
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriphase photocatalytic water-gas-shift reaction for hydrogen production with enhanced interfacial diffusion at gas–liquid–solid interfaces†
Huige
Chen
ab,
Zhenhua
Li
a,
Chao
Zhou
 a,
Run
Shi
*a and
Tierui
Zhang
*ab
a,
Run
Shi
*a and
Tierui
Zhang
*ab
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: shirun@mail.ipc.ac.cn; tierui@mail.ipc.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 20th February 2024
Abstract
The exothermic characteristic of the water-gas-shift (WGS) reaction, coupled with the thermodynamic constraints at elevated temperatures, has spurred a research inclination towards conducting the WGS reaction at reduced temperatures. Nonetheless, the challenge of achieving efficient mass transfer between gaseous CO and liquid H2O at the photocatalytic interface under mild reaction conditions hinders the advancement of the photocatalytic WGS reaction. In this study, we introduce a gas–liquid–solid triphase photocatalytic WGS reaction system. This system facilitates swift transportation of gaseous CO to the photocatalyst's surface while ensuring a consistent water supply. Among various metal-loaded TiO2 photocatalysts, Rh/TiO2 nanoparticles positioned at the triphase interface demonstrated an impressive H2 production rate of 27.60 mmol g−1 h−1. This rate is roughly 2 and 10 times greater than that observed in the liquid–solid and gas–solid diphase systems. Additionally, finite element simulations indicate that the concentrations of CO and H2O at the gas–liquid–solid interface remain stable. This suggests that the triphase interface establishes a conducive microenvironment with sufficient CO and H2O supply to the surface of photocatalysts. These insights offer a foundational approach to enhance the interfacial mass transfer of gaseous CO and liquid H2O, thereby optimizing the photocatalytic WGS reaction's efficiency.
Keywords: Water-gas-shift; Photocatalysis; Triphase interface; Hydrogen evolution; TiO2.
1 Introduction
Hydrogen (H2) stands as a pivotal renewable chemical with extensive applications in contemporary society.1–3 Currently, the predominant method for H2 production involves the steam reforming of hydrocarbons derived from fossil fuels.4,5 Nevertheless, this steam reforming technique demands elevated temperatures and pressures, leading to substantial costs and energy expenditures. Furthermore, the H2 generated often retains elevated levels of CO, rendering it less suitable for subsequent industrial applications.5The water-gas-shift (WGS) reaction (CO + H2O → CO2 + H2) holds significant importance in the chemical industry, serving multiple functions such as H2 production, CO removal, and syngas adjustment.6–8 Over recent decades, considerable research has centered on the thermocatalytic WGS reaction.9–12 Typically, thermocatalytic WGS transpires at temperatures ranging from 180–230 °C for noble metal catalysts, while non-precious metal catalysts necessitate even higher temperatures, surpassing 270 °C.9,13–16 Nonetheless, the reaction's exothermic nature coupled with thermodynamic constraints at elevated temperatures has spurred increased exploration into the WGS reaction at lower temperatures.
Photocatalysis has garnered extensive attention across various chemical reactions, presenting numerous advantages over conventional thermocatalysis. A primary advantage of photocatalysis lies in its mild reaction conditions and reduced reliance on fossil fuels, as it directly utilizes solar energy to drive reactions.17,18 Utilizing metal oxide semiconductors like TiO2, CuOx, among others, in photocatalysis offers a compelling approach to lowering the WGS reaction temperature to ambient levels.19,20
Presently, the photocatalytic WGS reaction predominantly operates within a gas–solid (G–S) diphase system, employing gaseous CO and water vapor as reactants.19,21 However, at lower temperatures, the constrained saturated vapor pressure of water can impede its interfacial mass transfer, potentially becoming the rate-determining step. Furthermore, water molecules interconnected via hydrogen bonds exhibit superior water-splitting capabilities compared to their isolated counterparts.22 This suggests that liquid water might serve as a more effective reactant source than water vapor for low-temperature WGS reaction. Notably, under standard conditions (25 °C, 1 atm), CO molecules demonstrate limited solubility (∼1 mM) and a modest diffusion coefficient (2.03 × 10−5 cm2 s−1) in the aqueous phase.23,24 This implies that in liquid–solid (L–S) diphase photocatalytic systems, which has been widely investigated for decades, the WGS reaction might be hindered by the interfacial mass transfer of CO molecules. Effectively enhancing both the mass transfer and the chemical reaction between gaseous CO and liquid H2O at the photocatalytic interface presents a complex challenge.
Recently, the scientific community has delved into photocatalysis at gas–liquid–solid (G–L–S) triphase interfaces. This approach has demonstrated distinct advantages in various photocatalytic reactions involving both gas and water reactants, such as CO2 reduction, active oxygen generation, and ethylene purification.25–27 By facilitating the diffusion of CO and H2O to the photocatalyst surface via both gaseous and liquid phases, the WGS reaction at G–L–S interfaces holds promise in overcoming the constraints observed in diphase systems, potentially enhancing the overall H2 production efficiency under mild reaction conditions.
In this study, we introduce a triphase photocatalytic system tailored for the WGS reaction, leveraging metal-loaded TiO2 (M/TiO2, where M represents Rh, Au, Ag, Pt, and Pd) as the benchmark photocatalysts. To realize this setup, a hydrophobic porous layer served as the support, enabling the photocatalysts to float at the gas–water interface and fostering abundant G–L–S interfaces. Among tested photocatalysts, Rh/TiO2 demonstrated the highest H2 production rate of 27.60 mmol g−1 h−1. Remarkably, this rate surpasses that of both the corresponding L–S and G–S diphase systems by factors of approximately 2 and 10, respectively. Finite element simulations reveal consistent concentrations of CO and H2O at the G–L–S interface during the photocatalytic diffusion–reaction process, although a significant decline in CO is evident at the L–S interface. This observation aligns with the elevated H2 production rates observed in the triphase system when utilizing diluted CO (0.02 MPa) as the gaseous feedstock. Serving as a proof-of-concept, our results underscore the pivotal role of the G–L–S triphase interface in augmenting the interfacial mass transfer and reaction of gaseous CO and liquid H2O, thereby enhancing the efficiency of the photocatalytic WGS reaction.
2 Results and discussion
The structural characteristics and surface morphology of the Rh/TiO2 photocatalysts were systematically examined, as depicted in Fig. 1. The X-ray diffraction (XRD) profile of x wt% Rh/TiO2 (x = 1, 3, 5, 7, and 9) reveals the presence of both anatase TiO2 (JCPDS no. 73-1764) and rutile TiO2 (JCPDS no. 87-0710). Notably, no discernible signal corresponding to metallic Rh was observed across all Rh/TiO2 samples (Fig. 1a). This absence could be attributed to the small crystal dimensions and the relatively modest Rh content.28 The experimental loading content of Rh within TiO2 align closely with the theoretical values (Fig. S1†). 5 wt% Rh/TiO2 was utilized throughout unless otherwise stated. Upon examination via transmission electron microscopy (TEM) and high-resolution TEM, Rh nanoparticles, each having a diameter of approximately 2.0 ± 0.5 nm, were found to be uniformly dispersed over the TiO2 surface (Fig. 1b and S2†). The observed lattice spacings of 0.352 nm and 0.222 nm correspond to the d-spacing values of TiO2 (101) and Rh (111), respectively (Fig. 1c). Moreover, high-angle annular dark-field scanning TEM (HAADF-STEM) coupled with energy-dispersive X-ray spectroscopy (EDS) elemental mapping corroborates the distinct spatial distribution of Rh nanoparticles on TiO2 (Fig. 1d). | ||
| Fig. 1 Characterization for Rh/TiO2 photocatalysts. (a) XRD pattern for x wt% Rh/TiO2; (b) TEM image and (c) high-resolution TEM image of 5 wt% Rh/TiO2; (d) the HAADF-STEM image and EDS element maps of 5 wt% Rh/TiO2. | ||
Polytetrafluoroethylene gas diffusion layers (GDL) served as the porous substrates for the immobilization of the photocatalyst. Rh/TiO2 was anchored onto the GDL using a vacuum filtration method, denoted as Rh/TiO2-GDL. The structure of Rh/TiO2-GDL is evident in the cross-sectional scanning electron microscopy (SEM) image presented in Fig. 2a. The corresponding EDS mapping, highlighting Ti, C, and F elements, provides insights into the photocatalyst layer's thickness, which is approximately 1.4 ± 0.2 μm (Fig. 2b). The top-view SEM reveals a distinct cross-linked fibre network within the porous substrate, establishing abundant pores for efficient gas diffusion (Fig. 2c). The inset of Fig. 2c displays a water droplet contact angle of 126°, underscoring the inherent hydrophobic nature of the GDL porous substrate. After the immobilization of Rh/TiO2, the resultant photocatalyst layer exhibits a significantly reduced water contact angle of 19°, indicating a pronounced hydrophilic–hydrophobic transition within the Rh/TiO2-GDL layered structure (Fig. 2d).
 | ||
| Fig. 2 Structural characterization of the Rh/TiO2-GDL. (a) Cross-sectional SEM image and (b) corresponding EDS mapping of Rh/TiO2-GDL; top-view SEM images of (c) pristine GDL porous substrate and (d) immobilized Rh/TiO2 photocatalyst layer. Inserts in (c) and (d) show photographs of water droplets on each sample. | ||
As illustrated in Fig. 3a, both pristine TiO2 and all x wt% Rh/TiO2 samples exhibit pronounced absorption in the ultraviolet region, attributed to the characteristic edge adsorption of TiO2. The visible light absorption, corresponding to Rh nanoparticles, amplifies with increasing Rh loading. To further discern the influence of different metal species on the WGS reaction, other M/TiO2 (where M = Au, Ag, Pt, and Pd) with a consistent metal loading of 5 wt% were synthesized using analogous procedures. The XRD patterns exhibit characteristic peaks corresponding to anatase TiO2 (JCPDS no. 73-1764) and rutile TiO2 (JCPDS no. 87-0710), while characteristic peaks corresponding to metal are notably absent, possibly attributable to the small crystal dimensions (Fig. S3†). The TEM images confirm the successful loading of metal nanoparticles onto TiO2 for all M/TiO2 samples (Fig. S4†). The achieved metal content closely aligns with theoretical predictions (Fig. S5†), with all samples demonstrating robust optical absorption below 400 nm (Fig. S6†). Consequently, a 365 nm LED lamp was employed as the illumination source to assess the photocatalytic efficiency. The H2 production rates of Rh/TiO2 and the other M/TiO2 photocatalysts in the G–L–S triphase system are depicted in Fig. 3b. TiO2 modified with distinct metals showcases marked variations in H2 production rates under both CO (WGS) and Ar (pure water splitting) gas phase conditions. In an Ar environment, Pt/TiO2 achieves the highest H2 production rate of 5.95 mmol g−1 h−1, consistent with prior studies employing Pt as co-catalysts for photocatalytic water splitting.29–31 However, transitioning to a CO environment only results in a slight enhancement over Pt/TiO2, suggesting a minimal contribution of the WGS reaction to H2 generation. Conversely, Rh/TiO2 registers a five-fold enhancement in H2 production rate (25.77 mmol g−1 h−1) compared to its performance in an Ar condition, underscoring the pivotal role of CO in enhancing generating H2via the WGS pathway. A pronounced boost was also observed for Au/TiO2 (13.77 mmol g−1 h−1), with the H2 production trend across M/TiO2 as follows: Rh/TiO2 > Au/TiO2 > Pt/TiO2 > Ag/TiO2 > Pd/TiO2. The observed disparities in H2 production rates among various M/TiO2 samples in different gas conditions likely stem from the synergistic interplay between CO adsorption and proton reduction kinetics at the metal active sites.32,33
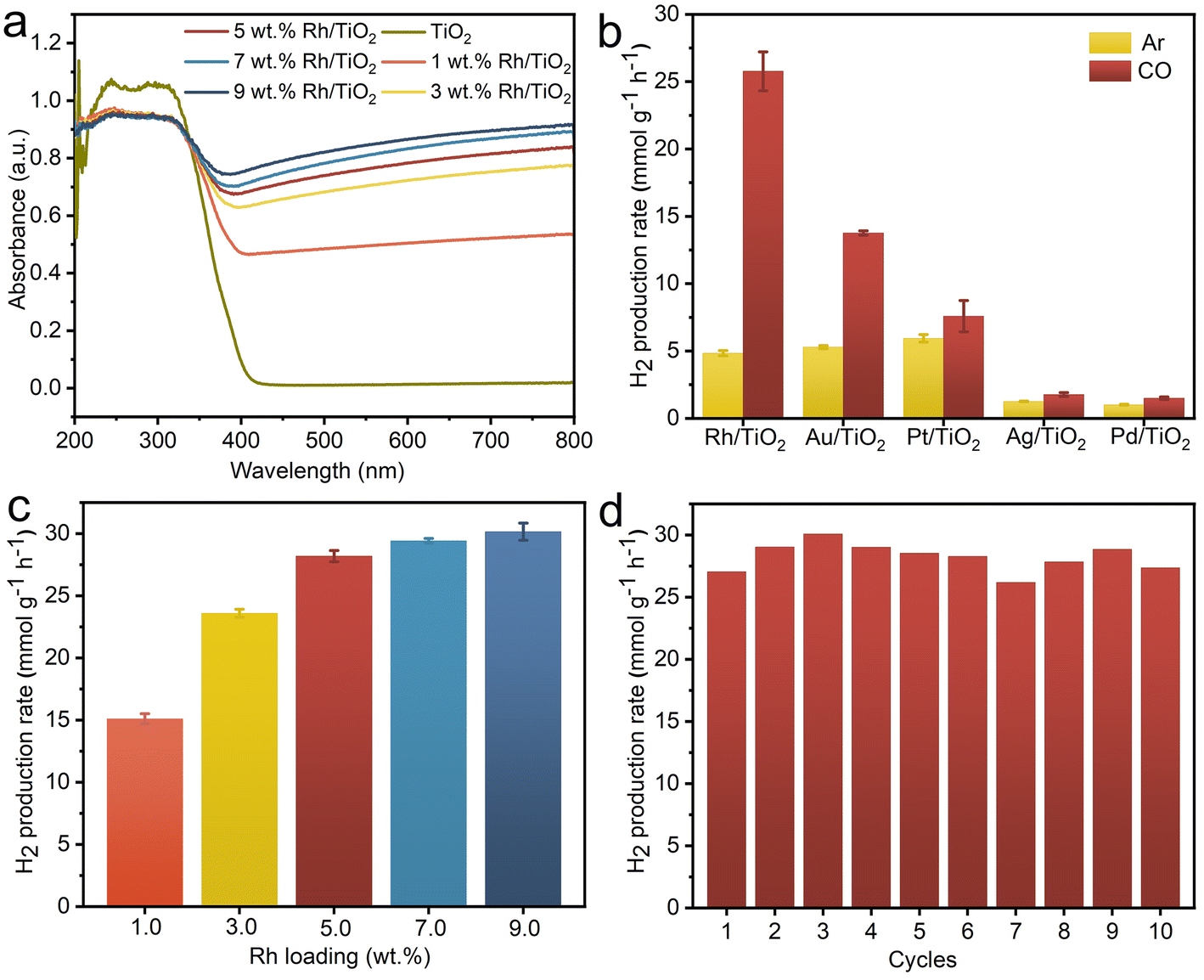 | ||
| Fig. 3 (a) Absorption spectra for TiO2 and x wt% Rh/TiO2; (b) triphase photocatalytic H2 production rate for 5 wt% M/TiO2 (M = Rh, Au, Ag, Pt, Pd) in Ar or CO atmosphere; (c) photocatalytic H2 production rate as a function of the loading amount of Rh in Rh/TiO2; (d) cycling tests for Rh/TiO2. Reaction conditions: gas phase = 0.2 MPa of CO, liquid phase = 10 mL of H2O, solid phase = 2 mg of photocatalyst immobilized on GDL (4 cm in diameter), light source = 365 nm LED (300 mW cm−2). Error bars represent the standard deviation from at least three independent measurements. | ||
The loading amount of Rh within Rh/TiO2 significantly impacts its photocatalytic WGS efficiency. As depicted in Fig. 3c, the H2 production rate escalates progressively from 15.10 to 28.17 mmol g−1 h−1 with Rh loadings escalating from 1 wt% to 5 wt%. However, augmenting the Rh content beyond 5 wt% only marginally enhances the H2 production rate. Consequently, 5 wt% Rh-loaded Rh/TiO2 was adopted for subsequent experiments. Subsequently, a photostability assessment was conducted on Rh/TiO2 within the G–L–S photocatalytic system. Under continuous irradiation for 5 hours, the H2 production rate shows a linear correlation with irradiation time, culminating in a cumulative yield of 111.52 mmol g−1 (Fig. S7†). Additionally, Fig. 3d underscores that the H2 production activity of the Rh/TiO2 photocatalyst remains consistent after 10 photocatalytic cycles (1 h for each cycle, 10 h in total). Notably, the structure and morphology of the Rh/TiO2 photocatalyst show no obvious change after the cycling test (Fig. S8 and S9†), signify its commendable photostability. The contact angle of water droplets on the GDL porous substrate did not show a significant change after the cycling experiment (Fig. S10†), indicating its stable hydrophobicity.
The photocatalytic WGS reaction was examined across various triphase and diphase systems using Rh/TiO2 as the photocatalyst. Schematic illustrations of the G–S and L–S diphase system and the G–L–S triphase system are provided in Fig. 4a–c. For the G–S system (Fig. 4a), which is a configuration frequently employed in conventional (photo)thermal WGS reactions, the CO reactant can directly diffuse to the photocatalyst from the gas phase. The water concentration within this gas phase is standardized at 0.95 mM, representing 74% of the relative humidity at room temperature. Fig. 4b showcases the L–S system where Rh/TiO2 is immobilized onto a quartz plate instead of the GDL porous substrate. Here, CO from the gas phase dissolves and undergoes a long-range diffusion process within the aqueous phase to reach the photocatalyst. In the G–L–S system, as depicted in Fig. 4c, gaseous CO traverses to the photocatalyst's surface via the hydrophobic GDL pores. Simultaneously, a high interfacial water concentration (equivalent to 55.6 M for pure water) is maintained at ambient conditions. This ensures an optimized diffusion–reaction process for both CO and H2O molecules during the photocatalytic WGS reaction.
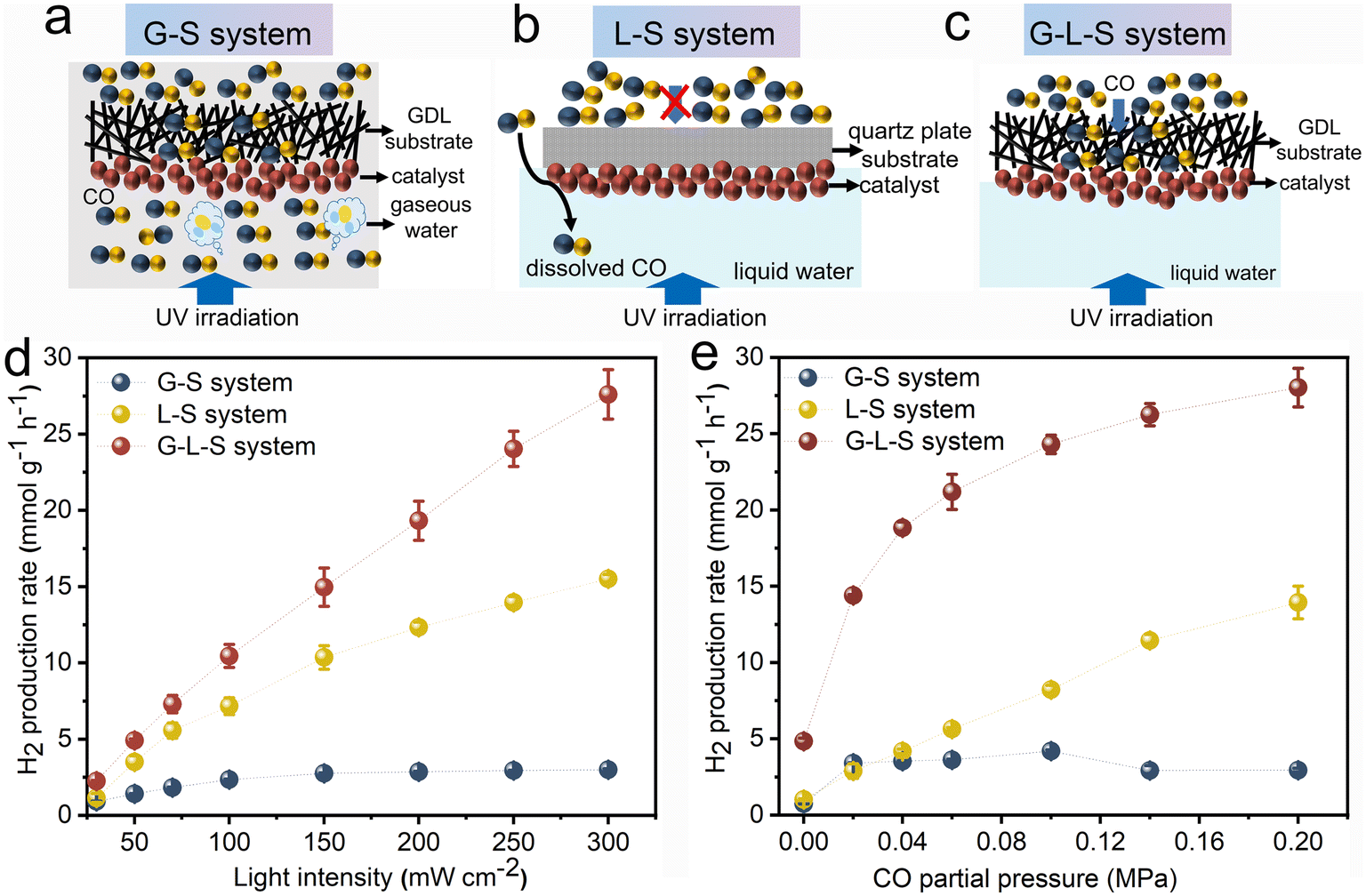 | ||
| Fig. 4 Schematic illustration of (a) G–S, (b) L–S, and (c) G–L–S systems; (d) light intensity-dependent photocatalytic H2 production rate over Rh/TiO2 at a CO partial pressure of 0.2 MPa; (e) CO partial pressure-dependent photocatalytic H2 production rate at a light intensity of 300 mW cm−2. Error bars represent the standard deviation from at least three independent measurements. | ||
The photocatalytic WGS reaction activities across the three systems were investigated using light intensity and CO partial pressure as variables. As depicted in Fig. 4d, the light intensity-dependent behaviour of the photocatalytic WGS reaction was evaluated at a constant CO partial pressure of 0.2 MPa. Within the G–S system, the WGS reaction performance remained largely insubstantial across the tested light intensities (30–300 mW cm−2). This minimal activity could be attributed to certain limiting factors. In the L–S system, the H2 production rate surged to 7.16 mmol g−1 h−1 at an intensity of 100 mW cm−2, but only experienced a twofold increase upon elevating the intensity to 300 mW cm−2. These findings imply that at higher light intensities, the rate-determining step potentially shifts to the mass transfer of CO, given the rapid depletion of reactants. Conversely, within the G–L–S system, the H2 production rate exhibited a nearly linear augmentation even under elevated light intensities. This behaviour suggests that the rate-limiting steps are more associated with the photo-generation and subsequent separation of charge carriers, rather than the mass transfer process. Notably, the H2 production rate within the triphase system reached 27.60 mmol g−1 h−1 at 300 mW cm−2, marking an approximately twofold and tenfold enhancement compared to the L–S and G–S diphase systems, respectively.
Subsequently, the impact of CO partial pressure on the photocatalytic WGS reaction was assessed with a consistent light intensity set at 300 mW cm−2, as illustrated in Fig. 4e. In the G–S system, escalating CO partial pressures did not yield any discernible increase in activity, aligning with prior observations. In contrast, the L–S system showcased a nearly proportional correlation between the H2 production rate and CO partial pressure, peaking at 13.93 mmol g−1 h−1 at 0.2 MPa. These findings indicate an increased solubility of CO molecules in the aqueous phase and an enhanced mass transfer, both of which correlate positively with elevated CO partial pressures in the connected gas phase. Within the G–L–S system, the H2 production rate witnessed a marked surge at modest CO partial pressures, outperforming the L–S system at a 10 times reduced CO partial pressure of 0.02 MPa (14.37 mmol g−1 h−1). This underscores the G–L–S system's advantage in selectively converting low-concentration CO, positioning potential applications like CO removal in fuel cells (for hydrogen purification) and mitigating CO poisoning risks in confined environments. However, the rate of enhancement decelerates at elevated CO partial pressures, indicative of a transition in the rate-determining step, potentially shifting from CO mass transfer to other processes, such as the photo-generation and subsequent separation of charge carriers.
A three-dimensional finite element method (FEM) simulation was employed to computationally elucidate the interfacial mass transfer of CO and H2O across various reaction systems. As depicted in the left panel of Fig. 5, three distinct models were constructed to represent the G–S (top), L–S (middle), and G–L–S (bottom) systems. In the G–L system, the simulation focused on the local H2O concentration, with an initial concentration set at 0.95 mM based on experimental data. For both the L–S and G–L–S systems, the simulation targeted the local CO concentration, with the initial concentration determined by the saturated solubility of CO within the aqueous phase (1.01 mM).23 The FEM simulation aimed to capture the diffusion–reaction dynamic process occurring at the reaction interface. To align with the optimized photocatalytic H2 production rates observed in the G–L–S system (approximately 10 mM s−1), the consumption rates of CO and H2O within the photocatalytic layer region were modulated, ranging from 5 to 15 mM s−1.
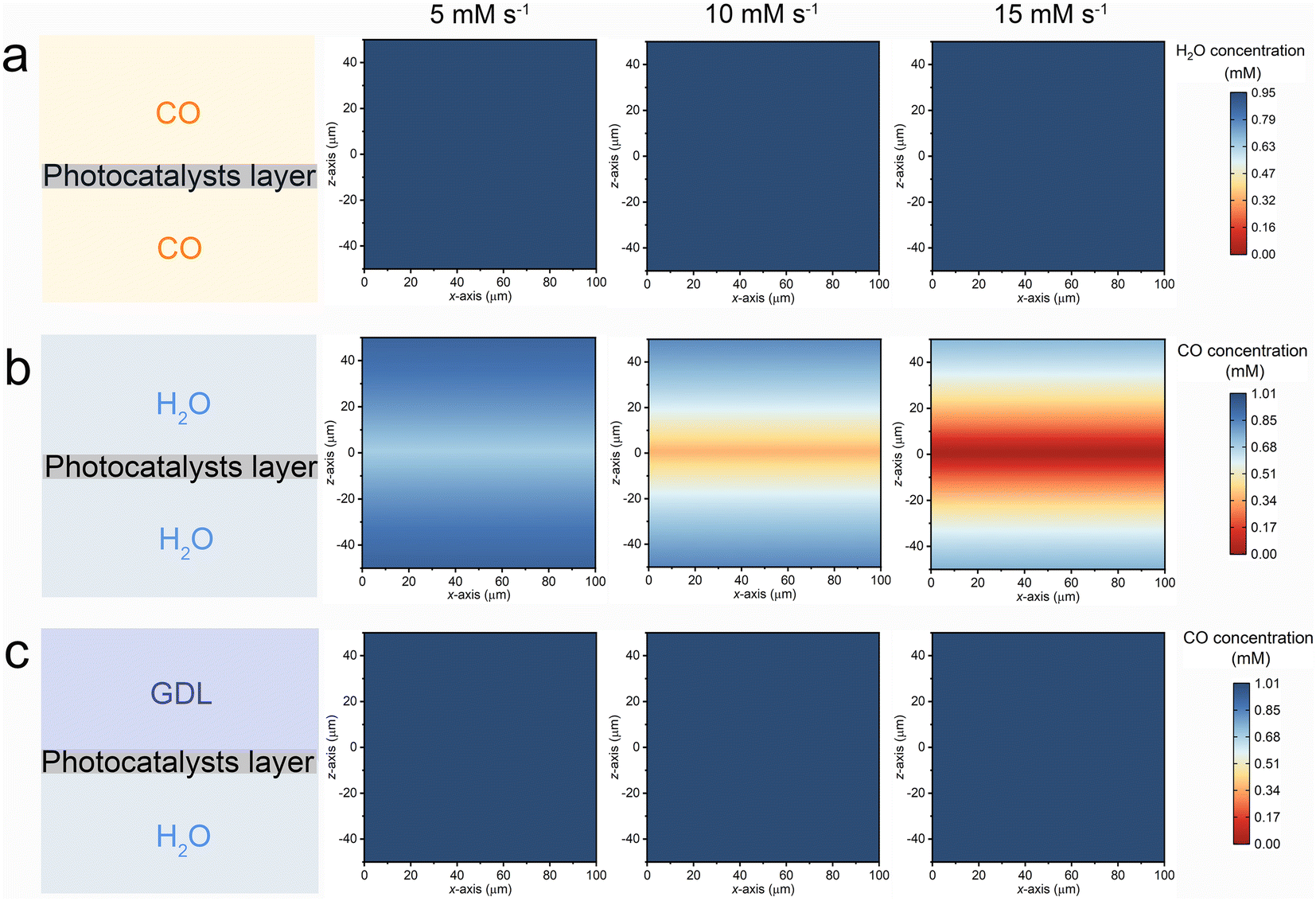 | ||
| Fig. 5 Schematic diagrams and FEM simulation results of (a) G–S, (b) L–S, and (c) G–L–S systems. The colour scale in (a) represents the water vapor concentration, in (b) and (c) represents the CO concentration. | ||
The xz diagrams depicting the simulated reactant concentrations for the G–S, L–S, and G–L–S systems are illustrated in Fig. 5a–c, respectively. In the G–S system (Fig. 5a), the local water vapor concentration within the photocatalyst layer exhibits minimal decline even at escalated H2O consumption rates. This observation implies that the subdued H2 production in the G–S setup might not be directly attributable to interfacial diffusion or mass transfer challenges of water molecules. Instead, it could be linked to the adverse impact of water vapor, characterized by a weak hydrogen bond network, on H2 production, as previously suggested.22 In the L–S system (Fig. 5b), where the photocatalyst layer is submerged within a bulk water phase, the local CO concentration surrounding the catalyst surface diminishes markedly with the rising CO consumption rate. However, the G–L–S configuration (Fig. 5c) showcases a consistent CO concentration even at elevated consumption rates. This stability underscores the function of the triphase interface in maintaining a robust supply of CO molecules within the local reaction microenvironment. Such consistent CO availability ensures efficient H2 production via the photocatalytic WGS pathway, even when subjected to diluted CO conditions.
3 Conclusions
In summary, we have developed a G–L–S triphase photocatalytic system utilizing M/TiO2 (where M = Rh, Au, Ag, Pt, and Pd) immobilized on a hydrophobic porous GDL for the WGS reaction under ambient conditions. Among the tested catalysts, Rh/TiO2 exhibited the most impressive photocatalytic H2 production rate, registering at 27.60 mmol g−1 h−1. This catalyst not only showcased commendable photostability but also displayed remarkable enhancements, even when utilizing gas sources with low CO concentrations. Crucially, the G–L–S configuration notably enhances the mass transfer of both CO and H2O to the photocatalyst surface, which appears to be the primary rate-determining step. Consequently, this design exhibited H2 production rates that were 10 and 2 times higher than those observed in conventional G–S and L–S diphase systems, respectively. FEM simulations provided insights into the interfacial mass transfer process of CO and H2O. These findings underscore the triphase system's distinct advantage in ensuring a consistent CO supply to the photocatalyst layer, especially under low-concentration conditions. Overall, our research underscores the promising potential of the G–L–S triphase photocatalytic system, highlighting its capability to optimize the diffusion–reaction interplay in the WGS process, thereby achieving superior H2 production efficiencies under mild conditions.4 Experimental section
4.1 Chemicals and materials
TiO2 (P25) was purchased from Degussa AG. RhCl3·3H2O was obtained from Bide Pharmatech Ltd. (China). HAuCl4·4H2O, H2PtCl6·6H2O, and Na2PdCl4 were purchased from Aladdin (Shanghai, China). AgNO3 was purchased from Sinopharm Chemical Reagent Corporation (Shanghai, China). NaBH4 was purchased from Guangdong Guanghua Chemical Factory Co., Ltd. Polytetrafluoroethylene GDL with a thickness of 30 μm and a diameter of 47 mm (Chuangwei Filter Media, China) was used as a porous support for photocatalyst immobilization. All chemicals were analytically pure and had not been further purified. Deionized water was used in the whole experiments.4.2 Photocatalyst preparation
Preparation of M/TiO2: M/TiO2 with varying weight loadings were synthesized using the NaBH4 reduction method as described by Zhang.34 Specifically, for the preparation of Rh/TiO2, 200 mg of TiO2 was dispersed in 100 mL of water using ultrasonication for 30 minutes. Subsequently, different volumes of an aqueous RhCl3·3H2O solution (with a concentration of 10 mg mL−1 for Rh) were introduced to the aforementioned dispersion and sonicated for another 30 minutes. Following this, a surplus NaBH4 solution (concentration: 2.5 mg mL−1) was meticulously added dropwise to the suspension with continuous stirring. After an additional hour of stirring, the samples underwent centrifugation, followed by rinsing with neutral water. They were then subjected to overnight vacuum freeze-drying. The resulting catalysts were denoted as x wt% Rh/TiO2 (x = 1, 3, 5, 7, and 9, representing the theoretical weight ratio of Rh loadings). A consistent synthesis approach was adopted for other M/TiO2 variants, employing appropriate aqueous solutions of HAuCl4·4H2O, AgNO3, H2PtCl6·6H2O, and Na2PdCl4 for the synthesis of Au/TiO2, Ag/TiO2, Pt/TiO2, and Pd/TiO2, respectively.Preparation of M/TiO2-GDL: taking Rh/TiO2 as an example, the ethanol dispersion of Rh/TiO2 (concentration: 0.1 mg mL−1) was subjected to sonication for approximately 30 minutes. Subsequently, 20 mL of this suspension was applied to the GDL surface via vacuum filtration, resulting in a circular photocatalyst layer with a diameter of 4 cm. After a 1 hour drying period in an oven, Rh/TiO2-GDL with a loading density of 0.16 mg cm−2 was obtained (Fig. S11a†). This layer was then utilized for both the G–L–S and G–S systems. For the L–S system, a 20 mL ethanol dispersion of Rh/TiO2 (concentration: 0.1 mg mL−1) was dripped onto the surface of a circular quartz substrate (4 cm in diameter), followed by a 1 hour drying process in an oven (Fig. S11b†).
4.3 Characterization
The structure and crystallization of the particles were provided by XRD (Bruker AXSD8 Advance, Germany). Sample morphologies were examined using SEM (S4800, Hitachi, Japan) and TEM (JEOL-2100F, Japan), both of which were equipped with EDS for elemental analysis. The diffuse reflection spectra of the photocatalyst powders were measured using an ultraviolet-visible spectrophotometer (Cary 7000, Agilent, USA) with an integrating sphere attachment. A contact angle system (OCA20, Dataphysics, Germany) was used to measure the contact angles, with the probe liquid being a 2.0 μL droplet of water.4.4 Photocatalytic measurements
Photocatalytic WGS reaction measurements: The photocatalytic activities of the synthesized samples were evaluated using a custom-built photocatalytic reactor (Fig. S12 and S13†). A 365 nm LED (PLS-LED100C, PerfectLight, China), paired with a λ < 400 nm filter (Xujiang Electromechanical Plant, Nanjing, China), served as the illumination source. In the G–S configuration, the Rh/TiO2-GDL was positioned within the reactor. CO (99.99%) was then bubbled through a water tank, producing a CO/water gas mixture under atmospheric conditions. The humidified CO was introduced into the reactor at a flow rate of 400 mL min−1 for 30 minutes, achieving a water vapor concentration of 0.95 mM (equivalent to 74% relative humidity). Subsequently, the reaction gas was introduced to elevate the pressure to 0.2 MPa. For the L–S system, the Rh/TiO2-immobilized quartz plate was submerged in 10 mL of water. For the G–L–S system, the Rh/TiO2-GDL was positioned above a 10 mL water surface. Both L–S and G–L–S systems were pressurized to 0.20 MPa with CO gas and then sealed. After a 1 hour irradiation period, the WGS reaction products were quantified using a gas chromatograph (GC-2014C, Shimadzu, Japan) equipped with a TCD detector, molecular sieve 5, and utilizing N2 as the carrier gas. For the photocatalytic cycling stability experiment, reaction products were quantitatively analysed using a gas chromatograph after 1 hour of irradiation. Subsequently, the gas within the photocatalytic reactor was entirely exhausted and replaced with CO for the subsequent cycle. This cycle was repeated ten times.Light intensity and CO partial pressure modulation: Experiments were conducted to assess the light intensity dependence in the photocatalytic WGS reaction, maintaining a constant CO partial pressure of 0.2 MPa. The varying light intensities were achieved by modulating the output power of the 365 nm LED. Additionally, tests at different CO partial pressures were performed under a steady light intensity of 300 mW cm−2. The CO partial pressures were adjusted by manipulating the CO/Ar volume ratio at a consistent total pressure of 0.2 MPa.
4.5 FEM simulation
The mass transfer and consumption behaviours of CO and H2O in the G–S, L–S, and G–L–S systems were analysed via Fick's second law using FEM simulations conducted in COMSOL 5.4. Fick's second law is as follows: | (1) |
 | (2) |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for financial support from the National Key R&D Program of China (2021YFA1500803), the National Natural Science Foundation of China (51825205, 52120105002, 22272190, 22209190, 22088102), the Beijing Natural Science Foundation (2222035), the CAS Project for Young Scientists in Basic Research (YSBR-004), the DNL Cooperation Fund, CAS (DNL202016), the China Postdoctoral Science Foundation (2021M703288, 2021T150665), the Young Elite Scientist Sponsorship Program by CAST (2021QNRC001), and the Youth Innovation Promotion Association of the CAS.References
- M. G. Rasul, M. A. Hazrat, M. A. Sattar, M. I. Jahirul and M. J. Shearer, The future of hydrogen: Challenges on production, storage and applications, Energy Convers. Manage., 2022, 272, 116326 CrossRef CAS.
- W. H. Chen and C. Y. Chen, Water gas shift reaction for hydrogen production and carbon dioxide capture: A review, Appl. Energy, 2020, 258, 114078 CrossRef CAS.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Perspectives on low-temperature electrolysis and potential for renewable hydrogen at scale, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Hydrogen production reactions from carbon feedstocks fossil fules and biomass, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, An overview of hydrogen production technologies, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- C. Ratnasamy and J. P. Wagner, Water gas shift catalysis, Catal. Rev.: Sci. Eng., 2009, 51, 325–440 CrossRef CAS.
- A. Ambrosi and S. E. Denmark, Harnessing the power of the water-gas shift reaction for organic synthesis, Angew. Chem., Int. Ed., 2016, 55, 12164–12189 CrossRef CAS PubMed.
- Y. X. Tong, L. Z. Song, S. B. Ning, S. X. Ouyang and J. H. Ye, Photocarriers-enhanced photothermocatalysis of water-gas shift reaction under H2-rich and low-temperature condition over CeO2/Cu1.5Mn1.5O4 catalyst, Appl. Catal., B, 2021, 298, 120551 CrossRef CAS.
- S. Y. Yao, X. Zhang, W. Zhou, R. Gao, W. Q. Xu, Y. F. Ye, L. L. Lin, X. D. Wen, P. Liu, B. B. Chen, E. Crumlin, J. G. Guo, Z. J. Zuo, W. Z. Li, J. L. Xie, I. Lu, C. Kiely, L. Gu, G. Shi, J. Rodriguez and D. Ma, Atomic-layered Au clusters on α-MoC as catalysts for the low-temperature water-gas shift reaction, Science, 2017, 357, 389–393 CrossRef CAS PubMed.
- Z. H. Zhang, X. Y. Chen, J. C. Kang, Z. Y. Yu, J. Tian, Z. M. Gong, A. P. Jia, R. You, K. Qian, S. He, B. T. Teng, Y. Cui, Y. Wang, W. H. Zhang and W. X. Huang, The active sites of Cu-ZnO catalysts for water gas shift and CO hydrogenation reactions, Nat. Commun., 2021, 12, 4331 CrossRef CAS PubMed.
- K. Xu, C. Ma, H. Yan, H. Gu, W. W. Wang, S. Q. Li, Q. L. Meng, W. P. Shao, G. H. Ding, F. R. Wang and C. J. Jia, Catalytically efficient Ni-NiOx-Y2O3 interface for medium temperature water-gas shift reaction, Nat. Commun., 2022, 13, 2443 CrossRef CAS PubMed.
- Z. H. Cui, S. Song, H. B. Liu, Y. T. Zhang, F. Gao, T. Ding, Y. Tian, X. B. Fan and X. G. Li, Synergistic effect of Cu+ single atoms and Cu nanoparticles supported on alumina boosting water-gas shift reaction, Appl. Catal., B, 2022, 313, 121468 CrossRef CAS.
- Z. H. Zhang, S. S. Wang, R. Song, T. Cao, L. F. Luo, X. Y. Chen, Y. X. Gao, J. Q. Lu, W. X. Li and W. X. Huang, The most active Cu facet for low-temperature water gas shift reaction, Nat. Commun., 2017, 8, 488 CrossRef PubMed.
- Y. J. Zhang, C. Q. Chen, X. Y. Lin, D. L. Li, X. H. Chen, Y. Y. Zhan and Q. Zheng, CuO/ZrO2 catalysts for water-gas shift reaction: Nature of catalytically active copper species, Int. J. Hydrogen Energy, 2014, 39, 3746–3754 CrossRef CAS.
- J. L. Santos, T. R. Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, Gold promoted Cu/ZnO/Al2O3 catalysts prepared from hydrotalcite precursors: Advanced materials for the WGS reaction, Appl. Catal., B, 2017, 201, 310–317 CrossRef CAS.
- L. Torrente-Murciano and F. R. Garcia-Garcia, Effect of nanostructured support on the WGSR activity of Pt/CeO2 catalysts, Catal. Commun., 2015, 71, 1–6 CrossRef CAS.
- A. Kubacka, M. Fernandez-García and G. Colón, Advanced nanoarchitectures for solar photocatalytic applications, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Titanium dioxide-based nanomaterials for photocatalytic fuel generations, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS.
- L. K. Zhao, Y. H. Qi, L. Z. Song, S. B. Ning, S. X. Ouyang, H. Xu and J. H. Ye, Solar-driven water-gas shift reaction over CuOx/Al2O3 with 1.1% of light-to-energy storage, Angew. Chem., Int. Ed., 2019, 58, 7708–7712 CrossRef CAS PubMed.
- F. Sastre, M. Oteri, A. Corma and H. García, Photocatalytic water gas shift using visible or simulated solar light for the efficient, room-temperature hydrogen generation, Energy Environ. Sci., 2013, 6, 2211–2215 RSC.
- N. Liu, M. Xu, Y. S. Yang, S. M. Zhang, J. Zhang, W. L. Wang, L. R. Zheng, S. Hong and M. Wei, Auδ--Ov-Ti3+ interfacial site: catalytic active center toward low-temperature water gas shift reaction, ACS Catal., 2019, 9, 2707–2717 CrossRef CAS.
- X. C. Ma, Y. L. Shi, J. Y. Liu, X. T. Li, X. F. Cui, S. J. Tan, J. Zhao and B. Wang, Hydrogen-bond network promotes water splitting on the TiO2 surface, J. Am. Chem. Soc., 2022, 144, 13565–13573 CrossRef CAS.
- K. S. Davidge, R. Motterlini, B. E. Mann, J. L. Wilson and R. K. Poole, Carbon monoxide in biology and microbiology: Surprising roles for the “detroit perfume”, Adv. Microb. Physiol., 2009, 56, 85–167 CrossRef CAS PubMed.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, Cambridge university press, Cambridge, 3rd edn, 2009 Search PubMed.
- H. N. Huang, R. Shi, Z. H. Li, J. Q. Zhao, C. L. Su and T. R. Zhang, Triphase photocatalytic CO2 reduction over silver-decorated titanium oxide at a gas-water boundary, Angew. Chem., Int. Ed., 2022, e202200802 CAS.
- X. Y. Xiong, Z. P. Wang, Y. Zhang, Z. H. Li, R. Shi and T. R. Zhang, Wettability controlled photocatalytic reactive oxygen generation and Klebsiella pneumoniae inactivation over triphase systems, Appl. Catal., B, 2020, 264, 118518 CrossRef CAS.
- L. J. Li, L. P. Xu, Z. F. Hu and J. C. Yu, Enhanced mass transfer of oxygen through a gas-liquid-solid interface for photocatalytic hydrogen peroxide production, Adv. Funct. Mater., 2021, 31, 2106120 CrossRef CAS.
- H. Song, X. G. Meng, Z. J. Wang, Z. Wang, H. L. Chen, Y. X. Weng, F. Ichihara, M. Oshikiri, T. Kako and J. H. Ye, Visible-light-mediated methane activation for steam methane reforming under mild conditions: A case study of Rh/TiO2 catalysts, ACS Catal., 2018, 8, 7556–7565 CrossRef CAS.
- Y. J. Chen, S. F. Ji, W. M. Sun, Y. P. Lei, Q. C. Wang, A. Li, W. X. Chen, G. Zhou, Z. D. Zhang, Y. Wang, L. R. Zheng, Q. H. Zhang, L. Gu, X. D. Han, D. S. Wang and Y. D. Li, Engineeringthe atomic interface with single platinum atoms for enhanced photocatalytic hydrogen production, Angew. Chem., Int. Ed., 2020, 59, 1295–1301 CrossRef CAS.
- Y. L. Chen, X. Q. Liu, L. Hou, X. R. Guo, R. W. Fu and J. M. Sun, Construction of covalent bonding oxygen-doped carbon nitride/graphitic carbon nitride Z-scheme heterojunction for enhanced visible-light-driven H2 evolution, Chem. Eng. J., 2020, 383, 123132 CrossRef CAS.
- C. C. Han, P. F. Su, B. H. Tan, X. G. Ma, H. Lv, C. Y. Huang, P. Wang, Z. F. Tong, G. Li, Y. Z. Huang and Z. F. Liu, Defective ultra-thin two-dimensional g-C3N4 photocatalyst for enhanced photocatalytic H2 evolution activity, J. Colloid Interface Sci., 2021, 581, 159–166 CrossRef CAS PubMed.
- H. F. Wei, H. Liu, L. Yu, M. Zhang, Y. L. Zhang, J. C. Fan, X. J. Cui and D. H. Deng, Alloying Pd with Cu boosts hydrogen production via room-temperature electrochemical water-gas shift reaction, Nano Energy, 2022, 102, 107704 CrossRef CAS.
- X. Zhang, M. T. Zhang, Y. C. Deng, M. Q. Xu, L. Artiglia, W. Wen, R. Gao, B. B. Chen, S. Y. Yao, X. C. Zhang, M. Peng, J. Yan, A. W. Li, Z. Jiang, X. Y. Gao, S. F. Cao, C. Yang, A. J. Kropf, J. N. Shi, J. L. Xie, M. S. Bi, J. A. van Bokhoven, Y. W. Li, X. D. Wen, M. Flytzani-Stephanopoulos, C. Shi, W. Zhou and D. Ma, A stable low-temperature H2-production catalyst by crowding Pt on alpha-MoC, Nature, 2021, 589, 396–401 CrossRef CAS PubMed.
- P. Wang, J. Q. Zhao, R. Shi, X. R. Zhang, X. D. Guo, Q. Dai and T. R. Zhang, Efficient photocatalytic aerobic oxidation of bisphenol A via gas-liquid-solid triphase interfaces, Mater. Today Energy, 2022, 23, 100908 CrossRef CAS.
- I. Amdur and L. M. Shuler, Diffusion coefficients of the systems CO-CO and CO-N2, J. Chem. Phys., 1963, 38, 188–192 CrossRef CAS.
- D. H. Shou, J. T. Fan, M. F. Mei and F. Ding, An analytical model for gas diffusion though nanoscale and microscale fibrous media, Microfluid. Nanofluid., 2014, 16, 381–389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3im00135k |
| This journal is © Institute of Process Engineering of CAS 2024 |