91
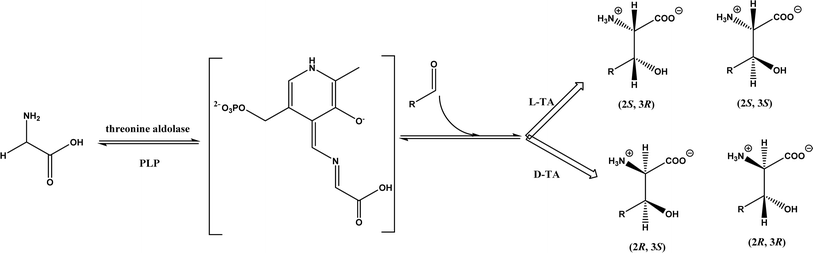 |
| | Fig. 8 Threonine aldolase (TA)-catalyzed aldol addition of glycine with aldehyde. | |
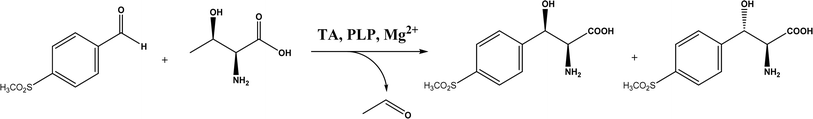 |
| | Fig. 9 L-p-Methylsulfonylphenylserine synthesis catalyzed by threonine aldolase (TA). | |
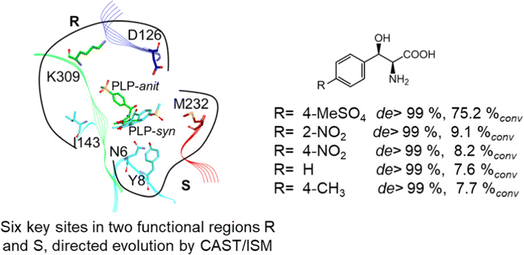 |
| | Fig. 10 Directed evolution of L-threonine aldolase leading to improved diastereoselectivity [Reprinted with permission from ref. 83 Copyright 2021 American Chemical Society]. | |
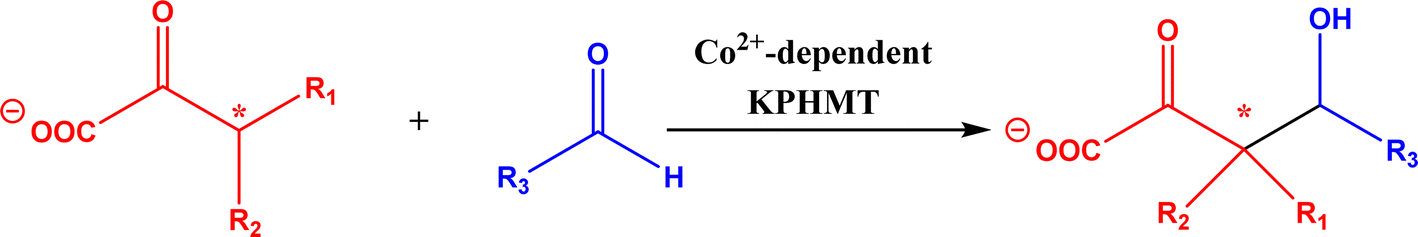
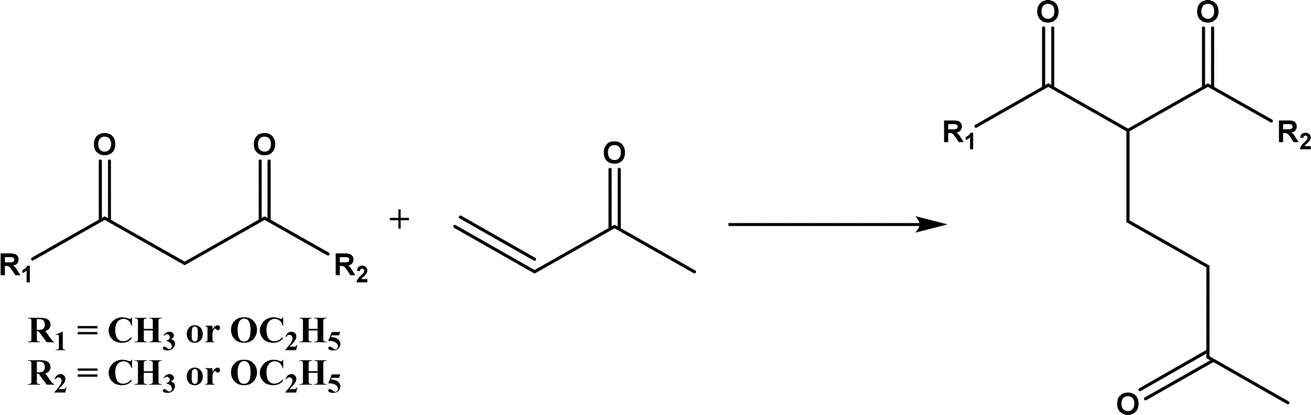
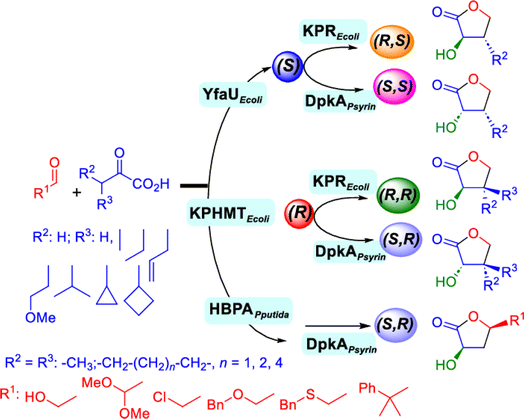
2.1.4 Other recent advances. Prior to the development of biosynthesis of L-sorbose and L-psicose using biocatalytic aldol addition in the Corynebacterium glutamicum strain, Yang et al.18 conducted the in vitro aldol addition of DHAP and five different aldehydes catalyzed by 1,6-diphosphate aldolases (FruA) or tagatose 1,6-diphosphate (TagA) aldolases, and noticed that some aldolases lost their stereoselectivity when L-glyceraldehyde was the acceptor, producing both L-sorbose and L-psicose. This group collaborated with other groups92 to further develop in vitro synthesis of 2-deoxy-D-ribose and rare ketoses (e.g., D-allulose, L-tagatose, D-sorbose, L-fructose, and D-xylulose) from aldol reaction of D-glyceraldehyde 3-phosphate (or DHAP) with various aldehydes catalyzed by 2-deoxy-D-ribose 5-phosphate aldolase, D-fructose 1,6-bisphosphate aldolase (FruA), or L-rhamnulose 1-phosphate aldolase (RhaD); D-glyceraldehyde 3-phosphate and DHAP were produced from starch and pyrophosphate by using six artificial ATP-free cascade enzymatic reactions. 2-Deoxy-D-ribose and rare ketoses could be produced with >80% yields from high concentrations of substrates. A thermophilic recombinant aldolase, knowns as rhamnulose 1-phosphate aldolase from Thermotoga maritima activated by Co2+ as a divalent metal ion cofactor, was identified to show a maximum activity at 95 °C and its half-life time was 44 h and 33 h respectively at 80 and 95 °C; this aldolase maintained 90% of its initial activity in 40% acetonitrile, almost 100% of its activity in 20% DMSO, 50% of the activity in 25% DMF, and about 40% of the activity in 10% isopropanol and THF.93 This aldolase could be suitable for aldol reactions conducted under extreme conditions.94The Clapés group95 employed Co2+-dependent 3-methyl-2-oxobutanoate hydroxymethyltransferase (KPHMT, EC 2.1.2.11) and its variants to catalyze aldol additions of 3,3-disubstituted 2-oxoacids to aldehydes (Fig. 11) forming 3,3,3-trisubstituted 2-oxoacids, which were further converted to 2-oxolactones, 3-hydroxy acids, and ulosonic acid derivatives carrying gem-dialkyl, gem-cycloalkyl, or spirocyclic quaternary centers. Many of these chiral precursors are important to the preparation of medicinal molecules. As a type of pyruvate-dependent aldolases, sialic acid aldolases [also referred as N-acetylneuraminate pyruvate lyases (NPL)] promoted the reversible reaction of pyruvate and aldose to sialic acids. When catalyzing the reaction of pyruvate with D-mannose (or D-galactose), recombinant sialic acid aldolase originated from freshwater snail Biomphalaria glabrata (sNPL) displayed a different diastereoselectivity from sialic acid aldolase from chicken (chNPL).96 In addition, the wild-type sNPL could catalyze the aldol reaction of pyruvate with different aliphatic aldehydes to produce 4-hydroxy-2-oxoates with 21–78% yields, while chNPL could not. The Clapés group97 converted various L-α-amino acids to 2-substituted 3-hydroxycarboxylic acid derivatives via a cascade enzymatic reaction method, which involved the oxidative deamination of L-α-amino acids to 2-oxoacid intermediates by L-α-amino acid deaminase from Cosenzaea myxofaciens, followed by the aldol addition reaction with formaldehyde to form (R)- or (S)-3-substituted 4-hydroxy-2-oxoacids (36–98% yields and 91–98% ee for each enantiomer) when mediated by metal-dependent carboligases known as 2-oxo-3-deoxy-L-rhamnonate aldolase (YfaU) and ketopantoate hydroxymethyltransferase (KPHMT), respectively. Similar cascade approach involving enzymatic aldol addition was used to prepare γ-hydroxy-α-amino acid derivatives,98 and (R)- or (S)-2-substituted 3-hydroxycarboxylic esters.99 Moreno and co-workers100 developed a two-step strategy for synthesizing 2-hydroxy-4-butyrolactone derivatives (Fig. 12): in the first step, different chiral aldol adducts were prepared from 2-oxoacids and aldehydes by using different aldolases including 3-methyl-2-oxobutanoate hydroxymethyltransferase (KPHMT), 2-keto-3-deoxy-l-rhamnonate aldolase (YfaU), and trans-o-hydroxybenzylidene pyruvate hydratase-aldolase from Pseudomonas putida (HBPA); in the second step, 2-oxogroup of the aldol adduct was reduced by ketopantoate reductase and Δ1-piperidine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase with promiscuous ketoreductase ability. This enzymatic tandem reaction approach produced two enantiomers of 2-hydroxy-4-butyrolactone (>99% ee), twenty one (2R, 3S), (2S, 3S), (2R, 3R), or (2S, 3R)-2-hydroxy-3-substituted-4-butyrolactones [with diastereomeric ratio (d.r.) ranging from 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 to 98:2], and six (2S, 4R)-2-hydroxy-4-substituted-4-butyrolactones (with d.r. ranging from 87:13 to 98:2). In addition, the diastereoselectivity of aldolases could be tuned via protein engineering.25,26 Mutants of L-threonine aldolase from Cellulosilyticum sp were constructed by the combinatorial active-site saturation test/iterative saturation mutation method to improve the syn addition diastereoselectivity from 37.2% to 99.4%, or to invert the reaction to anti addition with 97.2% diastereoselectivity.101
:40 to 98:2], and six (2S, 4R)-2-hydroxy-4-substituted-4-butyrolactones (with d.r. ranging from 87:13 to 98:2). In addition, the diastereoselectivity of aldolases could be tuned via protein engineering.25,26 Mutants of L-threonine aldolase from Cellulosilyticum sp were constructed by the combinatorial active-site saturation test/iterative saturation mutation method to improve the syn addition diastereoselectivity from 37.2% to 99.4%, or to invert the reaction to anti addition with 97.2% diastereoselectivity.101
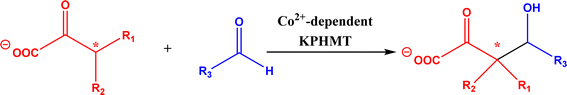 |
| | Fig. 11 Aldol addition of 3,3-disubstituted 2-oxoacids to aldehydes catalyzed by 3-methyl-2-oxobutanoate hydroxymethyltransferase (KPHMT). | |
 |
| | Fig. 12 Enzymatic tandem aldol addition and carbonyl reduction to synthesize homochiral 2-hydroxy-4-butyrolactone derivatives [Reprinted from ref. 92 which is an open-access publication licensed under CC-BY 4.0.]. | |
2.2. Non-aldolase enzymes
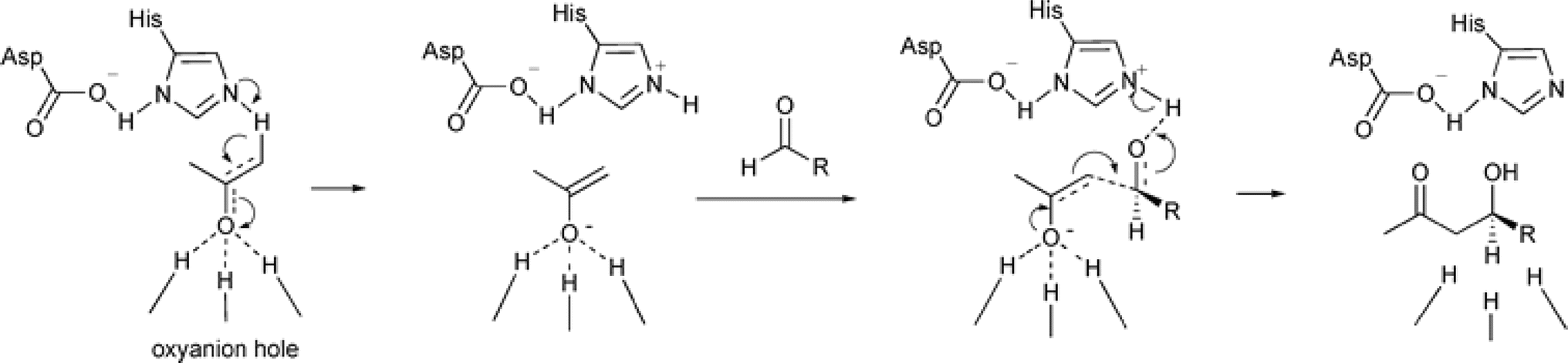
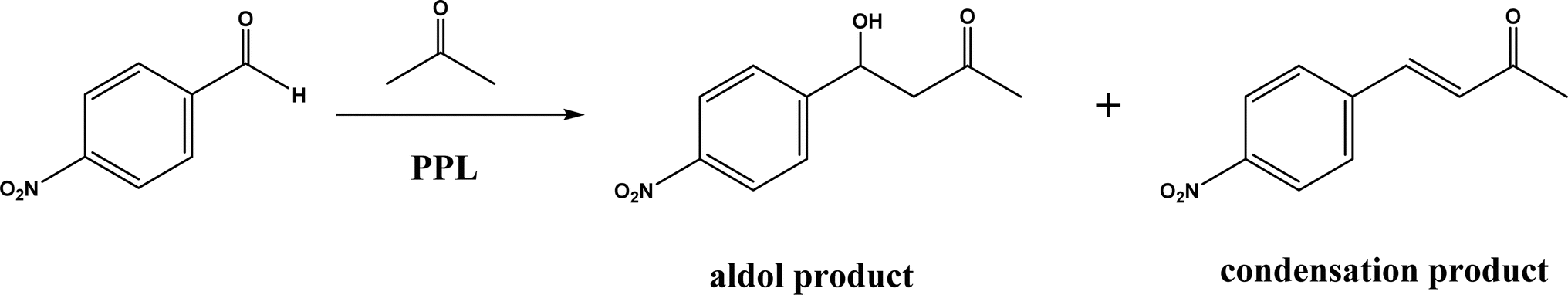
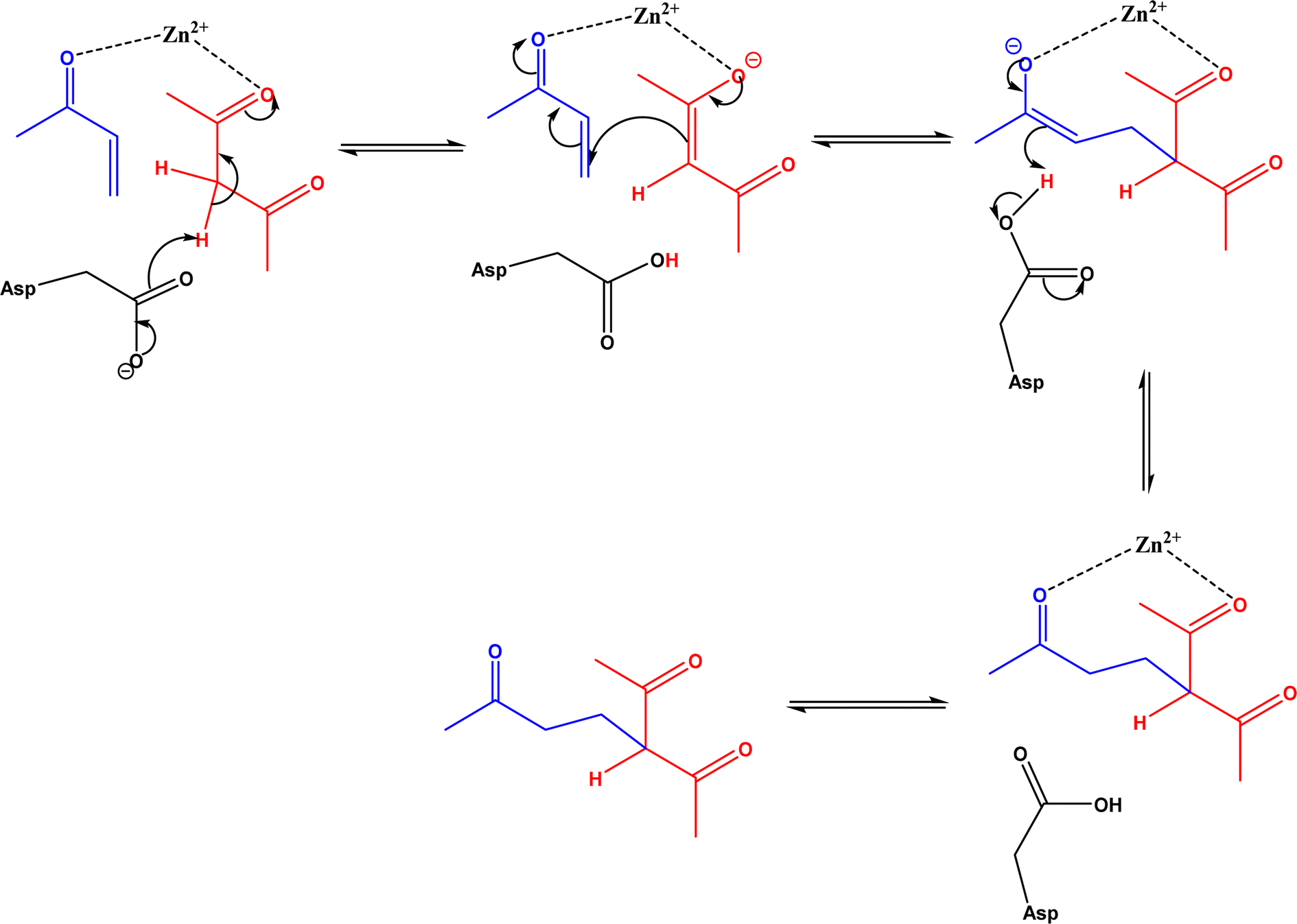
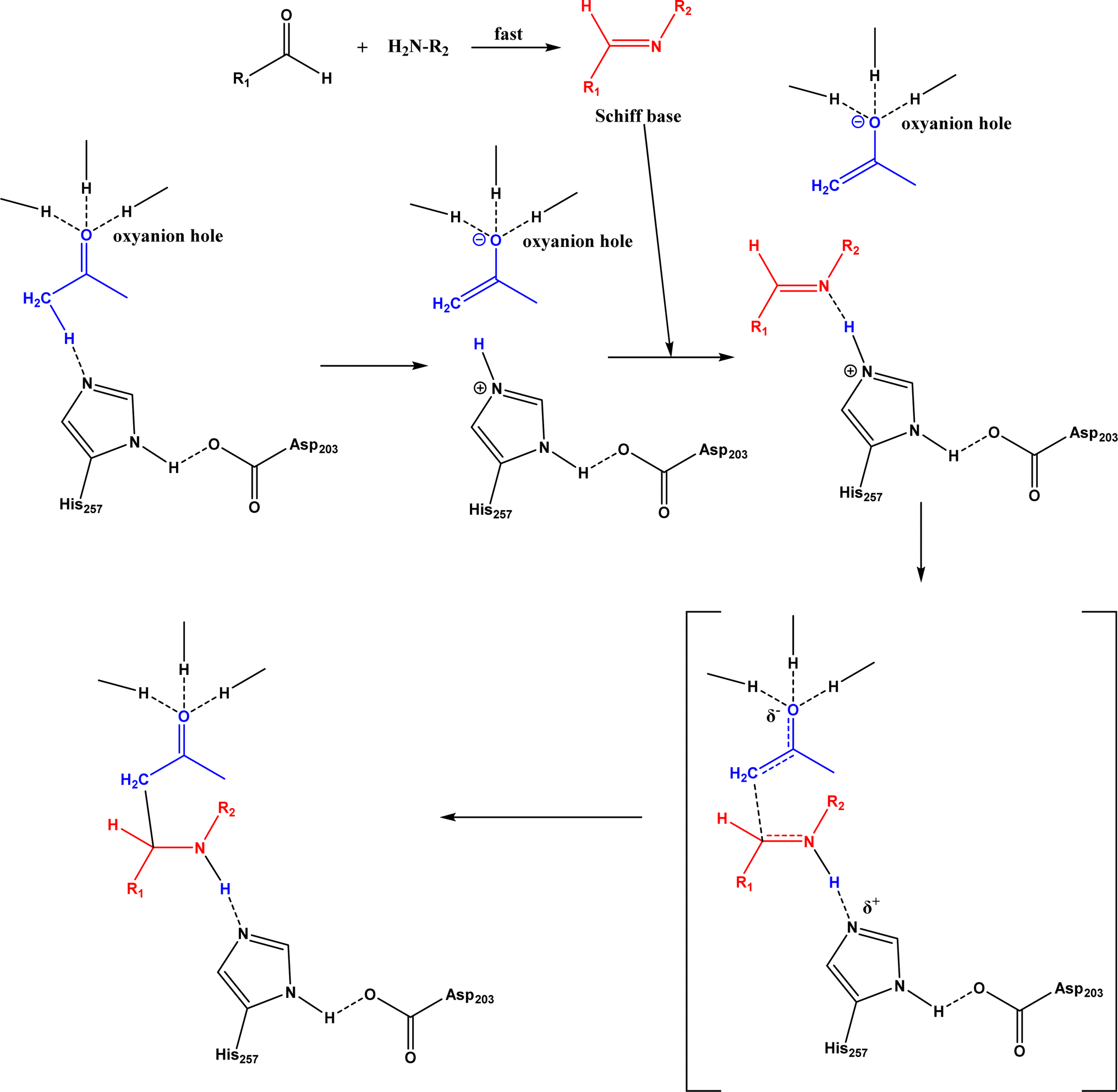
Non-aldolase biomolecular catalysts [such as lipases and proteases,24 and catalytic antibodies72] have been developed to overcome the issues with aldolases. Several lipases especially lipase from porcine pancreas (PPL) were able to catalyze the aldol reaction between benzaldehyde derivatives with acetone in the presence of 20 v% water, producing aldol products with yields up to 96.4% but relatively low enantiomeric excesses (ees, 9.4–43.6%).33 The mechanism is depicted in Fig. 13: acetone interactions with the Asp–His dyad and the oxyanion, proton transfer from acetone to His residue forming an enolate, proton transfer to aldehyde and C–C bonding formation with acetone, and the release of aldol adduct from the oxyanion hole.33 Alcalase (protease from Bacillus licheniformis) could catalyze the aldol addition of 4-nitrobenzaldehyde and acetone with 20% water at 45 °C (see Fig. 14) producing 68% aldol product (with 13% ee and 94% selectivity of aldol product vs. the condensation product).34 The Holtmann group35 conducted the aldol reaction of 4-nitrobenzaldehyde with acetone (see Fig. 15) in several deep eutectic solvents (DES), and found that bovine serum albumin (BSA) showed no specificity for aldol and olefin products; however, PPL favored the aldol product especially in more hydrophobic DES although the initial reaction rate was faster in hydrophilic DES (i.e., choline chloride/glycerol at 1:1.5 molar ratio). One drawback of DES in this application is the low solubility of 4-nitrobenzaldehyde in DES (0.2–1.3 M). The study did not report the configuration of asymmetric center.
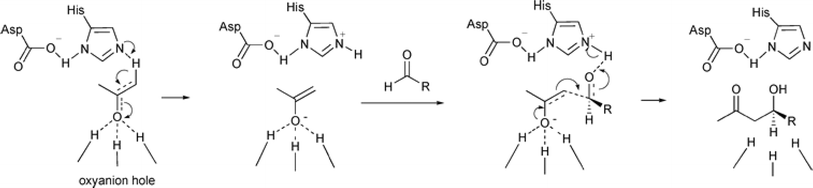 |
| | Fig. 13 Mechanism of lipase-catalyzed aldol reaction [Reprinted with permission from ref. 33 Copyright 2004 Royal Society of Chemistry]. | |
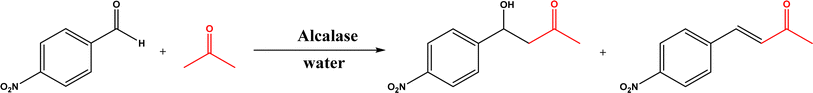 |
| | Fig. 14 Aldol addition and condensation of 4-nitrobenzaldehyde with acetone. | |
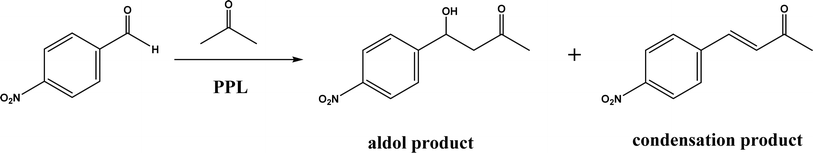 |
| | Fig. 15 Aldol addition and condensation of 4-nitrobenzaldehyde with acetone. | |
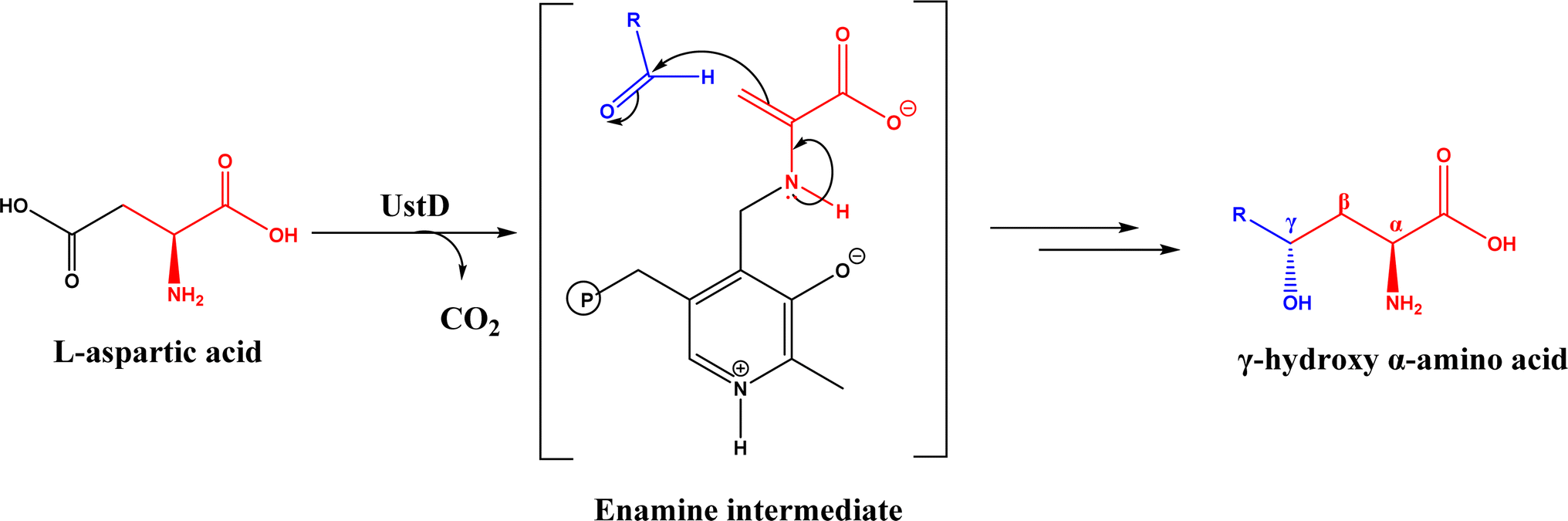
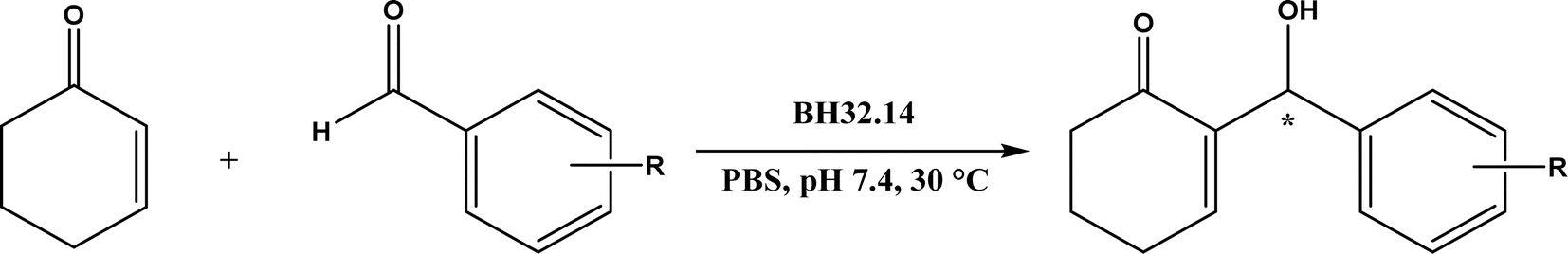
Nuclease p1 from Penicillium citrinum was found capable of catalyzing aldol reactions between benzaldehyde derivatives and cyclic ketones, resulting in higher ee and diastereomeric ratio under solvent-free condition than in organic solvents and water.102 UstD is a PLP-dependent enzyme that is engaged in the biosynthesis of Ustiloxin B (an inhibitor of microtubilin polymerization). In an aldol reaction shown in Fig. 16, UstD eliminates carboxyl group (C–C activation) from L-aspartic acid to form a nucleophilic enamine intermediate, which attacks the aldehyde to yield γ-hydroxy α-amino acid.103 The decarboxylation step produces CO2, which makes this aldol reaction irreversible. This mechanism is fundamentally different from classic Type I aldolase, where an enamine nucleophile is formed from the tautomerization of an imine. This enzyme UstD showed high stereoselectivities for aromatic and aliphatic aldehydes even on gram-scale.103,104
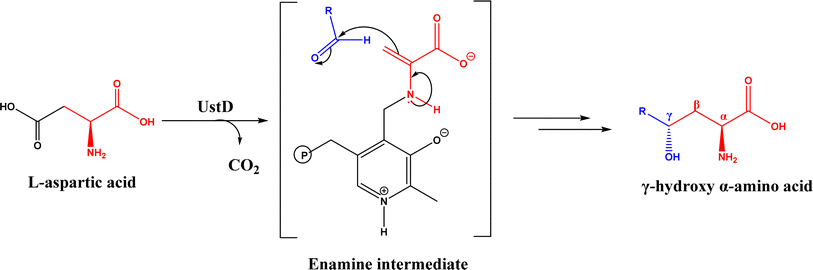 |
| | Fig. 16 Decarboxylative aldol reaction of L-aspartic acid with aldehyde catalyzed by UstD. | |
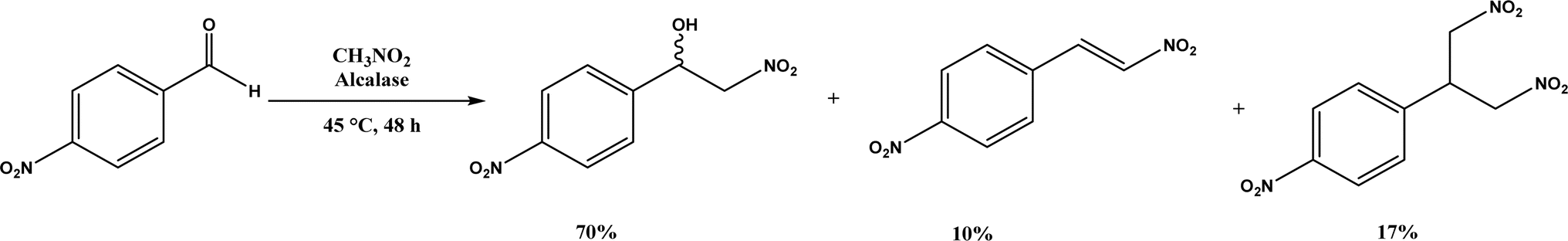
Henry Reaction, also known as nitroaldol addition, is the nucleophilic addition of nitroalkanes to aldehydes or ketones to synthesize β-nitro alcohols, which can be further manipulated to biologically active compounds. This reaction is usually promoted by base catalysts such as hydroxides, alkoxides, carbonates, bicarbonates, amines, and LiAlH4, etc.105 As an extension of Henry reaction, the addition of nitroalkanes to imines (called aza-Henry reaction) forms β-nitroamine derivatives.106 Strong base catalysts often produce byproducts from side reactions and chiral catalysts are required to generate enantioselective products. On the other hand, various enzymes (e.g., hydroxy nitrile lyases, transglutaminase, lipases, and D-aminoacylase) are mild catalysts to produce enantiopure β-nitro alcohols as detailed in a 2012 review.107 This section provides a more recent update, or studies that were not covered in the earlier review. Alcalase's active site was found capable of catalyzing the Henry reaction between 4-nitrobenzaldehyde and nitromethane at 45 °C forming racemic nitroalcohol with 70% yield and 72% selectivity (Fig. 17).34 Whole-cell baker's yeast is an affordable and effective catalyst for Henry reactions of substituted benzaldehydes and nitromethane in ethanol, resulting in 55–90% products (although enantioselectivities were not reported).108 Acylase from Aspergillus oryzae, various lipases, and BSA were evaluated in TX-100/H2O/[BMIM][PF6] microemulsions for their catalytic capabilities in Henry reaction of 4-nitrobenzaldehyde with nitromethane at 30 °C, and the reaction produced 62% yield in the absence of enzyme suggesting the catalytic role of this solvent system (without the solvent system and enzyme, the yield was 24%); the acylase gave the highest overall yield of 88% for this reaction, and 28–87% yields for other substituted benzaldehydes.36 Interestingly, gelatin and collagen proteins showed great potential as catalysts for Henry reactions of substituted benzaldehydes and nitromethane in DMSO or aqueous solution containing tetra-n-butylammonium bromide as the phase transfer catalyst (with up to 70–92% yields for those benzaldehyde derivatives containing electron-withdrawing –NO2 or –CN groups); among different gelatins, porcine skin type-A (PSTA) gelatin, bovine skin type-B (BSTB) gelatin, and cold-water fish skin (CWFS) gelatin showed high catalytic activities; the first-order rate constant increased in the order of chitosan < gelatin < bovine serum albumin (BSA) < collagen.37 CALB immobilized on hydrophobic PS-DVB (polystyrene-divinylbenzene) beads improved the enzymatic activity in water by 15–18 times when compared with the commercial Novozym 435; the Henry reaction of 4-nitrobenzaldehyde and nitromethane catalyzed by this new lipase preparation at 40 °C obtained 87% yield in water, 40% yield in [BMIM][Tf2N], and 22% yield in tert-butanol, but were all significantly higher than those catalyzed by Novozym 435 although no stereoselectivity was discussed.109 However, inhibited or thermally deactivated enzyme preparation still showed a considerable amount of catalytic activity, implying a different mechanism not related to the active site of lipase is in play. FT-IR spectra indicate that α-helix and β-turn structures not related to hydrogen bonds of CALB are significantly higher in new enzyme immobilization than in Novozym 435 (54% vs. 15%).
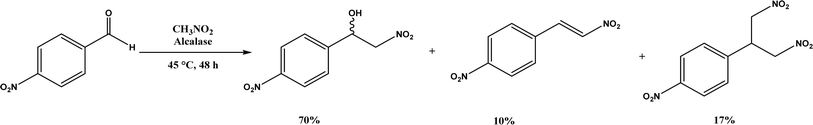 |
| | Fig. 17 Henry reaction between 4-nitrobenzaldehyde and nitromethane. | |
3. Knoevenagel condensation
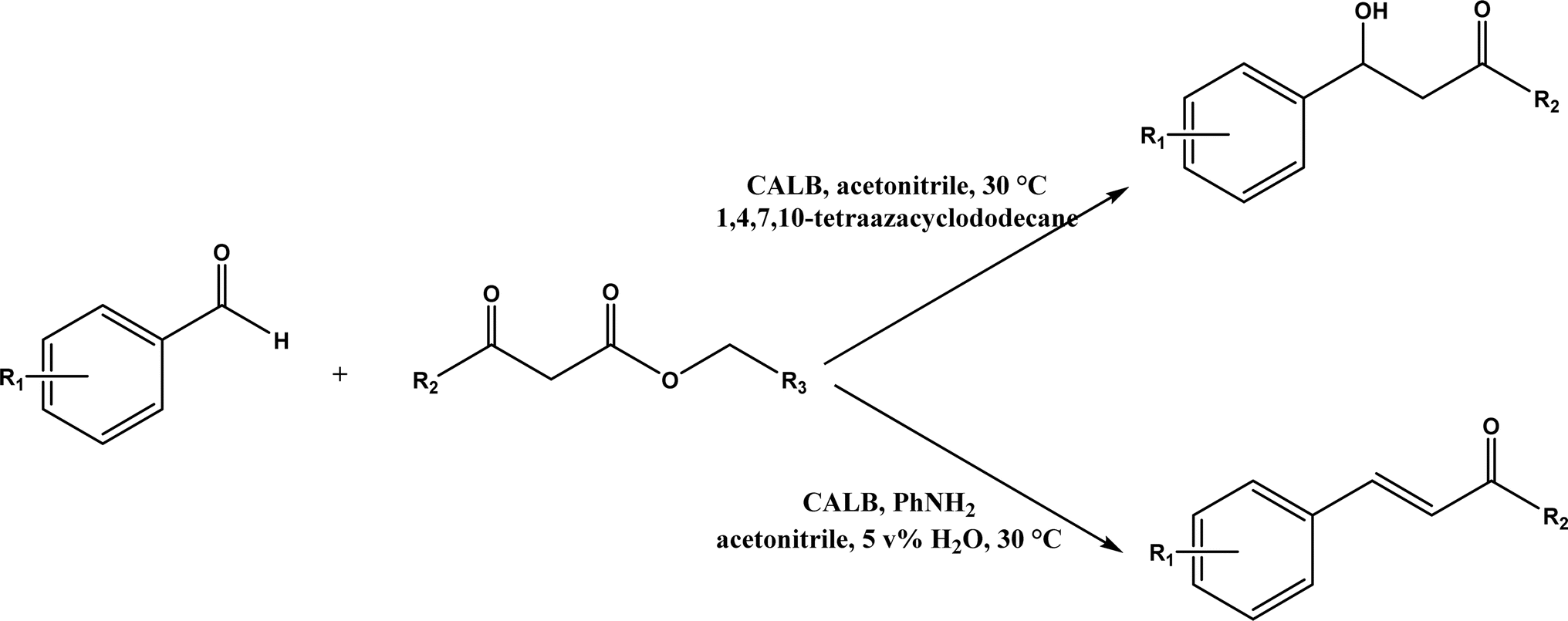
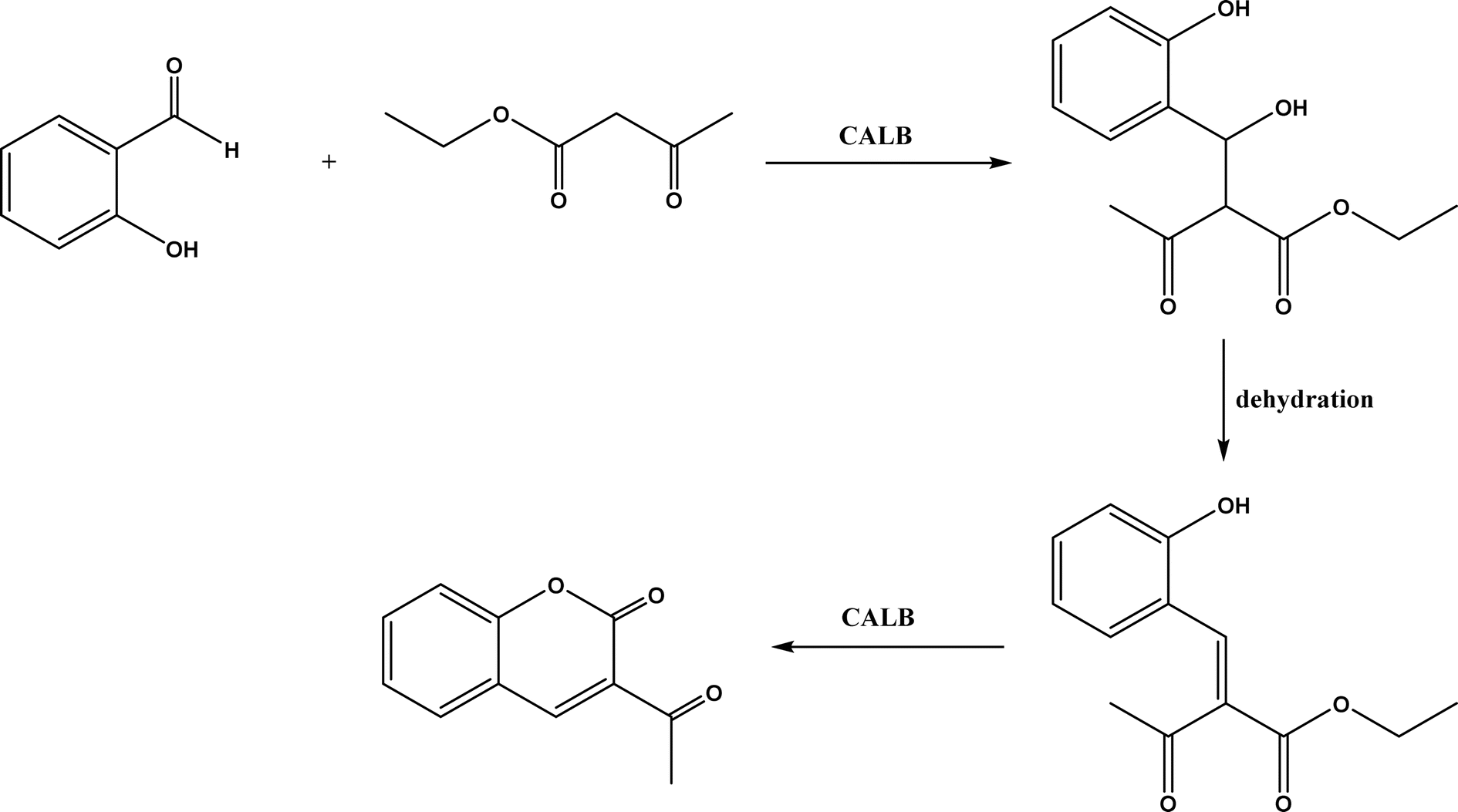
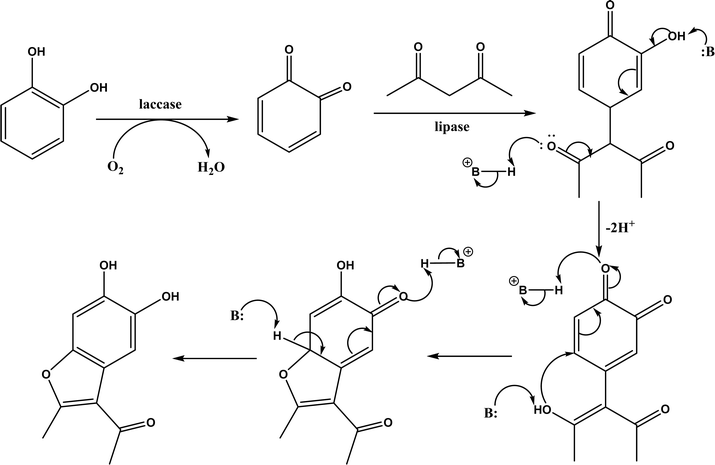
Knoevenagel condensation reaction is considered a variation of aldol condensation, which involved the nucleophilic addition of an activated methylene compound to a carbonyl group (aldehyde or ketone) followed by the dehydration (i.e., condensation) step to form an alkene. Knoevenagel condensation is highly valuable for preparing active pharmaceutical ingredients (APIs), and also precursors for other reactions such as Diels–Alder addition, Michael addition, oxidative coupling, and Nazarov cyclization.110–112 Knoevenagel condensation is traditionally catalyzed by various amines, but also by Lewis acids, zeolites, clays, amino acids, or ionic liquids (ILs).113–116 Alternatively, lipases and other enzymes have been investigated as efficient catalysts (‘catalytic promiscuity’) for Knoevenagel condensation (a few examples were discussed in reviews,12,117 but there is no systematic review on this). Immobilized lipase B from Candida antarctica (CALB) was reported to mediate decarboxylative aldol reactions of aromatic aldehydes and β-ketoesters at 30 °C in acetonitrile containing 1,4,7,10-tetraazacyclododecane as an additive to give 81–97% isolated yields, while the same reactions in acetonitrile with 5 v% water and a primary amine (e.g., aniline, p-toluidine and benzylamine) produced Knoevenagel products with 56–91% isolated yields (Fig. 18).38 However, the Bornscheuer group39 observed no promiscuous catalytic activity of CALB for the decarboxylative aldol addition and Knoevenagel reaction between 4-nitrobenzaldehyde and ethyl acetoacetate; what happened was the enzymatic hydrolysis of ethyl acetoacetate in the presence of water to form the corresponding acetoacetic acid, which reacted with 4-nitrobenzaldehyde to form the aldol and Knoevenagel products. In another study, CALB immobilized on chitosan-functionalized electrospun PMA-co-PAA membrane showed a better stability and recyclability than free enzyme, and produced up to 73% yield of 3-acetylcoumarin from Knoevenagel condensation and the cyclization of salicylaldehyde and acetoacetate (Fig. 19) in methanol/water (4:1, v/v) mixture.118 In a different study, CALB, Lipozyme RMIM (immobilized lipase from Rhizomucor miehei), Lipozyme TLIM (immobilized lipase from Thermomyces lanuginosus), and several “Amano” lipases including AK (from Pseudomonas fluorescen), DF (from Rhizopus oryzae), and AS (from Aspergillus niger) were evaluated in Knoevenagel–Michael cascade reactions of benzaldehyde and 1,3-cyclohexanedione in N,N-dimethylformamide (DMF) at 40 °C (Fig. 20), where ‘‘Amano’’ lipase DF gave a far better yield (89%) than other enzymes (9–29%); the extension of this reaction to other aromatic aldehydes and 1,3-cyclodiketones afforded 83–94% yields.119 However, a separate study demonstrated that RMIM produced higher yields than other lipases (including lipase DF, PPL and Novozym 435) in water during the Knoevenagel–Michael cascade reaction of 4-chlorobenzaldehyde with 4-hydroxycoumarin (Fig. 21).120
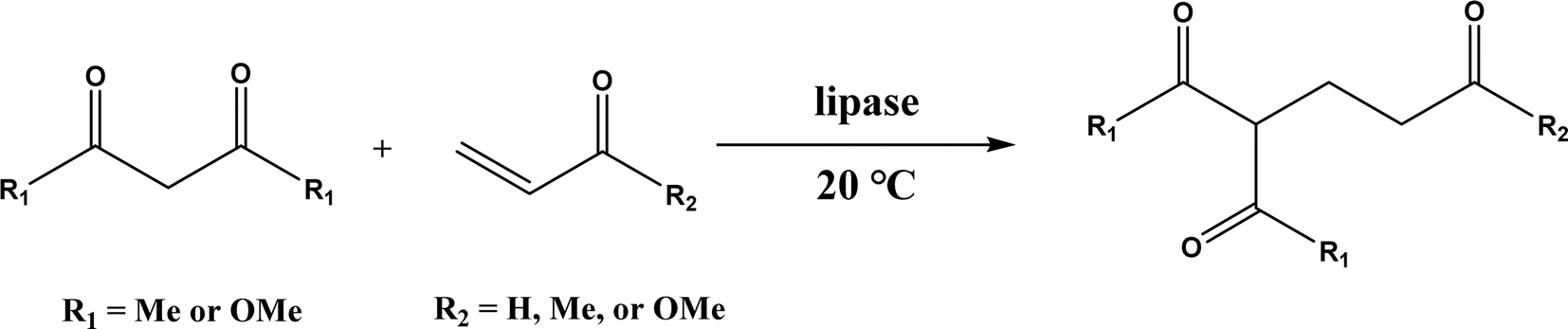
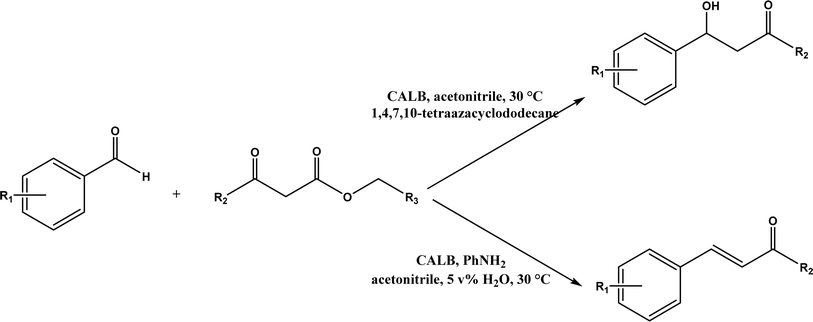 |
| | Fig. 18 Lipase-catalyzed decarboxylative aldol and Knoevenagel reactions. | |
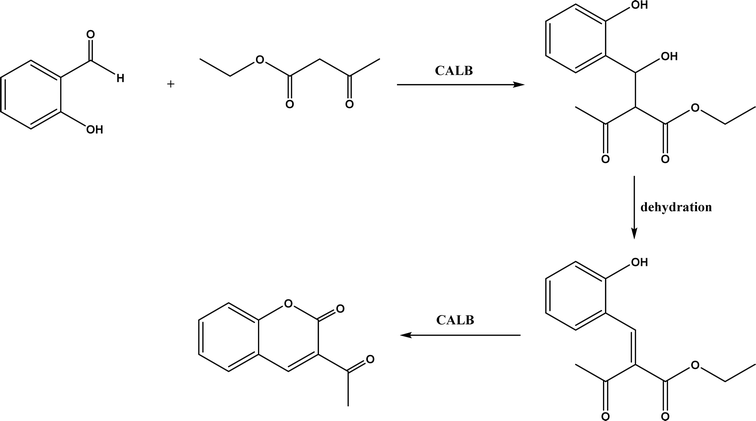 |
| | Fig. 19 CALB-catalyzed Knoevenagel condensation and the cyclization of salicylaldehyde and acetoacetate. | |
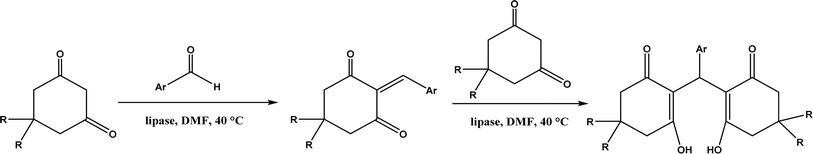 |
| | Fig. 20 Lipase-catalyzed Knoevenagel–Michael cascade reactions of aromatic aldehydes and 1,3-cyclodiketones. | |
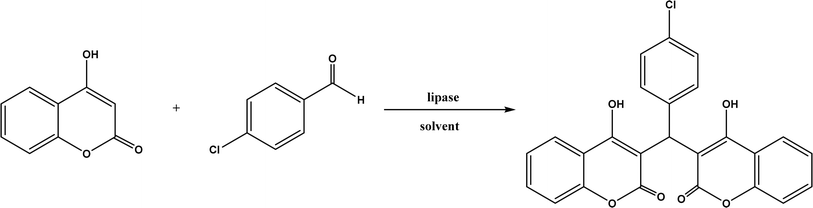 |
| | Fig. 21 Lipase-catalyzed Knoevenagel–Michael cascade reaction of p-chlorobenzaldehyde with 4-hydroxycoumarin. | |
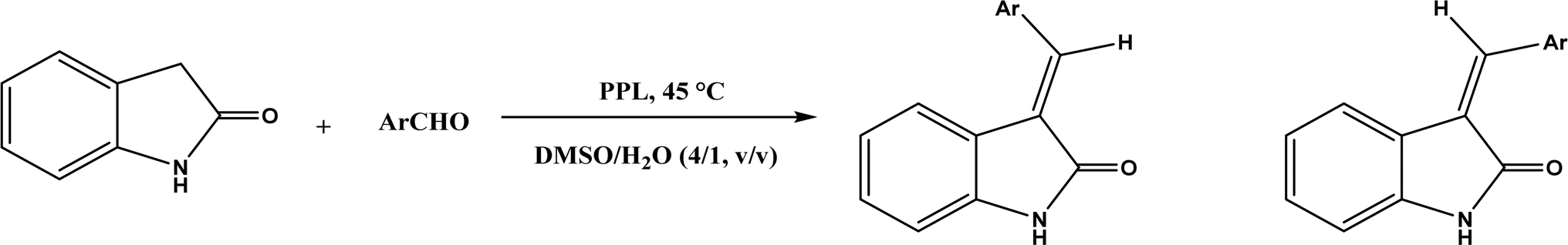
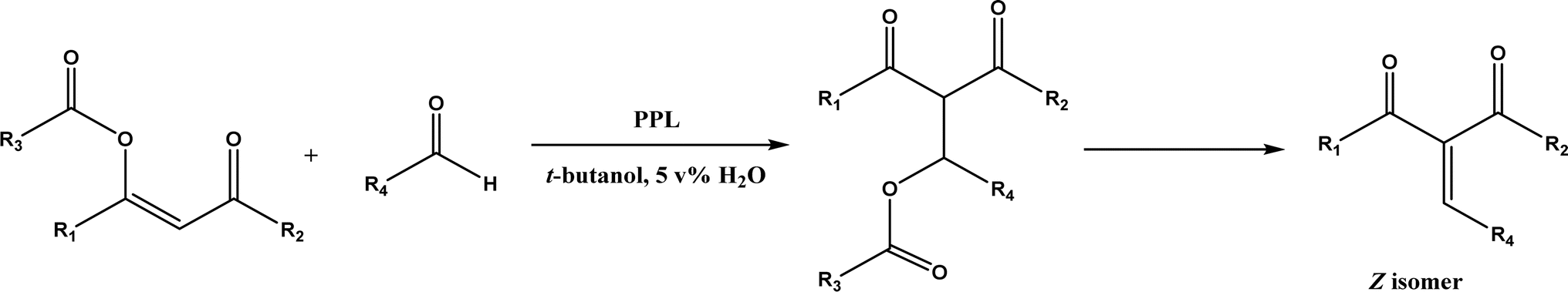
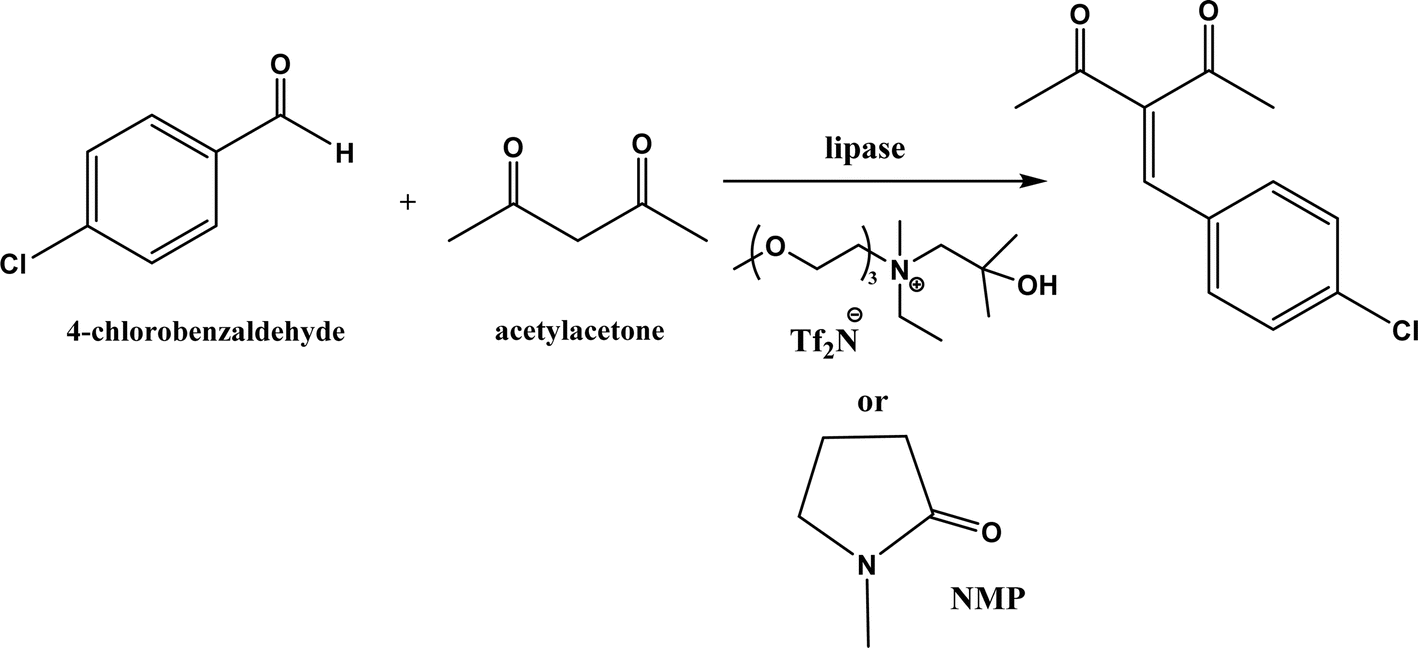
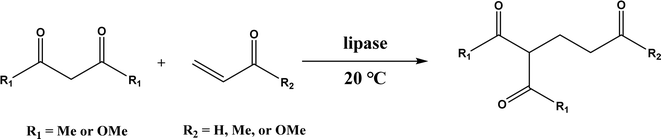
PPL displayed a higher catalytic activity than other lipases (including Novozym 435) for Knoevenagel reactions of aromatic aldehydes with 1,3-dihydroindol-2-one in DMSO with 20 v% water at 45 °C (Fig. 22), resulting in 75–97% yields and different E/Z ratios.121 In other Knoevenagel condensation studies, PPL also showed better performance than other lipases in tert-butanol with 20 v% water122 and ethanol.123 On the other hand, using in situ generated acetaldehyde from the enzymatic hydrolysis of vinyl carboxylates, chemoenzymatic tandem reaction (Fig. 23) catalyzed by Novozym 435 in tert-butanol or acetonitrile led to ethyl 2-aryoylbut-2-enoate compounds with up to 72% yields; PPL showed a lower activity than Novozym 435.113 Candida cylindracea lipase and Novozyme 435 enabled higher yields (up to 50%) than PPL and other lipases when catalyzing the esterification-Knoevenagel cascade reaction of cyanoacetic acid and benzaldehyde dimethyl acetal in toluene.124 Since Knoevenagel condensation product could react with activated methylene compound to form Michael addition byproduct, the Koszelewski group125 developed a method by using the enzymatic hydrolysis of enol carboxylates to generate active methylene compounds in situ for reacting with aldehydes catalyzed by PPL in tert-butanol with 5 v% water (Fig. 24); this hydrolysis–Knoevenagel cascade reaction produced target compounds with 11–86% yields and high E/Z selectivities (from 82:18 to mostly 99:1). The high selectivity was explained by the enol product preferably staying in one configuration in the active site of lipase, leading to the exclusive Z isomer. Wang and co-workers126 examined α-amylase from hog pancreas and PPL in different ILs and DES for Knoevenagel condensations of acetylacetone and 4-nitrobenzaldehyde (and other aromatic benzaldehydes later) at 50 °C, and found that α-amylase was most active in [HOEtMIM][NO3]/H2O (80:20, v/v) allowing 89% yield, while PPL was mostly active in choline chloride/glycerol (1:2, molar ratio) affording 93% yield. Interestingly, both enzymes were found highly active in nitrate-containing ILs among all ILs evaluated (with anions of BF4−, PF6− and NO3−) although NO3− is known enzyme-denaturing.127 Our group39 conducted Knoevenagel condensation of 4-chlorobenzaldehydes and acetylacetone (Fig. 25), and reported that porcine pancreas lipase (PPL) in water-mimicking ILs containing ammonium, imidazolium and benzimidazolium cations led to higher reaction rates (up to 3.22 mM per min per g lipase) and improved yields than tert-butanol, glymes, and [BMIM][Tf2N]. More fascinatingly, tertiary amides such as 1-methyl-2-pyrrolidone (NMP), N,N-dimethylformamide (DMF) and N,N-dimethylacetamide (DMA) enabled 8.2–11.1 times of increases in the initial reaction rate (up to 35.66 mM per min per g lipase) than dual-functionalized ILs, whose exact mechanism is under investigation although there is likely some synergistic effect of tertiary amides with the lipase.
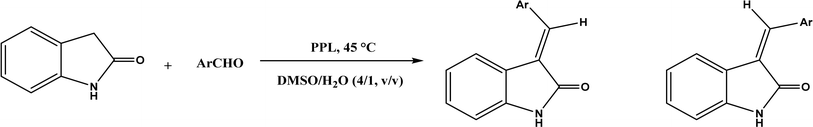 |
| | Fig. 22 PPL-catalyzed Knoevenagel reactions of aromatic aldehydes with 1,3-dihydroindol-2-one. | |
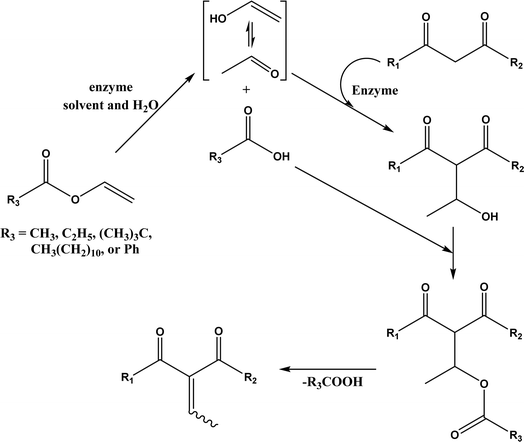 |
| | Fig. 23 Lipase-catalyzed Knoevenagel condensation using in situ generation of acetaldehyde (redrawn from ref. 113). | |
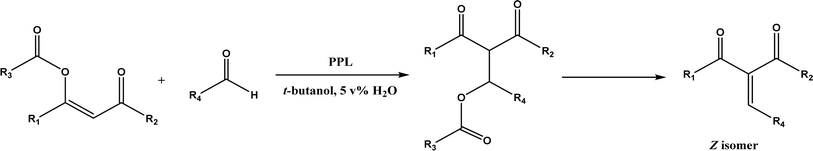 |
| | Fig. 24 Lipase-catalyzed tandem Knoevenagel reaction of enol carboxylates. | |
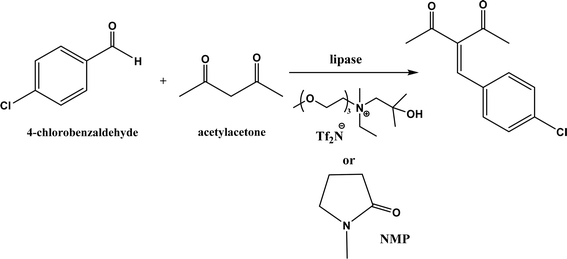 |
| | Fig. 25 Lipase-catalyzed Knoevenagel reaction between 4-chlorobenzaldehyde and acetylacetone in different solvents. | |
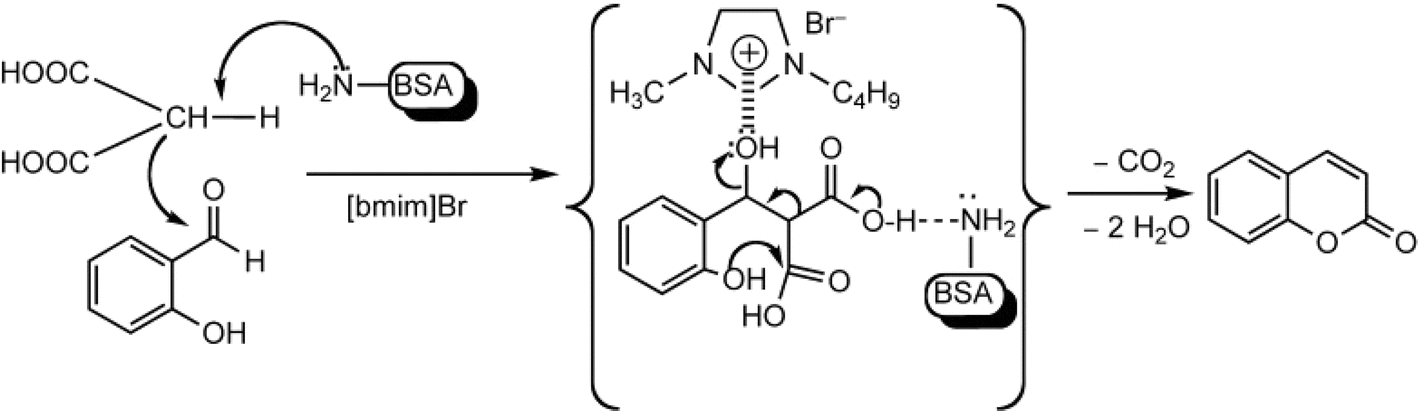
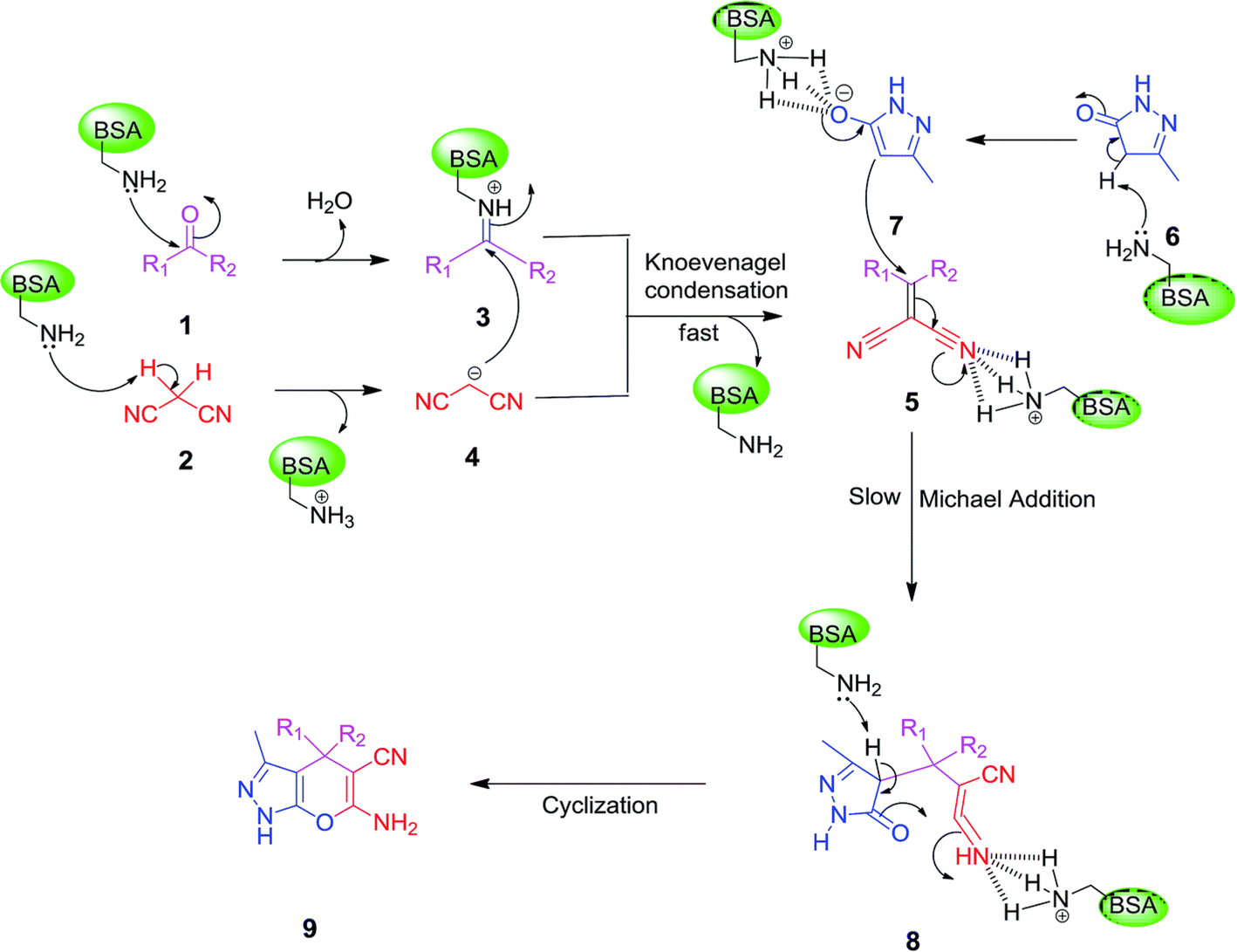
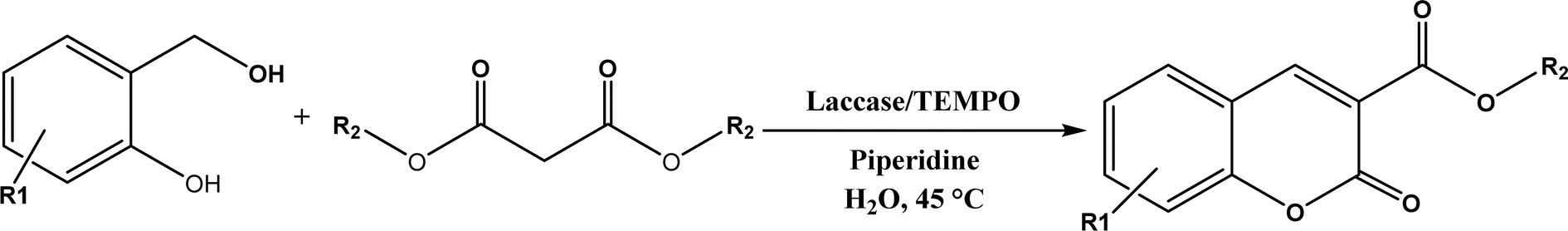
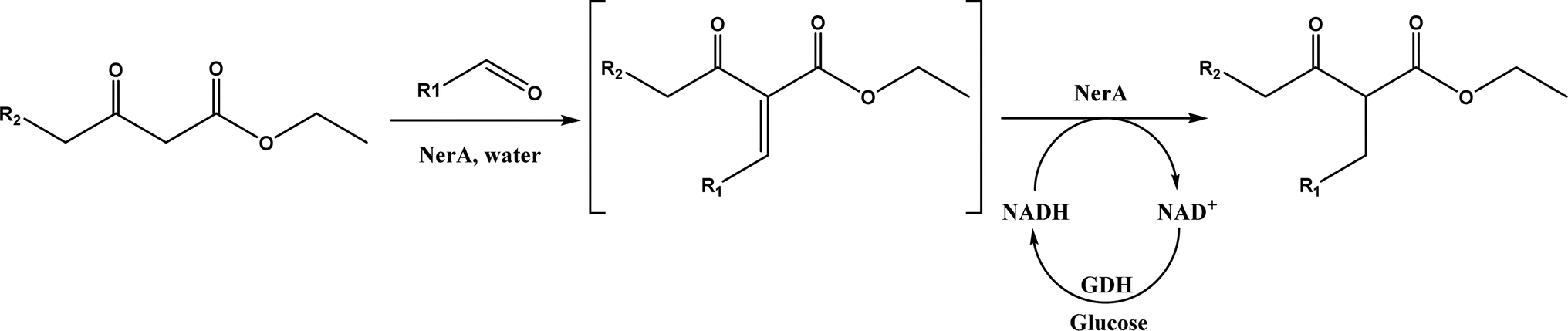
Other enzymes have also been investigated for Knoevenagel reactions. Alkaline protease from Bacillus licheniformis mediated Knoevenagel reactions between aromatic, hetero-aromatic, and α,β-unsaturated aldehydes with less reactive acetylacetone or ethyl acetoacetate in DMSO with 5 v% water at 45 °C, producing functionalized trisubstituted alkenes and α,β,γ,δ-unsaturated carbonyl compounds with 24–82% yields and various E/Z isomeric ratios.128 With an organic salt ([BMIM]Br), bovine serum albumin (BSA) showed a similar performance as PPL in catalyzing aldol condensations of benzaldehyde derivatives with different ketones, and Knoevenagel–Doebner condensations of benzaldehyde derivatives with activated methylene compounds with good yields; in the absence of BSA or [BMIM]Br, there was little product formed.129 It was rationalized that amino acid residues (e.g., lysine) in BSA and [BMIM]Br both played critical roles in the reaction as illustrated by Fig. 26. In addition, BSA was found capable of catalyzing three-component reaction of an aldehyde/ketone/isatin, malononitrile, and 3-methyl-1H-pyrazol-5-(4H)-one in the ethanol/water (3:7) mixture at room temperature to dihydropyrano[2,3-c]pyrazole and spiro[indoline-3,40-pyrano[2,3-c]pyrazole] derivatives with 72–98% yields; BSA outperformed lipases, trypsins, papain, and α-amylase,130 although for Knoevenagel condensations of benzaldehyde derivatives with acetylacetone (or its analogues) in the DMSO/water mixture, papain enabled better yields (42–86% yields),131 and papain immobilized in Cu3(PO4)2 nanoflowers exhibited higher activities (still moderate yields of 9–53%) than free enzyme.132 The reaction mechanism is described in Fig. 27 as three key steps: Knoevenagel condensation, Michael addition, and cyclization. A similar one-pot three-component condensation of aldehyde, cyanoacetamide, and 1,3-dicarnonyl compound followed same steps of Knoevenagel condensation, Michael addition, and intramolecular cyclization, where D-aminoacylase and acylase ‘Amano’, and Amano lipase M from Mucor javanicus exhibited considerably higher activities than BSA, immobilized penicillin G acylase, lipase AK ‘Amano’, and Candida rugosa lipase; 3,4-dihydropyridin-2-one derivatives were synthesized in 28–99% yields and varying diastereomeric ratios under optimum conditions.133 Li and co-workers134 pointed out that serine residues of lipases are not involved in Knoevenagel condensation, while unspecific residues of lipases, BSA or other proton acceptors could promote the reaction. Baker's yeast as the whole cell biocatalyst effectively mediated Knoevenagel condensations between aryl aldehydes and malononitrile (or ethyl cyanoacetate, or 2,4-thiazolidinedione) in ethanol at room temperature, leading to good yields in most cases.41 At pH 7.0, segments of RNA/DNA salts were discovered as efficient as PPL in catalyzing Knoevenagel condensations of benzaldehyde derivatives and activated methylene compounds; the catalytic rate was associated with a higher content of GC nucleosides in RNA/DNA while a higher catalytic turnover number is correlated with a longer strand of DNA.135 Directed evolution of an artificial retro-aldolase was able to optimize its catalytic activity relying on a reactive lysine in a hydrophobic pocket to promote Knoevenagel condensations of electron-rich aldehydes and activated methylene compounds (see an example in Fig. 28), becoming >105-fold more proficient than BSA, and >108-fold more proficient than primary and secondary amines.136 Laccase and its mediator 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) were co-immobilized in mesoporous silica as a hybrid catalyst to oxidize salicyl alcohols to salicylaldehydes in situ, followed by the Knoevenagel condensation and cyclization (transesterification) to form coumarin-3-carboxylates (Fig. 29) with 84–95% yields in citrate buffer (pH 4.5, 0.1 M); however, same reactions in organic solvents such as THF, DMF and acetonitrile led to no product, and 65% yield in [BMIM][PF6].137 A single ene-reductase (NerA) catalyzed the Knoevenagel condensation of β-ketoesters first followed by a reduction to produce saturated α-substituted β-ketoesters (70–95% yields) as valuable synthons for pharmaceuticals and agrochemicals using in situ generation of NADH via glucose with glucose dehydrogenase (GDH), and it was shown that amino acid residues at the surface of NerA promoted the Knoevenagel condensation (Fig. 30),138 which is different from an earlier study where CALB catalyzed decarboxylative aldol reactions of β-ketoesters.38
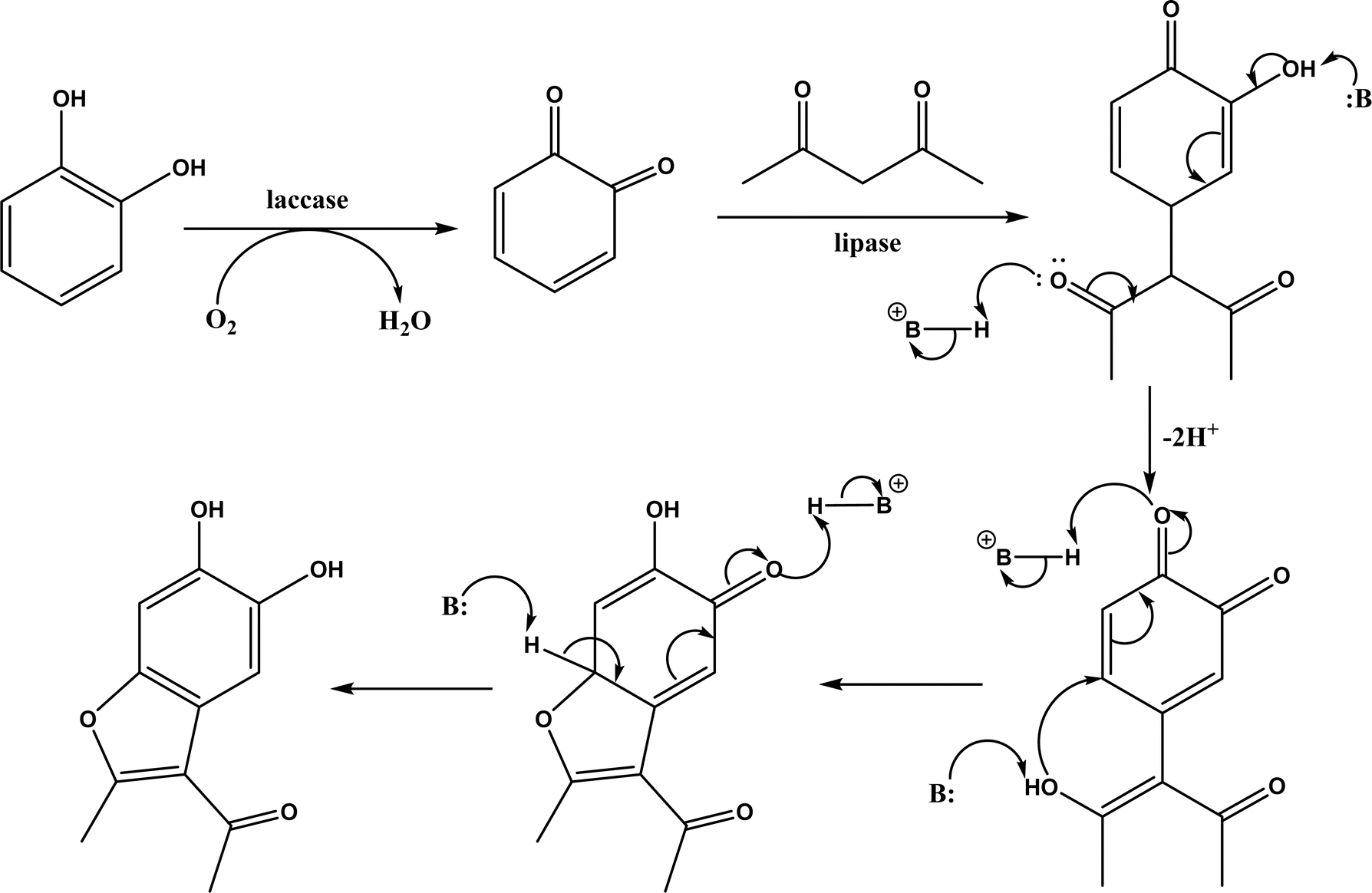
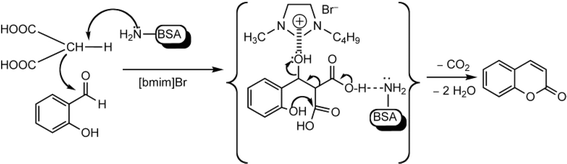 |
| | Fig. 26 Mechanism for the synthesis of coumarins via Knoevenagel condensation and cyclization [Reprinted with permission from ref. 129 Copyright 2011 Wiley-VCH Verlag GmbH&Co. KGaA]. | |
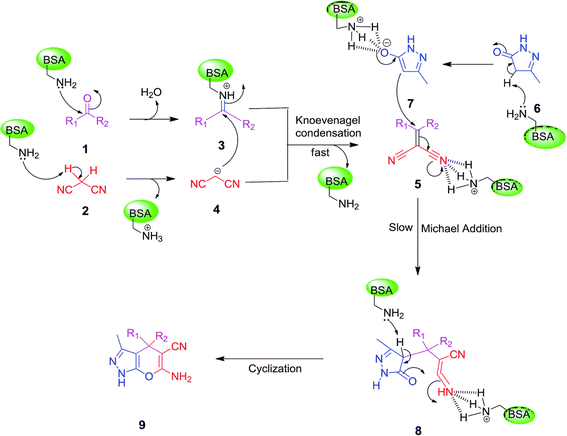 |
| | Fig. 27 Mechanism of three-component synthesis of dihydropyrano[2,3-c]pyrazole derivatives catalyzed by BSA in an aqueous ethanol [Reprinted with permission from ref. 130 Copyright 2016 Royal Society of Chemistry]. | |
 |
| | Fig. 28 Knoevenagel condensation of (E)-3-(4-(dimethylamino)phenyl)acrolein and ethyl 2-cyanoacetate. | |
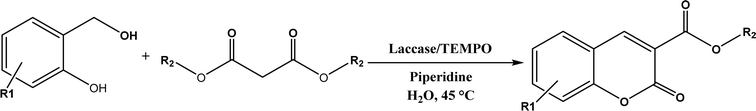 |
| | Fig. 29 One-pot synthesis of coumarin-3-carboxylates using laccase/TEMPO hybrid catalyst. | |
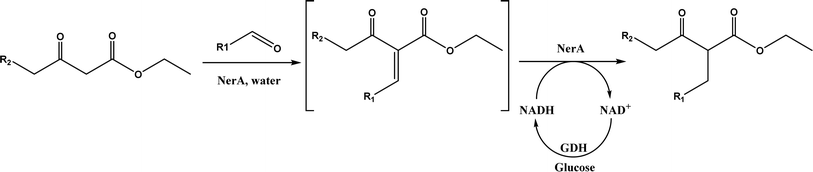 |
| | Fig. 30 Tandem Knoevenagel condensation–reduction reaction of β-ketoesters using ene-reductase (NerA) (GDH = glucose dehydrogenase). | |
4. Michael addition
Michael addition (1,4-addition) typically refers to the nucleophilic addition of a carbanion to unsaturated systems (α,β-unsaturated carbonyl compounds) in conjugation with an activating group.139 Many organocatalysts (e.g., chiral diamines, chiral crown ethers, chiral alkaloids, chiral amino acids, and chiral oxazolines) and organometallic catalysts (e.g., salts of amino acids, metal-diamine complexes, Schiff base-metal complexes, transition metal complexes, heterobimetallic complexes, and metal-N,N-dioxide complexes) have been extensively studied in asymmetric Michael addition reactions.140 However, there is no individual catalyst that can catalyze different Michael reactions.
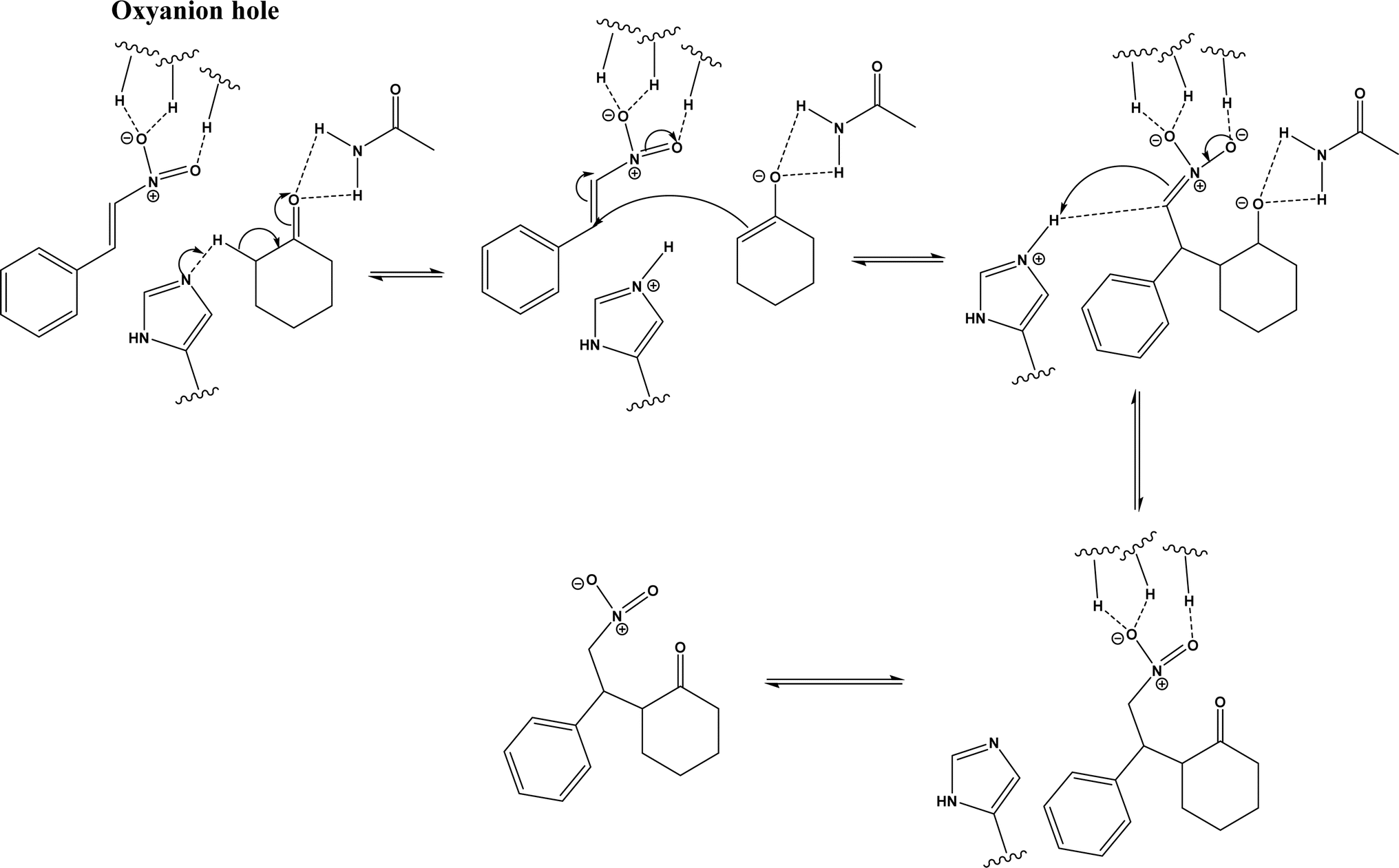
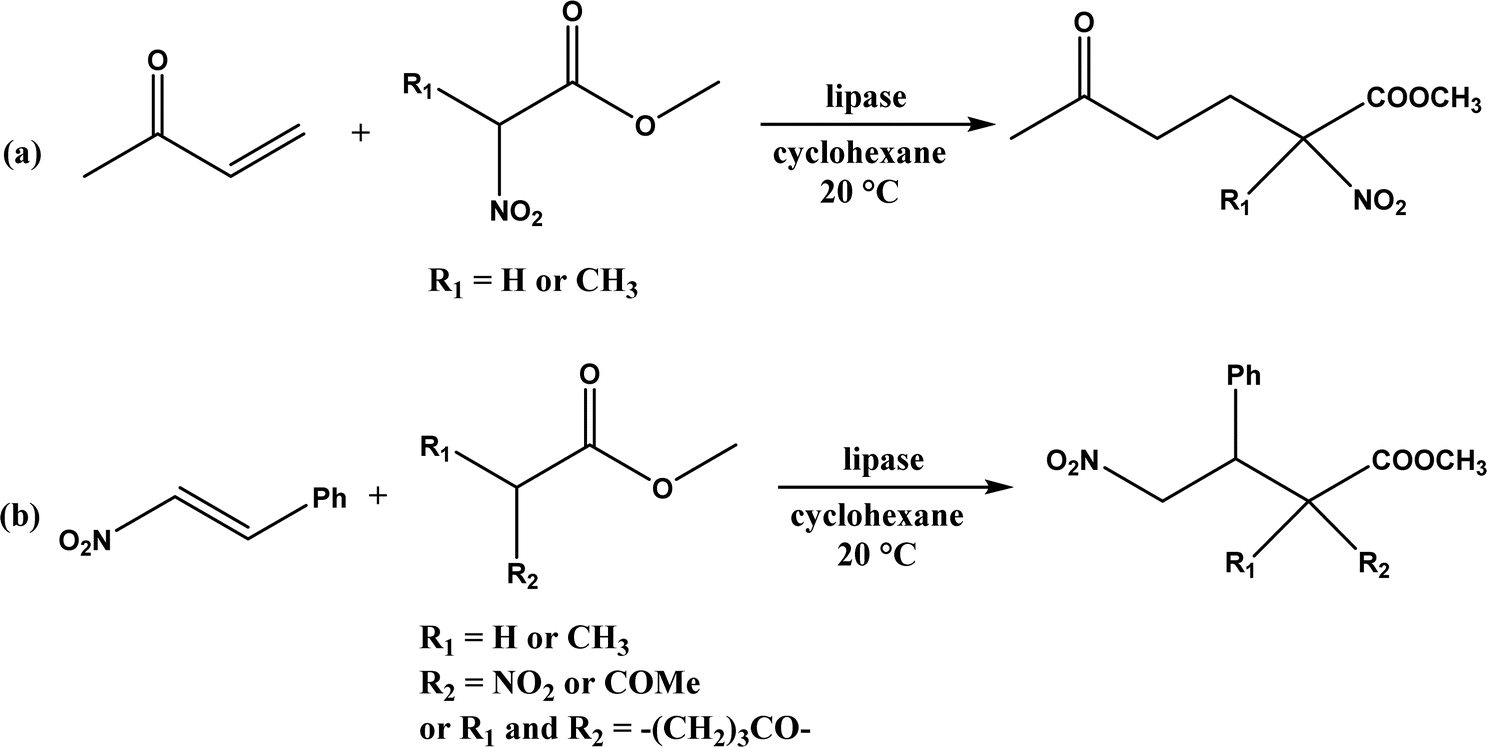
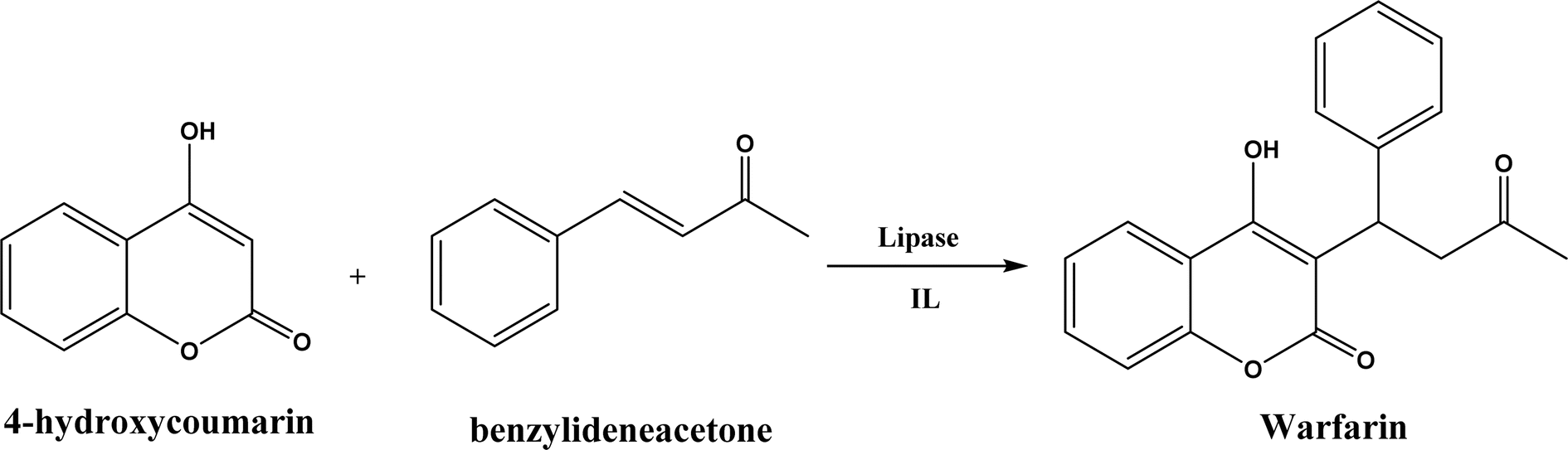
Several groups have reported catalytic promiscuity of lipases towards Michael addition. Svedendahl et al.42 improved the reaction specificity of lipase B from Candida antarctica (CALB) by substituting one amino acid (Ser105Ala) in the active site. They found that the lipase mutant exhibited much faster Michael addition rates (between 1,3-dicarbonyls and α,β-unsaturated carbonyl compounds, see Fig. 31) than the wild type at 20 °C. The Ragauskas group141 suggested that lipase from Pseudomonas cepacia (known as lipase PS) accelerated the regioselective addition reaction between laccase-generated o-quinones and 1,3-dicarbonyl compounds in aqueous medium at room temperature (Fig. 32), leading to a 30–70% increase in product yield. Cai et al.142 carried out the Michael addition of a wide range of 1,3-dicarbonyl compounds and cyclohexanone to aromatic and heteroaromatic nitroolefins and cyclohexanone catalyzed by various lipases (Fig. 33); they reported that lipozyme TLIM (immobilized lipase from Thermomyces lanuginosus) outperformed other lipases. Further, they found that DMSO (10/1, v/v, with water) was the best organic solvent in terms of generating a relatively high yield and ee. However, most yields were moderate (30–90%) and ees were relatively low (usually below 50%). The He143 and Hu groups144 conducted Michael additions of 4-hydroxycoumarin with α,β-unsaturated enones promoted by PPL in aqueous organic solvents (such as DMSO), obtaining moderate to high yields (up to 95%) but low enantioselectivities (up to 28% ee). However, Chen and co-workers43 reported that CALB alone could not catalyze Michael additions of aromatic nitroolefins and less-activated ketones (e.g., cyclohexanone instead of acetylacetone), but required co-catalyst acetamide to obtain products with 25–72% yields. Other primary (1°) amides showed similar or less activation effect; the role of acetamide can be elucidated by the following mechanism (Fig. 34): the activation of cyclohexanone by acetamide and the interaction of nitroolefin with oxyanion hole, proton transfer from cyclohexanone to His residue to form an enolate (which is stabilized by acetamide), nucleophilic attack of nitroolefin by enolate, proton transfer from His residue to the product, and the product release from active site. The Griengl group44 studied various lipases for Michael addition of β-ketoesters [methyl acetoacetate and methyl 2-(2-oxocyclopentyl)acetate] or nitroesters (methyl 2-nitropropanoate and methyl 2-nitroacetate) to 3-buten-2-one (or trans-β-nitrostyrene) in cyclohexane at 20 °C (Fig. 35), and identified several top-performing enzymes including Candida antarctica lipases A (CALA), CALB, and lipases from Mucor miehei, and Thermomyces lanuginosas. Methyl 2-nitroacetate was found the most active donor, leading to over 60–99% conversions of methyl vinyl ketone and trans-β-nitrostyrene in 20 h for selected lipases especially the CALB mutant; the alkene substrate requires electron withdrawing groups on it to act as the acceptor and strong nucleophilic CH-acidic donor to proceed with Michael addition. However, the enzymatic reaction between trans-β-nitrostyrene and acetylacetone failed. In contrast to other studies, this study44 reported no stereoselectivity for lipase-catalyzed Michael additions; it is suggested that the C–C-bond formation was due to the substrate activation by unique assembly of amino acids in the protein cavity. Hydroxy-functionalized ionic liquids (ILs) were evaluated as reaction media for the Michael addition synthesis of warfarin catalyzed by Candida rugosa lipase (Fig. 36), and it was found the hydroxy functionalization led to more hydrophilic (‘water-mimicking’) ILs and higher reaction yields while longer alkyl chains on ILs showed an opposite effect on the reaction; also, no stereoselectivity was observed in the reaction.45
 |
| | Fig. 31 Michael addition of 1,3-dicarbonyls to α,β-unsaturated carbonyl compounds. | |
 |
| | Fig. 32 Laccase/lipase catalytic Michael addition reaction of in situ-generated ortho-quinones (B: represents a base molecule such as water). | |
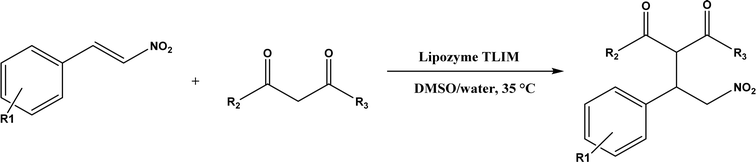 |
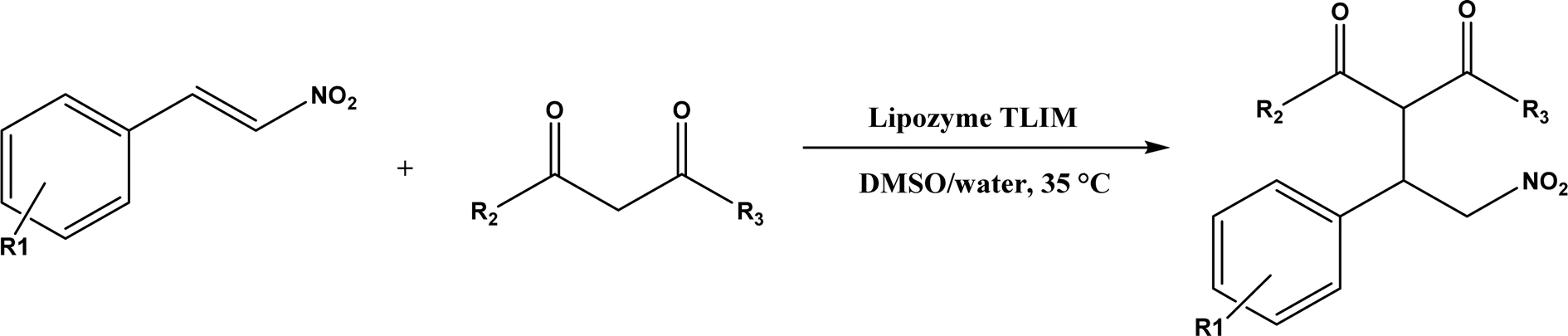
| | Fig. 33 Michael addition of aromatic nitroolefins and 1,3-dicarbonyl compounds catalyzed by Lipozyme TLIM. | |
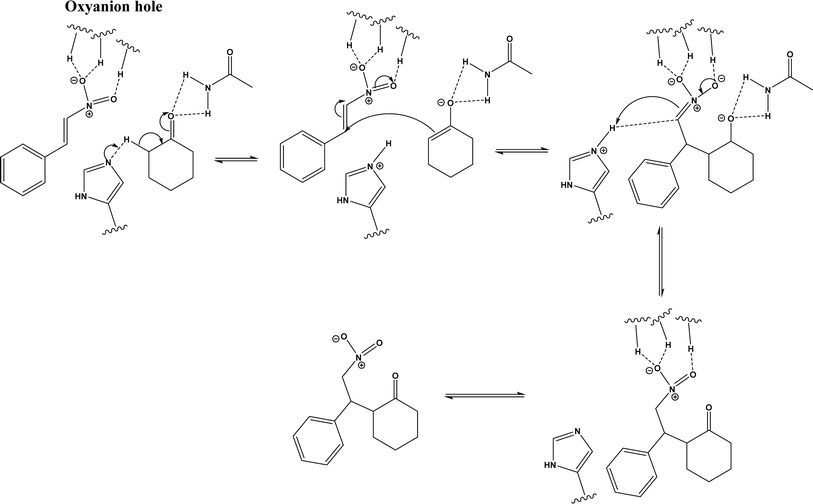 |
| | Fig. 34 Mechanism of lipase/acetamide-catalyzed Michael addition [redrawn from ref. 43]. | |
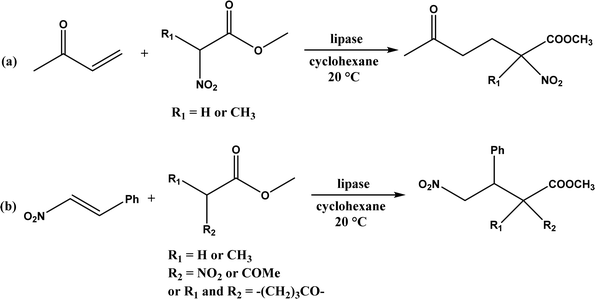 |
| | Fig. 35 Lipase-catalyzed Michael addition of (a) nitroesters to 3-buten-2-one, and (b) nitroesters or β-ketoesters to trans-β-nitrostyrene. | |
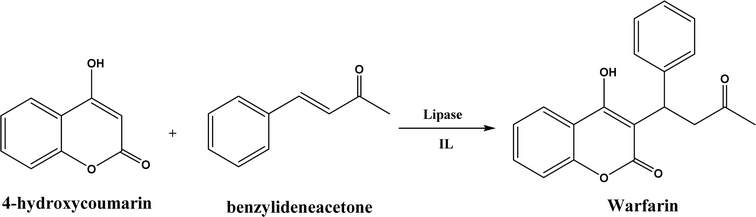 |
| | Fig. 36 Lipase-catalyzed Michael addition synthesis of warfarin in ILs. | |
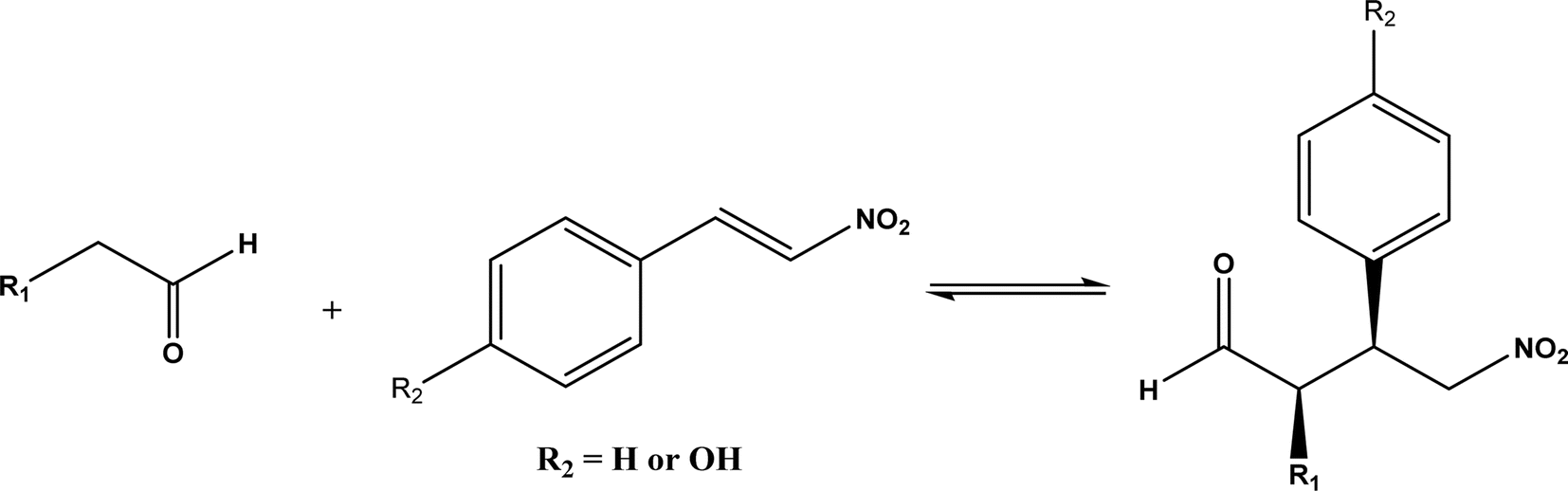
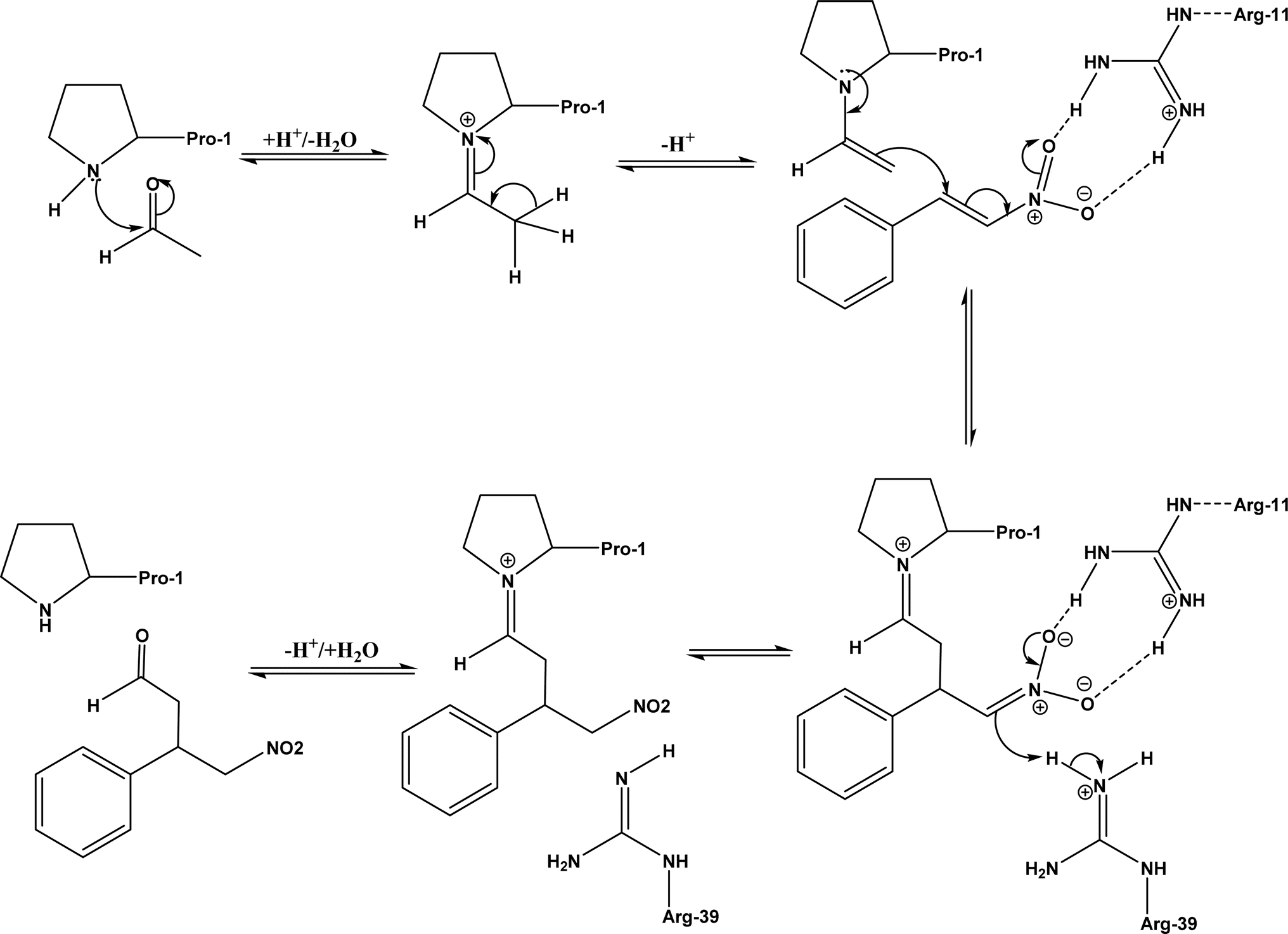
Other types of hydrolases, such as proteases and D-aminoacylase, are also capable of catalyzing the Michael addition. The Lin group145 screened various hydrolases for the Michael addition and reported that Bacillus subtilis protease, porcine pancreas lipase (PPL), and D-aminoacylase from Escherichia coli enabled moderate to high yields for the reactions of 1,3-dicarbonyl compounds with α,β-unsaturated compounds in 2-methyl-2-butanol and other organic solvents at 50 °C for 24 h. In another study,146 D-aminoacylase from Escherichia coli as a zinc-dependent acylase was found more active than other enzymes (e.g., Amano acylase from Aspergillus oryzae, CALB, Candida cylindracea lipase, and Amano lipase M) in catalyzing the Michael addition of 1,3-dicarbonyl compounds to methyl vinyl ketone (Fig. 37); tertiary alcohols (i.e., 2-methyl-2-butanol and tert-butanol) enabled much higher yields (up to 82.1%) than more hydrophobic (i.e., n-hexane, toluene, chloroform, and isopropyl ether) and hydrophilic solvents (i.e., THF and dioxane). The catalytic mechanism is described in Fig. 38: interactions of carbonyl groups from both substrates with Zn2+ near the active site, proton transfer from acetylacetone to Asp-366, nucleophilic attack of methyl vinyl ketone by acetylacetone to form an enolate, proton transfer from Asp to the enolate, and the release of final product from the active site. Wu et al.147 found that protease from Streptomyces griseus was able to catalyze Michael additions of a variety of malonates and enones in aqueous methanol, and achieved up to 84% yields and up to 98% ee under optimum conditions. Since proline and its derivatives have been used as organocatalysts for C–C bond formations including Michael addition,148 the Poelarends group149,150 noted that 4-oxalocrotonate tautomerase carries a catalytic amino-terminal proline, thus could catalyze the asymmetric Michael reaction between trans-nitrostyrenes and linear aldehydes ranging from acetaldehyde to octanal as donors (Fig. 39) in aqueous solutions (water, or water/ethanol = 9:1), giving 46–92% yields, good diastereoselectivities (from 85:15 to 93:7), and fair ees (23–89%); a larger aldehyde molecule caused a lower enantioselectivity and slower reaction. The mechanism includes several steps as shown in Fig. 40: the formation of iminium ion via nucleophilic attach of Pro-1 to carbonyl carbon of the aldehyde, the deprotonation of iminium ion to form enamine, Michael-type nucleophilic attack of trans-nitrostyrene by enamine (Arg-11 supports the correct substrate binding), proton transfer from Arg-39 to the reaction complex, and the release of final product from Pro-1.
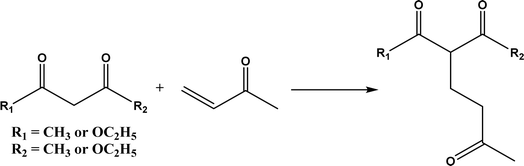 |
| | Fig. 37 D-Aminoacylase-catalyzed Michael addition of 1,3-dicarbonyl compounds to methyl vinyl ketone. | |
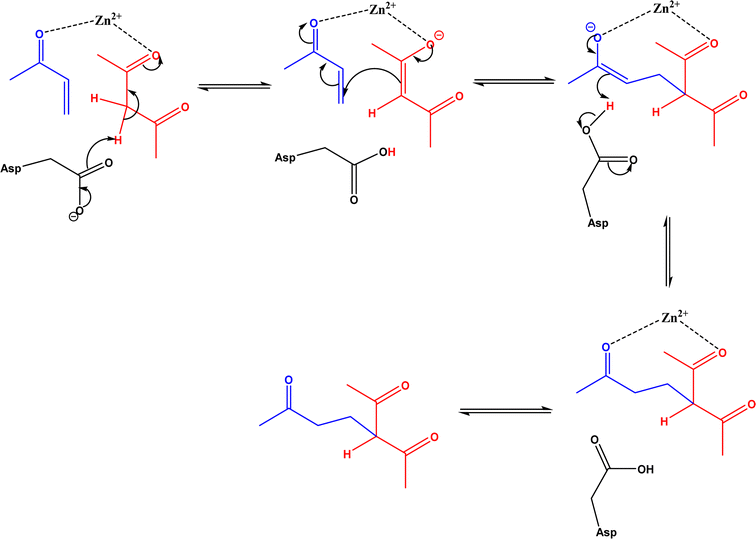 |
| | Fig. 38 Mechanism of zinc-dependent D-aminoacylase-catalyzed Michael addition.146 | |
 |
| | Fig. 39 Michael addition of trans-nitrostyrenes and linear aldehydes catalyzed by 4-oxalocrotonate tautomerase. | |
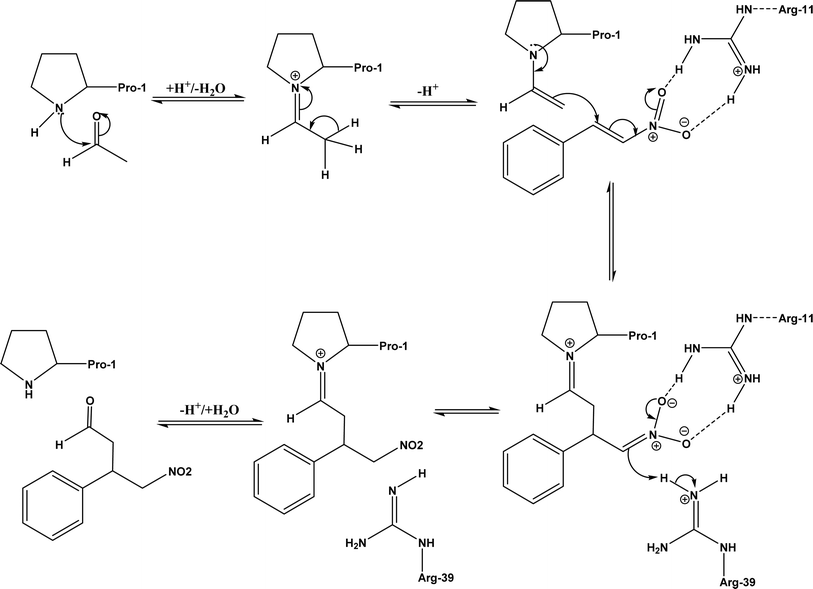 |
| | Fig. 40 Mechanism of Michael addition of trans-nitrostyrenes and acetaldehyde catalyzed by 4-oxalocrotonate tautomerase [redrawn from Scheme S1 in ref. 149]. | |
Duplex DNA, G-quadruplex DNA, and DNA/RNA-derived hybrid catalysts have been developed for asymmetric Diels–Alder, Michael addition, and Friedel–Crafts reactions in aqueous buffers or organic solvents.151–155 Our group conducted Michael addition in aqueous solutions of ionic liquids (ILs), deep eutectic solvents (DES), inorganic salts, glymes, glycols, and other organic solvents catalyzed by duplex DNA156 or G-quadruplex DNA-based catalysts,157 and found that the addition of glycerol, glyme, or DES enabled the reaction to be conducted at room temperature while maintaining up to 94–99% ees and mostly >70–97% yields.156
5. Friedel–Crafts alkylation and acylation
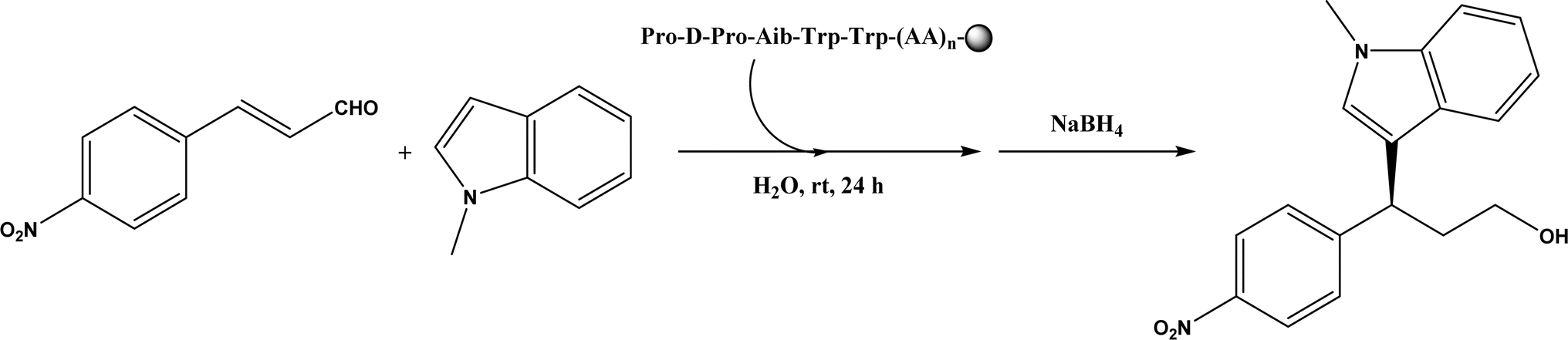
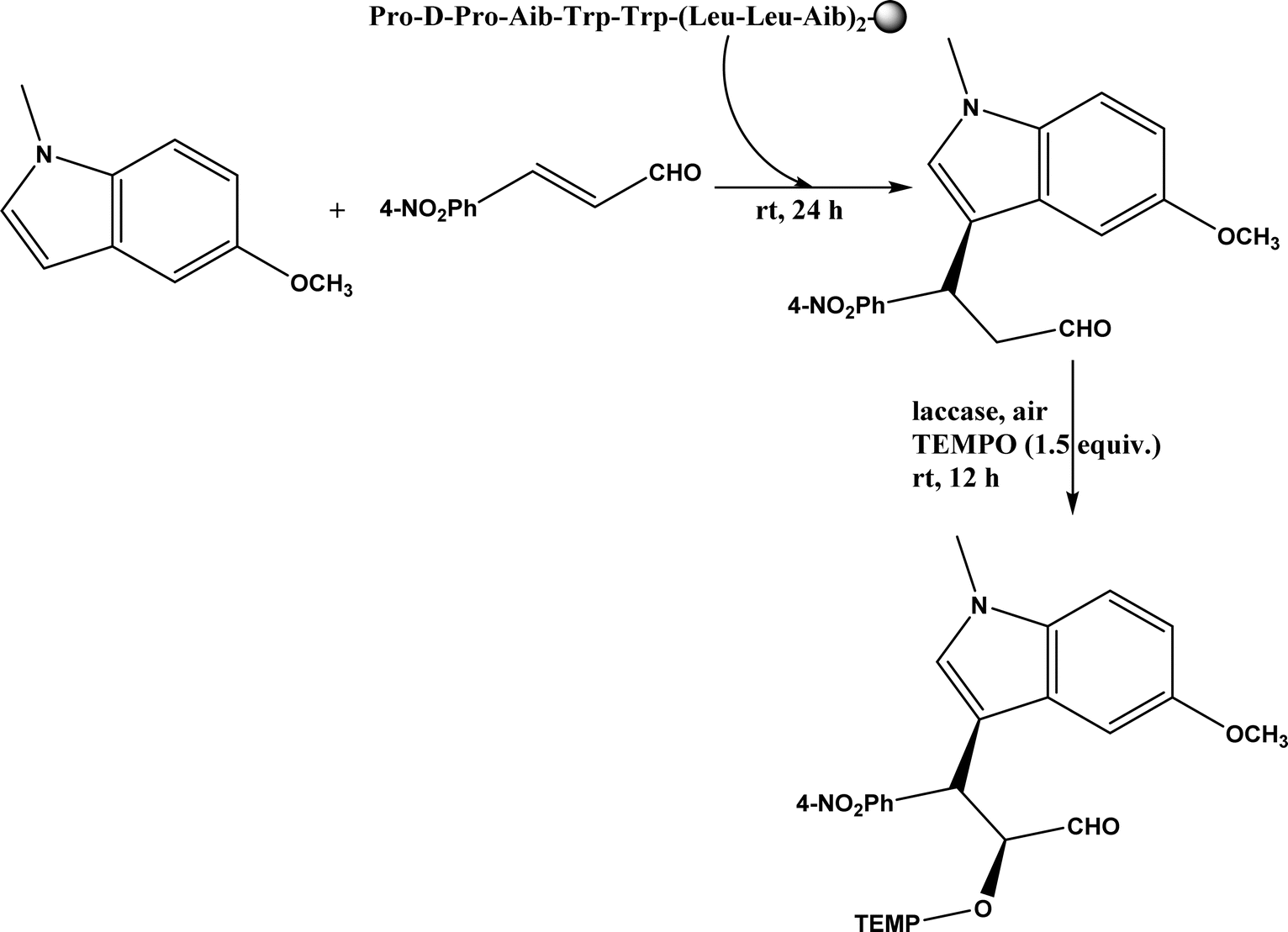
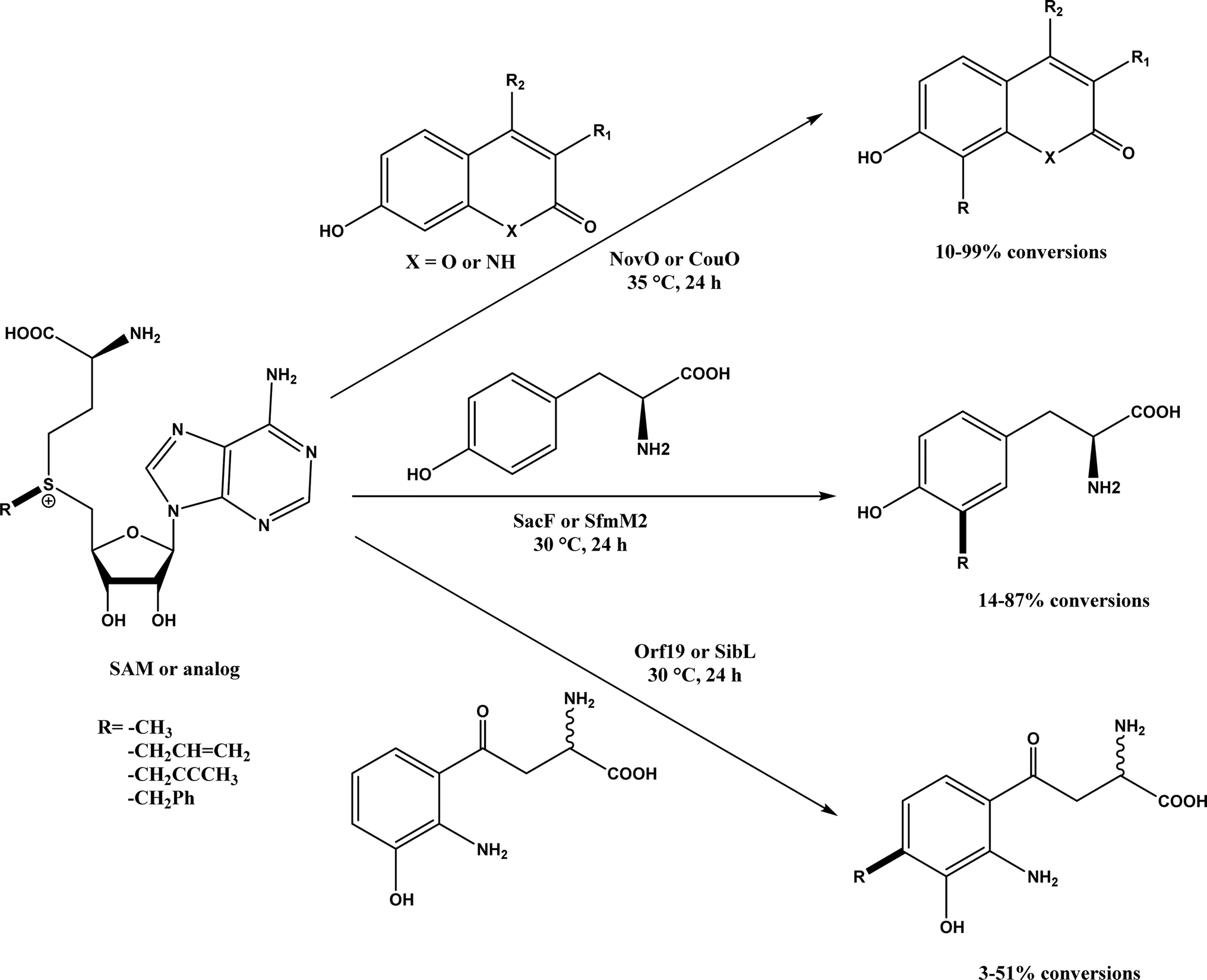
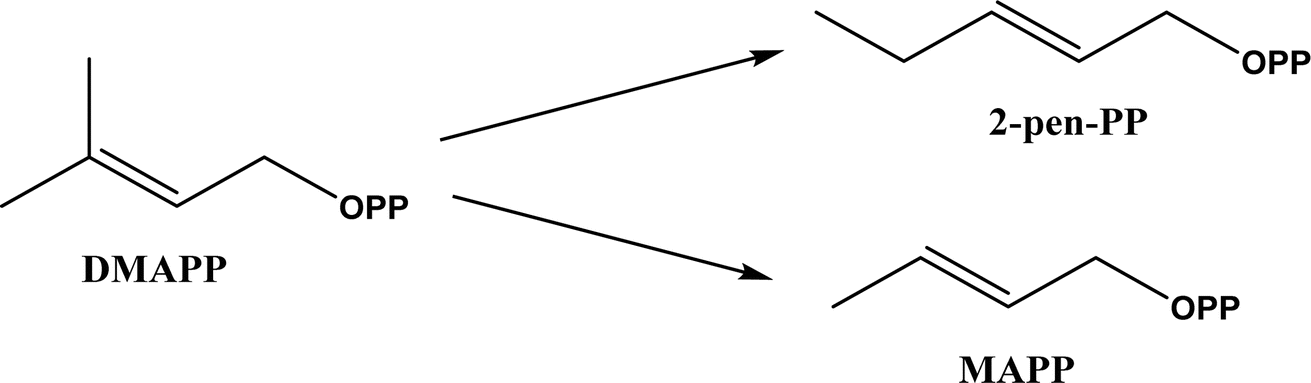
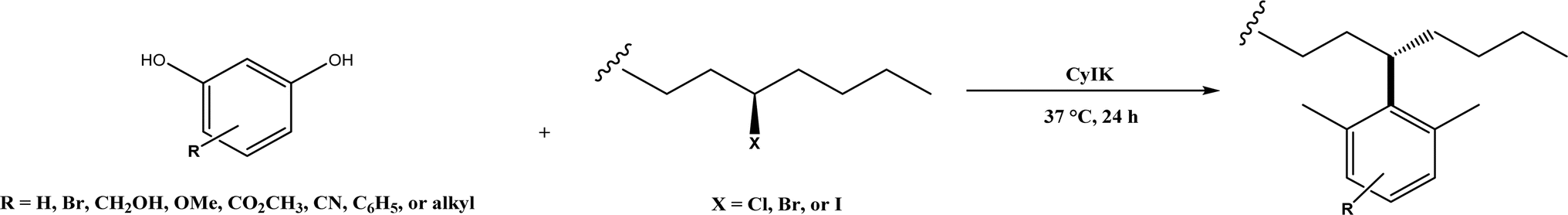
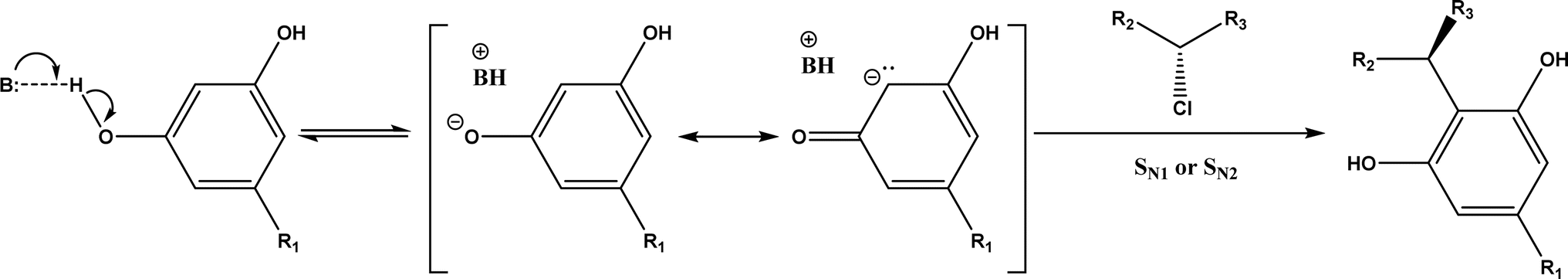
Friedel–Crafts alkylation and acylation represent an important category of C–C bond formation reactions, traditionally catalyzed by Lewis acids such as AlCl3, which leads to poor regioselectivity and multi-alkylation. Various biocatalysts pave a new avenue for regio- and chemoselective Friedel–Crafts. Recently, peptide catalysts supported on PEG-PS-resin were developed to catalyze the Friedel–Crafts alkylation shown in Fig. 41, and it was found that polyleucine in the form of –(AA)n–, such as (Leu-Leu-Aib)n where n = 1, 2 or 3, was able to form an α-helical structure and thus, along with β-turn motif D-Pro-Aib, could effectively facilitate alkylation reactions.158,159 Furthermore, the same group160 extended the peptide catalysts to synthesize oxygen-functionalized indole or pyrrole derivatives (often seen in the structures of antibiotics) through a tandem reaction of Friedel–Crafts-type alkylation of indole or pyrrole compounds followed by an α-oxyamination via laccase (an oxidative enzyme) in THF/H2O (1:2, v/v) mixture (see Fig. 42), leading to 70–79% syn products with 91–98% ee; the stereochemistry of the α-oxyamination step is primarily controlled by the peptide catalyst. In nature, methyltransferases catalyze the transfer of a methyl group in living cells such as DNA and RNA methylation; (S)-adenosyl-L-methionine (SAM, Fig. 43) is the most common methyl donor, which acts as the co-factor for the enzyme.161 Several methyltransferases originally found in bacteria such as NovO, CouO, SfmM2, and Orf19 from Streptomyces species, SibL from Streptosporangium sibiricum, and SacF from Pseudomonas fluorescens, could promote Friedel–Crafts alkylations of coumarins, naphthalenediols, and aromatic amino acids using SAM or non-natural SAM analogues (Fig. 43), resulting in excellent regioselectivity and various conversions.46–49 Dimethylallyl-tryptophan synthases (a type of as “aromatic prenyltransferases”) can catalyze Friedel–Crafts alkylations of various aromatic substrates (e.g., indoles, naphthalenes, flavonoids, and phenylpropanoids), but exhibit a high specificity for dimethylallyl diphosphate (DMAPP) as the alkyl donor;162–164 Liebhold and co-workers165 demonstrated that DMAPP can be modified by deleting or shifting one methyl group in DMAPP (Fig. 44) while still serving as alkyl donors for prenyltransferases, however, the double bond at β-position is important to keep for stabilizing the carbocation formed during the enzymatic alkylation on indoles. In another study, the cylindrocyclophane biosynthetic enzyme CylK was found capable of promoting a stereospecific Friedel–Crafts alkylation of resorcinol rings at their C-2-position (Fig. 45), resulting in high conversions (70–100%) and turn over numbers (>150) in most cases.166 Their DFT calculations point out a catalysis mechanism (Fig. 46) where CylK enables partial or full deprotonation of a hydroxyl group on the resorcinol, which acts as a nucleophile to initiate a concerted SN2 or stepwise SN1 reaction. α-Chymotrypsin from bovine pancreas (BPC) was found being able to catalyze Friedel–Crafts reactions between a broad range of isatins and indoles to produce 3-hydroxy-oxindoles in the presence of aprotic solvents such as 1,2-dichloroethane, or 3,3-bis(indol-3-yl)indolin-2-ones when methanol was used as the co-solvent (Fig. 47) although no stereoselectivity was specified, which enabled the synthesis of several pharmacologically active compounds.167 As relatively strong Brønsted acids, squalene hopene cyclases (SHCs) catalyze the regio- and stereoselective polycyclization of squalene, and could catalyze the intramolecular Friedel–Crafts alkylation of polyprenyl phenyl ethers, but showed a low catalytic activity and poor selectivity between the alkylation and hydration productions (see an example in Fig. 48); interestingly, variants of SHCs can be designed using site-directed and saturation mutagenesis to afford a high selectivity of alkylation (up to 100%) despite a moderate production formation of up to 29%.168,169
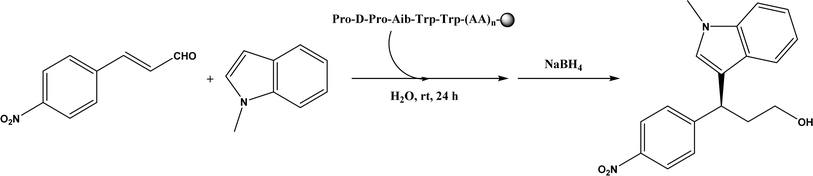 |
| | Fig. 41 Resin-supported peptide-catalyzed Friedel–Crafts alkylation (Aib = 2-aminoisobutyric acid; resin = –NH-CH2–CH2-PEG-PS). | |
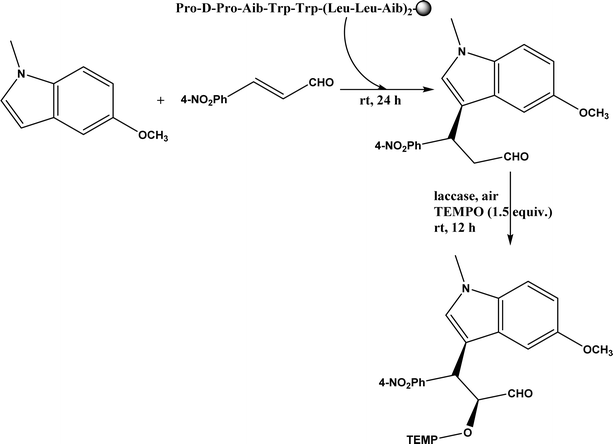 |
| | Fig. 42 One-pot sequential Friedel–Crafts-type alkylation and α-oxyamination (TEMPO = 2,2,6,6-tetramethylpiperidin-1-oxyl). | |
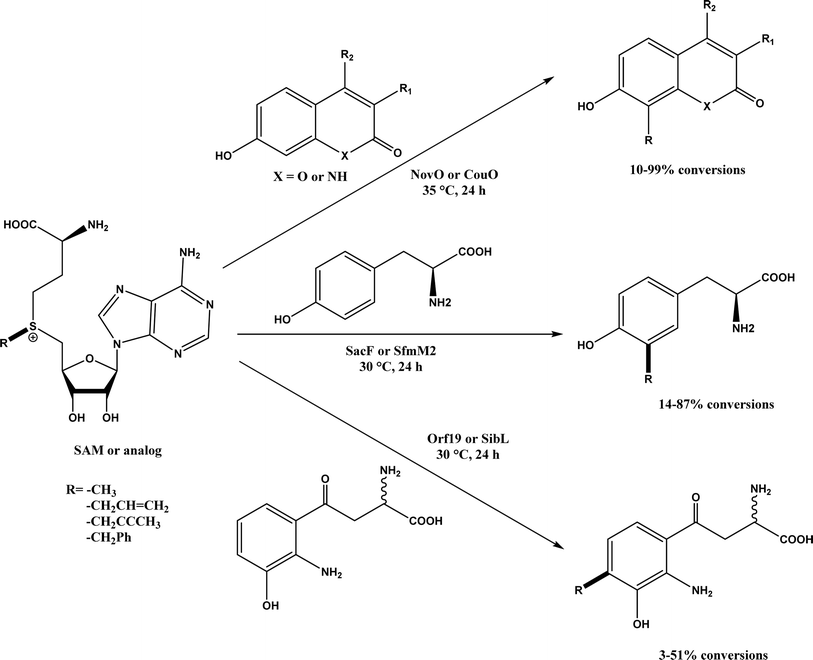 |
| | Fig. 43 Friedel–Crafts alkylation catalyzed by (S)-adenosyl-L-methionine (SAM) dependent methyltransferases. | |
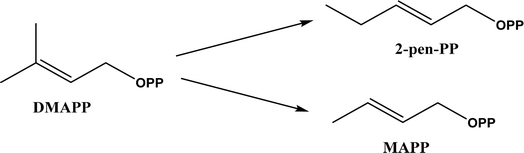 |
| | Fig. 44 Dimethylallyl diphosphate (DMAPP) and its analogues that can serve as alkyl donors for prenyltransferases. | |
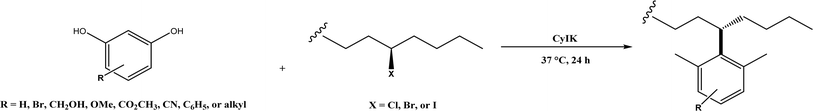 |
| | Fig. 45 CylK-mediated alkylation of resorcinols with alkyl halides. | |
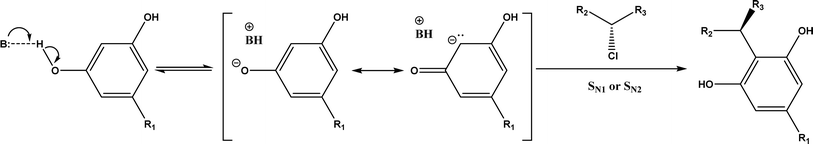 |
| | Fig. 46 Mechanism of resorcinol nucleophilic activation through hydrogen bonding or deprotonation. | |
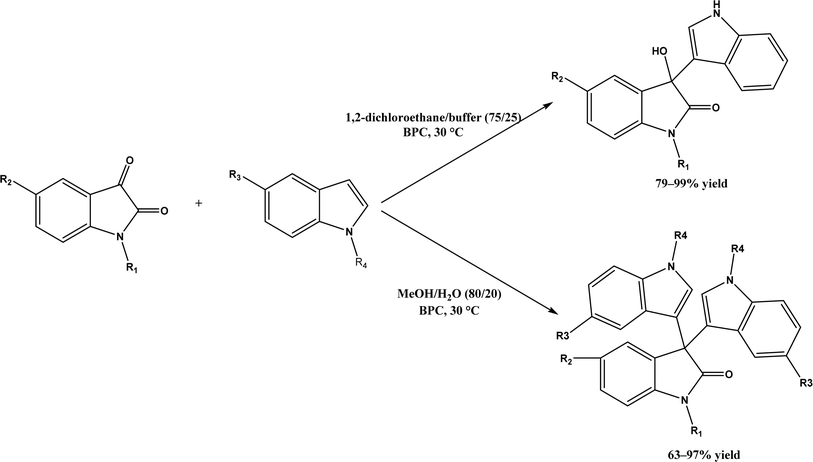 |
| | Fig. 47 Solvent-controlled Friedel–Crafts reactions between isatins and indoles catalyzed by α-chymotrypsin from bovine pancreas (BPC). | |
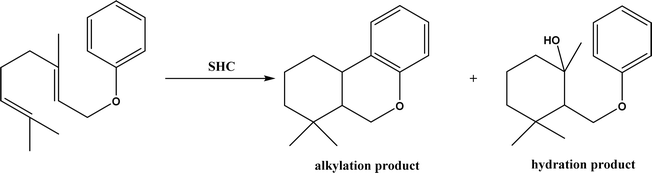 |
| | Fig. 48 Squalene-hopene cyclase (SHC)-catalyzed conversion of geranyl phenyl ether. | |
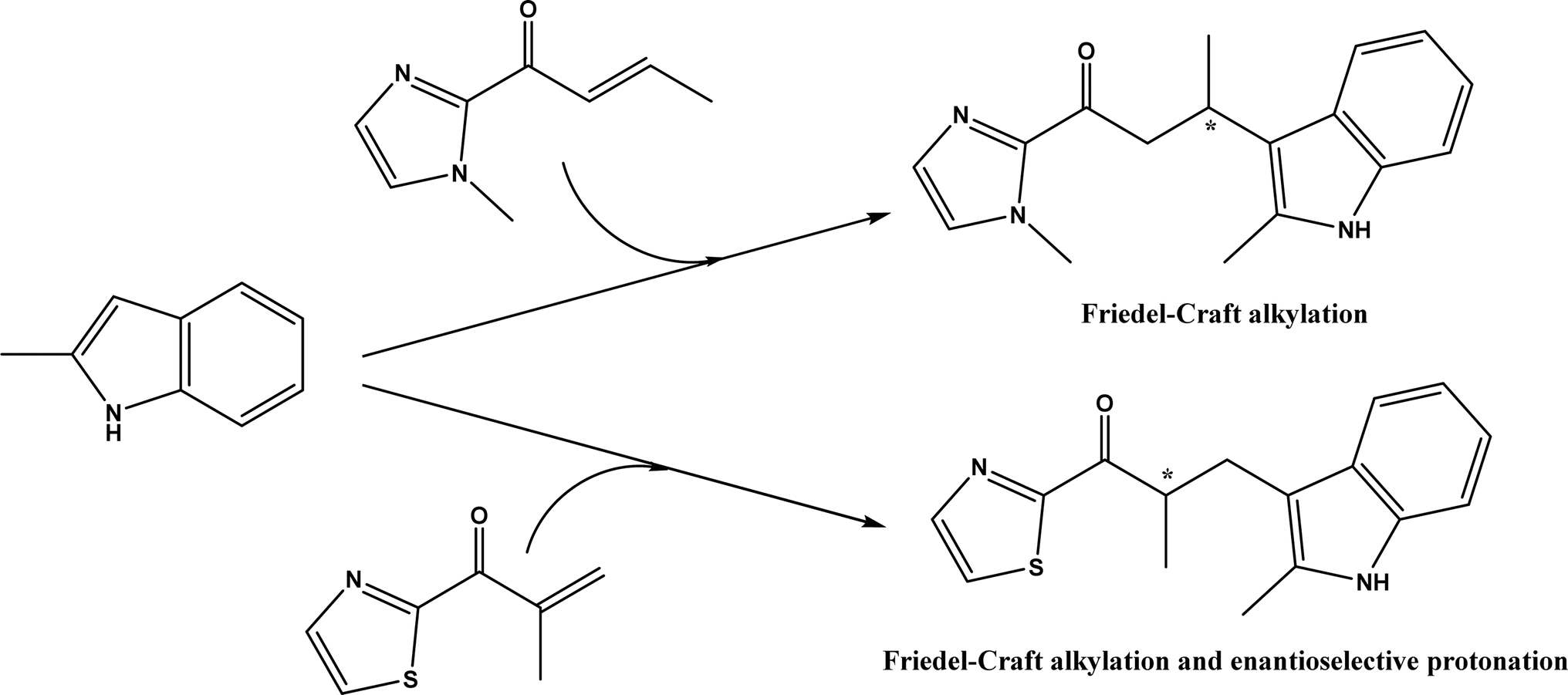
An artificial metalloenzyme was constructed by complexing Cu(II) with 1,10-phenanthroline as a ligand, which had a strong affinity with the transcription factor Lactococcal multidrug resistance Regulator (LmrR), a homodimeric protein.50 This LmrR metallozyme was used for the enantioselective Friedel–Crafts alkylation of indoles with α, β unsaturated 2-acyl-imidazoles to afford up to 92% ee, and the tandem Friedel–Crafts alkylation/enantioselective protonation reaction (Fig. 49). The protein mutation tailored the selectivity and activity of artificial metalloenzyme. This group170 further demonstrated that the protein's N19 and M89 positions are critical to the enzyme activity, and mutations at these locations indicate the importance of different side chains in the pocket of LmrR for controlling the reactivity and selectivity of mutants for both C–C bond formation and enantioselective protonation.
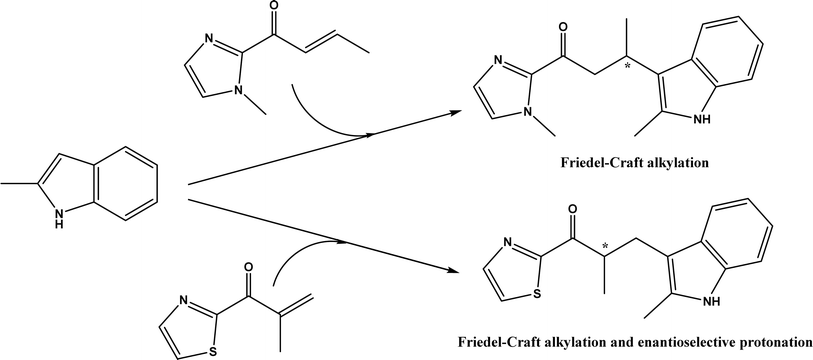 |
| | Fig. 49 Artificial metalloenzyme-catalyzed Friedel–Crafts and the tandem Friedel–Crafts/enantioselective protonation. | |
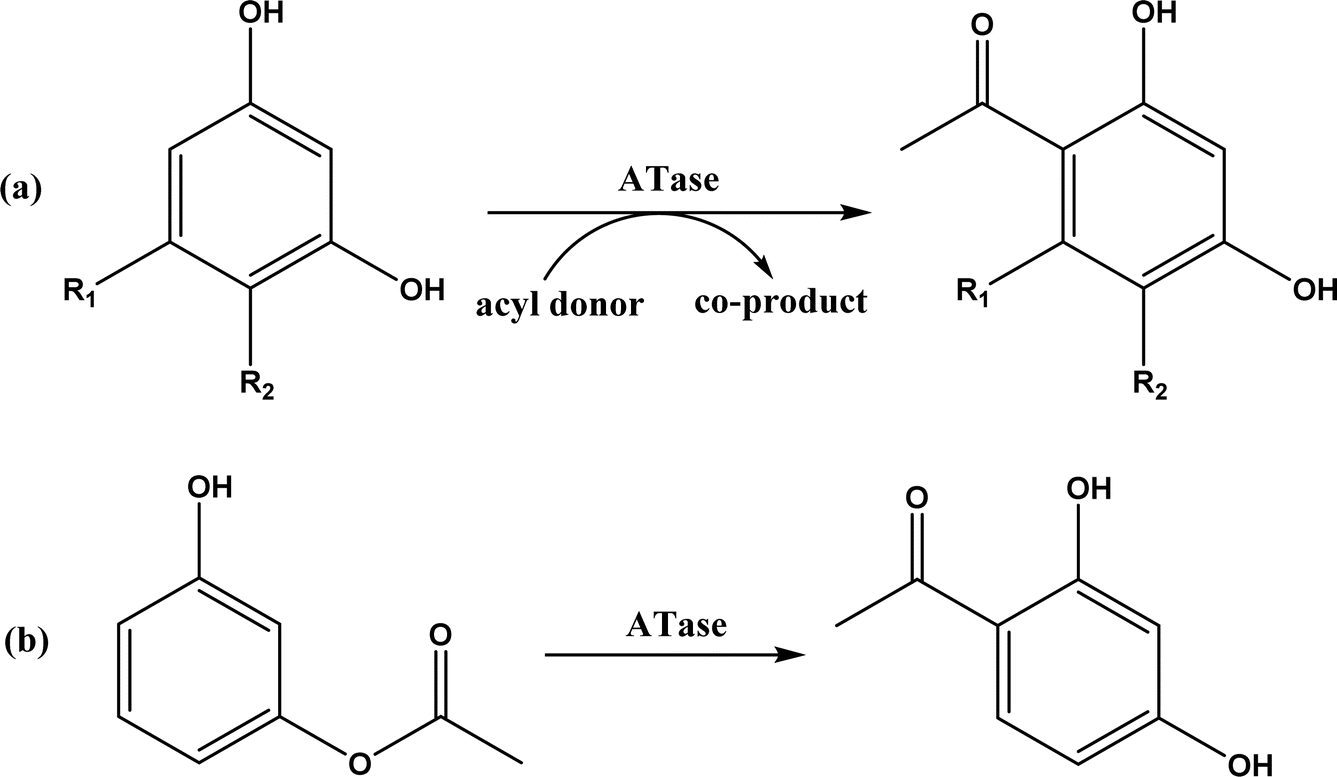
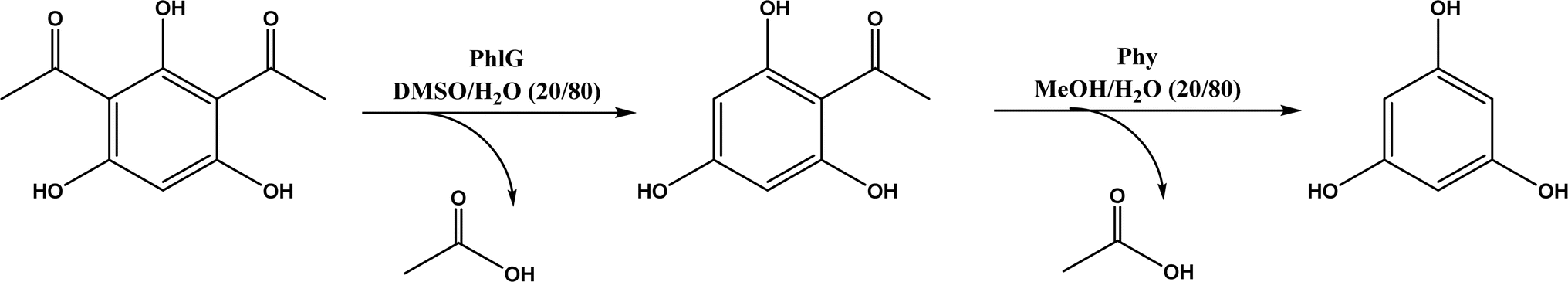
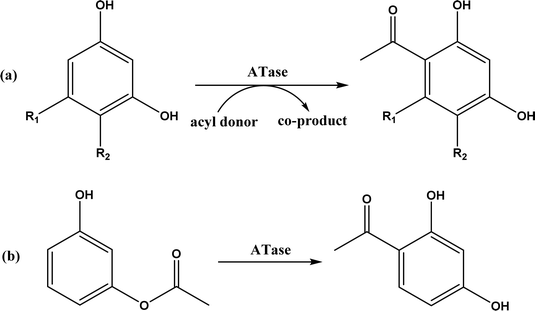
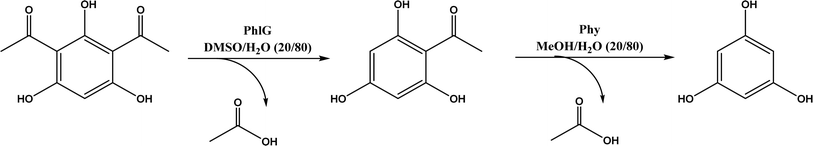
The multicomponent acyltransferase (ATase) catalyzes the in vivo reversible acetylation of monoacetylphloroglucinol. This activity can be extended to catalyze Friedel–Crafts acylation of resorcinols and Fries rearrangement of phenolic esters (Fig. 50). A mutant of ATase (known as PpATaseCH) showed five-fold higher activities than the wild type; polyketide 2,4-diacetylphloroglucinol (DAPG) and N-acetylimidazole were effective acyl donors leading to up to 99% product yields for regioselective Friedel–Crafts acylation.51 This group171 further discovered that the same enzyme (PpATaseCH) promoted the C–S bond cleavage prior to C–C bond formation, thus identified ethyl thioacetate as a suitable acetyl donor for the acylation of resorcinol derivatives (Fig. 50a), achieving up to 99% conversion and 88% isolated yield. On the other hand, reverse Friedel–Crafts acylation can be accomplished by a group of co-factor independent enzymes known as retro-Friedel–Crafts hydrolases, which requires substrates with a carbonyl group. Two of these enzymes, 2,6-diacetylphloroglucinol hydrolase (PhlG) from Pseudomonas fluorescens and phloretin hydrolase from Eubacterium ramulus (Phy), were selected to carry out the retro-Friedel–Crafts reactions shown in Fig. 51 in aqueous solutions containing organic solvents, resulting in 83% conversion in both reactions.172 However, attempts to form C–C bonds via Friedel–Crafts acylation by these two enzymes in different solutions of organic solvents all failed.
 |
| | Fig. 50 Acyltransferase-catalyzed Friedel–Crafts acylation (a) and Fries rearrangement (b). | |
 |
| | Fig. 51 PhlG and Phy-catalyzed retro-Friedel–Crafts reactions in nature. | |
6. Mannich reaction
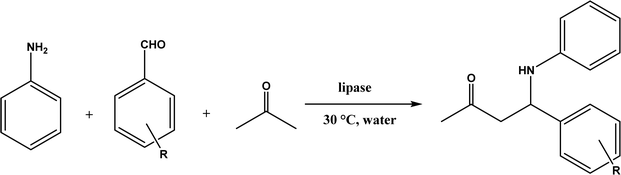
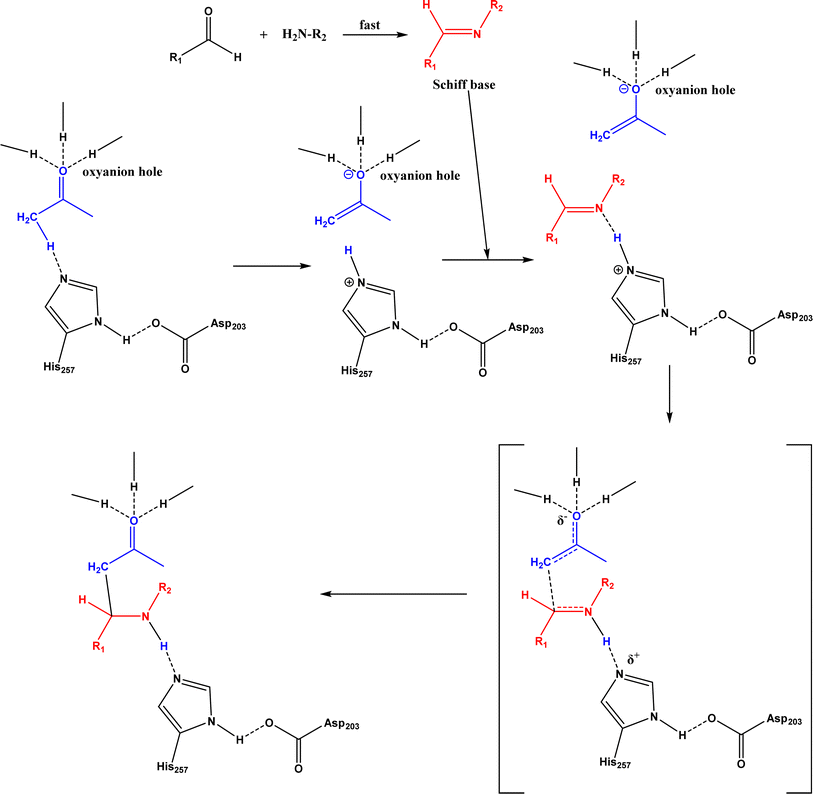
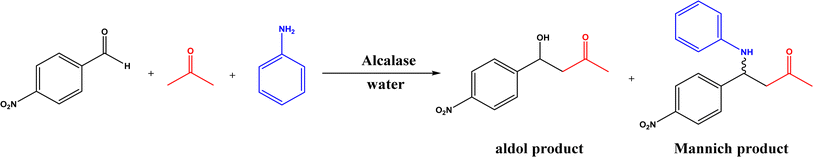
Mannich reaction is a three-component reaction involving a primary or secondary amine, an enolizable carbonyl compound, and a non-enolizable aldehyde to synthesize β-amino carbonyl compounds. This reaction usually competes with the aldol condensation. The Anilkumar group173 systematically reviewed the Mannich reaction catalyzed by various organo- and metal catalysts, along with two examples of enzyme catalysts (acylase from Aspergillus melleus174 and wheat germ lipase175). When Mannich reactions of substituted benzaldehyde, aniline, and acetone were catalyzed by various lipases (Fig. 52),52 it was found that Mucor miehei lipase led to the highest product yield (although the stereoselectivity was not specified), followed by Candida antarctica lipase B; in addition, neat organic solvents (i.e., toluene, dichloromethane, THF, DMF and acetone) resulted in the Schiff base product (>90%) instead of the Mannich product while the addition of water favored the Mannich reaction (e.g., 40–50% water mixing with acetone produced the highest yield). A lipase catalysis mechanism was described in Fig. 53:52 a quick formation of Schiff base between aldehyde and amine, the Schiff base forming a complex with the enolate anion (from ketone as being activated by the lipase) and the His residue, new C–C bond formation via electron transfer from Schiff base to enolate anion to form a new carbon–carbon bond, and the release of Mannich product from the oxyanion hole. In another study, trypsin from hog pancreas was found a more effective catalyst than lipases and α-amylase for Mannich reactions among 4-nitrobenzaldehyde, p-anisidine, and acetone; acetone and ethanol were shown better solvents than others while water was not necessary for the reaction.53 The Mannich reaction between 4-nitrobenzaldehyde, acetone and aniline (Fig. 54) was catalyzed by Alcalase, producing 51% aldol product and 46% Mannich product at 45 °C (no stereoselectivity was specified) with Alcalase-CLEA® while denatured Alcalase or no enzyme favored more aldol product.34
 |
| | Fig. 52 Lipase-catalyzed Mannich reaction in water. | |
 |
| | Fig. 53 Mechanism of lipase-catalyzed Mannich reaction (redrawn from ref. 52). | |
 |
| | Fig. 54 Mannich reaction between 4-nitrobenzaldehyde, acetone and aniline. | |
7. Morita–Baylis–Hillman (MBH) reaction
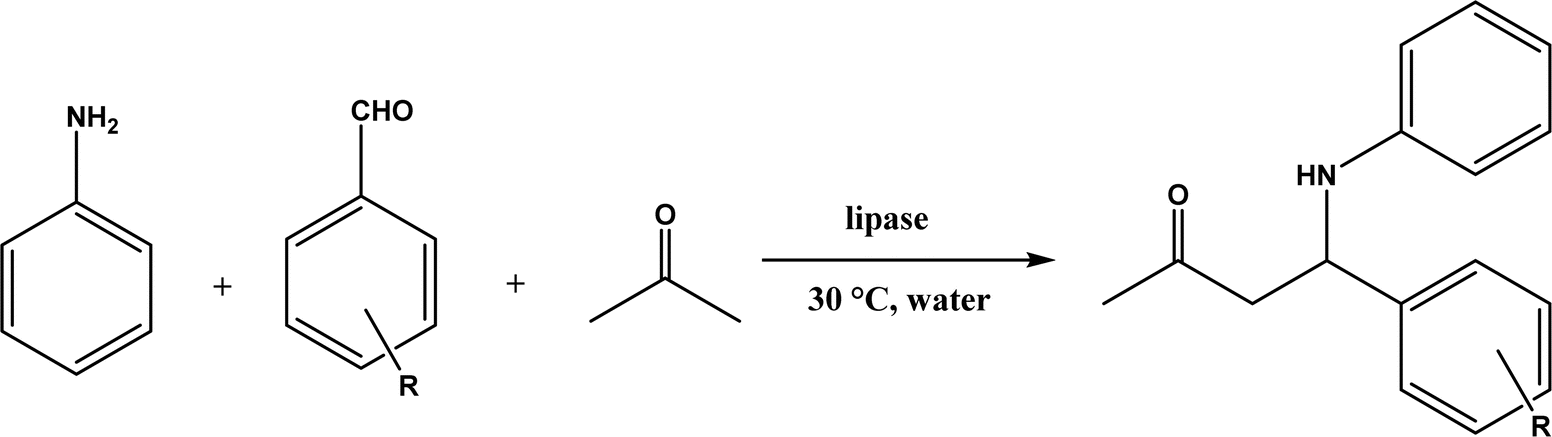
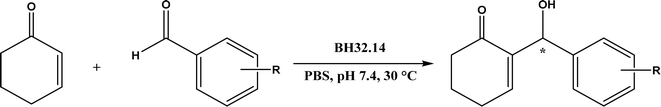
The MBH reaction, also known as Baylis–Hillman reaction, is a C–C coupling reaction between activated alkene (α-carbon of a conjugated carbonyl compound) and carbon electrophile, traditionally catalyzed by a tertiary amine or phosphine.176 The reaction mechanism typically begins with a Michael addition of the catalyst (nucleophile) at β-carbon of a conjugated carbonyl compound, continues with C–C bond formation with the electrophile, and ends with product release from the catalyst; the same mechanism is applicable to protein catalysts.177 Lipases and esterases could only achieve 10% conversion for the MBH reaction of 4-nitrobenzaldehyde and cyclohexenone although bovine serum albumin (BSA) enable 35% conversion.178 When the MBH reaction between 4-nitrobenzaldehyde and methyl vinyl ketone was catalyzed by Alcalase, a higher reaction temperature (30–60 °C) led to a higher conversion (up to 26%), but the reaction was non-specific protein catalysis because the denatured protease produced similar yields under the same conditions,34 which could be explained by the nonspecific catalytic role of the histidine residue because imidazole derivatives have been shown as effective catalysts for the MBH reaction.179 Other than the promiscuous activities of hydrolases for MBH reactions, Crawshaw and co-workers54 employed the directed evolution to optimize a primitive computationally designed protein for the MBH reaction (BH32), and found that BH32.14 variant acted as an efficient and enantioselective MBHase to promote the reactions between activated alkenes and aldehydes with 33–99% yields and 54–99% ees in most cases (Fig. 55); the likely catalytic mechanism involved a nucleophilic His23 and a multi-functional Arg124 to accelerate the MBH reaction.
 |
| | Fig. 55 Morita–Baylis–Hillman (MBH) reaction between activated alkene and aldehyde. | |
8. Diels–Alder reaction
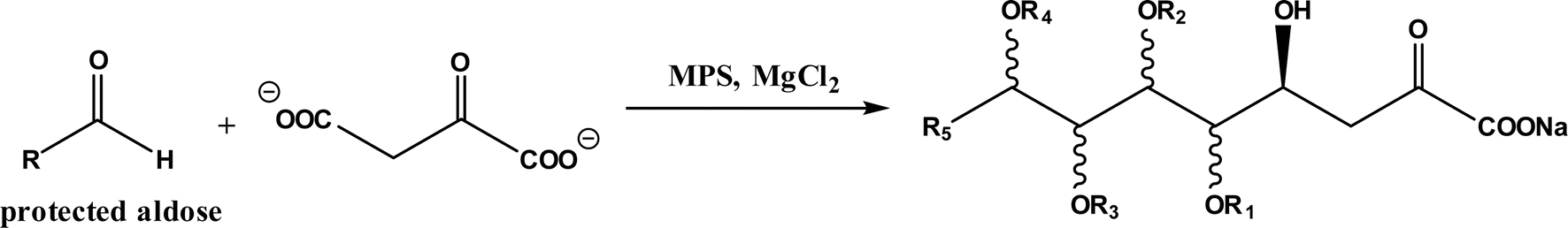
Diels–Alder reaction, known as [4 + 2] cycloaddition, yields a six-membered ring compound with regio- and stereoselectivity through reacting a conjugated diene with a substituted alkene (as dienophile), usually catalyzed by Lewis acids such as ZnCl2 and AlCl3. Many natural products are biosynthetically produced through Diels–Alder reactions catalyzed by enzymes, generally categorized as Diels–Alderases,182 for example, trans-decalin formation by Fsa2-family enzymes as shown in Fig. 56,180 and the biosynthesis of spinosyn A involving a cyclase SpnF to catalyze [4 + 2] cycloaddition as shown in Fig. 57.181 Several earlier studies have identified isolated enzymes for catalyzing Diels–Alder reactions such as a crude enzyme extract of solanapyrone synthase for cycloaddition of prosolanapyroneIII to the exo adduct solanapyrone A,183 crude lovastatin nonaketide synthase (LovB) for the cyclization of hexaketide triene esters,3 and riboflavin synthase for the cyclization of 6,7-dimethyl-8-ribityllumazine.184 Ose and co-workers4 determined the 1.70 Å resolution crystal structure of Mg2+-dependent fungal macrophomate synthase (MPS, a natural Diels–Alderase) in complex with pyruvate, and described the three-step catalytic mechanism for the Diels–Alder reaction of 2-pyrone and oxalacetate to form macrophomic acid (Fig. 58): decarboxylation of oxalacetate, Diels–Alder reactions of the enolate and 2-pyrones, and anti-elimination of water and decarboxylation. The C–C bond forming step was previously debated by Watanabe and co-workers55 whether it is Michael-aldol process or Diels–Alder reaction. Later, this second step was suggested by the Jorgensen group56 to be a stepwise Michael-aldol reaction instead of a Diels–Alder reaction (Fig. 59) based on the mixed quantum and molecular mechanics (QM/MM) in combination with Monte Carlo (MC) simulations and free-energy perturbation (FEP) computations. The free energy of Diels–Alder transition state was found over 20 kcal mol−1 higher than that of Michael and aldol transition states. Through site-directed mutagenesis, the Hilvert group186 identified three amino acid residues (Arg101, Asp70, and His73) of MPS are essential to oxaloacetate decarboxylation and trapping of the enolate with a 2-pyrone. Experimentally, it was found that MPS exhibited promiscuous aldolase activity for catalyzing aldol reactions between various aldehydes and oxaloacetate although enantioselectivities were generally low.187 However, a later study by the same group reported high aldolase activities and stereoselectivities of MPS when catalyzing the reaction between oxaloacetate and protected aldoses to synthesize protected 3-deoxysugar derivatives (28–84% yields and 8:1 to >19:1 dr) as illustrated in Fig. 60.188 A natural cofactor-independent Diels-Alderase, AbyU, is a homodimer consisting of two eight-stranded antiparallel β-barrels; this enzyme is found in abyssomicin C biosynthetic pathway to catalyze a Diels–Alder reaction step.189 AbyU maintained considerable catalytic activities at temperatures of up to 65 °C and in 3.0 M guanidinium hydrochloride (a protein denaturant), and >50% folding structures in up to 70% (v/v) acetonitrile and >70% folding in 80% (v/v) DMSO and methanol.190 The Baker group57 used de novo computational method to design the active site that is suitable for catalyzing a model Diels–Alder reaction between 4-carboxybenzyl trans-1,3-butadiene-1-carbamate and N,N-dimethylacrylamide (Fig. 61), searched 207 protein structures for backbone geometries that accommodate the active site and substrates, and narrowed down to test 50 enzymes, but only two of them showed measurable activities, which could be further improved via directed evolution. Despite its low efficiency, this method allows a rational design and search of enzyme structures for particular reactions. Quantum chemical calculations illustrated how enzyme active sites (of theozymes) accelerate the intramolecular Diels–Alder conversion of salvileucalin A to salvileucalin B; theozymes investigated contain common functional group arrays found in esters.191 Interestingly, RNA molecules were identified as efficient as DNA in catalyzing C–C bond formation in Diels–Alder reaction.192
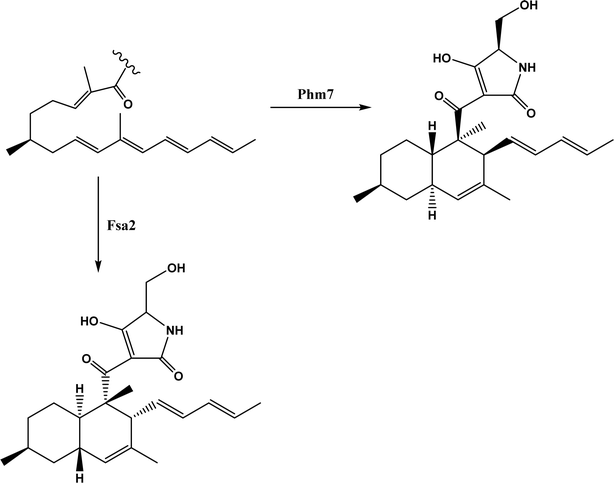 |
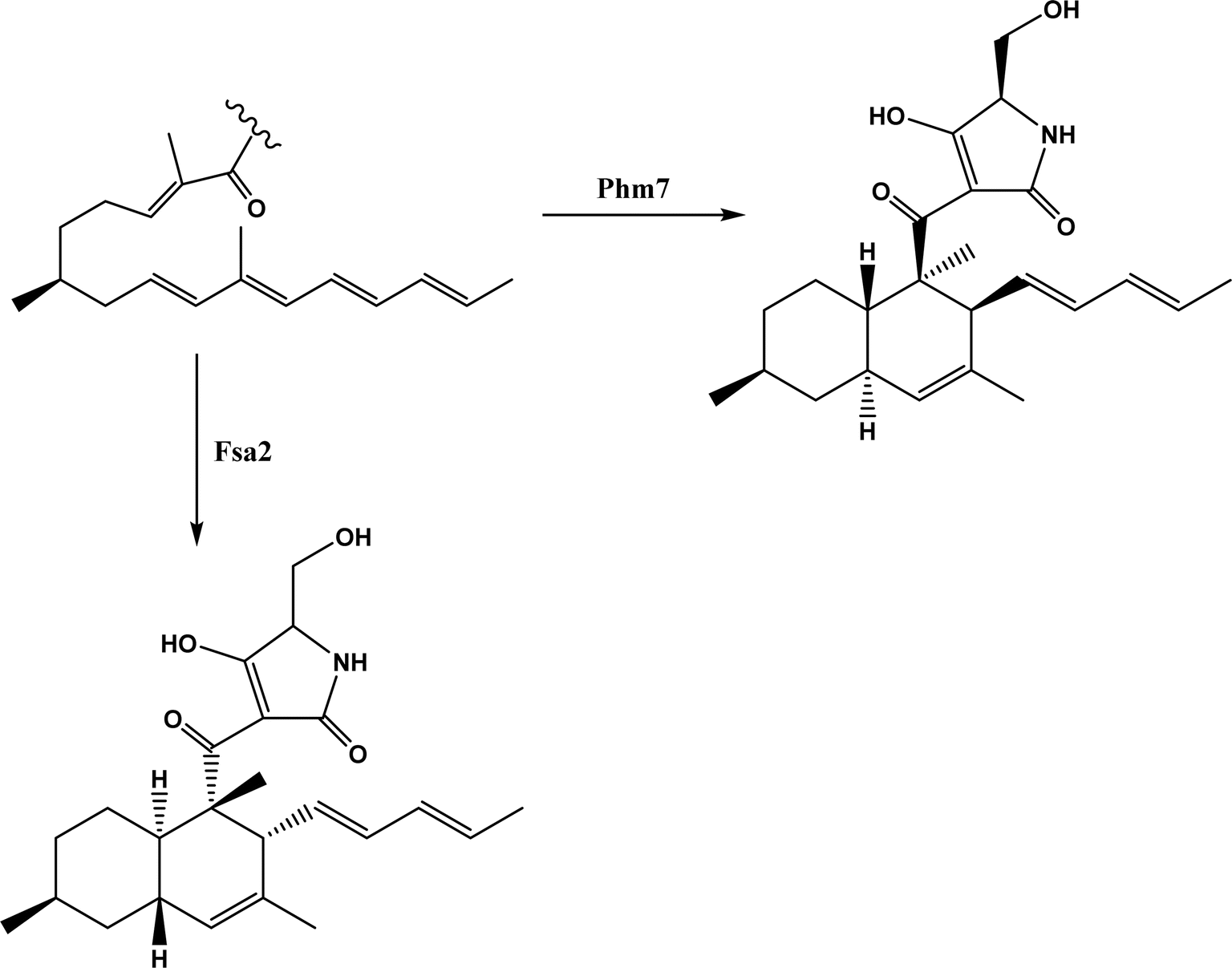
| | Fig. 56 Stereospecific [4 + 2] cycloaddition reactions catalyzed by decalin synthases Fsa2 and Phm7.180 | |
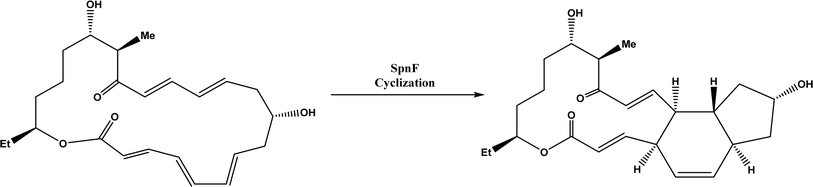 |
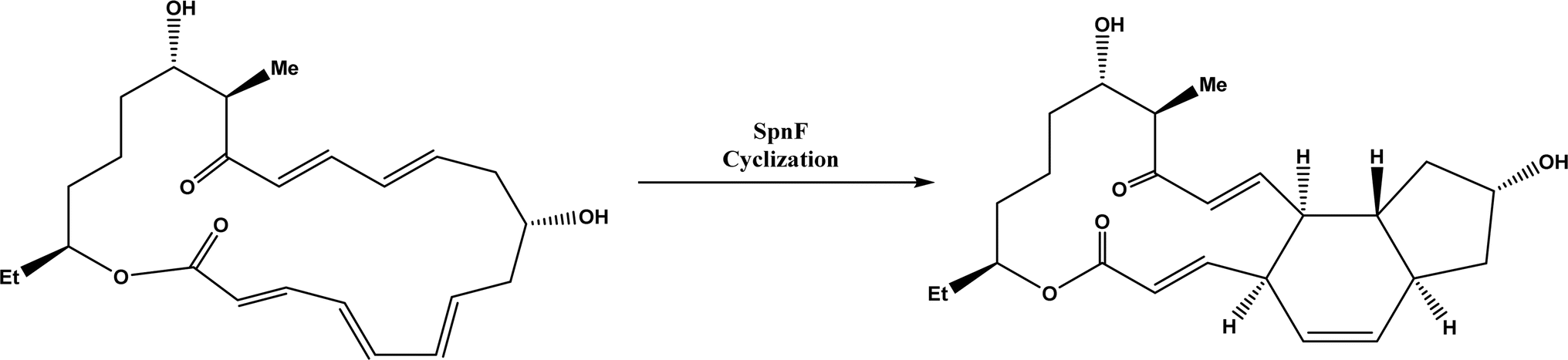
| | Fig. 57 Cyclase SpnF-catalyzed cyclization during the biosynthesis of spinosyn A.181 | |
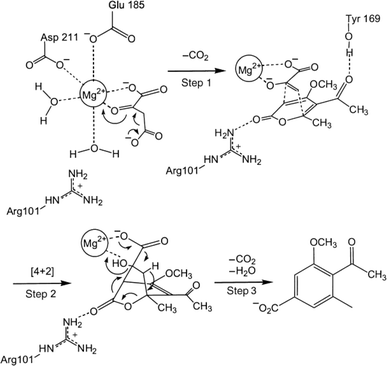 |
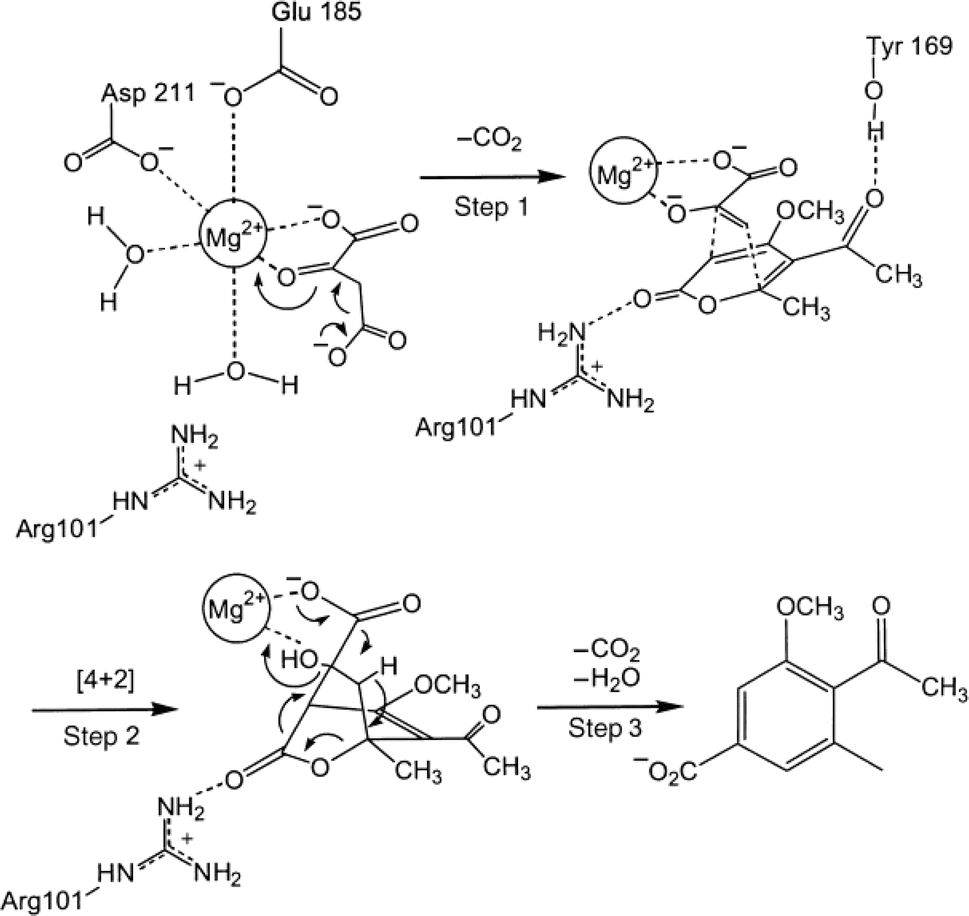
| | Fig. 58 Mechanism of macrophomate synthase-catalyzed Diels–Alder reaction of 2-pyrone and oxalacetate to synthesize macrophomic acid [Reprinted with permission from ref. 185 Copyright 2003 Wiley-VCH Verlag GmbH& Co.]. | |
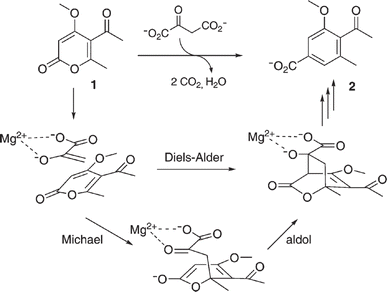 |
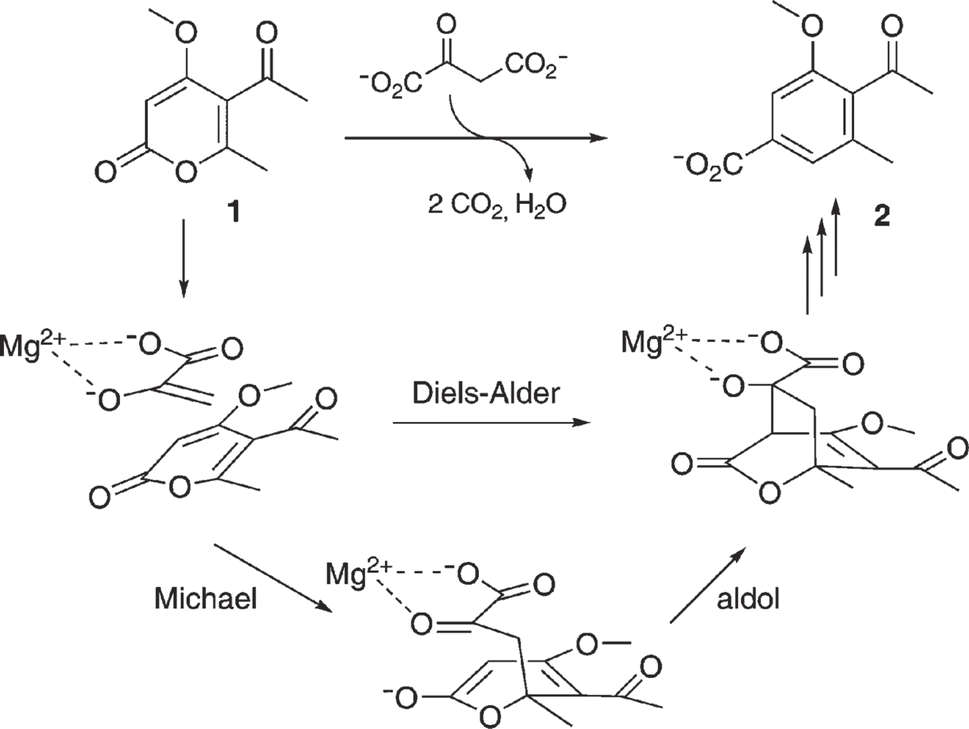
| | Fig. 59 Mechanism of MPS-catalyzed synthesis of macrophomate (2) from 2-pyrone (1) and oxaloacetate [Reprinted with permission from ref. 186 Copyright 2007 The Royal Society of Chemistry]. | |
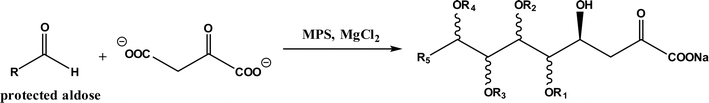 |
| | Fig. 60 MPS-catalyzed aldol reaction between oxaloacetate and protected aldoses. | |
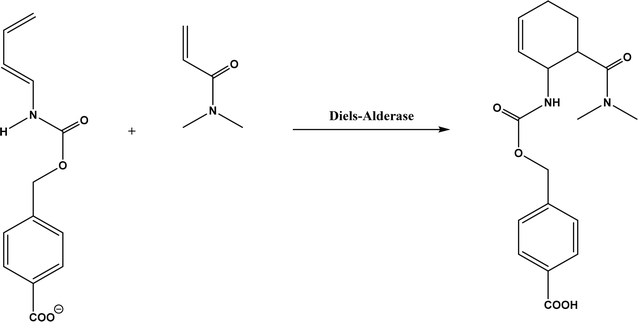 |
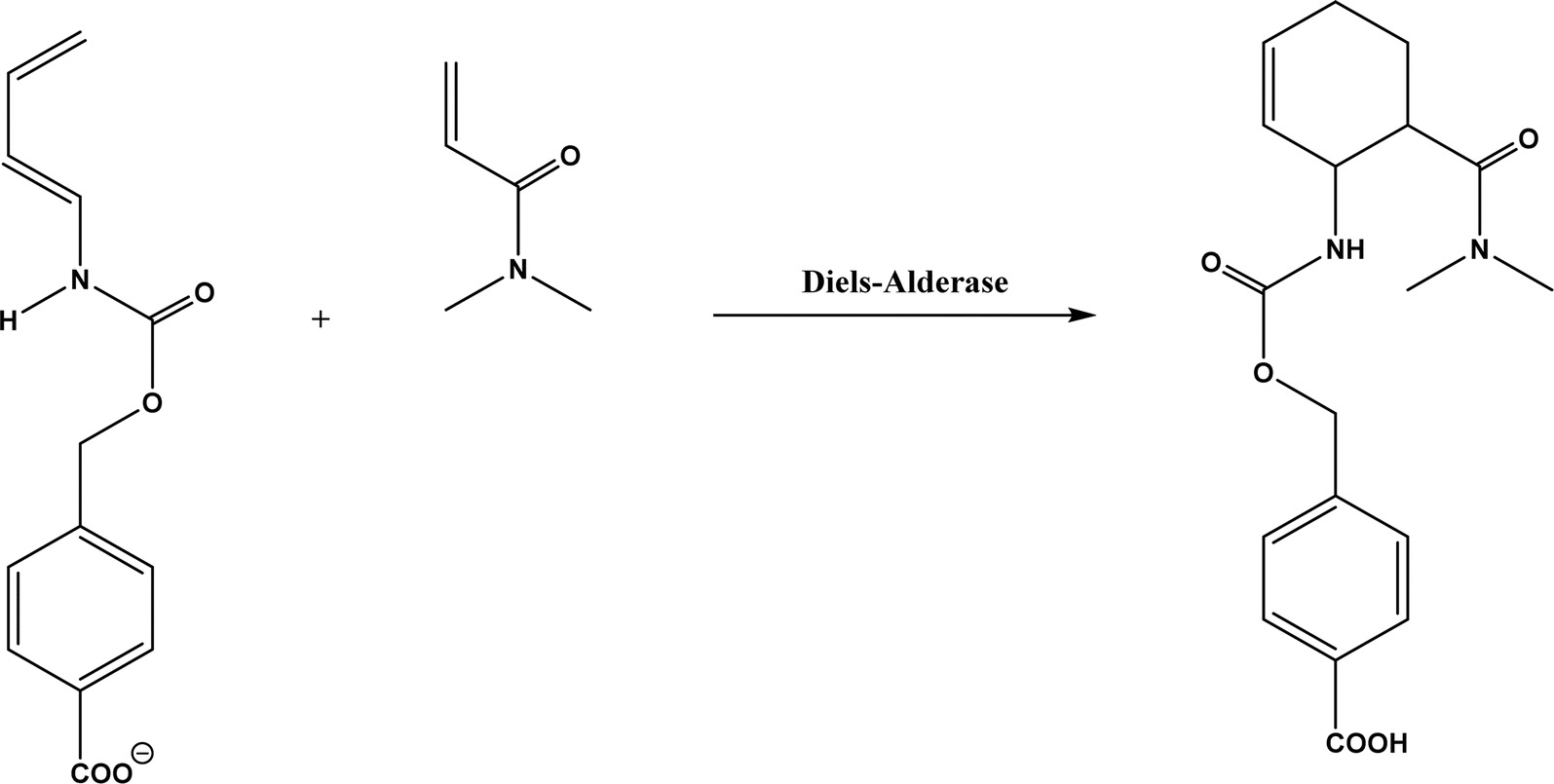
| | Fig. 61 Diels–Alder reaction between 4-carboxybenzyl trans-1,3-butadiene-1-carbamate and N,N-dimethylacrylamide. | |
Natural ribozymes catalyze the hydrolysis and transesterification of internucleotide bonds, but in vitro-selected ribozymes could facilitate the C–C bond formation through Diels–Alder reaction.193,194 In addition, some antibodies have been discovered for catalyzing Diels–Alder reactions.195–197 Topics on ribozymes and nucleic acid catalysis198 and antibody catalyzed cycloadditions199 have been covered by other reviews. Serganov and co-workers200 compared structural bases of these different biocatalysts: antibodies has a hydrophobic catalytic core, which is similar to natural Diels–Alderases; however, Diels–Alderases also has a co-factor Mg2+ cation to coordinate with carbonyl oxygens of the dienophile in addition to hydrogen bonding of the active site with substrates. The ribozyme has a wedge-shaped catalytic pocket to dictate the stereoseletivity, and its catalysis is accomplished through a combination of proximity, complementarity, and electronic effects.
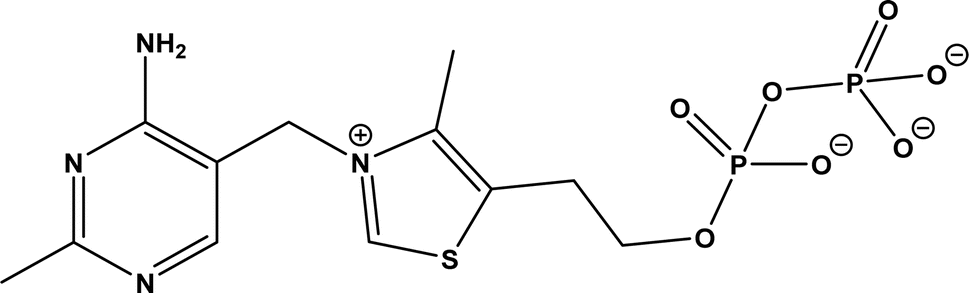
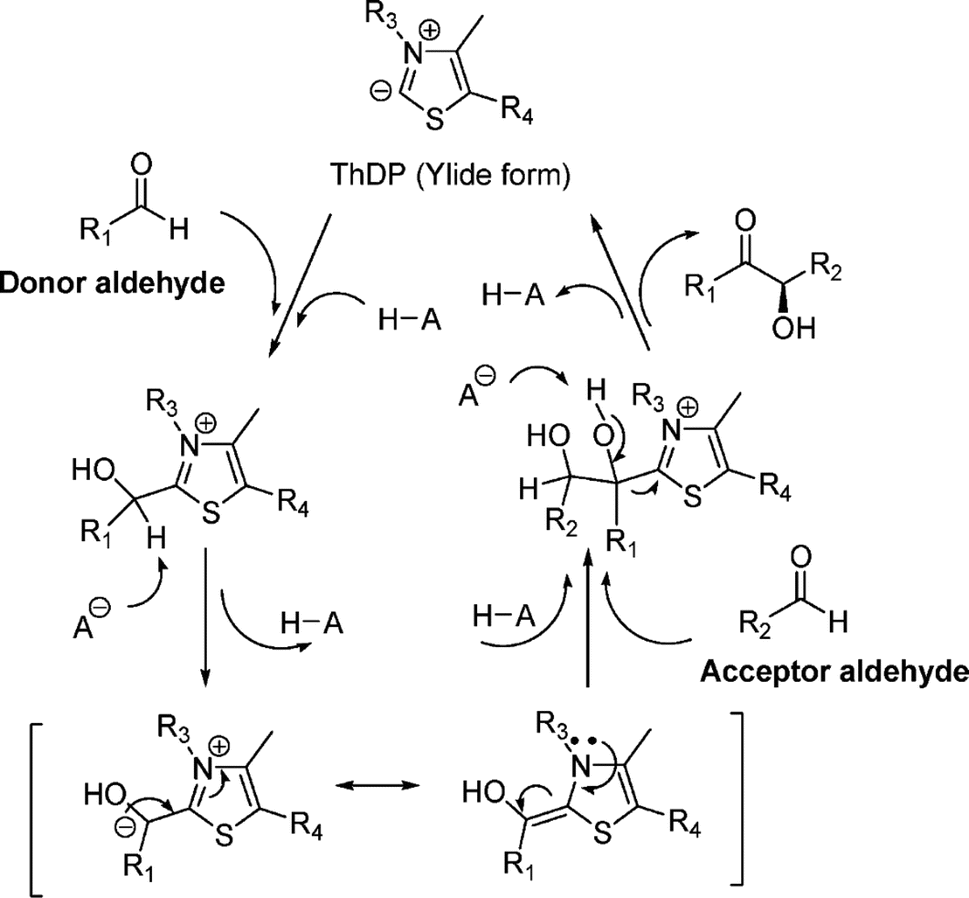
9. Acyloin condensations via thiamine diphosphate (ThDP)-dependent enzymes
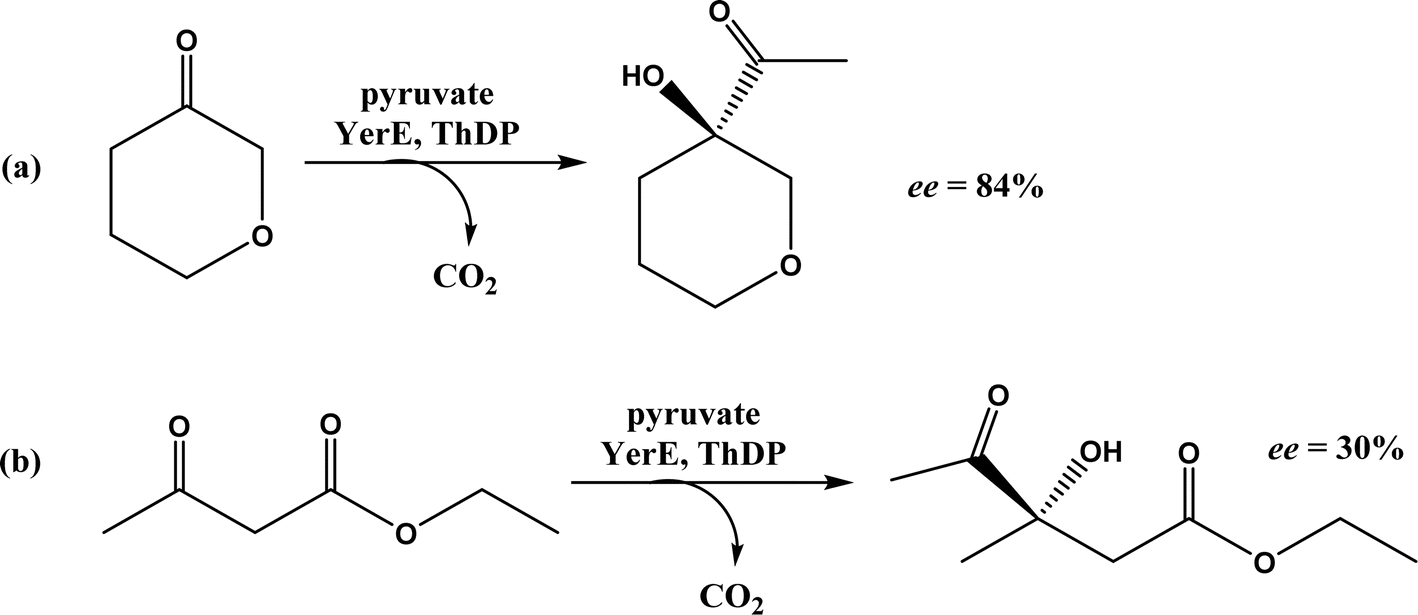
Thiamine (or thiamin) is better known as vitamin B1, a water-soluble vitamin. Its biologically active derivative, called thiamine diphosphate (ThDP) or thiamine pyrophosphate (TPP), is a cofactor of enzymes that are essential to many cellular metabolism cycles. ThDP is a natural thiazolium salt consisting of pyrimidine, thiazolium, and pyrophosphate units (Fig. 62). ThDP-dependent enzymes are known for their capabilities in forming C–C bonds via acyloin condensation; the general mechanism (Fig. 63) involves thiamine diphosphate cofactor reacting with an aldehyde (donor) to form an active zwitterion, which attack the acceptor aldehyde to yield (R)-α-hydroxyketone after the release of the cofactor.80 Applications of these enzymes in C–C bond formation and their specific catalytic mechanisms have been discussed in earlier reviews,80,201–205 which include several known enzymes such as acetohydroxy acid synthase (AHAS, EC 2.2.1.6), benzoylformate decarboxylase (BFD, EC 4.1.1.7),206 benzaldehyde lyase (BAL, EC 4.1.2.38), pyruvate decarboxylase (PDC, EC 4.1.1.1), phenylpyruvate decarboxylase (PhPDC, EC 4.1.1.43), keto acid decarboxylase (EC 4.1.1.72),207 and transketolase (TK, EC 2.2.1.1),208 as well as newer enzymes such as 1-deoxy-D-xylulose 5-phosphate synthase (DXPS, EC 2.2.1.7), flavoenzyme cyclohexane-1,2-dione hydrolase (CDH, EC 3.7.1.11), flavoenzyme YerE (the decarboxylation of pyruvate and the transfer of the activated acetaldehyde to aldehydes and ketones), Bacillus stearothermophilus acetylacetoin synthase (ketones as acceptors to form tertiary alcohols209), and ThDP-dependent PigD and MenD [for Stetter-type of 1,4 addition of aldehydes, or benzoin-condensation 1,2-addition210,211].
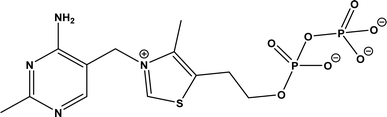 |
| | Fig. 62 Structure of thiamine diphosphate (ThDP). | |
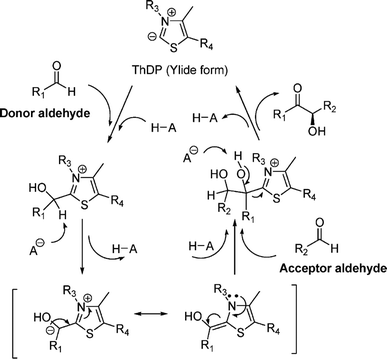 |
| | Fig. 63 Mechanism for ThDP-dependent lyase-catalyzed Umpolung carboligation of aldehydes [Reprinted with permission from ref. 80 Copyright 2011 American Chemical Society]. | |
A few recent updates beyond previous reviews are discussed here. Other than PigD for catalyzing the Stetter reaction of α-keto acids with α,β-unsaturated ketones (Michael acceptor substrates), two new ThDP-dependent enzymes, SeAAS from Saccharopolyspora erythraea and HapD from Hahella chejuensis were identified to have 39% and 51% similarity with PigD respectively in terms of their amino acid sequences, and thus could catalyze intermolecular Stetter reactions (1,4-carboligation in Fig. 64) and benzoin condensation (1,2-carboligation in Fig. 65) with high enantioselectivity.58 Benzaldehyde lyase (BAL) was evaluated in mixtures of deep eutectic solvents (DES) and water, and exhibited high activities (75–98% conversions) and good enantioselectivities (27–99% ee) for carboligation reactions of aldehydes conducted in a 60:40 (v/v) mixture of choline chloride/glycerol (1:2) with phosphate buffer (pH 8.0).59 As shown in Fig. 66, BAL promoted the enantioselective carboligation and diastereoselective condensations of benzaldehyde with a racemic aldehyde at the same time, leading to high diastereoselectivities (de up to 99%).212 YerE is a carbohydrate-modifying enzyme from Yersinia pseudotuberculosis, which catalyzed the carboligation of pyruvate to (R)-3-methylcyclohexanone to produce an (R,R)-tertiary alcohol with diastereomeric ratio (dr) >99:1, while the similar reaction with (R)-3-methylcyclohexanone yielded (S,S)-tertiary alcohol with dr >99:1; more interestingly, the YerE-catalyzed carboligation to non-chiral acceptors (with or without structural analogy to physiological carbohydrate substrates as shown in Fig. 67(a) and (b) respectively) led to corresponding 84% and 30% ees, which implies that the substrate structure dictates its interactions with the enzyme and the stereoselectivity of YerE.213 Along with MenD from E. coli, two other tricarboxylic acid (TCA) cycle-involving enzymes (with decarboxylation activity), SucA from E. coli and Kgd from Mycobacterium tuberculosis, were able to catalyze asymmetric mixed carboligation (1,2-addition) of α-ketoglutarate and different aldehydes to synthesize δ-hydroxy-γ-keto acids with moderate to excellent enantioselectivity (Fig. 68).214 Similar, C–C carboligation between substituted benzaldehyde and glyoxylic acid was catalyzed by variants of ThDP-dependent pyruvate decarboxylase to produce 2-hydroxyacetophenone (2-HAP) and its derivatives with 0.2–92.7% yields.215 Benzaldehyde lyase (BAL) from Pseudomonas fluorescens biovar I was evaluated for intramolecular benzoin reactions of dibenzaldehyde derivatives (Fig. 69), which require three-carbon linker to connect two benzaldehyde rings at 2,2′ positions via ether linkages; BAL also accommodated substituents (e.g., Cl, Br, and OCH3) at either 3 and 3′ or 5 and 5′ positions of benzaldehyde moieties, and a pyridine ring instead of benzaldehyde.216 This BAL was further found capable of catalyzing intramolecular stereoselective Stetter reaction of ethyl (E)-4-(2-formylphenoxy)but-2-enoate or it analogues to form chroman-4-one derivatives (as important intermediates for synthesizing medical molecules), resulting in yields >90% and enantiomeric ratios (er) >90:10 in most cases.217 In addition, BAL was used to promote hydroxymethylation of aldehydes followed by enzymatic reductive amination to form enantiomeric N-substituted 1,2-amino alcohols,218 and the coupling of formaldehyde with 3-hydroxypropanal.219 It was recently discovered52 that a subclass of (myco)bacterial ThDP-dependent enzymes (e.g., ErwE and MyGE) could extend the donor substrate range from achiral α-keto acids and simple aldehydes to customized chiral α-keto acids with a chain length from C4 to C8; as a result, enantioselectivity acyloin products were produced (Fig. 70) with 22–85% yields and >90% ees.
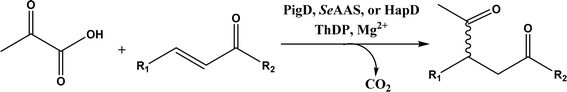 |
| | Fig. 64 1,4-Carboligation reactions of pyruvic acid with α,β-unsaturated ketones (Michael acceptors) catalyzed by PigD, SeAAS, or HapD. | |
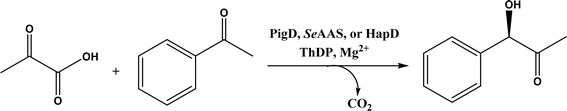 |
| | Fig. 65 1,2-Carboligation reaction of pyruvic acid with benzaldehyde (acceptor) catalyzed by PigD, SeAAS, or HapD. | |
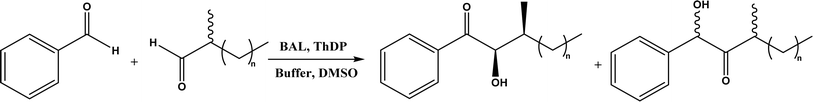 |
| | Fig. 66 BAL-catalyzed simultaneous enantioselective carboligation and kinetic resolution. | |
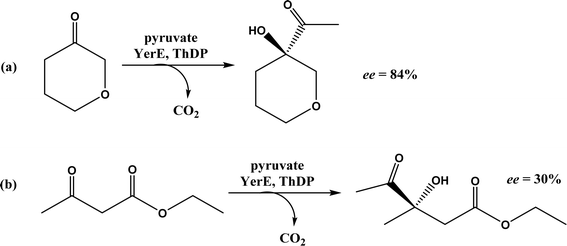 |
| | Fig. 67 YerE-catalyzed carboligation to non-chiral acceptor substrates [with or without structural analogy to physiological carbohydrate substrates as shown in (a) and (b) respectively]. | |
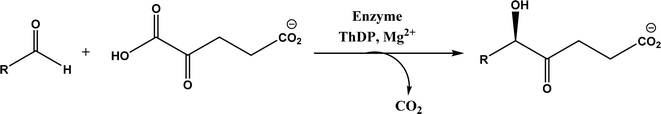 |
| | Fig. 68 Enzymatic 1,2-addition of α-ketoglutarate to aldehydes. | |
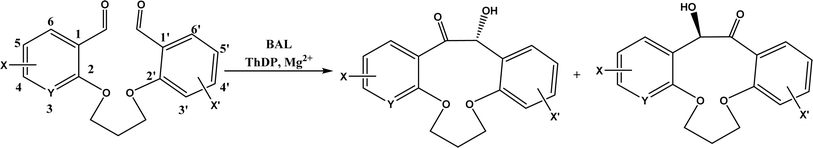 |
| | Fig. 69 BAL-catalyzed intramolecular benzoin reaction of dibenzaldehyde derivatives. | |
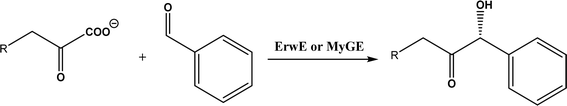 |
| | Fig. 70 Benzoin condensation reaction between 2-oxoalkanoate and benzaldehyde. | |
10. Oxidative and reductive C–C bond formation
In their 2011 review paper, the Dong group220 described a few examples of biological dehydrogenative C–C bond formations involving cytochrome P450 enzymes, redG, and dioxygenases, etc. In a more recent review (2018), Guengerich and Yoshimoto66 systematically surveyed enzymatic oxidation–reduction reactions and their mechanisms for forming (and breaking) C–C bonds, which covered cytochrome P450 and variants, nonheme iron mono- and dioxygenases, flavoproteins (such as berberine bridge enzyme), radical S-adenosylmethionine enzymes, and peroxidases, etc. Berberine bridge enzyme (BBE) promoted the oxidative intramolecular C–C bond formation using a non-natural racemic substrate that is the analogue of natural substrate (S)-reticuline (Fig. 71); the preparative scale synthesis was performed with 500 mg substrate in 70 v% toluene and buffer (pH 9, 50 mM) using BBE and catalase (to remove H2O2) at 40 °C for 24 h, resulting in 42% (S)-2 with >97% ee as the major product, 8% regioisomer 3 as the byproduct, and 50% (R)-1 with >97% ee as the remaining reactant.221 A nonheme iron enzyme, 2-oxoglutarate/Fe(II)-dependent dioxygenase (2-ODD), mediates the oxidative cyclization in the etoposide biosynthetic pathway; based on mechanistic probe design, in vitro biochemical assays, model studies, and LC-MS monitoring of 2-ODD catalyzed reactions, the reaction mechanism is likely the benzylic radical/carbocation intermediate initiating the C–C bond formation (Fig. 72), instead of previous known hydroxylated intermediate.53 Several studies have demonstrated oxidative biaryl coupling reactions catalyzed by cytochrome P450 or laccase.222–225
 |
| | Fig. 71 Berberine bridge enzyme (BBE)-catalyzed enantioselective oxidative C–C bond formation of the non-natural racemic substrate [Reprinted with permission from ref. 221 Copyright 2011 Wiley-VCH Verlag GmbH&Co. KGaA]. | |
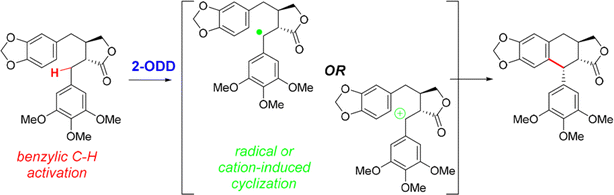 |
| | Fig. 72 Mechanism of nonheme iron enzyme 2-ODD catalyzed oxidative cyclization [Reprinted with permission from ref. 53 Copyright 2019 American Chemical Society]. | |
Reductases also showed potential for forming C–C bonds. Under photoexcitation, flavin-dependent ‘ene’-reductases (EREDs) can catalyze chemoselective and enantioselective cross-electrophile couplings (XECs) between various α-chloroamides and α-aryl-nitroalkanes to form C–C bonds. As illustrated by a model reaction between 2-chloro-N,N-dimethylacetamide and (1-nitroethyl)benzene in Fig. 73, the ‘ene’-reductase from Caulobacter segnis (CsER) selectively produced (S)-enantiomer with up to 92% yield and 90% ee while the ERED variant from Gluconobacter oxydans (GluER-T36A) preferred (R)-enantiomer with 51% yield and 80% ee.67 The reaction mechanism involves the formation of nitro radical anion by combining an in situ-generated nitronate with an alkyl radical, followed by the formation of nitrite and an alkyl radical from the nitro radial anion; the enantioselectivity is dictated by hydrogen atom transfer (HAT) controlled by the enzyme. For α,β-unsaturated aldehydes and ketones, the wild-type ene-reductases from the Old Yellow Enzyme (OYE) family favored the CC double bond reduction instead of carbocyclization (Fig. 74); however, single-site replacement of the critical proton donor Tyr residue (e.g., Tyr190 in OPR3, Tyr169 in YqjM) with a non-protic Phe or Trp led to more cyclization products; for example, YqjM Y169F-catalyzed the reaction in Fig. 74 showed 95% selectivity of cyclization, 94% de (trans/cis), >99% ee of (R,R)-product, and −29% ee of (S,R)-product.62
 |
| | Fig. 73 Phototenzymatic asymmetric cross-electrophile couplings catalyzed by flavin-dependent ‘ene’-reductases (i.e., CsER and GluER-T36A) (NADP+, nicotinamide adenine dinucleotide phosphate; GDH, glucose dehydrogenase; LED, light-emitting diode). | |
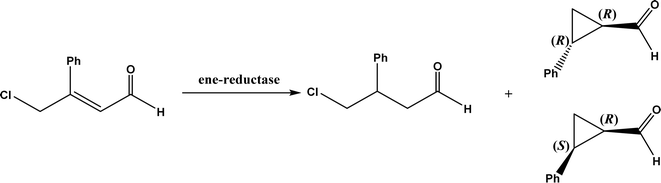 |
| | Fig. 74 Ene-reductase-catalyzed CC double bond reduction and carbocyclization of α,β-unsaturated aldehyde. | |
11. C–C bond formation through C1 resource utilization
The biotransformation of C1 resources such as CO2, CO, formaldehyde, and formate has become a new route for constructing C–C bonds. An earlier review79 surveyed the enzymatic conversions of formaldehyde to valuable chiral molecules by using aldolases and ThDP-dependent enzymes, and discussed the reaction mechanisms and enzyme discovery. Another review paper226 focused on light-driven C–H bond activation to form new C–C bonds using CO2 as the feedstock catalyzed by enzymes or molecular catalysts. A recent paper227 overviewed the CO2 conversions using carboxylases, formaldehyde transformations using C–C ligases, and CO and formate conversions via C–C ligases. Several more recent updates are discussed below. From CO2 and pyruvic acid, oxaloacetic acid and malate were derived phototenzymatically with malic enzyme using the photoreduction of a 1,1′-bis(p-sulfonatophenyl)-4,4′-bipyridinium salt as electron mediator and water-soluble tetraphenylporphyrin tetrasulfonate (H2TPPS) with triethanolamine (TEOA) as an electron donor.228,229 CO2 and succinyl coenzyme A (SCoA) can be converted to 2-oxoglutarate and CoA via light-driven carbon–carbon bond formation by using 2-oxoglutarate: ferredoxin oxidoreductase and photoexcited electrons from cadmium sulfide nanorods; electron transfer efficiency is highly dependent on how SCoA is bound at the enzyme's active site.230 The enzymatic fixation of CO2 was realized by enzymatic reductive carboxylation of crotonyl-CoA to (2S)-ethylmalonyl-CoA catalyzed by NADPH-dependent crotonyl-CoA carboxylase/reductase (Ccr), which was co-immobilized within a viologen-based redox hydrogel with the co-factor (NADPH) regeneration enzyme ferredoxin NADP+ reductase (FNR) for continuous NADPH recycling (Fig. 75); electrons were transferred from the electrode to FNR through a mediated electron transfer method (2,2′-viologen-modified hydrogel; see a review231 on viologens for enzymatic photoredox conversions of CO2); the reaction system achieved 92 ± 6% faradaic efficiency and at a rate of 1.6 ± 0.4 μmol cm−2 h−1.232 Formaldehyde can be produced from sustainable C1 feedstocks; formaldehyde could be converted to glycolaldehyde by formolase or its variants, and furthermore, glycolaldehyde was converted to erythrulose (C4 sugar) with 98% yield by another formolase variant.63 Alternatively, formaldehyde could be transformed to glycolaldehyde through glycolaldehyde synthase, followed by the conversions to ethylene glycol via alcohol or aldehyde dehydrogenases from Gluconobacter oxydans, to glycolic acid via acetaldehyde dehydrogenases, or to D-(−)-erythrose via 2-deoxy-D-ribose-5-phosphate aldolases (DERAs).233 In another study,64 CO2 was converted to a bis(boryl)acetal compound first, followed by selective enzymatic reactions to afford C3 (dihydroxyacetone, DHA) with up to 86% yield by using a formolase (FLS), or optically pure C4 (L-erythrulose) with 35% yield through a cascade reaction using FLS and D-fructose-6-phosphate aldolase (FSA) A129S variant (Fig. 76).
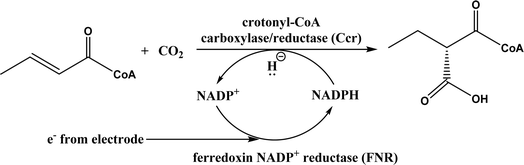 |
| | Fig. 75 Bioelectrocatalytic NADP+ cofactor regeneration coupled with enzymatic CO2 fixation. | |
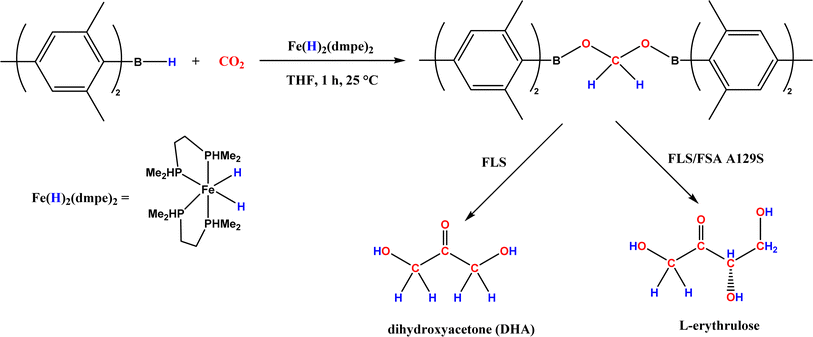 |
| | Fig. 76 Chemoenzymatic conversion of CO2 to C3 (dihydroxyacetone, DHA) and C4 (L-erythrulose) carbohydrates. | |
A chemoenzymatic route to convert CO2 to hexoses (e.g., glucose and D-allulose) was designed by the Ma group:234 chemical reduction of CO2 to ‘green’ methanol by ZnO–ZrO2 catalyst, methanol conversion to DHAP via multi-step strategy involving formolase, aldol condensation catalyzed by fructose-6-phosphate aldolases (FSAs), iso/epimerization, and dephosphorylation reactions. Similarly, ‘green’ methanol can be converted to L-alanine with 88% yield,235 or to starch at 22 nmol min−1 mg−1 of total catalyst and proteins (an 8.5-fold faster than starch formation via the Calvin cycle in maize),236 both through multi-enzyme cascade reactions under cell-free conditions.
12. Radical enzymes for C–C bond formation
Other than two electron mechanisms (involving nucleophile and electrophile), C–C bonds can be formed by free radical-mediated reactions such as those catalyzed by radical S-adenosylmethionine (SAM) enzymes. As discussed in a recent review,237 radical S-adenosylmethionine (SAM) enzymes are involved in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs), and O2-sensitive and hydrocarbon activating glycyl radical enzymes (GREs) include a subset known as X-succinate synthases [e.g., benzylsuccinate synthase (BSS), 4-isopropylbenzylsuccinate synthase (IBSS), hydroxybenzylsuccinate synthase (HBSS), naphthyl-2-methylsuccinate synthase (NMSS), and 1-methylalkylsuccinate synthase (MASS)]. More specifically, C–C bond forming radical SAM enzymes were surveyed in terms of SPASM–twitch subfamily, radical SAM enzymes with N-terminal cofactor binding domains, ThiH-like enzymes, and noncanonical radical SAM enzymes; additionally, three critical mechanistic factors (radical initiation, acceptor substrate activation, and radical quenching) were discussed in detail.238 In another review,239 mechanistic understandings are provided for C–C bond formation or cleavage reactions catalyzed by three enzymes: pyruvate-formate lyase (PFL), spore photoproduct lyase (SPL), and benzylsuccinate synthase (BSS). Our earlier sections also covered several examples of radical species during the C–C bond formation, such as radical S-adenosylmethionine enzymes for enzymatic redox reactions in C–C bond formation,66 the benzylic radical/carbocation intermediate initiating the C–C bond formation for a nonheme iron enzyme called 2-oxoglutarate/Fe(II)-dependent dioxygenase (2-ODD),61 and the formation of nitro radical anion by reacting nitronate with an alkyl radical during ‘ene’-reductase CsER-catalyzed cross-electrophile couplings (XECs) between alkyl halides and nitroalkanes.67 The Yang group65 suggested that cytochrome P450 could be engineered to have a fine control of the radical addition step and the halogen rebound step during stereoselective atom-transfer radical cyclization (ATRC), affording enantio- and diastereodivergent radical catalysis (Fig. 77); as indicated by molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) calculations, glutamine residue of P450 acts as hydrogen bond donor to interact with the carbonyl group of the substrate to facilitate the removal of bromine atom and control the stereoselectivity of ATRC.240 Spectroscopy and computational studies have revealed the C–C bond formation mechanism for radical SAM enzyme (cyclase),241 cytochrome P450,242 and benzylsuccinate synthase.243
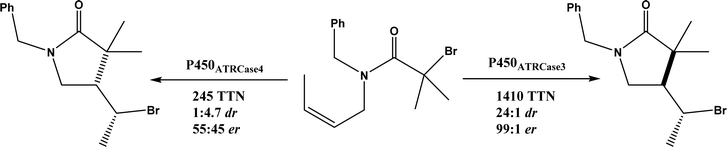 |
| | Fig. 77 Stereoselective atom-transfer radical cyclization (ATRC) with cytochrome P450 variants (TTN = total turnover number; dr = diastereomeric ratio; er = enantiomeric ratio). | |
13. Other C–C bond formation mechanisms
Two PLP-dependent enzymes, CndF and Fub7, induce C–O activation and catalyze γ-substitution providing a new route for stereoselective C–C bond formation.68,244 A chemoenzymatic method involving Fub7 (Fig. 78) afforded 5-alkyl-, 5,5-dialkyl-, and 5,5,6-trialkyl-L-pipecolic acids with diastereomeric ratio ranging from 50:50 to 95:5.244 CndF catalyzed the C–C coupling of O-acetyl-L-homoserine with 3-oxobutanoic acid to form (S)-2-amino-6-oxoheptanoate, which equilibrates with a cyclic Schiff base; a further reduction by a stereoselective imine reductase CndE gave (2S, 6S)-6-methyl pipecolate (Fig. 79).68 CndF is also capable of converting β-keto ethyl esters to enamine-containing pipecolates.
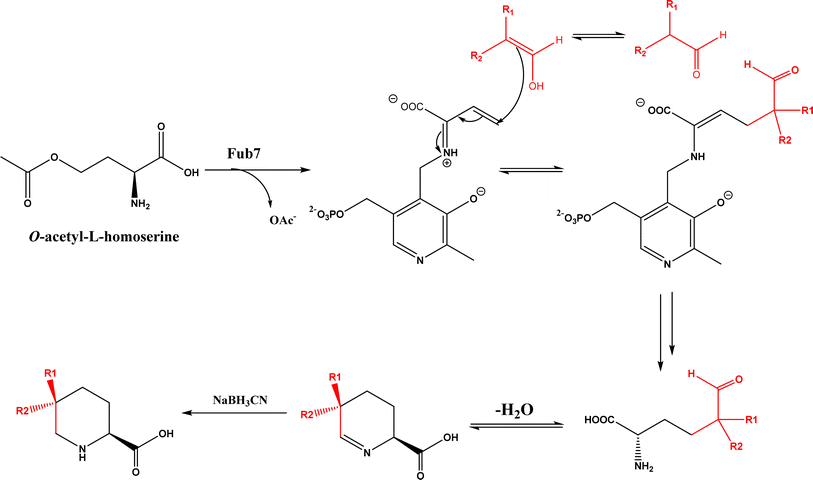 |
| | Fig. 78 Fub7-catalyzed C–C bond formation to prepare substituted L-pipecolic acids. | |
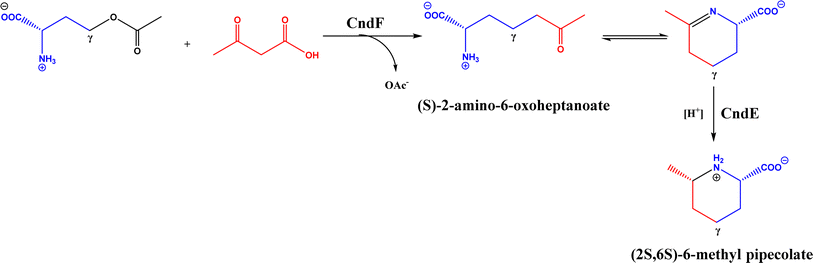 |
| | Fig. 79 Chemoenzymatic synthesis of (2S,6S)-6-methyl pipecolate using CndF. | |
Hydroxynitrile lyases (HNLs), or oxynitrilases (EC 4.1.2.x) catalyze the reversible enantioselective condensation of hydrocyanic acid (HCN) with aldehydes or ketones to produce cyanohydrins.70,80,81,245,246 Other enzymatic C–C bond formation mechanisms include intermolecular aryl coupling between 8-hydroxydihydrokalafungin molecules to actinorhodin (Fig. 80) catalyzed by NAD(P)H-dependent ActVA-ORF4 (NmrA-family dimerizing enzyme),247 sp3 C–H functionalization catalyzed by iron-based catalysts derived from cytochrome P450 (to become cytochrome P411),248 by trypsin,249 or by tyrosine phenol lyase,250 ketosynthase-catalyzed decarboxylative Claisen-like condensation,251 C-nucleoside synthase ForT-catalyzed C–C bond formation,252 carbon–carbon bond formation by deoxypodophyllotoxin synthase,253 cis-isoprenyl diphosphate synthase-catalyzed condensation conversions of isoprene units to produce isoprenoids or terpenoids,254 carboxymethylproline synthase (a member of crotonase family)-catalyzed C–C bond formation,255 and engineered SAM-dependent sterol methyltransferase for C-methylation of unactivated alkenes in mono-, sesqui- and diterpenoids to yield C11, C16 and C21 derivatives with high chemo- and regioselectivity.69
 |
| | Fig. 80 Enzymatic aryl coupling between 8-hydroxydihydrokalafungin molecules to synthesize actinorhodin. | |
14. Perspectives
Enzymes have shown unique and tailorable chemo-, regio- and/or stereoselectivity during the C–C bond formation through judicious engineering of enzyme structures and the optimization of reaction conditions. Enzymes discovered in the biosynthesis of C–C bond formation have a great potential to be evolved to become robust biocatalysts for asymmetric reactions in aqueous or nonaqueous environments. It is highly valuable to make carbon-based molecules through enzymatic conversions of C1 resources.
There have been some conflicting reports about the existence and extent of catalytic promiscuity of some enzymes, which require further experimental examinations. In addition, the catalytic mechanisms of enzymatic C–C bond formation are not well understood, and not fully backed by experimental and computational results. Aqueous reaction media are not always ideal for biocatalytic conversion due to insolubility of substrates resulting in low reaction efficiency; water-miscible organic co-solvents assist with the substrate dissolution, but may cause enzyme inactivation. There is still a great need to find and optimize non-aqueous solvents (e.g., ILs and DES) for enzymatic C–C formation reactions. Future efforts to address these issues will lead to more effective synthesis of stereoselective molecules with medicinal and biological significance, and a better utilization of C1 resources.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. [2244638].
References
- Z. Guan, L. Li and Y. He, RSC Adv., 2015, 5, 16801–16814 RSC.
- L. Poppe and J. Rétey, Angew. Chem., Int. Ed., 2005, 44, 3668–3688 CrossRef CAS PubMed.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519–11520 CrossRef CAS.
- T. Ose, K. Watanabe, T. Mie, M. Honma, H. Watanabe, M. Yao, H. Oikawa and I. Tanaka, Nature, 2003, 422, 185–189 CrossRef CAS PubMed.
- M. López-Iglesias, E. Busto, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2011, 353, 2345–2353 CrossRef.
- M. D. Patil, G. Grogan and H. Yun, ChemCatChem, 2018, 10, 4783–4804 CrossRef.
- P. Jakubec, D. M. Cockfield and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 16632–16633 CrossRef CAS PubMed.
- P. Chen, X. Bao, L.-F. Zhang, M. Ding, X.-J. Han, J. Li, G.-B. Zhang, Y.-Q. Tu and C.-A. Fan, Angew. Chem., Int. Ed., 2011, 50, 8161–8166 CrossRef CAS PubMed.
- T. Buyck, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12714–12718 CrossRef CAS.
- C. L. Windle, A. Berry and A. Nelson, Curr. Opin. Chem. Biol., 2017, 37, 33–38 CrossRef CAS PubMed.
- M. Müller, Adv. Synth. Catal., 2012, 354, 3161–3174 CrossRef.
- Y. Miao, M. Rahimi, E. M. Geertsema and G. J. Poelarends, Curr. Opin. Chem. Biol., 2015, 25, 115–123 CrossRef CAS PubMed.
- P. Clapés, in Organic Synthesis Using Biocatalysis, ed. A. Goswami and J. D. Stewart, Academic Press, 2016, pp. 285–337, DOI:10.1016/B978-0-12-411518-7.00010-X.
- N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed.
- B. T. Ueberbacher, M. Hall and K. Faber, Nat. Prod. Rep., 2012, 29, 337–350 RSC.
- A. L. Mattei, N. Bailly and A. Meissner, Trends Genet., 2022, 38, 676–707 CrossRef CAS.
- P. A. Storm, P. Pal, C. R. Huitt-Roehl and C. A. Townsend, ACS Chem. Biol., 2018, 13, 3043–3048 CrossRef CAS PubMed.
- J. Yang, J. Li, Y. Men, Y. Zhu, Y. Zhang, Y. Sun and Y. Ma, Appl. Environ. Microbiol., 2015, 81, 4284–4294 CrossRef CAS PubMed.
- Y. Zhu, T. Shiraishi, J. Lin, K. Inaba, A. Ito, Y. Ogura, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2022, 144, 16715–16719 CrossRef CAS PubMed.
- J. Liu, L. Harken, Y. Yang, X. Xie and S.-M. Li, Angew. Chem., Int. Ed., 2022, 61, e202200377 CrossRef CAS.
- A. Bolt, A. Berry and A. Nelson, Arch. Biochem. Biophys., 2008, 474, 318–330 CrossRef CAS PubMed.
- S.-H. Lee, S.-J. Yeom, S.-E. Kim and D.-K. Oh, Trends Biotechnol., 2022, 40, 306–319 CrossRef CAS PubMed.
- S. M. Dean, W. A. Greenberg and C.-H. Wong, Adv. Synth. Catal., 2007, 349, 1308–1320 CrossRef CAS.
- P. Clapés and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263–2283 CrossRef.
- G. J. Williams, S. Domann, A. Nelson and A. Berry, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 3143–3148 CrossRef CAS PubMed.
- G. J. Williams, T. Woodhall, L. M. Farnsworth, A. Nelson and A. Berry, J. Am. Chem. Soc., 2006, 128, 16238–16247 CrossRef CAS.
- W. Wang, S. Mazurkewich, M. S. Kimber and S. Y. K. Seah, J. Biol. Chem., 2010, 285, 36608–36615 CrossRef CAS PubMed.
- V. Laurent, E. Darii, A. Aujon, M. Debacker, J. L. Petit, V. Hélaine, T. Liptaj, M. Breza, A. Mariage, L. Nauton, M. Traïkia, M. Salanoubat, M. Lemaire, C. Guérard-Hélaine and V. de Berardinis, Angew. Chem., Int. Ed., 2018, 57, 5467–5471 CrossRef CAS PubMed.
- J. Yang, Y. Zhu, G. Qu, Y. Zeng, C. Tian, C. Dong, Y. Men, L. Dai, Z. Sun, Y. Sun and Y. Ma, Biotechnol. Biofuels, 2018, 11, 290 CrossRef CAS PubMed.
- C. Ren, J. Yang, Y. Zeng, T. Zhang, C. Tian, Y. Men and Y. Sun, Enzyme Microb. Technol., 2021, 147, 109784 CrossRef CAS PubMed.
- A. J. Rigual, J. Cantero, M. Risso, P. Rodríguez, S. Rodríguez, M. Paulino, D. Gamenara and N. Veiga, Mol. Catal., 2020, 495, 111131 CrossRef CAS.
- Q. Chen, X. Chen, J. Feng, Q. Wu, D. Zhu and Y. Ma, ACS Catal., 2019, 9, 4462–4469 CrossRef CAS.
- C. Li, X. Feng, N. Wang, Y. Zhou and X. Yu, Green Chem., 2008, 10, 616–618 RSC.
- M. López-Iglesias, E. Busto, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2011, 353, 2345–2353 CrossRef.
- S. Milker, M. Pätzold, J. Z. Bloh and D. Holtmann, Mol. Catal., 2019, 466, 70–74 CrossRef CAS.
- W.-J. Xia, Z.-B. Xie, G.-F. Jiang and Z.-G. Le, Molecules, 2013, 18, 13910–13919 CrossRef CAS PubMed.
- D. Kühbeck, B. B. Dhar, E.-M. Schön, C. Cativiela, V. Gotor-Fernández and D. D. Díaz, Beilstein J. Org. Chem., 2013, 9, 1111–1118 CrossRef.
- X.-W. Feng, C. Li, N. Wang, K. Li, W.-W. Zhang, Z. Wang and X.-Q. Yu, Green Chem., 2009, 11, 1933–1936 RSC.
- A. S. Evitt and U. T. Bornscheuer, Green Chem., 2011, 13, 1141–1142 RSC.
- H. Zhao and C. D. Campbell, J. Chem. Technol. Biotechnol., 2024, 99, 780–787 CrossRef CAS.
- U. R. Pratap, D. V. Jawale, R. A. Waghmare, D. L. Lingampalle and R. A. Mane, New J. Chem., 2011, 35, 49–51 RSC.
- M. Svedendahl, K. Hult and P. Berglund, J. Am. Chem. Soc., 2005, 127, 17988–17989 CrossRef CAS PubMed.
- X. Chen, G. Chen, J. Wang, Q. Wu and X. Lin, Adv. Synth. Catal., 2013, 355, 864–868 CrossRef CAS.
- G. A. Strohmeier, T. Sović, G. Steinkellner, F. S. Hartner, A. Andryushkova, T. Purkarthofer, A. Glieder, K. Gruber and H. Griengl, Tetrahedron, 2009, 65, 5663–5668 CrossRef CAS.
- Y. Fan, D. Cai, X. Wang and L. Yang, Molecules, 2018, 23, 2154 CrossRef PubMed.
- H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem., Int. Ed., 2009, 48, 9546–9548 CrossRef CAS PubMed.
- M. Tengg, H. Stecher, P. Remler, I. Eiteljörg, H. Schwab and M. Gruber-Khadjawi, J. Mol. Catal. B: Enzym., 2012, 84, 2–8 CrossRef CAS.
- M. Tengg, H. Stecher, L. Offner, K. Plasch, F. Anderl, H. Weber, H. Schwab and M. Gruber-Khadjawi, ChemCatChem, 2016, 8, 1354–1360 CrossRef CAS.
- J. C. Sadler, C.-w. H. Chung, J. E. Mosley, G. A. Burley and L. D. Humphreys, ACS Chem. Biol., 2017, 12, 374–379 CrossRef CAS PubMed.
- L. Villarino, S. Chordia, L. Alonso-Cotchico, E. Reddem, Z. Zhou, A. M. W. H. Thunnissen, J.-D. Maréchal and G. Roelfes, ACS Catal., 2020, 10, 11783–11790 CrossRef CAS PubMed.
- N. G. Schmidt, T. Pavkov-Keller, N. Richter, B. Wiltschi, K. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2017, 56, 7615–7619 CrossRef CAS PubMed.
- K. Li, T. He, C. Li, X. Feng, N. Wang and X. Yu, Green Chem., 2009, 11, 777–779 RSC.
- S. Chai, Y. Lai, H. Zheng and P. Zhang, Helv. Chim. Acta, 2010, 93, 2231–2236 CrossRef CAS.
- R. Crawshaw, A. E. Crossley, L. Johannissen, A. J. Burke, S. Hay, C. Levy, D. Baker, S. L. Lovelock and A. P. Green, Nat. Chem., 2022, 14, 313–320 CrossRef CAS PubMed.
- K. Watanabe, T. Mie, A. Ichihara, H. Oikawa and M. Honma, J. Biol. Chem., 2000, 275, 38393–38401 CrossRef CAS.
- C. R. W. Guimarães, M. Udier-Blagović and W. L. Jorgensen, J. Am. Chem. Soc., 2005, 127, 3577–3588 CrossRef PubMed.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS.
- E. Kasparyan, M. Richter, C. Dresen, L. S. Walter, G. Fuchs, F. J. Leeper, T. Wacker, S. L. A. Andrade, G. Kolter, M. Pohl and M. Müller, Appl. Microbiol. Biotechnol., 2014, 98, 9681–9690 CrossRef CAS PubMed.
- Z. Maugeri and P. Domínguez de María, J. Mol. Catal. B: Enzym., 2014, 107, 120–123 CrossRef CAS.
- J.-P. Steitz, L. Krug, L. Walter, K. Hernández, C. Röhr, P. Clapés and M. Müller, Angew. Chem., Int. Ed., 2022, 61, e202113405 CrossRef CAS PubMed.
- W.-C. Chang, Z.-J. Yang, Y.-H. Tu and T.-C. Chien, Org. Lett., 2019, 21, 228–232 CrossRef CAS PubMed.
- K. Heckenbichler, A. Schweiger, L. A. Brandner, A. Binter, M. Toplak, P. Macheroux, K. Gruber and R. Breinbauer, Angew. Chem., Int. Ed., 2018, 57, 7240–7244 CrossRef CAS PubMed.
- S. Güner, V. Wegat, A. Pick and V. Sieber, Green Chem., 2021, 23, 6583–6590 RSC.
- S. Desmons, K. Grayson-Steel, N. Nuñez-Dallos, L. Vendier, J. Hurtado, P. Clapés, R. Fauré, C. Dumon and S. Bontemps, J. Am. Chem. Soc., 2021, 143, 16274–16283 CrossRef CAS PubMed.
- Q. Zhou, M. Chin, Y. Fu, P. Liu and Y. Yang, Science, 2021, 374, 1612–1616 CrossRef CAS PubMed.
- F. P. Guengerich and F. K. Yoshimoto, Chem. Rev., 2018, 118, 6573–6655 CrossRef CAS PubMed.
- H. Fu, J. Cao, T. Qiao, Y. Qi, S. J. Charnock, S. Garfinkle and T. K. Hyster, Nature, 2022, 610, 302–307 CrossRef CAS PubMed.
- M. Chen, C.-T. Liu and Y. Tang, J. Am. Chem. Soc., 2020, 142, 10506–10515 CrossRef CAS PubMed.
- B. Aberle, D. Kowalczyk, S. Massini, A.-N. Egler-Kemmerer, S. Gergel, S. C. Hammer and B. Hauer, Angew. Chem., Int. Ed., 2023, 62, e202301601 CrossRef CAS PubMed.
- W.-D. Fessner, Curr. Opin. Chem. Biol., 1998, 2, 85–97 CrossRef CAS PubMed.
- P. Clapés, W.-D. Fessner, G. A. Sprenger and A. K. Samland, Curr. Opin. Chem. Biol., 2010, 14, 154–167 CrossRef PubMed.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1374 CrossRef CAS PubMed.
- A. K. Samland and G. A. Sprenger, Appl. Microbiol. Biotechnol., 2006, 71, 253–264 CrossRef CAS PubMed.
- P. Falcicchio, S. Wolterink-Van Loo, M. C. R. Franssen and J. van der Oost, Extremophiles, 2014, 18, 1–13 CrossRef CAS PubMed.
- C. C. Aragon, J. M. Palomo, M. Filice and C. Mateo, Curr. Org. Chem., 2016, 20, 1243–1251 CrossRef CAS.
- P. Clapés, in Green Biocatalysis, ed. R. N. Patel, John Wiley & Sons, Inc., Hoboken, New Jersey, 2016, pp. 267–306, DOI:10.1002/9781118828083.ch10.
- P. Clapés, in Biocatalysis in Organic Synthesis, ed. K. Faber, W.-D. Fessner and N. J. Turner, Georg Thieme Verlag KG, Stuttgart (Germany), 2015, vol. 2, pp. 31–92 Search PubMed.
- V. Hélaine, C. Gastaldi, M. Lemaire, P. Clapés and C. Guérard-Hélaine, ACS Catal., 2022, 12, 733–761 CrossRef.
- S. Desmons, R. Fauré and S. Bontemps, ACS Catal., 2019, 9, 9575–9588 CrossRef CAS.
- W.-D. Fessner, A. Schneider, H. Held, G. Sinerius, C. Walter, M. Hixon and J. V. Schloss, Angew. Chem., Int. Ed., 1996, 35, 2219–2221 CrossRef CAS.
- M. Brovetto, D. Gamenara, P. S. Méndez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- L. Wu, M. H. Tong, A. Raab, Q. Fang, S. Wang, K. Kyeremeh, Y. Yu and H. Deng, Appl. Microbiol. Biotechnol., 2020, 104, 3885–3896 CrossRef CAS PubMed.
- A. K. Samland, M. Rale, G. A. Sprenger and W.-D. Fessner, ChemBioChem, 2011, 12, 1454–1474 CrossRef CAS PubMed.
- A. K. Samland and G. A. Sprenger, Int. J. Biochem. Cell Biol., 2009, 41, 1482–1494 CrossRef CAS PubMed.
- S. Schneider, T. Sandalova, G. Schneider, G. A. Sprenger and A. K. Samland, J. Biol. Chem., 2008, 283, 30064–30072 CrossRef CAS PubMed.
- M. Schürmann and G. A. Sprenger, J. Biol. Chem., 2001, 276, 11055–11061 CrossRef PubMed.
- J. Blesl, M. Trobe, F. Anderl, R. Breinbauer, G. A. Strohmeier and K. Fesko, ChemCatChem, 2018, 10, 3453–3458 CrossRef CAS PubMed.
- S. F. Beaudoin, M. P. Hanna, I. Ghiviriga and J. D. Stewart, Enzyme Microb. Technol., 2018, 119, 1–9 CrossRef CAS PubMed.
- L. Xu, L. Wang, X. Xu and J. Lin, Catal. Sci. Technol., 2019, 9, 5943–5952 RSC.
- W. Zheng, H. Yu, S. Fang, K. Chen, Z. Wang, X. Cheng, G. Xu, L. Yang and J. Wu, ACS Catal., 2021, 11, 3198–3205 CrossRef CAS.
- W. Wang, J. Yang, Y. Sun, Z. Li and C. You, ACS Catal., 2020, 10, 1264–1271 CrossRef CAS.
- I. Oroz-Guinea, I. Sánchez-Moreno, M. Mena and E. García-Junceda, Appl. Microbiol. Biotechnol., 2015, 99, 3057–3068 CrossRef CAS PubMed.
- I. Oroz-Guinea, K. Hernández, F. Camps Bres, C. Guérard-Hélaine, M. Lemaire, P. Clapés and E. García-Junceda, Adv. Synth. Catal., 2015, 357, 1951–1960 CrossRef CAS.
- R. Marín-Valls, K. Hernández, M. Bolte, T. Parella, J. Joglar, J. Bujons and P. Clapés, J. Am. Chem. Soc., 2020, 142, 19754–19762 CrossRef PubMed.
- Z. Hu, C. Cheng, Y. Li, X. Qi, T. Wang, L. Liu and J. Voglmeir, ChemBioChem, 2022, 23, e202200074 CrossRef CAS PubMed.
- M. Pickl, R. Marín-Valls, J. Joglar, J. Bujons and P. Clapés, Adv. Synth. Catal., 2021, 363, 2866–2876 CrossRef CAS PubMed.
- C. J. Moreno, K. Hernández, S. J. Charnok, S. Gittings, M. Bolte, J. Joglar, J. Bujons, T. Parella and P. Clapés, ACS Catal., 2021, 11, 4660–4669 CrossRef CAS PubMed.
- R. Marín-Valls, K. Hernández, M. Bolte, J. Joglar, J. Bujons and P. Clapés, ACS Catal., 2019, 9, 7568–7577 CrossRef.
- C. J. Moreno, K. Hernández, S. Gittings, M. Bolte, J. Joglar, J. Bujons, T. Parella and P. Clapés, ACS Catal., 2023, 13, 5348–5357 CrossRef CAS PubMed.
- W. Zheng, Z. Pu, L. Xiao, G. Xu, L. Yang, H. Yu and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202213855 CrossRef CAS PubMed.
- H. Li, Y. He, Y. Yuan and Z. Guan, Green Chem., 2011, 13, 185–189 RSC.
- J. M. Ellis, M. E. Campbell, P. Kumar, E. P. Geunes, C. A. Bingman and A. R. Buller, Nat. Catal., 2022, 5, 136–143 CrossRef CAS PubMed.
- R. Zhang, J. Tan, Z. Luo, H. Dong, N. Ma and C. Liao, Catal. Sci. Technol., 2021, 11, 7380–7385 RSC.
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS.
- A. M. F. Phillips, Curr. Organocatal., 2016, 3, 222–242 CrossRef CAS.
- S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org Chem., 2012, 2012, 3059–3067 CrossRef CAS.
- P. Shrivas, N. Punyapreddiwar, A. Wankhade, S. Zodape and U. Pratap, Iran. J. Catal., 2017, 7, 337–340 CAS.
- D. F. Izquierdo, O. Barbosa, M. I. Burguete, P. Lozano, S. V. Luis, R. Fernandez-Lafuente and E. García-Verdugo, RSC Adv., 2014, 4, 6219–6225 RSC.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- K. Hackelöer, G. Schnakenburg and S. R. Waldvogel, Eur. J. Org Chem., 2011, 2011, 6314–6319 CrossRef.
- I. Walz, A. Bertogg and A. Togni, Eur. J. Org Chem., 2007, 2007, 2650–2658 CrossRef.
- D. Koszelewski, D. Paprocki, A. Madej, F. Borys, A. Brodzka and R. Ostaszewski, Eur. J. Org Chem., 2017, 2017, 4572–4579 CrossRef CAS.
- K. van Beurden, S. de Koning, D. Molendijk and J. van Schijndel, Green Chem. Lett. Rev., 2020, 13, 349–364 CrossRef CAS.
- X. Yuan, J. Liu, Y. Wang, X. Jie, J. Qin and H. He, Chem. Eng. J., 2023, 451, 138941 CrossRef CAS.
- V. Bhaskar, R. Chowdary, S. R. Dixit and S. D. Joshi, Bioorg. Chem., 2019, 84, 202–210 CrossRef CAS PubMed.
- A. Paul, A. Maji, A. Sarkar, S. Saha, P. Janah and T. K. Maity, Mini-Rev. Org. Chem., 2023, 20, 5–34 CrossRef CAS.
- C. Wang, N. Wang, X. Liu, P. Wan, X. He and Y. Shang, Fibers Polym., 2018, 19, 1611–1617 CrossRef CAS.
- L. Jiang, B. Wang, R.-R. Li, S. Shen, H.-W. Yu and L.-D. Ye, Chin. Chem. Lett., 2014, 25, 1190–1192 CrossRef CAS.
- Y. Fu, Z. Lu, K. Fang, X. He, H. Huang and Y. Hu, Bioorg. Med. Chem. Lett., 2019, 29, 1236–1240 CrossRef CAS PubMed.
- Y. Ding, X. Xiang, M. Gu, H. Xu, H. Huang and Y. Hu, Bioprocess Biosyst. Eng., 2016, 39, 125–131 CrossRef CAS PubMed.
- Z. Wang, C.-Y. Wang, H.-R. Wang, H. Zhang, Y.-L. Su, T.-F. Ji and L. Wang, Chin. Chem. Lett., 2014, 25, 802–804 CrossRef CAS.
- H. Bavandi, Z. Habibi and M. Yousefi, Bioorg. Chem., 2020, 103, 104139 CrossRef CAS PubMed.
- M. Wilk, D. Trzepizur, D. Koszelewski, A. Brodzka and R. Ostaszewski, Bioorg. Chem., 2019, 93, 102816 CrossRef CAS PubMed.
- D. Koszelewski and R. Ostaszewski, Chem.–Eur. J., 2019, 25, 10156–10164 CrossRef CAS PubMed.
- Y. Wang, H. Cheng, J. R. He, Q. X. Yao, L. L. Li, Z. H. Liang and X. Li, Catal. Lett., 2022, 152, 1215–1223 CrossRef CAS.
- H. Zhao, J. Chem. Technol. Biotechnol., 2016, 91, 25–50 CrossRef CAS PubMed.
- B.-H. Xie, Z. Guan and Y.-H. He, Biocatal. Biotransform., 2012, 30, 238–244 CrossRef CAS.
- N. Sharma, U. K. Sharma, R. Kumar, N. Katoch, R. Kumar and A. K. Sinha, Adv. Synth. Catal., 2011, 353, 871–878 CrossRef CAS.
- K. S. Dalal, Y. A. Tayade, Y. B. Wagh, D. R. Trivedi, D. S. Dalal and B. L. Chaudhari, RSC Adv., 2016, 6, 14868–14879 RSC.
- W. Hu, Z. Guan, X. Deng and Y.-H. He, Biochimie, 2012, 94, 656–661 CrossRef CAS PubMed.
- J. Yu, X. Chen, M. Jiang, A. Wang, L. Yang, X. Pei, P. Zhang and S. G. Wu, RSC Adv., 2018, 8, 2357–2364 RSC.
- Z.-Q. Liu, B.-K. Liu, Q. Wu and X.-F. Lin, Tetrahedron, 2011, 67, 9736–9740 CrossRef CAS.
- W. Li, R. Li, X. Yu, X. Xu, Z. Guo, T. Tan and S. N. Fedosov, Biochem. Eng. J., 2015, 101, 99–107 CrossRef CAS.
- W. Li, S. N. Fedosov, T. Tan, X. Xu and Z. Guo, ACS Catal., 2014, 4, 3294–3300 CrossRef CAS.
- X. Garrabou, B. I. M. Wicky and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 6972–6974 CrossRef CAS PubMed.
- M. Mogharabi-Manzari, M. Amini, M. Abdollahi, M. Khoobi, G. Bagherzadeh and M. A. Faramarzi, ChemCatChem, 2018, 10, 1542–1546 CrossRef CAS.
- X. Liu, X. Li, Z. Wang, J. Zhou, X. Fan and Y. Fu, ACS Sustainable Chem. Eng., 2020, 8, 8206–8213 CrossRef CAS.
- T. Tokoroyama, Eur. J. Org Chem., 2010, 2010, 2009–2016 CrossRef.
- S. C. Jha and N. N. Joshi, ARKIVOC, 2002, 167–196 Search PubMed.
- S. Witayakran and A. J. Ragauskas, Eur. J. Org Chem., 2009, 2009, 358–363 CrossRef.
- J. Cai, Z. Guan and Y. He, J. Mol. Catal. B: Enzym., 2011, 68, 240–244 CrossRef CAS.
- B. Xie, Z. Guan and Y. He, J. Chem. Technol. Biotechnol., 2012, 87, 1709–1714 CrossRef CAS.
- Y. Yuan, L. Yang, S. Liu, J. Yang, H. Zhang, J. Yan and X. Hu, Spectrochim. Acta, Part A, 2017, 176, 183–188 CrossRef CAS PubMed.
- J. Xu, F. Zhang, Q. Wu, Q. Zhang and X. Lin, J. Mol. Catal. B: Enzym., 2007, 49, 50–54 CrossRef CAS.
- J. Xu, F. Zhang, B. Liu, Q. Wu and X. Lin, Chem. Commun., 2007, 2078–2080, 10.1039/b700327g.
- L. Wu, L. Li, Y. Xiang, Z. Guan and Y. He, Catal. Lett., 2017, 147, 2209–2214 CrossRef CAS.
- B. List, Angew. Chem., Int. Ed., 2010, 49, 1730–1734 CrossRef CAS PubMed.
- E. Zandvoort, E. M. Geertsema, B.-J. Baas, W. J. Quax and G. J. Poelarends, Angew. Chem., Int. Ed., 2012, 51, 1240–1243 CrossRef CAS PubMed.
- Y. Miao, E. M. Geertsema, P. G. Tepper, E. Zandvoort and G. J. Poelarends, ChemBioChem, 2013, 14, 191–194 CrossRef CAS.
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083–2092 RSC.
- S. Park and H. Sugiyama, Angew. Chem., Int. Ed., 2010, 49, 3870–3878 CrossRef CAS.
- S. Park and H. Sugiyama, Molecules, 2012, 17, 12792–12803 CrossRef CAS.
- N. Duchemin, S. Aubert, J. V. de Souza, L. Bethge, S. Vonhoff, A. K. Bronowska, M. Smietana and S. Arseniyadis, JACS Au, 2022, 2, 1910–1917 CrossRef CAS.
- P. M. Punt, M. D. Langenberg, O. Altan and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 3555–3561 CrossRef CAS PubMed.
- H. Zhao and K. Shen, RSC Adv., 2014, 4, 54051–54059 RSC.
- H. Zhao and K. Shen, Biotechnol. Prog., 2016, 32, 891–898 CrossRef CAS PubMed.
- K. Akagawa, R. Suzuki and K. Kudo, Adv. Synth. Catal., 2012, 354, 1280–1286 CrossRef CAS.
- K. Akagawa, T. Yamashita, S. Sakamoto and K. Kudo, Tetrahedron Lett., 2009, 50, 5602–5604 CrossRef CAS.
- K. Akagawa, R. Umezawa and K. Kudo, Beilstein J. Org. Chem., 2012, 8, 1333–1337 CrossRef CAS.
- E. Abdelraheem, B. Thair, R. F. Varela, E. Jockmann, D. Popadić, H. C. Hailes, J. M. Ward, A. M. Iribarren, E. S. Lewkowicz, J. N. Andexer, P.-L. Hagedoorn and U. Hanefeld, ChemBioChem, 2022, 23, e202200212 CrossRef CAS PubMed.
- X. Yu and S.-M. Li, ChemBioChem, 2011, 12, 2280–2283 CrossRef CAS.
- L. L. Kang Zhou and S.-M. Li, J. Nat. Prod., 2015, 78, 929–933 CrossRef.
- X. Yu, A. Yang, W. Lin and S. Li, Tetrahedron Lett., 2012, 53, 6861–6864 CrossRef CAS.
- M. Liebhold, X. Xie and S. Li, Org. Lett., 2012, 14, 4882–4885 CrossRef CAS PubMed.
- E. E. Schultz, N. R. Braffman, M. U. Luescher, H. H. Hager and E. P. Balskus, Angew. Chem., Int. Ed., 2019, 58, 3151–3155 CrossRef CAS PubMed.
- J. Xue, J. Guo, Y. He and Z. Guan, Asian J. Org. Chem., 2017, 6, 297–304 CrossRef CAS.
- S. C. Hammer, J. M. Dominicus, P.-O. Syrén, B. M. Nestl and B. Hauer, Tetrahedron, 2012, 68, 7624–7629 CrossRef CAS.
- S. Henche, B. M. Nestl and B. Hauer, ChemCatChem, 2021, 13, 3405–3409 CrossRef CAS.
- R. B. Leveson-Gower, R. M. de Boer and G. Roelfes, ChemCatChem, 2022, 14, e202101875 CrossRef CAS PubMed.
- A. Żądło-Dobrowolska, N. G. Schmidt and W. Kroutil, ChemCatChem, 2019, 11, 1064–1068 CrossRef.
- E. Siirola and W. Kroutil, Top. Catal., 2014, 57, 392–400 CrossRef CAS.
- S. Saranya, N. A. Harry, K. K. Krishnan and G. Anilkumar, Asian J. Org. Chem., 2018, 7, 613–633 CrossRef CAS.
- Z. Guan, J. Song, Y. Xue, D. Yang and Y. He, J. Mol. Catal. B: Enzym., 2015, 111, 16–20 CrossRef CAS.
- L. Wu, Y. Xiang, D. Yang, Z. Guan and Y. He, Catal. Sci. Technol., 2016, 6, 3963–3970 RSC.
- D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed.
- S. Bjelic, L. G. Nivón, N. Çelebi-Ölçüm, G. Kiss, C. F. Rosewall, H. M. Lovick, E. L. Ingalls, J. L. Gallaher, J. Seetharaman, S. Lew, G. T. Montelione, J. F. Hunt, F. E. Michael, K. N. Houk and D. Baker, ACS Chem. Biol., 2013, 8, 749–757 CrossRef CAS PubMed.
- M. T. Reetz, R. Mondière and J. D. Carballeira, Tetrahedron Lett., 2007, 48, 1679–1681 CrossRef CAS.
- K. Asano and S. Matsubara, Synthesis, 2009, 3219–3226, DOI:10.1055/s-0029-1216944.
- N. Kato, T. Nogawa, R. Takita, K. Kinugasa, M. Kanai, M. Uchiyama, H. Osada and S. Takahashi, Angew. Chem., Int. Ed., 2018, 57, 9754–9758 CrossRef CAS PubMed.
- H. J. Kim, M. W. Ruszczycky, S. Choi, Y. Liu and H. Liu, Nature, 2011, 473, 109–112 CrossRef CAS.
- A. Ichihara and H. Oikawa, Curr. Org. Chem., 1998, 2, 365–394 CrossRef CAS.
- H. Oikawa, K. Katayama, Y. Suzuki and A. Ichihara, J. Chem. Soc., Chem. Commun., 1995, 1321–1322, 10.1039/C39950001321.
- R.-R. Kim, B. Illarionov, M. Joshi, M. Cushman, C. Y. Lee, W. Eisenreich, M. Fischer and A. Bacher, J. Am. Chem. Soc., 2010, 132, 2983–2990 CrossRef CAS PubMed.
- G. Pohnert, ChemBioChem, 2003, 4, 713–715 CrossRef CAS.
- J. M. Serafimov, H. C. Lehmann, H. Oikawab and D. Hilvert, Chem. Commun., 2007, 1701–1703, 10.1039/B703177G.
- J. M. Serafimov, D. Gillingham, S. Kuster and D. Hilvert, J. Am. Chem. Soc., 2008, 130, 7798–7799 CrossRef CAS PubMed.
- D. G. Gillingham, P. Stallforth, A. Adibekian, P. H. Seeberger and D. Hilvert, Nat. Chem., 2010, 2, 102–105 CrossRef CAS PubMed.
- M. J. Byrne, N. R. Lees, L.-C. Han, M. W. van der Kamp, A. J. Mulholland, J. E. M. Stach, C. L. Willis and P. R. Race, J. Am. Chem. Soc., 2016, 138, 6095–6098 CrossRef CAS PubMed.
- C. O. Marsh, N. R. Lees, L.-C. Han, M. J. Byrne, S. Z. Mbatha, L. Maschio, S. Pagden-Ratcliffe, P. W. Duke, J. E. M. Stach, P. Curnow, C. L. Willis and P. R. Race, ChemCatChem, 2019, 11, 5027–5031 CrossRef CAS.
- D. J. Tantillo, Org. Lett., 2010, 12, 1164–1167 CrossRef CAS PubMed.
- M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2008, 130, 2936–2937 CrossRef CAS PubMed.
- B. Seelig and A. Jäschke, Chem. Biol., 1999, 6, 167–176 CrossRef CAS.
- T. M. Tarasow, S. L. Tarasow and B. E. Eaton, Nature, 1997, 389, 54–57 CrossRef CAS PubMed.
- J. Xu, Q. Deng, J. Chen, K. N. Houk, J. Bartek, D. Hilvert and I. A. Wilson, Science, 1999, 286, 2345–2348 CrossRef CAS PubMed.
- J. Chen, Q. Deng, R. Wang, K. Houk and D. Hilvert, ChemBioChem, 2000, 1, 255–261 CrossRef CAS.
- M. Hugot, N. Bensel, M. Vogel, M. T. Reymond, B. Stadler, J.-L. Reymond and U. Baumann, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9674–9678 CrossRef CAS PubMed.
- S. Müller, B. Appel, D. Balke, R. Hieronymus and C. Nübel, F1000Research, 2016, 5, 1511 Search PubMed.
- M. R. Tremblay, T. J. Dickerson and K. D. Janda, Adv. Synth. Catal., 2001, 343, 577–585 CrossRef CAS.
- A. Serganov, S. Keiper, L. Malinina, V. Tereshko, E. Skripkin, C. Höbartner, A. Polonskaia, A. T. Phan, R. Wombacher, R. Micura, Z. Dauter, A. Jäschke and D. J. Patel, Nat. Struct. Mol. Biol., 2005, 12, 218–224 CrossRef CAS PubMed.
- M. Pohl, B. Lingen and M. Müller, Chem.–Eur. J., 2002, 8, 5288–5295 CrossRef CAS PubMed.
- A. S. Demir, P. Ayhan and S. B. Sopaci, Clean, 2007, 35, 406–412 CAS.
- F. Jordan, Nat. Prod. Rep., 2003, 20, 184–201 RSC.
- M. Müller, G. A. Sprenger and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef PubMed.
- S. Prajapati, F. R. von Pappenheim and K. Tittmann, Curr. Opin. Struct. Biol., 2022, 76, 102441 CrossRef CAS PubMed.
- M. Berheide, S. Kara and A. Liese, Catal. Sci. Technol., 2015, 5, 2418–2426 RSC.
- D. Gocke, C. L. Nguyen, M. Pohl, T. Stillger, L. Walter and M. Müller, Adv. Synth. Catal., 2007, 349, 1425–1435 CrossRef CAS.
- I. F. Fernández, L. Hecquet and W.-D. Fessner, Adv. Synth. Catal., 2022, 364, 612–621 CrossRef.
- P. P. Giovannini, P. Pedrini, V. Venturi, G. Fantin and A. Medici, J. Mol. Catal. B: Enzym., 2010, 64, 113–117 CrossRef CAS.
- M. Schapfl, S. Baier, A. Fries, S. Ferlaino, S. Waltzer, M. Müller and G. A. Sprenger, Appl. Microbiol. Biotechnol., 2018, 102, 8359–8372 CrossRef CAS PubMed.
- A. Kurutsch, M. Richter, V. Brecht, G. A. Sprenger and M. Müller, J. Mol. Catal. B: Enzym., 2009, 61, 56–66 CrossRef CAS.
- C. R. Müller, M. Pérez-Sáncheza and P. Domínguez de María, Org. Biomol. Chem., 2013, 11, 2000–2004 RSC.
- P. Lehwald, O. Fuchs, L. A. Nafie, M. Müller and S. Lüdeke, ChemBioChem, 2016, 17, 1207–1210 CrossRef CAS PubMed.
- M. Beigi, S. Waltzer, A. Fries, L. Eggeling, G. A. Sprenger and M. Müller, Org. Lett., 2013, 15, 452–455 CrossRef CAS PubMed.
- L. Wang, W. Song, B. Wang, Y. Zhang, X. Xu, J. Wu, C. Gao, J. Liu, X. Chen, J. Chen and L. Liu, ACS Catal., 2021, 11, 2808–2818 CrossRef CAS.
- K. Hernández, T. Parella, G. Petrillo, I. Usón, C. M. Wandtke, J. Joglar, J. Bujons and P. Clapés, Angew. Chem., Int. Ed., 2017, 56, 5304–5307 CrossRef PubMed.
- X. Chen, Z. Wang, Y. Lou, Y. Peng, Q. Zhu, J. Xu and Q. Wu, Angew. Chem., Int. Ed., 2021, 60, 9326–9329 CrossRef CAS PubMed.
- Y. Li, N. Hu, Z. Xu, Y. Cui, J. Feng, P. Yao, Q. Wu, D. Zhu and Y. Ma, Angew. Chem., Int. Ed., 2022, 61, e202116344 CrossRef CAS PubMed.
- Y. Li, P. Yao, S. Zhang, J. Feng, H. Su, X. Liu, X. Sheng, Q. Wu, D. Zhu and Y. Ma, Chem Catal., 2023, 3, 100467 CrossRef CAS.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- V. Resch, J. H. Schrittwieser, S. Wallner, P. Macheroux and W. Kroutil, Adv. Synth. Catal., 2011, 353, 2377–2383 CrossRef CAS.
- W. Hüttel and M. Müller, Nat. Prod. Rep., 2021, 38, 1011–1043 RSC.
- A. Präg, B. A. Grüning, M. Häckh, S. Lüdeke, M. Wilde, A. Luzhetskyy, M. Richter, M. Luzhetska, S. Günther and M. Müller, J. Am. Chem. Soc., 2014, 136, 6195–6198 CrossRef PubMed.
- L. E. Zetzsche, J. A. Yazarians, S. Chakrabarty, M. E. Hinze, L. A. M. Murray, A. L. Lukowski, L. A. Joyce and A. R. H. Narayan, Nature, 2022, 603, 79–85 CrossRef CAS PubMed.
- Z. Guo, P. Li, G. Chen, C. Li, Z. Cao, Y. Zhang, J. Ren, H. Xiang, S. Lin, J. Ju and Y. Chen, J. Am. Chem. Soc., 2018, 140, 18009–18015 CrossRef CAS PubMed.
- T. Katagiri and Y. Amao, Green Chem., 2020, 22, 6682–6713 RSC.
- Q. Yang, X. Guo, Y. Liu and H. Jiang, Int. J. Mol. Sci., 2021, 22, 1890 CrossRef CAS PubMed.
- Y. Amao, S. Ikeyama, T. Katagiri and K. Fujita, Faraday Discuss., 2017, 198, 73–81 RSC.
- T. Katagiri, K. Fujita, S. Ikeyama and Y. Amao, Pure Appl. Chem., 2018, 90, 1723–1733 CrossRef CAS.
- H. Hamby, B. Li, K. E. Shinopoulos, H. R. Keller, S. J. Elliott and G. Dukovic, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 135–140 CrossRef CAS PubMed.
- Y. Amao, Chem. Lett., 2017, 46, 780–788 CrossRef CAS.
- L. Castañeda-Losada, D. Adam, N. Paczia, D. Buesen, F. Steffler, V. Sieber, T. J. Erb, M. Richter and N. Plumeré, Angew. Chem., Int. Ed., 2021, 60, 21056–21061 CrossRef PubMed.
- J. Zhou, X. Tian, Q. Yang, Z. Zhang, C. Chen, Z. Cui, Y. Ji, U. Schwaneberg, B. Chen and T. Tan, Chem Catal., 2022, 2, 2675–2690 CrossRef CAS.
- J. Yang, W. Song, T. Cai, Y. Wang, X. Zhang, W. Wang, P. Chen, Y. Zeng, C. Li, Y. Sun and Y. Ma, Sci. Bull., 2023, 68, 2370–2381 CrossRef CAS PubMed.
- V. P. Willers, M. Döring, B. Beer and V. Sieber, Chem Catal., 2023, 3, 100502 CrossRef CAS.
- T. Cai, H. Sun, J. Qiao, L. Zhu, F. Zhang, J. Zhang, Z. Tang, X. Wei, J. Yang, Q. Yuan, W. Wang, X. Yang, H. Chu, Q. Wang, C. You, H. Ma, Y. Sun, Y. Li, C. Li, H. Jiang, Q. Wang and Y. Ma, Science, 2021, 373, 1523–1527 CrossRef CAS PubMed.
- B. Fu and E. P. Balskus, Curr. Opin. Biotechnol., 2020, 65, 94–101 CrossRef CAS PubMed.
- K. Yokoyama and E. A. Lilla, Nat. Prod. Rep., 2018, 35, 660–694 RSC.
- F. Himo, Biochim. Biophys. Acta, 2005, 1707, 24–33 CrossRef CAS PubMed.
- Y. Fu, H. Chen, W. Fu, M. Garcia-Borràs, Y. Yang and P. Liu, J. Am. Chem. Soc., 2022, 144, 13344–13355 CrossRef CAS PubMed.
- H. Pang, E. A. Lilla, P. Zhang, D. Zhang, T. P. Shields, L. G. Scott, W. Yang and K. Yokoyama, J. Am. Chem. Soc., 2020, 142, 9314–9326 CrossRef CAS PubMed.
- V. G. Dumas, L. A. Defelipe, A. A. Petruk, A. G. Turjanski and M. A. Marti, Proteins, 2014, 82, 1004–1021 CrossRef CAS PubMed.
- M. Szaleniec and J. Heider, Int. J. Mol. Sci., 2016, 17, 514 CrossRef PubMed.
- Y. Hai, M. Chen, A. Huang and Y. Tang, J. Am. Chem. Soc., 2020, 142, 19668–19677 CrossRef CAS PubMed.
- M. Liu, D. Wei, Z. Wen and J. Wang, Front. Bioeng. Biotechnol., 2021, 9, 653682 CrossRef PubMed.
- K. Koch, R. J. F. van den
Berg, P. J. Nieuwland, R. Wijtmans, H. E. Schoemaker, J. C. M. van Hest and F. P. J. T. Rutjes, Biotechnol. Bioeng., 2008, 99, 1028–1033 CrossRef CAS PubMed.
- M. Hashimoto, S. Watari, T. Taguchi, K. Ishikawa, T. Kumamoto, S. Okamoto and K. Ichinose, Angew. Chem., Int. Ed., 2023, 62, e202214400 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed.
- R. D. Shukla, B. Rai and A. Kumar, Eur. J. Org Chem., 2019, 2019, 2864–2868 CrossRef CAS.
- E. Busto, R. C. Simon and W. Kroutil, Angew. Chem., Int. Ed., 2015, 54, 10899–10902 CrossRef CAS PubMed.
- A. Chen, Z. Jiang and M. D. Burkart, Chem. Sci., 2022, 13, 4225–4238 RSC.
- W. Li, G. C. Girt, A. Radadiya, J. J. P. Stewart, N. G. J. Richards and J. H. Naismith, Open Biol., 2023, 13, 220287 CrossRef CAS PubMed.
- H. Tang, M.-H. Wu, H.-Y. Lin, M.-R. Han, Y.-H. Tu, Z.-J. Yang, T.-C. Chien, N.-L. Chan and W.-c. Chang, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2113770119 CrossRef CAS PubMed.
- C.-C. Chen, L. Zhang, X. Yu, L. Ma, T.-P. Ko and R.-T. Guo, ACS Catal., 2020, 10, 4717–4725 CrossRef CAS.
- E. T. Batchelar, R. B. Hamed, C. Ducho, T. D. W. Claridge, M. J. Edelmann, B. Kessler and C. J. Schofield, Angew. Chem., Int. Ed., 2008, 47, 9322–9325 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence