
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of interfacial local conformation of polymer chains on adhesion strength†
Tatsuki
Abe
 a,
Satoru
Yamamoto
a and
Keiji
Tanaka
*ab
a,
Satoru
Yamamoto
a and
Keiji
Tanaka
*ab
aCenter for Polymer Interface and Molecular Adhesion Science, Kyushu University, Fukuoka 819-0395, Japan
bDepartment of Applied Chemistry, Kyushu University, Fukuoka 819-0395, Japan. E-mail: k-tanaka@cstf.kyushu-u.ac.jp
First published on 3rd October 2024
Abstract
The aggregation states of polymer chains at solid interfaces are strongly related to their adhesion properties. In this study, we focused on sum-frequency generation (SFG) vibrational spectroscopy, which offers the best depth resolution at the sub-nanometer level among available techniques, and explored its potential as an imaging method. A poly(methyl methacrylate) (PMMA) thin film with a line-and-space pattern was prepared on a quartz substrate. Imaging of the interfacial line-and-space structure in the film was successfully achieved based on SFG signals arising from ester methyl groups as well as carbonyl groups. Once SFG imaging was established, it was applied to blend films of polystyrene (PS) and PMMA with different compositions on the quartz substrate, enabling the direct and non-destructive observation of wetting layers and phase-separated structures buried at the substrate interface for the first time. The interfacial adhesion strength between the blend films and the quartz substrate, evaluated using the surface and interfacial cutting analysis system, showed a clear correlation with the interfacial structure of the blend. This study, which enables the analysis of the relationship between the local orientation of polymer chains at the interface and adhesion strength, is expected to contribute greatly to the design of next-generation adhesives and adhesion technologies.
1. Introduction
The structure and properties of polymer chains at a buried solid interface are considered crucial for the adhesion properties of polymer-based adhesives.1–15 To better understand the emergence of adhesion strength, various theories have been proposed, including interlocking,16–19 chain diffusion,20–24 Coulomb interactions,25–27 van der Waals interactions,28–30 and hydrogen bonding31–33 at adhesion interfaces. These theories suggest that polymer chains directly contact the adherend surface, leading to both physical and, in some cases, chemical interactions. Thus, it is essential to understand and control the orientation of functional groups in polymer chains at the interface, the resulting local conformations, and the way these interfacial adsorbed chains interact with bulk polymer chains to form the interfacial layer. This understanding is key to optimizing adhesion performance.The interface is in a different energy state compared to the bulk because it is in contact with different media. Therefore, the aggregation state of molecular chains at the interface is unique compared to the bulk,34–44 necessitating direct evaluation. Interfacial selective spectroscopy is effective in analyzing the aggregation state of molecular chains at the interface. Specifically, techniques such as near edge X-ray absorption fine structure,45,46 energy loss near-edge structure,47,48 and soft X-ray absorption spectroscopy combined with etching using a gas cluster ion beam (GC-IB)49 are notable. Additionally, vibrational spectroscopy is also powerful, among which sum frequency generation (SFG) vibrational spectroscopy based on the second-order nonlinear optical effect50–57 is used for analyzing the orientation of functional groups at the interface, due to its excellent depth resolution.
Evaluating the in-plane aggregation states of polymers at interfaces buried within a material is not straightforward. So far, imaging techniques such as secondary ion mass spectrometry58,59 and scanning force microscopy observation using solvent etching,60,61 and electron microscopy observation combined with computer tomography have been reported. However, these techniques are fundamentally destructive tests. On the other hand, if imaging using the aforementioned SFG spectroscopy,62,63 which has not yet been applied to synthetic polymers by other research groups,64 were to become more commonly utilized, it would enable the non-destructive analysis of the aggregation states of polymer chains at the buried interfacial plane with a depth resolution of better than 1 nm.
In this study, we selected a blend system composed of typical amorphous polymers, polystyrene (PS) and poly(methyl methacrylate) (PMMA), prepared on a quartz substrate as a model of a hot-melt adhesive to examine the relationships between aggregation states at the outermost interface and adhesion strength. First, to demonstrate the capability of SFG imaging based on the orientational distribution of functional groups, a line pattern imposed onto a PMMA thin film was characterized. The interfacial ordering of side-chain functional groups of PMMA at the quartz interface, promoted by thermal annealing, was successfully visualized in both the carbonyl and the C–H regions. Then, the aggregation states of the PS/PMMA blend films with various mixing ratios at the quartz interface were examined based on SFG images. Finally, this information was combined with the interfacial adhesion strength measured by a surface and interfacial cutting analysis system (SAICAS),65–68 demonstrating that the orientation of functional groups at the interface is extremely important for adhesion phenomena.
2. Experimental
As a sample, PMMA with a number-average molecular weight (Mn) of 300k and a polydispersity index of 1.05, purchased from Polymer Source Inc. (Québec, Canada), was used. A thin film of PMMA with a thickness of approximately 100 nm was prepared on a quartz substrate by spin-coating from its toluene solution. Subsequently, a line-and-space (L&S) pattern with a half pitch of approximately 40 μm was imposed onto the PMMA film by irradiating vacuum ultraviolet light through a metal mask. The resulting structure was characterized using a digital microscope (VHX-700F, Keyence, Osaka, Japan). One of the PMMA films was not thermally annealed, while the other was annealed at 433 K for 24 h. To selectively obtain SFG signals from the quartz interface, the top surface of the patterned film was covered with another quartz substrate. Fig. 1 shows a schematic representation of the side view of the PMMA L&S film. To acquire SFG signals from carbonyl groups at the interface, CaF2 substrates covered with a 10 nm silicon oxide (SiOx) layer were used instead of quartz substrates to avoid the absorption of the excited infrared (IR) beams during SFG measurements.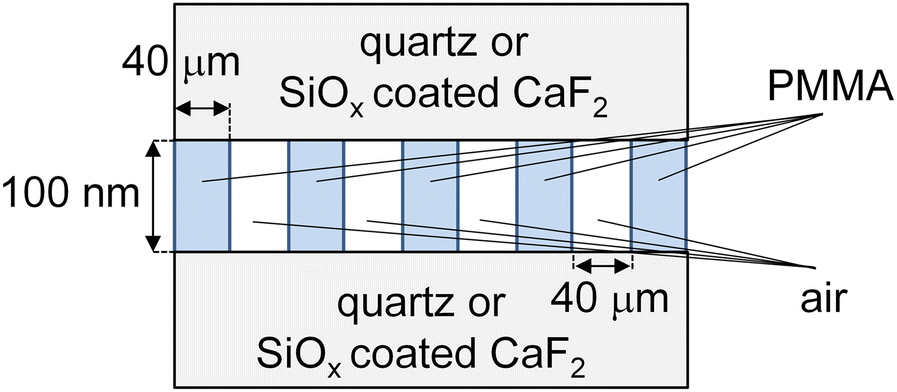 | ||
| Fig. 1 Side view of a PMMA L & S film. | ||
SFG spectroscopy and microscopy were conducted at room temperature using an SFG system (EKSPLA, Vilnius, Lithuania). A 532 nm-visible (VIS) beam was obtained from frequency-doubling fundamental output pulses from a picosecond Nd:YAG laser (PL2231-50, EKSPLA), and a tunable IR beam was generated from an optical parametric generation/amplification and difference frequency generation system with BBO and AgGaS2 crystals (PG500, EKSPLA). The incidence angles of s-polarized VIS and p-polarized IR beams were set to 60° and 55°, respectively. SFG signals for spectroscopy were collected by a photomultiplier tube (PMT) (R7899, EKSPLA) after being diffracted by a monochromator (MC) (MS2001, EKSPLA). For microscopy, signals passing through a telescope lens and an objective lens were gathered by a CCD camera with an image intensifier (iStar DH334T-18U-63, Oxford Instruments Andor Ltd, Belfast, United Kingdom). The magnification was 20× and the length scale of an obtained SFG image was calibrated using a gold grating with a line and space of 5 μm. In our current setup, the theoretical in-plane resolution is nearly equivalent to the diffraction limit. No corrections were applied to SFG images to account for the spatial distribution of the intensity of the excitation laser beams. The adhesion strength of the PMMA L&S film at the interface with a quartz substrate was examined using a SAICAS (NN-05, DAIPLA WINTES, Co. Ltd, Japan) at room temperature. Cutting was performed at room temperature using a diamond blade with a width of 1 mm. The cutting velocities were set to 2 nm s−1 perpendicular to the surface and 400 nm s−1 parallel to the surface. Once the blade reached the substrate interface, cutting continued only in the in-plane direction to measure the adhesion strength.
Then, an immiscible blend of PS and PMMA with various blend ratios at the quartz interface was characterized using SFG in conjunction with SAICAS. The Mn and PDI values of PS of PMMA, purchased from Tosoh (Tokyo, Japan) and Scientific Polymer Products Inc. (Ontario, NY, USA), respectively, were 1030k and 1100k, and 1.20 and 1.12. Films of the PS/PMMA blend were prepared by solvent-casting from toluene solutions onto CaF2 prisms covered with a 10 nm-thick SiOx layer. CaF2 plates, also coated with a 10 nm-thick SiOx layer, were then mounted on the films at 433 K for 48 h, creating a sandwich structure with the blend films. This procedure also served as the thermal annealing process for the films. For the interfacial adhesion tests using SAICAS, the blend films prepared on quartz substrates were used. The surface chemistry of quartz and SiOx should be identical. The cutting velocity was 50 nm s−1 perpendicular to the interface and 500 nm s−1 parallel to the interface. The peeling strength was determined by the force required in the parallel direction as the blade progressed during parallel cutting.
3. Results and discussion
Fig. 2(a) shows SFG spectra for the as-spun and annealed PMMA L&S films under the sp (s-polarized visible input and p-polarized IR input) polarization combination. This setup was used to amplify signal intensity in our SFG microscope by avoiding the insertion of a polarizer between the SF output and the detector. In this configuration, the specimen was positioned on a stage such that the angle between the lines in the pattern and the incident plane of the beams (ω) was set to 0°. For clarity, each SFG spectrum has been vertically shifted. The most intense peak observed at 2955 cm−1 could be attributed to the C–H symmetric stretching vibration of ester methyl groups (OCH3s).69 Peaks at 2912 and 2942 cm−1 were assignable to the symmetric and antisymmetric stretching vibrations of methylene groups (CH2s and CH2as).69 Although the types of SFG peaks observed were similar before and after the annealing treatment of the film, the intensity of each peak varied. Specifically, the intensity from CH2as and OCH3s increased after annealing. Thus, it is evident that the local conformation of PMMA chains at the quartz interface changed due to thermal annealing. | ||
| Fig. 2 (a) SFG spectra in the CH region with the sp polarization combination for a thin PMMA L&S film before (top) and after (bottom) annealing at 433 K for 24 h. SFG images for OCH3s and CH2as signals with the sp polarization combination for the (b and c) as-spun and (d and e) annealed films. (f) A digital microscopic image of the film. The angle formed with the line pattern (ω) is shown. | ||
Panels (b) and (c) of Fig. 2 show two-dimensional maps of SFG signals at 2955 and 2942 cm−1, respectively, for the as-spun PMMA L&S film. These correspond to the SFG images for OCH3s and CH2as signals, exhibiting horizontally aligned stripes with a period of approximately 40 μm. For comparison, Fig. 2(f) shows a digital microscopic image of the annealed PMMA L&S film. A similar stripe pattern aligned along the horizontal direction was observed. The width of a stripe in the SFG images is identical to that in the digital microscopic image. Additionally, when changing ω to 90°, the pattern in the SFG images rotated accordingly by 90° (not shown).
If successful imaging of the local conformation for interfacial chains is achieved, the signal intensity is expected to increase in proportion to the degree and/or the population of oriented functional groups at the interface. Panels (d) and (e) of Fig. 2 show corresponding SFG images to Fig. 2(b) and (c) but after annealing at 433 K for 24 h. The width of stripes shown in both panels was identical to that in Fig. 2(b) and (c). However, the intensity of stripe regions was enhanced after thermal annealing, resulting in a clearer contrast between the brighter and darker regions.
The tilt angle of ester methyl groups (θOCH3) relative to the substrate interface in the PMMA L&S films was determined from the peak intensity ratio obtained with the ssp and sps polarization combinations (not shown). Following the method by Chen et al.,70 the θOCH3 values for the as-spun and annealed films were calculated to be 48 and 61°, respectively, with the assumption of no angular distribution. These values are comparable to our recent results obtained from the combination of phase-sensitive SFG spectroscopy and full-atomistic molecular dynamics (MD) simulation.71 This angle of approximately 60° is effective for the formation of hydrogen bonds between side chain carbonyl groups of PMMA, which are directly connected to ester methyl groups, and silanol groups on the quartz surface, as discussed later.
When polymer chains undergo structural relaxation at the quartz interface through thermal annealing, as shown in Fig. 2(a), the orientation of functional groups is expected to differ from that before annealing.72–77Fig. 3(a) shows SFG spectra in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O region with the sp polarization combination for the PMMA L&S film before and after thermal annealing. A distinct peak at 1740 cm−1, attributed to the stretching vibration of free carbonyl groups, was observed for the as-spun film. However, after annealing, the peak shifted to the lower wavenumber position of 1728 cm−1. The red-shift of the peak after annealing, indicative of structural relaxation, can be explained by the formation of hydrogen bonding. This means that carbonyl groups bonded to SiOH groups at the quartz surface after annealing.69,78 Furthermore, the peak intensity was enhanced after thermal annealing, implying not only the enhanced orientation of carbonyl groups but also an increase in the number density of hydrogen-bonded carbonyl groups.79 To address the transient change in the local conformation of PMMA at the quartz interface during thermal annealing, MD simulations were conducted. The population of carbonyl groups orienting towards the quartz surface, associated with the formation of hydrogen bonds, increased with annealing, as shown in Fig. S1.† This MD result is in excellent accordance with SFG spectra shown in Fig. 3(a).
O region with the sp polarization combination for the PMMA L&S film before and after thermal annealing. A distinct peak at 1740 cm−1, attributed to the stretching vibration of free carbonyl groups, was observed for the as-spun film. However, after annealing, the peak shifted to the lower wavenumber position of 1728 cm−1. The red-shift of the peak after annealing, indicative of structural relaxation, can be explained by the formation of hydrogen bonding. This means that carbonyl groups bonded to SiOH groups at the quartz surface after annealing.69,78 Furthermore, the peak intensity was enhanced after thermal annealing, implying not only the enhanced orientation of carbonyl groups but also an increase in the number density of hydrogen-bonded carbonyl groups.79 To address the transient change in the local conformation of PMMA at the quartz interface during thermal annealing, MD simulations were conducted. The population of carbonyl groups orienting towards the quartz surface, associated with the formation of hydrogen bonds, increased with annealing, as shown in Fig. S1.† This MD result is in excellent accordance with SFG spectra shown in Fig. 3(a).
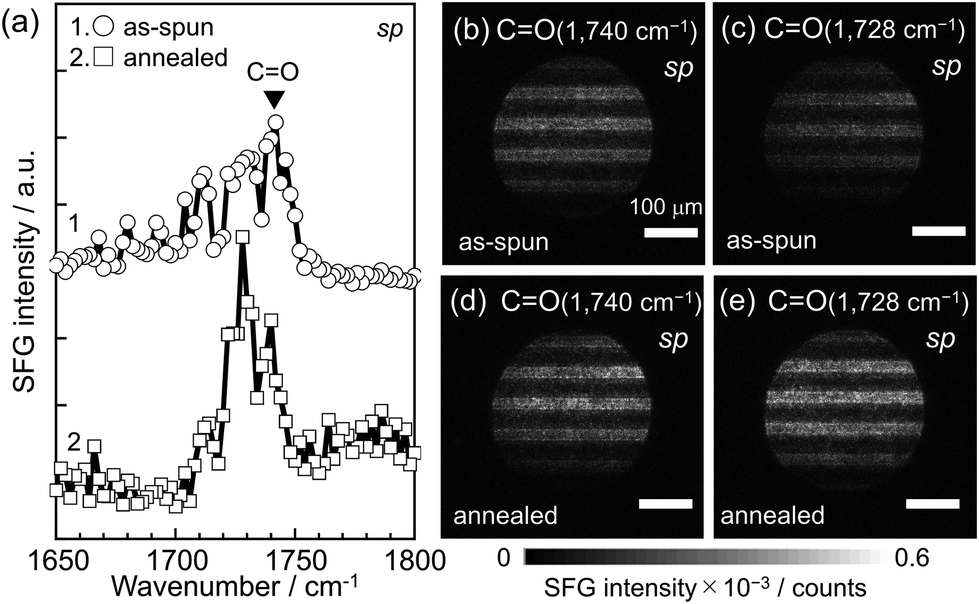 | ||
| Fig. 3 (a) SFG spectra in the CO region with the sp polarization combination for the thin PMMA L&S film before (top) and after (bottom) annealing at 433 K for 24 h. (b–e) SFG images for CO signals with the sp polarization combination obtained at IR wavenumber of (b and d) 1740 cm−1 and (c and e) 1728 cm−1 for the as-spun and annealed films, respectively. | ||
SFG imaging was then applied to this vibration region. Panels (b) and (c) of Fig. 3 show two-dimensional maps of SFG intensity at 1740 and 1728 cm−1, respectively, corresponding to the stretching of free and hydrogen-bonded carbonyl groups in PMMA L&S before thermal annealing. Similar stripe patterns, horizontally aligned with the identical width observed in Fig. 2, were noted, indicating that the signals in the SFG images can be attributed to PMMA. Panels (d) and (e) of Fig. 3 show SFG imaging at 1740 and 1728 cm−1 for the PMMA L&S after annealing. The contrast in the images became clearer at 1728 cm−1 than at 1740 cm−1, which is opposite to the trend observed before annealing. The integrated intensity of the SFG signals from hydrogen-bonded carbonyl groups was larger in Fig. 3(e) (i.e. 4.4 × 107) than in Fig. 3(c) (i.e. 2.1 × 107). This result is consistent with the SFG spectra shown in Fig. 3(a). Thus, it can be claimed that the formation of hydrogen bonds between carbonyl groups of the PMMA side chain and the substrate surface has been successfully visualized.
We now turn to an examination of how the formation of hydrogen bonds of carbonyl groups to the quartz surface impacts the adhesion strength. The angle between the cutting direction and the L&S structure corresponding to ω was set to 90°, meaning that the cutting blade penetrated perpendicular to the L&S structure. Fig. 4(a) shows a schematic representation of the cutting geometry for the SAICAS measurement. In the early stage of cutting, a diamond blade proceeds through the film diagonally (regime I). Once it reaches the substrate surface, the direction changes to horizontal against the substrate surface (regime II). The latter cutting mode corresponds to peeling, providing information on the adhesion strength between PMMA and quartz. Fig. 4(b) shows the horizontal force (Fh) profile for the PMMA L&S film before and after annealing. After thermal annealing, the Fh increased and the periodic fluctuation corresponding to the L&S structure was observed. Increased and decreased Fh regions correspond to interfacial peeling and the movement of the cutting blade on the substrate, respectively. Considering SFG results, the enhancement of interfacial adhesion strength can be attributed to the formation of hydrogen bonding by carbonyl groups, thereby changing the local conformation of PMMA at the adherend surface. Polymer chains in the as-spun film are frozen in an energetically unstable state, whereas side-chain carbonyl groups adopt an energetically preferable conformation after annealing. This conformation enables them to form a stronger bond with the adherend surface.
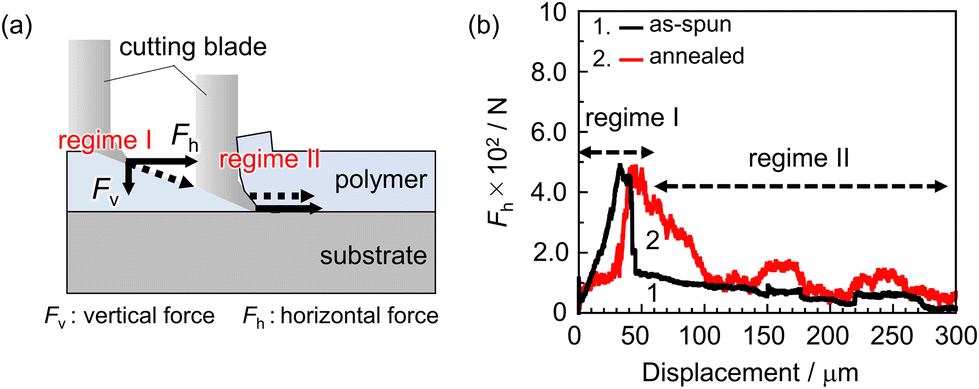 | ||
| Fig. 4 (a) Schematic representation of SAICAS measurement, showing cutting modes and detecting forces along the directions vertical and horizontal to the interface. (b) Horizontal force (Fh) profile for the PMMA L & S film before and after annealing, measured in the direction perpendicular to the line pattern. | ||
Once we established the mapping of functional groups at a buried interface using SFG with a sub-nanometer depth resolution, the technique was applied to examine the aggregation states of a polymer blend of PS and PMMA, which is in a phase-separated state in the bulk.60,61 On the other hand, it is widely accepted that a wetting layer, or at least an enriched layer, of a component might form at the free surface and the substrate interface to minimize the interfacial free energy, as is well established in block copolymers.80,81 To the best of our knowledge, it remains uncertain whether this blend system is in a phase-separated state even at the buried interface.
Panels (a) and (b) of Fig. 5 show SFG spectra for PS/PMMA blend films with different bulk compositions using the sp and pp polarization combinations. Since the film surface was covered with a SiOx-covered CaF2 plate, the spectra reflect the structural information at the substrate surface. The most prominent peak for the neat PMMA film was observed at 2955 cm−1, assignable to OCH3s.69 This peak was also observed for the PS/PMMA (25/75), (50/50), and (75/25) films, but not for the neat PS film. These results make it clear that ester methyl groups of PMMA were oriented at the SiOx interface due to the interaction through hydrogen bonding with silanol groups at the SiOx surface. As shown in Fig. S2,† both peaks attributed to CO and OH stretching vibrations shifted to lower wavenumber regions with increasing bulk PMMA ratio. This shift indicates the formation of hydrogen bonds between PMMA side chains and the substrate surface.82 In contrast, peaks in the range of 3000 to 3100 cm−1, assignable to stretching vibrations of phenyl groups,83,84 were observed for the PS neat film. These peaks were not observed for the PMMA film or the PS/PMMA (25/75), (50/50), and (75/25) films with the sp polarization combination, but were present for the PS/PMMA (75/25) film with the pp polarization combination. Thus, it can be claimed that the substrate interface of the PS/PMMA (25/75) and (50/50) films was covered with the wetting layer of PMMA.85–87 In the case of the PS/PMMA (75/25) film, although both components of PS and PMMA were present at the substrate interface, it is difficult to determine whether they are in a phase-separated or a homogeneous state. It is interesting to note that for the PS/PMMA (75/25) film, SFG signals attributed to phenyl groups were observed not with the sp polarization combination, but with the pp polarization combination. This result likely indicates that phenyl groups were predominantly tilted significantly toward the substrate interface in this blend film. This interpretation is also consistent with previous reports.83,84
 | ||
| Fig. 5 SFG spectra for PS/PMMA blend films with different bulk compositions with (a) sp and (b) pp polarization combinations. SFG images for PS/PMMA (c) (25/75), (d) (50/50), and (e) (75/25), respectively. All blend films were annealed at 433 K for 48 h under vacuum. | ||
To discuss the in-plane distribution of local conformation, SFG imaging was conducted. Panels (c–e) of Fig. 5 show two-dimensional mappings of SFG intensity at 2955 cm−1, corresponding to OCH3s, for the PS/PMMA (c) (25/75), (d) (50/50), and (e) (75/25) films at the SiOx interface. In the case of the PS/PMMA (50/50) and (25/75) films, homogeneous images were obtained. Thus, it is clear that PMMA was preferentially adsorbed onto the substrate surface, forming the wetting layer of PMMA formed at the SiOx surface, as discussed above. Since the degrees of polymerization for both the components were comparable, the conformational entropy loss of the components at the SiOx interface cannot be considered an important factor in this structural formation.88 Thus, the segregation of PMMA at the SiOx interface was likely driven by the surface energy difference between the components.
In the case of the PS/PMMA (75/25) film, a heterogeneous structure with domains approximately 20–40 μm in diameter appeared, as shown in Fig. 5(e). Considering that SFG signals in the black regions were finite, not zero, the white and black regions correspond to areas where ester methyl groups are more or less oriented, respectively. This indicates the presence of two phases with different aggregation states at the SiOx interface. Thus, it seems most likely that the PS/PMMA (75/25) blend is separated into PS-rich and PMMA-rich phases at the SiOx interface. It is noteworthy that the areal fraction of the white to black regions was 64% and was much larger than the bulk fed ratio of PMMA. This is simply because PMMA was preferentially segregated at the SiOx interface. To the best of our knowledge, this is the first report of a non-destructive structural analysis of a polymer blend at a buried interface with a solid substrate, made possible by the combination of SFG spectroscopy and microscopy.
We finally address the relationship between aggregation states and adhesive properties at the substrate interface. Fig. 6 shows the dependence of Fh, corresponding to peeling force at the SiOx interface, on the bulk blend composition, obtained through SAICAS measurements. The abscissa represents the bulk PMMA ratio. The Fh value increased with increasing bulk PMMA ratio because the extent and efficacy of hydrogen bonding between PMMA side chains and the SiOx surface increased with increasing bulk PMMA ratio, as shown in Fig. S2.† For example, for the PS/PMMA (25/75) and (50/50) blends, the SFG spectra and images appear similar, resulting in comparable peeling strength. Conversely, in the case of the PS/PMMA (75/25) blend film, although the interface was mainly covered with PMMA, the interfacial aggregation states were quite different from the others due to the phase-separated structure, specifically the presence of PS. This led to a lower peeling strength. As expected, the PS film exhibited the lowest peeling strength among samples employed owing to the lack of the formation of hydrogen bonding with the SiOx surface. Therefore, it can be concluded that interaction such as hydrogen bonding between polymer segments and the substrate surface are crucial factors in controlling adhesive properties.
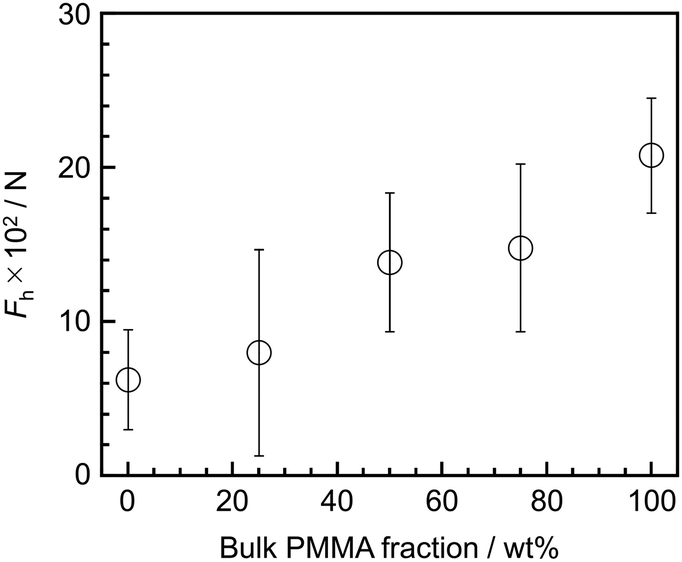 | ||
| Fig. 6 Bulk PMMA composition dependence of Fh for PS/PMMA blend films at the SiOx interface. | ||
4. Conclusions
We examined the effect of the local orientational distribution of functional groups at the solid interface on adhesion properties by applying SFG imaging in conjunction with SAICAS measurements. Initially, orientational imaging based on SFG signals from ester methyl and carbonyl groups was conducted for PMMA L&S films. The structural reorganization of PMMA chains, specifically ester methyl groups and carbonyl groups, induced by thermal annealing was visualized at the quartz interface. Then, the interfacial distribution of local conformation in PS/PMMA blends with various compositions was examined. In the case of the PS/PMMA (50/50) and (25/75) films, SFG images from ester methyl groups were obtained, indicating that PMMA was preferentially segregated at the quartz surface, namely forming a wetting layer of PMMA at the interface. In contrast, the PS/PMMA (75/25) film exhibited a heterogeneous SFG image from ester methyl groups, with SFG signals from aromatic CH vibrations also clearly observed. These results make it clear that a phase-separated structure was formed at the quartz interface. Furthermore, SFG spectroscopy revealed that hydrogen bonding between side chain carbonyl groups of PMMA and hydroxy groups on quartz was promoted with increasing PMMA fraction. The interfacial adhesion strength concurrently increased with PMMA fraction due to effective hydrogen bonding at the interface. Thus, it can be concluded that the interfacial aggregation states of polymer chains, specifically hydrogen bonding in this case, are crucial to control their adhesion properties. We believe that by further studying the molecular chain interactions at adhesive interfaces, particularly the interactions between polymers and adherend surfaces at the functional group level, next-generation adhesives and adhesion technologies will be established.Author contributions
Tatsuki Abe: data curation, formal analysis, methodology, writing – original draft. Satoru Yamamoto: data curation, formal analysis. Keiji Tanaka: project administration, conceptualization, funding acquisition, methodology, writing – review & editing, supervision. All authors discussed the results and contributed to the final manuscript.Data availability
The data supporting this article will be available upon request at https://www.cstf.kyushu-u.ac.jp/~tanaka-lab/english/.Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
This work was supported by JSPS KAKENHI for Scientific Research (B) (No. JP23H02014) to KT and for Early-Career Scientists (No. JP23K13794) to TA from the Ministry of Education, Culture, Sports, Science, and Technology, Japan. We are also thankful for the support from the JST-Mirai Program (JPMJMI18A2) (KT).References
- S. Wu, Polymer Interface and Adhesion, CRC Press, New York, 1982 Search PubMed.
- R. P. Wool, Polymer Interfaces: Structure and Strength, Hanser Publications, New York, 1994 Search PubMed.
- D. L. Allara, F. M. Fowkes, J. Noolandi, G. W. Rubloff and M. V. Tirrell, Mater. Sci. Eng., 1986, 83, 213–226 CrossRef CAS.
- H. R. Brown, Annu. Rev. Mater. Sci., 1991, 21, 463–489 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- A. J. Crosby, K. R. Shull, H. Lakrout and C. Creton, J. Appl. Phys., 2000, 88, 2956–2966 CrossRef CAS.
- H.-J. Kim, K.-J. Lee and Y. Seo, Macromolecules, 2002, 35, 1267–1275 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS.
- L. Ge, S. Sethi, L. Ci, P. M. Ajayan and A. Dhinojwala, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10792–10795 CrossRef CAS.
- E. P. Chan, E. J. Smith, R. C. Hayward and A. J. Crosby, Adv. Mater., 2008, 20, 711–716 CrossRef CAS.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- M. Aoki, A. Shundo, K. Okamoto, T. Ganbe and K. Tanaka, Polym. J., 2019, 51, 359–363 CrossRef CAS.
- G. Raos and B. Zappone, Macromolecules, 2021, 54, 10617–10644 CrossRef CAS.
- S. Yamamoto, R. Kuwahara and K. Tanaka, ACS Appl. Polym. Mater., 2022, 4, 6038–6046 CrossRef.
- M. Aizawa, H. Akiyama, Y. Matsuzawa and A. Shishido, Polym. J., 2024, 56, 401–408 CrossRef CAS.
- J. W. McBain and D. G. Hopkins, J. Phys. Chem., 1925, 29, 188–204 CrossRef CAS.
- T. Matsumoto, Y. Shimizu and T. Nishino, Macromolecules, 2021, 54, 7226–7233 CrossRef CAS.
- K. Hagita, T. Murashima, T. Miyata and H. Jinnai, Macromolecules, 2024, 57, 3862–3872 CrossRef CAS.
- K. Aragishi, Y. Takeda, Y. Suzuki and A. Matsumoto, Polym. J., 2024, 56, 529–540 CrossRef CAS.
- K. Jud, H. H. Kausch and J. G. Williams, J. Mater. Sci., 1981, 16, 204–210 CrossRef CAS.
- R. Schnell, M. Stamm and C. Creton, Macromolecules, 1998, 31, 2284–2292 CrossRef CAS.
- Y. M. Boiko and R. E. Prud'Homme, J. Appl. Polym. Sci., 1999, 74, 825–830 CrossRef CAS.
- K. Akabori, D. Baba, K. Koguchi, K. Tanaka and T. Nagamura, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3598–3604 CrossRef CAS.
- H. Kim, D. Kawaguchi, K. Tanaka and Y. Seo, Langmuir, 2018, 34, 11027–11033 CrossRef CAS PubMed.
- B. V. Derjaguin and V. P. Smilga, J. Appl. Phys., 1967, 38, 4609–4616 CrossRef.
- M. Erdmann, R. David, A. Fornof and H. E. Gaub, Nat. Nanotechnol., 2010, 5, 154–159 CrossRef CAS PubMed.
- S. Saeki, D. Kawaguchi, Y. Tsuji, S. Yamamoto, K. Yoshizawa and K. Tanaka, Langmuir, 2024, 40, 9725–9731 CrossRef CAS.
- G. Fourche, Polym. Eng. Sci., 1995, 35, 957–967 CrossRef CAS.
- C. Zhang, Y. Fujii and K. Tanaka, ACS Macro Lett., 2012, 1, 1317–1320 CrossRef CAS PubMed.
- Y. Tsuji, Y. Kitamura, M. Someya, T. Takano, M. Yaginuma, K. Nakanishi and K. Yoshizawa, ACS Omega, 2019, 4, 4491–4504 CrossRef CAS.
- J. L. Keddie, R. A. L. Jones and R. A. Cory, Faraday Discuss., 1994, 98, 219–230 RSC.
- T. Semoto, Y. Tsuji and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 11701–11708 CrossRef CAS.
- B. Cheng, J. Yu, T. Arisawa, K. Hayashi, J. J. Richardson, Y. Shibuta and H. Ejima, Nat. Commun., 2022, 13, 1892 CrossRef CAS PubMed.
- J. A. Forrest, K. Dalnoki-Veress and J. R. Dutcher, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 5705–5716 CrossRef CAS.
- C. J. Ellison and J. M. Torkelson, Nat. Mater., 2003, 2, 695–700 CrossRef CAS PubMed.
- K. Akabori, K. Tanaka, T. Nagamura, A. Takahara and T. Kajiyama, Macromolecules, 2005, 38, 9735–9741 CrossRef CAS.
- P. Gin, N. Jiang, C. Liang, T. Taniguchi, B. Akgun, S. K. Satija, M. K. Endoh and T. Koga, Phys. Rev. Lett., 2009, 109, 265501 CrossRef PubMed.
- Y. Fujii, Z. Yang, J. Leach, H. Atarashi, K. Tanaka and O. K. C. Tsui, Macromolecules, 2009, 42, 7418–7422 CrossRef CAS.
- S. Sun, H. Xu, J. Han, Y. Zhu, B. Zuo, X. Wang and W. Zhang, Soft Matter, 2016, 12, 8348–8358 RSC.
- N. G. Perez-de-Eulate, M. Sferrazza, D. Cangialosi and S. Napolitano, ACS Macro Lett., 2017, 6, 354–358 CrossRef CAS PubMed.
- D. Christie, R. A. Register and R. D. Priestley, ACS Cent. Sci., 2018, 4, 504–511 CrossRef CAS.
- W. Ren, Y. Hong, H. Wei, J. Xu, C. Zhang, X. Zhou and X. Wang, Macromolecules, 2023, 56, 1410–1418 CrossRef CAS.
- J. H. Merrill, R. Li and C. B. Roth, ACS Macro Lett., 2023, 12(1), 1–7 CrossRef CAS PubMed.
- Y. Yang, H. Shen, S. Ge, Z. Yao and B. Zuo, Adv. Mater. Interfaces, 2024, 2400239, DOI:10.1002/admi.202400239.
- L. R. Pattison, A. Hexemer, E. J. Kramer, S. Krishnan, P. M. Petroff and D. A. Fischer, Macromolecules, 2006, 39, 2225–2231 CrossRef CAS.
- P. K.-H. Ho, L.-L. Chua, M. Dipankar, X. Gao, D. Qi, A. T.-S. Wee, J.-F. Chang and R. H. Friend, Adv. Mater., 2007, 19, 215–221 CrossRef CAS.
- Y. Kamikawa, K. Amezawa and K. Terada, J. Phys. Chem. C, 2021, 125, 3890–3900 CrossRef CAS.
- T. Miyata, Y. K. Sato, Y. Kawagoe, K. Shirasu, H.-F. Wang, A. Kumagai, S. Kinoshita, M. Mizukami, K. Yoshida, H.-H. Huang, T. Okabe, K. Hagita, T. Mizoguchi and H. Jinnai, Nat. Commun., 2024, 15, 1898 CrossRef CAS PubMed.
- H. Yamane, M. Oura, D. Kawaguchi, K. Nitta, O. Sekizawa, T. Ishikawa, S. Yamamoto, K. Tanaka and T. Hatsui, Polym. J., 2024, 56, 215–220 CrossRef CAS.
- Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS.
- Z. Chen, Y. R. Shen and G. A. Somorjai, Annu. Rev. Phys. Chem., 2002, 53, 437–465 CrossRef CAS PubMed.
- Y. Oda, A. Horinouchi, D. Kawaguchi, H. Matsuno, S. Kanaoka, S. Aoshima and K. Tanaka, Langmuir, 2014, 30, 1215–1219 CrossRef CAS.
- H. Zhu, K. C. Jha, R. S. Bhatta, M. Tsige and A. Dhinojwala, Langmuir, 2014, 30, 11609–11618 CrossRef CAS PubMed.
- N. Dhopatkar, E. Anim-Danso, C. Peng, S. Singla, X. Liu, A. Joy and A. Dhinojwala, Macromolecules, 2018, 51, 5114–5120 CrossRef CAS.
- L. Kong, M. Zhang, Y. Zhang, W. Zhang, X. Zhou, L. Zhang and X. Wang, Polym. Chem., 2020, 11, 7429–7437 RSC.
- B. Li, J. S. Andre, X. Chen, B. Walther, R. Paradkar, C. Feng, C. Tucker, C. Mohler and Z. Chen, Anal. Chem., 2020, 92, 14145–14152 CrossRef CAS.
- Z. Chen, Langmuir, 2022, 38, 4483–4489 CrossRef CAS.
- J. P. Thomas, L. Zhao, M. Abd-Ellah, N. F. Heinig and K. T. Leung, Anal. Chem., 2013, 85, 6840–6845 CrossRef CAS.
- J. Bailey, R. Havelund, A. G. Shard, I. S. Gilmore, M. R. Alexander, J. S. Sharp and D. J. Scurr, ACS Appl. Mater. Interfaces, 2015, 7, 2654–2659 CrossRef CAS.
- K. Tanaka, A. Takahara and T. Kajiyama, Macromolecules, 1996, 29, 3232–3239 CrossRef CAS.
- A. Karim, T. M. Slawecki, S. K. Kumar, J. F. Douglas, S. K. Satija, C. C. Han, T. P. Russell, Y. Liu, R. Overney, J. Sokolov and M. H. Rafailovich, Macromolecules, 1998, 31, 857–862 CrossRef CAS.
- M. Flörsheimer, C. Brillert and H. Fuchs, Langmuir, 1999, 15, 5437–5439 CrossRef.
- S. A. Shah and S. Baldelli, Acc. Chem. Res., 2020, 53, 1139–1150 CrossRef CAS.
- T. Abe, H. Shimada, T. Hoshino, D. Kawaguchi and K. Tanaka, Polym. J., 2022, 54, 679–685 CrossRef CAS.
- N. Nagai, T. Imai, K. Terada, H. Seki, H. Okumura, H. Fujino, T. Yamamoto, I. Nishiyama and A. Hatta, Surf. Interface Anal., 2002, 34, 545–551 CrossRef.
- I. Narayama, D. Baba, A. Takahara and K. Tanaka, Appl. Surf. Sci., 2010, 257, 1145–1148 CrossRef CAS.
- F. Saito, I. Nishiyama and T. Hyodo, Surf. Coat. Technol., 2010, 205, 419–422 CrossRef CAS.
- B. Son, M.-H. Ryou, J. Choi, T. Lee, H. K. Yu, J. H. Kim and Y. M. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 526–531 CrossRef CAS.
- Y. Tateishi, N. Kai, H. Noguchi, K. Uosaki, T. Nagamura and K. Tanaka, Polym. Chem., 2010, 1, 303–311 RSC.
- J. Wang, C. Chen, S. M. Buck and Z. Chen, J. Phys. Chem. B, 2001, 105, 12118–12125 CrossRef CAS.
- D. Kawaguchi, K. Sasahara, M. Inutsuka, T. Abe, S. Yamamoto and K. Tanaka, J. Chem. Phys., 2023, 159, 244902 CrossRef CAS.
- S. Sugimoto, M. Inutsuka, D. Kawaguchi and K. Tanaka, ACS Macro Lett., 2018, 7, 85–89 CrossRef CAS.
- B. Zuo, M. Inutsuka, D. Kawaguchi, X. Wang and K. Tanaka, Macromolecules, 2018, 51, 2180–2186 CrossRef CAS.
- Y. Hong, Y. Li, F. Wang, B. Zuo, X. Wang, L. Zhang, D. Kawaguchi and K. Tanaka, Macromolecules, 2018, 51, 5620–5627 CrossRef CAS.
- H. K. Nguyen, S. Sugimoto, A. Konomi, M. Inutsuka, D. Kawaguchi and K. Tanaka, ACS Macro Lett., 2019, 8, 1006–1011 CrossRef CAS PubMed.
- C. Cai, M. S. Azam and D. Hore, J. Phys. Chem. C, 2021, 125, 22214–22222 CrossRef CAS.
- K. Uchida, K. Mita, S. Yamamoto and K. Tanaka, Polym. J., 2023, 55, 683–690 CrossRef CAS.
- M. Inutsuka, A. Horinouchi and K. Tanaka, ACS Macro Lett., 2015, 4, 1174–1178 CrossRef CAS.
- D. Rossi, Y. Dong, R. Paradkar, X. Chen, Y. Wu, C. Mohler, T.-C. Kuo and Z. Chen, Langmuir, 2024, 40, 12689–12696 CrossRef CAS PubMed.
- A. M. Mayes, T. P. Russell, P. Bassereau, S. M. Baker and G. S. Smith, Macromolecules, 1994, 27, 749–755 CrossRef CAS.
- K. Tanaka, A. Takahara and T. Kajiyama, Acta Polym., 1995, 46, 476–482 CrossRef CAS.
- S. Ye, S. Morita, G. Li, H. Noda, M. Tanaka, K. Uosaki and M. Osawa, Macromolecules, 2003, 36, 5694–5703 CrossRef CAS.
- K. S. Gautam, A. D. Schwab, A. Dhinojwala, D. Zhang, S. M. Dougal and M. S. Yeganeh, Phys. Rev. Lett., 2000, 85, 3854–3857 CrossRef CAS PubMed.
- B. Li, L. Chen, S. Zhang, Q. Tao, Y.-H. Ma, P. Hu, X. Lu, K. C. Chou and Z. Chen, Appl. Surf. Sci., 2023, 611, 155715 CrossRef CAS.
- H. Wang and R. J. Composto, J. Chem. Phys., 2000, 113, 10386–10397 CrossRef CAS.
- D. U. Ahn, Z. Wang, I. P. Campbell, M. P. Stoykovich and Y. Ding, Polymer, 2012, 53, 4187–4194 CrossRef CAS.
- Z. Zhang, Z. Wang and Y. Ding, Polymer, 2014, 55, 4150–4155 CrossRef CAS.
- T. Kajiyama, K. Tanaka and A. Takahara, Macromolecules, 1998, 31, 3746–3749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Local conformation of carbonyl groups by molecular dynamics simulation, SFG spectra in carbonyl and hydroxyl regions for PS/PMMA on SiOx. See DOI: https://doi.org/10.1039/d4py00951g |
| This journal is © The Royal Society of Chemistry 2024 |