
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous organic crystals crosslinked by free-radical reactions†
Krishanu
Samanta
a,
Jiashan
Mi
bc,
Albert D.
Chen
a,
Fangzhou
Li
d,
Richard J.
Staples
 e,
Aaron J.
Rossini
bc and
Chenfeng
Ke
*ad
e,
Aaron J.
Rossini
bc and
Chenfeng
Ke
*ad
aDepartment of Chemistry, Dartmouth College, 41 College Street, Hanover, NH 03755, USA. E-mail: cke@wustl.edu
bDepartment of Chemistry, Iowa State University, 2438 Pammel Drive, Ames, IA 50011, USA
cUS DOE Ames National Laboratory, Ames, Iowa, USA 50011
dDepartment of Chemistry, Washington University in St. Louis, One Brookings Drive, St. Louis, MO 63130, USA
eDepartment of Chemistry, Michigan State University, 578 S. Shaw Lane, East Lansing, MI 48824, USA
First published on 24th June 2024
Abstract
Two hydrogen-bonded crosslinked organic frameworks (HCOFs) were synthesized via free radical reactions utilizing butadiene and isoprene as crosslinkers. These HCOFs exhibit high crystallinity, enabling detailed structural characterization via single-crystal X-ray diffraction analysis. Subsequently, one of the olefin-rich HCOFs was converted to a hydroxylated framework through hydroboration–oxidation while maintaining the high crystallinity.
Porous materials1 are attractive for their diverse applications for substrate storage and separation,2 catalysis,3 ion transport,4 and sensing.5 Among them, porous framework materials like metal–organic frameworks (MOFs),6 covalent organic frameworks (COFs),7 and hydrogen-bonded organic frameworks (HOFs)8 have been extensively investigated for their adjustable pore characteristics via building block variation. Establishing a fundamental understanding of the structure–property relationship between these frameworks and their substrates hinges on atomic-level details discernable through single-crystal X-ray diffraction (SCXRD).9 Furthermore, achieving high chemical stability in these frameworks is crucial for their applications in various environments.10 However, enhancing chemical robustness and maintaining structural detail often presents a trade-off due to the reversible bond formation processes during the synthesis of these frameworks.
Recently, hydrogen-bonded crosslinked organic frameworks (HCOFs) have emerged, featuring high crystallinity and chemical stability.11 The synthesis of HCOFs involves the design of molecular building blocks with hydrogen bonding directing groups and reactive arms. These building blocks self-assemble via hydrogen bonds to form potentially porous networks. Unlike HOFs, these networks do not require high stability after solvent removal because the subsequent photo-crosslinkings with dithiol through the irreversible thiol-ene/yne reactions reinforce them as HCOFs. These HCOFs showed high performance in iodine/iodide removal for water purification,12 served as solid-state hosts for photo-switching,13 and facilitated boron trifluoride uptake for cationic vinyl ether polymerizations.14 However, the synthesis of HCOFs has been limited to thioether crosslinked variants. Diversifying the crosslinking methods could significantly broaden the potential applications for HCOFs.
In this study, we unveiled a new approach to synthesize single-crystalline HCOFs through free-radical crosslinking. By integrating styrene groups into the carboxylic acid-based monomer, we achieved co-crystallization of the monomer with triallyl-benzamide (TAB) through complementary hydrogen-bonding interactions. This self-assembly process produced porous co-crystals, which were subsequently subjected to free-radical reactions in the presence of butadiene and isoprene, resulting in HCOF-106 and HCOF-107 (Scheme 1). Interestingly, the reaction occurred among the diene crosslinker, the styrene units, and the allyl groups in these crosslinked HCOFs. The abundant olefin groups at the pore surfaces facilitated further modifications of HCOF-107, transforming its hydrophobic pore surface to a hydrophilic one through hydroboration–oxidation. This modification inversely affected the vapor sorption properties of these HCOFs, demonstrating the versatility of post-synthetic modification approach15 in tailoring material properties for specific vapors.
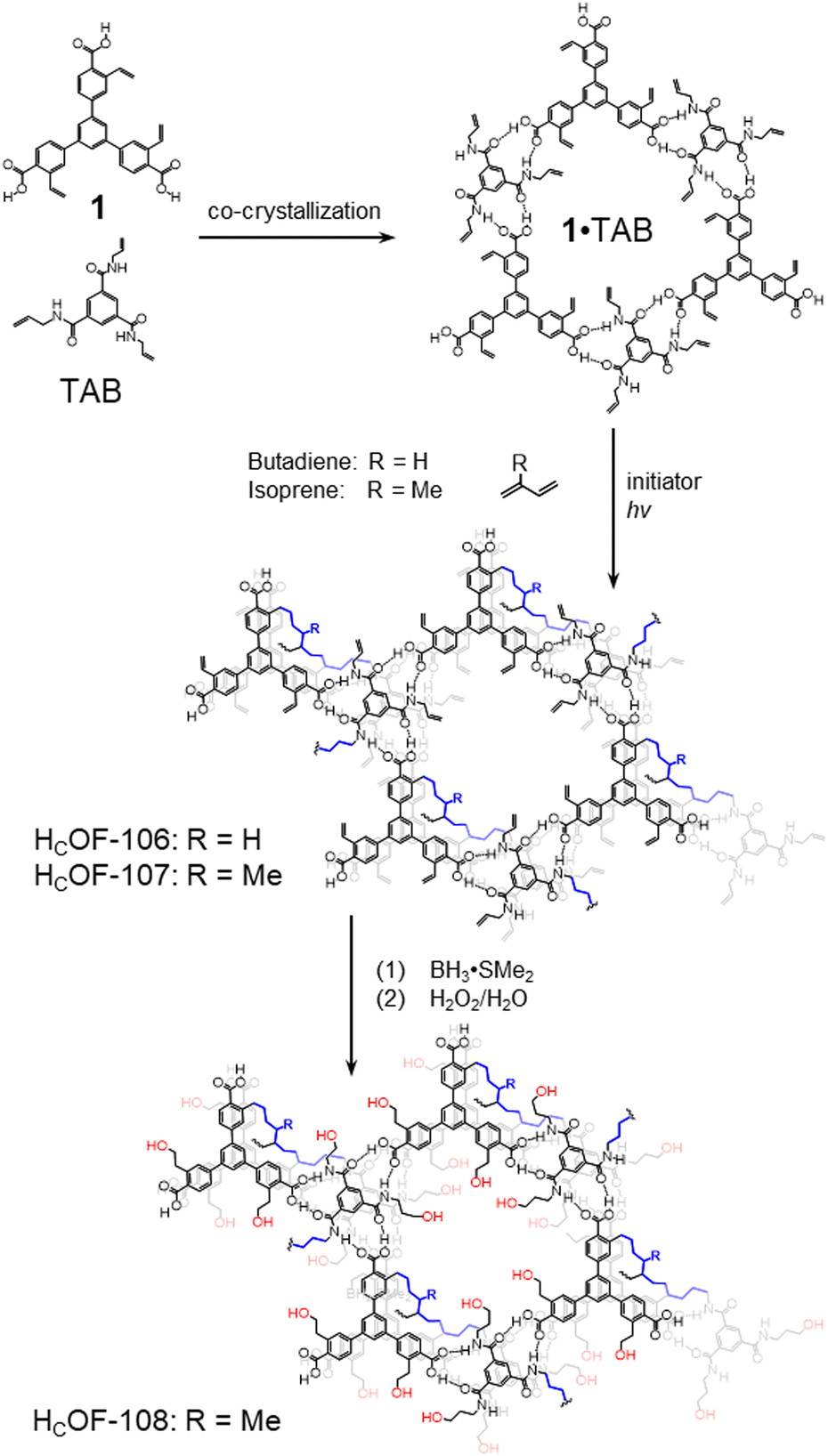 | ||
| Scheme 1 Synthesis of HCOF-106 and HCOF-107 through free-radical reactions with butadiene and isoprene. Post-modification of the single-crystalline HCOF-107 affords its hydroxylated derivative HCOF-108. | ||
We chose tris-(4-carboxyl phenyl)-benzene and TAB (Scheme 1) as the building blocks because they co-crystallized as a hydrogen-bonded network with large pores in the solid state as we reported recently.16 The styrene moieties are introduced to monomer 1 in four steps (Scheme S1, ESI†). Firstly, methyl-4-bromo-2-bromomethylbenzoate was converted to methyl-4-bromo-2-vinylbenzoate through the Wittig reaction.17 It was then reacted with 1,3,5-phenyltriboronic tris(pinacol)ester via Suzuki–Miyaura coupling followed by hydrolysis to generate monomer 1 in 49% overall yield.
Slow vapor diffusion of (cyclo)hexane into a dioxane solution of monomer 1 and TAB 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture for 7 days afforded needle-shaped co-crystals suitable for SCXRD analysis (Fig. 1). In the solid state, monomer 1 and TAB formed 1:1 co-crystals in the P
:1 mixture for 7 days afforded needle-shaped co-crystals suitable for SCXRD analysis (Fig. 1). In the solid state, monomer 1 and TAB formed 1:1 co-crystals in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S2, ESI†). In this 1·TAB co-crystal, the carboxylic acid groups of 1 form highly directional hydrogen bonds with the benzamide via a donor–acceptor to donor–donor–acceptor (DA–DDA) hydrogen bonding array.16 This array repeats among the three carboxylic acid groups of 1, forming a 2D hexagonal hydrogen bonding sheet along the b/c plane (Fig. 1a). These 2D hexagonal layers are stacked in a nearly eclipsed manner, forming 1D channels along the a-axis with pore aperture measured as 13.3 × 11.1 Å2 and 32% solvent-filled voids (Fig. 1a). Along the direction of the pores, monomer 1 and TAB are stacked with an alternative ABBA fashion, with the π–π distances measured as 3.7, 4.2, and 3.5 Å (Fig. 1b). Three sets of styrene groups of 1 in adjacent layers are packed close to each other with measured olefin-to-olefin distances as 3.83, 8.4, and 9.21 Å (Fig. 1b). In comparison, the closest olefin-to-olefin distance in the 2D layer is measured as 13.5 Å (Fig. S29a, ESI†). The various olefin distances in the 1·TAB co-crystal could result in different reactivities for free-radical crosslinking. We also measured the styrene to allyl group distances with the intra-2D-layer distances of 4.26–4.65 Å (Fig. S29c, ESI†) and inter-2D-layer distances of 3.63–7.86 Å (Fig. S29d, ESI†). Therefore, we chose butadiene and isoprene as the crosslinkers to connect these olefin groups in the 1·TAB co-crystal for HCOF synthesis. Their versatile reactivity for 1,2- or 1,4-addition may accommodate different olefin-to-olefin distances in the co-crystal.
space group (Table S2, ESI†). In this 1·TAB co-crystal, the carboxylic acid groups of 1 form highly directional hydrogen bonds with the benzamide via a donor–acceptor to donor–donor–acceptor (DA–DDA) hydrogen bonding array.16 This array repeats among the three carboxylic acid groups of 1, forming a 2D hexagonal hydrogen bonding sheet along the b/c plane (Fig. 1a). These 2D hexagonal layers are stacked in a nearly eclipsed manner, forming 1D channels along the a-axis with pore aperture measured as 13.3 × 11.1 Å2 and 32% solvent-filled voids (Fig. 1a). Along the direction of the pores, monomer 1 and TAB are stacked with an alternative ABBA fashion, with the π–π distances measured as 3.7, 4.2, and 3.5 Å (Fig. 1b). Three sets of styrene groups of 1 in adjacent layers are packed close to each other with measured olefin-to-olefin distances as 3.83, 8.4, and 9.21 Å (Fig. 1b). In comparison, the closest olefin-to-olefin distance in the 2D layer is measured as 13.5 Å (Fig. S29a, ESI†). The various olefin distances in the 1·TAB co-crystal could result in different reactivities for free-radical crosslinking. We also measured the styrene to allyl group distances with the intra-2D-layer distances of 4.26–4.65 Å (Fig. S29c, ESI†) and inter-2D-layer distances of 3.63–7.86 Å (Fig. S29d, ESI†). Therefore, we chose butadiene and isoprene as the crosslinkers to connect these olefin groups in the 1·TAB co-crystal for HCOF synthesis. Their versatile reactivity for 1,2- or 1,4-addition may accommodate different olefin-to-olefin distances in the co-crystal.
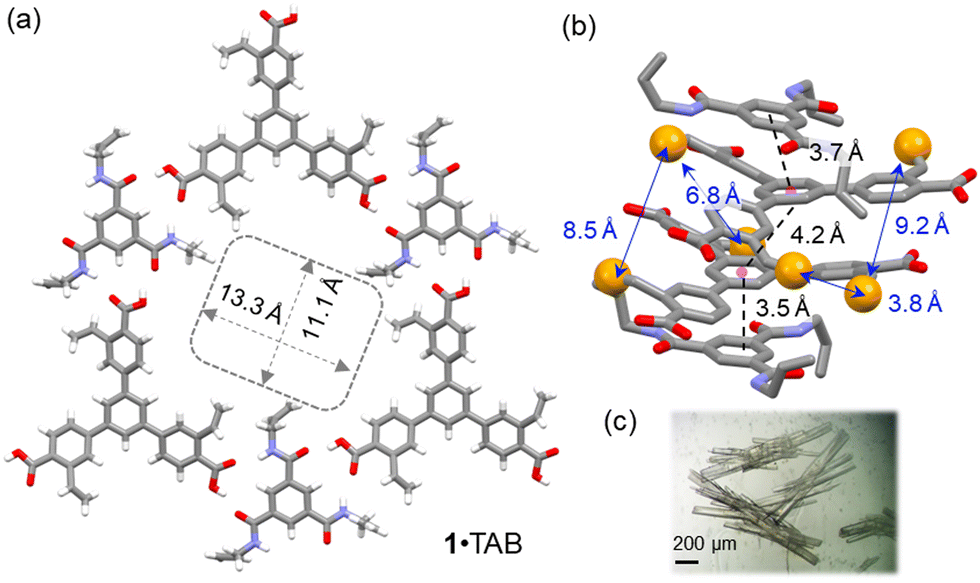 | ||
| Fig. 1 (a) Single-crystal structure of the 1·TAB co-crystal viewed along the b/c plane. (b) The ABBA-type alternative packing of 1 and TAB, viewed along the a-axis. The π–π stacking distances and distances between the terminal carbon atoms of the styrene moieties are highlighted. (c) An image of 1·TAB co-crystals. | ||
The 1·TAB co-crystals were washed extensively using hexane and then soaked in the butadiene hexane solution (15 w/w%) or neat isoprene, along with a photo-initiator 2,2-dimethoxy-2-phenylacetophenone (DMPA, 0.04 mol% to diene), for 24 h in the dark to allow extensive diffusion of dienes. The reaction vials were photo-irradiated for 48 h under the UV lamp. Interestingly, we didn’t observe significant amounts of polybutadiene or polyisoprene generated in the solution or the neat phase, and the diene conversion ratio for free-radical polymerization outside the co-crystals was too low to be detected by 1H NMR spectroscopy (Fig. S23 and S24, ESI†). The obtained crystals were washed to remove the unreacted dienes and soaked in boiling DMSO-d6. Upon cooling, a majority of the crystal samples remained insoluble (Fig. S12, ESI†). However, soluble residues account for unreacted or partially reacted monomer 1 and TAB were detected in the 1H NMR spectra (Fig. S13 and S14, ESI†). Using gravimetric analysis, approximately 60 wt% and 70 wt% of the co-crystals were estimated as crosslinked by butadiene and isoprene, affording HCOF-106 and HCOF-107, respectively (Scheme 1). In the FT-IR spectra, unreacted olefin groups attributed to 1 and TAB were observed (Fig. S15, ESI†). Compared to the solid-state 13C NMR spectrum of 1·TAB, carbon signals attributed to the styrene moieties and allyl groups were reduced (Fig. 2 and Table S5, ESI†). The addition of butadiene and isoprene crosslinkers was evident as the carbon signals for methylene units at around 25–40 ppm were found for both HCOF-106 and HCOF-107. Carbon signals for methyl groups originating from the isoprene were found at 15 ppm for HCOF-107 (Fig. 2).
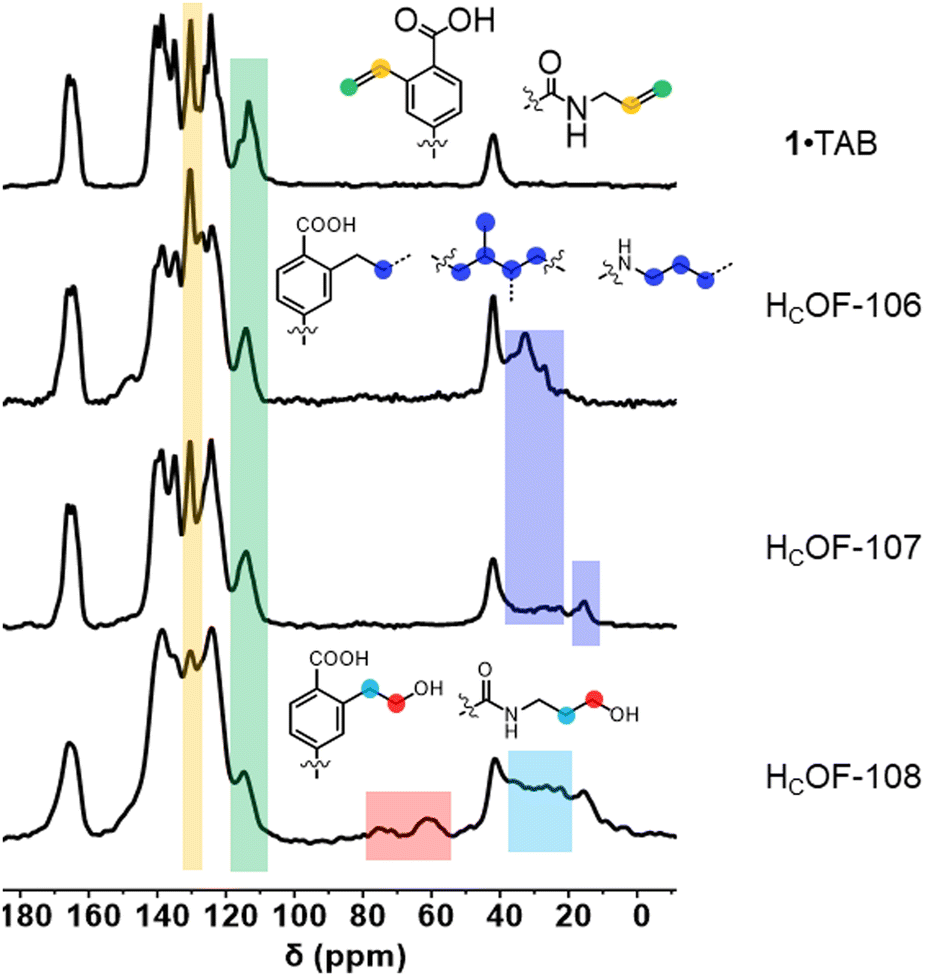 | ||
| Fig. 2 Stacked solid-state 13C NMR spectra of 1·TAB, HCOF-106, HCOF-107, and HCOF-108 from top to bottom, respectively. | ||
Powder X-ray diffraction (PXRD) analysis showed that HCOF-106 and HCOF-107 remained highly crystalline (Fig. S22, ESI†), with both suitable for SCXRD analysis. Compared to 1·TAB, HCOF-106 and HCOF-107 possess the same P space group and their unit cell volumes only expanded by 3% after the free-radical reaction (Table S2, ESI†). SCXRD analysis of HCOF-106 and HCOF-107 revealed that 50% of styrene units in 1 and 50% of allyl groups in TAB took part in crosslinking, resulting in residual olefins in the crystal lattice. To confirm this, we subjected HCOF-107 to the iodine value test following the Wijs method.18 Compared to the iodine value of 181 for the 1·TAB co-crystal, the iodine values of HCOF-107 decreased to 88, confirming the number of olefin groups decreased by ∼50% after crosslinking.
The hydrogen-bonding pattern remained unchanged in HCOF-106 and HCOF-107, along with the hexagonal pore structure (Fig. 3). After crosslinking, the pore apertures of HCOF-106 and HCOF-107 decreased to 11.2 × 10.5 Å2 and 11.2 × 10.9 Å2, respectively (Fig. S30c and S32c, ESI†). In HCOF-106, the butadiene reacted with two styrene groups present in consecutive layers via 1,4-addition, and one of the styrene reacted with allyl groups (Fig. 3a). A similar crosslinking pattern was also observed for HCOF-107 (Fig. 3b). These interlayer connections forms stable crosslinked frameworks. We suspect that the allyl groups of TAB might act as a radical chain transfer agent during the crosslinking, which enabled the subsequent addition of the second allyl group to the C2 position of the butadiene or isoprene. To confirm the reaction that took place between allyl groups and styrene/diene, we synthesized a control co-crystal using triethyl-benzamide (TEB) and 1. The 1·TEB co-crystal showed a nearly identical hydrogen-bonded network to 1·TAB (Table S2 and Fig. S33, ESI†). When the 1·TEB co-crystal was reacted with isoprene, the obtained crystals were largely dissolved in DMSO-d6 (Fig. S35, ESI†). 1H NMR spectrum of the dissolved sample showed that ∼40% of 1 remained unreacted (Fig. S36, ESI†), confirming that the allyl groups participated in the crosslinking, although the detailed reaction path remains ambiguous (Scheme S5, ESI†).
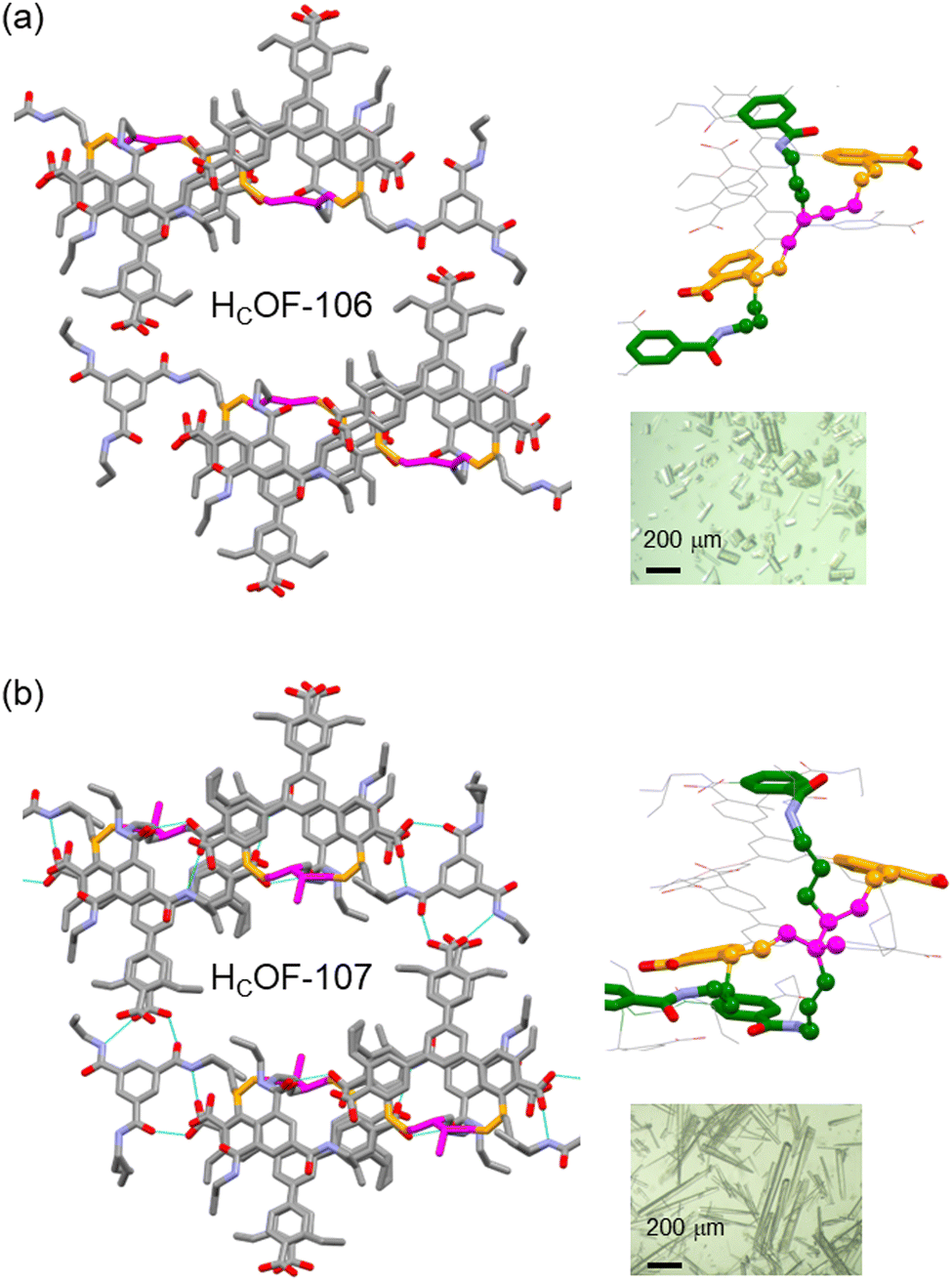 | ||
| Fig. 3 The SCXRD structure of (a) HCOF-106 and (b) HCOF-107 with highlighted crosslinkers (magenta) in the crystal lattices. The crosslinking connections between the layers in HCOF-106 and HCOF-107 are shown on the right. Inset: Optical images of these crystals. | ||
The rich olefin contents at the pore surface of these HCOFs encouraged us to post-synthetically convert the olefins to hydroxyl groups while maintaining the material's high crystallinity. This post-modification will enable us to convert the hydrophobic pore surface to a hydrophilic one. To illustrate this feasibility, we chose HCOF-107 as the model material. As shown in Scheme 1, single crystals of HCOF-107 were immersed in BH3·SMe2 for 24 h to allow extensive hydroboration, and the crystals turned pink after the reaction. 11B NMR spectra of HCOF-107-BH2 showed signals characteristic of boronic acid (Fig. S39, ESI†), suggesting that the –CBH2·SMe2 units are highly reactive and readily hydrolyzed or oxidized. HCOF-108 was obtained by reacting HCOF-107-BH2 with water and H2O2 to ensure full oxidation of any residual boronic esters. The HCOF-108 crystals retained high crystallinity, as shown by PXRD (Fig. 4a), but it is no longer suitable for SCXRD analysis. The 13C CP MAS NMR spectra showed that the carbon signals attributed to the residual alkene groups at 114 ppm decreased significantly (Fig. 2 and Table S5, ESI†), and a new carbon signal attributed to the oxidized –CH2OH group emerged at 64 ppm (Fig. 2). In addition, the carbon signals attributed to the saturated alkyl groups increased. These results showed successful hydroboration–oxidation of the olefin groups, generating HCOF-108 with hydrophilic pores.
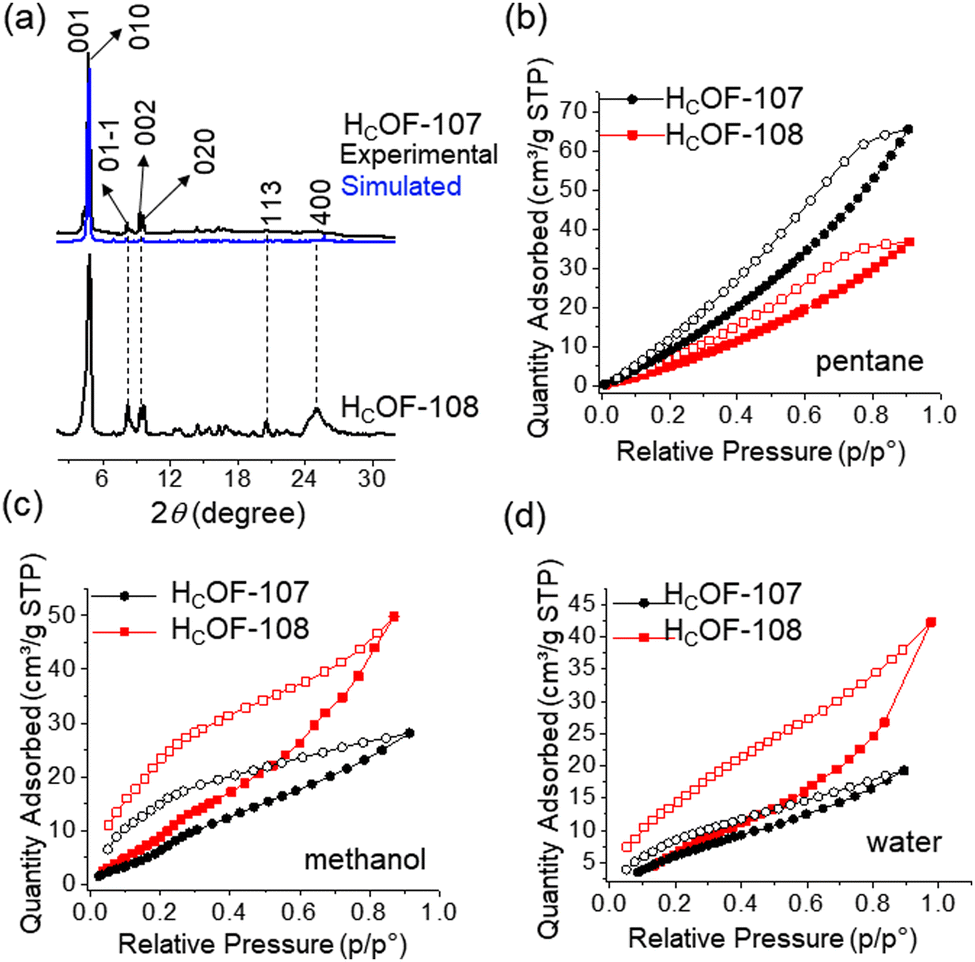 | ||
| Fig. 4 (a) Simulated (HCOF-107) and experimental PXRD patterns of HCOF-107 and HCOF-108. (b) Pentane, (c) methanol, and (d) water vapor sorption isotherms (295 K) for HCOF-107 (black) and HCOF-108 (red). | ||
To confirm the pore characteristic change, solvent vapor sorption analyses were performed for HCOF-107 and HCOF-108 using nonpolar solvent pentane and polar solvents methanol and water. As shown in Fig. 4(b)–(d), the sequence of the solvent vapor uptake values is inversed after the post-modification. HCOF-108 absorbed 50 cm3 g−1 of methanol and 36 cm3 g−1 of pentane, in contrast to HCOF-107, which absorbed 28 cm3 g−1 of methanol and 66 cm3 g−1 of pentane (Fig. S43d, ESI†). The inverted sorption feature highlights the benefit of post-modification.
In conclusion, our work has effectively demonstrated the application of free-radical crosslinking in creating hydrogen-bonded crosslinked organic frameworks, yielding two HCOFs through the use of butadiene and isoprene as crosslinkers and allyl/styrene-based monomers for hydrogen-bonded network formation. The structural analysis revealed that HCOF-106 and HCOF-107 feature crosslinked networks, with the crosslinking reactions occurring among styrene groups of the carboxylic acid-based monomers, the diene crosslinkers, and the allyl groups of the triallyl-benzamide monomers. Furthermore, we successfully converted the olefin-decorated pore surface of HCOF-107 to a hydroxylated HCOF-108 while preserving crystallinity. The opposite vapor sorption behaviors of the hydrophobic HCOF-107 and hydrophilic HCOF-108 emphasize their difference in the pore surface characteristics. The successful synthesis and post-synthetic modification of these HCOFs highlight the versatility and potential of utilizing new crosslinking methods to develop functional porous materials.
This work is supported by the National Science Foundation (NSF) CAREER award (DMR-1844920). Solid-state NMR experiments by A. J. R. were supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences. The Ames Laboratory is operated by Iowa State University for the DOE under contract no. DE-AC02-07CH11358. Funding for the SCXRD was provided by the NSF MRI program (1919565).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed; (b) R.-B. Lin, et al. , Chem. Soc. Rev., 2019, 48, 1362–1389 RSC; (c) Y. Jin, et al. , Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- (a) Z. Chen, et al. , Chemistry, 2022, 8, 693–716 CrossRef CAS; (b) Z. Wang, et al. , Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- S. Lin, et al. , Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Q. Xu, et al. , Angew. Chem., Int. Ed., 2020, 59, 4557–4563 CrossRef CAS PubMed.
- S. Wang, et al. , Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- (a) H. Furukawa, et al. , Science, 2013, 341, 1230444 CrossRef PubMed; (b) B. Li, et al. , Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- (a) S. Kandambeth, et al. , J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed; (b) S. B. Alahakoon, et al. , Chem. Soc. Rev., 2020, 49, 1344–1356 RSC.
- (a) Z. Zhang, et al. , Acc. Chem. Res., 2022, 55, 3752–3766 CrossRef CAS PubMed; (b) R.-B. Lin and B. Chen, Chemistry, 2022, 8, 2114–2135 CrossRef CAS.
- J. Han, et al. , Science, 2024, 383, 1014–1019 CrossRef CAS PubMed.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- J. Samanta, et al. , Acc. Mater. Res., 2022, 3, 1186–1200 CrossRef CAS.
- M. Zhang, et al. , Angew. Chem., Int. Ed., 2022, 61, e202214189 CrossRef CAS PubMed.
- R. Liang, et al. , Angew. Chem., Int. Ed., 2021, 60, 23176–23181 CrossRef CAS PubMed.
- F. Li, et al. , Angew. Chem., Int. Ed., 2023, 62, e202311601 CrossRef CAS PubMed.
- (a) E. Li, et al. , J. Am. Chem. Soc., 2018, 140, 15070–15079 CrossRef CAS PubMed; (b) E. Li, et al. , J. Am. Chem. Soc., 2020, 142, 15560–15568 CrossRef CAS PubMed; (c) E. Li, et al. , Angew. Chem., Int. Ed., 2022, 61, e202211780 CrossRef CAS PubMed.
- Y. Zhang, et al. , J. Am. Chem. Soc., 2024, 146, 15525–15537 CrossRef CAS PubMed.
- H. Wu, et al. , J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1792–1800 CrossRef CAS.
- J. W. McCutcheon, Ind. Eng. Chem., Anal. Ed., 1940, 12, 465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347283, 2347284, 2347291, and 2347292. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02454k |
| This journal is © The Royal Society of Chemistry 2024 |